

Abstract
Schaaf–Yang syndrome (SYS) is a complex neurodevelopmental disorder characterized by autism spectrum disorder, joint contractures, and profound hypothalamic dysfunction. SYS is caused by variants in MAGEL2, a gene within the Prader–Willi syndrome (PWS) locus on chromosome 15. In this review, we consolidate decades of research on MAGEL2 to elucidate its physiological functions. Moreover, we synthesize current knowledge on SYS, suggesting that while MAGEL2 loss‐of‐function seems to underlie several SYS and PWS phenotypes, additional pathomechanisms probably contribute to the distinct and severe phenotype observed in SYS. In addition, we highlight recent therapeutic advances and identify promising avenues for future investigation.
Pathogenic variants in MAGEL2 cause Schaaf–Yang syndrome (SYS). MAGEL2 has diverse physiological functions, particularly in the human hypothalamus. Loss of function likely explains several SYS phenotypes. In addition it has become evident that neomorphic effects of truncated MAGEL2 proteins likely contribute to the severity of this complex neurodevelopmental disorder. This review summarizes scientific knowledge about MAGEL2 physiology, pathophysiology, and SYS.
Plain language summary: https://onlinelibrary.wiley.com/doi/10.1111/dmcn.16065
Abbreviations
- AMPA
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole propionic acid
- ASD
autism spectrum disorder
- AVP
arginine vasopressin
- Bdnf
brain‐derived neurotrophic factor gene
- mTOR
mammalian target of rapamycin
- MUST
MAGEL2‐USP7‐TRIM27 protein complex
- POMC
proopiomelanocortin
- PWS
Prader–Willi syndrome
- SYS
Schaaf–Yang syndrome
- WASH
Wiskott–Aldrich syndrome protein and SCAR homologue
What this paper adds
Loss of function of MAGEL2 contributes to Schaaf–Yang syndrome phenotypes but does not fully explain them.
RNA interference or proteolysis‐based therapies to reduce the truncated protein are identified as a research priority.
MAGEL2 exerts pleiotropic functions in the human body, particularly because of its role in the hypothalamus, a brain region at the centre of organismal homeostasis that is critical for both individual and species success. 1 The MAGEL2 gene is part of the maternally imprinted set of Prader–Willi syndrome (PWS) genes on chromosome 15. 2 Pathogenic variants in MAGEL2 cause Schaaf–Yang syndrome (SYS), a neurodevelopmental disorder with a high prevalence of autism spectrum disorder (ASD, about 75%–85%), 3 joint contractures, and an overall more severe phenotype than PWS, profoundly disrupting the lives of patients, their primary caregivers, and families. 4 , 5 , 6 , 7
In this review, we comprehensively summarize current scientific knowledge about the role of MAGEL2 in both physiology and pathophysiology to dissect the complex phenotype of SYS (Figure 1). We simultaneously provide more clarity on the contribution of MAGEL2 loss‐of‐function in PWS.
FIGURE 1.
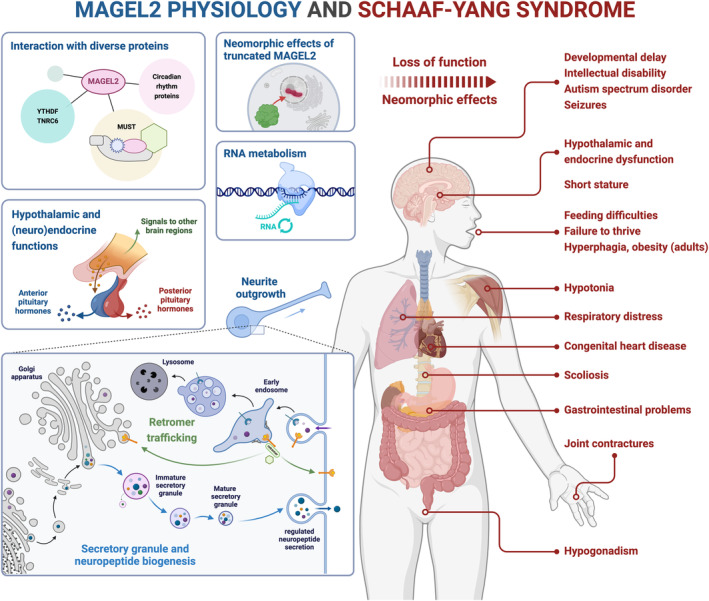
MAGEL2 physiology and SYS. MAGEL2 engages in diverse protein complexes and interactions to facilitate a range of processes spanning RNA metabolism, retromer trafficking, secretory granule and neuropeptide biogenesis, neurite outgrowth, and the fine‐tuning of hypothalamic and (neuro)endocrine activities. In addition to loss‐of‐function mechanisms, the emergence of neomorphic effects from truncated MAGEL2, which is encoded by typical SYS variants, probably contributes to the complex SYS phenotype. Abbreviations: MUST, MAGEL2‐USP7‐TRIM27 protein complex; SYS, Schaaf–Yang syndrome.
Leveraging insights from a translational research perspective, we emphasize that loss of MAGEL2 function alone cannot explain SYS phenotypes, highlight therapeutic advances, and propose directions for future research efforts towards improving the lives of individuals with lived experience as well as those of their families.
SYS
To date, more than 250 individuals with SYS have been identified. 3 The true prevalence may be greater because (1) many adults with disability have not undergone advanced genetic testing 3 and (2) variants in imprinted genes can be missed during trio‐exome sequencing. 8 , 9 Approximately 1 in 3500 hospitalized neonates has SYS if the data from the China Neonatal Genomes Project cohort are representative. 10 A study among Korean individuals subjected to whole‐exome sequencing because of developmental delay or intellectual disability found that 0.9% had SYS, revealing a significant contribution of SYS to developmental delay/intellectual disability phenotypes. 11 Although formal diagnostic criteria have been proposed, 9 SYS is generally diagnosed through molecular genetic testing and the identification of a heterozygous pathogenic variant in the paternally derived copy of MAGEL2. Approximately 50% of patients inherit their mutation from a clinically unaffected father (who himself carries the variant on his maternal allele), while the remainder are de novo. The penetrance is considered to be 100%. 3 The vast majority of mutations described so far are truncating or nonsense mutations, although some patients carry missense variants, which may not necessarily be SYS‐causing. 12 Considering the female‐protective model for neurodevelopmental disorders, the high penetrance of ASD even of female individuals with SYS is striking, and exploring SYS prevalence, phenotype, and severity based on sex should provide additional insight. 13
Phenotypes are highly variable in SYS
The phenotype of SYS (Figure 1) is highly variable among patients, but also over the course of their lives. In the prenatal period, the main findings include polyhydramnios and decreased fetal movements; however, the disease is unlikely to be identified during routine prenatal testing. 14 , 15 , 16 , 17 , 18 , 19 , 20 Infants present with generalized muscular hypotonia, respiratory distress with episodes of sleep apnoea, feeding difficulties with failure to thrive, and joint contractures. 3 , 20 The presence of joint contractures, and sometimes severe arthrogryposis multiplex congenita, which results from reduced or absent fetal movement, is the earliest distinctive feature of SYS. 6 , 21 , 22 In children and adolescents, mild to profound developmental delay, intellectual disability, seizures, ASD, and other cognitive or behavioural anomalies (e.g. impulsivity, compulsivity, manipulative behaviour, automutilation, heightened anxiety) become apparent. 3 , 5 Musculoskeletal problems that arise in addition to those already surfacing in infancy include scoliosis, kyphosis, and short stature, which may be disguised initially owing to the early onset of puberty in SYS. 3 , 21 Patients with SYS usually have gross hypothalamic dysfunction which is probably a result of MAGEL2 loss‐of‐function and manifests as several endocrinopathies (e.g. hypopituitarism, growth hormone deficiency, low gonadotropins with hypogonadism, etc.), temperature instability, and disrupted circadian rhythms (e.g. disturbed sleep cycle and daytime fatigue). 3 , 15 , 23 Non‐specific facial dysmorphisms and ocular anomalies (e.g. nystagmus 12 , 24 , 25 ) are noted alongside gastrointestinal problems (e.g. chronic constipation, gastroesophageal reflux disease). 16 , 26 , 27 Hyperphagia, food‐seeking behaviour, and obesity are hallmark features of PWS, but are present in only a minority of young patients with SYS. 16 , 26 , 27 In contrast to young patients, most adult patients do manifest these symptoms. 15 Some symptoms (e.g. hypotonia, feeding problems) become less prevalent and challenging in adult patients. 5 The life expectancy in SYS is reduced because of a greater risk of fatal complications, mostly during infancy and childhood, and Magel2‐null mice exhibit reduced embryonic viability. 3 , 28 A mortality rate of 17% in the SYS subcohort was reported in the China Neonatal Genomes Project project 10 with almost all deceased patients carrying paternally inherited variants. 29 Reports demonstrate that survival into adulthood is possible and, at the time of writing, the oldest patient with SYS in our registry is 41 years old (Christian P Schaaf, personal communication). 3
Distinct genotype–phenotype correlations
Phenotypes in SYS are highly variable, even among patients with the same genotype. 12 , 16 , 30 Genotype–phenotype correlations do exist, however, and it is conceivable that, depending on the precise location of frameshift or nonsense mutations, the variants would result in different truncated protein products, providing a potential foundation for the observed phenotypic differences. 6
Approximately 42% of all patients with SYS carry variants within the mutational hotspot located at nucleotides c.1990 to 1996. 9 , 16 , 20 Mutations in this area are almost exclusively responsible for early deaths of infants with SYS. 29 The region contains a sequence of seven cytosines 16 which probably presents a challenge to the DNA polymerase resulting in polymerase slippage and insertion (c.1996dupC) or deletion (c.1996delC) of a cytosine. 29 Although both mutations cause frameshifts in the same region, they result in dramatically different phenotypes, possibly because of the differences in amino acid sequence. 12 The c.1996delC variant causes severe arthrogryposis multiplex congenita and death in utero or shortly after birth. 10 , 22 Interestingly, this variant is predicted to result in a protein lacking the MAGE homology domain but with an additional change in the PAT1 domain, resulting in a novel DNA Pol3 gamma domain. 6 , 12 , 20 The most common SYS mutation, c.1996dupC, is associated with an infant mortality rate of about 24% due to respiratory failure. 10 , 31 Generally, this variant is associated with more severe phenotypes compared with all other pathogenic mutations except c.1996delC. Individuals with c.1996dupC have a higher prevalence of musculoskeletal problems, but also more profound levels of developmental delay/intellectual disability. For instance, the mean IQ of those with a c.1996dupC mutation was reported to be 14.2 compared with 53.2 in those with other variants. 6 , 12 , 20 Recently, an additional mutational hotspot at c.1912 was proposed. 10 Dissecting genotype–phenotype correlations further by taking into account the expected differences at the protein level, as well as sex‐specific variation, would provide additional insights to enhance clinicians' ability to effectively counsel patients.
