

Abstract
Background and purpose
X‐linked Charcot‐Marie‐Tooth disease type 1 (CMTX1) ranks as the second most prevalent hereditary neuropathy and, currently, has no definitive cure. Emerging preclinical trials offer hope for potential clinical studies in the near future. While it is widely accepted that experimental groups in these trials should be balanced for age and gender, there is a current shortfall in data regarding phenotype–genotype correlations. Our aim was to provide a more detailed understanding of these correlations to facilitate the formation of well‐matched patient groups in upcoming clinical trials.
Methods
We conducted a retrospective evaluation of CMTX1 patients from 13 designated reference centers in France. Data on genetics, clinical features, and nerve conduction were systematically gathered.
Results
We analyzed the genotype–phenotype correlations in 275 CMTX1 patients belonging to 162 families and carrying 87 distinct variants. Patients with variants affecting the transmembrane domains demonstrated significantly greater severity, as evidenced by a Charcot‐Marie‐Tooth Examination Score of 10.5, compared to 7.1 for those with intracellular domain variants and 8.7 for extracellular domain variants (p < 0.000). These patients also experienced an earlier age of onset, showed slower ulnar nerve conduction velocities and had more substantial loss of motor amplitude.
Conclusions
This study confirms the presence of a correlation between the mutated protein domain and the clinical phenotype. Patients with a variant in the transmembrane domains demonstrated a more severe clinical and electrophysiological profile. Consequently, the genotype could play a prognostic role in addition to its diagnostic role, and it will be essential to consider this in future clinical trials.
Keywords: Charcot‐Marie‐Tooth, clinical trial, CMTX, connexine 32, hereditary peripheral neuropathy
INTRODUCTION
X‐linked Charcot‐Marie‐Tooth disease (CMTX1) is caused by mutations in the GJB1 gene, which encodes the connexin 32 protein [1]. It is the second most prevalent form of Charcot‐Marie‐Tooth disease (CMT), accounting for approximately 10% of all CMT cases. This is preceded only by CMT1A, which makes up 62% of cases, and followed by CMT2A at 7% [2, 3]. Like other forms of CMT, CMTX1 results in length‐dependent sensory and motor neuropathy. However, due to its X‐linked inheritance pattern, males are generally more severely affected than females [4, 5, 6, 7, 8]. Unique features of CMTX1 include central nervous system manifestations such as stroke‐like symptoms [9, 10, 11, 12, 13], varied motor conduction velocities, and possible motor conduction blocks [6, 14, 15].
Connexin 32 is expressed both in the peripheral and central nervous systems. Like other connexins, it is a membrane protein situated in gap junction channels. This protein serves as a subunit that assembles in groups of six to form a hemichannel, also known as a connexon. These connexons are subsequently transported to the plasma membrane, where they pair with connexons from adjacent cells to create a gap junction channel. This channel structure facilitates direct cellular communication through layers of myelin, enabling the transit of small molecules, such as ions, while preventing the passage of larger proteins.
Over 400 variants of the GJB1 gene have been documented. The majority of these are missense variants, but nonsense and frameshift variants have also been identified [16]. Cases of complete deletion [17, 18] and variations in the non‐coding regions of the gene have also been reported but are rarer [19].
Current data on genotype–phenotype correlations in CMTX1 are limited. Although initial studies hinted at such a correlation [5], subsequent research across multiple cohorts has failed to consistently validate this relationship [8, 20, 21]. A recent large multicenter cohort study by Record et al. [22] found that patients with missense variants in the intracellular domain generally exhibited a milder phenotype compared to those with variants in the various transmembrane domains (TM 1, 2 and 3) and the second extracellular domain (EC 2).
Furthermore, no significant initial clinical or neurophysiological differences were observed between patients categorized as having variants of uncertain significance (VUS) and those deemed to have pathogenic or likely pathogenic variants. However, disease progression did vary significantly over 1 and 2 years of follow‐up, although these differences were no longer evident after 2 years. That suggests that patients with a VUS could potentially be reclassified as patients with likely pathogenic variants.
By conducting this multicenter study within a French cohort, we aimed to deepen our understanding of genotype–phenotype correlations. Our goal was to improve genetic counseling for CMTX1 patients and establish a foundation for upcoming clinical trials.
PATIENTS AND METHODS
Subjects and clinical assessment
We leveraged the same cohort as featured in our previous study [15], and extended it by incorporating data from an additional French center in Lille. Overall, clinical, genetic, and neurophysiological data were collated retrospectively from 13 reference centers specializing in neuromuscular diseases across France. These centers include Marseille, Lyon, Paris Pitié‐Salpêtrière, Paris Kremlin Bicêtre, Strasbourg, Angers, Saint‐Etienne, Nantes, Bordeaux, La Réunion, Limoges, Toulouse, and Lille.
Data elements gathered comprised information on sex, age, age at disease onset, results of clinical examinations, the use of walking aids (including orthoses, canes, crutches, and wheelchairs), history of stroke‐like events, orthopedic surgeries performed on the lower limbs, and the treatment of neuropathic pain with medications such as gabapentin, pregabalin, duloxetine, or transcutaneous electrical nerve stimulation.
Details of the methodologies used for the collection and interpretation of clinical and neurophysiological data are given in our previous publication [15].
Genetic data
We classified the variants according to the international criteria of the American College of Medical Genetics (ACMG), using Franklin Genoox, a computer software allowing automated classification and the classification reported in ClinVar on September 1, 2023.
In order to delineate the different domains of connexin 32, we used the segmentation performed by Bone et al. in 1997 [16]: N‐terminal domain: AA 1–19; first transmembrane domain (TM 1): AA 20–38; first extracellular domain (EC 1): AA 39–73; second transmembrane domain (TM 2): AA 74–92; intracellular domain: AA 93–130; third transmembrane domain (TM 3): AA 131–148; second extracellular domain (EC 2): AA 149–187; fourth transmembrane domain (TM 4): AA 188–207; and C‐terminal domain: AA 208–283.
