

Abstract
The syndrome of inappropriate antidiuretic hormone secretion (SIADH) is a common cause of euvolemic hyponatremia, resulting from non-osmotic release of antidiuretic hormone (ADH). SIADH is frequently associated with neurological conditions, including traumatic brain injury (TBI). TBI-associated SIADH usually develops within days to weeks and resolves within a few weeks. We present the case of a 74-year-old man who, after a fall resulting in TBI, initially had normal sodium levels. Fifteen days later, he developed moderate-to-severe hyponatremia (120 mmol/l) and significant neurological symptoms. Treatment with urea effectively normalized his sodium levels and resolved symptoms. However, recurrent hyponatremia persisted for over six months whenever urea treatment was discontinued. This unusual duration of TBI-associated SIADH underscores the importance of long-term follow-up in the management of post-traumatic hyponatremia.
LEARNING POINTS
While the syndrome of inappropriate antidiuretic hormone secretion (SIADH) is typically transient, it can persist after even minor head trauma, highlighting the importance of long-term follow-up in cases of post-traumatic hyponatremia.
Urea therapy is effective and well-tolerated for managing chronic hyponatremia in SIADH, offering a sustainable long-term treatment option.
Even mild hyponatremia can lead to subtle but impactful cognitive and motor symptoms.
Keywords: SIADH, hyponatremia, traumatic brain injury, persistent hyponatremia
INTRODUCTION
The syndrome of inappropriate antidiuresis (SIAD) is a well-recognized cause of hyponatremia, most commonly due to excessive and inappropriate secretion of antiduretic hormone (ADH) from the pituitary gland or ectopic sources, a condition known as the syndrome of inappropriate antidiuretic hormone secretion (SIADH)[1]. In a subgroup of patients with SIAD, vasopressin levels are suppressed due to gain-of-function mutations in vasopressin V2 receptors (V2R), a condition called nephrogenic syndrome of inappropriate antidiuresis (N-SIAD). This suppression can also result from direct stimulation of V2R, e.g., by certain drugs[2].
SIADH is commonly associated with neurological conditions, including traumatic brain injury (TBI), where it typically appears within days to weeks post-injury and resolves spontaneously over time[2].
The regulation of ADH can be disrupted in TBI, leading to inappropriate water retention despite normal or low serum osmolality[2]. Although SIADH is generally transient (less than one month)[3], cases have been reported in which it persists for a longer time period in TBI patients[4–7].
This report presents a case of SIADH following minor TBI that lasted for over six months, an unusual duration for this condition. This underscores the importance of considering prolonged hyponatremia in post-traumatic cases.
CASE PRESENTATION
A 74-year-old Caucasian man presented to the emergency department (ED) following an accidental fall down the stairs, resulting in a brief loss of consciousness. Initial findings included wrist-fracture and fractures of the right zygomatic and sphenoidal bones as revealed by a computed tomography (CT) scan (Fig. 1). Blood analysis at this time was normal, including natremia (140 mmol/l). Previous blood analyses also showed normal sodium levels. No further investigations were conducted regarding the etiology of the fall, as it was deemed clearly accidental. The patient underwent a wrist intervention for the fracture using an external fixator and was subsequently discharged promptly.
Figure 1.
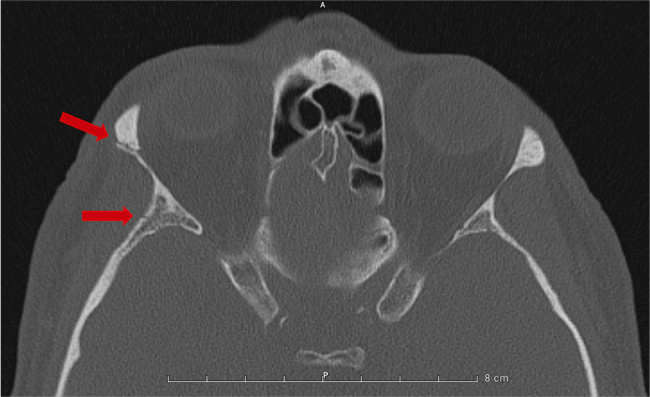
Axial computed tomography scan of the skull in bone window showing fractures. The top red arrow indicates a fracture of the right zygomatic bone, while the bottom red arrow points to a fracture of the right greater wing of the sphenoid bone.