MAGEL2 IN HEALTH AND DISEASE
MAGEL2 belongs to a family of closely related MAGE genes, whose protein products harbour the functional MAGE homology domain. The MAGE homology domain contains dynamic tandem winged‐helix motifs, is flexible, and is encoded by different sequences in different MAGEs to allow each MAGE protein to interact with distinct partners and confer specific functions. MAGEs are regulators of E3 RING ubiquitin ligases, which label substrates with ubiquitin chains to regulate processes such as proteasomal decay. MAGE proteins enhance the activity of E3 RING ligases, alter substrate specificity, and influence the subcellular localization of the ligase. Many MAGEs, including MAGEL2, are involved in neurogenesis and brain function. 32 MAGEL2 specifically is suggested to have evolved as a mammalian‐specific regulator of hypothalamic neuroendocrine function, fine‐tuning the hypothalamic regulation of physiological homeostasis and behaviour to adapt to environmental cues. 1
Gene expression, epigenetics, and RNA metabolism
The critical role of MAGEL2 in neurodevelopment is emphasized by its predominant expression in the developing brain in humans and mice, with the timing of expression coinciding with neurogenesis. 33 MAGEL2 is most highly expressed in the developing hypothalamus and pituitary at approximately 6 to 8 weeks gestational age. 25 High levels of hypothalamic expression are more deeply characterized in mice, which showed intense Magel2 expression at E12.5 in the ventral thalamus, anterior hypothalamus, supraoptic nucleus, paraventricular nucleus, and presumptive suprachiasmatic nucleus areas. 34 Interestingly, this is parallel to the timing of parvo‐ and magnocellular neuronal precursor generation in the developing paraventricular nucleus and supraoptic nucleus. 35 In the suprachiasmatic nucleus, Magel2 is expressed in a circadian fashion with peaks during the subjective day. 36 , 37 During adulthood, MAGEL2 expression is still highest in the hypothalamus, but mouse data show that Magel2 expression is generally lower in adult brain tissue than in the developing brain. 33 , 38 While predominantly present in the brain, MAGEL2 is expressed in other fetal tissues (e.g. kidney, liver, and lung) as well as in the placenta, possibly as part of peripheral nervous system structures. 33 , 38 Moreover, MAGEL2 is expressed in various cells of mesodermal origin, such as stem cells, chondrocytes, muscle cells, and pancreatic islets, and may contribute to the formation of muscle, bone, and fat. 39 , 40 Additionally, Magel2 is present in another tissue of mesenchymal origin, the cortex of the adrenal gland, which produces glucocorticoids, mineralocorticoids, and androgens. 39 , 41
MAGEL2 is maternally imprinted and, thus, expressed from the paternal allele. 3 Imprinting is an epigenetic phenomenon that is canonically regulated by DNA methylation. Although imprinted genes represent only a small subset of the mammalian genome, many of them have major effects on development and placental biology. 42 , 43 A group of paternally expressed genes including MAGEL2 are mainly expressed in both the placenta and hypothalamus and believed to have evolved owing to parent–infant co‐adaptation. 44 At early developmental stages, imprinting variance of porcine Magel2 has been reported with temporal and spatial variation in expression. 16 Compared with rats harbouring mutations in the paternal allele only, those with homozygous Magel2 mutations in both alleles, including the supposedly silent maternal allele, exhibited significant differences, which supports the hypothesis that switches between biallelic and monoallelic Magel2 expression occur during embryonic development. 45 For genetic counselling, it should be noted that if an SYS‐causing mutation is present in a family, several generations can remain phenotypically unaffected, as long as the mutation resides on the maternal allele. 16 To help families understand genetic risks, the father of a patient should always be tested for the MAGEL2 variant found in his offspring. Approximately half of the SYS variants identified so far are inherited from a carrier father, while the other half are found to be de novo. This has important implications for future family planning. The chance of recurrence for future children to inherit the paternal MAGEL2 variant is 50% if the father has been identified as a carrier. If the father is not a carrier, it is expected to be under 3% because of the possibility of germline mosaicism. In cases where the father does not carry the MAGEL2 variant identified in the proband, subsequent testing should be performed, using methylation‐sensitive methods to confirm that the variant is indeed located on the paternal allele of the index.
MAGEL2 expression may be regulated by NF1. 46 Additional transcriptional regulators of Magel2 have been identified in mice, most notably the oestrogen diethylstilbestrol, aristaless‐related homeobox (Arx), and the chromodomain helicase DNA‐binding 5 chromatin remodeller (Chd5). 47 , 48 , 50 Chd5‐null mice exhibit autism‐like phenotypes, potentially caused by downstream Magel2 dysfunction. 49
MAGEL2 itself may influence gene expression, even though transcriptomic analyses of neuronal tissue derived from patients with SYS, which has the greatest MAGEL2 expression, have not yet been published. In one patient with SYS, differentially methylated regions were found, predominantly in genes involved in macromolecule biosynthesis. 50 Apart from the influence of MAGEL2 mutations at the epigenetic level, the amino (N)‐terminal portion of MAGEL2 is probably involved in RNA metabolism through the regulation of messenger RNAs (mRNAs) modified by m6A methylation. In particular, YTHDF2, an m6A ‘reader’ regulating 5′ UTR methylation and translation initiation under stress, was identified as proximal to the N‐terminus of MAGEL2 (Figure 1). 51 , 52
MAGEL2 is part of regulatory complexes
Human MAGEL2 consists of 1249 amino acids. 53 Like other MAGE family members, it participates in at least one multi‐subunit protein complex, specifically referred to as the MAGEL2‐USP7‐TRIM27 (MUST) complex (Figure 2). 1 , 54 , 55 , 56 The MUST complex contains: (1) MAGEL2, an E3 ubiquitin ligase enhancer; 57 (2) TRIM27, an E3 RING ubiquitin ligase; and (3) USP7, a deubiquitinating enzyme. The MUST complex specifically functions as a regulator of the retromer‐dependent recycling pathway. 55 Retromer recycling is an essential cellular process that facilitates the trafficking of membrane proteins from endosomes to the plasma membrane or trans‐Golgi network, thus preventing lysosomal degradation (Figure 1). 58 , 59 The MUST complex proteins jointly control the K63‐linked ubiquitination of the Wiskott–Aldrich syndrome protein and SCAR homologue (WASH) K220 regulatory complex, which results in its activation, thus allowing WASH to promote actin nucleation on endosomes that is necessary for retromer functionality. USP7 buffers WASH ubiquitination levels, and MAGEL2 influences cargo selection among other functions (Figure 2). 32 , 55 , 56
FIGURE 2.
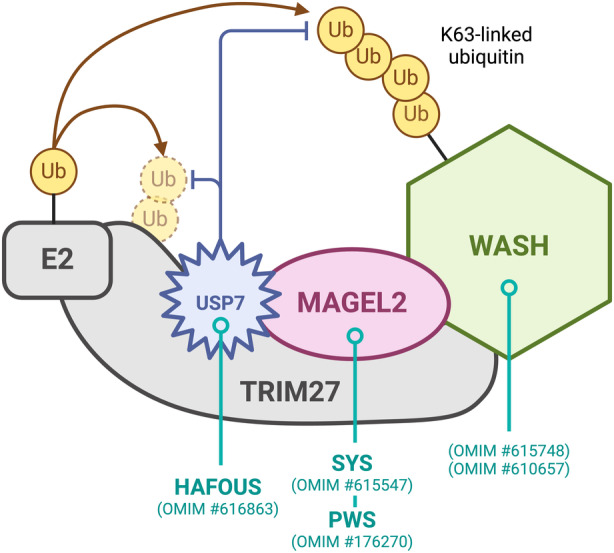
MUST complex. MAGEL2 acts as an E3 ubiquitin ligase enhancer and interacts with the E3 RING ubiquitin ligase TRIM27, and the deubiquitinating enzyme USP7, which buffers ubiquitination levels of substrates such as the WASH complex. 32 , 55 WASH ubiquitination by the MUST complex has been shown to be involved in retromer recycling. 56 The figure highlights several neurodevelopmental disorders linked to variants in genes encoding MUST proteins or their interaction partners. Abbreviations: MUST, MAGEL2‐USP7‐TRIM27; OMIM, Online Mendelian Inheritance in Man; PWS, Prader–Willi syndrome; SYS, Schaaf–Yang syndrome; Ub, ubiquitin; WASH, Wiskott–Aldrich syndrome protein and SCAR homologue.
The role of the MUST complex in retromer trafficking is tissue‐specific. To illustrate, retromer dysfunction has been reported in neurons derived from dental‐pulp stem cells of patients with PWS, who lack MAGEL2 entirely, and from a patient with SYS, while undifferentiated dental‐pulp stem cells were unaffected. 60 The critical role of retromer dysfunction in SYS pathology is emphasized by the fact that mutations in USP7 lead to a neurodevelopmental disorder (OMIM 616863) with phenotypes that partly overlap with those of SYS, 27 , 55 , 61 and mutations in WASH protein‐coding genes are associated with intellectual disability 62 , 63 (Figure 2). MAGEL2 has also been shown to interact with SUMO E3 ligases such as PIAS1, although the implications remain to be investigated. 64
MAGEL2 is involved in diverse cellular processes
Among the cargos of MAGEL2‐regulated retromer trafficking are components of the regulated secretory pathway such as proteins involved in hormone processing and secretory granule maturation. 60 The regulated secretion pathway is essential in secretory cells and requires the retrograde movement of lipids and proteins from immature secretory granules. In particular, MAGEL2‐regulated WASH is required for regulated secretion in the hypothalamus (Figure 1). 1 , 60 , 65 , 66 MAGEL2 deficiency in mice and cells derived from patients with PWS leads to decreased secretory granule and neuropeptide production, resulting in decreased levels of oxytocin, arginine vasopressin (AVP), somatostatin, and thyrotropin‐releasing hormone as well as decreased growth hormone, and luteinizing hormone. 60 In Magel2 tm1.1Mus mice, the intermediate form of several neuropeptides (oxytocin, AVP, and orexin‐A) accumulates, while mature neuropeptide production is impaired. 67 Overall, MAGEL2 functions as a tissue‐specific regulator of the core secretory machinery with loss‐of‐function leading to reduced secretory granule numbers, reduced bioactive hormone levels, and processing defects. 1
Furthermore, MAGEL2 may be involved in the mammalian target of rapamycin (mTOR) and autophagy pathways. mTOR is a serine/threonine kinase that acts downstream of growth factor signalling to modulate cellular homeostasis. 68 Magel2 could be required for normal levels of autophagy in both muscle tissue and the brain, with muscular atrophy being a result of increased autophagy. 39 , 69 mTORC1 is up‐regulated and autophagy is decreased in the brains of Magel2‐null mice, neurons derived from induced pluripotent stem cells of patients with SYS, and fibroblasts, although this last result was not recapitulated in a later study. HOTAIR, a regulator of mTOR, was found to be increased in those SYS fibroblasts. 70 , 71 Rapamycin shows therapeutic potential 70 , 72 but, to our knowledge, results have not been confirmed in follow‐up studies.
