

Abstract
Vaccines targeting the complex pre-erythrocytic stage of Plasmodium parasites may benefit from the inclusion of multiple antigens. However, discerning protective effects can be difficult because newer candidates may not be as protective as leading antigens like the circumsporozoite protein (CSP) in the conventional pre-clinical mouse model. We developed a modified mouse model challenge strategy that maximizes the contribution of T cells induced by novel candidate antigens at the sporozoite challenge time point and used this approach to test Plasmodium P36 and P52 vaccine candidates alone and in concert with non-protective doses of CSP. Co-administration of P36 and/or P52 with CSP achieved 80-100% sterile protection in mice, compared to only 7-30% protection for each individual antigen. P36 and P52 vaccination induced murine CD4+ and CD8+ T cell responses, but not antibody responses. This study adds P36 and P52 as promising vaccine antigens that may enhance the protection achieved by CSP vaccination.
Subject terms: DNA vaccines, Malaria
Introduction
Malaria is a potentially deadly parasitic disease caused by Plasmodium species that are spread across half of the globe by female Anopheles mosquitoes. Malaria causes an estimated 249 million annual cases and 608,000 deaths globally1. The emergence of drug and insecticide resistance highlights the urgent need for a highly effective malaria vaccine. RTS,S is a first-generation subunit malaria vaccine that targets the P. falciparum circumsporozoite protein (CSP) and is approved for use in humans, but RTS, S shows lower than desired efficacy and durability2. The newer R21 vaccine targets CSP and achieves improved efficacy of ~70%3–5, but this level of protection is still below WHO target threshold of 90% [https://www.who.int/publications/i/item/ 9789240057463]. Developing more efficacious vaccines may hinge on adding more protective antigens to boost the efficacy of CSP-containing vaccines. Although whole parasite vaccine (WPV) approaches have shown complete protection in pre-clinical and clinical trials6–17, WPV is hampered by complex vaccine production requirements and a need for direct venous administration. Therefore, to develop a more easily manufactured and simply administered vaccine, it is necessary to identify and credential additional novel protective candidate malaria antigens.
Complete protection against malaria can be achieved by targeting the parasite’s pre-erythrocytic stage6,18. Protective immune mechanisms at this stage include antibody responses that can neutralize the parasite and prevent hepatocyte infection, and CD8+ T cells that can kill infected hepatocytes to arrest Plasmodium growth during this pre-erythrocytic stage and confer sterile protection17,19,20. CD4+ T cells are also crucial for maintaining protective antibodies and CD8+ T cell responses14,17,21,22. Therefore, the selection of candidate antigens that utilize either or both arms of the immune system can more effectively protect the host. Early arresting radiation-attenuated sporozoites (RAS) and genetically attenuated parasites (GAPs) have achieved complete protection in pre-clinical and clinical models and rely on both humoral and cellular arms of the immune system6,10,16,23–26. Protective candidate antigens from the early pre-erythrocytic stage may be broadly protective across different Plasmodium species due to amino acid sequence conservation of immunogenic regions of candidate antigens27,28. The selected candidate antigen’s expression timing, concentration, and criticality of each antigen in the parasite life cycle all affect whether swift reactivation and targeting by the host immune cells will lead to protection. Based on these criteria, we evaluated P. yoelii P36 and P52 proteins as potential vaccine antigens.
P36 and P52 proteins are required by sporozoites (spz) for invasion of hepatocytes and establishment of the parasitophorous vacuole where the pre-erythrocytic parasite resides29. Parasites deficient in P36 and/or P52 lose their ability to develop in hepatocytes30–32. Abundant and early expression of P36 and P52 by spz make them good potential targets for the immune system. However, targeting these proteins in the spz stage is complicated for the host immune system as they are exposed to the immune system only after spz begin to invade host cells29. After spz invades host cells, P36 and P52 may be processed and presented by host cells for T-cell targeting. Because of their conserved functions and restricted exposure to the immune system, P52 and P36 are less variable27,33. Here, we hypothesized that targeting P36 and P52 with antibodies would block the invasion of hepatocytes. Furthermore, we hypothesized that after the invasion of hepatocytes, P36, P52, and CSP could singly or together be targeted by host T cells, and targeting these proteins in an orchestrated manner could help neutralize the parasite. Therefore, we screened vaccines based on individual or combined antigens for protective efficacy.
A pre-clinical limitation for credentialing new protective candidate antigens is the traditional mouse malaria models34,35. Plasmodium parasites that naturally infect mice (i.e., P. yoelii, P. berghei) complete their pre-erythrocytic stage in 2–2.5 days, as compared to 5-6 days for P. falciparum in humans. In mice, CD8+ T cells are critical to pre-erythrocytic vaccines but may not fully exert their protective effects in the short-duration pre-erythrocytic stage as it takes more than two days to recruit such cells to the liver from draining lymphoid organs6,36. This may be why some investigators have reported very high requirements for antigen-specific CD8+ T cells in mouse models of malaria37. Since both murine and human hosts have similar immune response kinetics, human T cells are much more activated, developed, and expanded during the 5-6 day human pre-erythrocytic Plasmodium infection as compared to the shorter infection in mouse models6. Humanized liver mice support P. falciparum infection and are being used to mimic the human liver stage of infection38, however humanized mice with human liver cells are immunocompromised, which eliminates their use for vaccine studies. Instead, we designed an alternative two-dose challenge strategy with P. yoelii (Py) using conventional inbred BALB/cJ mice to better evaluate protective outcomes of candidate antigens. In this two-dose challenge strategy, we maximize the participation of candidate antigens-specific T cells in the liver against Plasmodium at the challenge time point based on the T cells kinetics reported earlier by us and others6,39–42.
In this study, mice were vaccinated by subunit delivery of plasmid DNA encoding candidate antigen(s) by gene-gun (GG) to prime antigen-specific T-cell responses in the host. Four weeks later, we reactivated the peripherally GG-primed antigen-specific T cells with a low dose (2 × 103, 2 K) of radiation-attenuated sporozoites (RAS) of Py to initiate T cell recruitment to the liver. Four days after the RAS dose, at a time when T cells were expanded, we challenged mice with a high dose (1 × 104, 10 K) of wild-type (WT) Py spz to evaluate the protective potential of candidate antigens. We screened P36 and P52 antigens individually with this strategy and found them to be partially protective (~7% and 27% respectively). When P36 and/or P52 were mixed with a non-protective dose of CSP (20–30%), complete protection was reliably achieved (80-100%). Overall, this approach adds two much-needed antigens to the malaria vaccine pipeline and provides a path for identifying and credentialing candidate pre-erythrocytic antigens.
