

Abstract
Spinal cord injury remains a major cause of disability in young adults, and beyond acute decompression and rehabilitation, there are no pharmacological treatments to limit the progression of injury and optimize recovery in this population. Following the thorough investigation of the complement system in triggering and propagating cerebral neuroinflammation, a similar role for complement in spinal neuroinflammation is a focus of ongoing research. In this work, we survey the current literature investigating the role of complement in spinal cord injury including the sources of complement proteins, triggers of complement activation, and role of effector functions in the pathology. We study relevant data demonstrating the different triggers of complement activation after spinal cord injury including direct binding to cellular debris, and or activation via antibody binding to damage-associated molecular patterns. Several effector functions of complement have been implicated in spinal cord injury, and we critically evaluate recent studies on the dual role of complement anaphylatoxins in spinal cord injury while emphasizing the lack of pathophysiological understanding of the role of opsonins in spinal cord injury. Following this pathophysiological review, we systematically review the different translational approaches used in preclinical models of spinal cord injury and discuss the challenges for future translation into human subjects. This review emphasizes the need for future studies to dissect the roles of different complement pathways in the pathology of spinal cord injury, to evaluate the phases of involvement of opsonins and anaphylatoxins, and to study the role of complement in white matter degeneration and regeneration using translational strategies to supplement genetic models.
Keywords: complement, neuroinflammation, neuroplasticity, regeneration, spinal cord injury, targeted therapy
Introduction
The complement (C) system, a key component of the innate immune system, is now a well-recognized contributor to homeostasis, development, plasticity, and pathology in the central nervous system (CNS) (Alawieh et al., 2015a, 2018, 2020; Brennan et al., 2021; Holste et al., 2021; Stevens and Johnson, 2021). The early activation of C as an initiator of the inflammatory response within the injured CNS has positioned this system at the center of numerous prognostic and therapeutic investigations in conditions of ischemic, hemorrhagic, and traumatic insults to the CNS (Peterson and Anderson, 2014; Alawieh et al., 2015a, 2018, 2020; Brennan et al., 2021; Holste et al., 2021; Stevens and Johnson, 2021). Key to the significance of the C system is its dual function as both a recognition arm for the innate immune response as well as a robust immune effector capable of direct cell injury and activation of both the adaptive and innate immune system (Alawieh et al., 2015a; Brennan et al., 2021).
In spinal cord injury (SCI), the interplay between C activation and the neuroinflammatory response remains an active area of basic, clinical, and translational research. In addition to its normal role in homeostatic functions within the normal spinal cord (SC), the C system is well recognized for its role in exacerbating primary injury, amplifying the neuroinflammatory response, and limiting functional recovery after SCI. Eventually, translational efforts have been devoted to targeting different components or pathways of the C system to mitigate damage after SCI (Rebhun et al., 1991; Anderson et al., 2004, 2005; Qiao et al., 2006, 2010; Nguyen et al., 2008; Ankeny et al., 2009; Peterson and Anderson, 2014; Narang et al., 2017; Brennan et al., 2019). In this work, we start by reviewing complement biology and its role in general health and disease. We then report on the sources of complement activation proteins after SCI, the triggers of complement activation, and the role of effector functions in the pathology that can be targeted individually. Then, we provide a critical discussion of current inhibitory strategies used to understand the pathology and investigate translational approaches. We finally discuss the implications of our current understanding of the role of C in SCI on the future therapeutic possibilities.
Data Sources
To complete this review, two independent searches were performed in PubMed, Scopus, Embase, and Web of Science databases using the key (Complement AND “Spinal Cord Injury”) OR (Complement AND “Spinal Cord Trauma”). Original studies published between January 1990 and December 2023 were included in the review. The search key returned 327 studies on PubMed, 384 studies on Scopus, 522 studies on Embase, and 454 studies on Web of Science. After duplicates were removed, and papers were screened for original studies reporting C activation, deposition, or modulation in SCI, 27 original studies were included in this review (Additional Figure 1 (292KB, tif) ). Authors HS, AT, and AA performed the abstract and full-text screen.
Complement System Activation Pathways and Products
The C system involves a series of serum and membrane-bound proteins involved in the innate host defenses, clearance of dying cells, chemotaxis, and modulation of adaptive immune responses, among other homeostatic and physiologic functions (Peterson and Anderson, 2014). C proteins predominantly exit in an inactive form, and they are serially activated via a complex change of extracellular proteases in response to tissue damage or pathogens. Figure 1 describes the general mechanisms of C activation.
Figure 1.
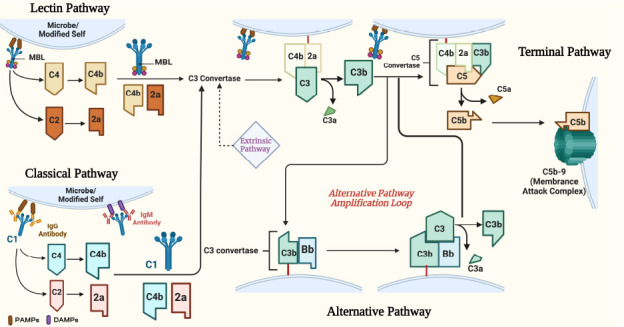
Overview of complement activation pathways.
Created with BioRender.com. C: Complement; DAMPs: damage-associated molecular patterns; MBL: mannose-binding lectins; PAMPs: pathogen associated molecular patterns.
The complement system is activated by one of three pathways: the classical, lectin, or alternative pathways (Figure 1). All three pathways converge at the cleavage of central C protein C3 into C3a and C3b leading to the downstream cleavage of C5 and assembly of the lytic membrane attack complex (MAC) (Figure 1; Alawieh et al., 2015a). The different pathways differ in their activation patterns. The classical complement pathway is initiated by the activation of C1q either by directly binding to pathogen or modified self surfaces, to acute phase reactants, or more commonly to the Fc region of antibodies bound to their target antigens. The binding triggers C1r and subsequently C1s zymogens activation leading to the cleavage of both C2 and C4, releasing C2a and C4a anaphylatoxins and creating the C3 convertase (C4b2a) that is capable of splitting and activating complement protein C3 into membrane-bound C3b and soluble anaphylatoxin C3a (Figure 1; Alawieh et al., 2015a).
The pattern of activation of the lectin pathway is similar and is initiated when soluble defense proteins like ficolins and mannose-binding lectins (MBL) bind to mannose-containing carbohydrate residues on pathogenic or stressed cellular surfaces (Alawieh et al., 2015a). The lectin pathway can also be activated by glycosylated patterns on the Fc regions of antibodies bound to their antigens (Figure 1). Once activated, two serine proteases, MASP-1 and MASP-2, cleave C2 and C4 in a similar fashion as in the classical pathway. The alternative pathway, on the other hand, can be activated by binding factor B to membrane-bound C3b, creating C3 convertase (C3bBb), which cleaves C3 in a similar manner to the classical and lectin pathways leading to the amplification of the effect of these pathways (Anderson et al., 2005). Additionally, the alternative pathway can be spontaneously activated when fluid-phase hydrolyzed C3 binds to surfaces not protected by complement regulators (Alawieh et al., 2015a).
All three pathways converge at the cleavage of the effector molecule (C3) leading to its breakdown into C3a and C3b. C3a is a soluble anaphylatoxin that binds to G-protein coupled receptor C3aR mediating predominantly immune cell chemotaxis and activation (Figure 1; Alawieh et al., 2015a). However, C3b binds covalently to nearby cell membranes and serves as a cell surface opsonin through interaction with complement receptors on phagocytic cells, contributes to the amplification of C3 activation via the alternative pathway, and leads to the assembly of the C5 convertase (Figure 1). Breakdown of C5 results in the release of C5a anaphylatoxin and C5b, which deposits on the cell surface and triggers the assembly of the lytic membrane attack complex (C5b-9, MAC) (Figure 1). Several endogenous regulators exist that protect the host from C-mediated attack by inhibiting the C3 convertase (e.g., Decay-accelerating factor, complement receptor 1-related gene/protein y [Crry], or membrane cofactor protein [CD46]), C1q activation (C1-Inhibitor), the alternative pathway convertase (Factor H) and the membrane attack complex (CD59, Clusterin) (Zipfel and Skerka, 2009)).