MAGEL2 function is critical for brain and body homeostasis
Brain, cognition, and behaviour
Magel2 deficiency leads to disruptions of neuronal activity with changes in the synaptic excitation/inhibition balance suppressing the activity of neurons in the hypothalamus and hippocampus of mice. 73 , 74 , 75 This is probably because of the selective loss of postsynaptic α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole propionic acid‐ (AMPA‐) dependent currents, a particularly interesting finding given the involvement of the retromer complex in AMPA receptor trafficking. 73 , 76 If similar mechanisms occur in the hypothalamus, it is conceivable that AMPA receptors may not be properly trafficked to the plasma membrane, explaining the loss of excitatory response. In addition, loss of Magel2 seems to delay the developmental excitatory‐to‐inhibitory GABA (γ‐aminobutyric acid) shift, in which oxytocin plays a prominent role. 74 , 77 , 78 Magel2 promotes neurite outgrowth (Figure 1) and both mutant mouse neurons and those derived from induced pluripotent stem cells of patients with SYS show impaired axonal outgrowth or dendrite formation, accompanied by alterations in the retinoid signalling pathway. 70 , 75 , 79 , 80 , 81 Magel2 is required for normal neurochemical balance as well, and Magel2‐null mice exhibit reduced serotonin, dopamine, and noradrenaline concentrations. 82 , 83 Recently, neuroinflammatory signals have also been reported in Magel2‐null mice and brains from patients with PWS. 84 , 85
Some of the alterations described above may contribute to developmental delay/intellectual disability in patients but also to other symptoms, including endocrine disturbances. Electroencephalogram patterns in infants with SYS can be reflective of a moderate depression of brain function, and other irregular activity, which, if present, generally normalizes within the first 2 years of life. 24 , 86 , 87 Patients with SYS present with varying levels of intellectual disability (mean Full‐scale ratio IQ: 27.0) although higher intelligence measures are reported in adult patients than young people with SYS. A sizeable portion of patients with SYS fail to reach developmental milestones at any age. Adult patients generally live in specialized institutions or at home with family but can often work in protected environments. It is, however, promising that patients have not shown regressions in skills after acquiring them. 6 , 7 , 15 Mouse models of Magel2‐deficiency recapitulate learning deficits. 88
The first behavioural concerns of parents are abnormal crying at birth, and difficulties categorizing their child's emotional expressions. 7 SYS adults may have a significantly greater risk for psychiatric disorders. 15 Among those, ASD is strikingly prevalent. Reports range from 50% prevalence 9 to 78% 6 or even 89% 7 in formally tested cohorts. Magel2‐deficient mice concordantly display behavioural abnormalities including a deficit in preference for social novelty, deficits in social memory, parenting behaviour, and separation‐induced vocalizations. 74 , 89 , 90 , 91 Impaired prosocial behaviour has also been reported in a novel rat model carrying a truncating mutation reflective of the SYS genotype. 45 Patients with SYS additionally exhibit significant deficits in adaptive‐functioning and can present with obsessive thoughts, stubbornness, and a paradoxical combination of heightened anxiety and reduced awareness of danger. 7 , 15 Increased anxiety may be recapitulated in mice, while another study reported modest reductions in anxiety. 82 , 89
(Neuro)endocrinology
As the key region of neuroendocrine function, the hypothalamus, where MAGEL2 is highly expressed, is at the centre of organismal homeostasis and adaptation to the environment. In MAGEL2‐related disorders such as PWS and SYS, this adaptive functionality seems to be disrupted. 1 , 38 In this section we focus on the involvement of MAGEL2 in (neuro)endocrinology and subsequently return to nonendocrine hypothalamic (dys‐)function. As part of the hypothalamic–pituitary axis, the hypothalamus produces releasing hormones to control the pituitary gland which in turn releases hormones to control other glands or tissues. 92 Pituitary hypoplasia has been reported, 93 , 94 and the endocrine system is clearly disrupted in patients with SYS, which is plausible considering the role of MAGEL2 in secretory granule and neuropeptide biogenesis.
Pituitary hormones can be separated into posterior pituitary (neurohypophysis) and anterior pituitary (adenohypophysis) hormones. Oxytocin and AVP, the two posterior pituitary hormones, are evolutionarily conserved mediators in the regulation of complex social cognition and behaviour. They are synthesized mostly in magnocellular neurons of the supraoptic nucleus and paraventricular nucleus, with axons projecting to the posterior pituitary where peptides are stored in secretory granules before being released into the blood stream. 1 , 95 Oxytocin also acts on the brain as a neuromodulator. 96 Oxytocin system function and oxytocin levels during prenatal development and around birth influence food intake, cognition, and behaviours related to autism, probably by stimulating and coordinating the maturation of neuronal networks. 96 , 97 , 98 Early alterations may therefore disturb oxytocin system maturation and have short‐ and long‐term pathological consequences that manifest as ASD, among other symptoms. 99
Patients with PWS, who lack the entire paternal MAGEL2 allele, exhibit altered oxytocin system anatomy and peptide levels, affecting physiological functions, social behaviours, and food intake. 1 , 100 , 101 , 102 , 103 , 104 Different Magel2‐null mouse lines have either reduced 105 or increased 88 oxytocin neuron numbers. Given the role of MAGEL2 in neuropeptide production, prohormone processing defects can be expected and are reported in neonatal Magel2‐null mice that present deficits in mature oxytocin levels accompanied by the accumulation of intermediate forms. Adult mutants, however, seem to harbour an increase in mature oxytocin levels. 67 , 88 Therefore, secretion may be impaired, which could be investigated with novel biological sensors for oxytocin release. 106 Neuromodulatory effects of oxytocin are mediated through oxytocin receptors whose expression varies depending on the brain region, sex, developmental age, and environment. Early‐life experience has long‐term effects on the expression of oxytocin receptors. 107 , 108 Magel2‐null mouse models show widespread downregulation of oxytocin receptors in neonates and in the adult nucleus accumbens, while other regions, such as the central amygdala, show increased oxytocin receptor expression in adult animals. 74 , 105 , 107
Oxytocin and AVP share a role in social fear and aggression through inhibitory synaptic transmission in the lateral septum, which harbours somatostatin neurons. This neuronal population is dysregulated in Magel2‐null mice, resulting in disrupted social fear extinction and discriminative social exploration. 109 , 110 Besides affecting social behaviours, AVP regulates the tonicity of body fluids, and AVP deficiency is known to result in diabetes insipidus, which has been reported in patients with SYS. 1 , 30 , 93 , 111
Hypothalamic steering hormones (thyrotropin‐releasing hormone, growth hormone‐releasing hormone, gonadotropin‐releasing hormone, corticotropin‐releasing hormone) control anterior pituitary hormone levels. Hypothyroidism, partly of central origin, has been described in patients with SYS, and mice concordantly show decreased hypothalamic levels of thyrotropin‐releasing hormone and lower circulating T4. 12 , 15 , 26 , 60 , 86 , 93 , 94 , 112 Several hallmark features of SYS, including short stature, hypoglycaemia, low bone mineral density, and body composition shifts from lean to fat mass, can be explained by the reduced growth hormone levels reported in patients. 25 , 26 , 86 , 93 , 94 , 113 Notably, mTOR is also a positive regulator of lipid synthesis, and fibroblasts from patients with SYS form more lipid droplets. 70 Since fetal growth is independent of pituitary growth hormone, 114 rapid growth retardation in patients with SYS onsets after birth. 115 Magel2‐null mice, and to some extent Magel2 Pmut rats, mimic alterations in hormone levels, body composition, growth restriction, and compromised bone health. 28 , 39 , 45 , 112 , 116 Deficiency in growth hormone in these mice has been suggested to be of hypothalamic origin, implying growth hormone‐releasing hormone disruption. 112 Hypogonadism, another common feature of SYS, may be explained by deficiencies in gonadotropin‐releasing hormone or luteinizing hormone/follicle‐stimulating hormone. Low levels of luteinizing hormone and follicle‐stimulating hormone have been reported in patients, but it is unclear whether hypogonadism is of primary or secondary aetiology; in PWS, both possibilities have been suggested. 15 , 26 , 93 , 117 Magel2‐deficient mice exhibit impaired fertility with low levels of gonadotropin‐releasing hormone and luteinizing hormone, irregular oestrous cycles in females, and decreased testosterone levels in males. 60 , 118 Finally, corticotropin‐releasing hormone stimulates release of adrenocorticotropic hormone, which in turn regulates the adrenal cortex. While adrenal insufficiency has been mentioned in patients with SYS, reports remain scarce, which is surprising given the additional information on local MAGEL2 expression. 25 , 39
The neurotransmitter dopamine plays its own role in inhibiting the release of prolactin from the anterior pituitary, while also being involved in behaviours such as motor function, sexual behaviour, and feeding. 83 Dopaminergic pathways mainly originate from the mediobasal hypothalamus, substantia nigra, and ventral tegmental area. 119 Interestingly, hyperprolactinaemia has been reported in patients with SYS, 25 , 120 and dopamine levels are reduced in brain tissue of Magel2‐null mice. 82 , 83 Dopaminergic dysfunction probably contributes to other aspects of SYS as well. Dopaminergic neurons within the paraventricular nucleus and periventricular nucleus, which are reduced in Magel2‐null mice, influence the release of pituitary hormones in addition to somatostatin, namely oxytocin, AVP, growth hormone, and thyroid‐stimulating hormone. 83 , 121 , 122 In humans, dopamine is particularly important in reward‐related behaviour, including overeating and hedonic feeding. The reward pathway or mesolimbic dopaminergic system consists of neurons in the ventral tegmental area that project to the nucleus accumbens, and are regulated by hormones such as leptin, insulin (both inhibitory), and ghrelin (activating). 83 , 123 Consequently, Magel2 is required for normal responses to changes in the caloric content of food. 83
In summary, loss of MAGEL2 function leads to gross neuroendocrine dysfunction, contributing to a broad range of SYS symptoms. Pathophysiological mechanisms probably include impaired secretory granule and neuropeptide biogenesis as well as electrophysiological changes in secretory neurons.
