

Abstract

We have developed photocatalytic oxidation of aromatic substrates using O2 as a terminal oxidant to afford only 2e–-oxidized products without the reductive activation of O2 in acidic water under visible-light irradiation. A RuII complex (1) bearing a pyrazine moiety as the active site in tetrapyrido[3,2-a:2′,3′-c:3″,2″-h:2‴,3‴-j]phenazine (tpphz) as a ligand was employed as a photocatalyst. The active species for the photocatalysis was revealed to be not complex 1 itself but the protonated form, 1-H+, protonated at the vacant diimine site of tpphz. Upon photoexcitation in the presence of an organic substrate, 1-H+ was converted to the corresponding dihydro-intermediate (2-H+), where the pyrazine moiety of the ligand received 2e– and 2H+ from the substrate. 2-H+ was facilely oxidized by O2 to recover 1-H+. Consequently, an oxidation product of the substrate and H2O2 derived from dioxygen reduction were obtained; however, the H2O2 formed was also used for oxidation of 2-H+. In the oxidation of benzyl alcohol to benzaldehyde, the turnover number reached 240 for 10 h, and the quantum yield was determined to be 4.0%. The absence of ring-opening products in the oxidation of cyclobutanol suggests that the catalytic reaction proceeds through a mechanism involving formal hydride transfer. Mechanistic studies revealed that the photocatalytic substrate oxidation by 1-H+ was achieved in neither the lowest singlet excited state nor triplet excited state (S1 or T1) but in the second lowest singlet excited state (S2), i.e., 1(π–π*)* of the tpphz ligand. Thus, the photocatalytic substrate oxidation by 1-H+ can be categorized into unusual anti-Kasha photocatalysis.
Introduction
Dioxygen (O2) is an abundant and clean oxidant; thus it has received considerable attention as one of the most important terminal oxidants in benchtop as well as industrial oxidative conversion of organic substrates.1 However, O2 in the triplet ground state is not thermodynamically reactive and difficult to be used under ambient conditions.2 On the other hand, enzymes use O2 as the terminal oxidant in an artful manner, in which a metal center such as iron is reduced at an appropriate timing through electron transfer (ET) from the corresponding reductases to reductively activate O2, followed by the O–O bond cleavage to form the active species.3,4 As an alternative approach to use O2 as the terminal oxidant, quinone derivatives have been employed as the responsible oxidant for the oxidation of organic substrates and the resultant reduced quinone, i.e., hydroquinone, is oxidized by O2 to form the original quinone and H2O2.5 For example, pyrroloquinoline quinone (PQQ), found in methanol dehydrogenase,6 has been reported to oxidize organic substrates such as benzyl alcohol,7 when PQQ coordinates to a metal ion such as Ca2+ for activation of PQQ itself, and the formed 2e–/2H+-reduced PQQ is oxidized by O2.8 It is proposed that the substrate oxidation by PQQ proceeds through a hydride-transfer mechanism;9 thus, the product selectivity for substrate oxidation by PQQ is high in spite of the reaction performed under aerobic conditions.
On the other hand, it is very difficult to activate O2 by an artificial system in the same manner with enzymes, and thus, photochemical pathways have been applied for artificial substrate oxidation with the use of a photosensitizer (PS) and O2 as the terminal oxidant.2 For example, flavin and the derivatives10,11 and other chromophores12,13 have been employed as PSs for photocatalytic oxidation of organic substrates. However, photoirradiation to a solution containing a PS under air or O2 atmosphere causes the formation of reactive oxygen species such as singlet oxygen, 1O2, and recently, several reports have revealed that 1O2 is the actual active species for the substrate oxidation.14,15 Formation of 1O2 usually causes decomposition of the PS and low product selectivity due to the high reactivity of 1O2.16
To efficiently carry out photocatalytic reactions using O2 as a terminal oxidant without formation of 1O2, faster ET from a substrate trapped in the close vicinity to a PS or photocatalyst in higher singlet excited states (Sn (n ≥ 2)) than the energy transfer to generate 1O2 should be required to oxidize organic substrates; the reduced PS or photocatalyst can be oxidized by O2 to recover the resting state. This strategy allows us to oxidize organic substrates by a photocatalyst in a high-energy singlet excited state without passing through intersystem crossing to the low-energy triplet excited state. However, the Sn (n ≥ 2) states usually decay to the lowest singlet excited states (S1) with very short lifetime by Kasha’s rule;17 thus, the Sn (n ≥ 2) states are generally difficult to be used for photocatalysis. As examples of photoprocesses breaking the Kasha’s rule, emission from a higher excited state has been well investigated.18−20 In addition, photoinduced ET proceeding from Sn (n ≥ 2) states has been reported so far.21,22 To the best of our knowledge, there are no reports of photocatalysis using higher singlet excited states.23
We have recently reported photocatalytic hydrogen evolution24 catalyzed by a RuII complex, [RuII(tpphz)(bpy)2](ClO4)2 (1; tpphz = tetrapyrido[3,2-a:2′,3′-c:3″,2″-h:2‴,3‴-j]phenazine, bpy = 2,2′-bipyridyl),25 with the use of Et3N as a sacrificial reductant. In the hydrogen evolution, a dihydro-intermediate, [RuII(H2tpphz)(bpy)2]2+ (2),26 was formed and photoirradiation of 2 induces the reaction with H+ from the solvent to form a H2 molecule and recover 1 (Figure 1).24 In addition, we have also reported that complex 2 is easily oxidized by the addition of a small amount of O2 to regenerate 1.24
Figure 1.

Proposed mechanism of the photocatalytic hydrogen evolution by 1 and recovery of 1 from 2 by O2 oxidation (red line).24
In this work, based on the facile recovery of 2 to 1 with O2, we have developed a photocatalytic oxidation system of organic substrates using 1 as a photocatalyst and O2 as a terminal oxidant in water. The point of the catalytic system described in this work has been revealed to be protonation of 1 at the vacant diimine site of the tpphz ligand. Photoirradiation to the protonated species, 1-H+, affords an emissive singlet π–π* excited state, which works as a reactive state for the oxidation of organic substrates and the resultant reduced species of 1-H+, that is 2-H+, is oxidized with use of O2 to recover 1-H+. Herein, we report photocatalytic oxidation of organic substrates in acidic water and detailed mechanistic insights into photocatalysis of 1-H+ for substrate oxidation based on various spectroscopies, including transient absorption spectroscopy.
Results and Discussion
Protonation of the tpphz Ligand in 1
We performed spectroscopic titration of 1 in Britton–Robinson (B.–R.) buffer27 by the addition of NaOH aq (0.2 M) to monitor the spectral change in the UV region, and the pKa value for the protonation/deprotonation process at the diimine site of the tpphz ligand in 1 was determined to be 2.99 ± 0.06 (Figure S1). Thus, complex 1 is protonated to be 1-H+ in an aqueous solution with a pH less than 3. To confirm the protonation of the diimine moiety of 1, the structure of 1-H+ was elucidated by a single-crystal X-ray diffraction analysis (Figure 2).
Figure 2.

ORTEP drawing for the crystal structure of [1-H+](OTf)3 with the 50% probability level. Counter anions and protons except that at N5 and hydrogen-bonded H2O molecule are omitted for clarity.
A single crystal of [1-H+](TfO)3 (TfO = trifluoromethane-sulfonate) was obtained from an aqueous solution of 1-H+, acidified by the addition of an excess amount of TfOH. The number of TfO– ion as the counteranion indicates that complex 1 is protonated to be a trication in the crystal. The bond angle of ∠C20–N5–C24 is 123.1(4)°, which is larger than that of ∠C15–N4–C19 (116.2(4)°) and the corresponding angles for [RuII(TPA)(tpphz)](BPh4)2 (116.8(8)° and 117.8(8)°).28 Expansion of the ∠Cα–N-Cα angle is typical for protonation at pyridine nitrogen,29 and thus, the diimine nitrogen, N5 in Figure 2, of the tpphz ligand is protonated in the crystal. As evidence of the monoprotonation at the diimine moiety of 1, one water molecule of crystallization resides in the distance of 2.69(1) Å from N5, indicating that N5 is protonated to form hydrogen bonding with the H2O molecule. The tpphz ligand in 1-H+ showed π–π stacking with the tpphz ligand in another 1-H+, and the separation between the two tpphz ligand was calculated to be 3.268 Å (Figure S2).
Redox Properties of 1 and 1-H+
Electrochemical studies of 1 and 1-H+ were performed in CH3CN containing tetrabutylammonium hexafluorophosphate (TBAPF6) as the electrolyte (Figure S3 and Table S1). Complex 1 exhibited one reversible oxidation wave at +0.94 V vs Fc+/Fc (Fc = ferrocene) and three reversible reduction waves at −1.36, −1.78, and −1.99 V vs Fc+/Fc (Figure S3a and Table S1). The oxidation wave can be assigned to be the RuIII/RuII process, whereas the reduction waves are ascribed to the processes of tpphz/tpphz•–, bpy/bpy•–, and bpy/bpy•–, respectively.30 Upon adding TfOH (0.5 mM) to the solution of 1 in CH3CN to form 1-H+, the oxidation wave derived from the RuIII/RuII process was observed at +0.97 V vs Fc/Fc+ without a significant shift relative to that before TfOH was added (Figure S3b and Table S1). In contrast, the reduction waves of 1-H+ exhibited large positive shifts relative to those before adding TfOH and were observed at −0.48, –0.83, and −1.09 V vs Fc+/Fc (Figure S3b and Table S1). The positive shift of the reduction potential for the tpphz/tpphz•– process of 1-H+ indicates the enhancement of oxidation activity of 1-H+ in the photoexcited state, since the excitation energy of 1 and 1-H+ can be considered to be almost the same for both based on the emission maxima of phosphorescence derived from the MLCT triplet states as the lowest excited states (See below).
Photocatalytic Oxidation of Benzyl Alcohol by 1-H+
A solution containing benzyl alcohol (BnOH; 100 mM) as a substrate and complex 1 (0.1 mM) as a photocatalyst in B.–R.-buffered D2O, whose pD was varied in the range of 5.0–1.5, was photoirradiated with white light (λ > 380 nm) under O2 (1 atm) for 10 h. The solution after photoreaction was analyzed by 1H NMR spectroscopy. The 1H NMR spectrum of the reaction mixture showed signals derived from benzaldehyde (PhCHO) as the sole oxidation product from BnOH (Figure S4a–e). The turnover number (TON) for the PhCHO formation for 10 h was plotted against the solution pD; the TON increased with lowering the solution pD and the inflection point was found to be around pD = 3 (Figure S4f), which matches the pKa value of 1 determined above. In addition, we performed photocatalytic oxidation of BnOH in H2O at pH 1.5 under the same conditions in D2O. The TON of PhCHO production for 18 h quantified by GC–MS was determined to be 320, which was almost the same as that in D2O as determined by 1H NMR measurements. Thus, photocatalysis of BnOH oxidation should be performed by 1-H+ as a photocatalyst with complete selectivity for the 2e–-oxidation reaction by using O2 as the oxidant (Scheme 1).
Scheme 1. Photocatalytic Oxidation of BnOH by 1-H+ Using O2 as the Terminal Oxidant.