Results
Two-dose challenge strategy and screening of rationally selected candidate antigens P36 and P52
CD8+ T cell responses have important roles in achieving sterile protection at the Plasmodium pre-erythrocytic stage17,19,20,43,44. Conventional pre-erythrocytic stage challenge strategies have identified relatively few protective candidate antigens such as CSP and TRAP (Thrombospondin-related anonymous protein). Some antigen screening efforts have found multiple immunogenic but non-protective targets35,45,46. It has been reported that protection in mouse models of malaria requires extremely high numbers of CD8+ T cells37. However, we recently showed that the short duration of the murine liver stage of infection could be a major reason for such a high threshold CD8+ T cell requirement against Plasmodium in the mouse model6. Based on the predicted CD8+ T cell kinetics against Plasmodium infection in longer duration liver stages as that seen in humans, we developed a modified mouse two-dose challenge strategy that makes use of expanded antigen specific CD8+ T cell to improve our ability to evaluate the contribution of candidate antigens in protection. Here, we primed the mice with desired candidate antigen(s) using two cartridges of plasmid DNA (0.5 µg plasmid DNA/cartridge) administered by gene-gun on days 0 and 2 to initiate peripheral CD8+ T cell responses. In some cases, administration consisted of fewer cartridges and/or dosing on only day 0 to reduce the immunogenicity of that antigen. Four weeks post-priming, we challenged those mice using a two-dose challenge strategy (Fig. 1a). For the two-dose challenge used herein, mice first received a low dose of 2 K P. yoelii-radiation attenuated sporozoites (Py-RAS) to reactivate and recruit the peripherally-primed antigen-specific CD8+ T cells to the liver. Four days after the Py-RAS dose, mice were challenged with a stringent high dose of 10 K P. yoelii-wild-type (Py-WT) spz. By this time, GG primed and Py-RAS reactivated antigen-specific CD8+ T cell responses peaked in the liver6. The 10 K wild-type challenge dose at this point is thus used to evaluate the protective efficacy of screening candidate antigen(s) specific CD8+ T cells that are present in the liver.
Fig. 1. Two-dose challenge to test for protection by CSP, P36, and P52 antigens.

a BALB/cJ mice were gene gun primed with DNA encoding the Py circumsporozoite protein (CSP)/P36 protein/P52 protein at days 0 and 2, and were subjected to two-dose challenge followed by blood smears to evaluate for sterile protection. Control group mice received empty vector DNA (VD) immunization via gene-gun, and/or a dose of Py-RAS, or CSP DNA immunization via gene-gun alone, and then were subjected to Py-WT challenge. b Sterile protection outcomes for controls and CSP, P36, and P52 immunized groups. c BALB/cJ mice were either (i) gene gun primed with DNA encoding the CSP protein at days 0 and 2 or (ii) gene gun primed with DNA encoding the CSP protein at day 0, and were subjected to two-dose challenge followed by blood smears to evaluate for sterile protection. d Sterile protection outcomes for control and Py-CSP-immunized groups. VD control group mice protection data acquired from panel b for comparison. e For P36/P52 and CSP co-administration studies, mice were gene gun primed on day 0 with a single cartridge of CSP and one or two cartridges of P36/P52, received two-dose challenge followed by blood smears. Sterile protection outcomes for CSP and P36 (f), CSP and P52, and CSP, P52 and P36 studies (g) compared with CSP protection data from panel d, and P36 or P52 protection data from panel b. h. BALB/cJ mice were gene gun primed 28 days earlier with CSP and P36 or CSP and P52 and were subjected to two-dose challenge. They were injected with CD8 depletion antibodies at indicated days followed by blood smears to evaluate for sterile protection. i. Protection data of CD8+ T cells depleted mice groups compared with protection data from panels f, g. N ≤ 5 mice/group for panels a–g. Data compiled from two or more independent experiments. N = 5 mice/group for panels h and i. Data were analyzed by Fisher Exact test: P < 0.05 is considered significant. *P < 0.05, **P < 0.01, ***P < 0.001 ****P < 0.0001. VD, vector DNA (control).
To assess the validity of this challenge strategy, we first immunized mice against the Py circumsporozoite protein (CSP) using a full gene gun dose (two cartridges per day on days 0 and 2 = ~2 µg CSP DNA). CSP is a dominant protective Py antigen in the BALB/cJ mouse model47–49. We found mice primed with this full CSP dose were all sterilely protected after the two-dose challenge (Fig. 1b). To determine if dose #1 (2K Py-RAS) of the two-dose challenge conferred any non-specific protection against challenge dose #2 (10 K Py-WT), we tested protection by challenging naïve control mice that were gene gun immunized with control vector DNA (VD) alone followed by the two-dose challenge. All (100%) control mice developed blood-stage infections (Fig. 1b). Next, we screened two rationally selected, early pre-erythrocytic stage candidate antigens, P36 and P52. Initially, mice were primed using four cartridges of plasmid DNA in these studies. Both P36 and P52 were partially protective when delivered as single antigen vaccines. P36 sterilely protected 1 of 15 mice (<7% efficacy), and P52 protected 4 of 15 mice (27% efficacy) (Fig. 1b). The observed low but partial protection encouraged further investigation into the ability of P36 and/or P52 to cooperate with CSP antigen-specific responses to as multi-antigen vaccines.
Co-administration of P36 and/or P52 with CSP enhances vaccine-induced protection
To evaluate P36 and P52 with CSP in multi-component antigens, we first needed to find an incompletely protective dose of CSP that could reliably highlight the added protective contribution of partner antigens. Dose de-escalation studies were performed to compare the standard dose of four cartridges (2 µg) of CSP plasmid DNA against fewer cartridges (i.e., less CSP). Two cartridges (1 µg) protected 45% of mice (Fig. 1c.i, d) and one cartridge (0.5 µg) protected only 20-30% mice (Fig. 1c.ii, d). Therefore, we used one cartridge of CSP DNA for immunization in subsequent multi-antigen vaccination studies. In these studies, we combined the single cartridge of CSP DNA with two cartridges of the partner antigen (either P36 and/or P52 encoding plasmid DNA) in initial multi-antigen studies described below; these gene gun vaccines were formulated with each antigen-coding plasmid on different aliquots of gold beads by highly parallel immunization to minimize antigenic competition. Mice were gene gun immunized with candidate antigen DNA followed by 2K Py-RAS dose after four weeks, and a 10 K Py-WT challenge dose four days post Py-RAS (Fig. 1e). Co-administration of a single cartridge of CSP with two cartridges of P36 on the same day sterilely protected 80% of mice (Fig. 1f). Co-administration of a single cartridge of CSP with two cartridges of P52 on the same day sterilely protected all mice (100%) (Fig. 1g). In contrast, co-administration of a single cartridge of each antigen (CSP + P36) and (CSP + P52) only partially protected mice (40% and 20%, respectively) (Fig. 1f, g). However, when mice received a single cartridge of each antigen (CSP, P36, and P52), 90% were sterilely protected (Fig. 1g). To ascertain the CD8+ T cell-dependent nature of protection in these immunized mice groups (CSP + P52 or CSP + P36) with this two-dose challenge strategy, we depleted CD8+ T cells in such mice after vaccination but just before challenging with 10 K Py-WT (Fig. 1h, S Fig. 1). All CD8+ T cell-depleted mice became infected in both vaccination groups (Fig. 1i), which highlights the requirement for CD8+ T cells in these vaccine regimens. The data thus suggests that three early pre-erythrocytic stage antigens, CSP, P36, and P52 can be combined in a synergistic manner to achieve sterile protection against malaria.