Metabolism, thermoregulation, and circadian rhythm
Albeit not a common feature in young patients with SYS, hyperphagia, food‐seeking behaviour, and obesity are highly prevalent in adult patients. 15 In patients with SYS, Magel2‐null mice, and Magel2 Pmut rats, body composition shifts towards greater fat mass. 26 , 28 , 45 In PWS, where these pathologies have been studied extensively, three pathomechanistic theories have emerged: (1) a deficit to generate satiety signals, (2) dysfunctional hypothalamic centres of energy homeostasis, and (3) abnormally high activation of reward pathways by food‐related stimuli. 28 , 124 , 125
The arcuate nucleus of the hypothalamus comprises two neuronal subpopulations that regulate food intake and bodyweight. 1 , 126 , 127 Neuropeptide Y and agouti‐related peptide neurons jointly stimulate food intake and reduce energy expenditure, while proopiomelanocortin (POMC) neurons have opposite effects. 126 , 128 POMC neurons are stimulated in the ‘fed state’ by circulating hormones such as leptin and insulin to promote the processing of POMC to the mature hormone α‐melanocyte‐stimulating hormone, which acts on melanocortin‐4 receptor‐positive neurons in the paraventricular nucleus to reduce food intake. In the ‘starved state’, agouti‐related peptide/neuropeptide Y neurons are activated by decreased leptin and insulin levels and increased ghrelin levels. 129 Agouti‐related peptide neuron function seems uncorrupted upon Magel2 loss. 130 On the other hand, Magel2‐null mice have fewer POMC neurons with fewer α‐melanocyte‐stimulating hormone projections to the paraventricular nucleus, and the remaining POMC neurons are unresponsive to leptin, leading to defective anorexigenic responses. 80 , 128 , 131 MAGEL2 has been shown to regulate the recycling of leptin receptors and therefore leptin receptor cell‐surface abundance in complex with Necdin, RNF31, and USP8. Thus, loss of MAGEL2 could well account for leptin resistance and increased susceptibility to obesity in mice and humans. This evidence also suggests the existence of an additional complex, similar to MUST. 1 , 28 , 54 , 126 , 132 , 133 Fasting levels of the orexigenic hormone ghrelin are even more strongly elevated in patients with SYS than in patients with PWS who are often morbidly obese. In general, it is curious that hyperphagia and obesity seem to be less prevalent and appear later in life in SYS. A few potential explanations have been discussed, particularly that severe intellectual disability in SYS may prevent patients from expressing their hunger. 10 , 12 , 18 , 30 , 94 , 113 , 134 This notion is corroborated by findings that overeating is observed only in adults with SYS who have higher degrees of independence and less frequently in individuals with c.1996dupC mutations which are associated with more severe intellectual disability. 6 , 15
Functional MAGEL2 is crucial in glucose metabolism as well, as evidenced by frequent reports of hypoglycaemic episodes in patients with SYS, which can often be treated with diazoxide. 10 , 12 , 18 , 30 , 94 , 113 , 134 Hypoglycaemia can be secondary to hormone deficiency, adrenal insufficiency, or hyperinsulinism. 113 Both growth hormone deficiency and hyperinsulinism have been shown in patients and could jointly impair the counterregulatory response to hypoglycaemia. Although MAGEL2 is expressed in the pancreas, its role in insulin metabolism is unclear. 113 Magel2‐null mice mimic increased insulin levels. 6 , 12 , 15 , 17 , 135 , 136 Evidence additionally points to dysfunctional glucose sensing mechanisms in the hypothalamus. 112 Gastrointestinal abnormalities include frequent chronic constipation and other complications, some of which may be connected to muscular hypotonia. 6 , 12 , 15 , 17 , 135 , 136
Thermoregulation, another hypothalamic function, may be impaired in patients with SYS who show temperature instability with excessive cold or sweating, sometimes leading to an initial diagnosis of Crisponi/cold‐induced sweating syndrome. 6 , 137 Dysregulated thyroid function may also contribute to this phenotype. Beyond its circadian expression pattern, MAGEL2 directly influences the generation of circadian rhythms in the suprachiasmatic nucleus, where dorsomedial AVP neurons express Magel2, at least in mice. 138 Magel2 has been shown to modulate the activity of circadian rhythm proteins. It induces the redistribution of Clock towards the cytoplasm, in contrast to the nucleus‐directed effect of Bmal1, interacts with Bmal1 and Per2, and probably promotes negative feedback regulation of the cellular circadian cycle. 139 In addition, as part of the MUST complex, MAGEL2 fine‐tunes CRY1 levels. 140 Loss of MAGEL2 function could explain the disturbed sleep cycles in patients and the blunted circadian rhythm in Magel2‐knockout mice.
Musculoskeletal and cardiovascular health
Alterations across multiple MAGEL2‐dependent pathways, most of which have already been mentioned, could combine to create a ‘perfect storm’ that causes a high incidence of musculoskeletal pathologies. 39 Hypotonia, as an almost universal and prominent early phenotype, probably contributes to several clinical manifestations, most importantly feeding difficulties, respiratory dysfunction, and joint contractures. 6 Magel2‐null mice exhibit reduced muscle strength, and signs of muscular atrophy. 39 The definite origin of hypotonia remains elusive. Newborn mice recapitulate impaired feeding with weak sucking, and other results support a key role for Magel2 in the initiation of feeding behaviour. The latter is probably dissociated from muscular functions and instead linked to oxytocin deficiency, which is required for modulating the sensory–motor reflexes necessary for sucking. 67 , 141 Respiratory dysfunction is another common, and sometimes lethal, SYS symptom. Both young and adult patients have been reported to have sleep apnoea, with data pointing to both central and peripheral aetiologies. 6 , 15 , 23 , 30 Mouse models do not recapitulate severe respiratory deficiency, but show Magel2 expression in the diaphragm, abdominal wall muscles, and fetal lung, while the novel rat model exhibits counterintuitive respiratory changes by showing reduced apnoea counts. 33 , 39 , 45 Joint contractures up to arthrogryposis multiplex congenita are considered the result of fetal akinesia or hypokinesia, and possibly linked to hypotonia. 22 Scoliosis is a common problem in humans with neuromuscular dysfunctions, 142 and has been reported both in patients with SYS and in Magel2‐null mice. 15 , 25 , 26 , 39
To close our investigation of MAGEL2 loss‐of‐function, we present congenital heart disease as a currently underappreciated feature of SYS. Several studies have shown alterations, particularly atrial septal defects, 10 , 12 , 20 , 30 , 143 pulmonary hypertension, 24 , 143 and patent foramen ovale. 20 , 24 , 143 Indeed, most reports of cardiovascular disease are from young children. Interestingly, Magel2 Pmut rats display alterations in cardiac structure and function. 45 Investigations into cardiovascular pathologies may be of particular interest and should also consider the connections between oxytocin and the heart. 144
We conclude that MAGEL2 loss‐of‐function probably contributes to a broad range of SYS phenotypes through disruptions in typical brain development, neuroendocrine functioning, metabolism, homeostatic mechanisms, and the musculoskeletal system. Numerous pathomechanistic pathways await clarification. Currently, the role of MAGEL2 in regulatory complexes such as MUST stands out, while its involvement in RNA metabolism remains underexplored in advanced models.
SYS beyond MAGEL2 loss‐of‐function
Up to this point, our review has focused on dissecting SYS phenotypes according to MAGEL2 loss‐of‐function. This is, as we will argue, a distorted and incomplete picture. Patients with PWS lack the entire copy of paternal MAGEL2, but have both overlapping and distinct and, importantly, less severe phenotypes than patients with SYS. 16 Other deletion cases leading to loss of paternal MAGEL2 while retaining other PWS genes do not recapitulate SYS severity, either. 145 , 146 It has been suggested that complete deletion of paternal MAGEL2 promotes leaky expression of the unaffected maternal allele. 16 There is evidence supporting this theory. 147 However, if leaky expression were to explain the reduced severity of complete MAGEL2 loss, the almost complete lack of SYS cases with missense mutations would be astounding. Since the promoter remains intact, these types of mutation are not predicted to cause leaky expression and should, thus, have similar severity as frameshift or nonsense cases. 147 These insights suggest an additional pathomechanism in SYS, apart from loss‐of‐function. Since MAGEL2 is a single‐exon gene, 16 mRNA transcripts containing premature stop codons should not be subject to nonsense‐mediated decay, thus leading to the presence of a truncated protein product. 12 , 16 , 148 , 149 Previously, dominant negative effects of truncated MAGEL2 have been suggested, 16 which are unlikely, given the lack of proof that MAGEL2 forms multimers, 150 and the probable absence of wild‐type protein. However, a harmful gain‐of‐function or neomorphic effect of truncated MAGEL2 could be a feasible explanation, both for the severity of SYS compared with other MAGEL2‐deficiencies, and for the varying phenotypes among patients with different predicted truncations. 12 , 16 , 29
To our knowledge, the presence of a truncated MAGEL2 protein has never been investigated in tissue from patients. It can, however, be detected in rats with a truncating Magel2 mutation, making the model superior for SYS research compared with knockouts. 45 Apart from the rat, a mouse model with a truncating mutation in Magel2 has been created but shows phenotypes similar to or less severe than those of Magel2‐null mice. 151 This also raises the possibility that the exact position and resulting protein product determine the neomorphic aspect of SYS pathophysiology, which is in line with genotype–phenotype observations. In another mouse model overexpressing the N‐terminal region of Magel2 only, offspring died during the fetal or neonatal period. 151 Recent studies have shown that truncated MAGEL2 proteins tend to localize to the nucleus, which is in contrast to a mainly cytoplasmic localization of wild‐type MAGEL2. This mislocalization is more pronounced in protein products resulting from variants associated with greater clinical severity, particularly c.1996delC. Truncated proteins may acquire new functions within the nucleus, or form aggregates that disrupt normal gene expression and other nuclear processes (Figure 1). 71 , 152
THERAPEUTIC ADVANCES FOR SYS
A cure for SYS does not yet exist. 3 Some symptomatic treatments are available and generally effective but do not ease the overall impact of SYS, leaving caregivers unsatisfied. 5 Recommendations for the clinical management of SYS have been reviewed elsewhere. 3 , 71 Therapies provided to patients include speech therapy, psychotherapy and psychiatric treatment, physical therapy, gastrointestinal medications and gastric‐ or nasogastric‐tube feeding, tonsillectomy or adenoidectomy, and continuous positive airway pressure. 5 Hormone replacement therapies target the pathomechanistic chain further upstream. Patients with SYS receiving recombinant human growth hormone, which is already approved by the US Food and Drug Administration for PWS, show significant improvements in height and body mass index. Parents also reported increased muscle strength and endurance, as well as improved cognitive and social skills. Recombinant human growth hormone should be considered as a therapeutic and investigated in prospective clinical trials. 115 , 153
The oxytocin system is implicated in ASD pathophysiology, and oxytocin has attracted considerable attention as a potential therapeutic for ASD and PWS with clinical trials yielding mixed results. 154 , 155 , 156 , 157 In Magel2‐knockout mice, oxytocin has been shown to improve feeding, assure normal oxytocin system anatomy, prevent deficits in social behaviour, learning, and memory, normalize thermosensory responses, and restore neurite outgrowth. 67 , 74 , 75 , 88 , 96 , 158 Early alterations in oxytocinergic function can have long‐term consequences on development. Similarly, oxytocin treatment during development can permanently rescue some hallmark SYS symptoms in mice and social impairments in ASD models. 75 , 99 , 107 , 155 Oxytocin is probably required in critical time windows, which must be considered for therapies. 99 , 157 Notably, oxytocin is not per se a prosocial drug and should be most effective when combined with behavioural therapy. 96 , 155 The SYS community places high hopes on oxytocin, and clinical trials are being considered.