Fifteen days after initial presentation the patient experienced significant phasic disorders characterized by speech difficulties, cognitive impairment, and gait instability, lasting 2–3 hours. A head angio-CT ruled out stroke or carotid dissection. Initial evaluation in the ED revealed moderate-to-severe hyponatremia with a sodium level of 120 mmol/l. Other laboratory values included a urea level of 26 mg/dl, creatinine at 0.7 mg/dl, osmolality at 249 mOsm/kg, and uric acid at 3.9 mg/dl, all of which indicate a normal renal profile (Table 1). Clinical examination indicated a state of euvolemia, with no signs of reduced extracellular fluid volume. Due to the presence of an external fixator on the wrist fracture, an initial brain magnetic resonance imaging (MRI) could not be performed. Hydric restriction was instituted but proved ineffective. Subsequently, treatment with urea 15 g bid was initiated, successfully normalizing natremia. Once the external fixator was removed, a brain MRI was performed, with particular attention given to the posterior pituitary for evidence of structural abnormalities or traumatic lesions affecting ADH release. The MRI findings were normal (Fig. 2). The etiology of the phasic disorders was retained as symptomatic hyponatremia, leading to the final diagnosis of SIADH due to minor head trauma. Notably, renal function remained unchanged throughout the treatment.
Table 1.
Key laboratory values at the time of SIADH diagnosis.
| Parameters | Values | Normal values |
|---|---|---|
| Urea (mg/dl) | 26 | 15–40 |
| Creatinine (mg/dl) | 0.7 | 0.7–1.2 |
| Glomerular filtration rate (CKD-EPI) (mL/min/1.73 m2) | 105 | >60 |
| Sodium (mmol/l) | 120 | 135–145 |
| Potassium (mmol/l) | 4.3 | 3.5–4.8 |
| Chloride (mmol/l) | 82 | 97–109 |
| Osmolality (mOsm/kg H2O) | 249 | 280–308 |
| Glucose (mg/dl) | 113 | 70–100 |
| Uric acid (mg/dl) | 3.9 | 3.4–7 |
| Urine analysis | ||
| Urea (mg/dl) | 696 | N/A |
| Creatinine (mg/dl) | 57 | N/A |
| Sodium (mmol/l) | 53 | N/A |
| Osmolality (mOsm/kg H2O) | 316 | 50–1400 |
| Endocrine biomarkers | ||
| TSH (mU/l) | 1.6 | 0.4–4 |
| T4 (ng/dl) | 1.08 | 0.8–1.8 |
| Cortisol (nmol/l) | 549 | 190–690 |
Abbreviations: SIADH, syndrome of inappropriate antidiuretic hormone secretion; TSH, thyroid stimulating hormone; T4, thyroxine; CKD-EPI, chronic kidney disease epidemiology collaboration equation.
Figure 2.
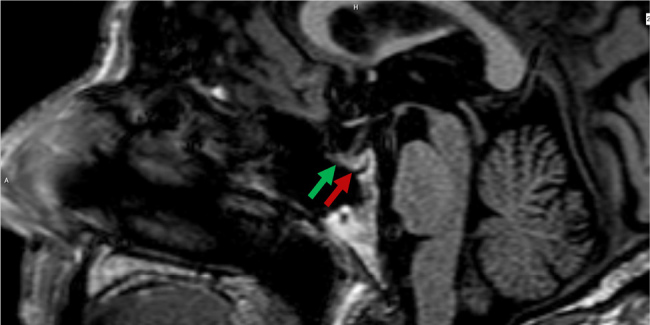
Sagittal T1-weighted magnetic resonance imaging with contrast of the sella turcica. The green arrow indicates the anterior pituitary (adenohypophysis), while the red arrow points to the posterior pituitary (neurohypophysis), which appears normally hyperintense due to typical T1 shortening.
During follow-up, repeated attempts to discontinue urea over the following months resulted in recurrent symptomatic hyponatremia, with sodium levels of 127 mmol/l and 125 mmol/l respectively (Fig. 3). Each recurrence was associated with ataxia and cognitive impairment, including difficulties in sustained attention, focused task performance, and short-term memory deficits. Six months after the initial trauma, the patient continued experiencing these symptoms with each recurrence. After the third attempt to discontinue urea seven months post-injury, natremia stabilized within normal limits (Fig. 3), and no new symptomatic episodes or biochemical abnormalities occurred during the subsequent one-year follow-up.
Figure 3.