CD4+ and CD8+ T cell responses against the candidate antigen proteins, P36 and P52
Since T cells play a critical role in protection at the pre-erythrocytic stage, we further examined T cells in the spleen and liver at the time point corresponding to the wild-type challenge in this two-dose challenge strategy. Mice GG primed with the protective cocktail of either CSP + P52 DNA or CSP + P36 DNA were given 2 K Py-RAS later to reactivate the P36- and P52-specific T cells. Four days after the Py-RAS dose, we harvested spleens and livers, and in vitro stimulated with P52 or P36 proteins to track the presence and activation of T cells by ELISPOT (Fig. 2a, b). Further, using fluorescent markers we looked for the CD4+ and CD8+ T cells identity of those responding cells against the P36 and P52 proteins (Figs. 2c, 3).
Fig. 2. Antigen specific T cell responses against P. yoelii P36 and P52 antigens.

a Mice were immunized with the protective cocktail of DNA encoded CSP and P36/P52 and 4-8 weeks later received 2 K Py-RAS followed by liver/spleen harvest for further cellular analysis by ELISPOT and flow-cytometry as shown. b Splenocyte ELISPOT data for in vitro stimulation with CSP peptide, P36 protein, and P52 protein. c Splenocytes from immunized mice groups depicted in panel a were stimulated overnight in vitro with the P36 (i and iii) or P52 (ii and iv) proteins and were tracked through flow cytometry to identify the activation of (i–ii) CD4+ and (iii–iv) CD8+ Tcm cells using the CD69 marker. A fluorescence minus one (FMO) control was used to gate on CD69 expressing CD8+ and CD4+ T cells (S Fig. 2, panel b). To quantify T cell activation following antigen stimulation, protein antigen was not added to unstimulated wells (UN). The frequency of T cell activation following either P36 or P52 protein stimulation was compared using the graphs at the bottom of each panel using the marker CD69. N = 5 mice per group. Data is representative of two independent experiments. Data are the mean ± SEM. Data were analyzed by the Mann-Whitney test. P < 0.05 is considered significant. *P < 0.05, **P < 0.01.
Fig. 3. Memory T cell recall responses against P. yoelii candidate antigens, P36 and P52.
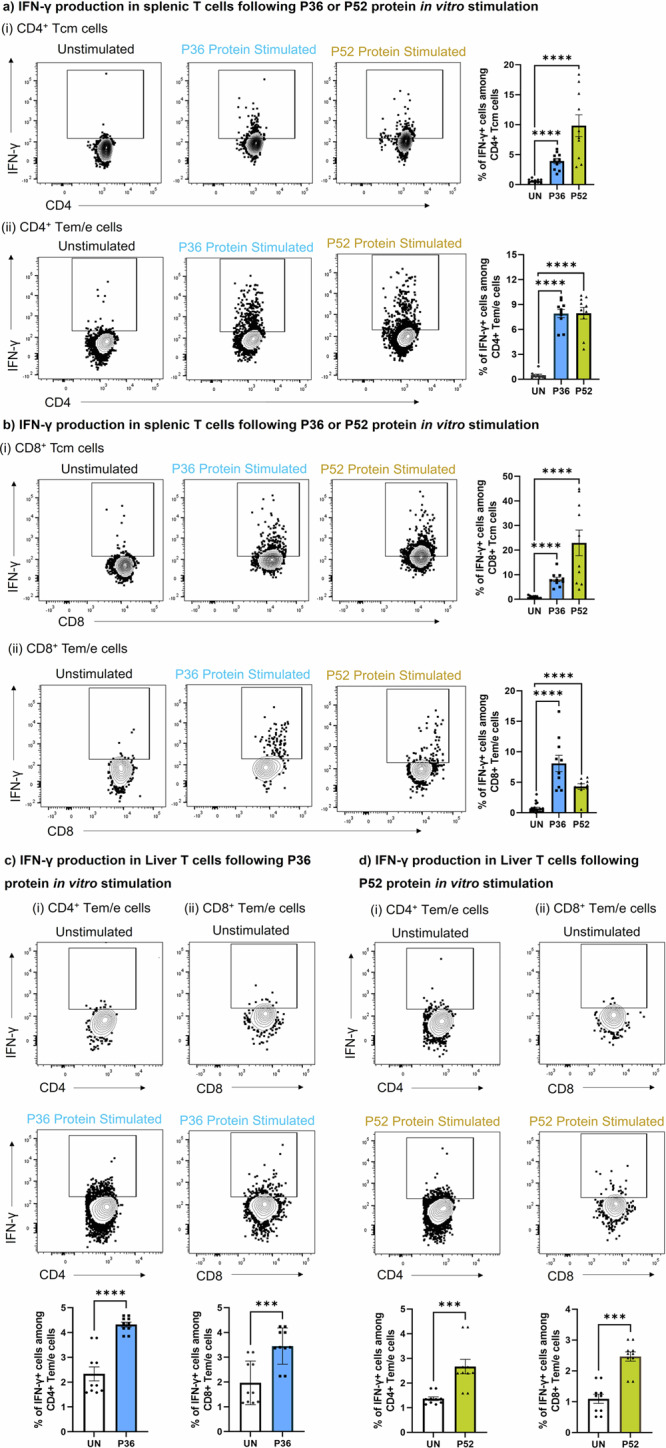
Splenocytes and liver-mononuclear cells from immunized mice groups depicted in panel a of Fig. 2 were stimulated overnight in vitro with the P36 or P52 proteins and were tracked through flowcytometry to identify the activation of CD4+ and CD8+ memory T cells using the IFN-γ marker. Memory populations (Tcm and Tem/e) among CD4+ and CD8+ T cells of spleen (a and b) and liver (c and d) were tracked for IFN-γ production following P36 or P52 protein stimulation. A fluorescence minus one (FMO) control was used to gate on IFN-γ expressing CD8+ and CD4+ T cells (S Fig. 2, panels c and d). To quantify T cell activation following antigen stimulation, protein antigen was not added to unstimulated wells (UN). Frequency of T cell activation following either P36 or P52 protein stimulation was compared using the graphs on right side or at bottom of each panel using the marker IFN-γ. N = 10 mice per group. Data is from two or three independent experiments. Data are the mean ± SEM. Data were analyzed by the Mann-Whitney test. P < 0.05 is considered significant. *P < 0.05, **P < 0.01, ***P < 0.001 ****P < 0.0001.