Interestingly, neonatal oxytocin increases hippocampal expression of brain‐derived neurotrophic factor (Bdnf), another therapeutic target. 159 BDNF is required for the development, maturation, and maintenance of neurons. It is also a critical regulator that functions downstream of the hypothalamic feeding circuits (leptin–POMC–melanocortin‐4 receptor pathway), thereby influencing energy homeostasis and behaviour. 84 , 160 , 161 , 162 , 163 , 164 Several neurodevelopmental disorders are associated with BDNF loss. 84 , 160 , 165 Adeno‐associated virus‐Bdnf gene therapy successfully improved body composition, energy expenditure, glucose metabolism, and behaviour in Magel2‐null mice while reversing neuroinflammation. A clear advantage of gene therapy is its action through an autoregulatory vector tied to central–peripheral feedback systems that reflect the body's physiological needs. 162 Melanocortin‐4 receptor agonists, such as setmelanotide, act within the same pathway, mimicking POMC activity. Treatment with setmelanotide is effective in reducing food intake and increasing energy expenditure in Magel2‐null mice, but translation into the clinic has proved difficult. 128 , 162 , 166 , 167
Given the current knowledge about potentially severe neomorphic effects of truncated MAGEL2, a viable treatment strategy could leverage recent advances in therapeutics that interfere with protein production at the RNA level (e.g. antisense oligonucleotides 168 ), or reduce protein levels directly (e.g. PROTACS 169 ). Thereby, the truncated protein load could be reduced, which may already offer substantial benefits. The imprinted nature of MAGEL2 offers a unique opportunity, since a functional copy of the gene is usually available on the silenced maternal allele. Genetic regions can be unsilenced, as studies targeting PWS genes have shown. 170 , 171 Another study is currently underway, 172 and it is tempting to speculate that combining an antisense oligonucleotide treatment to reduce truncated MAGEL2 with a therapeutic that reactivates the maternal copy of MAGEL2 should be close to a causal cure for SYS. Promising therapeutic strategies are summarized in Figure 3.
FIGURE 3.
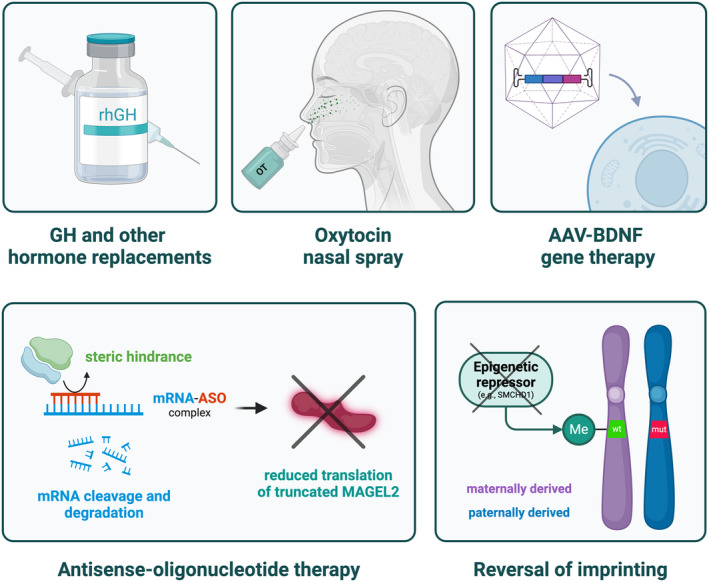
Therapeutic strategies for SYS. This figure summarizes some promising therapeutic advances with potential efficacy in addressing SYS, transcending symptomatic management. Notably, antisense oligonucleotides directed at truncated MAGEL2 could specifically address potential neomorphic effects. Abbreviations: AAV‐BDNF, adeno‐associated virus‐BDNF gene; ASO, antisense oligonucleotide; GH, growth hormone; Me, methylation signal; mRNA, messenger RNA; mut, mutation; OT, oxytocin; rhGH, recombinant human growth hormone; SYS, Schaaf–Yang syndrome; WT, wild type.
CONCLUSION
We have outlined the role of MAGEL2 in key processes at the DNA, RNA, protein, cellular, and systemic levels. MAGEL2 loss‐of‐function explains a broad range of symptoms in SYS and PWS, but incompletely accounts for the severity observed in SYS. We have reviewed available therapies as well as those in development, and proposed novel therapeutic avenues. Substantial effort should be directed towards the generation of a compound (e.g. antisense oligonucleotides or PROTAC) to reduce the amount of truncated MAGEL2 while further assessing the context‐dependent potential of other therapeutics already on the market.
Additional challenges remain to be overcome. Several findings need to be recapitulated in pathologically relevant model systems. Moreover, many therapeutics may need to surmount the blood–brain barrier to exert their effects on the central nervous system. Compounds such as oxytocin are probably not able to do that after a certain point in development. 74 , 154 Apart from drug delivery, an even greater challenge could be timing. If our current understanding of SYS as a neurodevelopmental disorder is correct, treatments may only be effective in critical periods when the formation of neural circuitry is still malleable. 99 , 157 Early intervention is probably favourable, but trials will need to investigate the ideal timepoints for starting SYS therapy, as well as the strategy‐dependent benefits of later treatment.
This work shows how concerted efforts among affected families and basic, translational, and clinical scientists can shape an increasingly comprehensive picture of a complex disease. The research presented herein serves dual objectives: it improves the understanding of human physiology and disease mechanisms, and it lays the groundwork for targeted therapies and, eventually, a cure for SYS.
FUNDING INFORMATION
None.
CONFLICT OF INTEREST STATEMENT
Tim Schubert and Christian Schaaf declare not conflict of interest in relationship to this article.
ACKNOWLEDGEMENTS
We thank Ferdinand Althammer, Jannis Bücking, Felix Franke, Annabel Kleinwächter, Johann Maaß, Moritz Wimmer, and Paul Schubert for proofreading the manuscript. The Foundation of Prader–Willi Research is acknowledged for continued support. We are grateful to all individuals with SYS and their families. They continue to be the inspiration for our work, and sharing part of this journey with them is a true honour. Open Access funding enabled and organized by Projekt DEAL.
Schubert T, Schaaf CP. MAGEL2 (patho‐)physiology and Schaaf–Yang syndrome. Dev Med Child Neurol. 2025;67:35–48. 10.1111/dmcn.16018
Plain language summary: https://onlinelibrary.wiley.com/doi/10.1111/dmcn.16065
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Hoyos Sanchez MC, Bayat T, Gee RRF, Fon Tacer K. Hormonal imbalances in Prader‐Willi and Schaaf‐Yang syndromes imply the evolution of specific regulation of hypothalamic neuroendocrine function in mammals. Int J Mol Sci 2023; 24: 13109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boccaccio I, Glatt‐Deeley H, Watrin F, Roëckel N, Lalande M, Muscatelli F. The human MAGEL2 gene and its mouse homologue are paternally expressed and mapped to the Prader‐Willi region. Hum Mol Genet 1999; 8: 2497–505. [DOI] [PubMed] [Google Scholar]
- 3. Schaaf CP, Marbach F. Schaaf‐Yang Syndrome. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle, 2021. [PubMed] [Google Scholar]
- 4. Schaaf CP, Gonzalez‐Garay ML, Xia F, et al. Truncating mutations of MAGEL2 cause Prader‐Willi phenotypes and autism. Nat Genet 2013; 45: 1405–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dötsch L, Matesevac L, Strong TV, Schaaf CP. Caregiver‐based perception of disease burden in Schaaf‐Yang syndrome. Mol Genet Genomic Med 2023; 11: e2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McCarthy J, Lupo PJ, Kovar E, et al. Schaaf‐Yang syndrome overview: Report of 78 individuals. Am J Med Genet A 2018; 176: 2564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thomason MM, McCarthy J, Goin‐Kochel RP, Dowell LR, Schaaf CP, Berry LN. Neurocognitive and neuroysal phenotype of youth with Schaaf‐Yang syndrome. J Autism Dev Disord 2020; 50: 2491–500. [DOI] [PubMed] [Google Scholar]
- 8. Aten E, Fountain MD, van Haeringen A, Schaaf CP, Santen GWE. Imprinting: the Achilles heel of trio‐based exome sequencing. Genet Med 2016; 18: 1163–4. [DOI] [PubMed] [Google Scholar]
- 9. Negishi Y, Kurosawa K, Takano K, Matsubara K, Nishiyama T, Saitoh S. A nationwide survey of Schaaf‐Yang syndrome in Japan. J Hum Genet 2022; 67: 735–8. [DOI] [PubMed] [Google Scholar]
- 10. Huang Z, Lu W, Zhang P, et al. Early onset critically ill infants with Schaaf‐Yang syndrome: a retrospective study from the China neonatal genomes project and literature review. Ann Transl Med 2023; 11: 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ahn H, Seo GH, Oh A, et al. Diagnosis of Schaaf‐Yang syndrome in Korean children with developmental delay and hypotonia. Medicine 2020; 99: e23864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Patak J, Gilfert J, Byler M, et al. MAGEL2‐related disorders: A study and case series. Clin Genet 2019; 96: 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yin J, Schaaf CP. Autism genetics ‐ an overview. Prenat Diagn 2017; 37: 14–30. [DOI] [PubMed] [Google Scholar]
- 14. Nunes S, Xavier M, Lourenço C, Melo M, Godinho C. Schaaf‐Yang syndrome: A real challenge for prenatal diagnosis. Cureus 2021; 13: e20414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marbach F, Elgizouli M, Rech M, et al. The adult phenotype of Schaaf‐Yang syndrome. Orphanet J Rare Dis 2020; 15: 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fountain MD, Aten E, Cho MT, et al. The phenotypic spectrum of Schaaf‐Yang syndrome: 18 new affected individuals from 14 families. Genet Med 2017; 19: 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mari A, Sartorio MUA, Degrassi I, et al. Late‐onset pyloric stenosis and intussusception with final diagnosis of food proteins' hypersensitivity in Schaaf‐Yang syndrome: A case report. JPGN Rep 2022; 3: e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Soden SE, Saunders CJ, Willig LK, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med 2014; 6: 265ra168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de Andrade G, de Oliveira Silva T, do Nascimento I, Boath A, da Costa Cunha K, Chermont AG. Schaaf‐Yang syndrome: A novel variant in MAGEL2 gene in the first Brazilian preterm neonate. Int J Case Rep Images 2020; 11: 01144Z01GA202. [Google Scholar]
- 20. Xu N, Shi W, Cao X, et al. Preimplantation genetic testing (PGT) and prenatal diagnosis of Schaaf‐Yang Syndrome: A report of three families and a research on genotype‐phenotype correlations. J Clin Med Res 2023; 12: 1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Juriaans AF, Kerkhof GF, Hokken‐Koelega ACS. The spectrum of the Prader‐Willi‐like pheno‐ and genotype: A review of the literature. Endocr Rev 2022; 43: 1–18. [DOI] [PubMed] [Google Scholar]
- 22. Mejlachowicz D, Nolent F, Maluenda J, et al. Truncating Mutations of MAGEL2, a Gene within the Prader‐Willi Locus, Are Responsible for Severe Arthrogryposis. Am J Hum Genet 2015; 97: 616–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Powell WT, Schaaf CP, Rech ME, Wrede J. Polysomnographic characteristics and sleep‐disordered breathing in Schaaf‐Yang syndrome. Pediatr Pulmonol 2020; 55: 3162–7. [DOI] [PubMed] [Google Scholar]
- 24. Alavanda C, Arslan Ateş E, Yavaş Abalı Z, Geçkinli BB, Turan S, Arman A. Two new cases with novel pathogenic variants reflecting the clinical diversity of Schaaf‐Yang syndrome. Clin Genet 2023; 104: 127–32. [DOI] [PubMed] [Google Scholar]
