

Abstract
Mitotic duration is tightly constrained, with extended mitosis characteristic of problematic cells prone to chromosome missegregation and genomic instability. We show that mitotic extension leads to the formation of p53-binding protein 1 (53BP1)–Ubiquitin–specific protease 28 (USP28)–p53 protein complexes that are transmitted to, and stably retained by, daughter cells. Complexes assemble through a Polo-like kinase 1-dependent mechanism during extended mitosis and elicit a p53 response in G1 that prevents proliferation of the progeny of cells that experienced an ~3-fold extended mitosis or successive less extended mitoses. The ability to monitor mitotic extension is lost in p53-mutant cancers and some p53-wildtype cancers, consistent with classification of TP53BP1 and USP28 as tumor suppressors. Cancers retaining the ability to monitor mitotic extension exhibited sensitivity to anti-mitotic agents.
One-Sentence Summary:
Time spent in mitosis is carefully monitored to halt the proliferation of potentially dangerous cells in a population.
Mitosis is a complex event that occurs within a tightly constrained time frame (1). Prolonged mitosis is a sign of problems that can lead to chromosome missegregation, a trigger event for genomic instability (2, 3). Identifying cells that have experienced extended mitosis is therefore an effective means to identify potentially problematic cells in a proliferative population. In human non-transformed immortalized Retinal Pigment Epithelial (RPE1) cells, when a threshold mitotic duration is surpassed, the resulting daughter cells arrest in G1 in a p53-dependent manner. Arrest of daughter cells is independent of the cause of the mitotic extension (4). In addition to p53 and its transcriptional target, the cell cycle kinase inhibitor p21, two additional factors—the scaffold protein 53BP1 and the deubiquitinase USP28—are required for daughter cell arrest after extended mitosis ((5–7), Fig. 1A,B; Fig. S1A,B). When spindle assembly is prolonged after centriole removal in the early mouse embryo or developing nervous system, the observed cell death, embryonic arrest, and microcephaly phenotypes are ameliorated by deletion of TP53 or USP28 genes, indicating that this pathway is operational during development (8–13). However, how mitotic extension is monitored and the physiological significance of mitotic extension-regulated control of cell proliferation remain unknown.
Fig. 1. Control of G1 progression and cell proliferation by memories of mitosis.
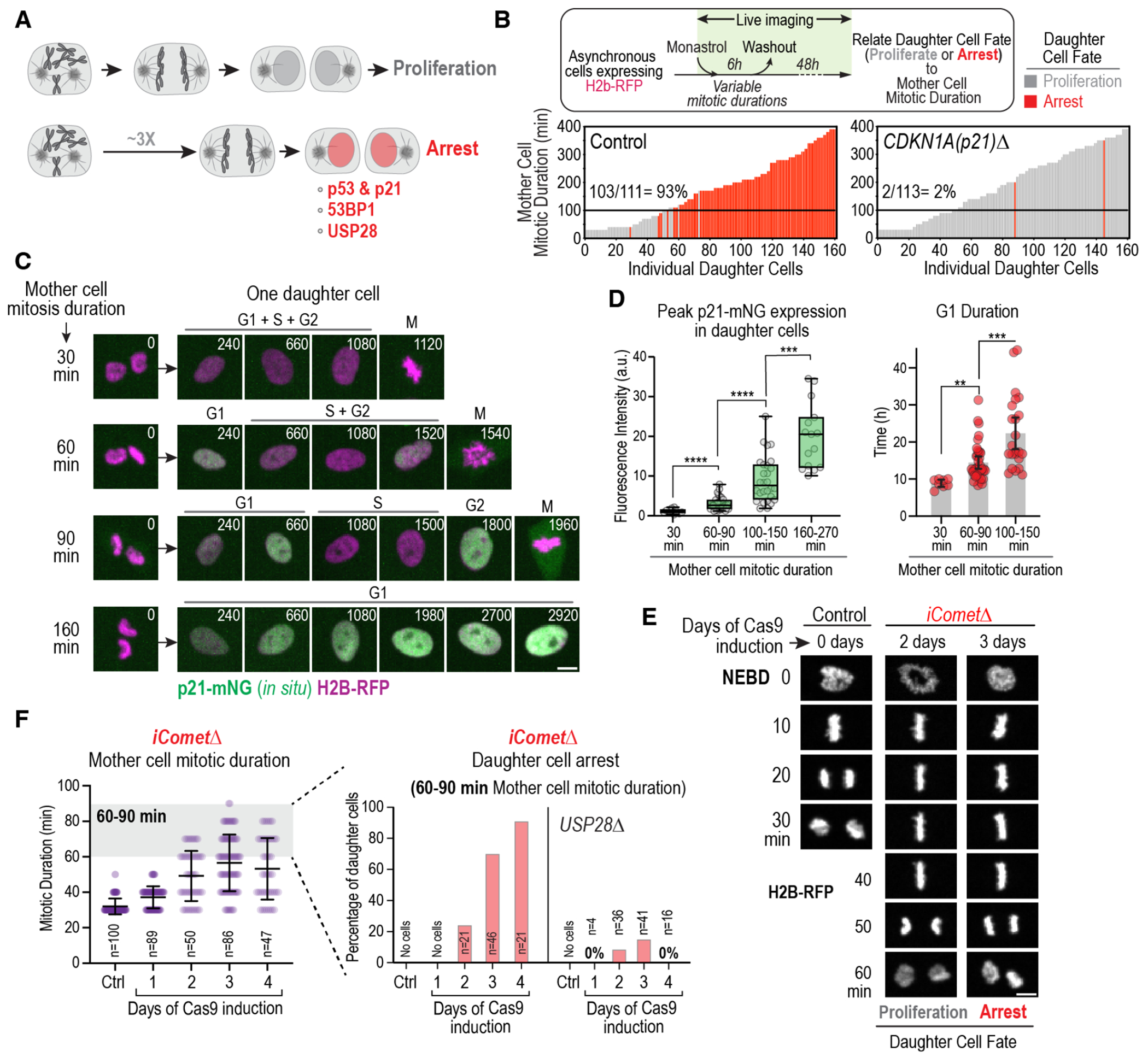
(A) Schematic highlighting that prolonging mitosis beyond a threshold (~3 times longer than normal mitosis) leads to G1 arrest of daughter cells that requires p53, p21, 53BP1 and USP28. (B) Plots of the fate of daughter cells as a function of mother cell mitotic duration for WT and CDKN1AΔ (p21 knockout) RPE1 cell lines. Mother cells with mitotic durations between 30 and 400 minutes were generated by treating an asynchronous cell population with a reversible inhibitor of spindle assembly for 6 hours. Mother cells were imaged in the presence of the inhibitor to measure mitotic duration. After inhibitor washout, mother cells completed mitosis and the resulting daughter cells were imaged for 48h to determine whether they divided again (grey), arrested (red) or underwent apoptosis (black, not observed for RPE1 cells). Each bar represents a single daughter cell, with bar height representing the mitotic duration of its mother, and color representing its fate. Percentage of arrested daughter cells produced by mothers that spent ≥100 minutes in mitosis is noted above the black lines. See also Fig. S1A,B. (C) Stills from timelapse movies monitoring in situ-tagged p21-mNeonGreen and H2B-RFP. Monastrol treatment and washout was used to prolong mitosis to different extents and p21-mNG expression was monitored in daughter cells. Panels show mother cells of different mitotic durations (left) and one of their daughters (right). (D) (left) Box-and-whiskers plot of peak daughter cell p21-mNG expression in G1, as a function of mother cell mitotic duration. (right) G1 duration of daughter cells produced by mother cells of the indicated mitotic durations. Mean and 95% CI are indicated. p-values in (D) are from t-tests (**: p<0.01; ***: p<0.001; ****: p<0.0001). See also Fig. S1C–F. (E) Panels from time-lapse movies of representative control and iCometΔ cells expressing H2B-RFP. NEBD: nuclear envelope breakdown. (F) (left) Mitotic duration at different days after induction of Comet knockout, which extends mitosis by slowing the disassembly of mitotic checkpoint complexes; mean and SD are indicated. (right) Frequency of daughter cells that arrest produced by mothers with 60-90 min mitotic duration on different days following induction of Comet knockout. The Comet knockout was also induced and analyzed in USP28Δ (shown here) and TP53sh (Fig. S2D) backgrounds. See also Fig. S2 and Fig. S3A–G. Scale bars in (C) & (E), 5 μm.
An analog memory of mitotic extension is transmitted to daughter cells
We monitored the response to mitotic extension in RPE1 (Fig. 1B) and two additional non-transformed cell lines (Fig. S1B) by treating asynchronously growing cells with the spindle assembly inhibitor monastrol for 6 hours. Since cells enter mitosis at various times, this treatment generates cells with mitotic durations between ~30 and ~400 minutes. After inhibitor washout, cells complete mitosis, and the resulting daughter cells were followed for 48 hours to determine whether they arrested, underwent apoptosis, or continued to proliferate (Fig. 1B, Fig. S1B). All three tested cell lines were sensitive to extended mitosis that exceeded a threshold of 90 to 110 minutes (Fig. 1B, Fig. S1B). To investigate the observed sharp transition in daughter cell fate as a function of mother cell mitotic duration we in situ-tagged the p53 effector p21, which is necessary for daughter cell G1 arrest (Fig. 1B), with mNeongreen (mNG). In the p21-mNG line, the threshold mother cell mitotic duration for daughter cell arrest was ~50% longer than in the parental line (Fig. S1C and D), possibly due to the fluorescent protein tag. We used monastrol to prolong mitosis and correlated mother cell mitotic duration with p21-mNG expression in daughter cells to determine whether: (1) p21 only accumulated in daughters of mothers whose mitotic duration surpassed the arrest threshold, or (2) increasing mitotic durations were encoded in progressively higher amounts of p21 in G1 that trigger arrest when a threshold is surpassed. After mitoses with a normal duration of ~30 min, p21 expression was low in most cells (Fig. 1C and D). Extending mitotic duration to sub-threshold times between 60 and 150 minutes led to progressive increases in p21 abundance (Fig. 1C and D; Fig. S1C). p21-mNG signal was only observed after transit into G1. For mitotic extensions below the 150-minute threshold, p21-mNG signal was detected in G1 and then abruptly dropped, possibly because of ubiquitin-dependent degradation in early S-phase (14); p21-mNG was then detected again in G2 prior to the next M-phase (Fig. 1C). Above the 150 min threshold, high p21-mNG signal in G1 persisted and no subsequent M-phase was observed (Fig. 1C). Using the drop in p21 signal as a marker for the G1-S transition revealed that G1 duration, like p21-mNG expression, progressively increased for sub-threshold mitotic durations between 30 and 150 minutes (Fig. 1D). A similar increase in G1 duration, but not in S+G2 duration, was observed using a cell cycle phase sensor in a cell line without tagged p21 (Fig. S1E and F). These results suggest that an analog memory of mitotic extension is transmitted to daughter cells, which leads to p21 expression in G1 that reflects the extent to which the mother’s mitosis was prolonged. The fact that the progressive increase in p21 abundance is converted into a sharp threshold in mother cell mitotic time for G1 arrest is consistent with a stoichiometric inhibition of G1 Cdk complexes by p21, exhibiting ultrasensitivity (15, 16). As the mechanism monitoring mitotic duration acts in an analog manner and is proportional to time, we refer to it as the mitotic stopwatch.