Recently, extrinsic pathways of C have been described including direct C activation by the coagulation and fibrinolytic pathways (Amara et al., 2008) and intracellular complement activation in T-cells (Howell et al., 2021; Figure 1).
Complement Activation Following Spinal Cord Injury
The pathophysiology of SCI is characterized by a potent neuroinflammatory response that follows the primary impact exacerbating original damage, promoting secondary injury, and limiting the extent of recovery (Alexander and Popovich, 2009). Within this robust neuroinflammatory response, the C cascade has been well recognized as an early trigger and potential optimal target for treatment. In a network-based analysis of the SCI interactome (Alawieh et al., 2015b), the C pathway was identified as a major upstream protein subnetwork that triggered the activation of the core “rich-club” of the network responsible for the overall pathological phenotype (Alawieh et al., 2015b). Consistent with this finding, early studies in human subjects demonstrated elevated serum complement activity, factor B levels, C4 and C5 levels in subjects with SCI although a correlation with disease severity or outcomes was not studied (Rebhun et al., 1991). Follow-up proteomics analysis of cerebrospinal fluid from patients with severe SCI demonstrated upregulation in both C4 and C2 levels (Sengupta et al., 2014) while proteomic studies on serum markers of patients with SCI demonstrated enrichment in the complement and coagulation pathways among differentially expressed genes (Zhao et al., 2022; Li et al., 2023). In translational models of SCI, almost all components of the C system have been shown to be present in the injury core and perilesional tissue (Additional Table 1 (117.5KB, pdf) ) except for the lectin pathway that is yet to be investigated in the context of SCI.
Sources of C activation products after spinal cord injury
Whereas evidence demonstrating local C activation in the injured SC is robust, the sources of C proteins remain unclear given the immune-privileged status of the normal SC. C proteins are among the most abundant serum proteins whose hematogenous source is predominantly hepatic production (Guo et al., 2023), but local production by both infiltrating immune cells and CNS resident cells has been well demonstrated in different disease models (Alawieh et al., 2015a).
As summarized in Figure 2, three potential sources of C proteins in the injured SC have been proposed including hematogenous and local production. The blood–spinal cord barrier (BSB) maintains the immune-privileged rendering it impermeable to serum-derived C proteins. A disruption of the BSB is likely to explain the rapid deposition of C proteins at the epicenter of the injured SC. Studies on BSB permeability following SCI using both macro and micromolecular tracers in rats demonstrated persistent permeability for 2–4 weeks after injury (Dusart and Schwab, 1994; Popovich et al., 1996). The permeability of BSB progresses cranio-caudally as time from injury elapses and can reach up to 30 mm away from the injury epicenter in rats over several days (Figure 2). Since BSB permeability in the perilesional SC occurs later in the course of the disease, complement immunoreactivity observed at all levels of the SC one day after injury suggests a different source contributing to complement deposition (Anderson et al., 2004). Different C proteins have been shown to be present at both the epicenter and perilesional areas of the SC after injury including C1q, C3/C3b, complement factor B (fB), and C5b-9 (Additional Table 1 (117.5KB, pdf) ; Anderson et al., 2004, 2005; Qiao et al., 2006, 2010; Ankeny et al., 2009; Li et al., 2009; Narang et al., 2017). C3 deposition was noted to peak around 3 days after SCI that corresponds with the timing of peak BSB permeability and immune cell infiltration (Qiao et al., 2006, 2010; Li et al., 2009).
Figure 2.
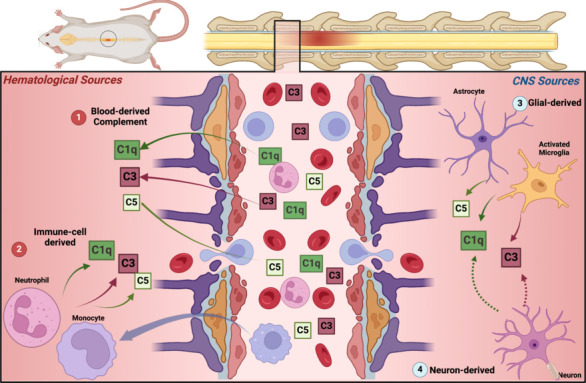
Sources of complement proteins after SCI.
Different C activation products are present in the spinal cord parenchyma after injury from a direct leak through disrupted BSB (1), local production by infiltrating immune cells (2), or production by CNS-resident cells including predominantly microglia and astrocytes (3). Neuronal sources of C proteins are possible (dashed lines) based on other disease models but have not been demonstrated in SCI literature (4). Created with BioRender.com. BSB: Blood–spinal cord barrier; C: complement; CNS: central nervous system; SCI: spinal cord injury.
Evidence of local expression of C in SCI comes from transcriptomics studies (Additional Table 1 (117.5KB, pdf) ). Hao et al. (2018) performed a whole-transcriptome analysis of alterations in gene expression after SCI in mice demonstrating that C3 was among the most upregulated genes in the SC tissue. Along with studies demonstrating upregulation of mRA expression of C1q, C3, C4, C6, MBL, fH, fP, and C5 (Nguyen et al., 2008; Zhao et al., 2019), these studies support local production of C in the SC after SCI. However, the source of local production is potentially related to C production by infiltrating immune cells or local CNS-resident cells (Figure 2 and Additional Table 1 (117.5KB, pdf) ). Nguyen et al. (2008) demonstrated that infiltrating polymorphonuclear cells had upregulation of C1q, C3, and C4 mRNA indicating that at least, in part, infiltrating immune cells contribute to the local C production. Although specific neuroglia or neuronal production of C in the SC has not been reported, indirect evidence supports this assumption. First, neurons, astrocytes, and microglia can produce the full complement of C proteins as shown in models of brain injury and ischemia (Barnum, 1999; Alawieh et al., 2015a). Additionally, immune cell infiltration tends to be more localized to white matter tracts whereas complement deposition was noted to affect both white and grey matter and localize to areas with absent infiltrates (Anderson et al., 2004). In models of dorsal rhizotomy and peripheral nerve transection, Liu et al. (1998) demonstrated expression of C3 mRNA in the dorsal horn even with intact PMBSB supporting CNS-origin of injury-induced C activation. Collectively, these findings support a multifactorial source of C protein access to the SC after injury including both serum-derived, hematogenous immune cell-derived, and local CNS-produced proteins (glial or neuronal) (Figure 2).
Different C activation products were reported after SCI in histological and molecular studies. C3a and C5a were noted to be persistently elevated in both the SC and the periphery for at least 7 days after SCI (Brennan et al., 2019). Complement opsonins C1q and C3 as well as C9 were noted to deposit on neurons and astroglial cells in the acute phase after injury with a peak around 3 days after injury (Anderson et al., 2004; Qiao et al., 2006). Interestingly, C1q and C3 were noted to co-localize with both IgG and IgM deposition in the spinal parenchyma (Qiao et al., 2006; Narang et al., 2017).