- 25. Gregory LC, Shah P, Sanner JRF, et al. Mutations in MAGEL2 and L1CAM are associated with congenital hypopituitarism and arthrogryposis. J Clin Endocrinol Metab 2019; 104: 5737–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCarthy JM, McCann‐Crosby BM, Rech ME, et al. Hormonal, metabolic and skeletal phenotype of Schaaf‐Yang syndrome: a comparison to Prader‐Willi syndrome. J Med Genet 2018; 55: 307–15. [DOI] [PubMed] [Google Scholar]
- 27. Fountain MD, Schaaf CP. Prader‐Willi Syndrome and Schaaf‐Yang Syndrome: Neurodevelopmental Diseases Intersecting at the MAGEL2 Gene. Diseases 2016; 4. 10.3390/diseases4010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bischof JM, Stewart CL, Wevrick R. Inactivation of the mouse Magel2 gene results in growth abnormalities similar to Prader‐Willi syndrome. Hum Mol Genet 2007; 16: 2713–9. [DOI] [PubMed] [Google Scholar]
- 29. Maaß JG, Brennenstuhl H, Schaaf CP. Morbidity and mortality in Schaaf‐Yang syndrome. Annals of Translational Medicine 2023; 11. 10.21037/atm-23-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rodriguez AM, Schain K, Jayakar P, Wright MS, Chowdhury S, Salyakina D. Report of two cases of Schaaf‐Yang syndrome: Same genotype and different phenotype. Clin Case Rep 2023; 11: e7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiao B, Ji X, Wei W, Hui Y, Sun Y. A Recurrent Variant in MAGEL2 in Five Siblings with Severe Respiratory Disturbance after Birth. Mol Syndromol 2020; 10: 286–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tacer KF, Potts PR. Cellular and disease functions of the Prader‐Willi Syndrome gene MAGEL2. Biochem J 2017; 474: 2177–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee S, Kozlov S, Hernandez L, et al. Expression and imprinting of MAGEL2 suggest a role in Prader‐willi syndrome and the homologous murine imprinting phenotype. Hum Mol Genet 2000; 9: 1813–9. [DOI] [PubMed] [Google Scholar]
- 34. Lee S, Walker CL, Wevrick R. Prader‐Willi syndrome transcripts are expressed in phenotypically significant regions of the developing mouse brain. Gene Expr Patterns 2003; 3: 599–609. [DOI] [PubMed] [Google Scholar]
- 35. Nakai S, Kawano H, Yudate T, et al. The POU domain transcription factor Brn‐2 is required for the determination of specific neuronal lineages in the hypothalamus of the mouse. Genes Dev 1995; 9: 3109–21. [DOI] [PubMed] [Google Scholar]
- 36. Panda S, Antoch MP, Miller BH, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002; 109: 307–20. [DOI] [PubMed] [Google Scholar]
- 37. Su AI, Cooke MP, Ching KA, et al. Large‐scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A 2002; 99: 4465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue‐based map of the human proteome. Science 2015; 347: 1260419. [DOI] [PubMed] [Google Scholar]
- 39. Kamaludin AA, Smolarchuk C, Bischof JM, et al. Muscle dysfunction caused by loss of Magel2 in a mouse model of Prader‐Willi and Schaaf‐Yang syndromes. Hum Mol Genet 2016; 25: 3798–809. [DOI] [PubMed] [Google Scholar]
- 40. Guo L, Qiao M, Wang C, Zheng R, Xiong Y‐Z, Deng C‐Y. Imprinting analysis of porcine MAGEL2 gene in two fetal stages and association analysis with carcass traits. Mol Biol Rep 2012; 39: 147–55. [DOI] [PubMed] [Google Scholar]
- 41. Dutt M, Wehrle CJ, Jialal I. Physiology, Adrenal Gland. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing, 2023. [PubMed] [Google Scholar]
- 42. Peters J. The role of genomic imprinting in biology and disease: an expanding view. Nat Rev Genet 2014; 15: 517–30. [DOI] [PubMed] [Google Scholar]
- 43. Isles AR. The contribution of imprinted genes to neurodevelopmental and neuropsychiatric disorders. Transl Psychiatry 2022; 12: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Keverne EB, Curley JP. Epigenetics, brain evolution and behaviour. Front Neuroendocrinol 2008; 29: 398–412. [DOI] [PubMed] [Google Scholar]
- 45. Reznik DL, Yang MV, Albelda de la Haza P, et al. Magel2 truncation alters select ysal and physiological outcomes in a rat model of Schaaf‐Yang syndrome. Dis Model Mech 2023; 16. 10.1242/dmm.049829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Akamine S, Sagata N, Sakai Y, et al. Early‐onset epileptic encephalopathy and severe developmental delay in an association with de novo double mutations in NF1 and MAGEL2. Epilepsia Open 2018; 3: 81–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mahfouz A, Lelieveldt BPF, Grefhorst A, et al. Genome‐wide coexpression of steroid receptors in the mouse brain: Identifying signaling pathways and functionally coordinated regions. Proc Natl Acad Sci U S A 2016; 113: 2738–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fulp CT, Cho G, Marsh ED, Nasrallah IM, Labosky PA, Golden JA. Identification of Arx transcriptional targets in the developing basal forebrain. Hum Mol Genet 2008; 17: 3740–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pisansky MT, Young AE, O'Connor MB, Gottesman II, Bagchi A, Gewirtz JC. Mice lacking the chromodomain helicase DNA‐binding 5 chromatin remodeler display autism‐like characteristics. Transl Psychiatry 2017; 7: e1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salles J, Eddiry S, Lacassagne E, et al. Patients with PWS and related syndromes display differentially methylated regions involved in neurodevelopmental and nutritional trajectory. Clin Epigenetics 2021; 13: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sanderson MR, Fahlman RP, Wevrick R. The N‐terminal domain of the Schaaf‐Yang syndrome protein MAGEL2 likely has a role in RNA metabolism. J Biol Chem 2021; 297: 100959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian S‐B. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 2015; 526: 591–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. UniProt Consortium . UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res 2023; 51: D523–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Florke Gee RR, Chen H, Lee AK, et al. Emerging roles of the MAGE protein family in stress response pathways. J Biol Chem 2020; 295: 16121–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hao YH, Fountain MD Jr, Fon Tacer K, et al. USP7 Acts as a Molecular Rheostat to Promote WASH‐Dependent Endosomal Protein Recycling and Is Mutated in a Human Neurodevelopmental Disorder. Mol Cell 2015; 59: 956–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hao YH, Doyle JM, Ramanathan S, et al. Regulation of WASH‐dependent actin polymerization and protein trafficking by ubiquitination. Cell 2013; 152: 1051–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Doyle JM, Gao J, Wang J, Yang M, Potts PR. MAGE‐RING protein complexes comprise a family of E3 ubiquitin ligases. Mol Cell 2010; 39: 963–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 2009; 10: 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang J, Fedoseienko A, Chen B, Burstein E, Jia D, Billadeau DD. Endosomal receptor trafficking: Retromer and beyond. Traffic 2018; 19: 578–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen H, Victor AK, Klein J, et al. Loss of MAGEL2 in Prader‐Willi syndrome leads to decreased secretory granule and neuropeptide production. JCI Insight 2020; 5. 10.1172/jci.insight.138576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wimmer MC, Brennenstuhl H, Hirsch S, et al. Hao‐Fountain syndrome: 32 novel patients reveal new insights into the clinical spectrum. Clin Genet 2024; 105: 499–509. [DOI] [PubMed] [Google Scholar]
- 62. Urreizti R, Cueto‐Gonzalez AM, Franco‐Valls H, et al. A De Novo Nonsense Mutation in MAGEL2 in a Patient Initially Diagnosed as Opitz‐C: Similarities Between Schaaf‐Yang and Opitz‐C Syndromes. Sci Rep 2017; 7: 44138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Elliott AM, Simard LR, Coghlan G, et al. A novel mutation in KIAA0196: identification of a gene involved in Ritscher‐Schinzel/3C syndrome in a First Nations cohort. J Med Genet 2013; 50: 819–22. [DOI] [PubMed] [Google Scholar]
- 64. Gur I, Fujiwara K, Hasegawa K, Yoshikawa K. Necdin promotes ubiquitin‐dependent degradation of PIAS1 SUMO E3 ligase. PLoS One 2014; 9: e99503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Štepihar D, Florke Gee RR, Hoyos Sanchez MC, Fon Tacer K. Cell‐specific secretory granule sorting mechanisms: the role of MAGEL2 and retromer in hypothalamic regulated secretion. Front Cell Dev Biol 2023; 11: 1243038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim T, Gondré‐Lewis MC, Arnaoutova I, Loh YP. Dense‐core secretory granule biogenesis. Physiology 2006; 21: 124–33. [DOI] [PubMed] [Google Scholar]
- 67. Schaller F, Watrin F, Sturny R, Massacrier A, Szepetowski P, Muscatelli F. A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted Magel2 gene. Hum Mol Genet 2010; 19: 4895–905. [DOI] [PubMed] [Google Scholar]
- 68. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149: 274–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ravenscroft G, Laing NG, Bönnemann CG. Pathophysiological concepts in the congenital myopathies: blurring the boundaries, sharpening the focus. Brain 2015; 138: 246–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Crutcher E, Pal R, Naini F, et al. mTOR and autophagy pathways are dysregulated in murine and human models of Schaaf‐Yang syndrome. Sci Rep 2019; 9: 15935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Castilla‐Vallmanya L, Centeno‐Pla M, Serrano M, et al. Advancing in Schaaf‐Yang syndrome pathophysiology: from bedside to subcellular analyses of truncated MAGEL2. J Med Genet 2023; 60: 406–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yang L, Peng X, Li Y, et al. Long non‐coding RNA HOTAIR promotes exosome secretion by regulating RAB35 and SNAP23 in hepatocellular carcinoma. Mol Cancer 2019; 18: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ates T, Oncul M, Dilsiz P, et al. Inactivation of Magel2 suppresses oxytocin neurons through synaptic excitation‐inhibition imbalance. Neurobiol Dis 2019; 121: 58–64. [DOI] [PubMed] [Google Scholar]
- 74. Bertoni A, Schaller F, Tyzio R, et al. Oxytocin administration in neonates shapes hippocampal circuitry and restores social ys in a mouse model of autism. Mol Psychiatry 2021; 26: 7582–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Reichova A, Schaller F, Bukatova S, Bacova Z, Muscatelli F, Bakos J. The impact of oxytocin on neurite outgrowth and synaptic proteins in Magel2‐deficient mice. Dev Neurobiol 2021; 81: 366–88. [DOI] [PubMed] [Google Scholar]
- 76. Temkin P, Morishita W, Goswami D, Arendt K, Chen L, Malenka R. The Retromer Supports AMPA Receptor Trafficking During LTP. Neuron 2017; 94: 74‐82.e5. [DOI] [PubMed] [Google Scholar]
