

Abstract
Nucleoside analogues have broad clinical utility as antiviral drugs. Key to their systemic distribution and cellular entry are human nucleoside transporters. Here we establish that the human concentrative nucleoside transporter 3 (hCNT3) interacts with antiviral drugs used in the treatment of coronavirus infections. We report high-resolution single-particle cryo-electron microscopy (cryo-EM) structures of bovine CNT3 complexed to antiviral nucleosides N4-hydroxycytidine, PSI-6206, GS-441524, and ribavirin all in inward-facing states. Notably, we found that the orally bioavailable antiviral molnupiravir arrests CNT3 in four distinct conformations, allowing us to capture cryo-EM structures of drug-loaded outward-facing and drug-loaded intermediate states. Our studies uncover the conformational trajectory of CNT3 during membrane transport of a nucleoside analogue antiviral drug, yield new insights into the role of interactions between the transport and the scaffold domains in elevator-like domain movements during drug translocation, and provide insights to the design of nucleoside-analogue antiviral prodrugs with improved oral bioavailability.
Introduction
There is a dire need for improved small-molecule therapeutics to treat viral diseases. Nucleoside analogue antivirals (NAAs) represent a large class of drugs against a variety of infectious agents, including hepatitis C virus (HCV), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and influenza viruses1,2. The mechanisms of action include inhibition of viral replication through polymerase inhibition, lethal mutagenesis, or nucleic acid chain termination1,2,4,5. NAA drugs are prodrugs, meaning they require cellular uptake and subsequent phosphorylation to transform into their active nucleotide triphosphate form.
The efficacy of NAAs relies on their systemic distribution and disposition. Remdesivir (an FDA-approved therapeutic for SARS-CoV-2)6, sofosbuvir (an FDA-approved therapeutic for HCV)7, and molnupiravir (MPV; currently under FDA Emergency Use Authorization for SARS-CoV-2)8, harbor protecting groups (PGs) at the 5’ position of their parent nucleosides: GS-441524 (GS4), PSI-6206, and N4-hydroxycytidine (NHC) (Supplementary Fig. 1). Pharmacokinetics studies show that serum esterases and phosphoramidases rapidly convert these drugs into the parent nucleoside forms, rendering them the dominant species in plasma9–13. In some cases, PGs are not required for optimal efficacy, as ribavirin (Supplementary Fig. 1) is an unprotected NAA with a rich history of clinical use against HCV5,14, GS4 has proven activity against feline coronavirus infection in vivo15, and NHC is a broad spectrum antiviral against various viruses including coronaviruses11,16 and influenza viruses17,18. There is a growing demand for rigorous testing of unprotected NAAs to assess their clinical potential12.
Regardless of 5’-hydroxyl protecting groups (5’-PGs), free NAAs are systemically distributed shortly after drug administration9–12. These xenobiotics are small, hydrophilic molecules, depending on two distinct carrier families for NAA membrane transport in humans – concentrative nucleoside transporters of the solute carrier family 28 (hCNTs; SLC28)19–22 and equilibrative nucleoside transporters of the solute carrier family 29 (hENTs; SLC29)23–25. Both hCNTs and hENTs play crucial roles in the pharmacokinetics and pharmacodynamics of NAAs, as they control NAA distribution and cellular entry into infected cells. This is supported by reported correlations between human nucleoside transporter (hNT) expression levels or polymorphisms and patient responsiveness to NAAs, ribavirin being a prime example26–30. Recent reports highlight the role of hNTs in SARS-CoV-2 therapeutic action31.
Human CNT subtype 3 (hCNT3), a sodium-coupled nucleoside symporter, is the most effective human transporter for cellular GS4 uptake32, consistent with its role in a broad range of NAA transport3. It exhibits high expression in epithelial cells23, wide substrate specificity, and a 2:1 sodium:nucleoside coupling stoichiometry, which contrasts the narrow nucleoside selectivity and 1:1 coupling ratios in subtypes 1 and 233. Therefore, understanding the nature of drug interaction with hCNT3 will prove critical for developing therapeutics with improved pharmacological properties.
There is a wealth of previous structural studies concerning the nucleoside recognition and the elevator alternating access mechanism by the CNT family, but they are mostly limited to bacterial CNTs19–22. Our understanding of antiviral drug recognition and transport by mammalian CNTs is relatively incomplete, with numerous questions remaining. Are NAA prodrugs efficiently translocated by mammalian CNT3? Does the conformational trajectory of drug loaded-carrier during translocation deviate from what is reported for apo-carrier? Do the subunits of the CNT3 trimer operate independently, or do they exhibit any cooperativity? Here, we address these open questions and delve into the mechanisms of NAA recognition and transport by a mammalian CNT3. We discovered the prodrug MPV exhibits severely altered transport properties compared to its parent NAA NHC, elucidated cryo-EM structures of numerous distinct MPV arrested CNT3 conformations, interrogated the energetic landscape of NAA translocation via CNT3, and explored the structural basis of subunit cooperativity between subunits within a CNT3 trimer. Our findings also present an idea that could be useful in the design of NAA prodrugs with improved pharmacological properties.
Results
Antiviral drug interaction at human CNT3
We expressed five hNT subtypes in Xenopus laevis oocytes (Extended Data Fig. 1a–c) and assessed the ability of cold NHC or GS4 to competitively inhibit [3H]-nucleoside uptake. Both antiviral nucleosides significantly inhibited [3H]-ribavirin uptake at hCNT1–3 and hENT1–2 (Fig. 1a). NHC and GS4 showed half-maximal inhibitory concentration (IC50) values of ~13 μM and ~400 μM, respectively, for hCNT3-mediated [3H]-ribavirin uptake (Fig. 1b). Unexpectedly, the prodrug MPV also showed an IC50 value of ~54 μM (Fig. 1b). These results confirm drug-interaction with these hNT subtypes, but their transportability remain ambiguous. Since CNTs are sodium-coupled symporters, co-transport of nucleoside and sodium ions is electrogenic, which can be measured with two-electrode voltage-clamp (TEVC)33. Using TEVC, we found that NHC evokes inward sodium currents in oocytes expressing hCNT3 to levels similar to uridine, suggesting it is a substrate for hCNT3 (Fig. 1c). Regarding GS4, because hCNT3 raw functional activity was lower in radiotracer uptake experiments either due to weaker surface expression or lower turnover rate (Extended Data Fig. 1d), we utilized bCNT3 to detect GS4-evoked inward sodium currents and found that GS4 is a substrate for bCNT3 (Fig. 1d). In line with this, GS4 was reported to be transported by hCNT332.
Figure 1 |. Nucleoside analogue antivirals interact with human CNT3.

a, Cold competition of 1.0 mM unlabeled NHC or GS4 against 1.0 μM [3H]-ribavirin uptake mediated by nucleoside transporter-expressing Xenopus laevis oocytes in 5 minutes (WT hCNT2, WT hCNT3, WT hENT1, WT hENT2) or 15 minutes (WT hCNT1). Negative controls were as follows: use of sodium-free ND-96 (96 mM choline chloride) media for all hCNT isoforms, +10 μM dilazep (ENT-specific inhibitor) for hENT1, +1.0 mM dilazep for hENT2. (n=6 biological replicates, individual replicates and mean ± s.e.m. shown). b, Cold-competition inhibition of WT hCNT3 mediated [3H]-ribavirin uptake by NHC, GS4, or MPV (15-minute uptake with 1.0 μM [3H]-ribavirin and co-applied cold inhibitor; n=3 biological replicates with individual replicates shown; IC50 fits ± s.e.m. shown in legend). c, Two-electrode voltage-clamp (TEVC) recordings of inward currents elicited by application of 100 μM uridine or 100 μM NHC to hCNT3 expressing oocytes (representative trace at left, summary of peak currents from n=3 biological replicates at right with individual measurements and mean ± s.e.m. shown; values are baseline corrected to leak current per oocyte). d, TEVC recordings of inward currents elicited by application of 100 μM uridine or 400 μM GS4 in WT bCNT3 expressing oocytes (representative trace at left, summary of peak currents from n=4 biological replicates at right; values are baseline corrected to leak current per oocyte).