To clarify the fate of 1-H+ after photocatalysis, photocatalytic BnOH oxidation was performed at a higher concentration (0.2 mM) of 1-H+ in D2O (pD = 1.5) (Figure 3). The 1H NMR signals of 1-H+ were intact after the photocatalytic oxidation of BnOH performed under O2 (1 atm) and white-light irradiation (λ > 380 nm) (Figure 3b), indicating the robustness of 1-H+ under the catalytic conditions. The amount of PhCHO formed was determined to be 11.2 mM based on the integration ratio of a signal due to the formyl group relative to that of 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt (DSS) as an internal standard. The TON was calculated to be 56 for 12 h. On the other hand, when photocatalytic oxidation of BnOH by 1-H+ was performed in B.–R.-buffered D2O (pD 1.5) under Ar atmosphere, only 0.2 mM of PhCHO, i.e., 1 mol eq against 1-H+, was observed and complex 1-H+ was fully converted to the dihydro-derivative, 2-H+ (Figure 3c). Therefore, the excited state of 1-H+, formed by photoirradiation, should accept 2H+ and 2e– from BnOH at the phenazine moiety of the tpphz ligand to afford PhCHO and complex 2-H+. In the presence of O2, complex 2-H+ obtained is easily oxidized to regenerate 1-H+ (Figure S5), similar to 1.24,31
Figure 3.
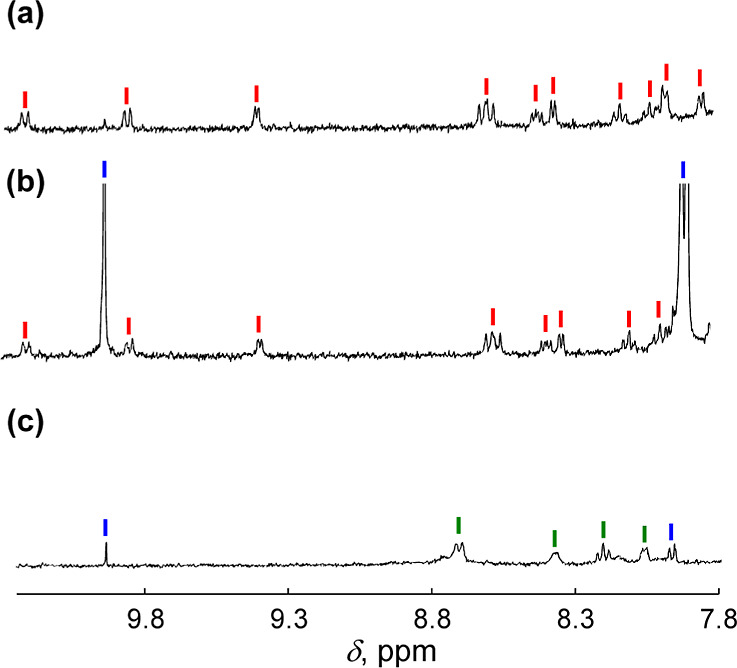
1H NMR spectrum of 1-H+ (0.2 mM) (a) and those of 1-H+ (0.2 mM) and BnOH (100 mM) after photoirradiation (λ > 380 nm) for 12 h under O2 (1 atm) (b) or Ar atmosphere (1 atm) (c). Solvent: B.-R.-buffered D2O (pD = 1.5). The 1H NMR signals of 1-H+, 2-H+ and PhCHO are indicated by red, green, and blue sticks, respectively.
At solution pD = 1.5, effects of the catalyst concentration on the photocatalytic efficiency were investigated. Photoirradiation (λ > 380 nm) was performed for 10 h to a B.–R.-buffered D2O containing BnOH (100 mM) and various concentrations of 1-H+ (5–50 μM) and the amounts of PhCHO as the product were quantified by 1H NMR spectroscopy. The time course of the PhCHO formation from the photocatalytic BnOH oxidation by 1-H+ (10 μM) is provided in Figure S6, and the reaction was almost saturated at 10 h.32 The TONs for 10 h obtained from the experiments performed with various concentrations of 1-H+ were plotted against the concentration of 1-H+ (Figure S8a), and the plot exhibits the tendency that with the lowering of the concentration of 1-H+, the TON increases.33 The TON reached 240, when the concentration of 1-H+ was 5 μM. The substrate concentration was also optimized; as shown in Figure S8b, the TON for the PhCHO formation for 10 h was saturated over 50 mM. Complex 1′-H+ (See Figure 1) was also used as a photocatalyst instead of 1-H+ for the BnOH oxidation under the same conditions; the TON was determined to be 60. Thus, introduction of CF3 groups as an electron-withdrawing group to the bpy ligand does not severely affect the activity of a photocatalyst for the photocatalytic oxidation of BnOH.
Substrate Scope for the Photocatalytic Oxidation by 1-H+
Other organic substrates were also oxidized by photocatalysis of 1-H+ (Table 1 and Figure S9); a solution of 1-H+ (10 μM) and 50 mM of an organic substrate in D2O (pD = 1.5) was photoirradiated for 10 h, and the products were characterized and quantified with 1H NMR spectroscopy. BnOH derivatives, 1-phenyl-ethanol and 4-methoxy-benzyl alcohol, were oxidized to afford the corresponding ketone or aldehyde selectively, and the TONs were estimated to be 152 and 100, respectively. Sodium ethylbenzene-sulfonate and sodium toluenesulfonate as water-soluble alkylbenzene derivatives were also used as substrates for the photocatalytic oxidation by 1-H+, obtaining modest amounts of 1-phenylethanol and benzyl alcohol derivatives as the sole products, respectively. Benzene was also photocatalytically oxidized by 1-H+ to phenol without any overoxidation; however, the TON was modest. Thus, an LED lamp centered at 365 nm was used instead of white light from a Xe lamp. In consequence, the TON for the phenol formation from benzene was improved from 6 to 23 (Table 1).35 There are a limited number of examples of photocatalytic systems by molecular catalysts to convert benzene to phenol using oxygen as the oxidant.36
Table 1. Summary of TONs for the Photocatalytic Substrate Oxidation by 1 under O2 (1 atm).

In kcal mol–1.
Reference (34).
TONs obtained using an LED lamp (λcenter = 365 nm) as a light source. Reaction conditions: [1-H+] = 10 μM, [substrate] = 50 mM, T = room temperature. Reaction time: 10 h. Solvent: B.–R.-buffered D2O (pD = 1.5). Light source: Xe lamp (λ > 380 nm).
Mechanistic Insights of the Photocatalytic Oxidation by 1-H+
Generally, photoirradiation to a solution of a RuII-tris(diimine) complex under a O2 atmosphere affords singlet oxygen (1O2) through energy transfer from the triplet excited state of the RuII-tris(diimine) complex to the triplet O2 (3O2).37 To confirm the hypothesis that the active species for the photooxidation of BnOH by 1-H+ is not 1O2 but the photoexcited state of 1-H+, we added NaN3 (200 mM) as a quencher of 1O238 to the reaction solution (Figure S10). Consequently, even when adding NaN3, the TON for the photocatalytic BnOH oxidation by 1-H+ was not lowered (Figure S10b); thus, 1O2 is not the active species for the photocatalytic oxidation (Scheme 2a). As a reference, we used [RuII(bpy)3]Cl2 as a PS instead of 1-H+; white-light irradiation to a solution containing BnOH (100 mM) and [RuII(bpy)3]Cl2 (0.2 mM) in B.–R.-buffered D2O (pD = 1.5) under O2 (1 atm) for 12 h afforded PhCHO as the product with TON of 33 (Figure S11a), which was almost the same with that using 1-H+ as a PS under the same conditions (Figure S10a). In contrast, when adding NaN3 (200 mM) to the same aqueous solution of [RuII(bpy)3]Cl2 and performing photocatalytic oxidation, PhCHO was scarcely obtained (Figure S11b). Therefore, the active species of the photocatalytic oxidation by [RuII(bpy)3]Cl2 should be 1O2, formed through energy transfer from 3{[RuII(bpy)3]2+}* to 3O2 (Scheme 2b).39
Scheme 2. Photoinduced Reactions of 1-H+ (a) and [RuII-(bpy)3]2+ (b) with O2 and BnOH.

