

Summary
Molecular de-extinction could offer avenues for drug discovery by reintroducing bioactive molecules that are no longer encoded by extant organisms. To prospect for antimicrobial peptides encrypted within extinct and extant human proteins, we introduce the panCleave random forest model for proteome-wide cleavage site prediction. Our model outperformed multiple protease-specific cleavage site classifiers for three modern human caspases, despite its pan-protease design. Antimicrobial activity was observed in vitro for modern and archaic protein fragments identified with panCleave. Lead peptides showed resistance to proteolysis and exhibited variable membrane permeabilization. Additionally, representative modern and archaic protein fragments showed anti-infective efficacy against A. baumannii in both a skin abscess infection model and preclinical murine thigh infection model. These results suggest that machine learning-based encrypted peptide prospection can identify stable, nontoxic antimicrobial peptides. Moreover, we establish molecular de-extinction through paleoproteome mining as a framework for antibacterial drug discovery.
Graphical Abstract
Introduction
The idea of reintroducing extinct organisms into extant environments has captured the public and scientific imagination, raising profound ethical and ecological questions1. Here, we introduce molecular de-extinction as an antibiotic discovery framework. Molecular de-extinction is the resurrection of extinct molecules of life: nucleic acids, proteins, and other compounds no longer encoded by living organisms. While the societal benefit of organismal de-extinction is still unknown and contentious, technical challenges like incomplete genomic coverage remain significant1,2. By synthesizing only isolated compounds, molecular de-extinction circumvents many of the ethical and technical problems posed by whole-organism de-extinction. Molecular de-extinction is motivated by the hypothesis that molecules that conferred benefits to extinct organisms could be beneficial in the current global environment. Such molecules could be of biomedical or economic utility by bolstering defenses against future challenges that resemble stressors from environments past, including climate change or infectious disease outbreaks. The present work proposes molecular de-extinction as a drug discovery framework for expanding the therapeutic search space through paleoproteome mining.
The global antibiotic resistance crisis, the threat of emerging pathogens, and the overuse of traditional antibiotic scaffolds necessitate new, computer-aided drug development paradigms3,4. Protein informatics is fertile ground for antibiotic discovery, as many peptides are known to modulate the host immune system, disrupt bacterial cell membranes, suppress biofilms, and promote wound healing5,6. Furthermore, 20n variants exist per n-length canonical amino acid sequence, presenting an enormous combinatorial space from which to select peptides with targeted activity. Antimicrobial peptides (AMPs) are an ancient class of host defense molecule found across the domains of life, representing an essential facet of innate immunity throughout evolution. Some AMPs are associated with collateral sensitivity in antibiotic-resistant bacteria and a low propensity to induce resistance5–7. The human cryptome is a subset of the proteome known to harbor AMPs that are released from precursor proteins by both host and pathogen proteases8,9. These bioactive encrypted peptides (EPs) can serve as natural templates for antibiotics and for bioinspired engineered therapies10,11.
Given the enormity of protein space, exhaustively searching for random AMPs is intractable. Instead, guided prospection can be constrained to sequences with desired physicochemical properties (e.g., charge and hydrophobicity)11, to the natural products of taxa known to synthesize antimicrobial compounds, or to other protein subspaces. The majority of antibiotics in use were derived from nature, including fungal and bacterial products12. AMP prospection has successfully mined diverse natural sources, including amphibian skin secretions13,14, snake venoms15, insect venoms16–20, and the gut microbiome21. By searching the human proteome for antimicrobial EPs, the present work builds on the tradition of guided prospection within natural resources. By proposing paleoproteome mining for AMP discovery, we contribute to a limited literature on the antimicrobial activity and resistance of isolates from ancient and extinct sources22–25 and on natural product discovery through paleobiotechnology26.
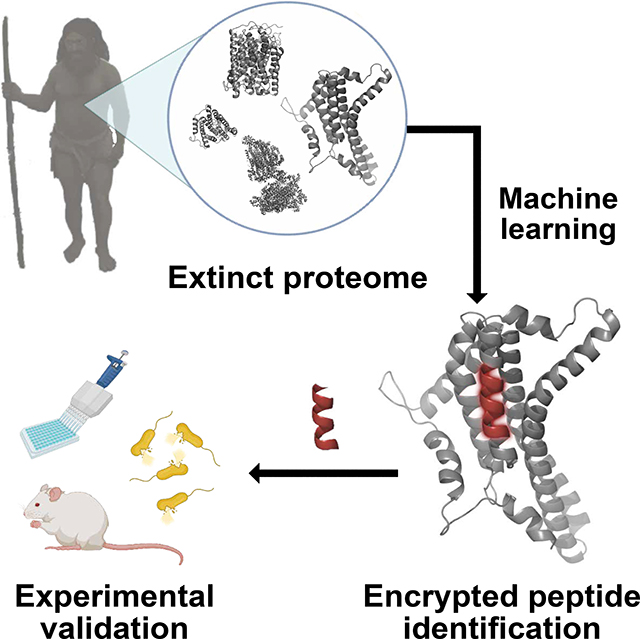
To mine extinct and extant human proteomes for potential encrypted peptides, we present the panCleave Python pipeline (https://gitlab.com/machine-biology-group-public/pancleave). This open-source machine learning (ML) tool leverages a pan-protease cleavage site classifier to perform computational proteolysis: the in silico digestion of human proteins into peptide fragments (Figs. 1, S1a). We experimentally validate panCleave for the prospection of antimicrobial EPs in modern human secreted proteins and in the archaic proteomes of our closest extinct relatives, Neanderthals and Denisovans (Fig. 1). Using panCleave, we discovered peptides encrypted within known precursor protein groups and rediscovered a known antimicrobial EP. An experimental comparison of peptide curation methods reveals low but ranging positive hit rates, highlighting challenges in contemporary antimicrobial activity prediction. No positive hits discovered by panCleave were rediscovered by seven human protease-specific cleavage predictors27, suggesting that panCleave cleavage products are distinctive. By discovering AMPs through computational paleoproteome mining, this work offers a proof-of-concept for molecular de-extinction as an antibiotic discovery framework. Furthermore, this study introduces previously uncharacterized antimicrobial subsequences encrypted in archaic human proteins.
Fig. 1. Computational-experimental framework for molecular de-extinction of antimicrobial peptides.

Panel (A) demonstrates the computational proteolysis pipeline, where user-defined proteins are processed into 8-residue subsequences that are classified as cleavage and non-cleavage sites. Input proteins are then tokenized at predicted cleavage sites, and the resulting fragments can be filtered by user-defined curation methods. Curation methods can include machine learning-based activity prediction, human expert curation, or other methods. Successes in archaic and modern proteome mining are visualized in panel (B), where precursors were computationally digested to reveal encrypted antimicrobial subsequences. The pipeline concludes with in vitro (C) and in vivo (D) experimental validation of fragment bioactivity, including proteolytic degradation assays, MoA assays, and mouse weight monitoring as a proxy for host toxicity. Figure created with BioRender.com and the PyMOL Molecular Graphics System, Version 2.1 Schrödinger, LLC.
Results
Computational proteolysis for proteome-wide searches
The panCleave Python pipeline (Figs. 1, S1a) is a protein informatics tool that uses ML for computational proteolysis: the in silico fragmentation of human protein sequences into peptides. The development of this predictive tool was motivated by the hypothesis that protease-agnostic cleavage site prediction could facilitate biologically inspired prospection for encrypted host defense peptides. Prior cleavage site classifiers are specialized models that predict cleavage activity for only a subset of human proteases27–38. Theoretically, protease-specific models could be expected to lose generalizability on inputs from distantly related proteases with different substrate specificities. A pan-protease design facilitates proteome-wide searches, circumventing the need to hypothesize protease-substrate relationships. Obviating this need provides a unique advantage for bioinspired prospection and molecular de-extinction tasks by enabling preliminary explorations of under-characterized regions in modern and archaic proteomes.
Unlike prior cleavage site classifiers, panCleave was trained on all human protease substrates in the MEROPS Peptidase Database39. Training and testing data included substrates for 369 proteases representing 6 catalytic types, 31 clans, and 73 families. In comparison, DeepCleave features 43 proteases in its full dataset (20 caspases and 23 matrix metallopeptidases)29. Source code, training data (), and testing data () are available on GitLab (https://gitlab.com/machine-biology-group-public/pancleave). Detailed descriptions of data preparation and model selection processes are available in the STAR Methods. Substrate amino acid frequencies, length distributions, protease representation, and precursor protein functions for all training and testing data are characterized in Figs. S1b–S2e.
The performance of the panCleave random forest can be quantified on an aggregated, protease-agnostic level and a disaggregated, protease-specific level. On the complete independent test set comprising substrates from 182 proteases (), panCleave achieved an overall accuracy of 73.3%. Ten-fold cross-validation on the training set () yielded an average accuracy of 73.0%, indicating that panCleave did not overfit to the training data. Random forests can return the estimated probability of binary class membership (i.e., the probability that a subsequence is a cleavage site or non-cleavage site), which can be useful for providing a degree of confidence in model predictions40. Thresholding results in this way indicates increasing accuracy with increasing estimated probability: panCleave achieved 81.9% accuracy for predictions of 60% estimated probability or greater (62.8% of test set predictions) and a maximum accuracy of 96.6% for predictions of 90% estimated probability or greater (2.1% of predictions) (Fig. 2a). The area under the receiver operating characteristic curve was 80.8% and the average precision was 80.3% (Fig. 2b,c). Negative predictive value, positive predictive value, sensitivity, and specificity were 73.2%, 73.5%, 73.0%, and 73.6%, respectively.
Fig. 2. Model performance and antimicrobial peptide data distributions.

Panels describe panCleave random forest performance evaluation (a-h) and physicochemical distributions for positive hits (i–l). Optimized panCleave random forest performance is reported for independent test data (): (a) accuracy-probability threshold tradeoff curves, comparing accuracy per estimated probability of class membership; (b) the receiver operating characteristic curve; (c) precision-recall curve; (d) panCleave test accuracy for proteases with at least 100 test observations; (e) panCleave test accuracy by protease catalytic type; (f) accuracy of panCleave relative to pre-existing models for three caspases (panCleave in red); (g) positive hit rate by fragment curation method; and (h) positive hit rate by antimicrobial activity classifier. Panels i–l compare amino acid frequency (i), fragment length (j), normalized hydrophobicity (k), and net charge distributions (l) for MEPs, AEPs, and AMPs reported in DBAASP41. Hydrophobicity scores employ the Eisenberg and Weiss scale30. Note that DBAASP data were restricted to fragments of length 8–40 residues for length, hydrophobicity, and charge distributions, with null values excluded. DBAASP amino acid frequencies were computed by excluding noncanonical residues.
When disaggregating model accuracy by protease, panCleave performance ranged widely (Fig. 2d,e; Tables S1–S2). Among proteases with at least 100 test set observations, panCleave achieved greater than 80% accuracy on caspase-3 (C14.003; 99.2%), caspase-6 (C14.005; 98.6%), granzyme B (S01.010; 93.2%), legumain (C13.004; 90.6%), and cathepsin S (C01.034; 81.9%) (Fig. 2d; Table S1). Among protease clans, panCleave achieved greater than 70% accuracy on endopeptidase clan CD (type protease caspase-1 [C14.001]; 93.9%), endopeptidase/exopeptidase clan SB (type protease subtilisin Carlsberg [S08.001]; 88.6%), cysteine protease clan CA (type protease papain [C01.001]; 74.1%), and endopeptidase clan PA (type protease chymotrypsin A [S01.001]; 70.6%) (Table S1). The average accuracy was greatest for cysteine catalytic types (81.3%; 1858/2286 observations predicted correctly) and lowest for threonine catalytic types (34.6%; 18/52) (Fig. 2e).
When compared to pre-existing protease-specific models, panCleave outperformed for caspase-2 (C14.006; 100.0%), caspase-3 (C14.003; 99.15%), and caspase-1 (C14.001; 92.68%) (Fig. 2f; Table S2). However, pre-existing models outperformed for multiple matrix metallopeptidases (Table S2). While the pan-protease design of panCleave does not preclude the possibility of high or state-of-the-art accuracy for specific proteases, the use of panCleave for protease-specific applications should be guided by the reported disaggregated accuracies (Fig. 2d,e,f; Tables S1–S2).
The random forest algorithm provides in-built feature importance calculations, which lends interpretability to the model. An analysis of feature importance based on mean decrease in impurity was conducted using functions provided by scikit-learn for random forest classifiers. As expected, results suggest that the most significant features are those corresponding to the residues most proximal to the cleavage site (i.e., the P1 and P1’ positions in the eight residue P4:P4’ flanking site) (Fig. S2f,g). However, relative feature importances are known to vary across importance scoring methods. Thus, we avoid overinterpretation of these results.