For specific analyses, we categorized the domains based on their cellular localization: intracellular localization encompassing the N‐terminal, intracellular domain, and C‐terminal (N‐terminal domain + intracellular domain + C‐terminal domain); transmembrane regions including the first, second, third, and fourth transmembrane domains (TM 1 + TM 2 + TM 3 + TM 4); and extracellular regions covering the first and second extracellular domains (EC 1 + EC 2).
In our analyses, we grouped variants classified as pathogenic and likely pathogenic under the ‘pathogenic’ category. Similarly, we combined benign and likely benign variants into a single ‘benign’ category for evaluation.
Statistical analysis
Continuous variables were compared using Student's t‐test or the non‐parametric Wilcoxon rank sum test, depending on the conditions of the application. Analysis of variance, followed by Dunnett's post hoc test, were used to compare multiple continuous variables. Categorical variables were compared using the chi‐squared test or Fisher's test, depending on the application conditions.
Standard protocol approvals, registrations, and patient consents
This study was approved by the Ethics Committee of La Timone (reference PADS22‐172) and conducted in compliance with the Declaration of Helsinki.
Anonymized data not published within this article will be made available by request from any qualified investigator.
RESULTS
A total of 275 patients were identified, comprising 146 females and 129 males from 162 CMTX1‐affected families. These patients carried a range of 87 distinct variants, which included 73 (84%) missense variants, two (2%) nonsense variants, six (7%) frameshift insertion/deletions, (6%) five mutations in the 5′ untranslated region (5'UTR) promoter region, and one (1%) complete coding sequence deletion.
We identified variants distributed across all functional domains of connexin 32 (Figure 1). The 10 most frequently occurring variants accounted for 37% (60 out of 162) of the families in our cohort. These variants were, in descending order of frequency: p.Arg22* (9% of families), p.Arg22Gln (4% of families), p.Val95Met (4% of families), p.Arg164Trp (4% of families), p.Asn205Ser (4% of families), p.Val13Met (3% of families), p.Arg75Trp (3% of families), p.Arg107Trp (3% of families), p.Arg164Gln (3% of families), and p.Arg215Trp (3% of families).
FIGURE 1.
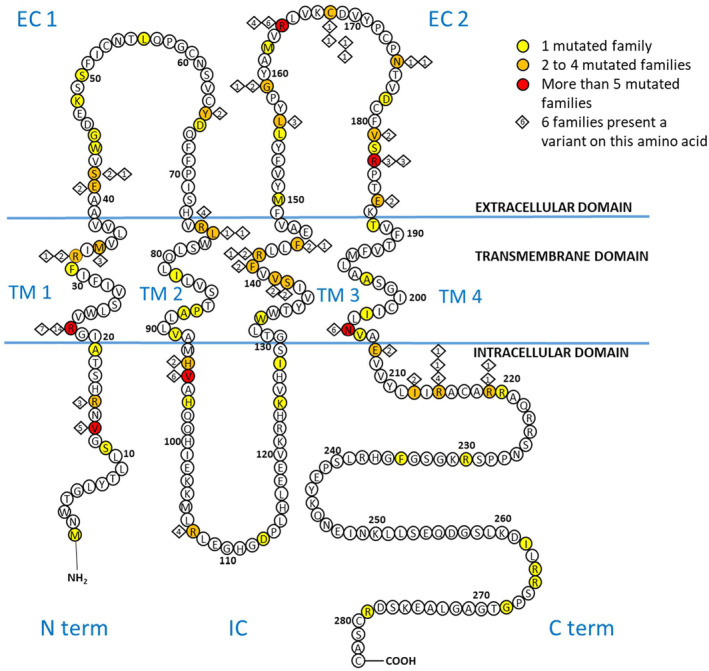
Schema c diagram of gap junction protein beta 1 and GJB variants in our cohort, from Bone et al. [16] and Barbat du Closel [15]. All reported missense, nonsense and frameshift GJB1 mutations are indicated with a colored circle. The five variants in the non‐coding regions and the complete coding sequence deletion (c.1_852del) are not indicated. C term, C‐terminal domain; EC, extracellular domain; IC, intracellular domain; N term, N‐terminal domain; TM, transmembrane.
The primary clinical data have been published in our earlier study. The inclusion of an additional 13 patients does not materially affect the statistical significance of our findings.
Genotype–phenotype correlation by structural domain of the GJB1 gene
To explore the correlation between mutated functional domains and phenotypic presentation, our analysis focused solely on missense variants (230 patients, 134 families, 73 variants). This was to rule out any severity effects attributable to the variant type rather than the functional domain itself.
We identified missense variants across all nine functional domains of connexin 32. The second extracellular domain (EC2) was the most represented, featuring 17 variants (23%) and accounting for 78/230 patients (34%), from 34/134 families (25.4%). Conversely, the N‐terminal domain was least represented, with five variants (7%) affecting 15 patients (7%), from 10/134 families (11.2%).
Figure 2 illustrates the relationship between CMTES (a) and Overall Neuropathy Limitation Score (ONLS) (b) across each functional domain of connexin 32. Interestingly, the intracellular domain, which included 24 patients with seven missense variants, exhibited milder symptoms. The mean CMTES was 6.4, versus 9.3 in patients with missense mutations in other domains (p = 0.004), the ONLS was 2.0, versus 2.9 (p = 0.012); and the average age of disease onset was 25 years, versus 18 years for other domains (p = 0.024). The median nerve motor conduction velocity (MCV) was 42 m/s, versus 39 m/s (p = 0.183) and the ulnar nerve MCV was 44 m/s, versus 42 m/s (p = 0.183). The sum of compound muscle action potentials (CMAPs) was 12.1, versus 10.7 in other domains (p = 0.470). Detailed clinical and neurophysiological data for each of the nine functional domains are provided in Table S1.