Memory of mitotic extension is integrated across cell cycles to control proliferation
The results above indicate that a memory of mitotic duration is transmitted to immediately produced daughter cells. We assessed if a memory of mitotic extension can also be transmitted in a multigenerational manner across successive cell cycles. For this purpose, we developed a means to extend average mitotic duration from ~30 min to ~60 min, well below the ~100 min threshold for immediate daughter cell G1 arrest (Fig. 1E). We achieved this through inducible inactivation of the gene encoding the mitotic checkpoint complex disassembly factor Comet, which accelerates but is not essential for anaphase onset (iCometΔ; Fig. 1E; Fig. S2A and B; (17, 18)). Although it delays anaphase onset, Comet deletion does not perturb spindle assembly or chromosome alignment and segregation (Fig. 1E; Fig. S2C). Despite mother cells transiting mitosis in comparable time intervals and without visible segregation errors, the frequency of daughter cell arrest following sub-threshold mitotic durations of 60-90 minutes increased progressively between days 2 and 4 after induction of Comet deletion, as cells experienced successive sub-threshold extended mitoses (Fig. 1E and F; Fig. S2B). Daughter cell arrest resulting from iCometΔ was greatly reduced in cell lines lacking USP28 or p53 (Fig. 1F; Fig. S2D). Thus, a mitotic stopwatch-dependent memory of sub-threshold mitotic extension is transmitted and integrated across sequential cell cycles to control cell proliferation. An orthogonal perturbation by chemical inhibition of PLK4, which leads to a sub-threshold mitotic extension by delaying spindle assembly (19), coupled with imaging of p21-mNG, provided additional support for this conclusion (Fig. S3A through G).
Mitotic stopwatch complexes transmit memory of mitotic extension
To understand the molecular basis for the transmission of an analog memory of extended mitosis to daughter cells, we conducted biochemical analysis of 53BP1 and USP28 in extracts prepared from mitotically arrested and asynchronously cycling (primarily interphase) cells. Although USP28 was soluble in both, 53BP1 was soluble in mitotic but largely insoluble in interphase extracts (Fig. 2A and B; Fig. S4A and B); low solubility of 53BP1 in interphase is consistent with its chromatin association (20, 21). Immunoprecipitation of 53BP1 from cells held in different cell cycle states revealed co-immunoprecipitation of 53BP1, USP28 and p53 specifically from mitotic cells (Fig. 2C; Fig. S4B through D). Neither an interaction between 53BP1 and USP28 nor increased 53BP1 solubility was detected in cells with DNA damage, despite the increased abundance of p53, indicating that complex formation in mitosis is not a secondary consequence of DNA damage (Fig. 2D; Fig. S4A and B). Cell cycle arrest observed when mitosis is extended by centriole depletion also does not exhibit any of the molecular signatures of the DNA damage-associated p53 response (19). Immunoprecipitation of 53BP1 from mitotically-arrested cells lacking USP28 or p53 revealed that 53BP1 independently interacts with the other two complex components (Fig. 2E), which is consistent with the C-terminal tandem BRCT domain of 53BP1 interfacing with p53 (22, 23) and a point mutation (G1560K) in the Tudor domain of 53BP1 disrupting interaction with USP28 (24). Introducing the G1560K mutation by base editing selectively disrupted the interaction of 53BP1 with USP28 in mitotically arrested cells (Fig. 2F, Fig. S4E), and inhibited stopwatch function to a similar extent as did TP53BP1 knockout (Fig. 2G). Thus, stopwatch complex formation is required to limit the proliferation of daughter cells in response to mitotic extension.
Fig. 2. Mitosis-specific formation of stopwatch complexes transmits memory of extended mitotic duration to daughter cells.
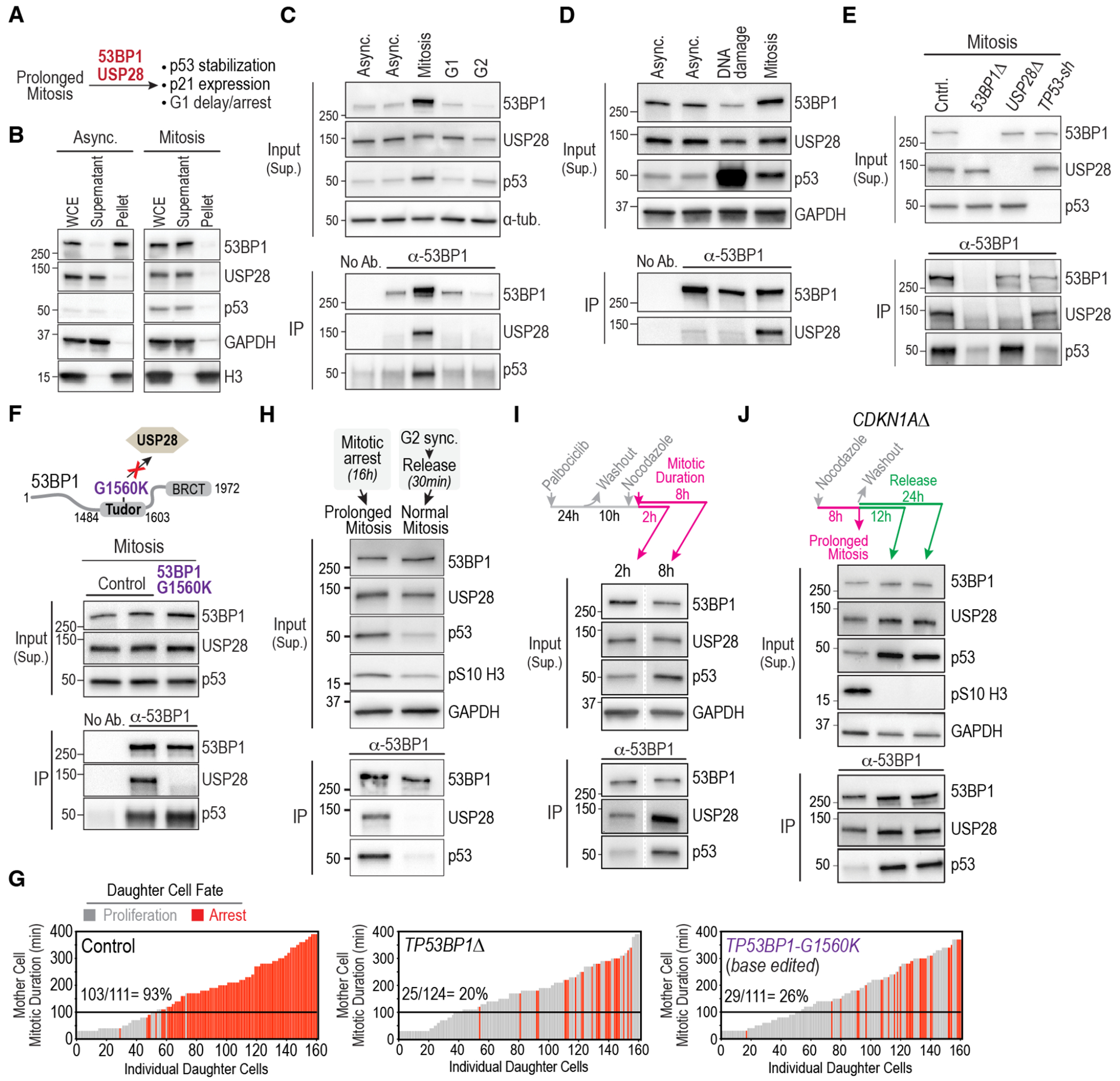
(A) Schematic highlighting the requirement for 53BP1 and USP28 for mitotic stopwatch function. (B) Immunoblots monitoring solubility of 53BP1 and USP28 in asynchronous and mitotic cell extracts. WCE: Whole Cell Extract. GAPDH and histone H3 are soluble and chromatin-bound insoluble controls, respectively. See also Fig. S4A. (C) – (E) Analysis of 53BP1 immunoprecipitates from: (C) Asynchronous cells or cells treated to arrest at indicated cell cycle stages; (D) Asynchronous cells and cells treated to induce DNA damage or prolong mitosis. (E) Cells of the indicated genotypes treated to prolong mitosis. Treatment details are indicated in Fig. S4B. Inputs are soluble supernatants; IP: immunoprecipitate; “No Ab.” is a beads-only control. α-tubulin and GAPDH serve as loading controls. See also Fig. S4C,D. (F) (top) Schematic showing the location of the G1560K point mutation in 53BP1’s Tudor domain that disrupts its interaction with USP28 (24) and was introduced by base editing of the endogenous TP53BP1 locus; see Fig. S4E). (bottom) Analysis of 53BP1 immunoprecipitates from cells treated to prolong mitosis as described in Fig. S4B. (G) Functional analysis of the mitotic stopwatch for the indicated engineered mutant lines. The control graph is the same as in Fig. 1B. (H) Analysis of 53BP1 immunoprecipitates from cells treated to prolong mitosis or following release from synchronization at the G2-M boundary using a CDK1 inhibitor into an unperturbed mitosis. Treatment details outlined in Fig. S5A. Anti-pS10 H3, which monitors mitosis-specific phosphorylation of histone H3 on Ser10, was used as a marker for mitosis and GAPDH served as a loading control. (I) Analysis of 53BP1 immunoprecipitates from synchronized cells held in mitosis for ~2 or ~8h using the protocol schematized on the top. GAPDH serves as a loading control. Lanes shown are from a single exposure of the same immunoblot. See also Fig. S5B. (J) Analysis of the stability of stopwatch complexes following their formation in prolonged mitosis. CDKN1AΔ cells were employed to avoid the G1 arrest observed following release from extended mitosis (Fig. 1B) and were treated as indicated in the schematic above the blot. See also Fig. S5D. 12h and 24h after release from mitosis represents late G1/S and G2 phases of the daughter cells’ cell cycle. GAPDH serves as a loading control. Progression of daughter cells into the next mitosis was confirmed by live imaging (see Fig. S5E).