Triggers of C activation after spinal cord injury
C activation contributes to dual recognition and effector functions in the innate immune response after SCI, and several studies have investigated the triggers for C activation after SCI. Antibody-dependent activation of the classical pathway has been well documented by studies demonstrating co-localization of C1q and C3 with immunoglobulins on endothelial, neuronal, and astroglial cells as well as studies demonstrating suppression of C activation in antibody or B-cell deficient mice (Ankeny et al., 2009; Narang et al., 2017). In the work of Ankeny et al. (2009), B-cell depletion resulted in a reduction of C activation after SCI and improved functional recovery. When the sera of mice with SCI containing IgG and IgM were introduced into the normal murine SC in vivo, injury and complement deposition were reconstituted suggesting that autoantibodies are triggering C activation in these models (Ankeny et al., 2009). However, these autoantibodies were not specifically isolated or characterized in terms of their target antigens. Given their early deposition after injury, these antibodies are more likely to be natural antibodies already present in the serum rather than produced by secondary immune responses (Arevalo-Martin et al., 2018). In a more recent work by Narang et al. (2017), the role of natural IgM antibodies was further explored. Natural IgM antibodies are not autoreactive but rather bind to modified self-antigens or “eat-me” signals on the surface of stressed or damaged cells and trigger C and immune cell activation (Narang et al., 2017). These antibodies are produced by B1-type B-cells and are genetically encoded rather than being elicited by infection or specific exposure. Depletion of B1 cells or deficiency in the Rag1 gene required for recombination and antibody production resulted in protection in SCI and reduced C activation in vivo (Narang et al., 2017). A panel of these natural antibodies has been identified including two specific antibodies B4-IgM (recognizes post-translational modification of annexin IV) and C2-IgM (binds cell surface phospholipids) that were found to reinstate injury and C activation in Rag1–/–mice supporting the role of these natural IgM antibodies in post-SCI C activation and subsequent pathology (Figure 3 and Additional Table 2 (150.1KB, pdf) ). Despite these findings, more recent work by Ulndreaj et al. (2021) showed that preferential knockout of the entire complement of IgM antibodies while sparing IgG antibodies following SCI resulted in worsening outcomes. Worse outcomes in IgM-KO mice in the chronic phase of SCI were secondary to the development of auto-reactive IgG that resulted in robust local activation of C3.
Figure 3.
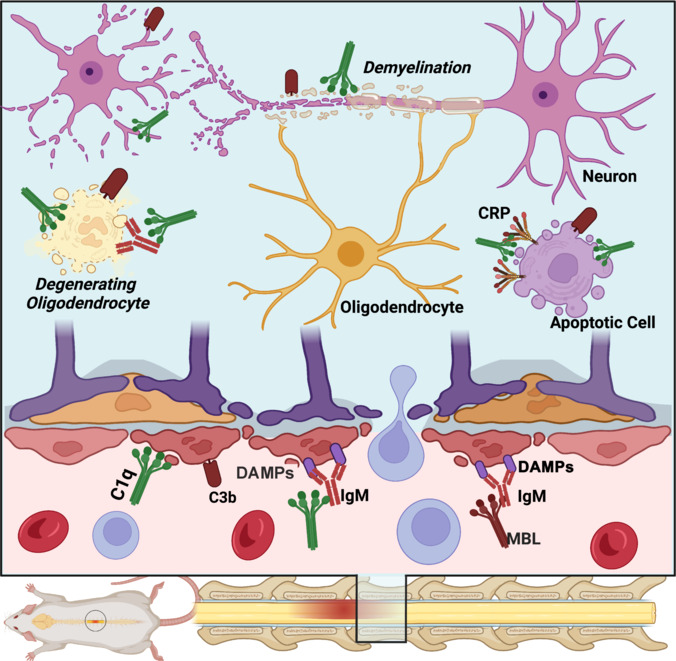
Triggers of complement activation after spinal cord injury.
Complement pathways are activated by either the direct binding of opsonins to stressed and degenerating cells, or by antibodies that recognize DAMPs. An alternative mechanism also involves binding to CRP (an acute phase reactant). These mechanisms involve the classical and alternative pathways of C. The lectin pathway has not yet been investigated. Created with BioRender.com. C: Complement; CRP: C-reactive protein; DAMPs: damage-associated molecular patterns; MBL: mannose-binding lectins.
Deposition of C opsonins, specifically C1q and C3b, was also noted on apoptotic bodies, degenerating myelin, and stressed neurons (Liu et al., 1998; Anderson et al., 2004, 2005; Ankeny et al., 2009; Narang et al., 2017; Zhao et al., 2019; Figure 3). Based on these findings, the triggers of C activation after SCI appear to mimic those reported in models of traumatic and ischemic brain injury (Alawieh et al., 2018). These triggers include direct binding of C opsonins to apoptotic bodies and degeneration products, binding to IgG/IgM antibodies that target post-injury damage-associated molecular patterns, or activation via acute phase reactants such as C-reactive protein (Figure 3).
Effector mechanisms of complement activation
The exact mechanisms of complement-dependent pathological processes after SCI remain poorly understood. Whereas preclinical studies have focused on correlating the inhibition of different complements with histological outcomes and functional recovery (Figure 4), only a few studies investigated C-dependent pathophysiological mechanisms in detail and will be reviewed here.
Figure 4.
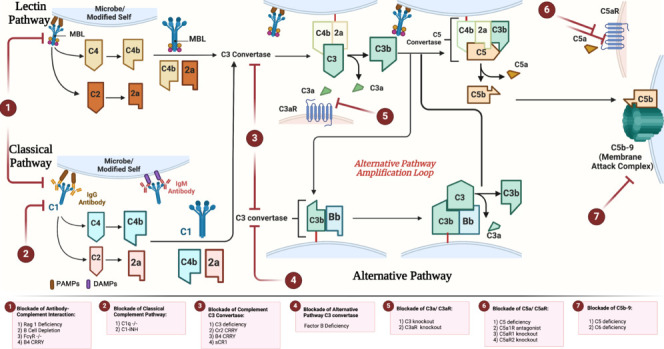
Inhibitors of complement activation after spinal cord injury.
The figure shows the different pathways of C activation and the sites of action of different endogenous and exogenous inhibitors. (1) Inhibitors of the antibody-complement axis include genetic models of Rag1 deficiency, B-cell depletion, and Fc-gamma receptor in addition to the fusion protein B4-Crry that inhibits the bind of B4IgM to modified annexin IV. (2) Inhibitors of the classical pathway include genetic deficiency in C1q or use of C1-inhibitor (C1-INH). (3) Inhibitors of C3 convertase from all pathways include the fusion proteins B4-Crry and CR2-Crry, soluble CR1 (sCR1), and C3 deficiency. (4) Inhibition of the alternative pathway C3 convertase via factor B deficiency. (5) Inhibition of C3a-C3aR1 interaction via C3aR1 deficiency. (6) Inhibition of C5a-C5aR interaction using C5aR1 antagonist (PMX-205), C5aR1 deficiency or C5aR2 deficiency. (7) Inhibition of membrane attack complex includes C5 deficiency and C6 deficiency. Created with BioRender.com. C: Complement; DAMPs: damage-associated molecular patterns; MBL: mannose-binding lectins; PAMPs: pathogen associated molecular patterns.
Inhibition or deficiency at the level of C1q, C3, or fB resulted in a significant reduction in myeloperoxidase activity in the SC and neutrophil infiltration (Li et al., 2005; Qiao et al., 2006, 2010; Galvan et al., 2008; Tei et al., 2008; Narang et al., 2017). Proposed mechanisms include binding of opsonins (C1q and C3) to their corresponding receptors or via anaphylatoxin-dependent mechanisms. The latter has been investigated in more details (Beck et al., 2010; Li et al., 2014; Brennan et al., 2015, 2019; Biggins et al., 2017). The role of C5a in neutrophil activation after SCI was investigated using genetic deficiency in one of two C5a receptors (C5aR1 and C5aR2) or using a competitive antagonist of C5a at C5aR1 (PMX205) (Beck et al., 2010; Brennan et al., 2015; Biggins et al., 2017). Deficiency of C5a receptors or use of PMX-205 did not affect neutrophil infiltration in vivo despite having an impact on macrophage and astrocyte activation (Beck et al., 2010; Brennan et al., 2015; Biggins et al., 2017). At the same time, the genetic deficiency in C3aR1 resulted in increased neutrophil infiltration after SCI compared to wild-type (WT) mice, an effect that persisted when bone marrow chimera was generated that included transplantation of C3aR1–/– neutrophils in WT mice. However, when WT neutrophils were transplanted into C3aR1–/– mice, there was no effect on neutrophil infiltration (Brennan et al., 2019). Suppression of C3a-C3aR1 signaling reduced cellular responsiveness to CXCL1/CXCR2 signaling leading to inhibition of the PI3K/AKT pathway required for neutrophil chemotaxis. These findings implicate C3a-C3aR1 signaling in the inhibition of bone marrow mobilization of neutrophils after SCI suggesting a possible protective role for C3a release. Furthermore, C3 deficiency or inhibition and C1q deficiency have been consistently associated with reduced neutrophil activation which could be attributed to an opsonin-dependent effect given that neutrophils express C3b/d receptors (CR1, CR3) and C1qR (Li et al., 2005; Qiao et al., 2006, 2010; Galvan et al., 2008; Tei et al., 2008; Narang et al., 2017). This mechanism is yet to be explored.