FIGURE 2.
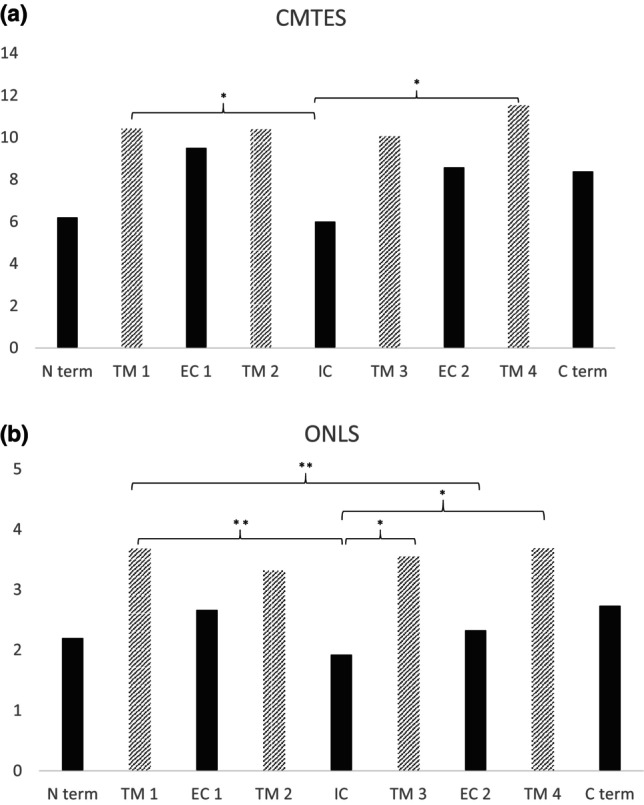
(a) Charcot‐Marie‐Tooth Examination Score (CMTES) and (b) Overall Neuropathy Limitation Score (ONLS) of patients carrying a missense variant, by their mutated protein domain. Transmembrane domains (TM) are depicted with stripes. * Significance at <0.05, ** <0.01. C term, C‐terminal domain; EC, extracellular domain; IC, intracellular domain; N term, N‐terminal domain.
When categorizing the domains into three groups, we identified 22 variants in 64 patients from 42/134 families (31%) in the intracellular domains, 25 variants in 72 patients from 47/134 families (35%) in the transmembrane domains, and 26 variants in 94 patients from 45/134 families (34%) in the extracellular domains (Table 1). Gender distribution and age at evaluation were comparable among these groups. Patients with a missense variant in the transmembrane domains exhibited significantly more severe symptoms than those with variants in either the intracellular or extracellular domains. This was evident in terms of CMTES (10.5 vs. 7.1 and 8.7; p < 0.000), ONLS (3.6 vs. 2.3 and 2.4, p < 0.000), age of disease onset (13 years vs. 22 and 20 years; p < 0.000), and utilization of orthoses (60% vs. 47% and 41%; p = 0.045). There was also a difference among the three groups concerning the conduction velocities of the ulnar (p = 0.025) and median nerves (p = 0.017), which were slower in patients with a missense variant in transmembrane domains, as well as the sum of CMAPs (p = 0.002), which were lower in patients with a missense variant in transmembrane domains. These results are graphically represented in Figure 3.
TABLE 1.
Phenotype of missense variants causing X‐linked Charcot‐Marie‐Tooth disease, stratified by structural domain of connexin 32.
| Intracellular | Transmembrane | Extracellular | p value | |
|---|---|---|---|---|
| Patients, % (n) | 27 (64) | 31 (72) | 41 (94) | |
| Families, % (n) | 31 (42) | 35 (47) | 34 (45) | |
| Variants, % (n) | 30 (22) | 34 (25) | 36 (26) | |
| Male/Female | 30/34 | 35/37 | 41/53 | |
| Age at the evaluation, years | 46 | 48 | 44 | |
| CMTES, mean (±SD) | 7.1 (±0.6) | 10.5 (±0.5) | 8.7 (±0.5) | <0.000 1 (44) |
| ONLS, mean (±SD) | 2.3 (±0.2) | 3.6 (±0.2] | 2.4 (±0.2) | <0.000 1 (44) |
| Age of onset, mean (±SD) years | 22 (±1.8) | 13 (±1.1) | 20 (±1.4) | <0.000 1 (44) |
| Walking aids (cane or walker), % (n) | 11 (7) | 17 (12) | 16 (12) | |
| Orthosis, % (n) | 47 (30) | 60 (43) | 41 (31) | 0.045 |
| Orthopedic surgery of the lower limbs, % (n) | 16 (10) | 17 (12) | 16 (12) | |
| Treatment for neuropathic pain, % (n) | 15 (9) | 24 (16) | 17 (12) | |
| MNVC median nerve, mean (±SD) m/s | 42 (±1.4) | 37 (±1.2) | 38 (±1.4) | 0.017 1 (44) |
| MNCV ulnar nerve, mean (±SD) m/s | 46 (±1.4) | 40 (±1.4) | 43 (±1.4) | 0.025 1 (44) |
| Sum of the CMAPs of the median and ulnar nerves (±SD) mV | 13.1 (±1.2) | 8.1 (±0.9) | 12.0 (±0.9) | 0.002 1 (44) |
| Motor conduction block ≥30% and/or temporal dispersion, % (n) | 50 (18) | 48 (16) | 29 (13) | |
| Distal latency >4 ms, % (n) | 61 (20) | 75 (24) | 53 (23) | 0.037 |
Wilcoxon rank sum test, Pearson’s chi‐squared test or Fisher’sexact test, dedending on the type of variable studied.