Although the stopwatch mechanism can detect relatively small mitotic extensions, it is insensitive to normal mitotic duration. Consistent with this observation, formation of complexes containing 53BP1, USP28 and p53 was observed in cells held in mitosis but not in cells released from synchronization at the G2/M boundary into an unperturbed mitosis (Fig. 2H; Fig. S5A). Comparing stopwatch complex formation following release from G1 arrest into the microtubule polymerization inhibitor nocodazole revealed that stopwatch complexes could be detected in cells held for 2 hours in mitosis and were present in increased amounts in cells held in mitosis for 8 hours (Fig. 2I; Fig. S5B). A progressive increase in stopwatch complex abundance was also observed following treatment of asynchronous cells with nocodazole for 4 to 16 hours (Fig. S5C).
The mitotic stopwatch mechanism monitors mildly extended mitoses and integrates these extensions over subsequent cell cycles to trigger proliferation arrest (Fig. 1E and F). This property requires transmission of a molecular memory of extended mitotic duration across cell cycles. Such a memory is unlikely to reside in p21 protein or mRNA, as p21 protein is degraded during S-phase entry (Fig. 1C) and p21 mRNA lifetime is relatively short (25, 26). We thus tested whether, once formed during extended mitosis, stopwatch complexes were stable enough to transmit the memory of extended mitosis across cell cycles. To assess the stability of stopwatch complexes as cells progress through G1, S, and G2, we used cells lacking p21 because they do not arrest in G1 after release from extended mitosis (Fig. 1B). Cells lacking p21 were held in mitosis and then released to allow daughter cells to progress through the cell cycle and eventually enter the subsequent mitosis (Fig. S5D and E). This analysis revealed that amounts of USP28 co-immunoprecipitated with 53BP1 were comparable during extended mitosis and when cells were in G1/S or G2, 12 or 24 hours after release from extended mitosis (Fig. 2J). More p53 was detected in the 53BP1 immunoprecipitates and p53 abundance was higher overall at the post-release timepoints, indicating that 53BP1-USP28 complexes are not stoichiometrically bound to p53 during prolonged mitosis. By comparison, cells held in G1 or G2 without a prior extended mitosis did not contain stopwatch complexes (Fig. 2C). Collectively, these data indicate that mitotic stopwatch complexes are only formed if mitosis is extended beyond its normal duration, increase progressively in abundance during extended mitosis, are inherited by daughter cells, and are sufficiently stable to transmit the memory of extended mitosis.
PLK1 kinase activity is required for mitotic stopwatch complex formation and function
To address how mitotic stopwatch complexes form during extended mitosis, we performed a focused screen with chemical inhibitors targeting protein kinases that act in mitosis or after DNA damage (Fig. 3A); CDK1, MPS1, and Aurora B were not tested because they are essential to maintain an extended mitotic state in response to defective spindle assembly. Kinase inhibitors were added at the same time as the reversible spindle assembly inhibitor monastrol used to extend mitosis (Fig. 3A). This screen identified the mitotic kinase PLK1 as essential for the mitotic stopwatch (Fig. 3A). By contrast, the kinases that act after DNA damage did not contribute to stopwatch function (Fig. 3A; Fig. S6A–C). Inhibition of Aurora A had only a minor effect on stopwatch function (Fig. S7A), and BET bromodomain inhibition, which is a secondary consequence of commonly used PLK1 inhibitors including BI2536 (27), had no effect (Fig. S7B). A chemically distinct and PLK1-specific inhibitor also suppressed stopwatch function (Fig. S7C), and introduction of a mutation conferring partial PLK1 inhibitor resistance restored stopwatch function in the presence of the inhibitor, confirming the specific requirement for PLK1 activity (Fig. S7D). 53BP1 becomes soluble during mitosis (Fig. 2C,H) and was equally soluble in cells held in mitosis with nocodazole, nocodazole & PLK1i, or PLK1i only, indicating that 53BP1 release from chromatin during mitotic entry does not require PLK1 activity. Instead, PLK1 activity appears to be required for the association of USP28 and p53 with 53BP1 to form stopwatch complexes when mitosis is extended (Fig. 3B; Fig. S7E). After release into G1, cells in which PLK1 was inhibited during extended mitosis behaved like cells experiencing normal-length mitosis with 53BP1 becoming insoluble upon exit from mitosis; by contrast, 53BP1 remained soluble if PLK1 was not inhibited during the extended mitosis and stopwatch complexes were formed (Fig. S7F). This analysis also confirmed that p21 accumulation is only observed after the transition to G1 (Fig. S7F), as observed with in situ-tagged p21 (Fig. 1C).
Fig. 3. PLK1 kinase activity is central to the formation of stopwatch complexes that are transmitted to daughter cells.
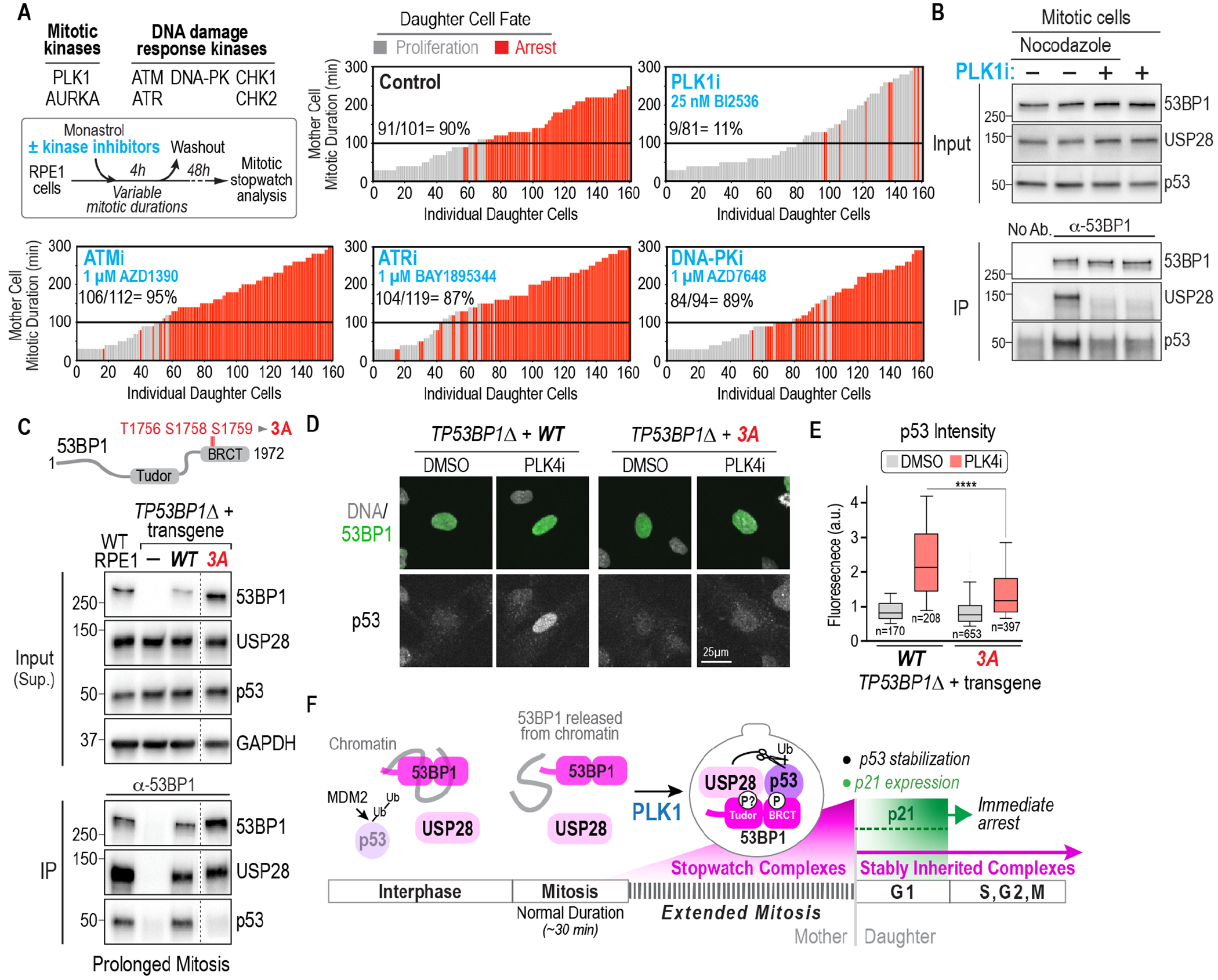
(A) (top left) Experimental approach and list of inhibited mitotic and DNA damage kinases. Plots show results of functional analysis of the mitotic stopwatch for the indicated conditions. See also Fig. S6 and Fig. S7A–D. (B) Analysis of 53BP1 immunoprecipitates from cells treated to prolong mitosis with and without PLK1 inhibition. For treatment details, see Fig. S7E. (C) Analysis of PLK1 activity-dependent mitotic phosphorylation sites in the 53BP1 BRCT region. Schematic above indicates the 3 sites mutated to non-phosphorylatable alanine. Immunoblot below shows analysis of 53BP1 immunoprecipitates from cells treated to prolong mitosis, comparing WT RPE1 and TP53BP1Δ RPE1 cells after introduction of transgenic WT and phospho-site mutant forms of 53BP1. See also Fig. S8A–H. Lanes shown are from a single exposure of the same immunoblot; the full blot is shown in Fig. S8D. (D) Analysis of p53 levels by immunostaining following 4-day treatment with PLK4i, which delays spindle assembly (Fig. S3A). Transgene expression is heterogenous; thus, cells with comparable transgene expression are shown. (E) Quantification of p53 fluorescence signal in nuclei, comparing DMSO and PLK4i treatment, for the indicated conditions. Due to heterogeneity of transgene expression in TP53BP1Δ cells, 53BP1 signal intensity was used to first select cells with comparable expression (Fig. S8H) and p53 signal was then quantified. 10th-90th percentile of measured values, normalized to the average value in DMSO-treated TP53BP1Δ +WT condition, are plotted. p-value is from a t-test (****: p<0.0001). (F) Schematic summary of mitotic extension being encoded by PLK1 activity-dependent formation of stopwatch complexes that are stably inherited by daughter cells where, depending on their abundance, they either trigger immediate proliferation arrest or impart a memory of prolonged mitosis into the subsequent cell cycle.