Activation and recruitment of monocytes/macrophages after SCI have been more directly linked to the role of C anaphylatoxins. Deficiency in C5 or use of C5aR1A (PMX-205) reduced macrophage infiltration acutely after SCI (Li et al., 2014). A similar effect was demonstrated with C5aR1–/– (Brennan et al., 2015) in both acute and chronic phases of SCI. Yet, C5aR2–/– did not impact macrophage infiltration or activation in SCI, suggesting that C5a-C5aR1 interaction is implicated in propagating complement-dependent macrophage activation. Inhibiting C3 generation using C3–/– or fB–/– also reduced macrophage infiltration after SCI and a contributing role of opsonins could not be ruled out.
The interplay between astrocyte activation and C after SCI followed a similar pattern to those of infiltrating macrophages. Astrocytes carry both opsonin receptors as well as receptors for anaphylatoxin and a role of both C activation products in astrocytosis is likely. C3 deficiency reduced acute astrocyte activation, and C3 activation products were found to localize to astrocyte cell bodies following injury (Guo et al., 2023). When the role of C5a-C5aR1 signaling was investigated, acute inhibition of C5aR1 using PMX-205 resulted in suppression of astrocyte activation in the subacute phase. Chronic suppression of astrocytosis was only present with genetic deficiency of C5 or C5aR1 or with continued PMX-205 treatment through the subacute and chronic phases (Beck et al., 2010; Brennan et al., 2015, 2019; Biggins et al., 2017). The impact of C5a-C5aR1 signaling on astrocyte activation was noted to be dependent on STAT3 phosphorylation downstream of C5aR1 signaling. Notably, inhibition of astrocyte activation in the acute phase was associated with better histological and functional outcomes, but chronic suppression resulted in worse outcomes likely due to a homeostatic role for astrocytes beyond the acute phase (Brennan et al., 2015, 2019). Guo et al. (2023) studied the interplay between neurite growth in-vitro and C-dependent astrocyte activation and demonstrated that C3 deficiency in astrocytes protected neurite formations in cultured dorsal root ganglion neurons. In fact, studies from stroke and neurodegenerative models have implicated C-dependent phagocytosis of nerve endings by astrocytes and microglia in the chronic degenerative response after injury (Alawieh et al., 2020; Dejanovic et al., 2022), a mechanism likely relevant and unexplored in SCI.
Increased proinflammatory cytokine production in the injured SC remains a key part of the pathology. Inhibition of both C opsonins or anaphylatoxins has been shown to suppress cytokine production from both infiltrating and local immune cells after SCI (Beck et al., 2010; Qiao et al., 2010; Li et al., 2014; Brennan et al., 2015, 2019; Biggins et al., 2017; Narang et al., 2017). These include interleukin-1β, tumor necrosis factor α, monocyte chemoattractant protein-1, interleukin-8, interleukin-6, macrophage colony-stimulating factor, and RANTES (Regulated on Activation, Normal T Cell Expressed and Secreted) among others. This proinflammatory environment involving C activation and cytokine upregulation persisted for at least 2–4 weeks after the initial insult (Anderson et al., 2004; Brennan et al., 2015, 2019).
The role of the MAC in CNS trauma is less prominent (Qiao et al., 2010; Alawieh et al., 2018; Su et al., 2020). Neurons are generally protected from MAC-mediated attack given the high expression of CD59 (MAC inhibitor) on the surface. Emerging evidence has shown that neuronal CD59 expression is likely activity-dependent and could be downregulated after stress, and that sublytic MAC formation could exacerbate excitotoxicity after SCI (Alawieh et al., 2015a). CNS resident cells like oligodendrocytes or neurons can tolerate sublethal levels of MAC complex deposition due to soluble and surface regulators and can clear MAC via endocytosis. In SCI models, C5 deficiency but not C6 deficiency conferred protection from SCI, indicating that the MAC is not a prominent effector in this pathological response (Li et al., 2014; Su et al., 2020). Significant immunoreactivity of Factor H (alternative pathway inhibitor) and clusterin (MAC inhibitor) was found in neurons and oligodendrocytes at least 20 mm away from the injury epicenter starting at 1 day and persisting for at least 6 weeks after injury (Anderson et al., 2004, 2005). Consistent with these findings, CD59–/– increased the susceptibility of neurons to MAC-mediated attack leading to worse functional outcomes (Qiao et al., 2010). Although sublethal MAC can play a role in cellular signaling and homeostasis, it is challenging to ascertain a role for the terminal pathway in SCI.
Complement opsonins exhibit a homeostatic role in mitigating the propagation of the inflammatory response by clearance of pro-inflammatory antigens. Early-phase complement components like C1q and C3 are involved in cell and myelin debris opsonization for phagocytosis and subsequent clearance (Kopper and Gensel, 2018). On histopathological examination, C proteins localized to oligodendrocytes as well as degenerating myelin (Anderson et al., 2004). Studies in demyelinating disease demonstrated a role for macrophages and microglia phagocytosis of myelin often in C1q, C3b/d, or antibody-dependent manner (Linzey et al., 2022). In these models, C1q plays a more homeostatic role with the clearance of myelin debris, whereas the alternative pathway exacerbates injury in relapsing disease (Linzey et al., 2022). Similarly, in SCI myelin debris clearance can be performed through either antibody-dependent or antibody-independent mechanisms (Anderson et al., 2004; Kopper and Gensel, 2018). Acute inhibition of C3 or the alternative pathway reduced the extent of demyelination whereas chronic suppression of C5a signaling limited the extent of remyelination in the chronic phase suggesting a biphasic role of C activation in myelin regeneration after SCI.
A summary of these functions of C following SCI is illustrated in Figure 5.
Figure 5.
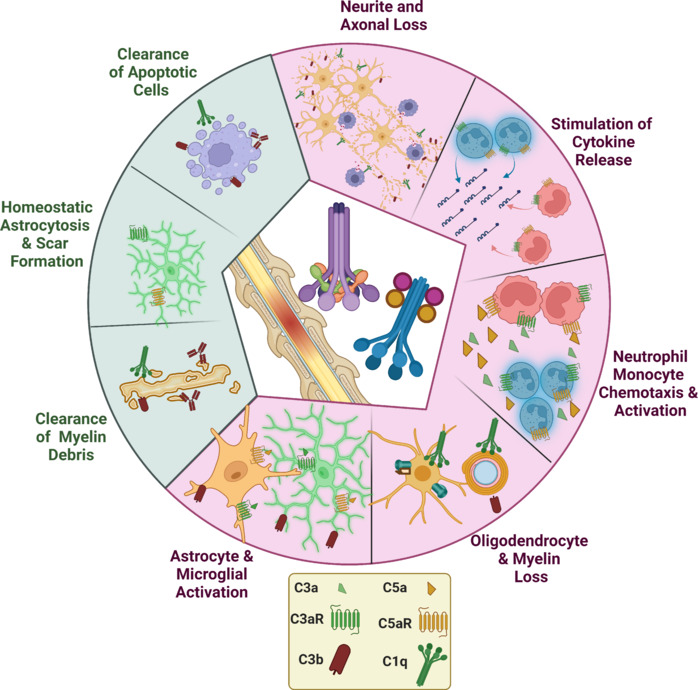
Summary of the effects of complement in spinal cord injury.
Green section demonstrates protective/reparatory functions whereas the red section corresponds to pathological functions of complement. Created with BioRender.com.
Complement Modulation in Preclinical Models
Preclinical studies examining the role of C in SCI pathology adopted a translational route focusing on the impact of inhibiting different C components on disease pathology. These studies targeted different C pathways and used both genetic and pharmacological approaches for C modulation as summarized in Figure 4 and Additional Table 2 (150.1KB, pdf) . Although the use of inhibitors allows for a more translational approach toward C modulation, genetic deficiency models still provide a good resource to elucidate the contribution of different pathways in disease processes. However, the genetic models come with the limitation that genetic deficiency in C components alters the CNS neurodevelopment and can lead to compensatory mechanisms and aberrant wiring (Lu-Culligan and Iwasaki, 2020), an effect that should be recognized when interpreting these studies.