Structure determination and transporter architecture
The bCNT3 ortholog exhibits excellent biochemical stability in detergent buffer. bCNT3 is 79% identical to hCNT3, with the residues corresponding to nucleoside coordination20,21 absolutely conserved between the orthologs (Supplementary Fig. 2). Functionally, bCNT3 mediates sodium-dependent uptake of [3H]-ribavirin when expressed in oocytes, with its uptake activity inhibited by the presence of cold NHC or GS4 (Extended Data Fig. 1d). We first obtained six cryo-electron microscopy (cryo-EM) reconstructions of bCNT3 in lipid nanodiscs including drug-free and drug-bound states (NHC, GS4, PSI-6206, and ribavirin), in the presence of NaCl (Fig. 2, Extended Data Figs. 2–3 and Supplementary Table 1). The reconstructions are of high quality, ranging from 2.31 Å to 3.23 Å resolution, and feature layers of lipid densities (Fig. 2a, Extended Data Figs. 2–3).
Figure 2 |. Cryo-EM structures of inward-facing state Bos taurus CNT3 in lipid nanodiscs.
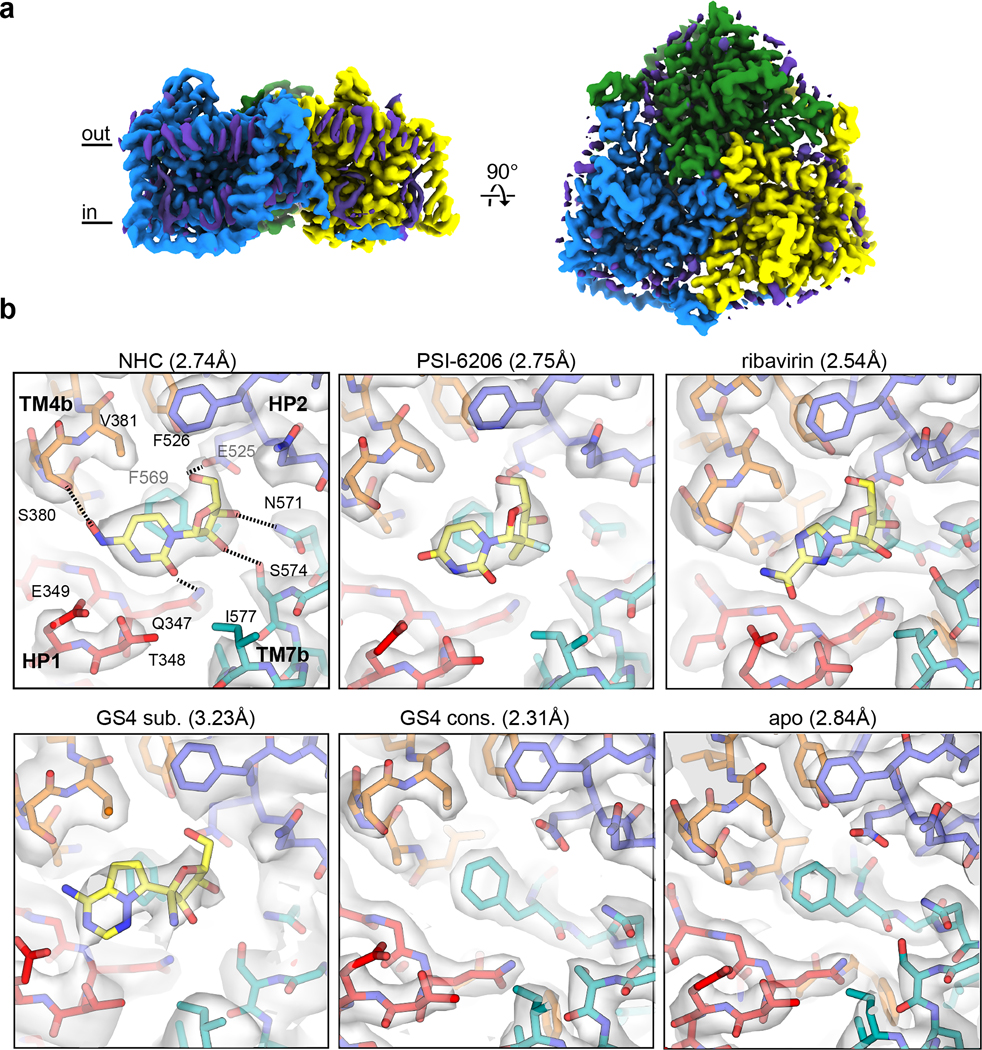
a, Representative cryo-EM reconstruction of trimeric Bos taurus CNT3 in the IFS conformation (2.31 Å “GS4 consensus” - unsharpened map). Protein cryo-EM densities colored by protomer, and non-protein lipid cryo-EM densities are depicted in purple. b, Transporter-drug interactions in additional drug-bound and drug-free states. For better comparison, cryo-EM maps shown here are low passed to 2.84 Å (NHC, PSI-6206, ribavirin, GS4 consensus and apo) or without low pass filter (GS4 subset), followed by uniform map sharpening with −50 e−/Å2 post-hoc B-factor applied. The true resolutions for these cryo-EM reconstructions are shown in parenthesis. Cryo-EM densities are shown at map threshold values of 6σ for NHC, PSI-6206, ribavirin, GS4 sub., apo, or 7σ for GS4 cons.
For consistency with previous CNT studies19–21, we preserve the numbering for conserved structural elements. The architecture is divided into two parts - a “scaffold domain”, which serves as a rigid trimerization interface that also surrounds the mobile “transport domain” (Supplementary Fig. 3a,b). In the scaffold domain, transmembrane helices 3 and 6 (TM3 and TM6) participate in the trimerization and TM6 serves as a hydrophobic barrier to nucleoside transport (Supplementary Fig. 3a,b)19,21. The nucleoside binding site is in the transport domain, at the interface of TM4b, TM7b, helical hairpin 1 (HP1), and HP2. A feature unique to eukaryotic CNTs are three additional TMs (denoted as TM1–3Eu) and two interfacial helices (IH1–2Eu) at their N-terminus22, which we term the “eukaryotic scaffold domain” (Supplementary Fig. 3a,b). Part of this motif is domain swapped, where IH1Eu and TM1Eu contact the neighboring protomer. Well-ordered lipids occupy the crevices between the eukaryotic scaffold and transport domains (Fig. 2a). Deletion of this eukaryotic scaffold domain reduces transport activity by 94–97% but does not abolish function34. This domain has also been implicated in membrane trafficking and protein-protein interaction35.
Antiviral drug recognition in the IFS
All six aforementioned cryo-EM structures adopt nearly identical conformations with global Cα r.m.s.d. values of ~0.2 Å within the protomer (Extended Data Fig. 2). Considering the nucleoside binding pockets are below TM6 and open to the intracellular solution, they represent inward-facing states (IFS) (Supplementary Fig. 3a,b). This apparent propensity for bCNT3 adopting the IFS state after detergent extraction, in the presence of NaCl, is consistent with previous structural studies on CNTs19–22. The quality of the maps enabled unambiguous modeling of nucleoside drugs for NHC, PSI-6206, and ribavirin including the assignments of the nucleobase ring orientation and the sugar pucker of ribose as 3’-endo (“north”), consistent with previous studies on CNTs (Fig. 2b, Supplementary Figs. 3–5, Supplementary Table 2)20,36. While GS4 is a low-affinity CNT3 substrate (Fig. 1a,b) and the initial reconstruction showed no clear drug density (“GS4 consensus”; Fig. 2b), after extensive image classification (Extended Data Fig. 2), a subset of particles yielded a reconstruction with a noticeable drug density, enabling GS4 modeling (“GS4 subset”; Fig. 2b).
The poses for all four drugs are similar, as side chains of HP1 and TM4 interact with the nucleobase portion of the drug, while those of HP2 and TM7 interact with the ribose portion of the drug (Fig. 2b). Central to ligand binding is F569, in an unwound portion of TM7, that forms a π-π interaction with the nucleobase ring19–21. NHC is a cytidine analogue, with the only difference being an N4-hydroxyl group. This substituent interacts with S380 in the bCNT3-NHC structure, and likely contributes to its high affinity for CNT3 (Fig. 1a–c, Extended Data Fig. 1d). The anti-HCV nucleoside PSI-6206 is a uridine analogue, featuring a fluorine at the epimeric position analogous to the 2’-hydroxyl group and a methyl group at the opposite epimeric position at 2’ of its ribose. This 2’ methyl substituent appears to alter the drug’s interaction with TM7, as it pushes the ribose moiety away from TM7 and F569 (Supplementary Fig. 3c,e). The broad-spectrum NAA GS4 features a cyano group at the 1’ position, at the epimeric position opposite to nucleobase. Although this bulky group points away from TM7, it appears to affect the ribose interactions with bCNT3, as its ribose is shifted compared to NHC (Supplementary Fig. 3d,e), likely explaining its low CNT3 affinity and low-level evoked sodium currents in bCNT3 (Fig. 1b,d).