To determine if PLK1 stimulates complex formation by phosphorylating stopwatch complex subunits, we focused on 53BP1 because it has PLK1 activity-dependent phosphorylation sites and putative PLK1 docking sites, one of which has been validated (28, 29). Because USP28 and p53 interface with the Tudor and BRCT repeat regions of 53BP1, respectively (23, 24, 30), we mutated 23 previously identified PLK1 activity-dependent phosphorylation sites in 53BP1 that span this region (28) along with 12 additional high-scoring candidate PLK1 sites predicted by GPS 5.0 (31) The 35 phosphorylation sites targeted for mutation were grouped into 7 clusters; we additionally mutated the validated PLK1-docking site in the 53BP1 N-terminus (Fig. S8A and B). Transgenes mutating the grouped sites to alanine were introduced into TP53BP1Δ cells and stopwatch complex formation was analyzed after prolonged mitosis (Fig. S8C through E). Disrupting 3 phosphosites in 53BP1 that were sensitive to PLK1 inhibition in vivo and are located in a disordered loop of the BRCT1 repeat that interfaces with p53 (23) reduced interaction with p53 in cells experiencing extended mitosis (Fig. 3C; Fig. S8C through G). Mutation of these sites also compromised stopwatch function, monitored by using PLK4 inhibition to extend mitosis and measuring p53 levels by immunostaining (Fig. 3D and E; Fig. S8H). A second mutant targeting a cluster of sites between the Tudor and BRCT domains, reduced interaction with both USP28 and p53 and compromised stopwatch function (Fig. S8B and D); however, this mutant also impaired nuclear localization of 53BP1 (Fig. S8E). These results indicate that PLK1 activity directly contributes to stopwatch complex formation during prolonged mitosis.
Collectively, the results above indicate that mitotic entry releases 53BP1 from chromatin through a PLK1 activity-independent mechanism. Subsequently, if mitosis is prolonged, the degree of mitotic extension is encoded in the formation of progressively greater numbers of stopwatch complexes (Fig. 3F). Stopwatch complex formation involves the PLK1-regulated interaction of 53BP1’s Tudor and BRCT domains with USP28 and p53, respectively. For the p53 interaction, a set of mitotic PLK1 target sites in 53BP1 appear to have an important role. Incorporation of 53BP1 into stopwatch complexes prevents it from reassociating with chromatin when daughter cells exit mitosis (Fig. 2J; Fig. S7F); instead, soluble stopwatch complexes constitute a stable mark of mitotic extension that is transmitted to daughter cells. In G1, stopwatch complexes stabilize p53, likely via USP28-mediated deubiquitination, leading to synthesis of an amount of p21 that reflects the magnitude of the mitotic extension (Fig. 3F). If p21 concentration is insufficient to halt daughter cells in G1, stopwatch complexes persist, transmitting a memory of extended mitosis that enables the detection of cells that experience sequential moderately prolonged mitoses (Fig. 1E,F; Fig. S2,S3).
Frequent inactivation of the mitotic stopwatch in p53-wildtype cancers
Difficulty in executing events such as spindle assembly or chromosome-spindle attachment extends mitosis via the spindle checkpoint and is associated with elevated rates of chromosome missegregation (32), which in turn lead to aneuploidy and genomic instability. The mitotic stopwatch monitors mitotic duration and halts the proliferation of cells that experience either one highly delayed mitosis or successive moderately delayed mitoses, making it well suited to act as a fidelity filter that suppresses proliferation of potentially dangerous cells in a population. Consistent with this notion, the genes encoding the three stopwatch complex subunits are tumor suppressor genes ((33); Fig. S9). Classification of USP28 as a tumor suppressor gene is particularly notable as, unlike TP53BP1 and TP53, it is not an integral component of the DNA damage response (34). As stopwatch function relies on p53 (4), the stopwatch is predicted to be inactive in the ~50% of human cancers that harbor p53 mutations. This expectation was confirmed in three p53-mutant cancer-derived cell lines (Fig. 4A and B; Fig. S10A and B). A more interesting question, related to the physiological function of the stopwatch, is its status in the ~50% of cancers that express wildtype p53 (35, 36). To determine the status of the stopwatch in p53-wildtype cancers, we surveyed 15 cell lines from pediatric and adult cancers that were annotated as expressing wildtype p53 (Fig. 4A). For all tested cell lines, expression of functional p53 was confirmed by monitoring cell proliferation after treatment with an inhibitor that stabilizes p53 by preventing its ubiquitination by MDM2 (Fig. S10A). Analysis of stopwatch function partitioned the 15 p53-wildtype cell lines into 3 groups: 5 with a functional stopwatch, 2 with a partially compromised stopwatch, and 8 with an inactive stopwatch (Fig. 4A and B; Fig. S10C through E). In two neuroblastoma-derived p53-wildtype cancer cell lines, stopwatch activation led to cell death instead of arrest (Fig. 4A; Fig. S10C), possibly because of higher expression or activity of the cell death machinery. Of the 5 cell lines with a functional stopwatch, 3 appeared to have a higher temporal threshold for stopwatch activation (Fig. 4A and B; Fig. S10C).
Fig. 4. The mitotic stopwatch is compromised in cancers and influences efficacy of anti-mitotic agents.
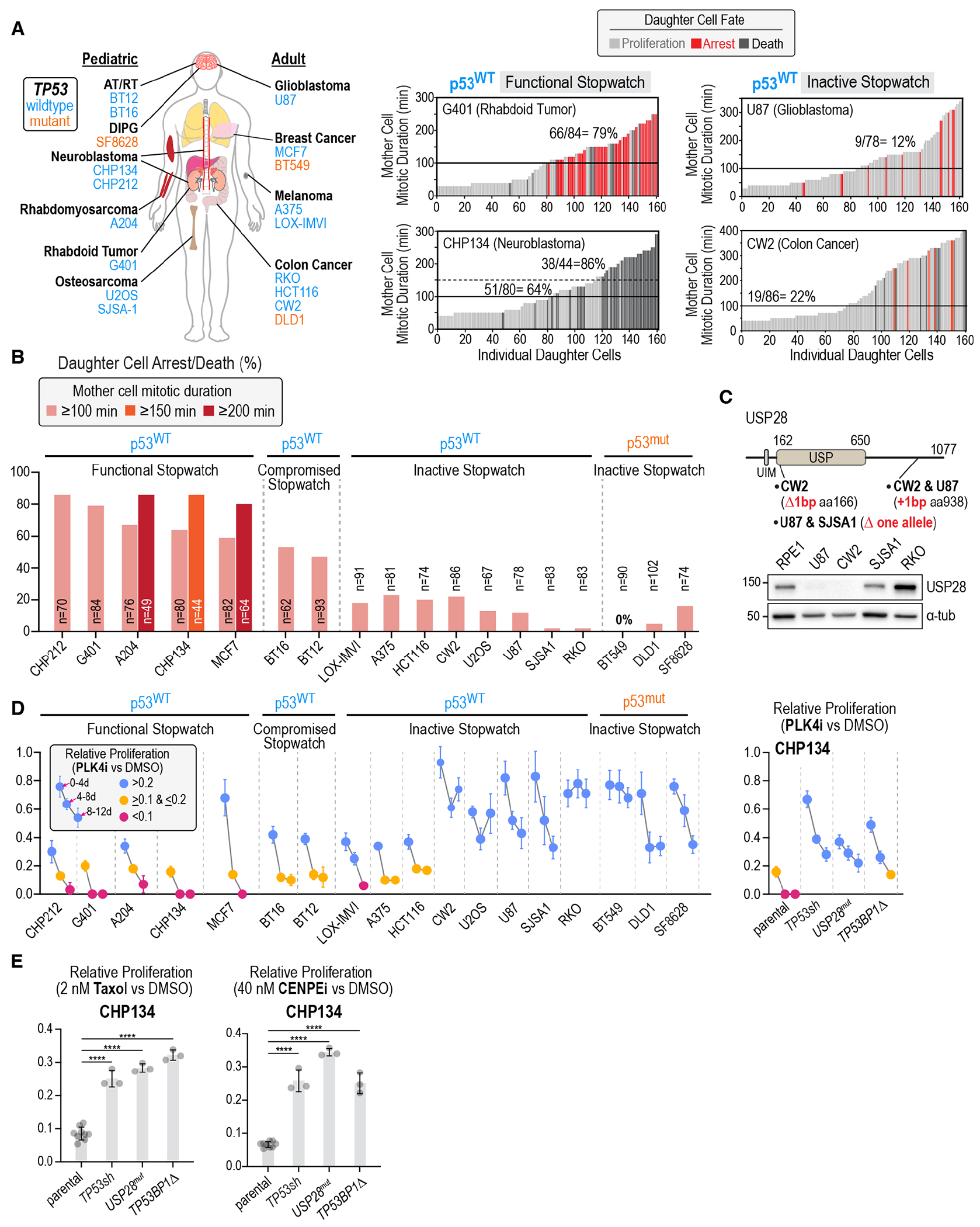
(A) (left) Schematic of diverse tissue-of-origin cancer-derived cell lines annotated as expressing wildtype (blue) or mutant (orange) p53; for experimental confirmation of p53 status see Fig. S10A. (right) Representative plots analyzing mitotic stopwatch function in p53-wildtype cancer-derived cell lines. See also Fig. S10B–E. (B) Plot of daughter cell arrest/death for mother cell mitotic durations above the indicated thresholds in the p53-wildtype and p53-mutant cancer cell lines. (C) (top) USP28 schematic showing mutations identified in p53-wildtype cancer lines that lack stopwatch function. (bottom) USP28 immunoblot for the indicated cell lines; α-tubulin is a loading control. See also Fig. S10F,G and Fig. S11. (D) (left) Mean relative proliferation in PLK4i versus DMSO plotted for successive 4-day intervals in a passaging assay for 15 p53-wildtype and 3 p53-mutant cancer lines. See also Fig. S12. p53-wildtype lines are grouped based on the functionality of their mitotic stopwatch. (right) Mean relative proliferation in PLK4i of CHP134 neuroblastoma cells and derived isogenic clonal lines with mutations/knockdown of stopwatch complex components. Error bars are the SD (n=3). See also Fig. S13A–C. (E) Mean relative proliferation of parental CHP134 cells and derived lines knocked down or mutated for stopwatch components in 2 nM Taxol (left) or 40 nM CENPEi (right); n=9 for controls and 3 for other conditions. Error bars are the SD. p-values are from pair-wise t-tests comparing derived cell lines to the parental line (****: p<0.0001). See also Fig. S13D.