Abbreviations: CMAP, compound muscle action potential; CMTES, Charcot‐Marie‐Tooth Examination Score version 2; MNCV, motor nerve conduction velocity; ONLS, Overall Neuropathy Limitation Score; SD, standard deviation.
FIGURE 3.
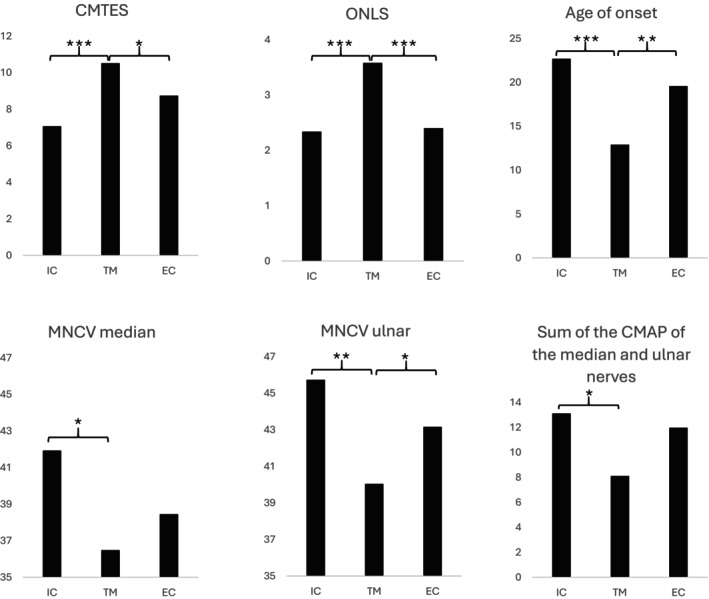
Phenotype of missense variants causing X‐linked Charcot‐Marie‐Tooth disease, by structural domain of the connexin 32. Intracellular domain (IC), including N‐terminal domain: AA 1‐19; IC: AA 93‐130 and C‐terminal domain: AA 208‐283; transmembrane domain (TM), including first TM (TM 1): AA 20‐38, second TM (TM 2): AA 74‐92, third TM (TM 3): AA 131‐148 and fourth TM (TM 4): AA 188‐207. Extracellular domain (EC), including first EC (EC 1): AA 39‐73 and second EC (EC 2): AA 149‐187. * Significance at <0.05, ** <0.01, *** <0.001. CMAP, compound muscle action potential; CMTES, Charcot‐Marie‐Tooth Examination Score; MNCV, motor nerve conduction velocity; ONLS, Overall Neuropathy Limitation Score.
Mutation types and clinical correlations
Apart from the 73 missense variants reported in this cohort, two nonsense (affecting 28 patients from 15 families) and six frameshift variants (affecting nine patients from seven families) were also found. Moreover, we identified five variants in the non‐coding regions such as the 5'UTR (affecting eight patients from five families) and one complete coding sequence deletion affecting a single patient (c.1_852del).
On statistical analysis, no significant differences were observed in the clinical and neurophysiological parameters among these different types of mutations (Table 2).
TABLE 2.
Phenotype of variants causing X‐linked Charcot‐Marie‐Tooth disease, stratified by type of mutation.
| Missense | Nonsense | Frameshift | 5'UTR | |
|---|---|---|---|---|
| Patients, % (n) | 84 (230) | 10 (27) | 3 (9) | 3 (8) |
| Families, % (n) | 83 (134) | 9 (15) | 4% (7) | 3 (5) |
| Variants, % (n) | 84 (73) | 3 (2) | 7 (6) | 6 (5) |
| Male/Female | 106/124 | 14/13 | 3/6 | 5/3 |
| Age at evaluation, years | 46 | 48 | 43 | 38 |
| CMTES, mean | 9 | 11 | 10 | 7 |
| ONLS, mean | 2.8 | 3.5 | 2.9 | 2.3 |
| Age of onset, mean years | 19 | 15 | 15 | 20 |
| MNVC median nerve, mean m/s | 39 | 39 | 38 | 42 |
| MNCV ulnar nerve, mean m/s | 43 | 45 | 35 | 47 |
| Sum of the CMAP of the median and ulnar nerves, mV | 10.8 | 9.4 | 7 | 11 |
Abbreviations: 5′UTR, 5′ untranslated region; CMAP, compound muscle action potential; CMTES, Charcot‐Marie‐Tooth Examination Score version 2; MNCV, motor nerve conduction velocity; ONLS, Overall Neuropathy Limitation Score; SD, standard deviation.
Subgroup analysis
For a more focused analysis, we grouped nonsense variants, frameshift variants and the complete coding sequence deletion, as these should cause loss of function. This subgroup was then compared with the missense variants alone. Again, there were no statistically significant differences in the clinical and neurophysiological parameters between these two groups.
Case Study: Complete deletion
A 27‐year‐old male patient presented with a complete deletion (c.1_852del) in the GJB1 gene. His CMTES was 15 (breakdown: 3 + 1 + 1 + 3 + 4 + 2 + 1) and his ONLS was 3. The patient's symptoms characterized by muscle cramps appeared at age 20 years. Although he used orthoses, he did not require any walking assistance and had not undergone orthopedic surgery on his lower limbs. While the patient displayed tremor in the upper limbs, there were no signs of deafness, scoliosis, or stroke‐like symptoms. His median nerve MCV was 27 m/s and his ulnar nerve MCV was 33 m/s.
Comparatively, his clinical profile did not significantly differ from 16 male patients with missense variants, aged between 22 and 32 years. These findings indicate a lack of significant correlation between the type of mutation and clinical presentation, adding a layer of complexity to our understanding of genotype–phenotype relationships in neuromuscular diseases involving connexin 32 mutations. This could be valuable for future studies aiming to elucidate the underlying mechanisms of variability in clinical outcomes among patients.