Modern encrypted peptides display antimicrobial activity in vitro
Eight of 80 (10.0%) modern secreted protein fragments were active against one or more pathogens in at least one of the conditions tested (Fig. 3; Table S3). Importantly, none of the tested sequences have yet been reported as AMPs or as AMP subsequences in the Database of Antimicrobial Activity and Structure of Peptides (DBAASP)41. The modern encrypted peptides (MEPs) identified by panCleave were found to have similar content of aliphatic (Ala, Gly, Ile, Leu, Pro, and Val), aromatic (Phe, Trp, and Tyr), and basic (His, Arg, and Lys) amino acid residues as AMPs described in the DBAASP41 (Fig. 2i–l). Relative to EPs recently identified by a non-ML scoring function11, MEPs identified here had 18% fewer basic residues and similar aliphatic and aromatic residue content, yet almost five-fold more acidic residues and 25% more polar residues11. Interestingly, the MEPs displayed more acidic (Asp and Glu) and polar (Asn, Cys, Gln, Met, Ser, and Thr) amino acid residues than DBAASP AMPs (63% vs 26%, respectively). MEPs had notably higher median values for net charge and normalized hydrophobicity relative to archaic encrypted peptides (AEPs) (Fig. 2k,l), yet lower median values than recently reported EPs11 (Table S4). MEPs had slightly higher median length than AEPs and short AMPs (8–40 residues) reported in the DBAASP41 (Fig. 2j), with MEPs being enriched in glycine and arginine relative to AEPs (Fig. 2i). The physicochemical properties of each individual MEP are described in Table S4 and Fig. S3a–c.
Fig. 3. Antimicrobial activity, resistance to enzymatic degradation, and mechanism of action of modern and archaic EPs.

(a) Antimicrobial activity of the EPs. Briefly, a fix number of 106 bacterial cells per mL−1 was used in all the experiments. The modern and archaic EPs were two-fold serially diluted ranging from 128 to 2 μmol L−1 in a 96-wells plate and incubated at 37 °C for one day. After the exposure period, the absorbance of each well was measured at 600 nm. Untreated solutions were used as controls and minimal concentration values for complete inhibition were presented as a heat map of antimicrobial activities (μmol L−1) against nine pathogenic bacterial strains. All the assays were performed in three independent replicates and the heat map shows the mode obtained within the two-fold dilutions concentration range studied. (b) Schematic of the resistance to enzymatic degradation experiment, where peptides were exposed for a total period of six hours to fetal bovine serum that contains several active proteases. Aliquots of the resulting solution were analyzed by liquid chromatography coupled to mass spectrometry. (c) Modern and (d) archaic peptides had different degradation behaviors. In summary, archaic peptides are more resistant to enzymatic degradation than modern peptides. Experiments were performed in two independent replicates. (e) Schematic showing the behavior of 1-(N-phenylamino)naphthalene (NPN) the fluorescent probe used to indicate membrane permeabilization caused by the EPs. (f) Modern and (g) archaic EPs fluorescence values relative to the untreated control showing that MEPs are more efficient at permeabilizing the outer membrane of A. baumannii cells than polymyxin B (PMB) and archaic EPs. (h) Schematic of how 3,3′-dipropylthiadicarbocyanine iodide [DiSC3-(5)], a hydrophobic fluorescent probe, was used to indicate membrane depolarization caused by the EPs. (i) Modern and (j) archaic EPs fluorescence values relative to the untreated control showing that archaic peptides are much stronger depolarizers of the cytoplasmic membrane of A. baumannii cells than polymyxin B (PMB) and modern EPs. Experiments were performed in three independent replicates. Figure created with BioRender.com and the PyMOL Molecular Graphics System, Version 2.1 Schrödinger, LLC.
The mean estimated probability of flanking cleavage sites for MEPs did not differ substantially from that of inactive modern fragments (0.800 vs 0.803, respectively). To assess how many positive hits would have been missed if existing protease-specific predictors had been employed, we ran all MEP precursor proteins against the trypsin, ArgC, chymotrypsin, GluC, LysC, AspN, LysN, and LysargiNase models provided by DeepDigest27. With the exception of CALR-GWT20, no MEPs were predicted to be cleavage products of their respective precursors. However, CALR-GWT20 was identified by the LysargiNase model, the only DeepDigest model that is trained on yeast instead of human data.
The EP CBPZ-GSK24 from carboxypeptidase Z (UniProt ID: CBPZ_HUMAN) demonstrated the strongest antimicrobial activity in vitro, inhibiting Pseudomonas aeruginosa PA01 (8 μmol L−1), Pseudomonas aeruginosa PA14 (4 μmol L−1), Escherichia coli AIC221 (4 μmol L−1), Escherichia coli AIC222 (2 μmol L−1), and Acinetobacter baumannii ATCC19606 (16 μmol L−1). Fragment A7E2T1-SPR29 of uncharacterized protein A7E2T1_HUMAN also displayed activity against E. coli AIC221 (64 μmol L−1), E. coli AIC222 (64 μmol L−1), and A. baumannii ATCC19606 (8 μmol L−1). CALR-GWT20, encrypted in calreticulin (UniProt ID: CALR_HUMAN), displayed antimicrobial activity against colistin-resistant E. coli AIC222 at 128 μmol L−1 and A. baumannii ATCC19606 at 64 μmol L−1. Fragment XDH-AVA32, a subsequence of xanthine dehydrogenase/oxidase (UniProt ID: XDH_HUMAN), was active at 32 μmol L−1 against both E. coli AIC221 and AIC222 strains. ISK5-GKI32, part of the serine protease inhibitor kazal-type 5 (UniProt ID: ISK5_HUMAN), was also active at 128 μmol L−1 against both E. coli strains. LYSC-AVA39, encrypted in lysozyme C (UniProt ID: LYSC_HUMAN), displayed activity at 128 μmol L−1 against P. aeruginosa PA14 and both E. coli strains. Fragment CO7A1-AIG15 from human long-chain collage (UniProt ID: CO7A1_HUMAN) displayed activity at 32 μmol L−1 against P. aeruginosa PA14, while the protachykinin-1 (UniProt ID: TKN1_HUMAN) fragment TKN1-SSI27 was active at 64 μmol L−1 against A. baumannii ATCC19606.
Archaic EPs display antimicrobial activity in vitro
Six of 69 (8.7%) archaic protein fragments displayed in vitro antimicrobial activity (Fig. 3; Table S3). None of these fragments are reported as AMPs nor AMP subsequences in DBAASP41. The amino acid residue frequencies of AEPs differed from those of MEPs, recently described EPs11, and known natural and synthetic AMPs described in the DBAASP41 (Fig. 2). Similar to recently described EPs11, AEPs presented 9% and 16% higher content of aromatic residues than MEPs and DBAASP AMPs, respectively. AEPs display 100% and 59% higher polar residue content than DBAASP AMPs and MEPs, respectively. AEPs presented the lowest frequency of basic residues across all groups analyzed, with frequencies 58%, 53%, and 62% lower than those of DBAASP AMPs, MEPs, and recently described EPs. AEPs also contain many acidic residues, similar to MEPs. Aliphatic amino acid content was similar across all analyzed groups.
AEPs had lower median values for net charge and normalized hydrophobicity relative to MEPs, recently describe encrypted peptides11, and AMPs reported in the DBAASP41 (Figs. 2k,l; Tables S4). AEPs were enriched in threonine, isoleucine, methionine, and phenylalanine (Fig. 2i) and demonstrated an extremely high median propensity to aggregate (Tables S4). Although AEPs differed from MEPs and other EPs (Fig. 2i), their properties were still within the range of some known AMPs due to their (i) high hydrophobic content, (ii) low net charge, (iii) disordered conformation, and (iv) considerable tendency to aggregate (Fig. S3a–c). Physicochemical properties of each individual AEP are described in Table S4. The mean estimated probability of flanking cleavage sites for AEPs did not differ substantially from that of inactive archaic fragments (0.633 vs 0.617, respectively). As with MEPs, AEP precursors were run against the eight neural networks provided by DeepDigest27 to assess how many positive hits would have been missed by existing protease-specific predictors. No AEPs were rediscovered by these eight models.
Fragment PDB6I34D-ALQ29 of chain D of the Neanderthal glycine decarboxylase protein displayed activity against the largest number of organisms, moderately inhibiting both P. aeruginosa and E. coli strains (MICs from 32 to 128 μmol L−1). Denisovan transmembrane protein fragments A0A0S2IB02-AYT38 and A0A343EQH0-NVK38 displayed selective activity against P. aeruginosa PA01 at 128 μmol L−1. Similarly, fragment A0A343AZS4-FMA25 from chain 1 of Denisovan NADH-ubiquinone oxidoreductase and fragment A0A343EQH4-LAM11 from Neanderthal ATP synthase subunit A displayed selective activity against A. baumannii ATCC19606 at 128 μmol L−1. Neanderthal adenylosuccinate lyase fragment A0A384E0N4-DLI09 moderately inhibited A. baumannii ATCC19606 (128 μmol L−1), methicillin-resistant Staphylococcus aureus ATCC BAA-1556 (128 μmol L−1), and Staphylococcus aureus ATCC12600 (128 μmol L−1).
Resistance to proteolytic degradation
Among MEPs, those selected for clustering strongly with known AMPs were highly resistant to serum proteases (Fig. 3). Up to 85% of the initial concentration of these peptides remained after six hours of continuous exposure to serum proteases. Shorter MEPs (8-residues long) were less susceptible to cleavage than longer MEPs (up to 24 residues), with ~35% of the initial concentration present after six hours of exposure to proteases versus 15–20%, respectively. On average, AEPs were more susceptible to proteolytic degradation than MEPs. An exception to this was the 9-residue-long A0A384E0N4-DLI09, the shortest AEP tested. This short peptide resisted degradation for two hours, decreasing to 80% of its initial concentration, with ~55% of its initial concentration remaining after six hours of exposure (Fig. 3). Our experimentally validated results (Fig. 3c,d) are consistent with the notion that sequence composition, cleavage site number, and length play a role in proteolytic degradation20,42.
Membrane permeabilization and depolarization assays
MEPs and AEPs were investigated with fluorescent probes to determine how they affect the bacterial membrane. Positive control polymyxin B (PMB) is a peptide antibiotic having known permeabilizing and depolarizing effects (Figs. 3, S3d–k). In both assays, A. baumannii cells (Figs. 3, S3d,e, S9g,h) and P. aeruginosa PA01 (Fig. S3f,i) were exposed to the most active MEPs (CALR-GWT20, CBPZ-GSK4, TKN1-SSI27, and A7E2T1-SPR29 for A. baumannii) and AEPs (A0A384E0N4-DLI09 and A0A343EQH4-LAM11 for A. baumannii; A0A343EQH0-NVK38 and A0A0S2IB01-AYT38 for P. aeruginosa) at their respective MICs (Figs. 3, S3d–i).
All MEPs except TKN1-SSI27 presented permeabilizing profiles similar to that of PMB. MEP TKN1-SSI27 initially demonstrated the slowest permeabilizing kinetics, yet progressively displayed the highest permeabilization efficiency (Figs. 3, S3d–k). The only peptide with an overall permeabilization efficacy lower than PMB was MEP CALR-GWT20. All MEPs initially displayed relatively slow depolarizing kinetics that increased over time. After 30 minutes, modern peptides had stronger depolarizing effects than PMB, which were maintained until the end of the experiment (Fig. 3). No significant differences were observed among their depolarizing activities.
AEPs permeabilized A. baumannii cells similarly to (A0A343EQH4-LAM11) or less than (A0A384E0N4-DLI09) PMB, but had much stronger depolarizing effects (Figs. 3, S3d–k). AEPs A0A343EQH0-NVK38 and A0A0S2IB01-AYT38 permeabilized P. aeruginosa cells (Figs. 3, S3d–i), with higher relative fluorescence over time, indicating that P. aeruginosa was more sensitive than A. baumannii to these two peptides. Notably, A0A343EQH0-NVK38 and A0A0S2IB01-AYT38 were more strongly depolarizing than PMB for P. aeruginosa cells (Fig. S3d–i).
Hemolytic and cytotoxic activities
Five AEPs and seven MEPs displaying antimicrobial activity were tested for their hemolytic and cytotoxic activities by exposing them to human red blood cells (RBCs) and human embryonic kidney (HEK293T) cells, respectively. HEK293T and red blood cells were selected as they are widely used to assess the toxicity of antimicrobial agents18,43,44. We estimated by non-linear regression the peptide dose that led to 50% hemolysis (HC50) and 50% cytotoxicity (CC50).