Consistent with bioinformatic analysis indicating that USP28 and TP53BP1 exhibit a high frequency of deleterious mutations in cancer ((33); Fig. S9), mutations in USP28 or TP53BP1 or both were present in 4 of the 8 cell lines lacking stopwatch function (Fig. 4C; Fig. S10F and G). Creating mutations that resembled cancer-associated mutations in the nontransformed RPE1 cell line revealed that introducing frameshift mutations in both alleles of either USP28 or TP53BP1 eliminated protein expression and abrogated stopwatch function (Fig. S11A through E), whereas heterozygous frameshift mutations reduced protein expression by ~50% and compromised stopwatch function (Fig. S11A through E). 3 other p53-wildtype cancer cell lines lacking stopwatch function had genetic alterations that dampen p53 signaling (Fig. S11F through H). In particular, mutations that truncate and hyperactivate the p53-antagonizing phosphatase WIP1 (37) were associated with loss of stopwatch function (Fig. 4B; Fig. S11F), and treatment with a WIP1 inhibitor (38) partially restored stopwatch activity (Fig. S11G). Collectively these results indicate that both p53-mutant and a significant proportion of p53-wildtype human cancers have compromised mitotic stopwatch function, which may enable them to tolerate problematic mitoses that are both a cause and a consequence of genomic instability.
Mitotic stopwatch status predicts sensitivity to anti-mitotic agents
As the stopwatch monitors mitotic duration to control proliferation, its functional status has the potential to influence the efficacy of agents that prolong mitosis and are used or in development as cancer therapeutics. To prolong mitosis to a moderate extent, we used the PLK4 inhibitor centrinone (19) which delays spindle assembly by causing loss of centrosomes (Fig. S12A–D). Monitoring the proliferation of the 3 p53-mutant and 15 p53-wildtype cancer cell lines revealed that p53-mutant lines continued to proliferate in PLK4i, albeit at reduced rates due to mitotic challenges caused by centrosome loss (Fig. 4D; Fig. S12E (19)). For the p53-wildtype cancer lines, proliferation in PLK4i was broadly inversely correlated with stopwatch status. p53-wildtype lines with a functional mitotic stopwatch exhibited rapid and penetrant cessation of proliferation in PLK4i (Fig. 4D; Fig. S12E); by contrast, those with a compromised or absent stopwatch exhibited improved proliferation in PLK4i (Fig. 4D). To extend this correlational analysis to an isogenic context, CHP134 neuroblastoma cells that have an intact stopwatch and are highly sensitive to PLK4i were engineered to delete or mutate the 3 stopwatch complex subunits. Inactivation of stopwatch components improved proliferation in PLK4i, relative to that of parental CHP134 cells (Fig. 4D; Fig. S13A–C), indicating that a functional stopwatch enhances the response to PLK4i treatment. Although PLK4 inhibitors have the potential to target specific cancer types (39, 40), they have not been used therapeutically. Thus, we assessed if stopwatch status impacted sensitivity to the widely used mitosis-targeting chemotherapy agent taxol and to a clinically-tested inhibitor of the mitotic kinesin CENPE (41–45). Low-dose taxol and CENPEi treatments extend mitosis by mildly perturbing microtubule dynamics and chromosome congression, respectively. In both CHP134 neuroblastoma cells and in a nontransformed model cell line, mutation of stopwatch complex subunits improved proliferation relative to the parental lines after low-dose taxol or CENPEi treatment (Fig. 4E, Fig. S13D). Thus, stopwatch status may influence the efficacy of therapeutic agents currently in use or being developed to target mitotic processes and could serve as a potential biomarker for their use in cancer treatment.
Conclusion
Defects in chromosome segregation during mitosis, which not only generate cells with incorrect numbers of chromosomes but also precipitate drastic chromosome rearrangements, are intermediates during the generation of nearly all cancers (46, 47). Given the hundreds of billions of cells that divide in an adult human every day, monitoring these mitoses to filter out potentially problematic cells poses a significant challenge. This work shows that the degree of mitotic extension is encoded via the PLK1 kinase-dependent assembly of mitotic stopwatch complexes that are transmitted to daughter cells. The ability of this mechanism to detect subtle repeated extensions of mitosis functions as a fidelity filter in a proliferative cell population and may explain its frequent inactivation in cancers, as well as the classification of stopwatch complex components as tumor suppressors. Compromised stopwatch function is likely important for the tolerance of problematic mitoses that are contributors to and a consequence of the aneuploidy and genomic instability that is characteristic of cancers. (48–66).
Materials and Methods
Chemical inhibitors
The chemical inhibitors and concentrations used in this study were: centrinone (PLK4i; LCR-263; 150 nM; synthesized by Sundia MediTech); monastrol (100 μM; Tocris Bioscience); MDM2 inhibitor (Mdm2i; 1 μM; R7112; Proactive Molecular Research); BI2536 (PLK1i; 100 nM; MedChem Express); BI6727 (PLK1i; 25 nM; SelleckChem); GSK461364 (PLK1i; 25 nM; SelleckChem); MK-8745 (AuroraAi; 10 μM; SelleckChem); JQ1 (BET/Bromodomain inhibitor; 10 μM; MedChem Express); CHIR124 (CHKi; 100 nM; Axon); CCT24533 (CHK2i; 1 μM; MedChem Express); CHK2 inhibitor II (CHK2i; 1 μM; Sigma-Aldrich); doxorubicin (Sigma-Aldrich); aphidicolin (1 μM; Cayman); BAY1895344 (ATRi; 1 μM; SelleckChem); AZD7648 (DNA-PKi; 1 μM; SelleckChem); AZD1390 (ATMi; 1 μM; Chemgood LLC); Palbociclib (CDK4/6i; 0.5 μM; SelleckChem); RO-3306 (CDK1i; 10 μM; Calbiochem); GSK2830371 (WIP1i; 10μM; Seleckchem); paclitaxel (Taxol; 2 nM; Sigma-Aldrich); nocodazole (0.1-0.2 μg/ml, Sigma-Aldrich); GSK923295 (CENPEi; 40 nM; SelleckChem).
Antibodies
The Cep192 antibody (raised against aa 1-211; used at 0.5 μg/ml) was previously described (19). The following antibodies were purchased from commercial sources, with their working concentrations indicated in parentheses: anti-53BP1 (1:5000; Novus Cat# NB100-304, RRID:AB_10003037), USP28 (1:1000; Abcam Cat# ab126604, RRID:AB_11127442), p53 (1:100; Millipore Cat# OP43F-100UG, RRID:AB_10683244), p53 (1:1000; Santa Cruz Biotechnology Cat# sc-126, RRID:AB_628082), p21 (1:1000; Cell Signaling Technology Cat# 2947 (also 2947S, 2947P), RRID:AB_823586), anti-α-tubulin (1:5000; DM1A; Sigma-Aldrich), anti-GAPDH (1:1000; Cell Signaling Technology Cat# 5174 (also 5174P, 5174S, 5174T), RRID:AB_10622025), anti-Histone H3.3 (1:1000; Millipore Cat# 09-838, RRID:AB_10845793); anti-γH2AX (Ser139; 1:2000; Millipore Cat# 05-636, RRID:AB_309864); IgG Rabbit (1:5000; Vector Laboratories Cat# I-1000-5). Secondary antibodies were purchased from Jackson ImmunoResearch and GE Healthcare.
Cell lines
All cell lines used in this study are described in Table S1. RPE1 (hTERT RPE-1), U2OS, HCT116, CHP212, G401, SJSA1, RKO, U87, MCF7, BT549, A204 and A375 were obtained from the American Type Culture Collection (ATCC); CHP134 and DLD1 were obtained from Sigma-Aldrich (ECACC general collection) and CW2 line from Cell Bank Riken. LOX-IMVI was obtained from the NCI-60 collection. BT12, BT16 and SF8628 were a gift from Nalin Gupta (UCSF). H1 ESCs were a gift from Karl Willert (UCSD). Cell lines were cultured as recommended at 37°C and 5% CO2, supplementing the growth media with 100 IU/ml penicillin and 100 μg/ml streptomycin. H1 cells were maintained in E8 media (48) on Matrigel precoated plates.
The RPE1 USP28Δ, TP53BP1Δ, TP53sh cell lines have been described (5, 39). Knock-out and frameshift mutations were generated using transient transfection (RPE1 USP28Δ; RPE1 TP53BP1Δ; RPE1 USP28fs/fs; RPE1 TP53BP1fs/fs; RPE1 CDKN1AΔ; CHP134 USP28mut; CHP134 TP53BP1Δ) or nucleofection of preassembled RNP complexes (RPE1 USP2wts/fs; RPE1 TP53BP1wt/fs). Knockout and frameshift mutations were introduced using gene-specific sequences that target Cas9 to USP28 (knockout targeting exon 4, TGAGCGTTTAGTTTCTGCAG; frameshift in exon 23, TGCTCTGGTAGGCATATACC), TP53BP1 (knockout targeting exon 3, CTGCTCAATGACCTGACTGA; frameshift in exon 19, GTTTCCCCTTCACAGACTGG) or CDKN1A (knockout targeting exon 2, ATGTCCGTCAGAACCCATG). For transient transfections, double-stranded oligonucleotides for specific guide RNAs were cloned into PX459 (a gift from Feng Zhang; Addgene plasmid # 48139; http://n2t.net/addgene:48139; RRID:Addgene_48139) (49). RPE1 cells were plated in 10 cm plates at 500,000 cells/plate the day before transfection. Cells were transfected with plasmid using Lipofectamine 3000 according to the manufacturer’s instructions (ThermoFisher). For nucleofection of RNP complexes 4 μl crRNA (100 μM) and 4 μl tracrRNA (100 μM) were incubated at 95°C for 5 min, cooled at room temperature for 15 min, incubated with 10 μl Cas9 (40 μM) for 20 min at room temperature, and nucleofected according to the manufacturer’s instructions (Lonza). For lentiviral infection, gRNAs were cloned into LentiCRISPR v2 (gift from Feng Zhang; Addgene plasmid # 52961; http://n2t.net/addgene:52961; RRID:Addgene_52961) (50). Two days after transfection or nucleofection, 150 nM centrinone was added to select for mutants; control RPE1 cells and CHP134 cells arrest and die, respectively, after 3-4 divisions in centrinone. After 10 days, centrinone-resistant RPE1 or CHP134 cells were plated at low density in 96 well plates (30-40 cells per plate) for single clone selection. For selecting CHP134 TP53BP1Δ and USP28mut mutants, centrinone-resistant CHP134 cells were plated at a density that supported direct picking of clones from 10 cm plates. Gene knock-out was assessed by genotyping of the sequence surrounding the CRISPR cut site (51) and/or by immunoblotting.