Ion binding sites
We assigned Na+ ions and their coordination in the 2.31-Å-resolution GS4 consensus maps (Extended Data Fig. 4). The first Na+ site, located ~7 Å from the nucleoside binding site with octahedral coordination (Extended Data Fig. 4), matches the single Na+ site in bacterial CNTs21 and is termed “Na+ site 1”. The 2:1 sodium:nucleoside ratio in hCNT333 and previous studies22,37 suggests an additional Na+ site in bCNT3, proximal to C602 (C608 in bCNT3). We observed an oblong cryo-EM density peak consistent with Na+ and water. The CMM server38 aided in the assignment of Na+ and water, exhibiting a square planar coordination (Extended Data Fig. 4). Intriguingly, this “Na+ site 2” is connected to Na+ site 1 via the unwound portion of TM4 (Extended Data Fig. 4). We previously showed that Na+ at site 1 does not directly interact with nucleoside but its occupancy stabilizes the nucleoside binding pocket, thereby enhancing its nucleoside affinity19,21. The arrangement and proximity of the Na+ site 2 to the nucleoside binding pocket resemble those of Na+ site 1, suggesting that the role of the Na+ site 2 is similar to that of the Na+ site 1 in modulating nucleoside affinity. Consistent with this, the CNT3 C602R variant, which has an altered stoichiometry of 1:1 for Na+/nucleoside, exhibits altered specificity for nucleoside drugs39.
MPV arrested OFS, INT1, INT2, and INT3 conformations
Previous bacterial CNT studies revealed elevator-type movements in apo states, representing the return path after nucleoside and sodium release19. However, elevator-type movements of a CNT with drug and sodium bound have yet to be elucidated.
Using TEVC, we found that MPV rapidly and reversibly reduces uridine-evoked inward currents in hCNT3 and bCNT3 indicating either that MPV transports through CNT3 very slowly or acts as an inhibitor (Extended Data Fig. 5a,b). We limited this competition TEVC experiment to MPV - ribavirin, GS4, and NHC are efficiently transported by CNT3 (Fig. 1a–d). This unanticipated result led us to posit that MPV might stabilize purified bCNT3 in conformational states different from IFS. We employed two approaches to introduce the drug into bCNT3 for cryo-EM grid preparation: throughout protein purification (“condition 1”), or just before freezing (“condition 2” - see Methods). In the “condition 1” dataset, using both symmetry expansion with 3D classification and 3D-variability analysis, we identified three distinct non-IFS conformers in the protomer: 1) an outward-facing state (OFS), 2) an intermediate conformer in which the nucleoside binding site is completely occluded (termed “INT1”), and 3) a state close to IFS (“INT3”) (Extended Data Fig. 6a,b and Supplementary Table 3). In the “condition 2” dataset, using symmetry expansion with 3D classification, we found two different non-IFS conformations within the protomer: 1) one nearly identical to INT1 in “condition 1” and 2) a state distinct from both INT1 and INT3 (termed henceforth “INT2”) (Extended Data Fig. 7a,b and Supplementary Table 3). All four conformations (OFS, INT1, INT2, and INT3) feature robust cryo-EM densities for MPV (Extended Data Fig. 8).
Using TM3 and TM6 as alignment references, rigid body motions of the transport domain are apparent from OFS to INT3 states (Fig. 3a–c). The OFS to INT1 transition shows a large downward movement of the transport domain, whereas from INT1 to INT2 and INT3 the transport domain rotates with a small downward shift (Fig. 3b). The INT1state features a cavity in which the solvent has no access to bound MPV from either side of the membrane, while the rotations from INT1 to INT2 and INT3 progressively expose this cavity to the intracellular solution (Fig. 3a). Hence, the elevator-type transport domain motions slide the nucleoside binding site across TMs 3 and 6 by ~11 Å, accompanied by a ~15° rotation (Fig. 3b,c).
Figure 3 |. Alternating access of molnupiravir-loaded bCNT3.

a, Surface representation of OFS, INT1, INT2, INT3, or IFS states. MPV or NHC shown as sticks with yellow highlight. Cavity outline highlighted with a red line. b, Rigid-body transitions of the transport domain (tube cartoon, colored) relative to the scaffold domain (cartoon, grey) in all five elucidated conformational states. Lines serve as an approximate reference for the transport domain tilt angle, with dotted lines representing the previous conformer in the trajectory, and solid lines representing the current conformer. c, Cryo-EM map to model for the TM7b-TM8 turn in stepwise transitions from OFS to INT1, INT1 to INT2, and INT2 to INT3 (maps low-pass filtered to 4.5 Å for comparison). d, Components of the extracellular gate (HP2 and TM4b) and the intracellular gate (HP1 and TM7b) in OFS, INT1, and IFS, with relative position of scaffold domain elements TM3 and TM6 shown for reference. e Interactions of F526 of HP2 with the 5’ protecting group of MPV and F289, F290 of the scaffold domain, in the OFS, INT1, INT2, INT3 and IFS states (top). The transport domain of the NHC structure modeled onto the conformers by structural superposition (bottom).
State-dependent solvent accessibility changes to the nucleoside pocket result from varied interactions between the transport and scaffold domains. In OFS, TM7b and HP1 pack against TMs 3 and 6, forming an intracellular gate (Fig. 3d). In IFS, TM4b and HP2 pack against TMs 3 and 6, forming an extracellular gate. These motions alternately form or break two gates, underlying the alternating access mechanism of the NAA-loaded carriers (Fig. 3d). The transport domain in INT1 features simultaneous engagement of both extracellular and intracellular gates, representing a drug and sodium bound fully-occluded state (Fig. 3a,d). The previous bacterial CNT study that showed apo-intermediate states midway between the OFS and IFS states19. In stark contrast, the drug-loaded intermediate states here are closer to IFS (Extended Data Fig. 9a,b).
Contribution of the switch of HP2 to transport dynamics
Our data indicates MPV as either a low turnover substrate or an inhibitor of CNT3 (Fig. 1b and Extended Data Fig. 5a,b). How does MPV stabilize various bCNT3 conformations? Alignment of the transport domains of non-IFS states to an IFS structure (NHC bound) reveals that the key structural difference is the side chain rotamer change of F526 on HP2. The 5’-isopropyl ester PG of MPV contacts F526, causing its reorientation, without affecting other side chains in the binding pocket (Extended Data Fig. 9c). Notably, this change in the F526 rotamer conformation impacts its local interaction network with the scaffold domain (Fig. 3e). In the OFS conformation, this 5’-PG of MPV is sandwiched between F526 and phenylalanine residues on the scaffold domain (F289, F290) (Fig. 3e), making the transport domain transition from OFS to INT1 (or IFS) difficult. However, the F526 alternate rotamer, when modelled, seems to slide more easily past F289 of the scaffold domain (Fig. 3e). The modeled alternate F526 rotamer would be poised to form optimal π-π interactions with F289 as it slides past it in the three intermediate states (Fig. 3e). These findings provide mechanistic insights into the transport mechanism of CNT3. When in its optimal side-chain conformation, F526 would mediate optimal interactions with the scaffold domain in the transient intermediate states, facilitating smoother transitions. When NAA bearing a bulky 5’-PG is loaded, F526 adopts a different rotamer, acting as a “steric switch” hindering efficient sliding of the transport domain (Fig. 3e). We found that non-aromatic side chain substitutions of F526 decreased uptake activity for [3H]-ribavirin, suggesting the need for an aromatic side chain at position 526 for nucleoside transport (Extended Data Fig. 9d).
To test the F526 steric switch hypothesis, we conducted all-atom molecular dynamics (MD) simulations (Fig. 4). We postulated that: 1) when MPV binds to CNT3 in OFS, a combination of the activated F526 steric switch and the bulky 5’-PG of MPV would hinder the OFS to INT transition; and 2) In the INT1 state, the activated F526 steric switch stabilize the INT1 state, making the INT1 to INT2 transition more difficult.
Figure 4 |. All-atom MD simulations of CNT3 transport dynamics.

a, Simulation systems for bCNT3 trimer with either NHC (left) or MPV (right) in the INT1-INT1-OFS state was embedded in a mixed lipid bilayer in 150 mM NaCl. b, Standard all-atom MD simulation of the OFS state with NHC-bound (top) or MPV bound (bottom). c, Standard all-atom MD simulation of the INT1 state with NHC-bound (top) or MPV bound (bottom). d, Steered MD (SMD) simulations of NHC from OFS to INT1 (top) and MPV from OFS to INT1 (bottom); RMSD time series calculated based on drug relative to the binding pocket in the OFS or INT1 states. e, Representative frames from SMD simulations of NHC bound bCNT3 from OFS to INT1. f, Representative frames from SMD simulations of MPV bound bCNT3 from OFS to INT1.