To gain mechanistic insights into the photocatalytic oxidation by 1-H+, the initial rates for the oxidation of BnOH and its derivative deuterated at the benzyl position (BnOH-d2) were determined and the concentrations of PhCHO and PhCDO as the products were obtained from the 1H NMR spectroscopy. The time course of the product formation exhibited linearity during the initial 8 h for both BnOH and BnOH-d2 oxidation (Figure 4). The slope of the time courses afforded the initial rates, v0, for the BnOH and BnOH-d2 oxidation as v0H = 0.174 ± 0.002 mM h–1 and v0D = 0.0532 ± 0.0008 mM h–1, respectively. Therefore, the kinetic isotope effect (KIE = v0H/v0D) was calculated to be 3.3. The significant KIE indicates that the rate-determining step (RDS) of the photocatalytic BnOH oxidation by 1-H+ involves C–H bond cleavage at the benzylic position.
Figure 4.

Time courses of the product formation for the photocatalytic oxidation of BnOH (black squares) and BnOH-d2 (red circles) by 1-H+ as the photocatalyst under photoirradiation (Xe lamp; λ > 380 nm) and O2 (1 atm) at 298 K in B.–R.-buffered D2O (pD = 1.5). Conditions: [1-H+] = 0.1 mM, [substrate] = 100 mM.
Photocatalytic oxidation of cyclobutanol (cBuOH) by 1-H+ was also performed. After photoirradiation (λ > 380 nm) for 12 h to a solution of cBuOH (100 mM) and 1-H+ (0.1 mM) in B.-R.-buffered D2O (pD 1.5), the 1H NMR spectrum of the reaction solution exhibited the signals of cyclobutanone (indicated by the blue line in Figure 5a) as the sole product accompanying the 1H NMR signals of 1-H+ (indicated by yellow lines in Figure 5a). This result suggests that the photocatalytic oxidation by 1-H+ as the catalysts proceeds through formal hydride transfer from the substrate (Scheme 3a).40 The TON for cyclobutanone formation for 12 h was determined to be 8. On the other hand, photocatalytic oxidation of cBuOH by [RuII(bpy)3]Cl2 as a PS under the same conditions afforded a complicated mixture of various products including ring-opening ones (Figure 5b). This indicates that 1O2 as the active species formed by the photoirradiation performs hydrogen-atom abstraction from cBuOH to induce ring-opening reactions (Scheme 3b).40,41
Figure 5.
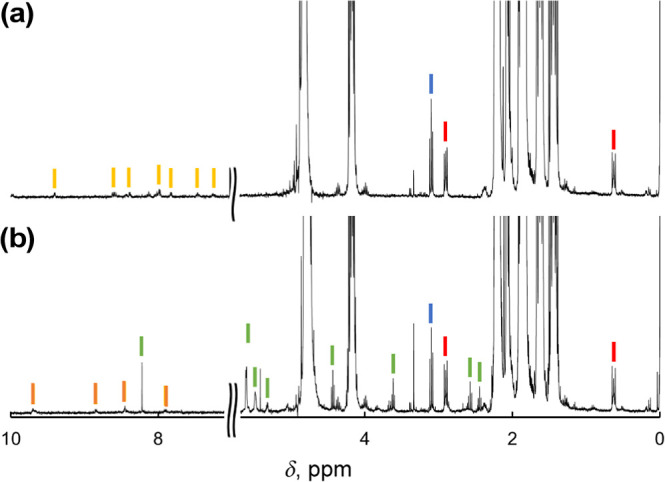
1H NMR spectra of the reaction mixtures for the photocatalytic oxidation of cBuOH by 1-H+ (a) or [RuII(bpy)3]Cl2 (b) under photoirradiation (Xe lamp; λ > 380 nm) and O2 (1 atm) in B.–R.-buffered D2O. Conditions: [1-H+] = 0.1 mM, [cBuOH] = 100 mM, T = room temperature. Reaction time: 12 h. The 1H NMR signals of 1-H+, [RuII(bpy)3]2+, cyclobutanone, and various ring-opening products are indicated by yellow, orange, blue, and green sticks, respectively. DSS was used as an internal standard, and its signals are indicated by red sticks.
Scheme 3. Photocatalytic Oxidation of cBuOH by 1-H+ (a) or [Ru(bpy)3]2+ (b) Using O2 as the Terminal Oxidant.

Elucidation of Photochemical Formation of 2-H+ from 1-H+
To clarify the dependence of photocatalytic BnOH oxidation by 1-H+ on irradiation wavelength, we have monitored the formation process of 2-H+ by photoirradiation to an aqueous solution of 1-H+ (25 μM) under Ar in the presence of BnOH (50 mM). At the solution pH of 7.0, photoirradiation with white light (λ > 380 nm) did not induce any spectral changes of 1 even after irradiation for 60 min (Figure S12a,d). In contrast, when the solution pH was lowered to be 1.3 by addition of TfOH (50 mM), white light irradiation (λ > 380 nm) only for 250 s fully converted 1-H+ to 2-H+, which was verified by disappearance of the π–π* transition of the tpphz ligand in 1-H+ observed around 350–390 nm24,30 (Figure S12b,e).42 Instead of white light, to generate the triplet MLCT excited states,30 light centered at 440 nm with a band-path filter was employed to irradiate an aqueous solution of 1-H+, whose pH was adjusted to be 1.3 by addition of TfOH; however, any spectral change was not observed after irradiation for 60 min (Figure S12c,f). Thus, the species in the MLCT excited states cannot perform photocatalytic oxidation of BnOH. When the same solution of 1-H+ was irradiated by light centered at 380 nm using another band-path filter, the spectral change from 1-H+ to 2-H+ was observed and completed within 1000 s (Figure 6).42 As mentioned above, the absorption band of 1-H+ around 350–390 nm is derived from the π–π* transition of the tpphz ligand;30 thus, the photocatalytic oxidation of BnOH by 1-H+ requires photoexcitation of the π–π* transition of 1-H+.
Figure 6.