5′ untranslated region domain
We identified a total of eight patients in our cohort, five male and three female patients, with a mean age of 38 years, who carried one of five distinct variants in the non‐coding 5'UTR region (c.‐16‐529 T > C, c.‐16‐459C > T, c.‐6G > A, c.‐103C > T, c.‐16‐581G > A). These patients represented 3% of the overall cohort (or 3% of the families) and accounted for 6% of the identified variants. Clinically, these individuals had an average CMTES of 6.9 and an ONLS of 2.6. None of the patients required any form of walking aid. However, three of them utilized ankle‐foot orthoses, and one had undergone orthopedic surgery on the lower limbs. The median nerve MCV was 42 m/s and CMAP was 5 mV, the ulnar nerve MCV was 47 m/s and CMAP was 8 mA.
Variant ACMG class
According to ClinVar, 31 variants (representing 36% of the total) were found in 156 patients from 95 families (58.6%) and were classified as either pathogenic or likely pathogenic. Another 25 variants, representing 29% of the dataset and identified in 70 patients from 35 families (21.6%), were categorized as VUS. Additionally, two variants (or 2% of the dataset) from two patients (two families) were deemed to be benign or likely benign. A total of 29 variants (33%) from 30 families (18.5%) were not listed (Figure 4a) in ClinVar.
FIGURE 4.
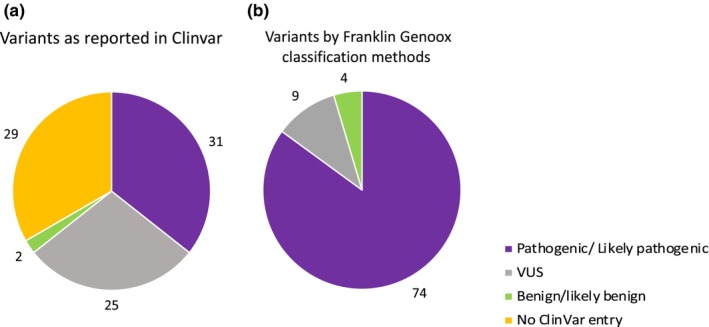
American College of Medical Genetics classification of variants reported on (a) ClinVar or (b) based on their interpretation on Franklin Genoox. VUS, variants of uncertain significance.
Using the Franklin Genoox classification, 74 variants (or 85% of the total), found in 250 patients from 150 families, were classified as either pathogenic or likely pathogenic. Meanwhile, nine variants (or 10% of the dataset), observed in 20 patients from seven families, were classified as VUS. Furthermore, four variants (or 5% of the dataset), found in five patients from five families, were considered benign or likely benign (Figure 4b). Among the 29 variants not listed in ClinVar, Franklin Genoox interpreted 23 variants as pathogenic and six variants as VUS. Additionally, 20 variants classified as VUS in ClinVar were deemed pathogenic by Franklin Genoox. Detailed information on each variant, along with its ACMG classification according to both ClinVar and Franklin Genoox, can be found in Table S2. Interestingly, there were no discernible differences in the physiological and neurophysiological data among the patient groups classified as either pathogenic, VUS, benign, or those not reported in ClinVar. Similarly, no differences were observed among the patient groups classified as pathogenic, VUS, or benign based on the Franklin Genoox classification. The primary data for these groups are presented in Table 3.
TABLE 3.
Phenotype of patients with X‐linked Charcot‐Marie‐Tooth disease, stratified by their American College of Medical Genetics classification based on interpretation on ClinVar or Franklin Genoox.
| ClinVar | Franklin by Genoox | ||||||
|---|---|---|---|---|---|---|---|
| Pathogenic | VUS | Benign | No entry | Pathogenic | VUS | Benign | |
| Patients, % (n) | 57 (156) | 25 (70) | 1 (2) | 17 (47) | 91 (250) | 7 (20) | 2 (5) |
| Variants, % (n) | 36 (31) | 29 (25) | 2 (2) | 33 (29) | 85 (74) | 10 (9) | 5 (4) |
| Male/Female | 78/79 | 26/43 | 0/2 | 23/24 | 120/140 | 6/4 | 3/2 |
| Age at evaluation, years | 43 | 47 | 82 | 44 | 45 | 39 | 69 |
| CMTES, mean | 10 | 8 | 12 | 9 | 9 | 6 | 9.8 |
| ONLS, mean | 3.1 | 2.2 | 3 | 2.7 | 2.9 | 2.1 | 2 |
| Age of onset, mean years | 16 | 22 | 36 | 18 | 18 | 16 | 23 |
| MNVC median nerve, mean m/s | 39 | 43 | 46 | 37 | 39 | 40 | 50 |
| MNCV ulnar nerve, mean m/s | 43 | 49 | 47 | 41 | 43 | 50 | 51 |
| Sum of the CMAP of the median and ulnar nerves, mV | 10.3 | 12.7 | 16.7 | 9.5 | 10.6 | 9.3 | 12.1 |
Abbreviations: CMAP, compound muscle action potential; CMTES, Charcot‐Marie‐Tooth Examination Score version 2; MNCV, motor nerve conduction velocity; ONLS, Overall Neuropathy Limitation Score; VUS, variants of uncertain significance.
Likely benign and benign variants
The two variants classified as benign or likely benign in ClinVar were also categorized as such in Franklin. One was p.Arg230Cys, a missense variant encoding the C‐terminal domain of the protein, found in only one patient in our cohort. This individual was a 73‐year‐old woman who also had an affected daughter, exhibiting a CMTES of 12. We did not have conduction study data for this patient. The other variant classified as benign was c.‐6G > A, a variant encoding the non‐coding 5'UTR domain of GJB1, also found in only one patient in our cohort. This patient was a 72‐year‐old man with a CMTES of 12. Here conduction study showed ulnar and median MCV of 47 m/s.