We chose the INT1-INT1-OFS trimer structure with MPV for our MD simulation studies. For comparison, we also generated a model in which MPV is replaced with NHC (without a 5’ substituent) and the “steric switch” is turned off (Fig. 4a). We performed two types of all-atom MD simulations: 1) standard MD without restraints and 2) steered MD (SMD) for OFS to INT1 transition (see Methods, Supplementary Data Table 4, and Supplementary Table 5).
We first monitored NAA-bound bCNT3 dynamics through ligand r.m.s.d. relative to the binding pocket in standard MD simulations. There are 5 replicas with each replica contains a trimer with 2 INT1 and 1 OFS conformers - 10 INT1 and 5 OFS in total. After 500 ns, out of 10 INT1 conformers, 6 with NHC and 8 with MPV remained bound (Fig. 4b). Notably, NHCs in INT1 (Fig. 4b) exhibit larger deviations than those of MPVs (Fig. 4b). Interestingly, 4 out of 10 INT1 states with NHC transitioned into an IFS-like conformation, releasing NHC into the cytoplasm (Supplementary Fig. 6). This is consistent with the ideas that the INT1 is a metastable state with greater stability when MPV is bound and is closer to the IFS than OFS in its energetic landscape. We also observed more MPVs (3/5 MPV) stay bound in the OFS conformer than NHC (1/5 NHC) (Fig 4c). The OFS to INT1 transition was not observed during the standard MD simulations.
Using SMD simulations, we monitored the conformational transitions of ligands and the transport domain (binding pocket) with r.m.s.d. per SMD frame, relative to the starting OFS and INT1 conformations. When NHC was bound and the steric switch off (Fig. 4d), r.m.s.d.s of both NHC and the binding pocket relative to the OFS reference increase linearly, and those relative to the INT1 decrease linearly with a minimum at ~14 ns. This illustrates that the transport domain transitions from OFS to INT1 with NHC bound during SMD runs (Fig. 4d,e). In contrast, in SMD runs in which the MPV OFS structure was utilized as the starting frame, MPV r.m.s.d. and the binding pocket r.m.s.d. relative to the references (OFS or INT1) did not change coherently, but fluctuated, suggesting an unsuccessful conformational transition (Fig. 4d,f). This result is consistent with our hypothesis that the steric switch, influenced by either prodrug or free NAA, alters the interface between the transport and scaffold domains, impacting the energetic landscape of the elevator-like movement (Supplementary Fig. 7).
Implications for rational drug modification
Our discovery concerning the role of F526 on HP2 as a steric switch suggest recommendations for drug modifications of existing NAAs. Decreasing the 5’-PG size in MPV may enhance its transportability through hCNT3 in intestinal epithelial apical membranes, potentially increasing its intestinal uptake and improving oral drug bioavailability. Previous in vitro studies on hCNT3 imply this strategy’s feasibility, as NAAs bearing modest 5’ modifications can still translocate efficiently40. Ribose hydroxyl modifications are an attractive strategy for enhancing NAA pharmacological properties – for example, recent efforts have been focused on ribose hydroxyl substituted GS4 analogues that have shown promise in preclinical studies11,41,42. In fact, a GS4 prodrug featuring the same 5’ isopropyl ester PG as MPV (obeldesivir) is currently in phase III clinical trials for COVID-19 treatment43. Therefore, the structural finding presented here has broad-reaching implications for drug modification of any NAA with a 5’-PG.
Comprehensive cryo-EM ensembles of transporter trimers
Our cryo-EM studies of bCNT3 reveal large elevator-like motions within the transport domain of the protomer. Do neighboring subunits “feel” these motions as cooperativity? Previous studies on excitatory amino acid transporters, another type of trimeric elevator-mechanism transporters, implicated subunit independence44–46, but it remains unclear for CNTs.
Conventional classification strategies to isolate oligomeric arrangements in trimeric elevator-type transporters led to incomplete cryo-EM ensembles that converged well in the form of high-resolution 3D reconstructions44,47. We devised an image processing scheme to identify intersecting particle pick locations for every possible combination of protomer conformers (classes from symmetry expansion) and obtained cryo-EM reconstructions for every possible trimeric arrangement from the MPV “condition “1 and “condition 2” datasets (Fig. 5, Extended Data Figs. 6, 7, 9 and 10, Methods). The 15 reconstructions reflect a total of 14 unique trimeric arrangements, and range in resolution from 3.00 Å to 6.92 Å. The lowest resolution trimer reconstructions are from rare states with low particle numbers (Fig. 5, Extended Data Figs. 6, 7 and 10).
Figure 5 |. Cryo-EM ensembles of bCNT3 trimers.

a, Cryo-EM reconstructions of each of the isolated trimeric states obtained from the ensemble analysis (cryo-EM surface colored by assigned conformer). b, Population distributions of trimer particles in the MPV “condition 1” and “condition 2” datasets. Observed relative frequencies represent relative particle distribution after classification and trimer identification. Expected relative frequencies (assuming a normal distribution) derived from raw symmetry expanded protomer populations before trimer identification (see Methods).
The population distributions of these trimeric arrangements conform to those calculated using raw frequencies of the protomer classes with a normal distribution (Fig. 5). Any form of bias (positive or negative cooperativity) would have led the distribution of trimeric arrangements to deviate from a normal distribution. This is therefore the first line of experimental evidence that protomer movements within a CNT3 trimer are independent of each other. Future biophysical studies would be needed to corroborate this observation.
State-dependent changes in the local lipid environment
Our findings on the independence of subunits within the trimer raise a question about how this is achieved given that the transport domain undergoes such substantial movements. We inspected structurally superposed OFS-INT1-INT1, INT1-INT1-INT1 and INT3-INT1-INT1 bCNT3 trimers (structural alignment based on two of the INT1 protomers). We found the scaffold domain is not static but exhibits considerable conformational changes that track with the transport domain movement (Fig. 6a,b). The eukaryotic scaffold domain appears to undergo the largest deformations, likely owing to the fact that TM3Eu directly leads into TM1 of the transport domain (Supplementary Fig. 2). From OFS to INT1, TM3Eu exhibits a downward displacement of ~3 Å, and a swinging motion of ~2 Å from INT1 to INT3 (Fig. 6b). This movement appears to be dissipated along the rest of the eukaryotic scaffold domain, as TM1–2Eu are relatively static, with IH2Eu serving as the hinge (Fig. 6a,b).
Figure 6 |. Scaffold domain and local lipid bilayer deformations during transport domain movements.

a, Structural overlay of OFS-INT1-INT1, INT1-INT1-INT1, and INT3-INT1-INT1 trimers, highlighting the changes in the scaffold domain during conformational transitions (transport domain shown as colored surfaces, scaffold domain shown as thick yellow cartoon; alignment targeted on the two INT1 protomers). b, Overlay of the eukaryotic scaffold domain from all five conformers reported in this study (alignment targeted to trimerization interface TM3 and TM6; ribbon depiction follows the same color scheme in Fig. 3 and Fig. 4; spheres shown for the Cα of select marker residues). c, OFS-INT1-INT1, INT1-INT1-INT1, and INT3-INT1-INT1 cryo-EM reconstructions overlayed (map alignment targeted to the two INT1 protomers), with non-protein densities shown in yellow. A lipid-like density sandwiched between TM3Eu and the transport domain is circled in red in each conformational state. d, Displacement of modeled phosphatidylcholine molecule in OFS, INT1 and INT3 conformational states, relative to the transport domain (shown as surface colored by surface electrostatics) and eukaryotic scaffold domain (shown as grey cartoon). e, Schematic for the role of the scaffold domain in assisting local membrane deformation during translocation of antiviral drugs. f, Current working model for the alternating access mechanism of antiviral drug transport by CNT3.
Notably, the well-ordered layers of lipids also track with the transport domain and eukaryotic scaffold domain movements (Fig. 6c,d). One well-defined lipid density, tentatively assigned as phosphatidylcholine, is located in the inner-leaflet in a crevice between the transport-domain and TM3Eu in the OFS-INT1-INT1, INT1-INT1-INT1 and INT3-INT1-INT1 reconstructions. This lipid undergoes substantial displacements from OFS to INT1 and INT1 to INT3 (Fig. 6c,d), which also correlate to transport domain movements. Therefore, the elevator-like motion of bCNT3 significantly deforms the local membrane environment. The membrane deformations are expected for elevator-type transport47,48, and the cost of hydrophobic mismatch would disfavor simple sliding of the transport-domain in and out of the membrane bilayer. The eukaryotic scaffold domain assists with this process by allowing efficient sliding of lipids located at crevices between the transport and scaffold domains. This buffers the surrounding bilayer environment from the local deformations induced by transport domain movements (Fig. 6e).