(a) UV–vis spectral change of 1-H+ in an aqueous solution containing TfOH (50 mM) by irradiation of light regulated with a band-path filter centered at 380 nm (indicated with yellow background). Irradiation time: 0 s (black) and 1000 s (red). (b) Time courses of the absorbance changes at 375 nm (black) and 391 nm (red), extracted from (a).
Quantum Yield Determination of Formation of 2-H+ through the Photocatalytic Oxidation of BnOH by 1-H+
The light intensity at 380 nm was determined with an actinometer method43 to be 2.34 × 108 einstein s–1. Based on the initial rate for formation of 2-H+ from 1-H+ by irradiation at 380 nm in the presence of BnOH (50 mM) (Figure S14a), the quantum yield (Φ) for the formation of 2-H+ at 380 nm was determined to be 3.3%. The Φ values for the formation of 2-H+ were also determined by varying the concentrations of BnOH under similar conditions. The Φ values obtained were highly dependent on [BnOH]; the Φ values reached 4.0% as the maximum at [BnOH] = 75 mM (Figure S14b). Fukuzumi and co-workers also reported the decrease of the Φ values for photocatalytic substrate oxidation by a flavin derivative upon increasing the substrate concentration.10a They attributed the decrease of the Φ values to acceleration of the back electron transfer (BET) due to formation of a dimer radical cation ([(substrate)2]•+) between the substrate and its radical cation.44 The dependence of the quantum yield on the substrate concentration is discussed in detail in the section regarding the transient absorption spectra of 1-H+ (see below).
Characteristics of Photoexcited States of 1-H+
Emission spectra of 1 and 1-H+ were measured to elucidate the properties of the excited states (Figure S15). The absorption spectra of 1 and 1-H+ are slightly different (Figure S15a); thus, the irradiation wavelengths were also adjusted to be appropriate for 1 and 1-H+, respectively. Here, both excitation at the π–π* transition (λexc = 361 nm for 1 and 356 nm for 1-H+) and MLCT transition (λexc = 444 nm for both 1 and 1-H+) were performed to measure the emission spectra. Emission spectra of 1 at pH 7 exhibited a broad emission band around 640 nm regardless of the irradiation wavelength (orange and gray lines in Figure S15b). The emission band can be assigned to phosphorescence from the 3MLCT* excited state.30 Irradiation at 444 nm, corresponding wavelength to the MLCT transition, to an acidic aqueous solution of 1-H+ at pH 1.3, adjusted by addition of TfOH, also exhibited broad emission at 620 nm (yellow line in Figure S15b), which is ascribable to phosphorescence from the 3MLCT* excited state. On the other hand, an emission spectrum of 1-H+ with irradiation at 356 nm showed an intense emission band around 380 nm (blue line in Figure S15b). To confirm that the emission was derived from 1-H+, we measured an excitation spectrum of 1-H+ by monitoring the emission at 403 nm; the excitation spectrum obtained almost matched to the absorption spectrum of 1-H+ (Figure S15c), and thus the intense emission around 380 nm can be ascribed to an excited state of 1-H+. The quantum yield for the emission at 380 nm from 1-H+ was determined using phenanthrene as an external standard45 to be 2.8%. In addition, the emission intensity of 1-H+ was not altered under Ar and O2 (1 atm) (Figure S16), suggesting that no energy transfer to 3O2 and thus the emission around 380 nm is derived from a singlet excited state of 1-H+. Therefore, the 1(π–π*)* state of the tpphz ligand in 1-H+ as the second lowest singlet excited state does not decay to the 1MLCT* state as the lowest singlet excited state; that is, the Kasha’s rule17 is broken here.
The fluorescence lifetime of 1-H+ at 384 nm was determined to be 235 ± 2 ps (Figure S17a). A fluorescence quenching experiment of 1-H+ was performed with BnOH as a quencher, and upon the increasing concentration of BnOH, the fluorescence intensity gradually decreased (Figure S17b). A Stern–Volmer (S–V) plot46 for the fluorescence at 389 nm of 1-H+ quenched by BnOH was provided to clarify that dependence of I0/I against the concentration of BnOH was not linear at the high concentration of BnOH (Figure S17c). In addition, the kQ value was determined to be (6.1 ± 0.1) × 1010 M–1 s–1 from the slope for the linear fitting against the S–V plot at 389 nm in the lower concentration range of BnOH in Figure S17c; the rate constant exceeds the diffusion-limited rate constant in water (∼1 × 1010 M–1 s–1).47 Therefore, the photocatalytic oxidation of BnOH probably proceeds in its adduct with 1-H+.
Adduct Formation by 1-H+ with BnOH
To confirm the adduct formation between 1-H+ and BnOH, BnOH was titrated to an aqueous solution of 1-H+ (50 μM) in D2O, whose pD was adjusted to 1.3 by the addition of TfOH at 298 K, and the 1H NMR spectra were measured (Figure 7a). Significant upfield shifts were observed for the 1H NMR signals of the protons at the adjacent carbons of the vacant diimine nitrogens in the tpphz ligand, as well as those at the adjacent carbons of the diimine nitrogens coordinating to the RuII center (please also see the signal assignment in Figure S18a). This suggests that the benzene ring of BnOH moves between both of the diimine moieties of the tpphz ligand in the 1H NMR time scale in the adduct with π–π stacked structures, as shown in Figure S18b. The chemical shift change observed for the protons at the adjacent carbons of the vacant diimine nitrogens in the tpphz ligand of 1-H+ (blue star in Figure 7a) was plotted against the concentration of BnOH, and the plot was curve-fitted based on eq S2 appropriate for 1:1 adduct formation (Figure 7b). The curve fitting allowed us to determine the association constant (K) of 1-H+ with BnOH to be 2.4 ± 0.7 M–1.
Figure 7.
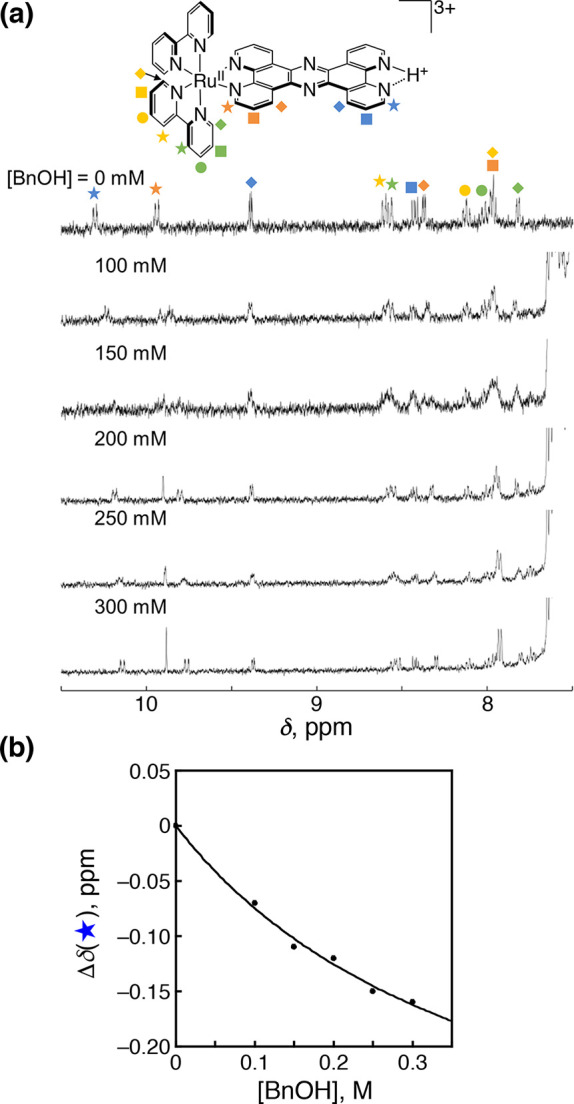
(a) 1H NMR spectral titration of 1-H+ (50 μM) in D2O, whose pD was controlled to be 1.3 by addition of TfOH, with BnOH at 298 K. (b) Plot of the chemical shift change for the most upfield-shifted signal, observed at 10.30 ppm upon [BnOH] = 0 M, against [BnOH] and its curve fitting based on eq S2.
Elucidation of Reaction of 2-H+ with O2
To confirm that O2 used for oxidation of 2-H+ turns to H2O2, we have performed quantification of H2O2 using titanium(IV)-oxo-5,10,15,20-tetra(4-pyridyl)porphyrinato (TiTPyP) as a probe compound.48 To a solution (0.2 mL) of TiTPyP (5 μM) in 0.05 M HClaq, the reaction solution, containing BnOH (50 mM) and 1-H+ (0.01 mM) in D2O (pD = 1.3 adjusted by addition of TfOH) and irradiated by white light (λ > 380 nm) for 10 h, was titrated, and then the solution was diluted to 2 mL with an acidic water. In the UV–vis spectrum of the diluted solution, the Soret band of TiTPyP, originally observed at 432 nm, bathochromically shifted to 450 nm (Figure S19a). Based on the titration curve (Figure S19b), the concentration of H2O2 was determined to be 0.17 mM, which does not match the concentration of PhCHO (4.68 mM) as the oxidation product from BnOH, determined by 1H NMR spectroscopy, in the reaction solution. This can be explained by the assumption that H2O2 is also used for oxidation of 2-H+, producing complex 1-H+ and two molecules of H2O.49
Then, we performed photocatalytic oxidation of BnOH by 1-H+ (0.01 mM) in the presence of H2O2 (7.0 mM) using white-light (λ > 380 nm) irradiation for 10 h. After the reaction, the concentrations of PhCHO and H2O2 in the solution were determined to be 1.3 and 5.6 mM, respectively, by 1H NMR spectroscopy and the TiTPyP method (Figure S21). Thus, the amounts of PhCHO obtained (1.3 mM) and H2O2 consumed (1.4 mM) were consistent, indicating that H2O2 also can function as the terminal oxidant for the photocatalytic oxidation of BnOH by 1-H+.
We have determined the rate constants of the reaction between 2-H+, photochemically formed under Ar in the presence of BnOH (50 mM), and H2O2 or O2 in the dark by monitoring the UV–vis spectral changes (Figures S22–S24). To an aqueous solution of 2-H+ (0.022 mM) containing TfOH (50 mM) and BnOH (50 mM) after white-light irradiation (λ > 380 nm) for 30 min, a large excess amount of H2O2 was added, and based on the absorbance change at 375 nm, the pseudo-first-order rate constants (kobs) were obtained (Figures S22a and S23). The kobs values were plotted against the initial H2O2 concentration (Figure 8a), and the slope of the linear fitting provided the second-order rate constant (k2) for the oxidation of 2-H+ by H2O2 as 0.039 ± 0.008 M–1 s–1. On the other hand, a similar experiment was performed with O2 as the oxidant (Figures S22b and S24), and the kobs values obtained also exhibited linear dependence against the concentration of O2 (Figure 8b). Based on the slope, k2 for the oxidation of 2-H+ by O2 was determined to be 53 ± 2 M–1 s–1. Thus, the reaction of 2-H+ by O2 is 1300 times faster than that by H2O2 in the dark, which contradicts the assumption that H2O2 formed by the reduction of O2 by 2-H+ is consumed by oxidation of another molecule of 2-H+. Therefore, to explain the reason that the amount of H2O2 detected in the reaction solution was much smaller than that of the oxidation product obtained, we assume that the photoexcitation of 2-H+ accelerates the reaction of 2-H+ with H2O2 under the photocatalytic reaction conditions.
Figure 8.

Plots of pseudo-first-order rate constants (kobs) for the reaction of 2-H+ (0.022 mM) with H2O2 (a) and O2 (b) against initial concentration of H2O2 or O2 added to the solution of 2-H+.
Femto-Second Transient Absorption Spectroscopy of 1-H+
A femto-second (fs)-transient absorption spectrum (TAS) of 1 in an aqueous solution at pH = 7 with photoexcitation at 440 nm, which is ascribed to the MLCT transition, exhibited a broad transient absorption band at 675 nm (Figure S25a). Analysis of the decay curve of the transient band at 675 nm with a single exponential indicates a lifetime of 1.78 ± 0.06 ns (Figure S25b). Comparison with TAS of similar RuII(tpphz) complexes reported in literature50 indicates that the band at 675 nm is attributed to the absorption of the 3MLCT* state. Photoexcitation at 375 nm, corresponding to the π–π* transition of the tpphz ligand, to the aqueous solution of 1 at pH = 7 also afforded almost the same fs-TAS (Figure S25c) as that observed by excitation at 440 nm. Therefore, the 1(π–π*)* state of the tpphz ligand in 1 immediately decays to the 1MLCT* state by internal conversion (IC); the 1MLCT* state also immediately decays to the 3MLCT* state through intersystem crossing (ISC) accelerated by heavy atom effect of the RuII center.
A fs-TAS of 1-H+ in an aqueous solution at pH = 1.3, adjusted by the addition of TfOH, was also measured with photoexcitation at 440 nm, also ascribed to the MLCT transition (Figure S26). A broad transient absorption band assignable to the absorption of the 3MLCT* state was observed at 740 nm. The decay of the absorbance at 740 nm was analyzed with a single exponential to determine the lifetime of the 3MLCT* state as (6.0 ± 0.2) × 102 ps (Figure S26b). In contrast, photoexcitation at 375 nm derived from the π–π* transition of the tpphz ligand exhibited a transient absorption band with two peaks at 580 and 735 nm (Figure 9a) and the analysis of the decay of the absorbance at 735 nm with double exponentials afforded lifetimes of (2.6 ± 0.1) × 102 ps (86%) and 12 ± 2 ps (14%). The former lifetime is matched to that of emission of 1-H+ observed at 380 nm determined by time-correlated single photon counting (see above), and thus, it corresponds to the singlet π–π* excited state of the tpphz ligand in 1-H+. In addition, in contrast to the case of 1, transition from the 1(π–π*)* of the tpphz ligand to 1MLCT*, which immediately transit to 3MLCT* through ISC, was not observed by fs-TAS. Thus, breaking of the Kasha’s rule17 in 1-H+ was also verified by the fs-TAS.
Figure 9.