IFN-γ ELISPOT demonstrated strong T cell responses against both the newly identified antigens P36 and P52, as well as against the co-administered CSP in spleen (Fig. 2b). IFN- γ responses of both the candidate antigens, P36 and P52 were comparable to the dominant antigen CSP. Further using flow cytometry, we looked for the activation (CD69, IFN- γ) and identity of T cells (CD4+ and CD8+) responding to the P36 and P52 antigens. We found that upon in vitro stimulation with either antigens, P36 or P52, both CD4+ (Fig. 2c.i and ii, S Fig. 2b.i) and CD8+ T cells (Fig. 2c.iii and iv, S Fig. 2b.ii) significantly upregulated the surface expression of early activation marker, CD69. Further, we tracked the antigen-specific reactivation of CD4+ and CD8+ memory T cells by intracellular staining for IFN-γ production (Fig. 3a, b, S Fig. 2c, d). For that, we characterized CD4+ and CD8+ T cells into central memory (Tcm) and effector memory/effector (Tem/e) populations (S Fig. 2a). We found that both memory populations in CD4+ and CD8+ T cells produced IFN- γ upon in vitro stimulation with P36 or P52 protein antigens (Fig. 3a, b). Control protein, HIV-I Env gp120 used to stimulate splenocytes of either mouse group cells did not elicit upregulation of CD69 (S Fig. 3a, b) or production of IFN-γ (S Fig. 3c–f). IFN-γ ELISPOT with both mice group cells also showed no stimulatory effect of HIV-I Env gp120 protein (S Fig. 3g). We also reciprocally tested the reactivity of proteins as well as identified immunogenic peptides after immunization of mice with single doses of CSP, P36, or P52 DNA. The IFN-γ responses tested by ELISPOT were antigen-specific (S Fig. 4a–d). We also tested immunization with 20 K PyRAS (a dose 10 times higher than used in our study) and found only CSP-specific responses were detectable (S Fig. 4a–d).
GG DNA immunization in mice is known to generate robust T cell responses against the target protein antigens in lymphoid organs50. However, for protection against pre-erythrocytic Plasmodium infection, T cells residency is required in the liver50,51. Therefore, candidate antigens, P36 and P52 specific T cells likely need to be present in the liver to achieve high rates of protection. We found that liver-recruited P36 (Fig. 3c) and P52 (Fig. 3d) antigen-specific CD4+ and CD8+ Tem/e cells produced IFN-γ upon in vitro stimulation. Thus, the data suggested that DNA immunization induced the P36 and P52 antigen-specific CD4+ and CD8+ T cells in the host, and those cells could be reactivated by an encounter with the parasite (Py-RAS).
Identification of candidate peptides using P36 and P52 peptide library
To identify T cell epitopes of P36 and P52 antigens, we screened overlapping peptide libraries for both proteins by IFN-γ ELISPOT. Mice were GG primed with DNA constructs as in Fig. 4a. T cell responses were boosted by repeated GG immunization two to four weeks after priming. Two weeks after the final booster dose, draining lymphoid organs near the site of immunization (spleen and inguinal lymph nodes) were harvested, and cells were used to screen the peptide library by IFN-γ ELISPOT using pools of 10 peptides, then pools of five peptides, and finally individual peptides based on the ELISPOT results (Fig. 4a). For P36, we identified one candidate peptide (Peptide 71) that responded robustly (VDRDILIYCNCSYNG) (Fig. 4b). For P52, responses were distributed across different pools and among them three pools (Pool A, Pool B and Pool C) of 10 peptides each showed better responses (Fig. 4c). For P52 Pool A, we identified responses to two overlapping candidate peptides (IKHVMKMSFKKMTKK and MKMSFKKMTKKIKGC) and subsequently used these as a pool of two peptides (hereafter called “Pool A*”). For P52 (Pools B and C), we down-selected to 5 peptides per pool (Table S1.a). Earlier, an immunogenic CD8+ T cell epitope of P52 was identified generating IFN-γ responses equivalent to CSP after recombinant adenoviruses immunization in BALB/c mouse, but did not provide any protection against Plasmodium challenge52. In our peptide library screening, we also found that peptide carrying pool responsive, but generated lower responses compared to the selected pools A, B and C. In further studies, we screened P36 Peptide 71 and P52 Pools A* and C for tracking peptide-specific responses as was previously done with full-length proteins (Fig. 4d).
Fig. 4. Identification and tracking of T cell responses against the P36 and P52 candidate peptides.

a Schematics of peptide pooling and screening strategy. For identifying the candidate immunogenic peptides from the P36 or 52 peptide library, BALB/cJ mice were gene gun primed with DNA encoding P36 or P52 on days 0 and 2. Mice were later boosted again with P36 or P52 on day 14 or 28. Finally, mice were sacrificed 14 days post-boosting and splenocytes were subjected to IFN-γ ELISPOT. b Individual mice ELISPOT data for an identified peptide from P36 peptide library (Peptide 71). c Individual mice ELISPOT data was compiled for three identified P52 peptide pools (Pool A, Pool B, Pool C). d BALB/cJ mice were gene-gun primed with protective cocktails of DNA encoding both CSP plus either P36 or P52 at day 0. Mice were later received a reactivation dose of 2 K Py-RAS followed by spleen harvest for IFN-γ ELISPOT of Peptide 71 and Pool A*, Pool C. e Number of IFN-γ positive spots per million splenocytes for P36 Peptide 71, P52 Pool A* and P52 Pool C. f, g Splenocytes from the mice of panel d were in vitro stimulated either with P36 Peptide 71 for CSP + P36 immunized group (f) or with P52 Pool A* and P52 Pool C for CSP + P52 immunized group (g) and Tem/e population among both CD4+ (i) and CD8+ T (ii) cells were tracked for IFN-γ production. For ELISPOT results are normalized against wells treated with equivalent volume of DMSO. For flow cytometry fluorescence minus one (FMO) control was used to gate on the IFN-γ expressing CD8+ and CD4+ T cells (S Fig. 2, panel d). To quantify T cell activation following antigen stimulation, peptide antigen(s) were not added to unstimulated wells. Frequency of T cells activation following either P36 or P52 antigen stimulation was compared with the column graph on right side of each panel using the marker IFN-γ. N = 3-4 mice per group for figures (b, c). N = 5 mice per group for panels d–g. Data are representative from two or three independent experiments. Data are the mean ± SEM. Data were analyzed by the Mann-Whitney test. P < 0.05 is considered significant. * P < 0.05, ** P < 0.01. Panel 4a made with Biorender.
Splenocytes from the immunized mice were used for antigen-specific ELISPOT and flow cytometry (Fig. 2a). IFN-γ ELISPOT demonstrated the presence of T cell responses against P36 Peptide 71 and against P52 Pools A* and C (Fig. 4e). Further we screened those candidates using flow cytometry by tracking antigen-specific IFN-γ production from CD4+ and CD8+ Tem/e cells following in vitro antigen stimulation. For P36 Peptide 71, CD4+ (Fig. 4f.i) and CD8+ Tem/e (Fig. 4f.ii) cells responded. For P52 Pools A* and C, significant responses were also noted for both CD4+ (Fig. 4g.i) and CD8+ Tem/e cells (Fig. 4g.ii).