Discussion
Nucleoside transporters are important players in NAA drug pharmacology, as they control their target access and disposition3. Our bCNT3 cryo-EM structures shed light on drug selectivity within the binding pocket and the intricate arrangement of the two Na+ binding sites, which aligns with the current “affinity control” model for sodium-coupled nucleoside transport by CNTs, wherein Na+ occupancy affects the binding pocket’s structure and nucleoside affinity3,19.
Interestingly, the architecture of the bCNT3 substrate and sodium binding sites bears vague resemblance to what has been reported in SLC1, an unrelated family of elevator-type transporters (Supplementary Fig. 8). How two unrelated protein families evolved to utilize similar mechanistic strategies for vastly different substrates poses interesting questions regarding transporter evolution.
The unique interactions of MPV with bCNT3 enables us to capture its cryo-EM structures in outward-facing and intermediate states, providing insights into its transport mechanism. Notably, INT1 exhibits a fully-occluded state, which is generally thought to be only transiently occupied during transporter isomerization49,50. We found that the F526 steric switch plays an important role in arresting this rare conformational state by MPV. During the transport of NHC, or transport of any NAA without a bulky 5’ substituent, F526 forms optimal interactions with the scaffold domain in the occluded intermediate states. In the case of MPV, or any NAA featuring a bulky 5’-PG group, the drug “flips on” this F526 switch, promoting a different rotamer conformation. This in turn rearranges the interface present between the transport and scaffold domains in the normally short-lived intermediate states. Our structural analysis and all-atom MD simulations suggested that the changes in the interface either stabilizes each state (e.g., INT1) or elevates the energetic barriers between the states (e.g., OFS to INT1), resulting in different energetic landscapes of the elevator-like motion with prodrug bound vs. free NAA bound (Supplementary Fig. 7).
Our findings suggest implications to drug design. Utilizing a smaller 5’-PG would keep the steric switch “turned off”, thereby enhancing the prodrug transportability through hCNT3 expressed in intestinal tissue, presumably improving its oral bioavailability. While previous in vitro studies of hCNT3 support the feasibility of this strategy40, because our structural studies were conducted using bCNT3, further research on hCNT3 is needed to further investigate this possibility.
Finally, we found independent subunit movements within the trimer (Fig. 5). Combined with observed local membrane deformation in various conformations, the scaffold domain of CNT3 appears to function beyond just a trimerization interface. We suggest that it acts as an “allosteric cushion”, dissipating the local membrane deformations arising from large transport domain movements. This appears to limit the footprint of membrane deformation and prevent the propagation of conformational changes from affecting neighboring subunits within the trimer (Fig. 6e,f).
Methods
Radiotracer uptake in transporter expressing Xenopus laevis oocytes
[3H]-ribavirin was purchased from Moravek (Brea, CA). Defolliculated Xenopus laevis oocytes were obtained from Xenopus 1 Corp. (Dexter, MI). Uptake assays were performed in a manner similar to a previous report51, with the following modifications. A total of 16–20 ng cRNA were injected per oocyte, with expression carried out at 17°C for 2–4 days. A reaction volume of 200 μL was used, and [3H]-ribavirin was used at full radioactivity in every experiment (6.5 Ci/mmol). Oocytes were batched by 5 per reaction vessel. For all ENT isoforms, oocytes were washed extensively in normal ND-96 buffer (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 5 mM HEPES, pH 7.4 w/ KOH) to terminate the uptake reaction, whereas sodium-free ND-96 buffer (NaCl replaced with choline chloride) was used for assay termination and washing for all CNT isoforms assessed.
Two-electrode voltage-clamp recording of transporter expressing Xenopus laevis oocytes
Oocytes expressing transporters of interest were studied using the two-electrode voltage clamp technique (TEVC). For the recording, oocyte membrane voltage was controlled using an OC-725C oocyte clamp (Warner Instruments). Data were filtered at 1–3 kHz and digitized at 20 kHz using pClamp 10.6 software (Molecular Devices) and a Digidata 1550 digitizer (Axon Instruments). Microelectrode resistances were 0.5–2 MΩ when filled with 3 M KCl. ND-96 buffer was used as external recording solution. All nucleosides and nucleoside analogue drugs were applied using a gravity-fed perfusion system. Recordings were performed in episodic stimulation recording mode - the voltage was initially held at 0 mV for 50 ms, followed by a step to −60 mV (bovine CNT3 ) or −90 mV (human CNT3) for 300 ms, then a step back to 0 mV for 50 ms, with a sweep happening every 2 s. All electrophysiological data analysis was done using Igor Pro 6.34A (Wavemetrics).
Expression, purification, and nanodisc reconstitution of Bos taurus CNT3
Full length WT Bos taurus CNT3 sequence (bCNT3; uniprot ID: F1MGR1) was synthesized in the pFastBac vector containing a FLAG tag (DYKDDDDK) at the N-terminus of the protein. Baculovirus was generated in Spodoptera frugiperda sf9 insect cells using standard protocols and amplified to P2. Insect cells were infected with 1% v/v high-titer P2 virus at a cell density of 1–1.5 × 106 mL-1. Protein expression was carried out at 27°C for 72 h, after which cells were harvested by centrifugation at 550 x g. Cell pellets were resuspended in ice cold lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1 mM phenylmethylsulphonyl fluoride, 10 μg mL−1 leupeptin, 10 μg mL−1 pepstatin, 1 μg mL−1 aprotinin, 2 mg/mL iodoacetamide) at ~1g cell pellet per 4 mL crude lysate. Resuspended cells were then subjected to three rounds of sonication on ice (30s, 1 pulse/s). A final concentration of 40 mM n-dodecyl-ß-D-maltoside (DDM) detergent was added to the disrupted cells and detergent extraction was carried out for 1 h at 4°C with gentle agitation. Insoluble material was then removed with centrifugation at 30,000 x g for 30 min. Clarified lysate was then applied to anti-FLAG M2 resin at 4°C with gentle agitation for 1 h. During resin binding a 10 mg/mL stock of PCEG synthetic lipid mixture was prepared in the following manner - chloroform stocks of 16:0–18:1 PC (Avanti #: 850457), 14:0 PE (Avanti #: 850745P), and 14:0 PG (Avanti #: 840445P), respectively, were combined in a 3:1:1 v/v ratio. The chloroform mixture was vortexed, then dried under gentle argon gas flow in a glass tube. The solid lipids were resuspended in 0.5 initial volume pentane, vortexed briefly, then dried again under argon gas flow. The solid lipid was then resuspended in 1.0 initial volume 20 mM Tris pH 8.0, 150 mM NaCl buffer and passed through an extruder loaded with a 0.2 μm Whatman filter ~30 times. At the conclusion of FLAG resin binding, resin was then collected with gentle centrifugation (550 x g) and resuspended in 1 column volume (CV) wash buffer (20 mM Tris pH 8.0, 150 mM NaCl, 1.0 mM DDM, 0.1mg/mL added PCEG lipids). The resuspended resin slurry was added to a gravity flow column and washed with 10 CV wash buffer. Protein was eluted from the column in wash buffer supplemented with 0.2 mg/mL FLAG peptide. Protein concentration was estimated with OD280 and combined with MSP2N2 protein and PCEG lipids at a final molar ratio of 1:3:200 bCNT3 trimer: MSP2N2 protein: PCEG lipids. The mixture was incubated on ice for 1 h and then transferred to a fresh tube containing degassed Bio-Bead SM-2 resin (Bio-Rad) at a final concentration of ~100 mg/mL beads. After 2 h incubation with gentle agitation, the beads were replaced, and incubation was carried out overnight at 4°C with gentle agitation. The reconstitution mixture was then removed from beads and spun down at 20,000 x g for 30 min prior to injection over a size-exclusion chromatography column (Superose 6 Increase 10/300 GS; Cytiva) pre-equilibrated in 20 mM Tris pH 8.0, 150 mM NaCl. Protein was usually prepared in the absence of drug, with the following exceptions: for the NHC structure, drug was present at 100 μM during lysis, resin binding, and resin washing; 1.0 mM was present in the resin elution buffer; 1.0 mM was present at all times during Bio-Bead treatment; SEC was performed in the absence of drug. For the GS-441524 structure, drug was present at 300 μM during lysis, resin binding, and resin washing; 1.0 mM was present in the resin elution buffer; 1.0 mM was present at all times during Bio-Bead treatment; SEC was performed in the absence of drug; an additional 2 mM drug was added to pooled fractions prior to concentration for grid freezing. For “MPV condition 1” protein drug was present at 1.0 mM during lysis; 0.5 mM during resin binding, resin washing, resin elution; 3 × 0.5 mM additions were added during Bio-Bead treatment; SEC was performed in the absence of drug.