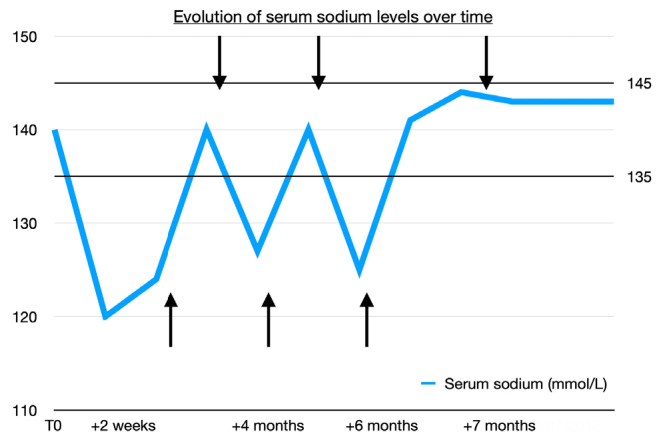
Evolution of serum sodium levels over time in a patient with post-traumatic syndrome of inappropriate antidiuretic hormone secretion. The upward arrows mark the initiation of oral urea therapy at 15 g bid, while downward arrows indicate attempts to discontinue treatment. T0 represents the initial measurement at the time of trauma. Upward arrows: initiation of treatment. Downward arrows: discontinuation attempts.
DISCUSSION
This 74-year-old patient developed SIADH following TBI, which persisted for more than six months, an unusually long duration for this condition. ADH (vasopressin), released by the posterior pituitary gland, can be secreted in dissociation of serum osmolality or circulating volume under pathological circumstances, leading to SIADH. This condition is characterized by low serum osmolality (<275 mOsm/kg), inappropriately high urine osmolality (>100 mOsm/l), high urine sodium concentration (>30 mmol/l), and a state of clinical euvolemia. It is essential to verify normal salt and water intake and exclude adrenal, thyroid, pituitary, or renal insufficiency, as well as diuretic use[2].
SIADH can result from various acquired or hereditary conditions, medications, and sometimes idiopathic causes. Malignancies and neurologic conditions, including head trauma, are among the leading causes[2]. The literature reports a broad range of prevalence rates for hyponatremia following TBI, with estimates around 13%[8]. In a significant proportion of cases, the hyponatremia is attributed to SIADH after ruling out other causes such as cerebral salt-wasting syndrome (CSWS), glucocorticoid deficiency, medications, and excessive fluid intake. In our case, these alternative causes were carefully excluded through comprehensive biological testing and clinical evaluation. Since CSWS can present with laboratory findings similar to SIADH, we assessed the effective circulating volume, urine output, and blood pressure. The absence of worsening renal function or signs of hypovolemia during treatment further ruled out CSWS. Post-traumatic SIADH typically appears in the first three weeks post-trauma[8] and is usually transient, resolving within three weeks[3]. In TBI, it is hypothesized that damage to the posterior pituitary or pituitary stalk can cause excessive ADH release, which may later resolve as the injury heals and inflammation decreases[8]. This disruption may occur because brain injuries can alter the excitatory and inhibitory inputs that regulate ADH release, which originate from osmoreceptors in the anterior hypothalamus and cardiovascular centers in the brainstem[9]. Such injuries can lead to ADH hypersecretion due to reduced tonic inhibitory inputs. This excess ADH subsequently impairs renal free water excretion, maintaining a persistent antidiuretic effect. As a result, urine osmolality remains inappropriately high, a hallmark of SIADH. Hyponatremia progresses until the body adapts by reducing the expression of V2R and aquaporin-2 water channels in the kidney[2]. Following this adaptation, hyponatremia typically stabilizes, although sodium levels may remain below normal until the underlying damage subsides.
Fluid restriction is the first-line treatment for chronic hyponatremia, though its effectiveness and patient tolerance are often limited[2]. Urea is the preferred second-line therapy and is suitable for long-term use to correct hyponatremia, while also reducing intracranial pressure and offering protection against osmotic demyelination syndrome[10,11]. Acting as an osmotic diuretic, urea promotes diuresis and decreases urinary sodium excretion[11].
On the CT scans performed at presentation and during the episode of phasic disorder, no intracranial traumatic lesions were found in our patient. In contrast, post-traumatic SIADH has been shown to correlate with more severe injury patterns on CT scans, as classified by the Marshall classification[3]. However, no correlation has been demonstrated between SIADH and the severity of the underlying TBI based on the Glasgow coma scale[3,8]. Regarding MRI, limited retrospective data suggested that the absence of the normal hyperintense signal of the posterior pituitary correlates with SIADH[4]. This was not observed in our patient, although it should be noted that the MRI was performed more than six weeks after the initial trauma due to the presence of an external fixator.