Lack of measurable antibody responses against the candidate antigens
Antibodies also play an important protective role against the pre-erythrocytic stage infection43,44. Candidate antigens P36 and P52 are highly expressed by the sporozoite stage of the parasite and can be targeted by antibodies preventing the invasion of the parasite into the hepatocyte34. Therefore, we looked for the presence of IgG responses against them after DNA immunization. Mice were GG immunized with protective cocktails of CSP + P36 or CSP + P52 plasmids (1 cartridge of CSP and 2 cartridges of P36 or P52) and after four weeks collected their blood for antibody measurement (Fig. 5a). We screened them for total IgG responses against the repeatless CSP and candidate antigens (P36 and P52) (Fig. 5b, c). There were no detectable IgG responses against P36 (Fig. 5b.i) or P52 (Fig. 5c.i). As expected, there were no detectable IgG responses against CSP in either group since the CSP antigen used herein to induce CD8+ T cell responses did not include the antibody-inducing repeat region (Fig. 5b.ii, c.ii). All IgG positive controls were reactive against their cognate antigens (S Fig. 5). However, it was recently reported that P36, P52, and CSP proteins from Py induce detectable antibody responses after full-length protein immunization34. Therefore, we also tested for antibody responses against P52, P36, and CSP in fully-protected CSP + P52 and CSP + P36 immunized mice four weeks after Py-WT spz challenge (Fig. 5d). Antibody responses were still undetectable against P52 and P36 in these mice, whereas CSP-specific Ab responses were detected (Fig. 5e). These data thus suggest that protection afforded by P36 and P52 is through T cell-based mechanisms.
Fig. 5. Lack of antibody responses to P52, P36, and repeatless CSP proteins.

a BALB/cJ mice were gene gun primed on day 0 with a single cartridge of repeatless CSP DNA and two cartridges of P36 or P52 DNA, and then bled for sera on day 28. b IgG serum ELISA against (i) P36 and (ii) CSP of mice given CSP/P36. Positive controls are shown in S Fig. 5.a, c. c IgG serum ELISA against (i) P52 and (ii) CSP of mice given CSP/P52. Positive controls are shown in S Fig. 5.b, c. d BALB/cJ mice were primed on day 0 with a single cartridge of CSP DNA and two cartridges of P36 or P52 DNA, and then subjected to a two-dose challenge as shown followed by blood collection for sera on day 62. e IgG serum ELISA for mice immunized as in panel d against (i) P36, (ii) P52, and (iii) CSP. Positive controls shown in S Fig. 5.c. N = 5 mice per group. Data is from an experiment. Data are the mean ± SD.
Immunogenicity and identification of candidate antigen peptides for P. yoelii P36 and P52 proteins in the C57BL/6 mouse model
To test the generalizable nature of these antigens, we evaluated immunogenicity of P36 and P52 antigens in the C57BL/6 mouse model. Mice were GG primed with DNA constructs depicted in Fig. 6a. Two weeks post-priming, spleens were harvested, and cells were used to screen P36 and P52 proteins and peptide libraries (5 peptides/pool), and finally individual peptides by IFN-γ ELISPOT (Fig. 6a–d). P52 protein was immunogenic in C57BL/6 mice, whereas P36 protein showed poor immunogenicity (Fig. 6b). This observation was confirmed for both P52 and P36 by screening the 5-peptide pools library. For P52, three pools induced IFN-γ secretion with pool #3 being the most robust (Fig. 6c). Individual peptide screening of pools #1 and #3 showed that pool #1 yielded a single responding peptide (peptide #1-A) “DKFYFYGTPYSSKDI” (Fig. 6d) and pool #3 yielded two responding overlapping peptides (peptide #3-A and 3-B), “NNVEEIAPLNDHYIS” and “EIAPLNDHYISIGDM” (Fig. 6d and Table S1.b). In silico screening of pool #3 responding peptides for MHC Class I peptides (https://services.healthtech.dtu.dk/services/NetMHCpan-4.1/) identified a common responding core peptide “IAPLNDHYI” predicted to have high affinity binding to H-2Db.
Fig. 6. Immunogenicity and identification of candidate antigen peptides of Plasmodium P36 and P52 proteins in C57BL/6 mouse model.

a. C57BL/6 mice were gene-gun primed with DNA encoding P36 or P52 on days 0 and 2. Finally, mice were sacrificed 14 days post priming and splenocytes were subjected to IFN-γ ELISPOT. b. IFN-γ ELISPOT data for candidate antigens P52 and P36 using respective proteins for in vitro stimulation. c. Screening result of responding 5-peptide pools of P52 protein antigen in mice by IFN-γ ELISPOT (Pool #1, Pool #2, Pool #3). d Screening result of responding individual peptide from the 5-peptide pools (1 and 3) of P52 protein antigen in mice by IFN-γ ELISPOT. N = 5 mice per group. Data is from an experiment. Data are the mean ± SEM.
The potent immunogenicity of P52 in two MHC backgrounds and protection in the BALB/cj model supports the evaluation of this class of antigens to potentially improve malaria vaccine protection.
Discussion
Clinical immunity against malaria can be acquired after years of frequent exposures to Plasmodium parasites53, but sterile immunity is difficult to achieve with natural exposures54. However, vaccines can help achieve sterile immunity effectively in a short period55–59. Spz neutralizing Abs and liver infiltrating CD8+ T cells of diverse antigen specificity are critical for sterile protection. Screening and identifying protective antigens using pre-clinal mouse models is a major starting point for malaria vaccine development. Unfortunately, despite sterile protective capabilities of WPV approaches in mice, it has been difficult to define antigens beyond the likes of CSP and TRAP. Among the identified antigens, only CSP is currently used for approved human vaccination, but CSP vaccines currently achieve sub-optimal protective efficacy, which leads us to consider adding other conserved antigens to increase protection. A hurdle for new antigen discovery is the several days difference in the duration of the liver stage between Plasmodium parasites of rodents and those of humans (2-2.5 versus 5-6 days, respectively). In addition, MHC alleles in inbred mouse models are restricted compared to the outbred MHC diversity of humans. Therefore, we need a robust screening strategy in mouse models to prepare a portfolio of candidate antigens for further credentialing and down-selection.
To identify candidate antigens that can be added to CSP vaccines, we leveraged the Py-BALB/cJ mouse model and modified doses and schedules to reduce the protection achieved by the immunodominantCSP protein. By adjusting the dosages and timing of antigens administered in a multi-antigen immunization regimen, we may be able to engineer an ideal vaccine-induced response that can help the host develop a wide pool of expanding T cells that can improve protective outcomes.