Cryo-EM sample preparation
Peak fractions from SEC were pooled and concentrated to a final concentration of 7–10 mg/mL prior to grid preparation. Drug stocks were added after sample concentration allowed to incubate on ice for ~1h to grid freezing (final concentrations reported): 2 mM NHC (2% DMSO final), 19.2 mM PSI-6206 (2% DMSO final), 25 mM GS-441524 (5% DMSO final), 2 mM ribavirin (2% DMSO final), 7.5 mM molnupiravir (“MPV condition 1” – 2% DMSO final), 15 mM molnupiravir (“MPV condition 2” – 2% DMSO final), and drug-free (2% DMSO final). Grids were prepared with a Leica EM GP2 Plunge Freezer operating at 4°C at 95% humidity. To address preferred orientation issues of nanodisc reconstituted bCNT3, fluorinated fos-choline-8 (Anatrace) was rapidly added to 3 μL protein to a final concentration of 3 mM, and rapidly aspirated 5 times (from 30 mM stock of fluorinated fos-choline-8 in 20 mM Tris pH 8, 150 mM NaCl). In under 10s from fluorinated fos-choline-8 addition, sample was quickly applied to a freshly glow-discharged Quantifoil Au R1.2/1.3 300 mesh grid, blotted with Whatman No. 1 filter paper for 3 s, followed by plunge-freezing in liquid ethane.
Cryo-EM data collection
The drug-free, PSI-6206, ribavirin, “MPV-condition 1”, and “MPV-condition 2” datasets were collected on a Titan Krios (ThermoFisher) transmission electron microscope operating at 300 kV equipped with a K3 camera (Gatan) in counting mode, with BioQuantum GIF energy filter with a slit width of 20eV, using Latitude S (Gatan) for image acquisition (Duke SMiF). The magnification for these datasets was 81,000x, with a physical pixel size of 1.08 Å/px. A total of 40 frames were acquired for each movie, with a 2.4s exposure time and a nominal dose rate of ~30 e−/px/s, for a total dose of ~60 e−/Å2 and a defocus range of −0.8 to −1.8 μm.
The NHC and GS-441524 datasets were collected on a Titan Krios (ThermoFisher) transmission electron microscope operating at 300kV equipped with a K3 camera (Gatan) in counting mode, with BioQuantum GIF energy filter with a slit width of 20eV, using Latitude S (Gatan) for image acquisition (NCI-NCEF). The magnification for these datasets was 81,000x, with a physical pixel size of 1.12 Å/px for the NHC dataset, or 1.08 Å/px for the GS-441524 dataset. A total of 40 frames were acquired for each movie, with a 4.4s or 3.3s exposure time for NHC and GS-441524, respectively. Nominal dose rate was 17.1 e−/px/s for the NHC dataset, and 17.7 e−/px/s for the GS-441524 dataset. Total dose was 60 e−/Å2 for the NHC dataset, and 50 e−/Å2 for the GS-441524 dataset. A defocus range of −1.00 to −2.25 μm was used for the NHC dataset, and a defocus range of −1.0 to −2.0 μm was used for the GS-441524 dataset.
Cryo-EM data processing – standard procedures
apo
A total of 2,622 movies were subjected to MotionCorr2 in Relion52, and CTFFIND4 in cryoSPARC53. Micrographs exhibiting a CTF estimated resolution under 6 Å were selected and subjected to template picking in cryoSPARC. After manual curation of pick locations, particles were extracted with a box size of 64 px with 4x Fourier binning (4.32 Å/px). A total of 2.2 M extracted particles were subjected to 2D classification. 2D classes featuring strong protein features were then re-extracted with a box size of 128px and 2x binning (2.16 Å/px), followed by a k=4 heterogenous refinement with C3 symmetry applied. The highest resolution class was then re-extracted with a box size of 256px with no binning (1.08 Å/px) and subjected to non-uniform refinement with C3 symmetry applied. The particle stack and angular priors were then transferred to Relion for 2 rounds of Bayesian polishing. The shiny particles were re-imported into cryoSPARC for a final round of non-uniform refinement with C3 symmetry applied, resulting in a 2.80 Å final reported resolution (FSC=0.143).
NHC
A total of 7,243 movies were subjected to MotionCorr2 in Relion52, and Gctf in Relion for CTF estimation. Micrographs featuring a rlnFigureOfMerit>0.07 were selected and subjected to template picking in Relion. Particles were then extracted with a box size of 64 px with 4x Fourier binning (4.48 Å/px). A total of 1.7 M extracted particles were subjected to 2D classification. 2D classes featuring strong protein features were then re-extracted with a box size of 128px and 2x binning (2.24 Å/px), followed by auto refine and no-image alignment classification (k=2/tau_fudge=20), with C3 symmetry applied. The highest resolution class was then re-extracted with a box size of 256px with no binning (1.12 Å/px) and subjected to another round of autorefine and no-alignment classification (k=2/tau_fudge=20). The highest resolution class, containing 175,676 particles, was subjected to CTF refinement and Bayesian polishing. The particle stack and angular priors were then transferred to cryoSPARC and subjected to a round of heterogenous refinement (k=3), with 147,332 particles in the best class. This particle stack was subjected to a final round of non-uniform refinement followed by local refinement, both with C3 applied, to a final resolution of 2.74 Å (FSC=0.143). After obtainment of numerous other bCNT3-IFS reconstructions, the cryo-EM map obtained for NHC appeared slightly dilated compared to all other reconstructions. A model-based pixel size calibration was performed, in which the map-to-model correlation (determined in Phenix54) was highest with a pixel size set to 1.11 Å. All maps for this structure are deposited with the pixel size set to 1.11 Å.
PSI-6206
A total of 3,820 movies were subjected to MotionCorr2 in Relion52, and CTFFIND4 in cryoSPARC53. Micrographs exhibiting a CTF estimated resolution under 6 Å were selected and subjected to template picking in cryoSPARC. After manual curation of pick locations, particles were extracted with a box size of 64 px with 4x Fourier binning (4.32 Å/px). A total of 2.8 M extracted particles were subjected to 2D classification. 2D classes featuring strong protein features were then re-extracted with a box size of 128px and 2x binning (2.16 Å/px), followed by a k=3 heterogenous refinement with C3 symmetry applied. The highest resolution class was then re-extracted with a box size of 256px with no binning (1.08 Å/px) and subjected to non-uniform refinement with C3 symmetry applied. The particle stack and angular priors were then transferred to Relion for 2 rounds of Bayesian polishing. The particle stack was then subjected to no-image alignment 3D classification (k=4/tau_fudge=20). A total of 82,592 particles contributed to the highest resolution class, and these particles were subjected to another round of Bayesian polishing. The shiny particles were re-imported into cryoSPARC for a final round of non-uniform refinement and local refinement with C3 symmetry applied, resulting in a 2.69 Å final reported resolution (FSC=0.143).
ribavirin
A total of 3,937 movies were subjected to MotionCorr2 in Relion52, and CTFFIND4 in cryoSPARC53. Micrographs exhibiting a CTF estimated resolution under 6 Å were selected and subjected to template picking in cryoSPARC. After manual curation of pick locations, particles were extracted with a box size of 64 px with 4x Fourier binning (4.32 Å/px). A total of 2.7 M extracted particles were subjected to 2D classification. 2D classes featuring strong protein features were then re-extracted with a box size of 256px and no binning (1.08 Å/px), followed by a non-uniform refinement with C3 symmetry applied. Two rounds of heterogenous refinements were then performed (both k=3, with C3 applied).The 247,980 particles from the highest resolution class was then subjected to non-uniform refinement with C3 applied, and then transferred to Relion for 2 rounds of Bayesian polishing. The shiny particles were re-imported into cryoSPARC for a final round of non-uniform refinement with C3 symmetry applied, resulting in a 2.61 Å final reported resolution (FSC=0.143).