(a) fs-TAS of 1-H+ in an aqueous solution at pH = 1.3 (λirr = 375 nm) and the decay curve for the transient absorbance of 1-H+ monitored at 675 nm with the double-exponential fitting curve. (b) fs-TAS of 1-H+ in an aqueous solution at pH = 1.3, containing BnOH (100 mM): λirr = 375 nm. The NIR absorption bands derived from [(BnOH)2]•+ are indicated by red arrows.
When BnOH (100 mM) was added to an aqueous solution of 1-H+, whose pH was controlled to be 1.3 by addition of TfOH, and the fs-TAS was measured with irradiation at 375 nm, a transient absorption band with two peaks at 580 and 735 nm was observed (Figure S27a), similar to that observed in the absence of BnOH. The lifetime of the excited state, obtained from the analysis of the absorbance decay at 735 nm, was (2.4 ± 0.1) × 102 ps (Figure S27b) and slightly shorter than that in the absence of BnOH. This is consistent with the fact that the addition of BnOH shortens the emission lifetime (see above), indicating that the 1(π–π*)* state reacts with BnOH. In the near-infrared (NIR) region of the fs-TAS of 1-H+ in the presence of BnOH (100 mM) (Figure 9b), a broad transient absorption band was observed in the region of 1200–1400 nm, not observed in the absence of BnOH (Figure S28). The transient absorption band in the NIR region can be assigned to a radical ion pair between 1e–-reduced 1-H+, (1-H)•, and 1e–-oxidized BnOH, BnOH•+, formed through photoinduced ET to the singlet excited state of 1-H+ from neutral BnOH in the adduct (Scheme 4).51 Formation of the radical ion pairs is rationalized by the dependence of the quantum yield of the photocatalysis on the substrate concentration as described above.44 Immediately after photoexcitation, the NIR band was observed in the fs-TAS spectrum (Figure 9b), indicating that the photoinduced ET in the adduct was very fast. The decay of the NIR band at 1315 nm was analyzed with double exponentials to afford a major and relatively long lifetime of (1.9 ± 0.2) × 102 ps as well as a minor and short lifetime of 8 ± 4 ps (Figure S29). The latter is probably ascribable to the photoinduced ET to afford the radical ion pair in the adduct, and the proportion of 1-H+ that forms the adduct is not so high in light of the equilibrium constant (2.4 ± 0.7 M–1) at 298 K. The former lifetime can be assigned to that of the radical ion pair. Additionally, the fact that the subsequent reaction after the photoinduced ET proceeded to afford the oxidation product from the substrate indicates that the BET to recover the ground state is slow enough.52
Scheme 4. Photoinduced ET from BnOH to Photoexcited 1-H+ in the Adduct.

TD-DFT calculations on the adduct of BnOH with 1-H+ suggested that an absorption observed at 348 nm is derived from the transition from a π orbital of the tpphz-H+ ligand with contribution from a filled 2p orbital of the oxygen atom of BnOH to a π* orbital of the ligand, as shown in Figure 10. This result suggested that photoinduced ET from π-stacked BnOH to the π–π* excited state of 1-H+ is plausible.
Figure 10.
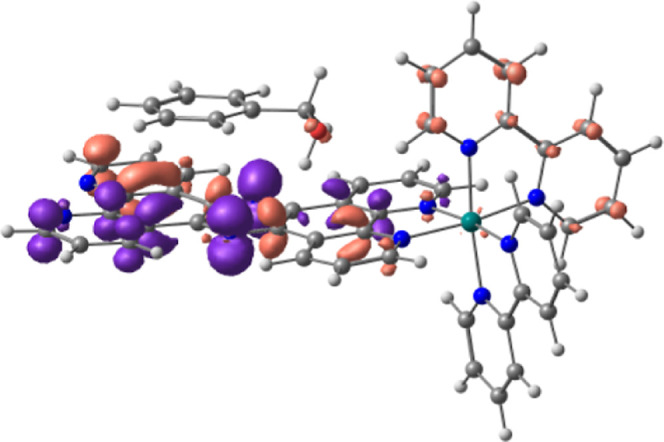
Charge transfer from BnOH to the protonated tpphz ligand of 1-H+ suggested by TD-DFT calculations at the B3LYP-D3 level of theory with GD3 for the adjustment of dispersion forces. Transition occurs from brown orbitals to purple ones.
Photodynamics of 1-H+
Based on the results of fs-TAS of 1-H+ (see above), the emission of 1-H+ at 380 nm is ascribable to the singlet π–π* excited state of its tpphz ligand, which is also responsible for the photocatalysis of substrate oxidation by 1-H+. Normally, a higher excited state such as the S2 state [1(π–π*)*] undergoes relaxation to form a lower excited state such as the S1 state (1MLCT*) through thermal IC and finally reaches the lowest triplet excited state of T1 (3MLCT*) through intersystem crossing, which is radiatively relaxed to the ground state (Figure 11a), according to Kasha’s rule.17 Not the lowest singlet excited state (S1), i.e., 1MLCT*, but a higher singlet excited state (S2), i.e., 1(π–π*)*, of 1-H+ performs the photocatalytic oxidation of organic substrates, that is, an example of anti-Kasha photocatalysis.17 A particularly unique feature of this system is that protonation at the vacant diimine site of the tpphz ligand in 1-H+ induces emission from the S21(π–π*)* state and oxidation of external organic substrates (Figure 11b). Probably, due to the protonation at the vacant diimine site of the tpphz ligand, spin density in the 1(π–π*)* excited state is biased toward the protonated diimine site, which retards the IC to 1MLCT*, since the spin density in the 1MLCT* mainly localizes at the RuII center and the other diimine site of the tpphz ligand bonded to the RuII center.53 Insufficient orbital overlapping in the excited electronic structures is well-known to be important for anti-Kasha chromophores.20a
Figure 11.

Jablonski diagrams for 1 (a) and energy diagram for photodynamics of 1-H+ (b). GS: Grand state. IC: Internal conversion. ISC: Intersystem crossing. Sub: Substrate. Pro: Product.
Plausible Reaction Mechanism of Photocatalytic Substrate Oxidation by 1-H+
Combining the data described above, we have proposed a plausible reaction mechanism for the photocatalytic oxidation of aromatic alcohols by 1-H+ (Figure 12). At first, catalyst 1-H+ forms an adduct with an aromatic substrate. Photoirradiation to the adduct affords an emissive π–π* excited state at the tpphz ligand of 1-H+, and in the photoexcited adduct, photoinduced intrasupramolecular ET proceeds from the substrate to the photoexcited 1-H+ to form a radical ion pair. Then, hydrogen atom transfer proceeds from the 1e–-oxidized substrate to 1e–-reduced 1-H+ and the process is the RDS.54 Subsequent proton transfer from a solvent H2O molecule to reduced 1-H+ affords the dihydro-intermediate, 2-H+, and nucleophilic attack of a H2O molecule to a carbocation derived from the substrate also proceeds to give the oxidized product from the substrate. Complex 2-H+ is immediately oxidized with O2 to reproduce 1-H+ and complete the catalytic cycle.
Figure 12.