Out of the seven tested MEPs, two (CBPZ-GSK24 and A7E2T1-SPR29) presented hemolytic activity at the concentration range tested, with HC50 values of 19.42 and 112 μmol L−1, respectively. Interestingly, CBPZ-GSK24 and A7E2T1-SPR29 contain a high frequency of tryptophan and arginine residues, respectively. These amino acid residues are commonly linked to increased hemolytic activity due to their hydrophobicity45 (in the case of tryptophan) and guanidyl group and long side chain (in the case of arginine), leading to increased interactions with the lipidic portion of the phospholipids that form the cell membrane bilayers46.
Those two MEPs also presented detectable CC50 values at 44.19 (for CBPZ-GSK24) and 12.78 μmol L−1 (for A7E2T1-SPR29). Additionally, the MEPs CALR-GWT20 and TKN1-SSI27 were not hemolytic (Table S3) but displayed CC50 values at 19.28 and 40.27 μmol L−1, respectively.
The three shorter AEPs tested (A0A384E0N4-DLI09, A0A343EQH4-LAM11, and PDB6I34D-ALQ29) did not elicit hemolysis. The two longer AEPs tested (A0A343EQH0-NVK38 and A0A0S2IB02-AYT38; 38-residues long each), which were also the best bacterial cell depolarizers, displayed HC50 values of 54.72 and 88.11 μmol L−1, respectively. None of the five AEPs tested showed cytotoxic effects, i.e., they had CC50 values higher than the maximum concentration tested of 128 μmol L−1.
Anti-infective efficacy in preclinical animal models
To assess whether modern and archaic EPs retain their in vitro antimicrobial activity in complex living systems, we probed their antimicrobial properties in two mouse models (Fig. 1): a skin abscess model and a preclinical murine thigh infection model.
For skin abscess experiments, we selected MEPs and AEPs with in vitro activity at concentrations lower than 64 μmol L−1 against A. baumannii. Bacterial loads of 106 cells in 20 μL of A. baumannii were administered to a skin abscess created on the back of each mouse (, Fig. 4a). A single dose of polymyxin B (control), MEP, or AEP was delivered as monotherapy to the infected area at MIC. Except for MEP A7E2T1-SPR39, all peptides demonstrated bactericidal effects in the skin abscess model (Fig. 4b). Activity levels were comparable to those of some of the most potent AMPs described to date in the literature using the same model, i.e., polybia-CP44 and PaDBS1R647. AEP A0A343EQH4-LAM11 and MEP CALR-GWT20 markedly reduced bacterial loads by 5–6 orders of magnitude against A. baumannii. No deleterious effects were observed in the animals (Fig. 4c).
Fig. 4. Anti-infective activity of modern and archaic EPs in pre-clinical animal models.

(a) Schematic of the skin abscess mouse model used to assess the anti-infective activity of the modern and archaic EPs with activity against A. baumannii cells. (b) Peptides were tested at their MIC in a single dose one hour after the establishment of the infection. Each group consisted of six mice (n = 6) and the bacterial loads used to infect each mouse derived from a different inoculum. (c) To rule out toxic effects of the peptides, mouse weight was monitored throughout the whole extent of the experiment. (d) Schematic of the neutropenic thigh infection mouse model in which bacteria is injected intramuscularly in the right thigh and modern and archaic EPs were administered intraperitoneally to assess their systemic anti-infective activity. Mice were euthanized six and eight days after the beginning of the experiment, i.e., two- and four-days post infection. Each group consisted of four mice (n = 4) and the bacterial loads used to infect each mouse derived from a different inoculum. (e) All EPs, except TKN1-SSI17, showed bacteriostatic activity inhibiting proliferation of bacteria. Peptides with bacteriostatic activity were able to maintain their effect during the entire experiment (eight days), except for A7E2T1-SPR39 that was effective for six days. (f) Mouse weight was monitored throughout the duration of the neutropenic thigh infection model (8 days total) to rule out potential toxic effects of cyclophosphamide injections, bacterial load, and the EPs. The antibiotic polymyxin B was used as positive control in both models. Statistical significance in b and e (day 6) was determined using one-way ANOVA, and in e (day 8) using Kruskal-Wallis test because of the non-normal distribution and unequal variance across groups; p values are shown for each of the groups, all groups were compared to the untreated control group; features on the violin plots represent median and upper and lower quartiles. Data in c and f are the mean plus and minus the standard deviation. Figure created with BioRender.com and the PyMOL Molecular Graphics System, Version 2.1 Schrödinger, LLC.
For the preclinical murine thigh infection with A. baumannii (, Fig. 4d), each peptide was injected at its MIC as a single intraperitoneal dose. The peptides used were active in vitro at concentrations lower than 64 μmol L−1 against A. baumannii. Two- and four-days post-treatment (6 and 8 days since the beginning of the experiment, respectively), all peptides tested presented bacteriostatic activity (Fig. 4e). In contrast, the PMB control displayed bactericidal activity and cleared the infection four days post-infection (day 8 since the beginning of the experiment). No significant changes in mouse weight were observed (Fig. 4f). As weight loss is a proxy for toxicity, these results suggest that the tested EPs are non-toxic.
Positive hit rates by peptide curation method
As panCleave is designed for general purpose cleavage site prediction rather than end-to-end bioactive EP detection, fragments were subsequently curated using various bioactivity prediction techniques. Modern and archaic protein fragment curation is described in the STAR Methods. We define a positive hit as a peptide that displays in vitro antimicrobial activity against at least one pathogen in at least one growth medium (MIC ≤128 μmol L−1). An experimental comparison of positive hit rates by MEP curation method revealed a range in success, with no single method exceeding 20%. Activity was observed for 5% (1/20) of randomly selected peptides; 6.7% (1/15) of peptides predicted to be antimicrobial by a peptide chemistry expert; 20% (3/15) of peptides predicted to be antimicrobial by ML model consensus; and 15% (3/20) of peptides identified by CD-HIT-2D48 (Fig. 2g). All ten sequences predicted to be inactive were indeed inactive against all pathogens tested experimentally (10/10, 100%). Though ML model consensus outperformed other curation methods, individual models ranged in positive hit rate from 6.7% (1/15) to 20% (3/15) (Fig. 2h). The results of exact tests indicated for low frequencies (Fisher, Boschloo, and Barnard) did not reveal statistically significant differences in hit rate across curation methods (at p ≤0.01), with the lowest p-value associated with CD-HIT-2D vs random selection using LB medium (under the Barnard exact test; p <0.087).
Discussion
This proof-of-concept study for ML-facilitated molecular de-extinction offers preliminary support for pharmacological prospection in paleoproteomes. We report previously uncharacterized antimicrobial subsequences encrypted within archaic human proteins, allowing access to bioactive peptides with unusual amino acid distributions. Results suggest that the de-extinction of archaic hominin peptides could expand the search space for protein therapies while remaining within the subspaces previously selected by human evolution. We hope that future work will refine the concept of molecular de-extinction and extend it beyond paleoproteome mining, perhaps to genomic mining for small molecules. Additionally, this work lends further support for computational EP prospection in the modern human proteome, which was previously proposed as an antibiotic discovery framework11. Future work might consider whether prospection within modern and archaic humans might minimize pharmacological risks such as toxicity, relative to mining evolutionarily distant or synthetic protein spaces.
This proof-of-concept does not make claims regarding the evolutionary history of the discovered EPs, whether they are (or were) proteolytically released from their precursors in vivo, nor whether they actively participate (or participated) in innate immune responses to bacterial infection. Rather, our results demonstrate the potential for therapeutics engineering to draw biological inspiration from the evolutionary processes that give rise to antimicrobial EPs in nature. However, we feel that future work in EP discovery could shed useful evolutionary insights. For example, given the essential role of AMPs in innate immunity, host defense peptides derived from archaic introgression may have been retained in the modern human proteome. The mammalian immunity protein lactoferrin contains antimicrobial motifs and displays evidence of positive selection across primates, perhaps entering the human population through convergent evolution or introgression from Neanderthals25. The maintenance of archaic AMPs in modern proteomes may merit further inquiry.
The modern and archaic peptides presented here may offer prototypes for antibiotic development. The observed membrane depolarization was unexpected11 and may have resulted from physicochemical differences between these peptides and human EPs mined with other computational methods11, which do not depolarize bacterial cytoplasmic membranes. If EPs operate via mechanisms of action independent of cytoplasmic membrane depolarization, antimicrobial EPs would be mechanistically distinct from AMPs from the DBAASP. EP diversity is, therefore, an intriguing area for future research.
While prior cleavage site classifiers favor protease-specific designs28–38, the panCleave random forest is trained on protease-agnostic data yet is highly accurate for multiple specific proteases (Fig. 2). To demonstrate the contributions of panCleave, we assessed how many positive hits would have been missed if existing protease-specific predictors had been employed instead. With the exception of fragment CALR-GWT20 from modern human calreticulin, no MEPs nor AEPs were predicted to be cleavage products of their respective precursors by eight protease-specific neural networks provided by DeepDigest27. This tool was selected for comparison due to its recent publication and accessibility as an open-source command line tool. Of note, CALR-GWT20 was rediscovered by the DeepDigest LysargiNase model, the only model in this suite that is not trained on human data. Instead, it is trained on yeast protease data and tested on E. coli protease data, making it unsuitable for human EP discovery. These results suggest that panCleave cleavage products may be distinct relative to those produced by existing protease-specific models. Further, the reasonable in silico performance of panCleave suggests that cleavage sites across the human proteome might share sufficient similarities to enable pan-protease model development, despite protease-substrate specificities. We hypothesize that algorithmic and data improvements could yield even better pan-protease models in the future, enabling higher quality proteome-wide searches.
Antimicrobial activity prediction remains challenging
The low accuracy of all peptide curation methods employed – including human expertise – suggests that the state-of-the-art in AMP activity prediction may be lacking (Fig. 2g). No method was found to significantly outperform random selection, though we caution against overinterpretation of significance tests given small sample sizes and low observed frequencies. Small sample sizes aside, the marked difference in curation method accuracy for predicted positives versus predicted negatives might suggest that more is currently understood about non-antimicrobial protein space than about AMP diversity. Low agreement and poor accuracy among popular, publicly available ML models for AMP discovery49–53 (Fig. 2h) may suggest that models often overfit to their training-testing distributions, rather than being generalizable across antimicrobial protein space. Furthermore, both CD-HIT48 and hierarchical k-means clustering (Fig. S3l,m,n) failed to identify insightful boundaries between peptides with and without antimicrobial activity. These results emphasize the importance of further characterizing antimicrobial protein space and highlight shortcomings in supervised and unsupervised learning for this research problem. Optimistically, while positive hit rates below 20% have been previously observed for ML-based AMP discovery54, recent deep learning models have enabled significantly higher hit rates21.
Rediscovery of a known antimicrobial motif
In addition to discovering antimicrobial EPs, the panCleave pipeline unintentionally uncovered a MEP containing a known bioactive subsequence. As lysozyme C is known to be bacteriolytic and to enhance immunoagent activity, it is unsurprising that a subsequence of this enzyme might itself display antimicrobial activity. A known EP of human lysozyme C is a subsequence of panCleave-generated MEP LYSC-AVA3955,56. The unintentional rediscovery of this antimicrobial motif in lysozyme C supports the use of the present pipeline for antimicrobial EP discovery. Similarly, all antimicrobial EPs discovered in the present work originate from proteins belonging to groups previously described in the EP literature. As peptide fragments were not curated based on their precursor protein, this further lends support for panCleave as an antimicrobial EP discovery tool.
Precedents for modern precursor proteins
Secreted proteins have previously been targeted for bioactive EP discovery11,57. A prior whole-proteome search found an overrepresentation of secreted and membrane-bound proteins among antimicrobial EP precursors, perhaps because AMPs are more likely to encounter pathogens in the extracellular environment11. As has been thoroughly reviewed, enzymes are common precursors for encrypted host defense peptides58. MEP precursor proteins identified in this study generally display catalytic activity (Table S5), with all MEP precursor groups having precedents in the EP literature.
Proteases across the tree of life not only catalyze EP release but also contain antimicrobial EPs58. In the present study, the MEP CBPZ-GSK24 is derived from the protease carboxypeptidase Z, which may participate in prohormone processing59.
Fragment CALR-GWT20 is derived from calreticulin, a calcium-binding chaperone protein that is highly conserved in multicellular life and is primarily localized to the endoplasmic reticulum. Calreticulin has been implicated in innate immune responses to bacterial infection in mammals, marine vertebrates, marine and terrestrial invertebrates, and plants60–63. Vasostatin is a well-characterized anti-angiogenesis and anti-tumor EP that is part of calreticulin64, lending precedent for the presence of bioactive subsequences in this precursor.