Two other variants were classified as benign or likely benign in Franklin, and conflicting in ClinVar. The first was p.Ile213Val, a missense variant encoding the C‐terminal domain of the protein, carried by two patients from two unrelated families: a 69‐year‐old woman, also with a symptomatic son, with a CMTES of 12, and a 54‐year‐old man with a CMTES of 8. The second was p.Phe235Cys, also a missense variant encoding the C‐terminal domain of the protein, found in only one patient, a 47‐year‐old man with an affected brother and sister, exhibiting a CMTES of 5, with conduction study showing axonal loss and slowed conduction velocities.
We considered all these variants as causative.
DISCUSSION
This study examines the genotype–phenotype relationships in a cohort of 275 patients from 162 distinct families, who collectively carried 87 different genetic variants. We observed that patients harboring a missense variant in the transmembrane domain exhibited significantly more severe symptoms compared to those with variants in the intracellular and extracellular domains. Importantly, we found no significant differences in clinical or neurophysiological parameters among the various types of mutations. VUS were clinically indistinguishable from those classified as pathogenic or benign.
Understanding these genotype–phenotype correlations serves a dual purpose: it aids in genetic counseling and informs the design of more homogeneous patient groups for clinical trials, particularly in light of promising preclinical studies [23]. While it is well established that clinical trial cohorts should be matched for age and gender [8], our findings suggest that it is equally imperative to consider the specific mutated protein domain when forming comparable patient groups.
Phenotype based on mutated protein domain in connexins
Connexins constitute a family of more than 20 highly conserved membrane proteins [24]. Each connexin protein shares a common topology, including an N‐terminal end, four transmembrane domains, a single intracellular loop, two extracellular loops, and a C‐terminal end. These domains are integral to their role as components of gap junctions [25].
In our study, patients with missense mutations in the transmembrane domains (TM 1 to TM 4) exhibited significantly more severe symptoms. They used more canes or foot drop orthoses and they had more difficulty buttoning shirts. This significance persisted even after adjustments for age and gender. The transmembrane segments of Cx32 are pivotal for its role in gap junctions. They constitute a hydrophobic α‐helical structure, crucial for selective molecular passage between adjacent cells. Mutations in these domains can adversely affect the protein's localization and function or lead to abnormal channel permeability.
Both the intracellular loop and the C‐terminal end, despite being inside the cell, contribute to connexin assembly into hexamers or connexons. They facilitate interactions between adjacent connexins at different assembly stages and are subject to post‐translational modifications such as phosphorylation, affecting channel dynamics. Patients with mutations in these domains displayed milder symptoms, aligning with the study by Record et al. [22], which found a less severe phenotype for patients with intracellular loop mutations compared to those with transmembrane mutations.
The N‐terminal domain also plays a role in connexin assembly but is primarily involved in subcellular localization.
One hypothesis for the milder symptoms in patients with mutations in the intracellular loop and the C‐terminal domains could be partial functional compensation by other intracellular domains.
Lastly, the extracellular loops modulate connexon–connexon interactions via disulfide bonds formed by cysteine residues [25].
Role of the 5'UTR region in Cx32 gene regulation and phenotypic impact in CMTX1 patients
The 5'UTR plays a pivotal role in regulating Cx32 gene expression. This region contains sequences that interact with a variety of transcriptional and translational regulatory factors, thereby influencing the expression levels of the Cx32 protein. In our study, we identified eight patients harboring mutations in the 5'UTR region. Intriguingly, the phenotypes of these patients were indistinguishable from those of other CMTX patients carrying mutations in the coding region. This observation aligns with previous findings [19, 22, 26], lending credence to the notion that these non‐coding variants are clinically significant.
Moreover, these non‐coding variants are not uncommon; they account for up to 11% of cases in the cohort studied by Tomaselli et al. [19]. Given their prevalence and phenotypic similarity to coding mutations, it is imperative to include these 5'UTR variants in systematic screenings when CMTX is suspected. Patients harboring such mutations should also be considered for inclusion in future clinical trials.
These data underscore the importance of the 5'UTR region in the clinical landscape of CMTX1 and suggest the necessity for its inclusion in both diagnostic processes and therapeutic investigations. We did not find any patients in our cohort with variants in the 3'UTR region, although it is reported that variants in this region can cause CMTX1 [19].
Implications of mutation types in clinical and neurophysiological parameters
Our study revealed no discernible differences in a range of clinical and neurophysiological parameters among patients harboring missense, nonsense, or frameshift mutations. This lack of disparity was also observed when comparing a combined group of nonsense and frameshift mutations against missense mutations. These findings lend support to the prevailing hypothesis that missense variants primarily contribute to disease via a loss‐of‐function mechanism, corroborating previous studies in the field [17, 22, 27].
Our data included a 27‐year‐old male patient with a complete deletion (c.1_852del). His clinical and electrophysiological features were congruent with those observed in age‐matched males with missense variants, providing additional validation to the loss‐of‐function hypothesis.
These results not only offer insights into the mechanistic underpinnings of the disease but also suggest the potential utility of gene therapy strategies aimed at functional replacement. Given the consistency in phenotypic expression irrespective of mutation type, gene therapy could emerge as a viable therapeutic avenue for this patient cohort.
This comprehensive analysis underscores the importance of understanding the types of mutations present in patients, not just for diagnostic purposes, but also for planning targeted therapeutic interventions.
Variants of uncertain significance
In our patient cohort, relying solely on ClinVar classifications for “pathogenic” or “likely pathogenic” would severely limit eligibility for clinical trials: only 156 of 275 patients carrying 31 out of 87 identified variants would qualify. This is particularly problematic considering that 25% of the variants in our cohort are classified as VUS on ClinVar. Such a high proportion of VUS poses significant logistical challenges for clinical trials, especially given the slow progression of the disease, which necessitates larger sample sizes to discern meaningful differences between treatment and control groups [22].