Identifying protective antigens is difficult because many antigens are immunogenic but ultimately non-protective in pre-clinical mouse models. Here, we used a two-dose challenge screening strategy to enhance our ability to detect antigens that enhance protection when combined with a CD8+ T cell-inducing CSP vaccine. The two-dose challenge strategy here differs somewhat from our previous report of two-dose challenge. In previous work, doses #1 and #2 were separated by 2-3 days and showed that CD8+ T cells increase during that time period6. In addition, we showed that single immunizations with low doses of RAS could protect against two-dose challenge6. Here, we extended the two-dose challenge interval to four days between doses #1 and #2 and increased the challenge dose 10-fold compared to our prior report. These changes were made to create a model whereby CSP-specific responses could participate in protection but CSP did not achieve complete protection alone. In doing so, we created the conditions whereby the protective effects of P36 and P52 as T cell antigens were more readily detected. The four-day interval between 2 K Py-RAS and 10 K Py-WT sporozoite challenge reported herein provided GG DNA-primed memory CD8+ T cells sufficient time to expand and recruit to the liver. Using this novel approach, we demonstrated that both P36 and P52 can be added to CSP T cell vaccines to achieve 80-100% sterile protection through both CD4+ and CD8+ T cell mechanisms. The strong immunogenicity of P52 in a second MHC-I background in C57BL/6 mice adds to its importance as a potential antigen. Although antibody responses against P52 and P36 antigens were not induced by our immunization strategy, a previous study34 reported that antibodies elicited by co-immunization of CSP and P52 recombinant proteins enhanced sterile protection over CSP alone, suggesting the broad importance of co-immunization in boosting protection across anti-malaria vaccine approaches. Together, these findings strongly suggest that designing a multi-antigen vaccine eliciting both cellular and humoral arms of the immune system is a rational next step in the fight against Plasmodium parasites.
Methods
Mice
Female BALB/cJ and C57BL/6 mice (strain #000651 and 000664, respectively; 4-6 weeks old) were obtained from Jackson Laboratories (Barr Harbor, ME), housed in an IACUC-approved animal facility at the University of Washington and used under an approved IACUC protocol 4317-01 (SCM). Infected mice were euthanized after detection of parasites in blood smears, while protected mice were euthanized after study completion. To euthanize mice, carbon dioxide inhalation was used as per approved IACUC guidelines at the University of Washington.
Plasmodium parasites
Wild-type P. yoelii (Py-WT) 17XNL spz were harvested 14-18 days after an infectious blood meal by salivary gland dissection from infected A. stephensi mosquitoes reared at the Brotman Insectarium, University of Washington and Center for Mosquito Production and Malaria Infection Research (CeMPMIR) at the Center for Global Infectious Disease Research (CGIDR), Seattle Children’s Research Institute. Following dissection, sporozoite purification was conducted prior to irradiation using the Accudenz gradient method60. Briefly, after layering one-part salivary gland spz suspended in Schneider’s media over three parts 17% (w/v) Accudenz and centrifuging as reported, the top one-third of the gradient was transferred into 1.6 mL tubes and centrifuged at 13,300 x g for 4 minutes. Pellets from these tubes were combined, diluted with at least four parts of Schneider’s medium, and the spz were counted using a hemocytometer. Py-WT spz were radiation attenuated (Py-RAS) with 10,000 rads (Rad Source, Suwanee, GA).
DNA cartridges
Plasmodium yoelii antigens P36 (PY01341), P52 (PY01340), and CSP (PY17XNL_000404050 without QGPGAP repeats) were cloned into vectors and then produced as vaccines. The antigen nucleotide sequences were codon optimized for mice (Mus musculus) and commercially synthesized (IDT). The antigens were then restriction-cloned into a vaccine vector (pUb.3) that contains an N-terminal ubiquitin tag. Plasmids were produced in E. coli HST08 and purified using an Endotoxin-free Maxiprep kit (Qiagen). Constructs were verified via Sanger Sequencing (Azenta Life Sciences). Vaccine plasmids plus 1:10 adjuvant plasmid (p7788 LT, encoding Escherichia coli heat-labile toxin) were loaded onto 0.8-1.5 µm gold beads (Technic Inc), and coated onto tubing to produce vaccine cartridges. Each cartridge contained approximately 500 ng of the vaccine plasmid and 50 ng of the adjuvant plasmid. The cartridges were quality controlled by verifying the DNA concentration. These vaccine cartridges were then used for particle-mediated epidermal delivery (PMED) using a PowderJect-style gene gun for needle-free delivery of plasmid DNA into trimmed abdominal skin.
Protein and peptide library production
Plasmodium yoelii proteins, P52, P36 and GP-120 were produced as described previously34. Instruments and reagents used to produce these proteins in mammalian cells were free of any bacterial contaminates. Overlapping peptide libraries for P52 and P36 proteins, 15 mers overlapped by 11 mers, were synthesized by TC Peptide Lab (San Diego, California, USA).
Vaccination and challenge
For sporozoite immunizations, Py-RAS was prepared by exposing Py-WT to 10,000 rads (Rad Source). For spz challenge, Py-WT was sequentially used four days after Py-RAS dosing. Immunization and challenge spz were administered intravenously (retro-orbital vein) in a volume of 100 μL per mouse. Dosages are indicated in separate experiments. For DNA vaccinations, plasmid DNA encoding the antigen of interest, was administered by gene gun either in a single shot or clustered on days 0 and 2.
Blood smear endpoints
From day 4 post-Py-WT challenge, blood collected from mice by tail prick was smeared on glass slides, fixed with methanol, and Giemsa stained for detection of blood-stage Plasmodium parasites. Blood smears were monitored for the presence of parasites under an oil-immersion lens (1000X total magnification); blood collection was stopped either at day 10 post-challenge or when parasites were detected in blood smears. Animals were humanely euthanized upon detection of parasites in blood or at the end of the study with carbon dioxide inhalation.
Quantification of antibody responses
IgG antibody responses against the protein antigens were quantitated by direct-immobilization ELISA as described previously34. Positive control antibodies used for detecting CSP, P36 and P52 were produced and characterized as described previously34,61,62.
IFN-γ ELISPOT
For ELISPOTs, peptides or protein (1 μg/ml final) were combined with 1 × 106 murine splenocytes and incubated for 18 h at 37 °C as reported previously and developed following manufacturer guidelines. The numbers of activated T cells were calculated based on the spot-forming units counted in each well after deducting the spots count from media control wells.
Protein or peptide antigen(s) stimulation and flow cytometry
Splenocytes were isolated and processed as previously described11, and 0.7 × 106 cells in triplicate were stimulated with 2 μg of antigen(s) for overnight. BFA was added 4 hours before processing cells for staining. Cells were blocked with normal mouse serum active as Fc block for 30 minutes. Surface staining was done by incubating cells for 20 minutes on ice with an antibody cocktail specific to cell surface markers (Table S2). Antibodies were diluted in staining buffer containing 50% Brilliant buffer by volume. Cells were then washed with staining buffer. Cells were then fixed and permeabilized using buffers used for intracellular staining as per the manufacturer guidelines. Intracellular staining was done by incubating cells for 30 minutes at room temperature in the dark. Cells were washed thrice with the permeabilization buffer, and then fixed with 1% formaldehyde reagent prepared in stain buffer. Stained cells were acquired on Fortessa instruments (BD Biosciences).