Targeting antibody-complement interactions
As discussed in Figure 2, antibody binding, specifically the binding of natural antibodies, is a major trigger of C activation after SCI. The volume of lesion as well as the extent of myelinated fiber loss was significantly reduced in mice lacking B-cells or those lacking Rag1 expression required to produce antibodies. The Basso, Beattie and Bresnahan score is one of the most common measures used to track recovery after SCI that is now being replaced with the CatWalk assay that provides a more sensitive and automated assessment. Following SCI, there is an immediate drop in the motor Basso, Beattie and Bresnahan score, and wild-type rodents exhibit a pattern of spontaneous recovery over 30 days after which the function plateaus at a significantly sub-baseline level. Depletion of antibodies in mice was associated with significant improvement in motor coordination and function as early as 7 days after SCI that persisted for up to 9 weeks compared to wild-type mice (Ankeny et al., 2009; Narang et al., 2017). Antibodies isolated from SCI mice were able to reconstitute histological damage and complement activation in both normal SC as well as in Rag1–/– mice (Narang et al., 2017) suggesting a pathological role of these antibodies. However, when SCI antibodies were given to C3–/– mice or FcγR–/–, the pathological effect was not observed. These findings support the idea that suppression of either antibody signaling (through FcγR) or antibody-associated C activation confer functional and histopathological protection after SCI (Ankeny et al., 2009; Narang et al., 2017; Additional Table 2 (150.1KB, pdf) ).
Narang et al. (2017) then isolated an antibody that targets post-translational modification of annexin IV, namely B4-IgM, which demonstrated a pathological role in SCI. The single chain variable fragment of this antibody (B4 scFv) was then fused to the inhibitor of the C3 convertase (Crry) to design a targeted fusion protein (B4-Crry) that inhibits both IgM binding and C activation after SCI (Narang et al., 2017). Administration of B4-Crry 30 minutes after SCI resulted in significant improvement in motor scores over 21 days, and a significant reduction in lesion volume and extent of demyelination. These findings were associated with acute reduction of both IgM and C3d deposition in the SC. Of note, this treatment was administered as a single dose in the acute setting (Narang et al., 2017).
Targeting the classical pathway
Following controlled impact to the thoracic spine, rodents lacking C1q (C1q–/–) or those treated with C1-Inhibitor acutely had significant improvement in their motor recovery over 7–28 days. Inhibition of C1q activation resulted in histological improvement in lesion volume and extent of demyelination, but the impact on C3 activation was not assessed in these studies (Additional Table 2 (150.1KB, pdf) ). However, the role of the classical pathway is also further supported by the role of antibody-dependent C activation in SCI as discussed earlier since this response is predominantly mediated via the classical pathway. The lectin pathway, although implicated in C-dependent pathology in the brain, has not been investigated in SCI and could also be part of the antibody-dependent pathological C activation.
Inhibitors of alternative pathway
Proteins of the alternative C pathway, specifically fB, were noted on neuron and glial cells after SCI (Anderson et al., 2004). One advantage of targeting the alternative pathway is its role in amplification of C activation while allowing for basal level of activation caused by other pathways, an effect that may be relevant in repair and regenerative functions. In mice unable to elicit the alternative pathway due to fB–/–, there was a histological reduction in the extent of injury expansion and myelin loss which corresponded with a better function on motor tasks up to 21 days after SCI (Qiao et al., 2010). The use of anti-fB antibodies to neutralize the effect of fB resulted in similar benefits when given at 1 and 13 hours after SCI (Qiao et al., 2010). Inhibition of fB was associated with a significant reduction in both C3 and C9 deposition in the spine without completely neutralizing C3 activity (Qiao et al., 2010).
Inhibitors of C3 activation
Compared to early studies using systemic complement depletion to study C-dependent role in SCI (Reynolds et al., 2003, 2004), recent work investigated direct inhibition of C3 activation via genetic C3–/–, using soluble complement receptor 1 (sCR1), or using targeted inhibitors like B4-Crry or CR2-Crry (Additional Table 2 (150.1KB, pdf) ). B4-Crry is a targeted inhibitor that recognizes post-translation modification of annexin IV, targets specifically to site of SCI, and inhibits both IgM deposition and activation of C3 convertase. CR2-Crry is a fusion protein of complement receptor 2 (CR2) that binds C3 activation products (C3b/d) and Crry that inhibits the C3 convertase. CR2-Crry targets specifically sites of C activation and interrupts C3 convertase activity locally (Qiao et al., 2010). All four approaches for C3 inhibition reported improved motor recovery in rodents starting at day 4 and up to 3 weeks after injury (Li et al., 2005; Qiao et al., 2010; Narang et al., 2017; Guo et al., 2023). Similarly, there was a reduction in injury expansion, overall lesion volume, and immune cell infiltrate across all these studies. Inhibition was associated with a significant reduction of C3b/d binding to the CNS-resident cells (Li et al., 2005; Qiao et al., 2010; Narang et al., 2017).
The use of tissue-targeted approaches for C modulation (CR2-Crry and B4-Crry) presents a novel translational approach in neurotrauma (Alawieh et al., 2018) as it allows for local suppression of C activity without overall systemic effects. These approaches address the key limitations in C inhibitory strategies that include target site bioavailability and risk of infection (Alawieh et al., 2015a, 2018). The half-life of these inhibitors is around 36 hours in the target tissue compared to minutes in serum which allows for persistent local effect even after a single dose of treatment (Qiao et al., 2010; Alawieh et al., 2015a; Narang et al., 2017). These inhibitors are currently in clinical development for different C-related indications (Kolev et al., 2023).
Downstream of C3 activation, the role of the terminal C pathway in SCI remains under investigation and does not appear to be a prominent player in the pathology, especially given its downstream location to the other prominent C effectors (Figure 4 and Additional Table 2 (150.1KB, pdf) ).
Inhibitors of anaphylatoxins C3a and C5a
We have discussed earlier the roles of C3a and C5a in the pathology of SCI, and approaches to target these effectors focus on their interaction with their corresponding receptors. The binding of C3a to C3aR1 was investigated by Brennan (Brennan et al., 2019) using C3aR1–/–. Loss of C3aR1 resulted in worsening neutrophil infiltrate acutely after SCI and worse histological and motor outcomes. The authors demonstrate that peripheral C3a/C3aR1 signaling inhibits the mobilization of bone-marrow neutrophils explaining the protective role. The translational relevance of these findings remains limited given the concerns with altered compensatory mechanisms in genetically deficient cells, but these findings do not support targeting this pathway for SCI treatment.
The C5a signaling access is more promising as a therapeutic target given the availability of a molecular inhibitor and findings from C5aR antagonist studies. Two receptors for C5a have been described. C5aR1 is the main G-protein coupled receptor mediating the traditional effects of C5a signaling. C5aR2 was more recently described and was initially believed to be a decoy receptor that limits C5aR1 signaling but has been associated with independent signaling via β-arrestins to modulate innate and adaptive immune responses (Li et al., 2019). Genetic deficiency in C5aR2 resulted in worsened motor function and histological outcomes after SCI, a phenotype that is rescued when C5aR1 antagonist (PMX-205) was administered (Biggins et al., 2017). These findings indicated that C5aR2 signaling downregulates C5a-C5aR1 signaling and is part of the protective mediators of C5a signaling after SCI. On the other hand, inhibition of C5aR1 signaling via PMX-205 improved histological and motor outcomes after SCI only when administered in the acute phase 1–7 days after injury. Both C5aR1 deficiency and prolonged administration of PMX-205 in the subacute phase resulted in the loss of the protective effect of C5aR1 inhibition (Biggins et al., 2017). When bone marrow chimera lacking C5aR1 were used, there was no pathological effect on outcome chronically. Therefore, these findings demonstrate that C5a-C5aR1 signaling is a potential acute target for SCI treatment but exhibits a delayed protective role that involves CNS-resident astrocytes (Biggins et al., 2017; Figure 5 and Additional Table 2 (150.1KB, pdf) ).