Serine protease inhibitors in diverse marine organisms have displayed antibacterial and antiviral innate immunity functions65–67. The observed antibacterial and antifungal activity of a kazal-type serine protease inhibitor in honeybee venom appears to act through microbial serine protease inhibition16. MEP ISK5-GKI32 is encrypted within serine protease inhibitor kazal-type 5, which is known to yield EPs with protease inhibition activity when cleaved by the protease furin68. The downregulation, deletion, and mutation of serine protease inhibitor kazal-type 5 are associated with inflammation, compromised skin-barrier function, atopic dermatitis, rosacea, and Netherton syndrome69–71. Assaying ISK5-GKI32 against skin microbes implicated in these conditions could be an area for future research.
Oxidoreductases are known to contain antimicrobial EPs in modern humans57,58,72, Bacillus73, Desulfocurvibacter74, Saccharomyces75, and Physcomitrella76. In the present study, MEP XDH-AVA32 is a subsequence of the oxidoreductase xanthine dehydrogenase, which catalyzes the oxidative metabolism of purines. CO7A1-AIG15 is contained within the collagen alpha-1(VII) chain (syn. long-chain collagen), whose Gene Ontology molecular functions include serine-type endopeptidase inhibitor activity and extracellular matrix structural functionality77.
MEP TKN1-SSI27 is contained within protachykinin-1, a neuropeptide implicated in antibacterial and antifungal humoral responses and defense responses to both Gram-negative and Gram-positive bacteria59. A7E2T1-SPR29 originates from the uncharacterized protein fragment A7E2T1_HUMAN, which shares 99.21% identity with both the Homo sapiens neuropeptide W preproprotein (BLAST E-value 4e-78) and prepro-Neuropeptide W polypeptide (BLAST E-value 1e-77)78. A7E2T1_HUMAN enables G protein-coupled receptor binding, according to Gene Ontology77.
Archaic precursors in the mitochondrial proteome
As publicly available Denisovan and Neanderthal data originate from the mitochondrial proteomes of these species, the AEP precursor proteins we identified are generally mitochondrial transmembrane proteins associated with transport, mitochondrial activity, and purine or ATP synthesis (Table S5). Precursor proteins were submitted to BLAST78 to assess similarity to modern human analogs (Table S5). On average, AEP precursor proteins shared 99.49% identity with a modern human protein (standard deviation <0.003). All AEP precursors identified here belong to protein groups with precedents in the literature on encrypted host defense peptides, lending support for the use of panCleave for archaic human AMP prospection.
As discussed above, host defense peptides are known to be encrypted in oxidoreductases from across the kingdoms of life. AEP A0A343AZS4-FMA25 originated from the Denisovan transmembrane protein NADH-ubiquinone oxidoreductase chain 1 (EC 7.1.1.2), while A0A343EQH0-NVK38 is a subsequence of the 347-residue Neanderthal NADH-ubiquinone oxidoreductase chain 2 (EC 7.1.1.2). AEP A0A0S2IB02-AYT38 is a subsequence of the Denisovan cytochrome C oxidase subunit 1 (EC 7.1.1.9), a transmembrane protein that participates in the respiratory chain by catalyzing the reduction of oxygen to water.
Precedents for lyases as precursor proteins include an AMP encrypted within the pterin-4-alpha-carbinolamine dehydratase of Mus musculus74. AEP A0A384E0N4-DLI09 is a subsequence of the Neanderthal adenylosuccinate lyase (syn. adenylosuccinase; EC 4.3.2.2), a coiled lyase involved in purine biosynthesis. AEP PDB6I34D-ALQ29 originates from chain D of the 984-residue Neanderthal lyase glycine decarboxylase.
The ATP synthase of the blowfly Sarconesiopsis magellanica is known to contain an encrypted AMP, and compounds excreted and secreted by this species have displayed antibacterial activity79. Likewise, the Neanderthal ATP synthase subunit A was found to contain AEP A0A343EQH4-LAM11.
Comparative performance evaluation for the machine learning model
Direct comparative performance analyses were not possible for the panCleave model due to data availability, necessitating indirect comparisons (i.e., comparing reported performance metrics rather than testing each model on the exact same evaluation dataset). As panCleave is the only pan-protease cleavage classifier for human proteins, its overall accuracy cannot be benchmarked against an existing model. On the other hand, no existing protease-specific model was known to release exact training data, and very few models released test data. Training and testing data for prior models would be necessary to cross-reference against the panCleave data splits in order to prevent data leakage (i.e., to ensure that testing instances were not present at training time), which could bias performance metrics in favor of any given model. The inability to directly compare panCleave with existing models highlights the importance of publicly releasing all training and testing data. Without the release of these data, the ML community is likely to face a significant reproducibility problem80–84.
Future questions for molecular de-extinction and evolutionary medicine
Although the potential for molecular de-extinction to support drug discovery is a new open question, this proof-of-concept study for ML-facilitated molecular de-extinction offers preliminary support for the value of pharmacological prospection in paleoproteomes. The influence of archaic genomes on contemporary innate immunity could guide future interest in this line of inquiry. Encounters among modern humans, Neanderthals, and Denisovans resulted in multiple known admixture events that produced reproductively viable hybrid offspring85,86. As Neanderthals and Denisovans evolved adaptive advantages in Eurasian environments over millennia, maintenance of introgressed alleles may have conferred fitness benefits to recently arrived modern humans87. An overrepresentation of introgressed alleles has been observed in innate immunity genes relative to other genomic regions88, resulting in a notable contribution to modern immune variation89,90. Of current interest, the Neanderthal OAS1 haplotype has been seen to provide protection against COVID-19 infection in individuals of European descent91, though an elevated risk of severe infection has also been linked to a 50 kb region of Neanderthal origin92. Given their essential role in innate immunity, archaic host defense peptides may have entered the modern proteome. The present work does not pursue this question, though it may merit further inquiry. These questions of evolutionary medicine can be extended outside the Homo lineage, as has been done for ancient extant marsupial and monotreme host defense peptides22.
Limitations of the study
The following limitations should be noted when interpreting the present work. The quality of the panCleave model was impacted by multiple data limitations. The negative dataset is likely to be noisy, as these observations were drawn randomly rather than through experimental validation. Positive training data may also be noisy, as the database from which they originate39 is aggregated across diverse experimental data sources. Testing panCleave against out-of-distribution data could further attest to overfitting and model quality. Further, future work could address optimal input length: though MEROPS reports cleavage sites as 8-residue P4:P4’ flanking sites, longer flanking sites might provide useful information for cleavage site prediction29.
The study design assumes that the extremely high similarity among the modern human, Neanderthal, and Denisovan proteomes is also suggestive of high conservation in protease activity (e.g., protease-substrate specificity). That is to say, we assume that a modern human protease with preference for a given amino acid sequence will also cleave Neanderthal or Denisovan proteins containing that subsequence. Though these assumptions leave claims of discovering naturally occurring archaic EPs unjustifiable, they do not undermine the present objective of bioinspired protein engineering. Relatedly, the panCleave model is designed for cleavage site prediction, not bioactive EP detection. Thus, the success of the present pipeline hinges both on panCleave and on subsequent bioactivity prediction. Assuming sufficient data availability, future models could be designed for end-to-end antimicrobial EP detection. Such a model might lend itself to deeper evolutionary insights than the current pipeline.
Finally, the low positive hit rates observed across curation methods must be improved to make EP prospection tractable. While the present EP discovery method is not intended to compete with traditional industrial-scale bioprospecting, future research should consider commercial viability and efficiency at scale.
STAR METHODS
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact upon reasonable request, Cesar de la Fuente-Nunez (cfuente@upenn.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Training data, testing data, and code used to develop the machine learning model are freely available on GitLab (https://gitlab.com/machine-biology-group-public/pancleave).
All data pertaining to the experimental validation of generated peptides are available in the Supplementary Data.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
Experimental model
Bacterial strains and growth conditions
Escherichia coli ATCC11775, Acinetobacter baumannii ATCC19606, Pseudomonas aeruginosa PA01, Pseudomonas aeruginosa PA14, Staphylococcus aureus ATCC12600, Staphylococcus aureus ATCC BAA-1556 (methicillin-resistant strain), Escherichia coli AIC221 [Escherichia coli MG1655 phnE_2::FRT (control strain for AIC 222)] and Escherichia coli AIC222 [Escherichia coli MG1655 pmrA53 phnE_2::FRT (polymyxin resistant; colistin-resistant strain)], and Klebsiella pneumoniae ATCC13883 were grown and plated on Luria-Bertani (LB) or Pseudomonas Isolation (Pseudomonas aeruginosa strains) agar plates and incubated overnight at 37 °C from frozen stocks. After incubation, one isolated colony was transferred to 5 mL of medium (LB) or basal medium with glucose (BM2), and cultures were incubated overnight (16 h) at 37 °C. The following day, inocula were prepared by diluting the overnight cultures 1:100 in 5 mL of the respective media and incubating them at 37 °C until bacteria reached logarithmic phase (OD600 = 0.3–0.5).
Human embryonic kidney and red blood cells
Human embryonic kidney (HEK293T) cells were obtained from the American Type Culture Collection (ATCC) and grown 37 °C in a humidified atmosphere containing 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1% antibiotics (penicillin and streptomycin) and 10% fetal bovine serum (FBS). Red blood cells were purchased from Zen-Bio from a certified healthy donor (blood type A−).
Skin abscess infection mouse model
A. baumannii ATCC19606 cells were grown in tryptic soy broth (TSB) medium to an OD600 = 0.5. Next, cells were washed twice with sterile PBS (pH 7.4, 13,000 rpm for 1 min) and resuspended to a final concentration of 2×105 colony-forming units (CFU) mL−1 for A. baumannii. Six-week-old female CD-1 mice from Charles River (stock number 18679700–022) were anesthetized with isoflurane for two minutes and had their backs shaved. A superficial linear skin abrasion was made with a needle to damage the stratum corneum and upper layer of the epidermis. An aliquot of 20 μL containing the bacterial load resuspended in PBS was inoculated over the scratched area (day 0, beginning of the experiment). One hour after the infection, peptides diluted in water at their MIC value were administered to the infected area (day 0, beginning of the experiment). Animals were euthanized and the area of scarified skin was excised two- and four-days post-infection (days 2 and 4 since the beginning of the experiment, respectively), homogenized using a bead beater for 20 minutes (25 Hz), and 10-fold serially diluted for CFU quantification. Each group had a total of six mice (), where three mice were infected with one inoculum and the remaining three mice were infected with another inoculum. Statistical significance was determined using one-way ANOVA; p values are shown for each of the groups, all groups were compared to the untreated control group. The skin abscess infection mouse model was approved by the University Laboratory Animal Resources (ULAR) from the University of Pennsylvania (Protocol 806763).
Thigh infection mouse model
The 4–6 weeks old female CD-1 mice from Charles River (stock number 18679700–022) were rendered neutropenic by two doses of cyclophosphamide (150 mg Kg−1) applied intraperitoneally with an interval of 72 h (days 0 and 3 since the beginning of the experiment, respectively). One day after the last dose of cyclophosphamide (day 4 since the beginning of the experiment), the mice were infected intramuscularly in their right thigh with a bacterial load of 106 CFU mL−1 of A. baumannii ATCC19606. The bacteria were grown in tryptic soy broth (TSB), washed twice with PBS (pH 7.4), and resuspended to the desired concentration. Two hours later, peptides resuspended in water were administered intraperitoneally. Prior to each injection, mice were anesthetized with isoflurane and monitored for respiratory rate and pedal reflexes11,18,44. Next, we monitored the establishment of the infection and euthanized the mice. The infected area was excised two- and four-days post-infection (days 6 and 8 since the beginning of the experiment, respectively), homogenized using a bead beater for 20 min (25 Hz), and 10-fold serially diluted for CFU quantification in MacConkey agar plates. The experiments were performed with 4 mice per group. Statistical significance in Fig. 4e (day 6) was determined using one-way ANOVA, and in Fig. 4e (day 8) using Kruskal-Wallis test because of the non-normal distribution and unequal variance across groups; p values are shown for each of the groups, all groups were compared to the untreated control group. The thigh infection mouse model was approved by the University Laboratory Animal Resources (ULAR) from the University of Pennsylvania (Protocol 807055).
Method details
Antibacterial assays
The 69 curated fragments were subjected to broth microdilution assays to assess in vitro antimicrobial activity. Minimum inhibitory concentration (MIC) values of the peptides were determined by using the broth microdilution technique93 with an initial inoculum of 5×106 cells in LB or BM2 medium supplemented with glucose in nontreated polystyrene microtiter 96-well plates. Peptides were added to the plate as solutions in water at concentrations ranging from 0 to 128 μmol L−1. The MIC was considered as the lowest concentration of peptide that completely inhibited the visible growth of bacteria after 24 h of incubation of the plates at 37 °C. Plates were read in a spectrophotometer at 600 nm. All assays were done as three independent replicates.