- 77. Leonzino M, Busnelli M, Antonucci F, Verderio C, Mazzanti M, Chini B. The Timing of the Excitatory‐to‐Inhibitory GABA Switch Is Regulated by the Oxytocin Receptor via KCC2. Cell Rep 2016; 15: 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ben‐Ari Y. Is birth a critical period in the pathogenesis of autism spectrum disorders? Nat Rev Neurosci 2015; 16: 498–505. [DOI] [PubMed] [Google Scholar]
- 79. Lee S, Walker CL, Karten B, et al. Essential role for the Prader‐Willi syndrome protein necdin in axonal outgrowth. Hum Mol Genet 2005; 14: 627–37. [DOI] [PubMed] [Google Scholar]
- 80. Maillard J, Park S, Croizier S, et al. Loss of Magel2 impairs the development of hypothalamic Anorexigenic circuits. Hum Mol Genet 2016; 25: 3208–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gordeeva O, Gordeev A, Khaydukov S. Expression dynamics of Mage family genes during self‐renewal and differentiation of mouse pluripotent stem and teratocarcinoma cells. Oncotarget 2019; 10: 3248–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mercer RE, Kwolek EM, Bischof JM, van Eede M, Henkelman RM, Wevrick R. Regionally reduced brain volume, altered serotonin neurochemistry, and abnormal ys in mice null for the circadian rhythm output gene Magel2. Am J Med Genet B Neuropsychiatr Genet 2009; 150B: 1085–99. [DOI] [PubMed] [Google Scholar]
- 83. Luck C, Vitaterna MH, Wevrick R. Dopamine pathway imbalance in mice lacking Magel2, a Prader‐Willi syndrome candidate gene. Behav Neurosci 2016; 130: 448–59. [DOI] [PubMed] [Google Scholar]
- 84. Bochukova EG, Lawler K, Croizier S, et al. A Transcriptomic Signature of the Hypothalamic Response to Fasting and BDNF Deficiency in Prader‐Willi Syndrome. Cell Rep 2018; 22: 3401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Queen NJ, Huang W, Zou X, Mo X, Cao L. AAV‐BDNF gene therapy ameliorates a hypothalamic neuroinflammatory signature in the Magel2‐null model of Prader‐Willi syndrome. Mol Ther Methods Clin Dev 2023; 31: 101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mizuno S, Yokoyama K, Yokoyama A, Nukata T, Ikeda Y, Hara S. Longitudinal analysis of electroencephalography pattern changes in an infant with Schaaf‐Yang syndrome and a novel mutation in melanoma antigen L2 (MAGEL2). Mol Genet Genomic Med 2022; 10: e1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Elia M, Rutigliano I, Sacco M, et al. EEG Patterns in Patients with Prader‐Willi Syndrome. Brain Sci 2021; 11. 10.3390/brainsci11081045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Meziane H, Schaller F, Bauer S, et al. An Early Postnatal Oxytocin Treatment Prevents Social and Learning Deficits in Adult Mice Deficient for Magel2, a Gene Involved in Prader‐Willi Syndrome and Autism. Biol Psychiatry 2015; 78: 85–94. [DOI] [PubMed] [Google Scholar]
- 89. Fountain MD, Tao H, Chen CA, Yin J, Schaaf CP. Magel2 knockout mice manifest altered social phenotypes and a deficit in preference for social novelty. Genes Brain Behav 2017; 16: 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Higgs MJ, Webberley AE, Allan AJ, Talat M, John RM, Isles AR. The parenting hub of the hypothalamus is a focus of imprinted gene action. PLoS Genet 2023; 19: e1010961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bosque Ortiz GM, Santana GM, Dietrich MO. Deficiency of the paternally inherited gene Magel2 alters the development of separation‐induced vocalization and maternal ys in mice. Genes Brain Behav 2022; 21: e12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Shahid Z, Asuka E, Singh G. Physiology, Hypothalamus. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing, 2023. [PubMed] [Google Scholar]
- 93. Enya T, Okamoto N, Iba Y, et al. Three patients with Schaaf‐Yang syndrome exhibiting arthrogryposis and endocrinological abnormalities. Am J Med Genet A 2018; 176: 707–11. [DOI] [PubMed] [Google Scholar]
- 94. Jobling R, Stavropoulos DJ, Marshall CR, et al. Chitayat‐Hall and Schaaf‐Yang syndromes:a common aetiology: expanding the phenotype of MAGEL2‐related disorders. J Med Genet 2018; 55: 316–21. [DOI] [PubMed] [Google Scholar]
- 95. Baribeau DA, Anagnostou E. Oxytocin and vasopressin: linking pituitary neuropeptides and their receptors to social neurocircuits. Front Neurosci 2015; 9: 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Grinevich V, Desarménien MG, Chini B, Tauber M, Muscatelli F. Ontogenesis of oxytocin pathways in the mammalian brain: late maturation and psychosocial disorders. Front Neuroanat 2014; 8: 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Auyeung B, Lombardo MV, Baron‐Cohen S. Prenatal and postnatal hormone effects on the human brain and cognition. Pflugers Arch 2013; 465: 557–71. [DOI] [PubMed] [Google Scholar]
- 98. Muscatelli F, Matarazzo V, Chini B. Neonatal oxytocin gives the tempo of social and feeding yss. Front Mol Neurosci 2022; 15: 1071719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Muscatelli F, Desarménien MG, Matarazzo V, Grinevich V. Oxytocin Signaling in the Early Life of Mammals: Link to Neurodevelopmental Disorders Associated with ASD. Curr Top Behav Neurosci 2018; 35: 239–68. [DOI] [PubMed] [Google Scholar]
- 100. Tauber M, Diene G. Chapter 26 ‐ Prader–Willi syndrome: Hormone therapies. In: Swaab DF, Buijs RM, Lucassen PJ, Salehi A, Kreier F, editors. Handbook of Clinical Neurology. Elsevier, 2021: 351–67. [DOI] [PubMed] [Google Scholar]
- 101. Swaab DF, Purba JS, Hofman MA. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader‐Willi syndrome: a study of five cases. J Clin Endocrinol Metab 1995; 80: 573–9. [DOI] [PubMed] [Google Scholar]
- 102. Höybye C, Barkeling B, Espelund U, Petersson M, Thorén M. Peptides associated with hyperphagia in adults with Prader–Willi syndrome before and during GH treatment. Growth Horm IGF Res 2003; 13: 322–7. [DOI] [PubMed] [Google Scholar]
- 103. Johnson L, Manzardo AM, Miller JL, Driscoll DJ, Butler MG. Elevated plasma oxytocin levels in children with Prader‐Willi syndrome compared with healthy unrelated siblings. Am J Med Genet A 2016; 170: 594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Martin A, State, Matthew , Anderson GM, et al. Cerebrospinal fluid levels of oxytocin in Prader–Willi syndrome: a preliminary report. Biol Psychiatry 1998; 44: 1349–52. [DOI] [PubMed] [Google Scholar]
- 105. Althammer F, Wimmer MC, Krabichler Q, et al. Analysis of the hypothalamic oxytocin system and oxytocin receptor‐expressing astrocytes in a mouse model of Prader‐Willi syndrome. J Neuroendocrinol 2022; 34: e13217. [DOI] [PubMed] [Google Scholar]
- 106. Qian T, Wang H, Wang P, et al. A genetically encoded sensor measures temporal oxytocin release from different neuronal compartments. Nat Biotechnol 2023; 41: 944–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gigliucci V, Busnelli M, Santini F, et al. Oxytocin receptors in the Magel2 mouse model of autism: Specific region, age, sex and oxytocin treatment effects. Front Neurosci 2023; 17: 1026939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bales KL, Perkeybile AM. Developmental experiences and the oxytocin receptor system. Horm Behav 2012; 61: 313–9. [DOI] [PubMed] [Google Scholar]
- 109. Dromard Y, Borie AM, Chakraborty P, et al. Disengagement of somatostatin neurons from lateral septum circuitry by oxytocin and vasopressin restores social‐fear extinction and suppresses aggression outbursts in Prader‐Willi syndrome model. Biol Psychiatry 2023; 95: 785–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Borie AM, Dromard Y, Guillon G, et al. Correction of vasopressin deficit in the lateral septum ameliorates social deficits of mouse autism model. J Clin Invest 2021; 131. 10.1172/JCI144450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hui C, Khan M, Khan Suheb MZ, Radbel JM. Arginine Vasopressin Disorder (Diabetes Insipidus). In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing, 2024. [PubMed] [Google Scholar]
- 112. Tennese AA, Wevrick R. Impaired hypothalamic regulation of endocrine function and delayed counterregulatory response to hypoglycemia in Magel2‐null mice. Endocrinology 2011; 152: 967–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Halloun R, Habib C, Ekhilevitch N, Weiss R, Tiosano D, Cohen M. Expanding the spectrum of endocrinopathies identified in Schaaf‐Yang syndrome ‐ A case report and review of the literature. Eur J Med Genet 2021; 64: 104252. [DOI] [PubMed] [Google Scholar]
- 114. Mehta A, Hindmarsh PC, Stanhope RG, et al. The role of growth hormone in determining birth size and early postnatal growth, using congenital growth hormone deficiency (GHD) as a model. Clin Endocrinol 2005; 63: 223–31. [DOI] [PubMed] [Google Scholar]
- 115. Hebach NR, Caro P, Martin‐Giacalone BA, et al. A retrospective analysis of growth hormone therapy in children with Schaaf‐Yang syndrome. Clin Genet 2021; 100: 298–307. [DOI] [PubMed] [Google Scholar]
- 116. Baraghithy S, Smoum R, Drori A, et al. Magel2 modulates bone remodeling and mass in Prader‐Willi syndrome by affecting oleoyl Serine levels and activity. J Bone Miner Res 2019; 34: 93–105. [DOI] [PubMed] [Google Scholar]
- 117. Emerick JE, Vogt KS. Endocrine manifestations and management of Prader‐Willi syndrome. Int J Pediatr Endocrinol 2013; 2013: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mercer RE, Wevrick R. Loss of magel2, a candidate gene for features of Prader‐Willi syndrome, impairs reproductive function in mice. PLoS One 2009; 4: e4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Narayanan NS, Guarnieri DJ, DiLeone RJ. Metabolic hormones, dopamine circuits, and feeding. Front Neuroendocrinol 2010; 31: 104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. D Hidalgo‐Santos A, Del Carmen DeMingo‐Alemany M, Moreno‐Macián F, et al. A novel mutation of MAGEL2 in a patient with Schaaf‐Yang syndrome and hypopituitarism. Int J Endocrinol Metab 2018; 16: e67329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. van Vulpen EH, Yang CR, Nissen R, Renaud LP. Hypothalamic A14 and A15 catecholamine cells provide the dopaminergic innervation to the supraoptic nucleus in rat: a combined retrograde tracer and immunohistochemical study. Neuroscience 1999; 93: 675–80. [DOI] [PubMed] [Google Scholar]
- 122. García‐Tornadú I, Rubinstein M, Gaylinn BD, et al. GH in the dwarf dopaminergic D2 receptor knockout mouse: somatotrope population, GH release, and responsiveness to GH‐releasing factors and somatostatin. J Endocrinol 2006; 190: 611–9. [DOI] [PubMed] [Google Scholar]