Complement in the Chronic Phase of Spinal Cord Injury
The effector roles of C activation in the early phase of SCI were discussed earlier, and targeting C activation, amplification, or C5a signaling in this phase has demonstrated promising functional results (Additional Table 2 (150.1KB, pdf) ). Beyond the acute phase, the role of C activation products remains poorly understood. This phase is characterized by stabilization of motor function following early spontaneous or treatment-induced recovery. C activation products including C3a and C5a persisted in the SC for several weeks after the initial insult (Brennan et al., 2015, 2019; Biggins et al., 2017). Flow cytometry studies of the injured spinal cord in rodents showed persistent neutrophil infiltration up to 6 months after injury whereas macrophage/monocyte and T-cell infiltration was noted for at least 180 days (Beck et al., 2010). The persistence of a chronic inflammatory response following SCI without progression of functional decline is interesting and raises the possibility that these effector functions are more involved in regenerative/repair processes. Studies from C5aR1 inhibition in the chronic phase illustrate some of these potential delayed roles of C. Mice with chronic C5aR1 inhibition had worse chronic outcomes and delayed maturation of the astrocytic scar (Brennan et al., 2015). Although the early formation of astrogliotic scar limits the growth and regeneration of neurites after injury (Alawieh et al., 2021; Guo et al., 2023), the presence of this scar is an evolutionary defensive mechanism against the expansion of secondary injury. Yet, this remains a hypothesis that warrants investigation.
Additionally, neuropathic pain is another sequel of SCI investigated in the chronic phase, showing an association between C3 and C1q gene expression and occurrence of neuropathic pain in rodent models. In a study on differential gene expression in rats with neuropathic pain, downregulation of C3 (Li et al., 2022), CR3, and C1q in the chronic phase after was more prominent in rats with neuropathic pain suggested that the complement opsonization pathway could play a role in synaptic pruning during the regenerative phase of SCI (Li et al., 2022). Yet, this hypothesis should still be experimentally validated.
An alternative hypothesis is that partial restoration of function and homeostasis leads to the upregulation of C inhibitors/regulators over time to balance the existing activity (Ankeny et al., 2009). Finally, a plateau in function on the motor scores (Basso, Beattie and Bresnahan or Basso Mouse Scale scores) may not necessarily indicate stability of deficits due to the limited sensitivity of these tests, and a more detailed analysis of motor function, coordination, and fine motor skills is warranted in the chronic phase. Notably, the Basso Mouse Scale might not be sensitive enough to detect small changes in locomotor function that may be caused by the chronic humoral response (Ankeny et al., 2009).
Future Perspectives
Cumulative evidence to date supports an early pathological role of C activation in the pathophysiology of SCI and suggests that C pathways could present a promising therapeutic target. However, the role of different C activation pathways has not yet been fully investigated. There remains a need to explore the role of the lectin pathway in SCI, and dissect the contribution of different C pathways to disease pathology using more novel approaches that include targeted inhibitors of the different pathways (Alawieh et al., 2018) or CNS-specific knockouts of different C proteins to help determine the optimal therapeutic target within the C activation cascade. Simultaneously, the published literature on SCI has not investigated the mechanisms of C-related pathology in SCI when compared to work in other disease models. Efforts by Brennan et al. on studying anaphylatoxin signaling helped answer some of these questions (Brennan et al., 2015, 2019; Biggins et al., 2017), but the roles of opsonins, their signaling, and microglia/astrocyte-complement-neuronal axis in SCI pathology and plasticity is still to be explored. A key point in recovery from SCI is the restoration of white matter connections and establishing new synaptic connections during the window of neuroplasticity after injury. Such a role for C has been explored in brain injury models (Alawieh et al., 2020), but the role of C in mediating phagocytosis, pruning, or survival of neurite growth and synaptic formation in the injured SC is unknown.
Clinical Implications and Limitations to Translation
From a clinical perspective, SCI remains a major cause of disability in young adults, and no pharmacological agents are currently approved for use to improve recovery in the acute setting. Management of SCI involves early decompression and stabilization, blood pressure augmentation, and prompt initiation of rehabilitation. The prognosis in patients with severe SCI is poor. Only 4.8% of patients with motor complete, sensory incomplete SCI (American Spinal Injury Association Impairment Scale B) will become ambulatory at 5 years post-injury (Kirshblum et al., 2021). While steroid administration after SCI exhibited significant neuroprotective potential in preclinical studies, current American Association of Neurological Surgeons and Congress of Neurological Surgeons guidelines recommend against the use of steroids for SCI due to high risks of complications (Hejrati et al., 2023). Therefore, there remains an unmet need to develop therapeutic strategies that can supplement the acute management of SCI.
Given the role of C as an early trigger of the neuroinflammatory response, it presents a favorable therapeutic target. In fact, there is a significant interest among both academic and industry sectors in the development of C inhibitors for various neurological and inflammatory disorders (Dalakas et al., 2020; Holers, 2023). By the end of 2023, there are eight different complement inhibitors approved by the FDA for various indications (West et al., 2024). In addition, over 10 different complement inhibitors are currently in clinical trials (Mastellos et al., 2024). These trials have demonstrated favorable safety profiles for C inhibitors. Despite the recent approval of C inhibitors in some CNS disorders including Myasthenia Gravis and age-related macular degeneration (Vu et al., 2022; Heier et al., 2023). Despite this outlook, there are currently no trials on C modulation in neurotrauma. A few complement inhibitors have been trialed in rodent models including sCR1, CR2-Crry, B4-Crry, and PMX-205. These inhibitors are in the very early stages of preclinical development, and replicating these findings across different research groups and using different SCI models is still needed. Additionally, these agents have not completed the necessary toxicology studies to date. Barriers to translation in preclinical studies of SCI include the need for more face validity of these studies to reflect the clinical disease and population. These studies should also address gender differences in outcomes and response to treatment, including both older and young animals, and follow chronic outcomes using more sensitive motor and behavioral tasks. Given the role of rehabilitation in human recovery from SCI, preclinical studies evaluating improved recovery from SCI should incorporate rehabilitative interventions to determine if the studied intervention is superior to spontaneous and rehabilitation-induced recovery.
An additional line of investigation relates to the role of C as a prognostic tool. Given the early role of C in SCI pathogenesis and the persistent C activity in the SC during recovery, C activation products could serve as potential injury biomarkers in patients with SCI for both prognostic purposes and determine eligibility for interventions addressing C activation.
Additional files:
Additional Figure 1 (292KB, tif) : Flow chart of literature review and selection.
Flow chart of literature review and selection.
Additional Table 1 (117.5KB, pdf) : Summary of studies reporting complement activation and deposition after SCI.
Summary of studies reporting complement activation and deposition after SCI
Additional Table 2 (150.1KB, pdf) : Summary of studies targeting complement activation after SCI.
Summary of studies targeting complement activation after SCI
Funding Statement
Funding: This work was supported by the Department of Veterans Affairs (VA Merit Award BX004256) (to AMA) and Emory Department of Neurosurgery Catalyst Grant, and Emory Medical Care Foundation Grant (to AMA and JG).
Footnotes
Conflicts of interest: Ali M. Alawieh holds intellectual property for targeted inhibitors of complement activation (US11806389B2 and WO2021247487A1).
C-Editors: Zhao M, Sun Y, Qiu Y; T-Editor: Jia Y
Data availability statement:
All relevant data are within the manuscript and its Additional files.