Resistance to proteolytic degradation assays
To evaluate the resistance to enzymatic degradation, EPs were incubated in fetal bovine serum (FBS)94. Peptides were exposed to an aqueous solution of 25% FBS at a concentration of 2 mg mL−1 for 6 h at 37 °C. Aliquots were collected after 0, 0.5, 1, 2, 4, and 6 h, and 10 μL of trifluoroacetic acid was added to each sample and incubated for 10 min at 4 °C. Samples were then processed in a Waters Acquity UPLCMS equipped with a photodiode array detector (190–400 nm data collection) and a Waters TQD triple quadrupole MSMS, with 5 μL injections. The column used was a Waters Acquity UPLC HSS C18, 1.8 μm (2.1 mm x 50 mm). The mobile phases used were A (100% water with 0.1%, v/v, formic acid) and B (100% acetonitrile with 0.1%, v/v, formic acid), Fisher optima grades. Measurements were made by ionization ESI +/− simultaneous over m/z 100–2000 Da. The percentage of remaining undamaged peptide was calculated by integrating the area under the curve related to the peptide at time point zero.
| Time (min) | A (%) | B (%) | Flow rate (mL min−1) |
|---|---|---|---|
|
| |||
| 0 | 95 | 5 | 0.5 |
| 0.5 | 95 | 5 | 0.5 |
| 2.5 | 5 | 95 | 0.5* |
| 3 | 5 | 95 | 0.5 |
| 3.25 | 5 | 95 | 0.5 |
linear gradient.
Membrane permeabilization assays
The membrane permeability of the peptides was determined by using the 1-(N-phenylamino)naphthalene (NPN) uptake assay11. NPN fluoresces weakly in extracellular environments and strongly when in contact with bacterial membrane lipids (Figs. 3e–g, S3d,e,f, and S3j), but only permeates the bacterial outer membrane when membrane integrity is compromised. A. baumannii ATCC19606 and P. aeruginosa PA01 were grown to an OD600 of 0.4, centrifuged (10,000 rpm at 4 °C for 10 min), washed, and resuspended in buffer (5 mmol L−1 HEPES, 5 mmol L−1 glucose, pH 7.4). Next, 4 μL of NPN solution (0.5 mmol L−1 – working concentration of 10 μmol L−1 after dilutions) was added to 100 μL of the bacterial solution in a white 96-well plate. The background fluorescence was recorded at and . Peptide solutions in water (100 μL solution at their MIC values) were added to the 96-well plate, and fluorescence was recorded as a function of time until no further increase in fluorescence was observed (20 min).
The relative fluorescence was calculated using a non-linear fit. The untreated control (buffer + bacteria + fluorescent dye) was used as baseline and the following equation was applied to reflect % of difference between the baseline and the sample:
Membrane depolarization assays
The ability of the peptides to depolarize the cytoplasmic membrane was determined by measurements of fluorescence of the membrane potential-sensitive dye, 3,3’-dipropylthiadicarbocyanine iodide [DiSC3-(5)]11, a potentiometric fluorophore that fluoresces in response to an imbalance of the cytoplasmic membrane transmembrane potential (Fig. 3h–j, S3g,h,i, and S3k). Briefly, A. baumannii ATCC19606 and P. aeruginosa PA01 were grown at 37 ºC with agitation until they reached mid-log phase (OD600 = 0.5). The cells were then centrifuged and washed twice with washing buffer (20 mmol L−1 glucose, 5 mmol L−1 HEPES, pH 7.2) and resuspended to an OD600 of 0.05 in the same buffer containing 0.1 mol L−1 KCl. The cells (100 μL) were then incubated for 15 min with 20 nmol L−1 of DiSC3(5) until the reduction of fluorescence stabilized, indicating the incorporation of the dye into the bacterial membrane. Membrane depolarization was then monitored by observing the change in the fluorescence emission intensity of the membrane potential-sensitive dye, DiSC3-(5) (, ), after the addition of the peptides (100 μL solution at MIC values).
The relative fluorescence was calculated using a non-linear fit. The untreated control (buffer + bacteria + fluorescent dye) was used as baseline and the following equation was applied to reflect % of difference between the baseline and the sample:
A machine learning-based protein informatics pipeline for computational proteolysis
The panCleave Python pipeline is a protein informatics tool that uses ML to perform computational proteolysis: the in silico digestion of human proteins into peptide fragments (Figs. 1, S1a). As the MEROPS database reports cleavage sites in terms of 8-residue P4:P4’ flanking sites39, the panCleave random forest model predicts cleavage sites within each 8-residue subsequence of a given protein. Thus, the minimum length for an input protein is also 8 amino acids. The domain model for this Python pipeline is visualized in Fig. S1a.
When presented with an 8-residue input, panCleave returns a binary label classifying the sequence as a cleavage site or non-cleavage site. Additionally, panCleave returns the estimated probability of class membership (i.e., the estimated probability that the assigned label for a given input is correct). Estimated probability reporting allows the user to filter predicted fragments by probability threshold, e.g., to bias fragment curation toward predictions with high estimated probability.
The panCleave pipeline performs the following procedure per input protein:
Sliding window: Every 8-residue contiguous subsequence of the protein sequence string is computed.
Encoding: Each subsequence is converted to a numerical feature vector using the ProtFP encoding method95.
Prediction: The label and estimated probability of class membership are computed per subsequence.
Fragmentation: The full protein string is tokenized at each predicted cleavage site, yielding a list of peptide fragments.
Additional utility functions are also provided, including prediction filtering functionality and FASTA file conversion. Pipeline source code, tutorial notebooks, and documentation are available on GitLab (https://gitlab.com/machine-biology-group-public/pancleave).
Model training and testing data
The panCleave random forest was trained and tested on all human protease substrates in the MEROPS Peptidase Database as of June 202039. Substrate sequences for all human proteases available in MEROPS encompassed 369 proteases representing 6 catalytic types (Cysteine, Metallo, Serine, Aspartic, Threonine, and Mixed), 31 clans, and 73 families. Protease representation and amino acid frequency distributions for the MEROPS dataset are visualized in Figs. S1b–f.
Model training and testing used a balanced dataset of 49,634 observations. As MEROPS reports cleavage sites as 8-residue P4:P4’ flanking sites, all observations are 8 residues in length. Cleavage site data were curated from MEROPS ( unique positive observations) and combined with 8-residue sequences generated from the human proteome and random protein space ( unique negative observations). Redundant sequences, sites containing non-canonical amino acids, and sites of length shorter than 8 residues were removed from the positive dataset. Negative observations were generated by three methods, each constituting one third of the negative dataset: randomly selected 8-residue contiguous subsequences of the human proteome, randomly generated sequences adhering to the amino acid frequencies of the human proteome, and randomly generated sequences with no amino acid frequency constraints. No sequences were present in both the positive and negative datasets.
Training and 10-fold cross-validation were performed using 80% of total observations (). The remaining 20% of observations were reserved as an independent test set (). The train-test split was stratified by label to ensure that each split maintained a label distribution representative of the entire dataset (50% positive observations and 50% negative observations). The complete training dataset, testing dataset, and Python code are available on GitLab and as supplemental files (https://gitlab.com/machine-biology-group-public/pancleave).
Hemolysis assays
We evaluated the lysis of human erythrocytes upon treatment with AEPs and MEPs to assess hemolytic activity96. Human red blood cells (RBCs) were obtained from ZenBio from certified healthy donor. The cells were collected using heparin as an anti-coagulant. Briefly, the RBCs were washed four times with PBS (pH 7.4) through centrifugation at 800 g for 10 min. Next, we mixed aliquots of 200-fold diluted cells (75 μL) with peptide solutions ranging from 2 to 128 μmol L−1 (75 μL) in round bottom 96-well plates and incubated the mixture for 4 h at room temperature. Following incubation, we centrifuged the plate at 1,300 g for 10 min to separate the cells and debris. Then, we transferred the supernatant (100 μL) from each well to new flat bottom 96-well plates for absorbance reading at 405 nm using a plate reader. The percentage of hemolysis was defined by normalizing the absorbance values obtained with those from the negative control samples (i.e., samples containing only PBS) and to those of the positive control samples (i.e., samples containing 1% SDS in PBS solution, v/v).
Cytotoxicity assays
Human embryonic kidney (HEK293T) cells from the American Type Culture Collection were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1% antibiotics (penicillin and streptomycin) and 10% fetal bovine serum (FBS). The cells were grown at 37 °C in a humidified atmosphere containing 5% CO2. HEK293T cells were seeded into cell treated 96-well plates at the density of 5×103 cells per well one day before the treatment with increasing concentrations of peptide (8–128 μmol L−1). After the incubation with each peptide, we performed the (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) tetrazolium reduction assay (MTT assay)96. Briefly, MTT reagent dissolved at 0.5 mg mL−1 in medium without phenol red was used to replace cell culture supernatants (100 μL per well), and the samples were incubated for 4 h at 37°C to obtain the insoluble formazan salts. The resulting salts were then solubilized in 0.04 mol L−1 HCl in anhydrous isopropanol and quantified by measuring the absorbance at 570 nm using a spectrophotometer.
Hyperparameter tuning and final model selection
Six classifiers were implemented using scikit-learn (https://scikit-learn.org/) and TensorFlow (https://www.tensorflow.org/): Gaussian Process (GP), K-Nearest Neighbor (KNN), Naive Bayes (NB), Random Forest (RF), Recurrent Neural Network (RNN), and Support Vector Machine (SVM). Each algorithm was trained and tested on 5 input representations: one-hot encoding, ProtFP95, ST-Scale97, Z-Scale98, and UniRep99. The resulting 30 candidate models each underwent Bayesian search hyperparameter tuning using the skopt Python package (https://scikit-optimize.github.io/optimize.github.io/) on the Stampede2 supercomputer (Texas Advanced Computing Center, The University of Texas at Austin, TX, USA). Hyperparameter tuning used 10-fold cross-validation on the training set.
Three tuned finalists were selected on the basis of superior 10-fold cross-validation: RF, RNN, and SVM, each trained on the ProtFP encoding. Finalists were assessed via three performance metrics, each computed using scikit-learn (https://scikit-learn.org/): test set accuracy, area under the receiver-operating characteristic curve (AUC-ROC), and average precision. Additionally, accuracy was assessed when thresholding the estimated probability of class membership at ≥50%, ≥60%, ≥70%, ≥80%, and ≥90%. The tradeoff between increases in accuracy and decreases in total valid observations at a given estimated probability threshold was quantified and visualized.
Among the 30 candidate classifiers, an RF trained on the ProtFP protein encoding95 was selected as the final model on the basis of marginally superior 10-fold cross-validation accuracy, AUC-ROC, average precision, and estimated probability thresholding. The final RF used 400 estimators (i.e., individual trees) and a Shannon information gain criterion. Under scikit-learn version 0.23.2, the model object is expressed with the following hyperparameter values:
RandomForestClassifier(bootstrap = True, ccp_alpha = 0.0, class_weight = None, criterion = “entropy”, max_depth = None, max_features = 3, max_leaf_nodes = None, max_samples = None, min_impurity_decrease = 0.0, min_impurity_split = None, min_samples_leaf = 5, min_samples_split = 2, min_weight_fraction_leaf = 0.0, n_estimators = 400, n_jobs = −1, oob_score = False, random_state = 5, verbose = 0, warm_start = False)
The final preserved model and a Jupyter Notebook that replicates training are available on GitLab, with all hyperparameter values listed in the repository README (https://gitlab.com/machine-biology-group-public/pancleave).
Modern protein fragment curation
The panCleave pipeline was run on all modern human proteins tagged with the keyword “secreted” in UniProt59 as of February 2021 (). Length distributions, amino acid frequencies, and PANTHER (http://www.pantherdb.org/)100 classification data characterizing the modern secreted protein dataset are visualized (Figs. S1b–S2e). The initial 80,729 unique cleavage products were reduced to 3,738 fragments by filtering such that peptide lengths were between 8 and 40 residues, flanking cleavage sites were of an estimated probability of 0.8 or higher (mean 0.803), and no fragments were subsequences of other fragments in the dataset.
Four curation methods were used to select panCleave-generated fragments for synthesis: 1) human expert curation; 2) ML model consensus using six publicly available AMP classifiers49–53; 3) clustering against an in-house database of experimentally validated AMPs using CD-HIT-2D, an algorithm for sequence alignment and comparison of protein databases48; and 4) random selection with no sampling bias. Twenty fragments were selected by each curation method ( total). In each case, fragment length was restricted to 8 to 40 amino acids.