Plausible reaction mechanism for the photocatalytic oxidation of aromatic alcohols by 1-H+ in the presence of O2.
Conclusions
Complex 1-H+, whose tpphz ligand is protonated at the vacant diimine site, catalyzes the oxidation of organic substrates under photoirradiation with the use of dioxygen as a terminal oxidant. In the photocatalytic cycle, photoexcited catalyst 1-H+ receives 2H+ and 2e– from an organic substrate through a formal hydride and proton transfer mechanism to afford complex 2-H+, in which the pyrazine moiety in the tpphz ligand is reduced to be the dihydro-pyrazine form. In addition, the photocatalysis by 1-H+ proceeds through the adduct formation of an aromatic substrate with the tpphz-H+ ligand by intermolecular π–π interaction. Since catalyst 1-H+ performs photocatalytic proton-coupled 2e–-oxidation through the second lowest singlet excited state, i.e., 1(π–π*)*, the photocatalysis by 1-H+ is classified as an anti-Kasha type.17 The results presented in this work indicate that using higher energy of the higher photoexcited state provides higher reactivity of the photocatalyst than that of the lowest excited state in the conventional matter.
Acknowledgments
This work was supported by Grants-in-Aid (nos. JP21H01947, JP21K18973, 24H00462, and 24K21245) from the Japan Society of Promotion of Science (JSPS) of Japan, a Grant-in-Aid for Transformative Research Areas (A) Green Catalysis Science for Renovating Transformation of Carbon-Based Resources (Green Catalysis Science) (JSPS KAKENHI grant nos. JP23H04902 to T.K. and JP23H04906 to M.F.) from MEXT, and the Cooperative Research Program of “Network Joint Research Center for Materials and Devices (MEXT)” and ENEOS Hydrogen Trust Fund.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c09962.
Experimental section, oxidation mechanism, and results of photocatalytic substrate oxidation and pH titration shown (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Liang Y.-F.; Jiao N. Oxygenation via C-H/C-C Bond Activation with Molecular Oxygen. Acc. Chem. Res. 2017, 50, 1640–1653. 10.1021/acs.accounts.7b00108. [DOI] [PubMed] [Google Scholar]; b Punniyamurthy T.; Velusamy S.; Iqbal J. Recent Advances in Transition Metal Catalyzed Oxidation of Organic Substrates with Molecular Oxygen. Chem. Rev. 2005, 105, 2329–2363. 10.1021/cr050523v. [DOI] [PubMed] [Google Scholar]
- Fukuzumi S.; Lee Y.-M.; Jung J.; Nam W. Thermal and Photocatalytic Oxidation of Organic Substrates by Dioxygen with Water as an Electron Source. Green Chem. 2018, 20, 948–963. 10.1039/C7GC03387G. [DOI] [Google Scholar]
- a Meunier B.; de Visser S. P.; Shaik S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]; b Denisov I. G.; Makris T. M.; Sligar S. G.; Schlichting I. Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005, 105, 2253–2278. 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- a Krebs C.; Galonić Fujimori D.; Walsh C. T.; Bollinger J. M. Non-Heme Fe(IV)-Oxo Intermediates. Acc. Chem. Res. 2007, 40, 484–492. 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang V. C.-C.; Maji S.; Chen P. P.-Y.; Lee H. K.; Yu S. S.-F.; Chan S. I. Alkane Oxidation: Methane Monooxygenases, Related Enzymes, and Their Biomimetics. Chem. Rev. 2017, 117, 8574–8621. 10.1021/acs.chemrev.6b00624. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Zhang R.; Jian R.; Zhang L.; Zhang M.-T.; Xia Y.; Luo S. Bio-Inspired Lanthanum-ortho-Quinone Catalysis for Aerobic Alcohol Oxidation: Semi-Quinone Anionic Radical as Redox Ligand. Nat. Commun. 2022, 13, 428. 10.1038/s41467-022-28102-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Afolabi P. R.; Mohammed F.; Amaratunga K.; Majekodunmi O.; Dales L.; Gill R.; Thompson D.; Cooper B.; Wood P.; Goodwin M.; Anthony C. Site-Directed Mutagenesis and X-ray Crystallography of the PQQ-Containing Quinoprotein Methanol Dehydrogenase and Its Electron Acceptor, Cytochrome cL. Biochemistry 2001, 40, 9799–9809. 10.1021/bi002932l. [DOI] [PubMed] [Google Scholar]; b Kasahara T.; Kato T. A New Redox-Cofactor Vitamin for Mammals. Nature 2003, 422, 832. 10.1038/422832a. [DOI] [PubMed] [Google Scholar]
- a Fukuzumi S.; Itoh S.; Komori T.; Suenobu T.; Ishida A.; Fujitsuka M.; Ito O. Photochemical Reactions of Coenzyme PQQ (Pyrroloquinolinequinone) and Analogues with Benzyl Alcohol Derivatives via Photoinduced Electron Transfer. J. Am. Chem. Soc. 2000, 122, 8435–8443. 10.1021/ja001351g. [DOI] [Google Scholar]; b Itoh S.; Kawakami H.; Fukuzumi S. Model Studies on Calcium-Containing Quinoprotein Alcohol Dehydrogenases. Catalytic Role of Ca2+ for the Oxidation of Alcohols by Coenzyme PQQ (4,5-Dihydro-4,5-dioxo-1H-pyrrolo[2,3-f]quinoline-2,7,9-tricarboxylic Acid). Biochemistry 1998, 37, 6562–6571. 10.1021/bi9800092. [DOI] [PubMed] [Google Scholar]
- a Itoh S.; Ohshiro Y.; Agawa T. Reaction of Reduced PQQ (PQQH2) and Molecular Oxygen. Bull. Chem. Soc. Jpn. 1986, 59, 1911–1914. 10.1246/bcsj.59.1911. [DOI] [Google Scholar]; b Itoh S.; Kawakami H.; Fukuzumi S. Catalysis by Calcium Ion of the Reoxidation of Reduced PQQ by Molecular Oxygen. Chem. Commun. 1997, 29–30. 10.1039/a606842a. [DOI] [Google Scholar]
- Chan S. I.; Chuankhayan P.; Reddy Nareddy P. K.; Tsai I.-K.; Tsai Y.-F.; Chen K. H.-C.; Yu S. S.-F.; Chen C.-J. Mechanism of Pyrroloquinoline Quinone-Dependent Hydride Transfer Chemistry from Spectroscopic and High-Resolution X-ray Structural Studies of the Methanol Dehydrogenase from Methylococcus capsulatus (Bath). J. Am. Chem. Soc. 2021, 143, 3359–3372. 10.1021/jacs.0c11414. [DOI] [PubMed] [Google Scholar]
- a Fukuzumi S.; Kuroda S.; Tanaka T. Flavin Analogue-Metal Ion Complexes Acting as Efficient Photocatalysts in the Oxidation of p-Methylbenzyl Alcohol by Oxygen under Irradiation with Visible Light. J. Am. Chem. Soc. 1985, 107, 3020–3027. 10.1021/ja00297a005. [DOI] [Google Scholar]; b Oka M.; Katsube D.; Tsuji T.; Iida H. Phototropin-Inspired Chemoselective Synthesis of Unsymmetrical Disulfides: Aerobic Oxidative Heterocoupling of Thiols Using Flavin Photocatalysis. Org. Lett. 2020, 22, 9244–9248. 10.1021/acs.orglett.0c03458. [DOI] [PubMed] [Google Scholar]
- a Mühldorf B.; Wolf R. C-H Photooxygenation of Alkyl Benzenes Catalyzed by Riboflavin Tetraacetate and a Non-Heme Iron Catalyst. Angew. Chem., Int. Ed. 2016, 55, 427–430. 10.1002/anie.201507170. [DOI] [PubMed] [Google Scholar]; b Zhang W.; Carpenter K. L.; Lin S. Electrochemistry Broadens the Scope of Flavin Photocatalysis: Photoelectrocatalytic Oxidation of Unactivated Alcohols. Angew. Chem., Int. Ed. 2020, 59, 409–417. 10.1002/anie.201910300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jung J.; Ohkubo K.; Prokop-Prigge K. A.; Neu H. M.; Goldberg D. P.; Fukuzumi S. Photochemical Oxidation of a Manganese(III) Complex with Oxygen and Toluene Derivatives to Form a Manganese(V)-Oxo Complex. Inorg. Chem. 2013, 52, 13594–13604. 10.1021/ic402121j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Prokop K. A.; Goldberg D. P. Generation of an Isolable, Monomeric Manganese(V)-Oxo Complex from O2 and Visible Light. J. Am. Chem. Soc. 2012, 134, 8014–8017. 10.1021/ja300888t. [DOI] [PubMed] [Google Scholar]
- a Ohkubo K.; Kobayashi T.; Fukuzumi S. Direct Oxygenation of Benzene to Phenol Using Quinolinium Ions as Homogeneous Photocatalysts. Angew. Chem., Int. Ed. 2011, 50, 8652–8655. 10.1002/anie.201102931. [DOI] [PubMed] [Google Scholar]; b Ohkubo K.; Suga K.; Morikawa K.; Fukuzumi S. Selective Oxygenation of Ring-Substituted Toluenes with Electron-Donating and -Withdrawing Substituents by Molecular Oxygen via Photoinduced Electron Transfer. J. Am. Chem. Soc. 2003, 125, 12850–12859. 10.1021/ja036645r. [DOI] [PubMed] [Google Scholar]