Depletion of immune cells
Immune cell depletion studies were conducted using the antibodies as depicted in Table S3. Antibodies were diluted in PBS and administered intraperitoneally on days −1, +1, +2 and +3 relative to 2 K Py-RAS dose of the two-dose challenge strategy.
Statistics
Data are presented as the mean ± SEM. All comparisons of cell frequency were by the non-parametric Mann-Whitney tests or Fisher Exact test. All statistical analyses were performed using Prism GraphPad Prism 9.1.2 Software (San Diego, CA). Flowcytometry data were analyzed using FlowJo software (v10.10.0).
Supplementary information
Acknowledgements
We thank the insectary teams at SCRI (Kappe and Kaushansky Laboratories) and UW (Pepper/Murphy) for support with sporozoites and Erik D. Layton for help with flow-cytometry data acquisition and suggestions. Funding: This work was partially funded by NIH/NIAID (1R01AI141857 to SCM). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author contributions
Conceptualization: N.Y., S.C.M. Funding acquisition: S.C.M. Investigation: N.Y. Methodology: N.Y., A.K., R.R., A.R. Project administration: N.Y., S.C.M. Supervision: S.C.M. Writing – original draft: N.Y., A.K., S.C.M. Writing – review & editing: N.Y., A.K., R.R., A.R., N.S., S.C.M.
Data availability
All relevant data generated, analyzed, and presented in this manuscript are available on request from the corresponding author.
Competing interests
NY and SCM have an ownership interest in the provisional patent filed for P36 and P52 proteins (Application # PCT/US24/16044, published). ACK, RAR, AR, and NS have no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41541-024-01040-6.
References
- 1.World Health Organization. World Malaria Report 2022. World Health Organization, Geneva (2023).
- 2.RTS,S Clinical Trials Partnership. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet386, 31–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Datoo, M. S. et al. Efficacy of a low-dose candidate malaria vaccine, R21 in adjuvant Matrix-M, with seasonal administration to children in Burkina Faso: a randomised controlled trial. Lancet397, 1809–1818 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Datoo, M. S. et al. Efficacy and immunogenicity of R21/Matrix-M vaccine against clinical malaria after 2 years’ follow-up in children in Burkina Faso: a phase 1/2b randomised controlled trial. Lancet Infect. Dis.22, 1728–1736 (2022). [DOI] [PubMed] [Google Scholar]
- 5.Datoo, M. S. et al. Safety and efficacy of malaria vaccine candidate R21/Matrix-M in African children: a multicentre, double-blind, randomised, phase 3 trial. Lancet403, 533–544 (2024). [DOI] [PubMed] [Google Scholar]
- 6.Yadav, N. et al. More time to kill: A longer liver stage increases T cell-mediated protection against pre-erythrocytic malaria. iScience26, 108489 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schofield, L. et al. Gamma interferon, CD8+ T cells and antibodies required for immunity to malaria sporozoites. Nature330, 664–666 (1987). [DOI] [PubMed] [Google Scholar]
- 8.Weiss, W. R., Sedegah, M., Beaudoin, R. L., Miller, L. H. & Good, M. F. CD8+ T cells (cytotoxic/suppressors) are required for protection in mice immunized with malaria sporozoites. Proc. Natl Acad. Sci USA85, 573–576 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss, W. R. & Jiang, C. G. Protective CD8+ T lymphocytes in primates immunized with malaria sporozoites. PLoS One7, e31247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yadav, N., Parmar, R., Patel, H., Patidar, M. & Dalai, S. K. Infectious sporozoite challenge modulates radiation attenuated sporozoite vaccine-induced memory CD8(+) T cells for better survival characteristics. Microbiol. Immunol.66, 41–51 (2022). [DOI] [PubMed] [Google Scholar]
- 11.Patel, H. et al. Parasite load stemming from immunization route determines the duration of liver-stage immunity. Parasite Immunol.41, e12622 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel, H. et al. Frequent inoculations with radiation attenuated sporozoite is essential for inducing sterile protection that correlates with a threshold level of Plasmodia liver-stage specific CD8(+) T cells. Cell Immunol.317, 48–54 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Lyke, K. E. et al. Attenuated PfSPZ Vaccine induces strain-transcending T cells and durable protection against heterologous controlled human malaria infection. Proc. Natl Acad. Sci. USA114, 2711–2716 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roestenberg, M. et al. Protection against a malaria challenge by sporozoite inoculation. N. Engl. J. Med.361, 468–477 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Roestenberg, M. et al. Long-term protection against malaria after experimental sporozoite inoculation: an open-label follow-up study. Lancet377, 1770–1776 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Hoffman, S. L. et al. Protection of humans against malaria by immunization with radiation-attenuated Plasmodium falciparum sporozoites. J. Infect. Dis.185, 1155–1164 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Seder, R. A. et al. Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science341, 1359–1365 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Fernandez-Ruiz, D. et al. Liver-resident memory CD8(+) T cells form a front-line defense against malaria liver-stage infection. Immunity45, 889–902 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Epstein, J. E. et al. Live attenuated malaria vaccine designed to protect through hepatic CD8+ T cell immunity. Science334, 475–480 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Ewer, K. J. et al. Protective CD8+ T-cell immunity to human malaria induced by chimpanzee adenovirus-MVA immunisation. Nat. Commun.4, 2836 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishizuka, A. S. et al. Protection against malaria at 1 year and immune correlates following PfSPZ vaccination. Nat. Med.22, 614–623 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mordmuller, B. et al. Sterile protection against human malaria by chemoattenuated PfSPZ vaccine. Nature542, 445–449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nussenzweig, R. S., Vanderberg, J., Most, H. & Orton, C. Protective immunity produced by the injection of X-irradiated sporozoites of Plasmodium berghei. Nature216, 160–162 (1967). [DOI] [PubMed] [Google Scholar]
- 24.Dodoo, D. et al. Measuring naturally acquired immune responses to candidate malaria vaccine antigens in Ghanaian adults. Malar. J.10, 168 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gwadz, R. W., Cochrane, A. H., Nussenzweig, V. & Nussenzweig, R. S. Preliminary studies on vaccination of rhesus monkeys with irradiated sporozoites of Plasmodium knowlesi and characterization of surface antigens of these parasites. Bull. World Health Org.57, 165–173 (1979). [PMC free article] [PubMed] [Google Scholar]
- 26.Clyde, D. F., Most, H., McCarthy, V. C. & Vanderberg, J. P. Immunization of man against sporozite-induced falciparum malaria. Am. J. Med Sci.266, 169–177 (1973). [DOI] [PubMed] [Google Scholar]
- 27.Ouattara, A. et al. An in silico analysis of malaria pre-erythrocytic-stage antigens interpreting worldwide genetic data to suggest vaccine candidate variants and epitopes. Microorganisms10, 10.3390/microorganisms10061090 (2022). [DOI] [PMC free article] [PubMed]