- 123. Palmiter RD. Is dopamine a physiologically relevant mediator of feeding ys? Trends Neurosci 2007; 30: 375–81. [DOI] [PubMed] [Google Scholar]
- 124. Igarashi M, Narayanaswami V, Kimonis V, et al. Dysfunctional oleoylethanolamide signaling in a mouse model of Prader‐Willi syndrome. Pharmacol Res 2017; 117: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Resnick JL, Nicholls RD, Wevrick R, Prader‐Willi Syndrome Animal Models Working Group. Recommendations for the investigation of animal models of Prader‐Willi syndrome. Mamm Genome 2013; 24: 165–78. [DOI] [PubMed] [Google Scholar]
- 126. Costa RA, Ferreira IR, Cintra HA, Gomes LHF, da Guida L C. Genotype‐Phenotype Relationships and Endocrine Findings in Prader‐Willi Syndrome. Front Endocrinol 2019; 10: 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Myers SE, Davis A, Whitman BY, Santiago JV, Landt M. Leptin concentrations in Prader‐Willi syndrome before and after growth hormone replacement. Clin Endocrinol 2000; 52: 101–5. [DOI] [PubMed] [Google Scholar]
- 128. Mercer RE, Michaelson SD, Chee MJS, Atallah TA, Wevrick R, Colmers WF. Magel2 is required for leptin‐mediated depolarization of POMC neurons in the hypothalamic arcuate nucleus in mice. PLoS Genet 2013; 9: e1003207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Baldini G, Phelan KD. The melanocortin pathway and control of appetite‐progress and therapeutic implications. J Endocrinol 2019; 241: R1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Oncul M, Dilsiz P, Ates Oz E, et al. Impaired melanocortin pathway function in Prader‐Willi syndrome gene‐Magel2 deficient mice. Hum Mol Genet 2018; 27: 3129–36. [DOI] [PubMed] [Google Scholar]
- 131. Pravdivyi I, Ballanyi K, Colmers WF, Wevrick R. Progressive postnatal decline in leptin sensitivity of arcuate hypothalamic neurons in the Magel2‐null mouse model of Prader‐Willi syndrome. Hum Mol Genet 2015; 24: 4276–83. [DOI] [PubMed] [Google Scholar]
- 132. Wijesuriya TM, De Ceuninck L, Masschaele D, et al. The Prader‐Willi syndrome proteins MAGEL2 and necdin regulate leptin receptor cell surface abundance through ubiquitination pathways. Hum Mol Genet 2017; 26: 4215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Schmidt O, Teis D. The ESCRT machinery. Curr Biol 2012; 22: R116‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Tong W, Wang Y, Lu Y, et al. Whole‐exome Sequencing Helps the Diagnosis and Treatment in Children with Neurodevelopmental Delay Accompanied Unexplained Dyspnea. Sci Rep 2018; 8: 5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Bayat A, Bayat M, Lozoya R, Schaaf CP. Chronic intestinal pseudo‐obstruction syndrome and gastrointestinal malrotation in an infantwith schaaf‐yang syndrome ‐ Expanding the phenotypic spectrum. Eur J Med Genet 2018; 61: 627–30. [DOI] [PubMed] [Google Scholar]
- 136. Chumpitazi B, Nurko S. Pediatric gastrointestinal motility disorders: challenges and a clinical update. Gastroenterol Hepatol 2008; 4: 140–8. [PMC free article] [PubMed] [Google Scholar]
- 137. Angius A, Uva P, Oppo M, et al. Exome sequencing in Crisponi/cold‐induced sweating syndrome‐like individuals reveals unpredicted alternative diagnoses. Clin Genet 2019; 95: 607–14. [DOI] [PubMed] [Google Scholar]
- 138. Kozlov SV, Bogenpohl JW, Howell MP, et al. The imprinted gene Magel2 regulates normal circadian output. Nat Genet 2007; 39: 1266–72. [DOI] [PubMed] [Google Scholar]
- 139. Devos J, Weselake SV, Wevrick R. Magel2, a Prader‐Willi syndrome candidate gene, modulates the activities of circadian rhythm proteins in cultured cells. J Circadian Rhythms 2011; 9: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Carias KV, Zoeteman M, Seewald A, Sanderson MR, Bischof JM, Wevrick R. A MAGEL2‐deubiquitinase complex modulates the ubiquitination of circadian rhythm protein CRY1. PLoS One 2020; 15: e0230874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Vaidyanathan R, Schaller F, Muscatelli F, Hammock EAD. Colocalization of Oxtr with Prader‐Willi syndrome transcripts in the trigeminal ganglion of neonatal mice. Hum Mol Genet 2020; 29: 2065–75. [DOI] [PubMed] [Google Scholar]
- 142. Murphy RF, Mooney JF 3rd. Current concepts in neuromuscular scoliosis. Curr Rev Musculoskelet Med 2019; 12: 220–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Chen X, Ma X, Zou C. Phenotypic spectrum and genetic analysis in the fatal cases of Schaaf‐Yang syndrome: Two case reports and literature review. Medicine 2020; 99: e20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Gutkowska J, Jankowski M, Lambert C, Mukaddam‐Daher S, Zingg HH, McCann SM. Oxytocin releases atrial natriuretic peptide by combining with oxytocin receptors in the heart. Proc Natl Acad Sci U S A 1997; 94: 11704–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Buiting K, Di Donato N, Beygo J, et al. Clinical phenotypes of MAGEL2 mutations and deletions. Orphanet J Rare Dis 2014; 9: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Kanber D, Giltay J, Wieczorek D, et al. A paternal deletion of MKRN3, MAGEL2 and NDN does not result in Prader‐Willi syndrome. Eur J Hum Genet 2009; 17: 582–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Matarazzo V, Muscatelli F. Natural breaking of the maternal silence at the mouse and human imprinted Prader‐Willi locus: A whisper with functional consequences. Rare Dis 2013; 1: e27228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hug N, Longman D, Cáceres JF. Mechanism and regulation of the nonsense‐mediated decay pathway. Nucleic Acids Res 2016; 44: 1483–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Brogna S, Wen J. Nonsense‐mediated mRNA decay (NMD) mechanisms. Nat Struct Mol Biol 2009; 16: 107–13. [DOI] [PubMed] [Google Scholar]
- 150. Negishi Y, Ieda D, Hori I, et al. Schaaf‐Yang syndrome shows a Prader‐Willi syndrome‐like phenotype during infancy. Orphanet J Rare Dis 2019; 14: 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ieda D, Negishi Y, Miyamoto T, et al. Two mouse models carrying truncating mutations in Magel2 show distinct phenotypes. PLoS One 2020; 15: e0237814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Centeno‐Pla M, Alcaide‐Consuegra E, Gibson S, et al. Subcellular localisation of truncated MAGEL2 proteins: insight into the molecular pathology of Schaaf‐Yang syndrome. J Med Genet 2024. 10.1136/jmg-2024-109898. [DOI] [PubMed] [Google Scholar]
- 153. Juriaans AF, Kerkhof GF, Garrelfs M, Trueba‐Timmermans D, Hokken‐Koelega ACS. Schaaf‐Yang syndrome: Clinical phenotype and effects of 4 years of growth hormone treatment. Horm Res Paediatr 2023; 97: 148–56. [DOI] [PubMed] [Google Scholar]
- 154. Peñagarikano O. Oxytocin in animal models of autism spectrum disorder. Dev Neurobiol 2017; 77: 202–13. [DOI] [PubMed] [Google Scholar]
- 155. Young LJ, Barrett CE. Neuroscience. Can oxytocin treat autism? Science 2015; 347: 825–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Ooi YP, Weng S‐J, Kossowsky J, Gerger H, Sung M. Oxytocin and Autism Spectrum Disorders: A Systematic Review and Meta‐Analysis of Randomized Controlled Trials. Pharmacopsychiatry 2017; 50: 5–13. [DOI] [PubMed] [Google Scholar]
- 157. Althammer F, Muscatelli F, Grinevich V, Schaaf CP. Oxytocin‐based therapies for treatment of Prader‐Willi and Schaaf‐Yang syndromes: evidence, disappointments, and future research strategies. Transl Psychiatry 2022; 12: 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Da Prato LC, Zayan U, Abdallah D, et al. Early life oxytocin treatment improves thermo‐sensory reactivity and maternal ys in neonates lacking the autism‐associated gene Magel2. Neuropsychopharmacology 2022; 47: 1901–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Bakos J, Lestanova Z, Strbak V, Havranek T, Bacova Z. Neonatal manipulation of oxytocin prevents lipopolysaccharide‐induced decrease in gene expression of growth factors in two developmental stages of the female rat. Neuropeptides 2014; 48: 281–6. [DOI] [PubMed] [Google Scholar]
- 160. Han JC. Rare Syndromes and Common Variants of the Brain‐Derived Neurotrophic Factor Gene in Human Obesity. Prog Mol Biol Transl Sci 2016; 140: 75–95. [DOI] [PubMed] [Google Scholar]
- 161. Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell 1994; 77: 627–38. [DOI] [PubMed] [Google Scholar]
- 162. Queen NJ, Zou X, Anderson JM, et al. Hypothalamic AAV‐BDNF gene therapy improves metabolic function and ys in the Magel2‐null mouse model of Prader‐Willi syndrome. Mol Ther Methods Clin Dev 2022; 27: 131–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Ranadive SA, Vaisse C. Lessons from extreme human obesity: monogenic disorders. Endocrinol Metab Clin North Am 2008; 37: 733–51, x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Colmers WF, Wevrick R. Leptin signaling defects in a mouse model of Prader‐Willi syndrome: An orphan genetic obesity syndrome no more? Rare Dis 2013; 1: e24421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Gray J, Yeo GSH, Cox JJ, et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain‐derived neurotrophic factor (BDNF) gene. Diabetes 2006; 55: 3366–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Bischof JM, Van Der Ploeg LHT, Colmers WF, Wevrick R. Magel2‐null mice are hyper‐responsive to setmelanotide, a melanocortin 4 receptor agonist. Br J Pharmacol 2016; 173: 2614–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. NCT02311673. https://www.clinicaltrials.gov/study/NCT02311673?cond=NCT02311673&rank=1 (accessed 10 April 2024).
- 168. Dhuri K, Bechtold C, Quijano E, et al. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J Clin Med Res 2020; 9. 10.3390/jcm9062004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Ottis P, Crews CM. Proteolysis‐Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem Biol 2017; 12: 892–8. [DOI] [PubMed] [Google Scholar]
- 170. Cruvinel E, Budinetz T, Germain N, Chamberlain S, Lalande M, Martins‐Taylor K. Reactivation of maternal SNORD116 cluster via SETDB1 knockdown in Prader‐Willi syndrome iPSCs. Hum Mol Genet 2014; 23: 4674–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Kim Y, Lee H‐M, Xiong Y, et al. Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader–Willi syndrome. Nat Med 2016; 23: 213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. FPWR . Targeting SMCHD1 to address the underlying cause of PWS and SYS. https://www.fpwr.org/fpwr‐funded‐projects/targeting‐smchd1‐to‐address‐the‐underlying‐cause‐of‐pws‐and‐sys (accessed 10 April 2024).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.