Most of these variants remain in the VUS category mainly because they are unique to single families and lack sufficient functional evidence to support their pathogenicity.
On a positive note, the application of computational classification tools, such as Franklin Genoox, has the potential to ameliorate this issue. In our cohort, such tools increased the number of variants classified as pathogenic from 31 to 74 out of 87, thereby expanding the pool of eligible patients for clinical trials. Moreover, our data show that patients with variants classified as VUS have clinical profiles similar to those with pathogenic variants. This finding aligns well with previous work, notably the study conducted by Record et al. [22]. We can suggest that patients carrying VUS, if the variant segregates in the family and if the clinical presentation is compatible, could be reasonably included in clinical trials without introducing significant bias. While VUS classifications can represent a barrier to research, strategies such as computational reclassification and broader clinical inclusion criteria could mitigate this issue and facilitate the conduct of meaningful clinical trials.
Variants classified as benign or likely benign
Our patients with variants classified as benign or likely benign did exhibit a clinical phenotype and a conduction study consistent with CMTX1. These variants may have been misclassified due to their location: either in the non‐coding 5'UTR domain or in the C‐terminal domain. Variants in the C‐terminal domain, especially beyond amino acid 215, may be located in regions less critical for the protein's essential functions [28], which could explain why these variants are classified as benign. However, we did not find a significant difference in our cohort between the 12 patients carrying a variant beyond amino acid 215 and others. As for variants in non‐coding domains, the significant lack of data could explain why these variants are classified as benign.
Conclusions
Our study, which encompasses a cohort of 275 patients from 162 unrelated families carrying 87 distinct variants, reaffirms the existence of a clear genotype–phenotype correlation. Specifically, we found that patients harboring missense variants in the transmembrane domains demonstrated a more severe clinical and electrophysiological profile compared to those with mutations in either the intracellular or extracellular domains. Considering these findings, it is crucial for upcoming clinical trials to account for the specific mutated protein domain, along with traditional factors such as age and sex, when creating comparison groups. This level of granularity will ensure the formation of more homogeneous groups, thereby increasing the reliability and validity of trial outcomes. Interestingly, our data also suggest that patients with VUS, or those with mutations in the non‐coding 5'UTR domain, do not show clinical disparities compared to other groups. Therefore, it would be prudent to consider including these patients in future trials, thus potentially enlarging the pool of eligible participants. By adopting a more nuanced approach to patient selection, based on these genotype–phenotype correlations, we can design more effective and scientifically rigorous clinical trials. This, in turn, will accelerate our progress toward identifying novel therapeutic interventions for this debilitating conditio
AUTHOR CONTRIBUTIONS
Luce Barbat du Closel: Conceptualization; methodology; data curation; investigation; project administration; writing – review and editing; writing – original draft. Nathalie Bonello‐Palot: Conceptualization; methodology; validation; writing – review and editing. Emilien Delmont: Validation; formal analysis; investigation. Yann Péréon: Investigation; validation. Andoni Echaniz‐Laguna: Investigation; validation. Jean Philippe Camdessanché: Investigation; validation. Aleksandra Nadaj Pakleza: Investigation; validation. Jean‐Baptiste Chanson: Investigation; validation. Simon Frachet: Investigation; validation. Laurent Magy: Investigation; validation. Julien Cassereau: Investigation; validation. Pascal Cintas: Investigation; validation. Ariane Choumert: Investigation; validation. Perrine Devic: Investigation; validation. Sarah Léonard Louis: Investigation; validation. Céline Tard: Investigation; validation. Guilhem Solé: Investigation; validation. Emmanuelle Salort‐Campana: Investigation; validation. Françoise Bouhour: Investigation; validation; writing – review and editing. Philippe Latour: Investigation; validation. Tanya Stojkovic: Investigation; validation; writing – review and editing. Shahram Attarian: Conceptualization; methodology; investigation; supervision; validation; project administration; writing – review and editing; visualization.
CONFLICT OF INTEREST STATEMENT
The authors report no disclosures relevant to the article.
Supporting information
Data S1.