RPE1 p21-mNeonGreen cells were generated using CRISPR/Cas9 in combination with rAAV mediated delivery of the repair construct as previously described (52). The gRNA, designed to cut close to the stop codon of CDKN1A(p21) (TTTGAGGCCCTCGCGCTTCC), was cloned into PX459 ((49); gift from Feng Zhang; Addgene plasmid # 48139; http://n2t.net/addgene:48139; RRID:Addgene_48139). The repair construct, cloned into pSEPT, contained NeonGreen for C-terminal fusion to p21 linked through a P2A sequence to the neomycin resistance gene Tn5 aminoglycoside phosphotransferase; this cassette was flanked on either side by homology arms corresponding to the regions up and down stream of the gRNA target site. The left homology arm contained 926 bp upstream of the stop codon and the right homology arm contained 684 bp downstream of the gRNA including 6bp overlapping with the gRNA sequence.
The following transgenes were stably integrated into the genome using lentiviral constructs (see Table S2): H2B-mRFP (EF1alpha promoter); TRE3pro-Cas9 (Edit-R Inducible Lentiviral Cas9; Dharmacon); 3FLAG-APOBEC-1-Cas9(D10A)-UGi (Base Editor; (53)). Cell lines for inducible knock-out of Comet or induction of a double strand break at the control AAVS1 locus were generated as previously described (39). In brief, cell lines were generated by sequential lentiviral integration of inducible Cas9 and a Comet sgRNA expressing plasmid based on the lentiGuide-Puro plasmid ((50); gift from Feng Zhang; Addgene plasmid # 52963; http://n2t.net/addgene:52963; RRID: Addgene_52963). The Comet gRNA (AAGTGCTTAAGCTGTTCATA) targets exon 3 and the AAVS1 gRNA (GGGCCACTAGGGACAGGAT) targets the intron between exon 1 and 2 of PPP1R12C. Cas9 expression was induced with 1 μg/ml doxycycline.
For base editing, Cas9 from the lentiCas9-Blast plasmid ((50); gift from Feng Zhang; Addgene plasmid # 52962 ; http://n2t.net/addgene:52962 ; RRID:Addgene_52962) was replaced with 3FLAG-APOBEC-1-Cas9(D10A)-UGI from the pLenti-FNLS-P2A-Puro plasmid ((53); gift from Lukas Dow; Addgene plasmid # 110841; http://n2t.net/addgene:110841; RRID:Addgene_110841). After lentiviral integration of 3FLAG-APOBEC-1-Cas9(D10A)-UGI, a specific gRNA (TATGTCCTTTCACCACTCCT) designed to facilitate mutation of 53BP1 Gly1560 to Lys was integrated using the lentiGuide-Puro plasmid (50)).
Viral particles were generated by transfecting the lentiviral construct into HEK-293T cells using Lenti-X Packaging Single Shots (Clontech). 48 hours after transfection, virus-containing culture supernatant was harvested and added to the growth medium of cells in combination with 2.5-8 μg/ml polybrene (EMD Millipore). Populations of each cell line were selected by FACS or antibiotics (Blasticidin, 5 μg/ml; Neomycin, 400 μg/ml; Puromycin, 10 μg/ml for RPE1, 0.5 μg/ml for CHP134). Single clones were isolated in 96-well plates.
The RPE1 cell line expressing an S-phase marker was generated using the lentiviral CSII-EF mVenus-hGeminin(1-110) construct (54). Virus production and cell transfection was done as described above. mVenus-hGeminin (1-110) expressing cells were sorted by FACS. The BI6727/BI2536-resistant mutation R136G in PLK1 was generated by chemical transfection of a specific gRNA (TGTTGGAGCTCTGCCGCCGG) expressing plasmid (pX459) and a repair oligonucleotide (GCGCTCCAACTGCCCCGCAGGCAGTTCCAGTTCCCCAGCAGCGACACTCACCCCGCGGCGGCAGAGCTCCAACACCACGAACACGAAGTCGTTGTCCTCG). The gRNA targets a region close to the mutation site. pSpCas9(BB)-2A-Puro (PX459) V2.0 was a gift from Feng Zhang ((49); Addgene plasmid # 62988; http://n2t.net/addgene:62988; RRID:Addgene_62988). Positive clones were selected with 25 nM BI6727 two days after the chemical transfection.
Phosphosite selection and transgene expression
23 PLK1 activity-dependent phosphorylation sites in the vicinity of the 53BP1 Tudor and BRCT regions were selected based on phosphoproteomic data (Kettenbach et al.; (28)). 12 additional high-scoring candidate PLK1 sites predicted by GPS 5.0 (31) were added to this list and the 35 sites were grouped into 7 regional clusters (Fig. S8A). In total, we tested nine 53BP1 transgenes (WT control, 7 transgenes with regional clusters of PLK1 activity-dependent or high-confidence prediction sites mutated, and 1 transgene with a previously identified PLK1 docking site in the 53BP1 N-terminus mutated (29)). Cluster mutants were synthesized (gBlocks, IDT) and cloned into a lentiviral vector along with wildtype 53BP1. Gene expression was under the control of a UbC promoter. TP53BP1Δ RPE1 cells were infected with lentiviral constructs and stable transgene expressing cells were selected with 400 μg/ml neomycin for 7 days.
Immunofluorescence
For immunofluorescence, ~10,000 cells per well were seeded into 96-well plates one day before fixation. Cells were fixed in 100 μl ice-cold methanol for 7 minutes at −20°C. Cells were washed twice with washing buffer (PBS containing 0.1% Triton X-100) and blocked with blocking buffer (PBS containing 2% BSA, 0.1% Triton X-100 and 0.1% sodium azide) overnight. After blocking, cells were incubated for 1-2 hours with primary antibody in fresh blocking buffer (concentrations as indicated above). Cells were washed three times with washing buffer, prior to 1-hour incubation with the secondary antibody and DNA staining with Hoechst 33342 dye. Finally, cells were washed three times with washing buffer.
Centrosome depletion:
The efficacy of centrinone (150nM) was monitored after 8 days treatment with the CEP192 antibody. Images were acquired on a CV7000 spinning disk confocal system (Yokogawa Electric Corporation) equipped with a 40X (0.95 NA) and a 2560x2160 pixel sCMOS camera (Andor). Image acquisition was performed using CV7000 software.
53BP1 transgene activity:
To test the activity of wildtype and mutant versions of 53BP1, transgene-expressing TP53BP1Δ RPE1 cells were treated for 4 days with centrinone (150 nM) or DMSO. On the third day ~7,000 cells were plated into a 96 well imaging plate while maintaining centrinone or DMSO treatment. Cells were fixed on day 4 and stained with Hoechst DNA dye and antibodies for 53BP1 and p53. Images were acquired on a CQ1 spinning disk confocal system (Yokogawa Electric Corporation) equipped with a 40X (0.95 NA) objective and a 2000x2000 pixel sCMOS camera (ORCA-Flash4.0V3, Hamamatsu Photonics). Image acquisition was performed using CellPathfinder software from Yokogawa. Signal intensities were quantified using Yokogawa Pathfinder software. In images of fixed cells, nuclei were detected using Hoechst DNA dye signal and cells selected for measurement based on the background-subtracted nuclear 53BP1 signal as follows: i) a cutoff was imposed so only cells with nuclear 53BP1 signal >5-fold the nuclear signal in 53BP1Δ cells were considered; ii) the measured nuclear integrated fluorescence intensity values for 53BP1 were normalized relative to the average of the DMSO-treated TP53BP1Δ+WT condition. iii) cells with 53BP1 signal ≥0.65 and ≤2.2 following normalization were selected for all conditions, thereby ensuring that cells with comparable 53BP1 expression are being assessed; iv) background-subtracted p53 nuclear fluorescence was measured in the cells selected in step iii).
Live cell imaging
Live cell imaging was performed on the CQ1 spinning disk confocal system (Yokogawa Electric Corporation) equipped with a 40X 0.95 NA U-PlanApo objective and a 2560x2160 pixel sCMOS camera (Andor) or 2000x2000 pixel sCMOS camera (ORCA-Flash4.0V3, Hamamatsu Photonics) at 37°C and 5% CO2. Image acquisition and data analysis were performed using CQ1 software and ImageJ, respectively.
Mitotic stopwatch assay:
All imaged cell lines were engineered to express H2B-RFP (see Table S1). One day before imaging, cells were seeded into 96-well cycloolefin plates at 2,000-4,000 cells/well. On the day of the experiment, asynchronous cells were first imaged for 4-6h in 100 μM monastrol. Images of H2B-RFP were acquired with 5 x 2 μm z-sections in the RFP channel (25% power, 150 ms) at 10-minute intervals. During monastrol treatment, cells enter at different times into mitosis and delay in prometaphase. This generates a set of mother cells that experience different mitotic durations prior to monastrol washout. After monastrol washout, the mother cells completion of mitosis and the fate of the resulting daughter cells was imaged at 10-minute intervals; daughter were tracked for 48h-72h. The fate of the daughter cells was classified into ‘arrest’, ‘death’ or ‘proliferate’. Inhibitors were added during prolonged mitosis and washed out together with monastrol, unless noted otherwise.
Measurement of mitotic duration:
Mitotic duration was measured in cell lines expressing H2B-RFP (see Table S1). To induce prolonged mitosis cells were either treated with doxycycline to induce Comet knockout (see generation of inducible knockout mutants above) or centrinone to deplete centrosomes. Doxycycline and centrinone were administered 1-4 days before imaging as indicated for each experiment. To determine the consequences of centrosome depletion on the length of mitosis in different cancer cell lines, cells were treated for 3 cell cycles with 150 nM centrinone to deplete centrosomes or with DMSO as a control. Mitotic duration was quantified as the time from nuclear envelope breakdown (NEBD) until chromosome decondensation. Prior to this analysis, the cell cycle duration for all analyzed cancer cell lines was measured by H2B-RFP live imaging of each cell line and quantifying time from NEBD of a mother cell to NEBD of its daughters. For all experiments, cells were seeded one day before imaging into 96-well cycloolefin plates at 5,000 - 10,000 cells/well. Images of H2B-RFP were acquired with 5 x 2 μm z-sections in the RFP channel (25% power, 150 ms) at 4-5-minute intervals.
Monitoring p21 expression:
To measure p53 activation after prolonged mitosis, the coding sequence for mNeonGreen was fused to the p53 target and effector p21 (encoded by CDKN1A) at both endogenous CDKN1A loci in RPE1 cells. Mitosis was prolonged using temporal monastrol or centrinone treatment as described above. H2B-RFP was used to track cells and mark the nuclear area throughout the experiment. Cells were seeded one day before imaging into 96-well cycloolefin plates at 4,000 cells/well. Images of p21-mNeonGreen (GFP channel, 35% power, 150 ms) and H2B-RFP (RFP channel, 25% power, 150 ms) were acquired with 5 x 2 μm z-sections at 10-minute intervals during monastrol treatment and at 20-minute intervals after monastrol washout or during centrinone treatment. Expression of p21 was quantified in the nucleus at each timepoint using Fiji software (55). The normalized signal intensity was determined by drawing a rectangle around the nucleus (w x h) and quantifying the average p21-mNG signal. The average background was determined by drawing a rectangle 2 pixels larger in width and height and quantifying the average p21-mNG signal in the intervening region. The total intensity was the average intensity in the smaller rectangle minus the background intensity multiplied by the area of the smaller rectangle.