- a Insinska-Rak M.; Sikorski M. Riboflavin Interactions with Oxygen-A Survey from the Photochemical Perspective. Chem.–Eur. J. 2014, 20, 15280–15291. 10.1002/chem.201403895. [DOI] [PubMed] [Google Scholar]; b Jia M.-Z.; Cui J.-W.; Rao C.-H.; Chen Y.-R.; Yao X.-R.; Zhang J. Switchable ROS Species Regulation Facilitates the Selective Oxidation of Benzyl Alcohols Enabled by an Organic Photocatalyst. ACS Sustainable Chem. Eng. 2022, 10, 9591–9599. 10.1021/acssuschemeng.2c02771. [DOI] [Google Scholar]
- Nikitas N. F.; Tzaras D. I.; Triandafillidi I.; Kokotos C. G. Photochemical Oxidation of Benzylic Primary and Secondary Alcohols Utilizing Air as the Oxidant. Green Chem. 2020, 22, 471–477. 10.1039/C9GC03000J. [DOI] [Google Scholar]
- Kearns D. R. Physical and Chemical Properties of Singlet Molecular Oxygen. Chem. Rev. 1971, 71, 395–427. 10.1021/cr60272a004. [DOI] [Google Scholar]
- a Demchenko A. P.; Tomin V. I.; Chou P.-T. Breaking the Kasha Rule for More Efficient Photochemistry. Chem. Rev. 2017, 117, 13353–13381. 10.1021/acs.chemrev.7b00110. [DOI] [PubMed] [Google Scholar]; b del Valle J. C.; Catalán J. Kasha’s Rule: A Reappraisal. Phys. Chem. Chem. Phys. 2019, 21, 10061–10069. 10.1039/C9CP00739C. [DOI] [PubMed] [Google Scholar]
- Veys K.; Escudero D. Anti-Kasha Fluorescence in Molecular Entities: Central Role of Electron-Vibrational Coupling. Acc. Chem. Res. 2022, 55, 2698–2707. 10.1021/acs.accounts.2c00453. [DOI] [PubMed] [Google Scholar]
- a Behera S. K.; Park S. Y.; Gierschner J. Dual Emission: Classes, Mechanisms, and Conditions. Angew. Chem., Int. Ed. 2021, 60, 22624–22638. 10.1002/anie.202009789. [DOI] [PubMed] [Google Scholar]; b Chang Y.-C.; Tang K.-C.; Pan H.-A.; Liu S.-H.; Koshevoy I. O.; Karttunen A. J.; Hung W.-Y.; Cheng M.-H.; Chou P.-T. Harnessing Fluorescence versus Phosphorescence Branching Ratio in (Phenyl)n-Bridged (n = 0–5) Bimetallic Au(I) Complexes. J. Phys. Chem. C 2013, 117, 9623–9632. 10.1021/jp402666r. [DOI] [Google Scholar]; c Tseng H.-W.; Shen J.-Y.; Kuo T.-Y.; Tu T.-S.; Chen Y.-A.; Demchenko A. P.; Chou P.-T. Excited-State Intramolecular Proton-Transfer reaction Demonstrating Anti-Kasha Behavior. Chem. Sci. 2016, 7, 655–665. 10.1039/C5SC01945A. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Dunlop D.; Ludvíková L.; Banerjee A.; Ottosson H.; Slanina T. Excited-State (Anti)Aromaticity Explains Why Azulene Disobeys Kasha’s Rule. J. Am. Chem. Soc. 2023, 145, 21569–21575. 10.1021/jacs.3c07625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hsu C.-C.; Lin C.-C.; Chou P.-T.; Lai C.-H.; Hsu C.-W.; Lin C.-H.; Chi Y. Harvesting Highly Electronically Excited Energy to Triplet Manifolds: State-Dependent Intersystem Crossing Rate in Os(II) and Ag(I) Complexes. J. Am. Chem. Soc. 2012, 134, 7715–7724. 10.1021/ja2107788. [DOI] [PubMed] [Google Scholar]; b Röhrs M.; Escudero D. Multiple Anti-Kasha Emissions in Transition-Metal Complexes. J. Phys. Chem. Lett. 2019, 10, 5798–5804. 10.1021/acs.jpclett.9b02477. [DOI] [PubMed] [Google Scholar]; c Franz J.; Oelschlegel M.; Zobel J. P.; Hua S.-A.; Borter J.-H.; Schmid L.; Morselli G.; Wenger O. S.; Schwarzer D.; Meyer F.; González L. Bifurcation of Excited-State Population Leads to Anti-Kasha Luminescence in a Disulfide-Decorated Organometallic Rhenium Photosensitizer. J. Am. Chem. Soc. 2024, 146, 11272–11288. 10.1021/jacs.4c00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mataga N.; Chosrowjan H.; Taniguchi S.; Shibata Y.; Yoshida N.; Osuka A.; Kikuzawa T.; Okada T. Ultrafast Charge Separation from the S2 Excited State of Directly Linked Porphyrin-Imide Dyads: First Unequivocal Observation of the Whole Bell-Shaped Energy-Gap Law and Its Solvent Dependencies. J. Phys. Chem. A 2002, 106, 12191–12201. 10.1021/jp021522z. [DOI] [PubMed] [Google Scholar]; b Mataga N.; Taniguchi S.; Chosrowjan H.; Osuka A.; Kurotobi K. Observations of the Whole Bell-Shaped Energy Gap Law in the Intra-molecular Charge Separation (CS) from S2 State of Directly Linked Zn-Porphyrin-Imide Dyads: Examinations of Wider Range of Energy Gap (−ΔGCS) for the CS Rates in Normal Regions. Chem. Phys. Lett. 2005, 403, 163–168. 10.1016/j.cplett.2004.12.093. [DOI] [Google Scholar]
- a Hayes R. T.; Walsh C. J.; Wasielewski M. R. Competitive Electron Transfer from the S2 and S1 Excited States of Zinc meso-Tetraphenylporphyrin to a Covalently Bound Pyromellitimide: Dependence on Donor-Acceptor Structure and Solvent. J. Phys. Chem. A 2004, 108, 2375–2381. 10.1021/jp037176i. [DOI] [Google Scholar]; b Fujitsuka M.; Cho D. W.; Tojo S.; Inoue A.; Shiragami T.; Yasuda M.; Majima T. Electron Transfer from Axial Ligand to S1- and S2-Excited Phosphorus Tetraphenylporphyrin. J. Phys. Chem. A 2007, 111, 10574–10579. 10.1021/jp076303y. [DOI] [PubMed] [Google Scholar]; c Rogozina M. V.; Ionkin V. N.; Ivanov A. I. Dynamics of Charge Separation from Second Excited State and Following Charge Recombination in Zinc-Porphyrin–Acceptor Dyads. J. Phys. Chem. A 2013, 117, 4564–4573. 10.1021/jp402734h. [DOI] [PubMed] [Google Scholar]
- Tomin V. I.; Dubrovkin J. M. Kinetics of Anti-Kasha Photoreactions. Direct Excitation of a Higher Excited State. ChemistrySelect 2017, 2, 8354–8361. 10.1002/slct.201701518. [DOI] [Google Scholar]
- Sawaki T.; Ishizuka T.; Namura N.; Hong D.; Miyanishi M.; Shiota Y.; Kotani H.; Yoshizawa K.; Jung J.; Fukuzumi S.; Kojima T. Photocatalytic Hydrogen Evolution Using a Ru(II)-Bound Heteroaromatic Ligand as a Reactive Site. Dalton Trans. 2020, 49, 17230–17242. 10.1039/D0DT03546G. [DOI] [PubMed] [Google Scholar]
- Bolger J.; Gourdon A.; Ishow E.; Launay J.-P. Stepwise Syntheses of Mono- and Di-nuclear Ruthenium tpphz Complexes [(bpy)2Ru-(tpphz)]2+ and [(bpy)2Ru(tpphz)Ru(bpy)2]4+ {tpphz = tetrapyrido[3,2- a:2’,3′-c:3”,2”-h:2”,3′”-j]phenazine. J. Chem. Soc., Chem. Commun. 1995, 1799–1800. 10.1039/C39950001799. [DOI] [Google Scholar]
- Similar dihydrogenation at the pyrazine moiety of the tpphz ligand has been recently reported to a hetero-dinuclear Ru–Pd complex having a tpphz moiety as a bridging ligandPfeffer M. G.; Müller C.; Kastl E. T. E.; Mengele A. K.; Bagemihl B.; Fauth S. S.; Habermehl J.; Petermann L.; Wächtler M.; Schulz M.; Chartrand D.; Laverdière F.; Seeber P.; Kupfer S.; Gräfe S.; Hanan G. S.; Vos J. G.; Dietzek-Ivanšić B.; Rau S. Active Repair of a Dinuclear Photocatalyst for Visible-Light-Driven Hydrogen Production. Nat. Chem. 2022, 14, 500–506. 10.1038/s41557-021-00860-6. [DOI] [PubMed] [Google Scholar]
- Britton H. T. S.; Robinson R. A. CXCVIII.-Universal Buffer Solutions and the Dissociation Constant of Veronal. J. Chem. Soc. 1931, 1456–1462. 10.1039/JR9310001456. [DOI] [Google Scholar]
- Sawaki T.; Ishizuka T.; Kawano M.; Shiota Y.; Yoshizawa K.; Kojima T. Complete Photochromic Structural Changes in Ruthenium(II)-Diimine Complexes, Based on Control of the Excited States by Metalation. Chem.–Eur. J. 2013, 19, 8978–8990. 10.1002/chem.201300437. [DOI] [PubMed] [Google Scholar]
- Milani B.; Anzilutti A.; Vicentini L.; Sessanta o Santi A.; Zangrando E.; Geremia S.; Mestroni G. Bis-Chelated Palladium(II) Complexes with Nitrogen-Donor Chelating Ligands Are Efficient Catalyst Precursors for the CO/Styrene Copolymerization Reaction. Organometallics 1997, 16, 5064–5075. 10.1021/om9703954. [DOI] [Google Scholar]
- a Bolger J.; Gourdon A.; Ishow E.; Launay J.-P. Mononuclear and Binuclear Tetrapyrido[3,2-a:2’,3’-c:3”,2”-h:2’’’,3’’’-j]phenazine (tpphz) Ruthenium and Osmium Complexes. Inorg. Chem. 1996, 35, 2937–2944. 10.1021/ic951436w. [DOI] [Google Scholar]; b Sun Y.; Lutterman D. A.; Turro C. Role of Electronic Structure on DNA Light-Switch Behavior of Ru(II) Intercalators. Inorg. Chem. 2008, 47, 6427–6434. 10.1021/ic800560x. [DOI] [PubMed] [Google Scholar]; c Chiorboli C.; Bignozzi C. A.; Scandola F.; Ishow E.; Gourdon A.; Launay J.-P. Photophysics of Dinuclear Ru(II) and Os(II) Complexes Based on the Tetrapyrido[3,2-a:2’,3’-c:3”,2”-h:2’’’-3’’’-j]phenazine (tpphz) Bridging Ligand. Inorg. Chem. 1999, 38, 2402–2410. 10.1021/ic981284f. [DOI] [Google Scholar]