- 28.Tucker, K. D. et al. Identification, selection and immune assessment of liver stage CD8 T cell epitopes from Plasmodium falciparum. Front. Immunol.12, 684116 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arredondo, S. A. et al. The micronemal Plasmodium Proteins P36 and P52 act in concert to establish the replication-permissive compartment within infected hepatocytes. Front. Cell Infect. Microbiol8, 413 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.VanBuskirk, K. M. et al. Preerythrocytic, live-attenuated Plasmodium falciparum vaccine candidates by design. Proc. Natl Acad. Sci. USA106, 13004–13009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Schaijk, B. C. et al. Gene disruption of Plasmodium falciparum p52 results in attenuation of malaria liver stage development in cultured primary human hepatocytes. PloS One3, e3549 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishino, T., Chinzei, Y. & Yuda, M. Two proteins with 6-cys motifs are required for malarial parasites to commit to infection of the hepatocyte. Mol. Microbiol58, 1264–1275 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Garzon-Ospina, D., Buitrago, S. P., Ramos, A. E. & Patarroyo, M. A. Identifying potential Plasmodium vivax Sporozoite Stage vaccine candidates: an analysis of genetic diversity and natural selection. Front. Genet. 9, 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vigdorovich, V. et al. Coimmunization with preerythrocytic antigens alongside circumsporozoite protein can enhance sterile protection against Plasmodium Sporozoite infection. Microbiol. Spectr.11, e0379122 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Speake, C. et al. Identification of novel pre-erythrocytic malaria antigen candidates for combination vaccines with circumsporozoite protein. PLoS One11, e0159449 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soulard, V. et al. Plasmodium falciparum full life cycle and Plasmodium ovale liver stages in humanized mice. Nat. Commun.6, 7690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmidt, N. W., Butler, N. S., Badovinac, V. P. & Harty, J. T. Extreme CD8 T cell requirements for anti-malarial liver-stage immunity following immunization with radiation attenuated sporozoites. PLoS Pathog.6, e1000998 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minkah, N. K., Schafer, C. & Kappe, S. H. I. Humanized mouse models for the study of human malaria parasite biology, pathogenesis, and immunity. Front Immunol.9, 807 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whitmire, J. K., Eam, B. & Whitton, J. L. Tentative T cells: memory cells are quick to respond, but slow to divide. PLoS Pathog.4, e1000041 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Osborn, J. F. et al. Central memory CD8+ T cells become CD69+ tissue-residents during viral skin infection independent of CD62L-mediated lymph node surveillance. PLoS Pathog.15, e1007633 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kohlmeier, J. E. et al. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity29, 101–113 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Danahy, D. B. et al. Polymicrobial sepsis impairs bystander recruitment of effector cells to infected skin despite optimal sensing and alarming function of skin resident memory CD8 T cells. PLoS Pathog.13, e1006569 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen, S., Mc, G. I. & Carrington, S. Gamma-globulin and acquired immunity to human malaria. Nature192, 733–737 (1961). [DOI] [PubMed] [Google Scholar]
- 44.Cohen, S., Butcher, G. A. & Crandall, R. B. Action of malarial antibody in vitro. Nature223, 368–371 (1969). [DOI] [PubMed] [Google Scholar]
- 45.Murphy, S. C., Kas, A., Stone, B. C. & Bevan, M. J. A T-cell response to a liver-stage Plasmodium antigen is not boosted by repeated sporozoite immunizations. Proc. Natl Acad. Sci.110, 6055–6060 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Müller, K. et al. Low immunogenicity of malaria pre-erythrocytic stages can be overcome by vaccination. EMBO Mol. Med13, e13390 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li, S. et al. Priming with recombinant influenza virus followed by administration of recombinant vaccinia virus induces CD8+ T-cell-mediated protective immunity against malaria. Proc. Natl Acad. Sci. USA90, 5214–5218 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodrigues, E. G., Zavala, F., Eichinger, D., Wilson, J. M. & Tsuji, M. Single immunizing dose of recombinant adenovirus efficiently induces CD8+ T cell-mediated protective immunity against malaria. J. Immunol.158, 1268–1274 (1997). [PubMed] [Google Scholar]
- 49.Bruna-Romero, O., Gonzalez-Aseguinolaza, G., Hafalla, J. C., Tsuji, M. & Nussenzweig, R. S. Complete, long-lasting protection against malaria of mice primed and boosted with two distinct viral vectors expressing the same plasmodial antigen. Proc. Natl Acad. Sci. USA98, 11491–11496 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olsen, T. M., Stone, B. C., Chuenchob, V. & Murphy, S. C. Prime-and-trap malaria vaccination to generate protective CD8(+) liver-resident memory T cells. J. Immunol.201, 1984–1993 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Lefebvre, M. N. & Harty, J. T. You shall not pass: Memory CD8 T cells in liver-stage malaria. Trends Parasitol.36, 147–157 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mishra, S. et al. Identification of non-CSP antigens bearing CD8 epitopes in mice immunized with irradiated sporozoites. Vaccine29, 7335–7342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doolan, D. L., Dobaño, C. & Baird, J. K. Acquired immunity to malaria. Clin. Microbiol Rev.22, 13–36 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pohl, K. & Cockburn, I. A. Innate immunity to malaria: The good, the bad and the unknown. Front. Immunol.13, 914598 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Richie, T. L. et al. Sporozoite immunization: innovative translational science to support the fight against malaria. Expert Rev. Vaccines22, 964–1007 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gibbins, M. P. et al. Importance of the Immunodominant CD8(+) T Cell Epitope of Plasmodium berghei Circumsporozoite Protein in Parasite- and Vaccine-Induced Protection. Infect. Immun.88, 10.1128/IAI.00383-20 (2020). [DOI] [PMC free article] [PubMed]
- 57.Lu, C. et al. Design and assessment of TRAP-CSP fusion antigens as effective malaria vaccines. PLoS One15, e0216260 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilder, B. K. et al. Anti-TRAP/SSP2 monoclonal antibodies can inhibit sporozoite infection and may enhance protection of anti-CSP monoclonal antibodies. NPJ Vaccines7, 58 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Venkatraman, N. et al. Safety and immunogenicity of heterologous prime-boost immunization with viral-vectored malaria vaccines adjuvanted with Matrix-M. Vaccine35, 6208–6217 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Kennedy, M. et al. A rapid and scalable density gradient purification method for Plasmodium sporozoites. Malar. J.11, 1–10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sack, B. K. et al. Model for in vivo assessment of humoral protection against malaria sporozoite challenge by passive transfer of monoclonal antibodies and immune serum. Infect. Immun.82, 808–817 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carbonetti, S. et al. A method for the isolation and characterization of functional murine monoclonal antibodies by single B cell cloning. J. Immunol. Methods448, 66–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data generated, analyzed, and presented in this manuscript are available on request from the corresponding author.