In severe trauma, TBI-induced SIADH symptoms can be masked by severe head injury, potentially leading to a missed diagnosis. Outside of severe cases, manifestations can be quite subtle. Generally, the severity of hyponatremia influences the range of clinical symptoms, which may vary from subtle to life-threatening (e.g. coma, seizures, cardiorespiratory distress)[2]. Even mild hyponatremia (<132 mmol/l), often considered asymptomatic, can cause subtle neurological symptoms that may go undetected, such as gait and attention impairments, significantly increasing the risk of falls and fractures[2]. Our patient initially exhibited speech impediment, cognitive impairment, and gait disturbance associated with moderate-to-severe hyponatremia. Upon relapse with moderate hyponatremia at more than six months following the initial trauma, symptoms included balance issues, difficulties with focused task performance, and short-term memory deficits. These manifestations align with the spectrum of neurological and cognitive symptoms that can occur with hyponatremia.
As noted earlier, TBI-induced SIADH is typically transient, with cases extending beyond six months being exceptional. Our case illustrates a rare instance of prolonged SIADH, as documented in a few reports[4–7]. This underscores the importance of considering long-term follow-up for post-traumatic hyponatremia disorders.
CONCLUSION
While TBI-induced SIADH is common and generally resolves within weeks, cases extending beyond six months are rare. Our case illustrates a prolonged course of SIADH following minor head trauma, highlighting the importance of long-term follow-up in managing post-traumatic hyponatremia.
Footnotes
Conflicts of Interests: The Authors declare that there are no competing interests.
Patient Consent: Written informed consent for the collection of data and publication of this report was obtained from the patient.
REFERENCES
- 1.Adrogué HJ, Madias NE. The Syndrome of Inappropriate Antidiuresis. N Engl J Med. 2023;389:1499–1509. doi: 10.1056/NEJMcp2210411. [DOI] [PubMed] [Google Scholar]
- 2.Spasovski G, Vanholder R, Allolio B, Annane D, Ball S, Bichet D, et al. Clinical practice guideline on diagnosis and treatment of hyponatraemia. Nephrol Dial Transplant. 2014;29:i1–39. doi: 10.1093/ndt/gfu040. [DOI] [PubMed] [Google Scholar]
- 3.Léveillé E, Aljassar M, Beland B, Saeedi RJ, Marcoux J. Determinants of hyponatremia following a traumatic brain injury. Neurol Sci. 2022;43:3775–3782. doi: 10.1007/s10072-022-05894-3. [DOI] [PubMed] [Google Scholar]
- 4.van der Voort S, de Graaf J, de Blok K, Sekkat M. Persevering syndrome of inappropriate antidiuretic hormone secretion after traumatic brain injury. Neth J Med. 2020;78:290–293. [PubMed] [Google Scholar]
- 5.Dick M, Catford SR, Kumareswaran K, Hamblin PS, Topliss DJ. Persistent syndrome of inappropriate antidiuretic hormone secretion following traumatic brain injury. Endocrinol Diabetes Metab Case Rep. 2015;2015:150070. doi: 10.1530/EDM-15-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang CH, Chuang CH, Lee CT, Liao JJ. Recurrent Hyponatremia After Traumatic Brain Injury. Am J Med Sci. 2008;335:390–393. doi: 10.1097/MAJ.0b013e318149e6f1. [DOI] [PubMed] [Google Scholar]
- 7.Kumar PD, Nartsupha C, Koletsky RJ. Delayed Syndrome of Inappropriate Antidiuretic Hormone Secretion 1 Year after a Head Injury. Ann Intern Med. 2001;135:932–933. doi: 10.7326/0003-4819-135-10-200111200-00027. [DOI] [PubMed] [Google Scholar]
- 8.Agha A, Thornton E, O’Kelly P, Tormey W, Phillips J, Thompson CJ. Posterior Pituitary Dysfunction after Traumatic Brain Injury. J Clin Endocrinol Metab. 2004;89:5987–5992. doi: 10.1210/jc.2004-1058. [DOI] [PubMed] [Google Scholar]
- 9.Wong LL, Verbalis JG. Systemic diseases associated with disorders of water homeostasis. Endocrinol Metab Clin North Am. 2002;31:121–140. doi: 10.1016/s0889-8529(01)00007-x. [DOI] [PubMed] [Google Scholar]
- 10.Wendt R, Fenves AZ, Geisler BP. Use of Urea for the Syndrome of Inappropriate Secretion of Antidiuretic Hormone: A Systematic Review. JAMA Netw Open. 2023;6:e2340313. doi: 10.1001/jamanetworkopen.2023.40313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soupart A, Coffernils M, Couturier B, Gankam-Kengne F, Decaux G. Efficacy and Tolerance of Urea Compared with Vaptans for Long-Term Treatment of Patients with SIADH. Clin J Am Soc Nephrol. 2012;7:742. doi: 10.2215/CJN.06990711. [DOI] [PubMed] [Google Scholar]