Selection by a human expert entailed fully manual curation of 15 peptides predicted to be antimicrobial and 5 peptides predicted to be inactive. Consensus prediction used six publicly available ML-based AMP models: amPEPpy (https://github.com/tlawrence3/amPEPpy)49, iAMPpred (http://cabgrid.res.in:8080/amppred/)50, Macrel (https://www.big-data-biology.org/software/macrel/biology.org/software/macrel/)51, and three models available from AxPEP (https://app.cbbio.online/ampep/home)52,53. A positive consensus vote by at least three of these six models was required for selecting the 15 peptides predicted to be active. A negative consensus vote by all six models was required for selecting the 5 peptides predicted to be inactive. Random selection used no biasing criteria. The CD-HIT-2D clustering algorithm (http://weizhong-lab.ucsd.edu/cdhit-web-server/cgi-bin/index.cgilab.ucsd.edu/cdhit-web-server/cgi-bin/index.cgi)48 was used to rank fragments by percent similarity to an in-house dataset of experimentally validated AMPs, and the top 20 hits were selected as predicted AMPs for experimental validation.
Archaic protein fragment curation
The panCleave pipeline was run on all Neanderthal and Denisovan proteins available in UniProt59 and NCBI (https://www.ncbi.nlm.nih.gov/protein/) as of February 2021 ( and , respectively). Six Neanderthal proteins (9.1%) and one Denisovan protein (3.8%) were identical to proteins in the modern proteome and were excluded. Results were filtered such that all fragments were between 8 and 40 residues in length and no fragments were subsequences of other fragments in the dataset. This filtering process yielded 249 unique Neanderthal cleavage products and 167 unique Denisovan cleavage products. No sequences were shared between modern human and Neanderthal panCleave results, nor between modern humans and Denisovans. There were 127 fragments common to both Neanderthals and Denisovans, leaving 289 non-redundant archaic fragments in total.
Archaic fragments were removed if present as subsequences of any protein in the modern human proteome. Archaic sequences were cross-referenced against all annotated and non-annotated modern human proteins () and all isoforms () available in UniProt as of February 2021. Subsequently, 73 archaic-only fragments remained (73/289, 25.3%), each with flanking cleavage sites of an estimated probability of 0.6 or higher (mean = 0.618). Of these, four were not selected for chemical synthesis because of their high hydrophobicity and aggregation propensity (i.e., WIGGQPVSYPFIIIG, VVAGVFLLIRFHPLA, LYDYGRWLVVVTGWTLFVGVYVVIE, and MTMYTTMTTLTLTSLIPPILTTLINPN), leaving 69 archaic-only fragments to be tested in vitro. All peptides used in the experiments were purchased from AAPPTec (Louisville, KY; USA).
Quantification and statistical analysis
Reproducibility of the experimental assays
Unless otherwise stated, all assays were performed in three independent biological replicates as indicated in each figure legend and Experimental Models and Methods details sections. The values obtained for hemolytic and cytotoxic activities were estimated by non-linear regression based on the screen of peptides in a gradient of concentrations and represent the hemolytic and the cytotoxic concentration values needed to lyse and kill 50% of the cells present in the experiment. For the cytotoxic activity assays, two technical replicates were performed within each of the three biological replicates. In the skin abscess and thigh infection mouse models, we used six and four mice per group, respectively, following established protocols approved by the University Laboratory of Animal Resources (ULAR) of the University of Pennsylvania.
Statistical tests
In the mouse experiments, the statistical significance was determined using one-way ANOVA followed by Dunnett’s test. In Fig. 4e (day 8) of the thigh infection model, the statistical significance was determined using Kruskal-Wallis test because of the non-normal distribution and unequal variance across groups. All the p values are shown for each of the groups, all groups were compared to the untreated control group.
Statistical analysis
All calculation and statistical analyses of the experimental data were conducted using GraphPad Prism v.9.1 and computational data were performed in Python. Statistical significance between different groups was calculated using the tests indicated in each figure legend. No statistical methods were used to predetermine sample size.
Supplementary Material
Key resources table
| Reagent or Resource | Source | Identifier |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Acinetobacter baumannii | American Type Culture Collection | ATCC 19606 |
| Escherichia coli | American Type Culture Collection | ATCC 11775 |
| Escherichia coli | Escherichia coli MG1655 phnE_2::FRT | AIC221 |
| Escherichia coli |
Escherichia coli MG1655 pmrA53 phnE_2::FRT (polymyxin-resistant; colistin-resistant strain) |
AIC222 |
| Klebsiella pneumoniae | American Type Culture Collection | ATCC 13883 |
| Pseudomonas aeruginosa | PA01 | |
| Pseudomonas aeruginosa | PA14 | |
| Staphylococcus aureus | American Type Culture Collection | ATCC 12600 |
| Staphylococcus aureus | American Type Culture Collection | ATCC BAA-1556 (methicillin-resistant strain) |
|
| ||
| Cell lines and red blood cells | ||
|
| ||
| Human embryonic kidney (HEK293T) cells | American Type Culture Collection | ATCC CRL-3216 |
| Red blood cells | Zen-Bio | SER-10MLRBC |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: CD-1 | Charles River | 18679700–022 |
|
| ||
| Chemicals | ||
|
| ||
| Luria-Bertani broth | BD | 244620 |
| Tryptic soy broth | Sigma | T8907–1KG |
| Agar | Sigma | 05039 |
| MacConkey agar | RPI | M42560–500.0 |
| Phosphate buffer saline | Sigma | P3913–10PAK |
| Ammonium sulfate [(NH4)2SO4] | Chem Cruz | 7783–20-2 |
| Dipotassium hydrogen phosphate (K2HPO4) | Sigma | SLBR8555V |
| Monobasic potassium phosphate (KH2PO4) | Macron | 164500 |
| Iron (II) sulfate (FeSO4) | Amresco | 387 |
| Magnesium sulfate (MgSO4) | Amresco | 1333C215 |
| Glucose | Sigma | G5767 |
| 1-(N-phenylamino)naphthalene | Sigma | 104043 |
| 3,3’-dipropylthiadicarbocyanine iodide | Sigma | 43608 |
| HEPES | Fisher | BP310–100 |
| Potassium chloride (KCl) | Sigma | P3911 |
| Fetal Bovine Serum (FBS) | ThermoFisher | 10437–028 |
|
| ||
| Software and Algorithms | ||
| Python 3 | https://www.python.org/ | |
| scikit-learn | https://scikit-learn.org/ | |
Highlights:
Machine learning guides bioinspired prospection for encrypted antimicrobial peptides.
Modern and extinct human proteins harbor antimicrobial subsequences.
Archaic encrypted peptides display in vitro and in vivo activity with low host toxicity.
Paleoproteome mining offers a framework for antibiotic discovery.
Acknowledgments:
Cesar de la Fuente-Nunez holds a Presidential Professorship at the University of Pennsylvania and acknowledges funding from the Procter & Gamble Company, United Therapeutics, a BBRF Young Investigator Grant, the Nemirovsky Prize, Penn Health-Tech Accelerator Award, and the Dean’s Innovation Fund from the Perelman School of Medicine at the University of Pennsylvania. Research reported in this publication was supported by the Langer Prize (AIChE Foundation), the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM138201, and the Defense Threat Reduction Agency (DTRA; HDTRA11810041, HDTRA1-21-1-0014, and HDTRA1-23-1-0001). Jacqueline R. M. A. Maasch acknowledges support from the University of Pennsylvania GAPSA-Provost Fellowship for Interdisciplinary Innovation and the Open Knowledge Foundation Frictionless Data for Reproducible Research Fellowship, funded by the Alfred P. Sloan Foundation. Computational resources included the Stampede2 supercomputer (Texas Advanced Computing Center, The University of Texas at Austin, TX, USA). We thank Dr. Mark Goulian for kindly donating the following strains: Escherichia coli AIC221 [Escherichia coli MG1655 phnE_2::FRT (control strain for AIC 222)] and Escherichia coli AIC222 [Escherichia coli MG1655 pmrA53 phnE_2::FRT (polymyxin resistant)]. We thank Dr. Charles W. Ross III, Director: Automated Synthesis and Characterization, Center Director MS, BCRC, HTE-Analytical of the University of Pennsylvania Chemistry Department for providing chromatographic/mass spectral method development, analyses, and data interpretation as needed. We thank Dr. Gui-Shuang Ying from the Center for Preventive Ophthalmology and Biostatistics (CPOB) Consulting Service for advice on our statistical analyses. We thank Dr. Karen Pepper for editing the manuscript and de la Fuente Lab members for insightful discussions. Figures created with BioRender.com are attributed as such. Molecules were rendered using the PyMOL Molecular Graphics System, Version 2.1 Schrödinger, LLC.
Footnotes
Declaration of interests:
Cesar de la Fuente-Nunez provides consulting services to Invaio Sciences and is a member of the Scientific Advisory Boards of Nowture S.L. and Phare Bio. The de la Fuente Lab has received research funding or in-kind donations from United Therapeutics, Strata Manufacturing PJSC, and Procter & Gamble, none of which were used in support of this work. Cesar de la Fuente-Nunez is on the Advisory Board of Cell Reports Physical Science. An invention disclosure associated with this work has been filed.