- Once complex 2-H+ was formed by photochemical reduction of 1-H+ with use of BnOH as a reductant under Ar, only 200 μL of O2 was injected to the headspace of the reaction vessel and then complex 1-H+ was immediately recovered from 2-H+ (Figure S5).
- The reason why the reaction was saturated at 10 h is probably because the catalyst 1-H+ was decomposed due to the photoirradiation (see Figure S7).
- As shown in Figure S2, a couple of complex 1-H+ tends to stack together through π–π interaction of the tpphz-H+ ligands. Thus, the reason why complex 1-H+ shows larger TON for the photocatalytic BnOH oxidation under the diluted conditions is probably that complex 1-H+ forms a dimeric structure in the excited state, disturbing the photocatalytic oxidation.
- Luo Y.-R.Handbook of Bond Dissociation Energy in Organic Compounds; CRC Press LLC: Boca Raton, 2003. [Google Scholar]
- Benzene was probably oxidized through electron transfer using the strong oxidizing ability of the photoexcited 1-H+ to afford benzene radical cation, which undergoes a nucleophilic attack of a water molecule to form phenol as the product.
- a Wang Z.; Hisahiro E. Recent Trends in Phenol Synthesis by Photocatalytic Oxidation of Benzene. Dalton Trans. 2023, 52, 9525–9540. 10.1039/D3DT01360J. [DOI] [PubMed] [Google Scholar]; b Han J. W.; Jung J.; Lee Y.-M.; Nam W.; Fukuzumi S. Photocatalytic Oxidation of Benzene to Phenol Using Dioxygen as an Oxygen Source and Water as an Electron Source in the Presence of a Cobalt Catalyst. Chem. Sci. 2017, 8, 7119–7125. 10.1039/C7SC02495A. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ohkubo K.; Fujimoto A.; Fukuzumi S. Visible-Light-Induced Oxygenation of Benzene by the Triplet Excited State of 2,3-Dichloro-5,6-dicyano-p-benzoquinone. J. Am. Chem. Soc. 2013, 135, 5368–5371. 10.1021/ja402303k. [DOI] [PubMed] [Google Scholar]
- a Demas J. N.; Harris E. W.; McBride R. P. Energy Transfer from Luminescent Transition Metal Complexes to Oxygen. J. Am. Chem. Soc. 1977, 99, 3547–3551. 10.1021/ja00453a001. [DOI] [Google Scholar]; b Abdel-Shafi A. A.; Worrall D. R.; Ershov A. Y. Photosensitized Generation of Singlet Oxygen from Ruthenium(II) and Osmium(II) Bipyridyl Complexes. Dalton Trans. 2004, 30–36. 10.1039/b310238f. [DOI] [PubMed] [Google Scholar]
- Li M. Y.; Cline C. S.; Koker E. B.; Carmichael H. H.; Chignell C. F.; Bilski P. Quenching of Singlet Molecular Oxygen (1O2) by Azide Anion in Solvent Mixtures. Photochem. Photobiol. 2001, 74, 760–764. . [DOI] [PubMed] [Google Scholar]
- A proposed mechanism for oxidation of BnOH by 1O2 is shown in Scheme S1.
- a Pestovsky O.; Bakac A. Reactivity of Aqueous Fe(IV) in Hydride and Hydrogen Atom Transfer Reactions. J. Am. Chem. Soc. 2004, 126, 13757–13764. 10.1021/ja0457112. [DOI] [PubMed] [Google Scholar]; b Rocek J.; Radkowsky A. E. The Role of Chromium(IV) in Oxidations by Chromic Acid. The Oxidative Cleavage of Cyclobutanol. J. Am. Chem. Soc. 1968, 90, 2986–2988. 10.1021/ja01013a059. [DOI] [Google Scholar]; c Ghosh M.; Nikhil Y. L. K.; Dhar B. B.; Sen Gupta S. Mechanism of Alcohol Oxidation by FeV(O) at Room Temperature. Inorg. Chem. 2015, 54, 11792–11798. 10.1021/acs.inorgchem.5b01937. [DOI] [PubMed] [Google Scholar]
- A proposed mechanism of the ring-opening reaction of cBuOH by 1O2 is shown in Scheme S2.
- The assignment of this spectral change to the reaction from 1-H+ to 2-H+ is supported by TD-DFT calculations (Figure S13). In the simulated UV–vis spectra, the red shift of the MLCT band of 2-H+ due to the dihydrogenation is reproduced.
- Lee J.; Selinger H.-H. Quantum Yield of the Ferrioxalate Actinometer. J. Chem. Phys. 1964, 40, 519–523. 10.1063/1.1725147. [DOI] [Google Scholar]
- Sharma N.; Jung J.; Ohkubo K.; Lee Y.-M.; El-Khouly M. E.; Nam W.; Fukuzumi S. Long-Lived Photoexcited State of a Mn(IV)-Oxo Complex Binding Scandium Ions That is Capable of Hydroxylating Benzene. J. Am. Chem. Soc. 2018, 140, 8405–8409. 10.1021/jacs.8b04904. [DOI] [PubMed] [Google Scholar]
- Hadley S. G.; Keller R. A. Direct determination of the singlet → triplet intersystem crossing quantum yield in naphthalene, phenanthrene, and triphenylene. J. Phys. Chem. 1969, 73, 4356–4359. 10.1021/j100846a056. [DOI] [Google Scholar]
- Lakowicz J. R.Principles of Fluorescence Spectroscopy; Springer-Verlag: New York, 2006. [Google Scholar]
- Eigen M. Proton Transfer, Acid-Base Catalysis, and Enzymatic Hydrolysis. Part I: ELEMENTARY PROCESSES. Angew. Chem., Int. Ed. Engl. 1964, 3, 1–19. 10.1002/anie.196400011. [DOI] [Google Scholar]
- Matsubara C.; Kawamoto N.; Takamura K. Oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato-titanium(IV): An Ultra-high Sensitivity Spectrophotometric Reagent for Hydrogen Peroxide. Analyst 1992, 117, 1781–1784. 10.1039/an9921701781. [DOI] [Google Scholar]
- The TONs for the PhCHO production catalyzed by 1-H+ were compared between in the presence and absence of MnO2 as a catalyst of disproportionation of H2O2 to H2O and O2 and they were almost identical (see Figure S20). Thus, the rate-determining step does not involve the recovery process from 2-H+ to 1-H+, and complex 2-H+ is smoothly oxidized either by O2 or H2O2.
- a Tschierlei S.; Presselt M.; Kuhnt C.; Yartsev A.; Pascher T.; Sundström V.; Karnahl M.; Schwalbe M.; Schäfer B.; Rau S.; Schmitt M.; Dietzek B.; Popp J. Photophysics of an Intramolecular Hydrogen-Evolving Ru–Pd Photocatalyst. Chem.–Eur. J. 2009, 15, 7678–7688. 10.1002/chem.200900457. [DOI] [PubMed] [Google Scholar]; b Majewski M. B.; de Tacconi N. R.; MacDonnell F. M.; Wolf M. O. Long-Lived, Directional Photoinduced Charge Separation in RuII Complexes Bearing Laminate Polypyridyl Ligands. Chem.–Eur. J. 2013, 19, 8331–8341. 10.1002/chem.201203786. [DOI] [PubMed] [Google Scholar]
- The excitation energy of the 1(π–π*)* state of the tpphz ligand in 1-H+ is calculated to be 3.26 eV based on the UV–vis and emission spectra of 1-H+ and thus the reduction potential of the 1(π–π*)* state is estimated to be +2.78 V vs SCE using ERed of 1-H+ (−0.48 V vs SCE). The driving force, −ΔGET, for the photoinduced ET to the singlet excited state of 1-H+ from BnOH (EOx = +2.33 V vs SCE) is calculated to be 0.45 eV. The −ΔGBET value for the BET in the radical ion pair is 2.81 eV.
- The reorganization energy (λ) of this system is probably small, since the redox center is located at the π-expanded ligand. Therefore, the driving force of the BET, −ΔGBET, is presumed to be large (2.81 eV; see ref (47)) enough to place the BET in the Marcus inverted region. SeeMarcus R. A. On the Theory of Electron-Transfer Reactions. VI. Unified Treatment for Homogeneous and Electrode Reactions. J. Chem. Phys. 1965, 43, 679–701. 10.1063/1.1696792. [DOI] [Google Scholar]
- Chiorboli C.; Rodgers M. A. J.; Scandola F. Ultrafast Processes in Bimetallic Dyads with Extended Aromatic Bridges. Energy and Electron Transfer Pathways in Tetrapyridophenazine-Bridged Complexes. J. Am. Chem. Soc. 2003, 125, 483–491. 10.1021/ja0284916. [DOI] [PubMed] [Google Scholar]
- Here, the nucleophilicity of the reduced tpphz ligand is assumed to be not so high, since the reduced tpphz ligand is protonated at the terminal diimine moiety and thus formally neutral. In addition, the nitrogen of the central pyrazine moiety in the tpphz ligand is sterically hindered by peri-C–H bonds (Please see Figure S30 and Table S5 in Supporting Information). Thus, the nucleophilic attack by a solvent H2O molecule to the benzyl cation should be preferred over the intrasupramolecular nucleophilic attack of the sterically crowded pyrazine nitrogen of the reduced tpphz ligand due to the high concentration of H2O (∼55 M).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.