To measure G1 duration after prolonged mitosis, cells were engineered to express the cell cycle marker mVenus-hGeminin and the nuclear marker H2B-RFP. Mitosis was prolonged using monastrol treatment as described above. H2B-RFP was used to track cells and mark the nuclear area throughout the experiment. Cells were seeded one day before imaging into 96-well cycloolefin plates at 4,000 cells/well. Images of mVenus-hGeminin (GFP channel, 35% power, 150 ms) and H2B-RFP (RFP channel, 25% power, 150 ms) were acquired with 5 x 2 μm z-sections at 10-minute intervals during monastrol treatment and at 20-minute intervals after monastrol washout. G1 duration was determined as the time between chromosome decondensation and the start of mVenus-hGeminin expression (S-phase).
Proliferation and viability assays
For proliferation analysis, cells were seeded into 6 well plates in triplicate at 25,000-150,000 cells/well and treated with the indicated inhibitors or DMSO as a control. At 72-hour or 96-hour intervals, cells were harvested, counted and, for passaging assays, re-plated at 25,000-150,000 cells/well. Cell counting was performed using a TC20 automated cell counter (Bio-Rad).
DNA damage analysis
ATM and ATR inhibitors prevent the formation of γH2AX foci at DNA damage sites, whereas DNA-PK inhibition at low concentrations of doxorubicin leads to an accumulation of γH2AX foci (56–58). To assess inhibitor efficacy, 4-8,000 cells per well were seeded into 96-well plates one day prior treatment with either doxorubicin or aphidicolin, at the indicated concentrations, along with ATM and ATR inhibitors for 2 hours (for ATMi & ATRi analysis), or with DNA-PKi for 24 hours. Cells were prepared for immunostaining as described above and stained with a γH2AX-antibody to visualize DNA damage sites and Hoechst 33342 for DNA staining. Images were acquired on a CV7000 spinning disk confocal system (Yokogawa Electric Corporation) equipped with a 40x, 0.95 NA U-PlanApo objective and a 2,560 x 2160-pixel sCMOS camera (Andor Technology). Quantification was performed using CV7000 analysis software to count the number of γH2AX foci within the nucleus for each cell. Each condition was analyzed in triplicate.
Immunoblotting
For immunoblotting, cells were cultured in 10 cm plates, harvested at 50-80% confluence and lysed by sonication in RIPA buffer (Cell Signaling Technology; 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin) plus protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Cell extracts were stored at −80°C until use. Extract concentrations were normalized based on protein concentration (Bio-Rad Protein Assay) and 20-30 μg protein was loaded per lane on Mini-PROTEAN gels (Bio-Rad) and transferred to PVDF membranes using a TransBlot Turbo system (Bio-Rad). Blocking and antibody incubations were performed in TBS-T + 5% non-fat dry milk. Detection was performed using HRP-conjugated secondary antibodies (GE Healthcare) with SuperSignal West Femto (Thermo Fisher Scientific) substrates. Membranes were imaged on a ChemiDoc MP system (Bio-Rad).
Immunoprecipitation and cell lysate fractionation
For analysis of cells at particular cell cycle stages, cells were arrested in G1 by treatment with 0.5 μM Palbociclib, in G2 by treatment with 10 μM RO-3306, and in Mitosis by treatment with 100-200 ng/ml (0.33 or 0.66 μM) Nocodazole or 100 nM BI2536 for 14-20 hours. DNA damage was induced using 1μM doxorubicin treatment for 16 hours.
For immunoprecipitation assays, ~5-20 million cells were harvested and washed with PBS. Cells were resuspended in lysis buffer (20 mM Tris/HCl pH 7.5, 50-200 mM NaCl, 0.5% Triton X-100, 5 mM EGTA, 1 mM dithiothreitol, 2 mM MgCl2 and EDTA-free protease inhibitor cocktail (Roche)) and lysed in an ice-cold sonicating water bath for 5 minutes. All immunoprecipitation assays were performed using 50 mM NaCl unless otherwise noted. After 15-minute centrifugation at 15,000 x g and 4 °C, soluble lysates were collected, and protein concentrations were quantified. Equal amounts of lysates were incubated with anti-53BP1 for 2 hours at 4°C and subsequently with Protein A magnetic beads (Thermo Fisher Scientific) for 1 hour at 4 °C. The beads were washed five times with lysis buffer and resuspended in SDS sample buffer. For immunoblotting, equal volumes of samples were run on Mini-PROTEAN gels (Bio-Rad) and transferred to PVDF membranes using a TransBlot Turbo system (Bio-Rad). Blocking and antibody incubations were performed in TBS-T plus 5% nonfat dry milk. Immunoblotting was performed as described above.
For immunoprecipitation assays following DNA damage, cells were treated for 16 hours with 1 μM doxorubicin. The lysis buffer contained 200 mM NaCl to increase the solubility of 53BP1. As control for 53BP1 and USP28 interaction, mitotic cells were harvested as described above and treated with the same lysis buffer containing 200 mM NaCl. Immunoprecipitation assays were performed as described above.
For immunoprecipitation assays in G1 phase following prolonged mitosis, cells were first arrested in mitosis with 200 ng/ml Nocodazole with and without 100 nM of the PLK1 inhibitor BI2536 for 14h. To allow cells to exit mitosis and enter G1 phase, mitotic cells were directly washed of the plate, washed three times with PBS and replated onto 15 cm dishes. Plated cells were allowed to exit mitosis and enter G1 phase. After 6 hours G1 phase cells were harvested, and immunoprecipitation assays were performed as described above.
For immunoprecipitation assays in G1/S and G2 phase following prolonged mitosis, RPE1 CDKN1AΔ cells were first arrested in mitosis with 100 ng/ml Nocodazole for 8h. To allow cells to exit mitosis and enter G1 phase, mitotic cells were washed off of the plate, washed three times with PBS and replated onto 15 cm dishes. After 12 hours (G1/S phase) and 24 hours (G2 phase) cells were harvested, and immunoprecipitation assays were performed as described above. To monitor cell cycle progression after release from 8h Nocodazole arrest, cells harvested by mitotic shakeoff were plated after washing out the nocodazole into 96 well imaging plates. After 12 hours 100 ng/ml Nocodazole and 0.5 μM SiR-DNA was added to the cells. Cells were imaged between 14.5 and 36.5 hours after nocodazole washout.
For immunoprecipitation assays in normal mitosis, cells were first arrested in G2 with 9 μM RO-3306 for 14h. Cells were washed 3 times with PBS and, after 30 minutes, mitotic cells were washed of the plate and collected in ice-cold PBS and immunoprecipitation assays were performed as described above.
For cell lysate fractionation, cells were cultured on 15 cm dishes. To induce DNA damage, cells were treated for 16h with doxorubicin (1 μM), and to arrest cells in mitosis cells were treated for 16h with Nocodazole (100 ng/ml or 0.33 μM). Four plates were harvested for each condition. Control and doxorubicin treated cells were harvested with trypsin and washed with PBS. Mitotic cells were directly washed of the plates. All cells were washed once with PBS and resuspended in 5 ml PBS. Suspended cells were counted. Cells were divided into aliquots of 1.5 million cells, pelleted and resuspended with 200 μl lysis buffer containing different amounts of sodium chloride (50 mM, 100 mM, 150 mM, and 200 mM). Cells were sonicated for 5 min to generate the whole cell extract. 100 μl whole cell extract of each sample was collected for immunoblotting. The remaining 100 μl WCE was centrifuged for 15 minutes at 15,000 x g at 4 °C to generate the supernatant and pellet fractions. Pellets were resuspended in lysis buffer to equal the total volume of the supernatant. 40μl 4x sample buffer was added to each sample. Equal amounts of whole cell extract, supernatant and pellet fractions were analyzed by immunoblotting.
Number of experiments/cells/clones analyzed
For single cell tracing experiments shown in Fig. 1B; 2G; 3A; 4A,B; S1B; S6A,B; S7A–D; S10B–E; S11D,G; S13C ~160 cells per condition were tracked from 2-8 independent wells. Control RPE1 cells were analyzed in 3 independent experiments (Fig 1B; S11D; biological replicates). Two independent 53BP1-G1560K mutant clones were measured with similar outcomes (only one clone is shown in Fig. 2G). Two independent clones were analyzed for USP28 fs/fs; TP53BP1 fs/fs; USP28 wt/fs; TP53BP1 wt/fs mutants (Fig. S11D).
For single cell tracing experiments shown in Fig. 1D (92 cells); 1F (control: 88 cells; USP28Δ: 97 cells ); S1E (30min: 41 cells; 40-60min: 85 cells; 70-90min: 81 cells); S1F (30 cells); S2C (0d: 100 cells; 1d: 89 cells; 2d: 50 cells; 3d: 86 cells; 4d: 47 cells); S2D (TP53sh: 143 cells); S3D (1d: 88 cells; 2d: 88 cells; 3d: 90 cells); S3E (Ctrl: 9 cells; 1d: 8 cells; 2d: 10 cells); S5E (between 810 and 1256 per timepoint); S12D, the indicated number of cells were monitored in 2-4 independent wells.
Results shown by immunoblots in Fig. 2B,C,D,E,F,H,I,J; Fig. 3B,C; Fig. S4A,C,D; Fig. S5C; Fig. S7F; Fig. S8D,F were confirmed by complementary experiments. Solubility of 53BP1, USP28 and p53 in different cell cycle stages is shown in Fig. 2B,C,D and Fig. S4A,C. Each immunoprecipitation experiment included positive (prolonged mitosis) and negative (beads only or beads with control IgG) controls; note that controls are not always shown for brevity. 53BP1-USP28-p53 complex formation upon prolonged mitosis (nocodazole or monastrol) is shown in nine independent experiments (Fig. 2C,D,E,F,H,I,J; Fig. S4C,D; biological replicates). Independent interaction of 53BP1 with USP28 and p53 (Fig. 2E) is supported by results of the 53BP1-G1560K mutant presented in Fig. 2F, analysis of a G1560K, N1845R and D1861R mutant transgenes shown in Fig. S8F and analysis of phosphorylation site mutants shown in Fig. 3C. Increased mitotic stopwatch complex formation upon progressive extension of mitotic duration shown in Fig. 2I is supported by results shown in Fig. S5C. Immunoprecipitation analysis of the stability of mitotic stopwatch complexes in the following cell cycle shown in Fig. 2J was conducted twice and is consistent with the results shown in Fig. S7F (biological replicates). Disruption of USP28 interaction in 53BP1-G1560K is shown in two independent experiments (Fig. 2F; Fig. S8F; biological replicates) and is consistent with inactivation of the mitotic stopwatch in this mutant (Fig. 2G). PLK1 activity-dependent interaction of 53BP1 with p53 shown in Fig. 3B was analyzed twice (biological replicates) and is supported by mutant analysis shown in Fig. 3C–F and loss of mitotic stopwatch activity in PLK1-inhibited cells shown in Fig. 3A & Fig. S7C,D. No mitotic stopwatch complex formation upon doxorubicin treatment (Fig. 2D) is supported by the finding that inhibition of DNA damage kinases does not impair mitotic stopwatch function (Fig. 3A).