References
- Alawieh A, Elvington A, Tomlinson S. Complement in the homeostatic and ischemic brain. Front Immunol. 2015;6:417. doi: 10.3389/fimmu.2015.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alawieh A, Sabra M, Sabra Z, Tomlinson S, Zaraket FA. Molecular architecture of spinal cord injury protein interaction network. PLoS One. 2015;10:e0135024. doi: 10.1371/journal.pone.0135024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alawieh A, Langley EF, Weber S, Adkins D, Tomlinson S. Identifying the role of complement in triggering neuroinflammation after traumatic brain injury. J Neurosci. 2018;38:2519–2532. doi: 10.1523/JNEUROSCI.2197-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alawieh AM, Langley EF, Feng W, Spiotta AM, Tomlinson S. Complement-dependent synaptic uptake and cognitive decline after stroke and reperfusion therapy. J Neurosci. 2020;40:4042–4058. doi: 10.1523/JNEUROSCI.2462-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alawieh A, Chalhoub RM, Mallah K, Langley EF, York M, Broome H, Couch C, Adkins D, Tomlinson S. Complement drives synaptic degeneration and progressive cognitive decline in the chronic phase after traumatic brain injury. J Neurosci. 2021;41:1830–1843. doi: 10.1523/JNEUROSCI.1734-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JK, Popovich PG. Neuroinflammation in spinal cord injury: therapeutic targets for neuroprotection and regeneration. Prog Brain Res. 2009;175:125–137. doi: 10.1016/S0079-6123(09)17508-8. [DOI] [PubMed] [Google Scholar]
- Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, Lambris JD, Huber-Lang M. Interaction between the coagulation and complement system. Adv Exp Med Biol. 2008;632:71–79. doi: 10.1007/978-0-387-78952-1_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AJ, Robert S, Huang W, Young W, Cotman CW. Activation of complement pathways after contusion-induced spinal cord injury. J Neurotrauma. 2004;21:1831–1846. doi: 10.1089/neu.2004.21.1831. [DOI] [PubMed] [Google Scholar]
- Anderson AJ, Najbauer J, Huang W, Young W, Robert S. Upregulation of complement inhibitors in association with vulnerable cells following contusion-induced spinal cord injury. J Neurotrauma. 2005;22:382–397. doi: 10.1089/neu.2005.22.382. [DOI] [PubMed] [Google Scholar]
- Ankeny DP, Guan Z, Popovich PG. B cells produce pathogenic antibodies and impair recovery after spinal cord injury in mice. J Clin Invest. 2009;119:2990–2999. doi: 10.1172/JCI39780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arevalo-Martin A, Grassner L, Garcia-Ovejero D, Paniagua-Torija B, Barroso-Garcia G, Arandilla AG, Mach O, Turrero A, Vargas E, Alcobendas M, Rosell C, Alcaraz MA, Ceruelo S, Casado R, Talavera F, Palazon R, Sanchez-Blanco N, Maier D, Esclarin A, Molina-Holgado E. Elevated autoantibodies in subacute human spinal cord injury are naturally occurring antibodies. Front Immunol. 2018;9:2365. doi: 10.3389/fimmu.2018.02365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnum SR. Inhibition of complement as a therapeutic approach in inflammatory central nervous system (CNS) disease. Mol Med. 1999;5:569–582. [PMC free article] [PubMed] [Google Scholar]
- Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, Anderson AJ. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain. 2010;133:433–447. doi: 10.1093/brain/awp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins PJC, Brennan FH, Taylor SM, Woodruff TM, Ruitenberg MJ. The alternative receptor for complement component 5a, C5aR2, conveys neuroprotection in traumatic spinal cord injury. J Neurotrauma. 2017;34:2075–2085. doi: 10.1089/neu.2016.4701. [DOI] [PubMed] [Google Scholar]
- Brennan FH, Gordon R, Lao HW, Biggins PJ, Taylor SM, Franklin RJ, Woodruff TM, Ruitenberg MJ. The complement receptor C5aR controls acute inflammation and astrogliosis following spinal cord injury. J Neurosci. 2015;35:6517–6531. doi: 10.1523/JNEUROSCI.5218-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan FH, Jogia T, Gillespie ER, Blomster LV, Li XX, Nowlan B, Williams GM, Jacobson E, Osborne GW, Meunier FA, Taylor SM, Campbell KE, MacDonald KP, Levesque JP, Woodruff TM, Ruitenberg MJ. Complement receptor C3aR1 controls neutrophil mobilization following spinal cord injury through physiological antagonism of CXCR2. JCI Insight. 2019;4:e98254. doi: 10.1172/jci.insight.98254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan FH, Coulthard LG, Alawieh AM, Reiner O, Pekna M. Editorial: complement in the development and regeneration of the nervous system. Front Immunol. 2021;12:694810. doi: 10.3389/fimmu.2021.694810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalakas MC, Alexopoulos H, Spaeth PJ. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat Rev Neurol. 2020;16:601–617. doi: 10.1038/s41582-020-0400-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejanovic B, et al. Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nat Aging. 2022;2:837–850. doi: 10.1038/s43587-022-00281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusart I, Schwab ME. Secondary cell death and the inflammatory reaction after dorsal hemisection of the rat spinal cord. Eur J Neurosci. 1994;6:712–724. doi: 10.1111/j.1460-9568.1994.tb00983.x. [DOI] [PubMed] [Google Scholar]
- Galvan MD, Luchetti S, Burgos AM, Nguyen HX, Hooshmand MJ, Hamers FP, Anderson AJ. Deficiency in complement C1q improves histological and functional locomotor outcome after spinal cord injury. J Neurosci. 2008;28:13876–13888. doi: 10.1523/JNEUROSCI.2823-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Fan X, Nagy LE, Tomlinson S, Yuan G. Editorial: new insights into the role of complement system in liver diseases. Front Immunol. 2023;14:1284944. doi: 10.3389/fimmu.2023.1284944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao M, Ji XR, Chen H, Zhang W, Zhang LC, Zhang LH, Tang PF, Lu N. Cell cycle and complement inhibitors may be specific for treatment of spinal cord injury in aged and young mice: transcriptomic analyses. Neural Regen Res. 2018;13:518–527. doi: 10.4103/1673-5374.226405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heier JS, et al. Pegcetacoplan for the treatment of geographic atrophy secondary to age-related macular degeneration (OAKS and DERBY): two multicentre, randomised, double-masked, sham-controlled, phase 3 trials. Lancet. 2023;402:1434–1448. doi: 10.1016/S0140-6736(23)01520-9. [DOI] [PubMed] [Google Scholar]
- Hejrati N, Aarabi B, Neal CJ, Ugiliweneza B, Kurpad SN, Shaffrey CI, Guest JD, Toups EG, Harrop JS, Fehlings MG. Trends in the use of corticosteroids in the management of acute spinal cord injury in North American Clinical Trials Network Sites. J Neurotrauma. 2023;40:1938–1947. doi: 10.1089/neu.2022.0409. [DOI] [PubMed] [Google Scholar]
- Holers VM. Complement therapeutics are coming of age in rheumatology. Nat Rev Rheumatol. 2023;19:470–485. doi: 10.1038/s41584-023-00981-x. [DOI] [PubMed] [Google Scholar]
- Holste K, Xia F, Garton HJL, Wan S, Hua Y, Keep RF, Xi G. The role of complement in brain injury following intracerebral hemorrhage: a review. Exp Neurol. 2021;340:113654. doi: 10.1016/j.expneurol.2021.113654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell MC, Green R, McGill AR, Kahlil RM, Dutta R, Mohapatra SS, Mohapatra S. Activation of intracellular complement in lungs of patients with severe COVID-19 disease decreases T-cell activity in the lungs. Front Immunol. 2021;12:700705. doi: 10.3389/fimmu.2021.700705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirshblum S, Snider B, Eren F, Guest J. Characterizing natural recovery after traumatic spinal cord injury. J Neurotrauma. 2021;38:1267–1284. doi: 10.1089/neu.2020.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev M, Barbour T, Baver S, Francois C, Deschatelets P. With complements: C3 inhibition in the clinic. Immunol Rev. 2023;313:358–375. doi: 10.1111/imr.13138. [DOI] [PubMed] [Google Scholar]