GS-441524 (consensus)
A total of 12,707 movies were subjected to MotionCorr2 in Relion52, and Gctf in Relion. Micrographs featuring a rlnFigureOfMerit>0.07 were selected and subjected to template picking in Relion. Particles were extracted with a box size of 64 px with 4x Fourier binning (4.32 Å/px). A total of 3.6 M extracted particles were subjected to 2 rounds of 2D classification. 2D classes featuring strong protein features were then re-extracted with a box size of 256px and no binning (1.08 Å/px). The stack of 993,579 particles were imported to cryoSPARC, split by optics group, and subjected to non-uniformed refinement (C3 applied) with both local and global CTF refinement enabled. The particles were then subjected to 2 rounds of heterogenous refinements (k=6 then k=3, C3 applied). The resulting 498,109 particles were then subjected to non-uniformed refinement (C3), transferred to Relion for one round of Bayesian polishing, then re-imported to cryoSPARC for a final round of non-uniform refinement (C3, local+global CTF refinement enabled) followed by a local refinement, for a final resolution of 2.31 Å (FSC=0.143).
GS-441524 (subset)
A particle stack of 993,579 identified from early processing on the GS-441524 dataset (see above) was subjected to one round of heterogenous refinement in cryoSPARC (k=4, C3 applied). 556,379 particles corresponding to the highest resolution class were subjected to non-uniform refinement (C3), then transferred to Relion for Bayesian polishing. The polished particles were then symmetry expanded (C3 operator) with the relion_particle_symmety_expand tool in Relion, resolution in a total of 1.7M expanded particles. A local angular search was then performed, with a single protomer masked, followed by signal subtraction. 2 rounds of no-image alignment classification were then performed with the protomer mask (k=3/tau_fudge=20, then k=3/tau_fudge=40), resulting in 290,664 protomer particles in the highest resolution class. The mask was then updated to only cover the transport domain within the protomer, and a no-image alignment classification was run with k=5/tau_fudge=64. The highest resolution class consisted of 177,264 protomer particles, and also featured some weak signal in the nucleoside binding site. The mask was then updated again, to a sphere only covering the nucleoside binding site. A no-image alignment using this mask was performed with k=3/tau_fudge=128. The extremely high tau_fudge value was necessary to sort for high frequency signal in the binding cavity and allowed us to identify a small subset of particles that featured much stronger signal consistent with the size and shape of purine nucleoside. The particles corresponding to this presumably higher-occupancy GS-441524 class were reverted to the original images, and also subjected to remove duplicates to revert symmetry expansion, resulting in 38,774 particles. Angular priors were kept, ensuring protomers featuring the stronger nucleoside signal remained placed on the same protomeric position within the trimer in angular space. The stack was then transferred to cryoSPARC for a final local refinement (C1 symmetry), with a final reported resolution of 3.23 Å (FSC=0.143). This subset of 38,774 particles features 1215 particle overlap (3.1%) with the 498,109 particle “consensus” set identified in a distinct processing scheme.
MPV condition 1
A total of 4,715 movies were subjected to MotionCorr2 in Relion52, and CTFFIND4 in cryoSPARC53. Micrographs exhibiting a CTF estimated resolution under 4 Å were selected and subjected to template picking in cryoSPARC. After manual curation of pick locations, particles were extracted with a box size of 64px with 4x Fourier binning (4.32 Å/px). A total of 2.6 M extracted particles were subjected to 2D classification. 2D classes featuring strong protein features were then re-extracted with a box size of 128px and 2x binning (2.16 Å/px), followed by heterogenous refinement (k=3, C3 applied). The 439,382 particles corresponding to the highest resolution class were then subjected to non-uniform refinement with C3 applied. The particle stack was then transferred to Relion and subjected to 2 rounds of Bayesian polishing. After re-imported to cryoSPARC, non-uniform refinement was performed with C3 symmetry applied, resulting in a 2.80 Å resolution (FSC=0.143) consensus reconstruction.
For 3D-variability analysis in cryoSPARC, the 439,382 shiny particles in Relion were subjected to an autorefine run in C1 symmetry, with relaxation to C3 enabled. This resulted in a single protomer featuring very weak signal in the transport domain. This C1 reconstruction was then subjected to Bayesian polishing and transferred to cryoSPARC, where it was then subjected to 3D-variability analysis, with the protomer featuring weak signal masked (2-components, C1, 5 Å LPF, 20 Å HPF, clustering output, k=5). Volumes from the clustering output were apparent 2:1 heterotrimers, composed of two INT1 protomers, and either INT1, INT3, or OFS conformers at the protomeric position that was masked. These volumes were used as references for rounds of heterogenous refinements to obtain a high-quality INT1-INT1-OFS heterotrimer reconstruction (three rounds, k=4, with one OFS-INT1-INT1 reference, one INT3-INT1-INT1 reference, and two INT1 homotrimer references as decoys). This classification approach resulted in 118,113 particles that yielded a high-quality OFS-INT1-INT1 reconstruction, with a final reported resolution of 3.20 Å.
MPV condition 2
A total of 3,735 movies were subjected to MotionCorr2 in Relion52, and CTFFIND4 in cryoSPARC53. Micrographs exhibiting a CTF estimated resolution under 6 Å were selected and subjected to template picking in cryoSPARC. After manual curation of pick locations, particles were extracted with a box size of 64 px with 4x Fourier binning (4.32 Å/px). A total of 2.0 M extracted particles were subjected to two rounds of 2D classification. 2D classes featuring strong protein features were then re-extracted with a box size of 256px and no binning (1.08 Å/px), followed by a non-uniform refinement with C3 symmetry applied. Heterogenous refinement was then performed (k=3, with C3 applied). The 108,187 particles from the highest resolution class were then subjected to non-uniform refinement with C3 applied, and then transferred to Relion for 2 rounds of Bayesian polishing. The shiny particles were re-imported into cryoSPARC for a final round of non-uniform refinement with C3 symmetry applied, resulting in a 2.99 Å final reported resolution (FSC=0.143) for the consensus reconstruction.
Cryo-EM data processing – ensemble analysis
MPV condition 1
The 439,382 shiny particles in Relion (consensus set), immediately after autorefine with C3 applied, were symmetry expanded (C3 operator) with the relion_particle_symmety_expand tool in Relion, resolution in a total of 1.3M expanded particles. Signal subtraction was then performed with a single protomer mask, followed by one round of no-image alignment classification (k=6/tau_fudge=40). The resulting classes were assigned by conformational state, resulting in 866.6k protomer particles adopting an INT1 state and 251.0k protomer particles adopting an INT3 state. One class, with 251.0k protomer particles, featured extremely weak signal in the transport domain. Based on the 3D variability result from this particle set, we presumed this class represents the OFS state, and the weak signal was resultant from inaccurate angular assignment for trimers containing the OFS conformer into the initial consensus reconstruction.
Having assigned 3 distinct conformers within the asymmetric unit of bCNT3 in this condition, there are a total of 11 possible arrangements of these three conformers within a bCNT3 trimer: OFS-OFS-OFS, OFS-OFS-INT1, OFS-OFS-INT3, INT1-INT1-OFS, INT1-INT1-INT1, INT1-INT1-INT3, INT3-INT3-OFS, INT3-INT3-INT1, INT3-INT3-INT3, OFS-INT1-INT3 (clockwise), OFS-INT1-INT3 (counterclockwise). We were then interested in identifying intersecting instances of conformers on grids to obtain cryo-EM reconstructions for every possible trimeric arrangement. Using the Remove Duplicates tool in cryoSPARC (minimum separation distance set to 0 Å), we were able to find X/Y positions on the grid corresponding to each of these trimeric arrangements. Whereas obtaining reconstructions of the homotrimers was straightforward, obtaining reconstructions of heterotrimers was less straightforward - we noticed the subtle asymmetric features identified from symmetry expansion often get flatted when subjecting isolated heterotrimer particles to C1 refinements with global angular searches. In order to keep overlap of assigned conformers at each position of the trimer, angular priors have to be selectively kept from a particular conformer of interest. For example – in a INT1-INT1-INT3 homotrimer, the INT3 protomer features an INT1 neighbor at both of its flanking trimeric positions. Therefore, selectively keeping priors from the INT3 subset metadata ensures overlap of each assigned conformer in the trimer in angular space. To achieve this, we used the “Score field” in the cryoSPARC remove duplicates tool (set to “alignments3D/error”) and edited the _rlnMaxValueProbDistribution column to an arbitrary high value (arbitrary low estimated error) for a particular conformer particle subset. This forced the Remove Duplicates tool to drop protomer metadata of the conformer of interest into the “rejected” stack, which ensured priors were retained from the conformer of interest for subsequent refinements. For the case of INT1-INT1-INT3, metadata for protomer particle subset INT1 was edited to contain high _rlnMaxValueProbDistribution values, which forced the priors from INT3 into the “rejected” stack after using remove duplicates to find X/Y pick instances of INT1-INT1-INT3 on the grid. For the OFS-INT1-INT3 heteromer, selectively retaining priors from INT1 was insufficient to ensure overlap in conformers in the trimer, as the three conformations could be arranged in two possible directions in the trimer (clockwise or counterclockwise). In order to break ambiguity in this case, we selectively retained priors from INT1, and performed an additional round of classification in Relion, using a mask that covered the INT1 protomer and one of the neighboring protomers (k=2/tau_fudge=40).