Maasch and Torres et al. described a machine learning model that identifies antimicrobial peptides within extinct human proteins. Molecular de-extinction shows promise in drug discovery by reintroducing unique antimicrobials with anti-infective efficacy in pre-clinical mouse models. This study demonstrates the potential of paleoproteome mining as a framework for drug discovery.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Sandler R. (2017). De-extinction: Costs, benefits and ethics. Nat Ecol Evol 1, 0105. 10.1038/s41559-017-0105. [DOI] [PubMed] [Google Scholar]
- 2.Lin J, Duchêne D, Carøe C, Smith O, Ciucani MM, Niemann J, Richmond D, Greenwood AD, MacPhee R, Zhang G, et al. (2022). Probing the genomic limits of de-extinction in the Christmas Island rat. Current Biology 32, 1650–1656.e3. 10.1016/j.cub.2022.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de la Fuente-Nunez C, Torres MDMD, Mojica FJFJ, and Lu TKTK (2017). Next-generation precision antimicrobials: towards personalized treatment of infectious diseases. Curr Opin Microbiol 37, 95–102. 10.1016/j.mib.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torres MDT, and de la Fuente-Nunez C. (2019). Toward computer-made artificial antibiotics. Curr Opin Microbiol 51, 30–38. 10.1016/j.mib.2019.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Mookherjee N, Anderson MA, Haagsman HP, and Davidson DJ (2020). Antimicrobial host defence peptides: functions and clinical potential. Nat Rev Drug Discov 19, 311–332. 10.1038/s41573-019-0058-8. [DOI] [PubMed] [Google Scholar]
- 6.Fjell CD, Hiss JA, Hancock REW, and Schneider G. (2011). Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov 11. 10.1038/nrd3591. [DOI] [PubMed] [Google Scholar]
- 7.Lázár V, Martins A, Spohn R, Daruka L, Grézal G, Fekete G, Számel M, Jangir PK, Kintses B, Csörgo B, et al. (2018). Antibiotic-resistant bacteria show widespread collateral sensitivity to antimicrobial peptides. Nat Microbiol 3, 718–731. 10.1038/s41564-018-0164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pizzo E, Cafaro V, Di Donato A, and Notomista E. (2018). Cryptic Antimicrobial Peptides: Identification Methods and Current Knowledge of their Immunomodulatory Properties. Curr Pharm Des 24, 1054–1066. 10.2174/1381612824666180327165012. [DOI] [PubMed] [Google Scholar]
- 9.Gaglione R, Pizzo E, Notomista E, de la Fuente-Nunez C, and Arciello A. (2020). Host Defence Cryptides from Human Apolipoproteins: Applications in Medicinal Chemistry. Curr Top Med Chem 20, 1324–1337. 10.2174/1568026620666200427091454. [DOI] [PubMed] [Google Scholar]
- 10.Cesaro A, Torres MDT, Gaglione R, Dell’Olmo E, Di Girolamo R, Bosso A, Pizzo E, Haagsman HP, Veldhuizen EJA, de la Fuente-Nunez C, et al. (2022). Synthetic Antibiotic Derived from Sequences Encrypted in a Protein from Human Plasma. ACS Nano 16, 1880–1895. 10.1021/acsnano.1c04496. [DOI] [PubMed] [Google Scholar]
- 11.Torres MDT, Melo MCR, Flowers L, Crescenzi O, Notomista E, and de la Fuente-Nunez C. (2022). Mining for encrypted peptide antibiotics in the human proteome. Nat Biomed Eng 6, 67–75. 10.1038/s41551-021-00801-1. [DOI] [PubMed] [Google Scholar]
- 12.Nothias L-F, Knight R, and Dorrestein PC (2016). Antibiotic discovery is a walk in the park. Proceedings of the National Academy of Sciences 113, 14477–14479. 10.1073/pnas.1618221114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C, Sutherland D, Hammond SA, Yang C, Taho F, Bergman L, Houston S, Warren RL, Wong T, Hoang LMN, et al. (2022). AMPlify: attentive deep learning model for discovery of novel antimicrobial peptides effective against WHO priority pathogens. BMC Genomics 23, 77. 10.1186/s12864-022-08310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanhoye D, Bruston F, Nicolas P, and Amiche M. (2003). Antimicrobial peptides from hylid and ranin frogs originated from a 150-million-year-old ancestral precursor with a conserved signal peptide but a hypermutable antimicrobial domain. Eur J Biochem 270, 2068–2081. 10.1046/j.1432-1033.2003.03584.x. [DOI] [PubMed] [Google Scholar]
- 15.de Barros E, Gonçalves RM, Cardoso MH, Santos NC, Franco OL, and Cândido ES (2019). Snake Venom Cathelicidins as Natural Antimicrobial Peptides. Front Pharmacol 10. 10.3389/fphar.2019.01415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim BY, Lee KS, Zou FM, Wan H, Choi YS, Yoon HJ, Kwon HW, Je YH, and Jin BR (2013). Antimicrobial activity of a honeybee (Apis cerana) venom Kazal-type serine protease inhibitor. Toxicon 76, 110–117. 10.1016/j.toxicon.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 17.Pedron CN, Araújo I, da Silva Junior PI, Dias da Silva F, Torres MDT, and Oliveira Junior VX (2019). Repurposing the scorpion venom peptide VmCT1 into an active peptide against Gram-negative ESKAPE pathogens. Bioorg Chem 90, 103038. 10.1016/j.bioorg.2019.103038. [DOI] [PubMed] [Google Scholar]
- 18.Silva ON, Torres MDT, Cao J, Alves ESF, Rodrigues LV, Resende JM, Lião LM, Porto WF, Fensterseifer ICM, Lu TK, et al. (2021). Repurposing a peptide toxin from wasp venom into antiinfectives with dual antimicrobial and immunomodulatory properties. Proceedings of the National Academy of Sciences 118, e2025351118. 10.1073/pnas.2025351118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torres MDT, Pedron CN, Araújo I, Silva PI, Silva FD, and Oliveira VX (2017). Decoralin Analogs with Increased Resistance to Degradation and Lower Hemolytic Activity. ChemistrySelect 2, 18–23. 10.1002/slct.201601590. [DOI] [Google Scholar]
- 20.Torres MDT, Pedron CN, da Silva Lima JA, da Silva PI, da Silva FD, and Oliveira VX (2017). Antimicrobial activity of leucine-substituted decoralin analogs with lower hemolytic activity. Journal of Peptide Science 23, 818–823. 10.1002/psc.3029. [DOI] [PubMed] [Google Scholar]
- 21.Ma Y, Guo Z, Xia B, Zhang Y, Liu X, Yu Y, Tang N, Tong X, Wang M, Ye X, et al. (2022). Identification of antimicrobial peptides from the human gut microbiome using deep learning. Nat Biotechnol 40, 921–931. 10.1038/s41587-022-01226-0. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Wong ESW, Whitley JC, Li J, Stringer JM, Short KR, Renfree MB, Belov K, and Cocks BG (2011). Ancient Antimicrobial Peptides Kill Antibiotic-Resistant Pathogens: Australian Mammals Provide New Options. PLoS One 6, e24030. 10.1371/journal.pone.0024030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergeijk D.A. van, Augustijn HE, Elsayed SS, Willemse J, Carrión VJ, Urem M, Grigoreva LV, Cheprasov MY, Grigoriev S, Wintermans B, et al. (2022). Taxonomic and metabolic diversity of Actinobacteria isolated from faeces of a 28,000-year-old mammoth. bioRxiv, 2022.12.22.521380. 10.1101/2022.12.22.521380. [DOI] [Google Scholar]
- 24.Paun VI, Lavin P, Chifiriuc MC, and Purcarea C. (2021). First report on antibiotic resistance and antimicrobial activity of bacterial isolates from 13,000-year old cave ice core. Sci Rep 11, 514. 10.1038/s41598-020-79754-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barber MF, Kronenberg Z, Yandell M, and Elde NC (2016). Antimicrobial Functions of Lactoferrin Promote Genetic Conflicts in Ancient Primates and Modern Humans. PLoS Genet 12, e1006063. 10.1371/journal.pgen.1006063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klapper M, Hübner A, Ibrahim A, Wasmuth I, Borry M, Haensch VG, Zhang S, Al-Jammal WK, Suma H, Fellows Yates JA, et al. (2023). Natural products from reconstructed bacterial genomes of the Middle and Upper Paleolithic. Science (1979). 10.1126/science.adf5300. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Gao Z, Ren X, Sheng J, Xu P, Chang C, and Fu Y. (2021). DeepDigest: Prediction of Protein Proteolytic Digestion with Deep Learning. Anal Chem 93, 6094–6103. 10.1021/acs.analchem.0c04704. [DOI] [PubMed] [Google Scholar]
- 28.Li F, Leier A, Liu Q, Wang Y, Xiang D, Akutsu T, Webb GI, Smith AI, Marquez-Lago T, Li J, et al. (2020). Procleave: Predicting Protease-specific Substrate Cleavage Sites by Combining Sequence and Structural Information. Genomics Proteomics Bioinformatics 18, 52–64. 10.1016/j.gpb.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li F, Chen J, Leier A, Marquez-Lago T, Liu Q, Wang Y, Revote J, Smith AI, Akutsu T, Webb GI, et al. (2020). DeepCleave: a deep learning predictor for caspase and matrix metalloprotease substrates and cleavage sites. Bioinformatics 36, 1057–1065. 10.1093/bioinformatics/btz721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang M, Zhao X-M, Tan H, Akutsu T, Whisstock JC, and Song J. (2014). Cascleave 2.0, a new approach for predicting caspase and granzyme cleavage targets. Bioinformatics 30, 71–80. 10.1093/bioinformatics/btt603. [DOI] [PubMed] [Google Scholar]
- 31.Piippo M, Lietzén N, Nevalainen OS, Salmi J, and Nyman TA (2010). Pripper: prediction of caspase cleavage sites from whole proteomes. BMC Bioinformatics 11, 320. 10.1186/1471-2105-11-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wee LJK, Tan TW, and Ranganathan S. (2007). CASVM: web server for SVM-based prediction of caspase substrates cleavage sites. Bioinformatics 23, 3241–3243. 10.1093/bioinformatics/btm334. [DOI] [PubMed] [Google Scholar]
- 33.Ozols M, Eckersley A, Platt CI, Stewart-McGuinness C, Hibbert SA, Revote J, Li F, Griffiths CEM, Watson REB, Song J, et al. (2021). Predicting Proteolysis in Complex Proteomes Using Deep Learning. Int J Mol Sci 22, 3071. 10.3390/ijms22063071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ayyash M, Tamimi H, and Ashhab Y. (2012). Developing a powerful In Silico tool for the discovery of novel caspase-3 substrates: a preliminary screening of the human proteome. BMC Bioinformatics 13, 14. 10.1186/1471-2105-13-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar S, Ratnikov BI, Kazanov MD, Smith JW, and Cieplak P. (2015). CleavPredict: A Platform for Reasoning about Matrix Metalloproteinases Proteolytic Events. PLoS One 10, e0127877. 10.1371/journal.pone.0127877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu S, Imai K, Sawasaki T, and Tomii K. (2014). ScreenCap3: Improving prediction of caspase-3 cleavage sites using experimentally verified noncleavage sites. Proteomics 14, 2042–2046. 10.1002/pmic.201400002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verspurten J, Gevaert K, Declercq W, and Vandenabeele P. (2009). SitePredicting the cleavage of proteinase substrates. Trends Biochem Sci 34, 319–323. 10.1016/j.tibs.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 38.Song J, Li F, Leier A, Marquez-Lago TT, Akutsu T, Haffari G, Chou K-C, Webb GI, and Pike RN (2018). PROSPERous: high-throughput prediction of substrate cleavage sites for 90 proteases with improved accuracy. Bioinformatics 34, 684–687. 10.1093/bioinformatics/btx670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, and Finn RD (2018). The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res 46, D624–D632. 10.1093/nar/gkx1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niculescu-Mizil A, and Caruana R. (2005). Predicting good probabilities with supervised learning. In Proceedings of the 22nd international conference on Machine learning - ICML ‘05 (ACM Press; ), pp. 625–632. 10.1145/1102351.1102430. [DOI] [Google Scholar]
- 41.Pirtskhalava M, Amstrong AA, Grigolava M, Chubinidze M, Alimbarashvili E, Vishnepolsky B, Gabrielian A, Rosenthal A, Hurt DE, and Tartakovsky M. (2021). DBAASP v3: database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res 49, D288–D297. 10.1093/nar/gkaa991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Böttger R, Hoffmann R, and Knappe D. (2017). Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS One 12, e0178943. 10.1371/journal.pone.0178943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nim S, O’Hara DM, Corbi-Verge C, Perez-Riba A, Fujisawa K, Kapadia M, Chau H, Albanese F, Pawar G, De Snoo ML, et al. (2023). Disrupting the α-synuclein-ESCRT interaction with a peptide inhibitor mitigates neurodegeneration in preclinical models of Parkinson’s disease. Nat Commun 14, 2150. 10.1038/s41467-023-37464-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torres MDT, Pedron CN, Higashikuni Y, Kramer RM, Cardoso MH, Oshiro KGN, Franco OL, Silva Junior PI, Silva FD, Oliveira Junior VX, et al. (2018). Structure-function-guided exploration of the antimicrobial peptide polybia-CP identifies activity determinants and generates synthetic therapeutic candidates. Commun Biol 1, 221. 10.1038/s42003-018-0224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bobone S, and Stella L. (2019). Selectivity of Antimicrobial Peptides: A Complex Interplay of Multiple Equilibria. In Antimicrobial Peptides, pp. 175–214. 10.1007/978-981-13-3588-4_11. [DOI] [PubMed] [Google Scholar]
- 46.Rice A, and Wereszczynski J. (2017). Probing the disparate effects of arginine and lysine residues on antimicrobial peptide/bilayer association. Biochimica et Biophysica Acta (BBA) - Biomembranes 1859, 1941–1950. 10.1016/j.bbamem.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fensterseifer ICM, Felício MR, Alves ESF, Cardoso MH, Torres MDT, Matos CO, Silva ON, Lu TK, Freire MV, Neves NC, et al. (2019). Selective antibacterial activity of the cationic peptide PaDBS1R6 against Gram-negative bacteria. Biochimica et Biophysica Acta (BBA) - Biomembranes 1861, 1375–1387. 10.1016/j.bbamem.2019.03.016. [DOI] [PubMed] [Google Scholar]
- 48.Huang Y, Niu B, Gao Y, Fu L, and Li W. (2010). CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682. 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lawrence TJ, Carper DL, Spangler MK, Carrell AA, Rush TA, Minter SJ, Weston DJ, and Labbé JL (2021). amPEPpy 1.0: a portable and accurate antimicrobial peptide prediction tool. Bioinformatics 37, 2058–2060. 10.1093/bioinformatics/btaa917. [DOI] [PubMed] [Google Scholar]