- Kopper TJ, Gensel JC. Myelin as an inflammatory mediator: myelin interactions with complement, macrophages, and microglia in spinal cord injury. J Neurosci Res. 2018;96:969–977. doi: 10.1002/jnr.24114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Liu X, Wang J, Wang F, Zhu Z, Tang T, Wang J, Zhou Z, Gao M, Liu S. Identification of immunodiagnostic blood biomarkers associated with spinal cord injury severity. Front Immunol. 2023;14:1101564. doi: 10.3389/fimmu.2023.1101564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Li J, Zhu Y, Fan G. Ephedra sinica inhibits complement activation and improves the motor functions after spinal cord injury in rats. Brain Res Bull. 2009;78:261–266. doi: 10.1016/j.brainresbull.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Li L, Xiong ZY, Qian ZM, Zhao TZ, Feng H, Hu S, Hu R, Ke Y, Lin J. Complement C5a is detrimental to histological and functional locomotor recovery after spinal cord injury in mice. Neurobiol Dis. 2014;66:74–82. doi: 10.1016/j.nbd.2014.02.008. [DOI] [PubMed] [Google Scholar]
- Li LM, Zhu Y, Fan GY. Effects of recombinant sCR1 on the immune inflammatory reaction in acute spinal cord injury tissue of rats. Chin J Traumatol. 2005;8:49–53. [PubMed] [Google Scholar]
- Li XX, Lee JD, Kemper C, Woodruff TM. The complement receptor C5aR2: a powerful modulator of innate and adaptive immunity. J Immunol. 2019;202:3339–3348. doi: 10.4049/jimmunol.1900371. [DOI] [PubMed] [Google Scholar]
- Li Y, Fang SC, Zhou L, Mo XM, Guo HD, Deng YB, Yu HH, Gong WY. Complement receptor 3 pathway and NMDA receptor 2B subunit involve neuropathic pain associated with spinal cord injury. J Pain Res. 2022;15:1813–1823. doi: 10.2147/JPR.S366782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linzey M, DiSano K, Welsh N, Pachner A, Gilli F. Divergent complement system activation in two clinically distinct murine models of multiple sclerosis. Front Immunol. 2022;13:924734. doi: 10.3389/fimmu.2022.924734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Persson JK, Svensson M, Aldskogius H. Glial cell responses, complement, and clusterin in the central nervous system following dorsal root transection. Glia. 1998;23:221–238. [PubMed] [Google Scholar]
- Lu-Culligan A, Iwasaki A. The role of immune factors in shaping fetal neurodevelopment. Annu Rev Cell Dev Biol. 2020;36:441–468. doi: 10.1146/annurev-cellbio-021120-033518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastellos DC, Hajishengallis G, Lambris JD. A guide to complement biology, pathology and therapeutic opportunity. Nat Rev Immunol. 2024;24:118–141. doi: 10.1038/s41577-023-00926-1. [DOI] [PubMed] [Google Scholar]
- Narang A, Qiao F, Atkinson C, Zhu H, Yang X, Kulik L, Holers VM, Tomlinson S. Natural IgM antibodies that bind neoepitopes exposed as a result of spinal cord injury, drive secondary injury by activating complement. J Neuroinflammation. 2017;14:120. doi: 10.1186/s12974-017-0894-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HX, Galvan MD, Anderson AJ. Characterization of early and terminal complement proteins associated with polymorphonuclear leukocytes in vitro and in vivo after spinal cord injury. J Neuroinflammation. 2008;5:26. doi: 10.1186/1742-2094-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SL, Anderson AJ. Complement and spinal cord injury: traditional and non-traditional aspects of complement cascade function in the injured spinal cord microenvironment. Exp Neurol. 2014;258:35–47. doi: 10.1016/j.expneurol.2014.04.028. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Horner PJ, Mullin BB, Stokes BT. A quantitative spatial analysis of the blood-spinal cord barrier. I. Permeability changes after experimental spinal contusion injury. Exp Neurol. 1996;142:258–275. doi: 10.1006/exnr.1996.0196. [DOI] [PubMed] [Google Scholar]
- Qiao F, Atkinson C, Song H, Pannu R, Singh I, Tomlinson S. Complement plays an important role in spinal cord injury and represents a therapeutic target for improving recovery following trauma. Am J Pathol. 2006;169:1039–1047. doi: 10.2353/ajpath.2006.060248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao F, Atkinson C, Kindy MS, Shunmugavel A, Morgan BP, Song H, Tomlinson S. The alternative and terminal pathways of complement mediate post-traumatic spinal cord inflammation and injury. Am J Pathol. 2010;177:3061–3070. doi: 10.2353/ajpath.2010.100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebhun J, Madorsky JG, Glovsky MM. Proteins of the complement system and acute phase reactants in sera of patients with spinal cord injury. Ann Allergy. 1991;66:335–338. [PubMed] [Google Scholar]
- Reynolds DN, Smith SA, Zhang YP, Lahiri DK, Morassutti DJ, Shields CB, Kotwal GJ. Vaccinia virus complement control protein modulates inflammation following spinal cord injury. Ann N Y Acad Sci. 2003;1010:534–539. doi: 10.1196/annals.1299.099. [DOI] [PubMed] [Google Scholar]
- Reynolds DN, Smith SA, Zhang YP, Mengsheng Q, Lahiri DK, Morassutti DJ, Shields CB, Kotwal GJ. Vaccinia virus complement control protein reduces inflammation and improves spinal cord integrity following spinal cord injury. Ann N Y Acad Sci. 2004;1035:165–178. doi: 10.1196/annals.1332.011. [DOI] [PubMed] [Google Scholar]
- Sengupta MB, Basu M, Iswarari S, Mukhopadhyay KK, Sardar KP, Acharyya B, Mohanty PK, Mukhopadhyay D. CSF proteomics of secondary phase spinal cord injury in human subjects: perturbed molecular pathways post injury. PLoS One. 2014;9:e110885. doi: 10.1371/journal.pone.0110885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Johnson MB. The complement cascade repurposed in the brain. Nat Rev Immunol. 2021;21:624–625. doi: 10.1038/s41577-021-00621-z. [DOI] [PubMed] [Google Scholar]
- Su D, Hooshmand MJ, Galvan MD, Nishi RA, Cummings BJ, Anderson AJ. Complement C6 deficiency exacerbates pathophysiology after spinal cord injury. Sci Rep. 2020;10:19500. doi: 10.1038/s41598-020-76441-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tei R, Kaido T, Nakase H, Sakaki T. Protective effect of C1 esterase inhibitor on acute traumatic spinal cord injury in the rat. Neurol Res. 2008;30:761–767. doi: 10.1179/174313208X284241. [DOI] [PubMed] [Google Scholar]
- Ulndreaj A, Vidal PM, Forgione N, Hong J, Fehlings MG. IgM immunoglobulin influences recovery after cervical spinal cord injury by modulating the IgG autoantibody response. eNeuro. 2021;8 doi: 10.1523/ENEURO.0491-19.2021. ENEURO.0491-19.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu T, Meisel A, Mantegazza R, Annane D, Katsuno M, Aguzzi R, Enayetallah A, Beasley KN, Rampal N, Howard JF. Terminal complement inhibitor ravulizumab in generalized myasthenia gravis. NEJM Evid. 2022;1:EVIDoa2100066. doi: 10.1056/EVIDoa2100066. [DOI] [PubMed] [Google Scholar]
- West EE, Woodruff T, Fremeaux-Bacchi V, Kemper C. Complement in human disease: approved and up-and-coming therapeutics. Lancet. 2024;403:392–405. doi: 10.1016/S0140-6736(23)01524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Zhou X, Qiu J, Xin D, Li T, Chu X, Yuan H, Wang H, Wang Z, Wang D. Exosomes derived from bone marrow mesenchymal stem cells inhibit complement activation in rats with spinal cord injury. Drug Des Devel Ther. 2019;13:3693–3704. doi: 10.2147/DDDT.S209636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Tong D, Wang M, Xu C, Gong X, Wang Z, Li C. Peptidomics analysis reveals serum biomarkers in spinal cord injury patients. Crit Rev Eukaryot Gene Expr. 2022;32:1–9. doi: 10.1615/CritRevEukaryotGeneExpr.2021039575. [DOI] [PubMed] [Google Scholar]
- Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Flow chart of literature review and selection.
Summary of studies reporting complement activation and deposition after SCI
Summary of studies targeting complement activation after SCI
Data Availability Statement
All relevant data are within the manuscript and its Additional files.