Immunoblots in Fig. 4C; Fig. S1A; Fig. S11B,F and Fig. S13B were done once to confirm protein expression of knocked out or depleted genes or to confirm annotated genetic mutations in cancer cell lines. Two independent clones were blotted for USP28 fs/fs; TP53BP1 fs/fs; USP28 wt/fs; TP53BP1 wt/fs mutants with similar outcomes (Fig. S11D).
For immunostaining experiments in Fig. 3E; Fig. 6C; Fig. S8H; Fig. S12B the indicated number of cells were quantified from triplicate wells unless noted otherwise. For proliferation (passaging) assays shown in Fig. 4B,D,E; Fig. S3B; Fig. S13D cells were analyzed in triplicate for each condition (technical replicates).
Statistics
For pairwise comparisons, t-tests were performed in Fig. 1D, 3E, 4E, S1E, S13D.
Materials Availability Statement
All materials (cell lines, plasmids) generated for this article will be made available upon request.
Supplementary Material
Acknowledgments:
We thank Midori Ohta and Aleesa Schlientz for feedback on the manuscript, Rebecca Green for help with the model figure, and members of the Oegema and Desai labs for discussion.
Funding:
Provide complete funding information, including grant numbers, complete funding agency names, and recipient’s initials. Each funding source should be listed in a separate paragraph.
National Institutes of Health grant GM074207 (KO)
National Institutes of Health grant GM074215 (AD)
German Research Foundation grant ME 4713/1-1 (FM)
Ludwig Institute for Cancer Research (salary and other support for KO and AD)
Okinawa Institute of Science and Technology (FM)
JSPS KAKENHI grant 23K05773 (FM)
Footnotes
Competing interests: Authors declare that they have no competing interests.
Data and materials availability:
All data and materials are available in the main text or the supplementary materials or upon request.
References and Notes:
- 1.Araujo AR, Gelens L, Sheriff RS, Santos SD, Positive Feedback Keeps Duration of Mitosis Temporally Insulated from Upstream Cell-Cycle Events. Mol Cell 64, 362–375 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Umbreit NT et al. , Mechanisms generating cancer genome complexity from a single cell division error. Science 368, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shoshani O. et al. , Chromothripsis drives the evolution of gene amplification in cancer. Nature 591, 137–141 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uetake Y, Sluder G, Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol 20, 1666–1671 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meitinger F. et al. , 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol 214, 155–166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lambrus BG et al. , A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol 214, 143–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fong CS et al. , 53BP1 and USP28 mediate p53-dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bazzi H, Anderson KV, Acentriolar mitosis activates a p53-dependent apoptosis pathway in the mouse embryo. Proc Natl Acad Sci U S A 111, E1491–1500 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao C. et al. , Gradual centriole maturation associates with the mitotic surveillance pathway in mouse development. EMBO Rep 22, e51127 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Insolera R, Bazzi H, Shao W, Anderson KV, Shi SH, Cortical neurogenesis in the absence of centrioles. Nat Neurosci 17, 1528–1535 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phan TP, Holland AJ, Time is of the essence: the molecular mechanisms of primary microcephaly. Genes Dev 35, 1551–1578 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phan TP et al. , Centrosome defects cause microcephaly by activating the 53BP1-USP28-TP53 mitotic surveillance pathway. EMBO J 40, e106118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marjanovic M. et al. , CEP63 deficiency promotes p53-dependent microcephaly and reveals a role for the centrosome in meiotic recombination. Nat Commun 6, 7676 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barr AR et al. , DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat Commun 8, 14728 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrell JE Jr., Ha SH, Ultrasensitivity part II: multisite phosphorylation, stoichiometric inhibitors, and positive feedback. Trends Biochem Sci 39, 556–569 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang HW, Chung M, Kudo T, Meyer T, Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature 549, 404–408 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corbett KD, Molecular Mechanisms of Spindle Assembly Checkpoint Activation and Silencing. Prog Mol Subcell Biol 56, 429–455 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Kim DH et al. , TRIP13 and APC15 drive mitotic exit by turnover of interphase- and unattached kinetochore-produced MCC. Nat Commun 9, 4354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong YL et al. , Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 348, 1155–1160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez-Vidal A, Vignard J, Mirey G, Around and beyond 53BP1 Nuclear Bodies. Int J Mol Sci 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zimmermann M, de Lange T, 53BP1: pro choice in DNA repair. Trends Cell Biol 24, 108–117 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derbyshire DJ et al. , Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. EMBO J 21, 3863–3872 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joo WS et al. , Structure of the 53BP1 BRCT region bound to p53 and its comparison to the Brca1 BRCT structure. Genes Dev 16, 583–593 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuella-Martin R. et al. , Functional interrogation of DNA damage response variants with base editing screens. Cell 184, 1081–1097 e1019 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porter JR, Fisher BE, Batchelor E, p53 Pulses Diversify Target Gene Expression Dynamics in an mRNA Half-Life-Dependent Manner and Delineate Co-regulated Target Gene Subnetworks. Cell Syst 2, 272–282 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scoumanne A, Cho SJ, Zhang J, Chen X, The cyclin-dependent kinase inhibitor p21 is regulated by RNA-binding protein PCBP4 via mRNA stability. Nucleic Acids Res 39, 213–224 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciceri P. et al. , Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat Chem Biol 10, 305–312 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kettenbach AN et al. , Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal 4, rs5 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Vugt MA et al. , A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol 8, e1000287 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuella-Martin R. et al. , 53BP1 Integrates DNA Repair and p53-Dependent Cell Fate Decisions via Distinct Mechanisms. Mol Cell 64, 51–64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C. et al. , GPS 5.0: An Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins. Genomics Proteomics Bioinformatics 18, 72–80 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cimini D. et al. , Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol 153, 517–527 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davoli T. et al. , Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knobel PA et al. , USP28 is recruited to sites of DNA damage by the tandem BRCT domains of 53BP1 but plays a minor role in double-strand break metabolism. Mol Cell Biol 34, 2062–2074 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khoo KH, Verma CS, Lane DP, Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 13, 217–236 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Olivier M, Hollstein M, Hainaut P, TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2, a001008 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kleiblova P. et al. , Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J Cell Biol 201, 511–521 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gilmartin AG et al. , Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat Chem Biol 10, 181–187 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Meitinger F. et al. , TRIM37 controls cancer-specific vulnerability to PLK4 inhibition. Nature 585, 440–446 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yeow ZY et al. , Targeting TRIM37-driven centrosome dysfunction in 17q23-amplified breast cancer. Nature 585, 447–452 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qian X. et al. , Discovery of the First Potent and Selective Inhibitor of Centromere-Associated Protein E: GSK923295. ACS Med Chem Lett 1, 30–34 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tischer J, Gergely F, Anti-mitotic therapies in cancer. J Cell Biol 218, 10–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dumontet C, Jordan MA, Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 9, 790–803 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernabeu E, Cagel M, Lagomarsino E, Moretton M, Chiappetta DA, Paclitaxel: What has been done and the challenges remain ahead. Int J Pharm 526, 474–495 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Penna LS, Henriques JAP, Bonatto D, Anti-mitotic agents: Are they emerging molecules for cancer treatment? Pharmacol Ther 173, 67–82 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Garribba L, Santaguida S, The Dynamic Instability of the Aneuploid Genome. Front Cell Dev Biol 10, 838928 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li R, Zhu J, Effects of aneuploidy on cell behaviour and function. Nat Rev Mol Cell Biol 23, 250–265 (2022). [DOI] [PubMed] [Google Scholar]
- 48.Chen G. et al. , Chemically defined conditions for human iPSC derivation and culture. Nat Methods 8, 424–429 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ran FA et al. , Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanjana NE, Shalem O, Zhang F, Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brinkman EK, Chen T, Amendola M, van Steensel B, Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaulich M, Dowdy SF, Combining CRISPR/Cas9 and rAAV Templates for Efficient Gene Editing. Nucleic Acid Ther 25, 287–296 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zafra MP et al. , Optimized base editors enable efficient editing in cells, organoids and mice. Nat Biotechnol 36, 888–893 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakaue-Sawano A, Kobayashi T, Ohtawa K, Miyawaki A, Drug-induced cell cycle modulation leading to cell-cycle arrest, nuclear mis-segregation, or endoreplication. BMC Cell Biol 12, 2 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schindelin J. et al. , Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Durant ST et al. , The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci Adv 4, eaat1719 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fok JHL et al. , AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat Commun 10, 5065 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kurose A. et al. , Effects of hydroxyurea and aphidicolin on phosphorylation of ataxia telangiectasia mutated on Ser 1981 and histone H2AX on Ser 139 in relation to cell cycle phase and induction of apoptosis. Cytometry A 69, 212–221 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Lara-Gonzalez P, Pines J, Desai A, Spindle assembly checkpoint activation and silencing at kinetochores. Semin Cell Dev Biol 117, 86–98 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Groot CO et al. , A Cell Biologist’s Field Guide to Aurora Kinase Inhibitors. Front Oncol 5, 285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Filippakopoulos P. et al. , Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ember SW et al. , Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem Biol 9, 1160–1171 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rudolph D. et al. , BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res 15, 3094–3102 (2009). [DOI] [PubMed] [Google Scholar]
- 64.Gilmartin AG et al. , Distinct concentration-dependent effects of the polo-like kinase 1-specific inhibitor GSK461364A, including differential effect on apoptosis. Cancer Res 69, 6969–6977 (2009). [DOI] [PubMed] [Google Scholar]
- 65.Chan KF, Koukouravas S, Yeo JY, Koh DW, Gan SK, Probability of change in life: Amino acid changes in single nucleotide substitutions. Biosystems 193-194, 104135 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Barretina J. et al. , The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and materials are available in the main text or the supplementary materials or upon request.