- 50.Meher PK, Sahu TK, Saini V, and Rao AR (2017). Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci Rep 7, 42362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santos-Júnior CD, Pan S, Zhao X-M, and Coelho LP (2020). Macrel: antimicrobial peptide screening in genomes and metagenomes. PeerJ 8, e10555. 10.7717/peerj.10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhadra P, Yan J, Li J, Fong S, and Siu SWI (2018). AmPEP: Sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci Rep 8, 1697. 10.1038/s41598-018-19752-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yan J, Bhadra P, Li A, Sethiya P, Qin L, Tai HK, Wong KH, and Siu SWI (2020). Deep-AmPEP30: Improve Short Antimicrobial Peptides Prediction with Deep Learning. Mol Ther Nucleic Acids 20, 882–894. 10.1016/j.omtn.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Das P, Sercu T, Wadhawan K, Padhi I, Gehrmann S, Cipcigan F, Chenthamarakshan V, Strobelt H, dos Santos C, Chen P-Y, et al. (2021). Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat Biomed Eng 5, 613–623. 10.1038/s41551-021-00689-x. [DOI] [PubMed] [Google Scholar]
- 55.González R, Albericio F, Cascone O, and Iannucci NB (2010). Improved antimicrobial activity of h-lysozyme (107–115) by rational Ala substitution. Journal of Peptide Science, n/a-n/a. 10.1002/psc.1258. [DOI] [PubMed] [Google Scholar]
- 56.González R, Mendive-Tapia L, Pastrian MB, Albericio F, Lavilla R, Cascone O, and Iannucci NB (2016). Enhanced antimicrobial activity of a peptide derived from human lysozyme by arylation of its tryptophan residues. Journal of Peptide Science 22, 123–128. 10.1002/psc.2850. [DOI] [PubMed] [Google Scholar]
- 57.Bosso A, Pirone L, Gaglione R, Pane K, Del Gatto A, Zaccaro L, Di Gaetano S, Diana D, Fattorusso R, Pedone E, et al. (2017). A new cryptic host defense peptide identified in human 11-hydroxysteroid dehydrogenase-1 β-like: from in silico identification to experimental evidence. Biochimica et Biophysica Acta (BBA) - General Subjects 1861, 2342–2353. 10.1016/j.bbagen.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 58.Bosso A, Di Maro A, Cafaro V, Di Donato A, Notomista E, and Pizzo E. (2020). Enzymes as a Reservoir of Host Defence Peptides. Curr Top Med Chem 20, 1310–1323. 10.2174/1568026620666200327173815. [DOI] [PubMed] [Google Scholar]
- 59.The UniProt Consortium (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–D515. 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yin X, Wu H, Mu L, Han K, Xu H, Jian J, Wang A, and Ye J. (2020). Identification and characterization of calreticulin (CRT) from Nile tilapia (Oreochromis niloticus) in response to bacterial infection. Aquaculture 529, 735706. 10.1016/j.aquaculture.2020.735706. [DOI] [Google Scholar]
- 61.Liu X, Xu N, and Zhang S. (2013). Calreticulin is a microbial-binding molecule with phagocytosis-enhancing capacity. Fish Shellfish Immunol 35, 776–784. 10.1016/j.fsi.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 62.Qiu Y, Xi J, Du L, and Poovaiah BW (2012). The function of calreticulin in plant immunity. Plant Signal Behav 7, 907–910. 10.4161/psb.20721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qiu Y, Xi J, Du L, Roje S, and Poovaiah BW (2012). A dual regulatory role of Arabidopsis calreticulin-2 in plant innate immunity. The Plant Journal 69, 489–500. 10.1111/j.1365-313X.2011.04807.x. [DOI] [PubMed] [Google Scholar]
- 64.Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, Nakhasi H, Teruya-Feldstein J, Wirth P, Gupta G, et al. (1998). Vasostatin, a Calreticulin Fragment, Inhibits Angiogenesis and Suppresses Tumor Growth. Journal of Experimental Medicine 188, 2349–2356. 10.1084/jem.188.12.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Augustin R, Siebert S, and Bosch TCG (2009). Identification of a kazal-type serine protease inhibitor with potent anti-staphylococcal activity as part of Hydra’s innate immune system. Dev Comp Immunol 33, 830–837. 10.1016/j.dci.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 66.Liu Y, Liu T, Hou F, Wang X, and Liu X. (2016). Lvserpin3 is involved in shrimp innate immunity via the inhibition of bacterial proteases and proteases involved in prophenoloxidase system. Fish Shellfish Immunol 48, 128–135. 10.1016/j.fsi.2015.09.039. [DOI] [PubMed] [Google Scholar]
- 67.Ponprateep S, Phiwsaiya K, Tassanakajon A, and Rimphanitchayakit V. (2013). Interaction between Kazal serine proteinase inhibitor SPIPm2 and viral protein WSV477 reduces the replication of white spot syndrome virus. Fish Shellfish Immunol 35, 957–964. 10.1016/j.fsi.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 68.Deraison C, Bonnart C, Lopez F, Besson C, Robinson R, Jayakumar A, Wagberg F, Brattsand M, Hachem JP, Leonardsson G, et al. (2007). LEKTI Fragments Specifically Inhibit KLK5, KLK7, and KLK14 and Control Desquamation through a pH-dependent Interaction. Mol Biol Cell 18, 3607–3619. 10.1091/mbc.e07-02-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, Bonafé J-L, Wilkinson J, Taïeb A, Barrandon Y, et al. (2000). Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet 25, 141–142. 10.1038/75977. [DOI] [PubMed] [Google Scholar]
- 70.Yamasaki K, Di Nardo A, Bardan A, Murakami M, Ohtake T, Coda A, Dorschner RA, Bonnart C, Descargues P, Hovnanian A, et al. (2007). Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med 13, 975–980. 10.1038/nm1616. [DOI] [PubMed] [Google Scholar]
- 71.Li Y, Li Y, Li W, Guo X, Zhou S, and Zheng H. (2020). Genetic polymorphisms in serine protease inhibitor Kazal-type 5 and risk of atopic dermatitis. Medicine 99, e21256. 10.1097/MD.0000000000021256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wagener J, Schneider JJ, Baxmann S, Kalbacher H, Borelli C, Nuding S, Küchler R, Wehkamp J, Kaeser MD, Mailänder-Sanchez D, et al. (2013). A Peptide Derived from the Highly Conserved Protein GAPDH Is Involved in Tissue Protection by Different Antifungal Strategies and Epithelial Immunomodulation. Journal of Investigative Dermatology 133, 144–153. 10.1038/jid.2012.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xin H, Ji S, Peng J, Han P, An X, Wang S, and Cao B. (2017). Isolation and characterisation of a novel antibacterial peptide from a native swine intestinal tract-derived bacterium. Int J Antimicrob Agents 49, 427–436. 10.1016/j.ijantimicag.2016.12.012. [DOI] [PubMed] [Google Scholar]
- 74.Brand GD, Magalhães MTQ, Tinoco MLP, Aragão FJL, Nicoli J, Kelly SM, Cooper A, and Bloch C. (2012). Probing Protein Sequences as Sources for Encrypted Antimicrobial Peptides. PLoS One 7, e45848. 10.1371/journal.pone.0045848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Branco P, Francisco D, Monteiro M, Almeida MG, Caldeira J, Arneborg N, Prista C, and Albergaria H. (2017). Antimicrobial properties and death-inducing mechanisms of saccharomycin, a biocide secreted by Saccharomyces cerevisiae. Appl Microbiol Biotechnol 101, 159–171. 10.1007/s00253-016-7755-6. [DOI] [PubMed] [Google Scholar]
- 76.Fesenko I, Azarkina R, Kirov I, Kniazev A, Filippova A, Grafskaia E, Lazarev V, Zgoda V, Butenko I, Bukato O, et al. (2019). Phytohormone treatment induces generation of cryptic peptides with antimicrobial activity in the Moss Physcomitrella patens. BMC Plant Biol 19, 9. 10.1186/s12870-018-1611-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carbon S, Douglass E, Good BM, Unni DR, Harris NL, Mungall CJ, Basu S, Chisholm RL, Dodson RJ, Hartline E, et al. (2021). The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res 49, D325–D334. 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Altschul S. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Díaz-Roa A, Gaona MA, Segura NA, Suárez D, Patarroyo MA, and Bello FJ (2014). Sarconesiopsis magellanica (Diptera: Calliphoridae) excretions and secretions have potent antibacterial activity. Acta Trop 136, 37–43. 10.1016/j.actatropica.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 80.Haibe-Kains B, Adam GA, Hosny A, Khodakarami F, Shraddha T, Kusko R, Sansone S-A, Tong W, Wolfinger RD, Mason CE, et al. (2020). Transparency and reproducibility in artificial intelligence. Nature 586, E14–E16. 10.1038/s41586-020-2766-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hutson M. (2018). Artificial intelligence faces reproducibility crisis. Science (1979) 359, 725–726. 10.1126/science.359.6377.725. [DOI] [PubMed] [Google Scholar]
- 82.Beam AL, Manrai AK, and Ghassemi M. (2020). Challenges to the Reproducibility of Machine Learning Models in Health Care. JAMA 323, 305. 10.1001/jama.2019.20866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Littmann M, Selig K, Cohen-Lavi L, Frank Y, Hönigschmid P, Kataka E, Mösch A, Qian K, Ron A, Schmid S, et al. (2020). Validity of machine learning in biology and medicine increased through collaborations across fields of expertise. Nat Mach Intell 2, 18–24. 10.1038/s42256-019-0139-8. [DOI] [Google Scholar]
- 84.McDermott MBA, Wang S, Marinsek N, Ranganath R, Foschini L, and Ghassemi M. (2021). Reproducibility in machine learning for health research: Still a ways to go. Sci Transl Med 13. 10.1126/scitranslmed.abb1655. [DOI] [PubMed] [Google Scholar]
- 85.Browning SR, Browning BL, Zhou Y, Tucci S, and Akey JM (2018). Analysis of Human Sequence Data Reveals Two Pulses of Archaic Denisovan Admixture. Cell 173, 53–61.e9. 10.1016/j.cell.2018.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Villanea FA, and Schraiber JG (2018). Multiple episodes of interbreeding between Neanderthal and modern humans. Nat Ecol Evol 3, 39–44. 10.1038/s41559-018-0735-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Racimo F, Sankararaman S, Nielsen R, and Huerta-Sánchez E. (2015). Evidence for archaic adaptive introgression in humans. Nat Rev Genet 16, 359–371. 10.1038/nrg3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Deschamps M, Laval G, Fagny M, Itan Y, Abel L, Casanova J-L, Patin E, and Quintana-Murci L. (2016). Genomic Signatures of Selective Pressures and Introgression from Archaic Hominins at Human Innate Immunity Genes. The American Journal of Human Genetics 98, 5–21. 10.1016/j.ajhg.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Quach H, Rotival M, Pothlichet J, Loh Y-HE, Dannemann M, Zidane N, Laval G, Patin E, Harmant C, Lopez M, et al. (2016). Genetic Adaptation and Neandertal Admixture Shaped the Immune System of Human Populations. Cell 167, 643–656.e17. 10.1016/j.cell.2016.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liston A, Humblet-Baron S, Duffy D, and Goris A. (2021). Human immune diversity: from evolution to modernity. Nat Immunol 22, 1479–1489. 10.1038/s41590-021-01058-1. [DOI] [PubMed] [Google Scholar]
- 91.Zhou S, Butler-Laporte G, Nakanishi T, Morrison DR, Afilalo J, Afilalo M, Laurent L, Pietzner M, Kerrison N, Zhao K, et al. (2021). A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat Med 27, 659–667. 10.1038/s41591-021-01281-1. [DOI] [PubMed] [Google Scholar]
- 92.Zeberg H, and Pääbo S. (2020). The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 587, 610–612. 10.1038/s41586-020-2818-3. [DOI] [PubMed] [Google Scholar]
- 93.Wiegand I, Hilpert K, and Hancock REW (2008). Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc 3, 163–175. 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- 94.Powell MF, Stewart T, Otvos L, Urge L, Gaeta FC, Sette A, Arrhenius T, Thomson D, Soda K, and Colon SM (1993). Peptide Stability in Drug Development. II. Effect of Single Amino Acid Substitution and Glycosylation on peptide Reactivity in Human Serum. Pharm Res 10, 1268–1273. [DOI] [PubMed] [Google Scholar]
- 95.van Westen GJ, Swier RF, Cortes-Ciriano I, Wegner JK, Overington JP, IJzerman AP, van Vlijmen HW, and Bender A. (2013). Benchmarking of protein descriptor sets in proteochemometric modeling (part 2): modeling performance of 13 amino acid descriptor sets. J Cheminform 5, 42. 10.1186/1758-2946-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cesaro A, Torres M, and de la Fuente-Nunez C. (2022). Methods for the design and characterization of peptide antibiotics. In Methods in Enzymology (Academic Press; ), pp. 303–326. 10.1016/bs.mie.2021.11.003. [DOI] [PubMed] [Google Scholar]
- 97.Yang L, Shu M, Ma K, Mei H, Jiang Y, and Li Z. (2010). ST-scale as a novel amino acid descriptor and its application in QSAM of peptides and analogues. Amino Acids 38, 805–816. 10.1007/s00726-009-0287-y. [DOI] [PubMed] [Google Scholar]
- 98.Sandberg M, Eriksson L, Jonsson J, Sjöström M, and Wold S. (1998). New Chemical Descriptors Relevant for the Design of Biologically Active Peptides. A Multivariate Characterization of 87 Amino Acids. J Med Chem 41, 2481–2491. 10.1021/jm9700575. [DOI] [PubMed] [Google Scholar]
- 99.Alley EC, Khimulya G, Biswas S, AlQuraishi M, and Church GM (2019). Unified rational protein engineering with sequence-based deep representation learning. Nat Methods 16, 1315–1322. 10.1038/s41592-019-0598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mi H, Ebert D, Muruganujan A, Mills C, Albou L-P, Mushayamaha T, and Thomas PD (2021). PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res 49, D394–D403. 10.1093/nar/gkaa1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Training data, testing data, and code used to develop the machine learning model are freely available on GitLab (https://gitlab.com/machine-biology-group-public/pancleave).
All data pertaining to the experimental validation of generated peptides are available in the Supplementary Data.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.