Barbat du Closel L, Bonello‐Palot N, Delmont E, et al. Phenotype–genotype correlation in X‐linked Charcot‐Marie‐Tooth disease: A French cohort study. Eur J Neurol. 2025;32:e16523. doi: 10.1111/ene.16523
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Bergoffen J, Scherer SS, Wang S, et al. Connexin mutations in X‐linked Charcot–Marie–tooth disease. Science. 1993;262(5142):2039‐2042. [DOI] [PubMed] [Google Scholar]
- 2. Murphy SM, Laura M, Fawcett K, et al. Charcot–Marie–tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry. 2012;83(7):706‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fridman V, Bundy B, Reilly MM, et al. CMT subtypes and disease burden in patients enrolled in the inherited neuropathies consortium natural history study: a cross‐sectional analysis. J Neurol Neurosurg Psychiatry. 2015;86(8):873‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. La BN. Maladie de Charcot–Marie‐tooth. Presse Médicale févr. 2009;38(2):200‐209. [DOI] [PubMed] [Google Scholar]
- 5. Hahn AF, Bolton CF, White CM, et al. Genotype/phenotype correlations in X‐linked dominant Charcot–Marie–tooth disease. Ann N Y Acad Sci. 1999;883:366‐382. [PubMed] [Google Scholar]
- 6. Dubourg O, Tardieu S, Birouk N, et al. Clinical, electrophysiological and molecular genetic characteristics of 93 patients with X‐linked Charcot–Marie–tooth disease. Brain J Neurol. 2001;124(Pt 10):1958‐1967. [DOI] [PubMed] [Google Scholar]
- 7. Kleopa KA, Zamba‐Papanicolaou E, Alevra X, et al. Phenotypic and cellular expression of two novel connexin32 mutations causing CMT1X. Neurology. 2006;66(3):396‐402. [DOI] [PubMed] [Google Scholar]
- 8. Panosyan FB, Laura M, Rossor AM, et al. Cross‐sectional analysis of a large cohort with X‐linked Charcot–Marie–tooth disease (CMTX1). Neurology. 2017;89(9):927‐935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Al‐Mateen M, Craig AK, Chance PF. The central nervous system phenotype of X‐linked Charcot–Marie–tooth disease: a transient disorder of children and young adults. J Child Neurol. 2014;29(3):342‐348. [DOI] [PubMed] [Google Scholar]
- 10. Hardy DI, Licht DJ, Vossough A, Kirschen MP. X‐linked Charcot–Marie–tooth disease presenting with stuttering stroke‐like symptoms. Neuropediatrics. 2019;50(5):304‐307. [DOI] [PubMed] [Google Scholar]
- 11. Tziakouri A, Natsiopoulos K, Kleopa KA, Michaelides C. Transient, recurrent central nervous system clinical manifestations of X‐linked Charcot–Marie–tooth disease presenting with very long latency periods between episodes: is prolonged sun exposure a provoking factor? Case Rep Neurol Med. 2020;2020:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stancanelli C, Taioli F, Testi S, et al. Unusual features of central nervous system involvement in CMTX associated with a novel mutation of GJB1 gene. J Peripher Nerv Syst déc. 2012;17(4):407‐411. [DOI] [PubMed] [Google Scholar]
- 13. Vivekanandam V, Hoskote C, Rossor AM, Reilly MM. CNS phenotype in X linked Charcot‐ Marie‐Tooth disease. J Neurol Neurosurg Psychiatry. 2019;90(9):1068.1. [DOI] [PubMed] [Google Scholar]
- 14. Gutierrez A, England JD, Sumner AJ, et al. Unusual electrophysiological findings in X‐linked dominant Charcot–Marie–tooth disease. Muscle Nerve. 2000;23(2):182‐188. [DOI] [PubMed] [Google Scholar]
- 15. Barbat du Closel L, Bonello‐Palot N, Péréon Y, et al. Clinical and electrophysiological characteristics of women with X‐linked Charcot–Marie–tooth disease. Eur J Neurol. 2023;30(10):3265‐3276. [DOI] [PubMed] [Google Scholar]
- 16. Bone LJ, Deschenes SM, Balice‐Gordon RJ, Fischbeck KH, Scherer SS. Connexin32 and X‐linked Charcot–Marie–Tooth Disease. Connexin32 and X‐Linked Charcot–Marie–Tooth Disease. Neurobiology of disease. Vol 4; 1997:221‐230. [DOI] [PubMed] [Google Scholar]
- 17. Ainsworth PJ, Bolton CF, Murphy BC, Stuart JA, Hahn AF. Genotype/phenotype correlation in affected individuals of a family with a deletion of the entire coding sequence of the connexin 32 gene. Hum Genet. 1998;103(2):242‐244. [DOI] [PubMed] [Google Scholar]
- 18. Gonzaga‐Jauregui C, Zhang F, Towne CF, Batish SD, Lupski JR. GJB1/Connexin 32 whole gene deletions in patients with X‐linked Charcot–Marie–Tooth disease. Neurogenetics. 2010;11(4):465‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tomaselli PJ, Rossor AM, Horga A, et al. Mutations in noncoding regions of GJB1 are a major cause of X‐linked CMT. Neurology. 2017;88(15):1445‐1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yuan JH, Sakiyama Y, Hashiguchi A, et al. Genetic and phenotypic profile of 112 patients with X‐linked Charcot–Marie–tooth disease type 1. Eur J Neurol. 2018;25(12):1454‐1461. [DOI] [PubMed] [Google Scholar]
- 21. Gouvea SP, Tomaselli PJ, Barretto LS, et al. New novel mutations in Brazilian families with X‐linked Charcot–Marie–tooth disease. J Peripher Nerv Syst. 2019;24(2):207‐212. [DOI] [PubMed] [Google Scholar]
- 22. Record CJ, Skorupinska M, Laura M, et al. Genetic analysis and natural history of Charcot–Marie–tooth disease CMTX1 due to GJB1 variants. Brain. 2023; 146 (10): 4336‐4349. awad187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kagiava A, Karaiskos C, Lapathitis G, et al. Gene replacement therapy in two Golgi‐retained CMT1X mutants before and after the onset of demyelinating neuropathy. Mol Ther—Methods Clin Dev. 2023;30:377‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Willecke K, Eiberger J, Degen J, et al. Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem. 2002;383(5): 725‐37. doi: 10.1515/BC.2002.076/html [DOI] [PubMed] [Google Scholar]
- 25. Bruzzone R, White TW, Paul DL. Connections with Connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238(1):1‐27. [DOI] [PubMed] [Google Scholar]
- 26. Murphy SM, Polke J, Manji H, et al. A novel mutation in the nerve‐specific 5′UTR of the GJB1 gene causes X‐linked Charcot–Marie–tooth disease. J Peripher Nerv Syst. 2011;16(1):65‐70. [DOI] [PubMed] [Google Scholar]
- 27. Shy ME, Siskind C, Swan ER, et al. CMT1X phenotypes represent loss of GJB1 gene function. Neurology. 2007;68(11):849‐855. [DOI] [PubMed] [Google Scholar]
- 28. Castro C, Gómez‐Hernandez JM, Silander K, Barrio LC. Altered formation of Hemichannels and gap junction channels caused by C‐terminal Connexin‐32 mutations. J Neurosci. 1999;19(10):3752‐3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.