Having identified X/Y pick instances for all possible trimer combinations, and selectively retaining angular priors during the process to ensure proper overlap of conformers in the trimer, particles were re-extracted in cryoSPARC and subjected to masked local refinements with C1 applied (for heterotrimer compositions) or C3 applied (in cases of homotrimer compositions). For reconstructions reaching higher resolutions after the first round of local refinement, rounds of Bayesian polishing and CTF refinements were repeated to further improve the final resolution. The 11 obtained reconstructions range in resolution from 3.00 to 6.92 Å (FSC=0.143), with the lowest resolution reconstructions suffering from low signal-to-noise owing to extremely low particle numbers.
MPV condition 2
The 108,187 shiny particles in Relion (consensus set), immediately after autorefine with C3 applied, were symmetry expanded (C3 operator) with the relion_particle_symmety_expand tool in Relion, resolution in a total of 324.6k expanded particles. Signal subtraction was then performed with a single protomer mask, followed by one round of no-image alignment classification (k=4/tau_fudge=40). The resulting classes were assigned by conformational state, resulting in 108.3k protomer particles adopting an INT1 state and 102.8k protomer particles adopting an INT2 state. We then applied the aforementioned method (see above section) to identify the following trimeric compositions on the grid and obtain reconstructions for each case: INT1-INT1-INT1, INT1-INT1-INT2, INT1-INT2-INT2, and INT2-INT2-INT2. The four reconstructions range in resolution from 3.43 to 3.67 Å (FSC=0.143).
Model building and refinement
A model was initially built into a preliminary IFS-bCNT3 reconstruction, via manual docking a polyalanine model of the vcCNT crystal structure (PDB ID: 3TIJ) into the cryo-EM maps, followed by manual model building and adjustments in COOT63. Hydrogens were added to the model using the Molprobity server64, followed by a round of real-space refinement in Phenix62, with local grid search, global minimization, and ADP refinement enabled. This initial model was rigid body fit in all 6 IFS-bCNT3 structures, followed by further minor model adjustments as necessary, drug modeling when relevant, and subsequent real-space refinements in Phenix (one round with global minimization, and ADP refinement enabled). Lipids were built as fragments of phosphatidylcholine (Chemical ID: LBN), owing to its abundance in the nanodiscs. Sodium ions and ion coordinating waters were modeled in the highest resolution reconstruction (GS4 consensus) – the first site was modelled based on the clear cryo-EM density and precedence23. There was ambiguity in ion and water placement in the second sodium site, which was ultimately broken by checking both possibilities with the CMM server65. Sodium ions were also placed into other high-resolution reconstructions in cases where the map contained clear ion density.
For OFS, INT1, INT2, and INT3 conformers, an IFS protomer model was initially placed in the cryo-EM maps, followed by rigid body fitting of the transport domain and mobile elements of the scaffold domain. Rounds of manual model building in COOT and real-space refinement in Phenix were carried out. Model building was performed using the INT1 or INT2 homotrimeric (C3) reconstructions obtained from the ensemble analyses; the “highest resolution” OFS-INT1-INT1 reconstruction (obtained from 3D-variability and heterogenous refinement) for the OFS state; and the INT1-INT1-INT3 reconstruction from “condition 1” ensemble analysis for the INT3 conformer. A summary of deposited coordinates can be found in Extended Data Fig. 2 and Extended Data Fig. 4.
Model-map correlation analysis of cryo-EM ensembles
Refined coordinates from the highest resolution instance of each distinct protomer conformation were used as references for this analysis – OFS protomer from OFS-INT1-INT1 (highest resolution, MPV dataset 1, 3DVar), INT1 protomer from INT1-INT1-INT1 (ensemble analysis, MPV dataset 1), INT2 protomer from INT1-INT1-INT2 (ensemble analysis, MPV dataset 2), INT3 protomer from INT1-INT1-INT3 (ensemble analysis, MPV dataset 1). The reference models were trimmed to Cα, and residues 90–200 were removed (eukaryotic scaffold domain). All of the follow steps were performed in UCSF Chimera55. Conserved scaffold domain elements TM3 and TM6 (trimer) were rigid body fit into each of the reconstructions (unsharpened maps used). Reference protomers were then superposed to the placed scaffold domain at each protomeric position, with mmaker. Placed reference protomers were then converted to maps with molmap (output filtered to a resolution of 6 Å). Map correlation coefficients were then calculated between the experimental map and the placed reference volumes for each conformational state.
MD Simulation set up and protocol
All all-atom simulation systems and inputs were generated using CHARMM-GUI Membrane Builder56. Additionally, steered MD (SMD) systems and inputs were created using CHARMM-GUI Enhanced Sampler57. System information, including the membrane composition, total number of atoms and ions, and simulation time length, are shown in Supplementary Table 4 and Table 3. The CHARMM36(m) force field, TIP3P model, and CGenFF were utilized in each system for protein/lipids58, water, and ligands, respectively. The SHAKE algorithm was used to constrain bond lengths involving hydrogen, and the hydrogen mass repartitioning technique was employed to facilitate simulations with a 4-fs time step. van der Waals interactions were handled with a cutoff distance of 12 Å, and a force switch between 10 and 12 Å. Particle-mesh Ewald method was used for electrostatic interactions. The NPT ensemble (constant particle number, pressure, and temperature) was applied in each system at a pressure of 1 bar and temperature of 310 K, following default CHARMM-GUI’s six-step equilibration protocol59 using OpenMM60.
In standard MD systems, an additional weak positional restraint (0.12 kcal/mol/Å2) was applied on protein backbone and sodium ions located at the binding site during the first 5 ns of production, followed by another weak restraint (0.06 kcal/mol/ Å2) during the next 5 ns. After 10 ns, the production continued with no restraints. In SMD systems, dihedral restraints (4.78 kcal/mol/ Å2) were applied on TM6 helix of the OFS conformer throughout production to maintain its secondary structure. The backbone center of mass (COM) of the OFS transport domain was used as the collective variable with a spring constant of 47.8 kcal/mol/Å2 and was pulled down relative to the backbone COM of the IFS scaffold domain, at a speed of 1 Å/ns on the -z direction over a total translocation distance of 9 Å. The membrane normal is the z direction and the +z is the extracellular side. Due to the high energy barrier between the OFS and INT1 conformations, the actual pulling speed was lower and thus the SMD production continued to 15 ns.
RMSD calculations
All root-mean-square deviation (r.m.s.d.) results from our MD simulations were calculated on heavy atoms of either ligands or the bCNT3 nucleoside binding pocket backbone. In standard MD systems, the bCNT3 snapshots were superimposed by aligning the binding pocket backbone atoms to its initial coordinates to calculate ligand r.m.s.d.. In SMD systems, the bCNT3 snapshots were superimposed by aligning the TM3 backbone atoms to its OFS or INT1 coordinates to calculate both the ligand r.m.s.d. and the nucleoside binding pocket r.m.s.d..
Extended Data
Extended Data Fig. 1. Functional characterization, protein biochemistry, and cryo-EM sample preparation of bCNT3.

a, b, c, Time-dependent uptake of 1.0 .M [3H]-ribavirin at room temperature is linear within 30 min for hCNT1(a, background corrected uptake rate of 0.0036 ± 0.0002 pmole/min), hCNT2(b, background corrected uptake rate of 0.41 ± 0.01 pmole/min), hCNT3 (b, background corrected uptake rate of 0.87 ± 0.04 pmole/min), hENT1 (c, background corrected uptake rate of 0.056 ± 0.004 pmole/min) and hENT2 (c, background corrected uptake rate of 0.098 ± 0.007 pmole/min). Data shown in panels a, b, c are from the same series of experiments (n = 3 biological replicates, individual replicates shown) with the water-injected control measurements re-shown in each figure panel to convey signal-to-background for each transporter subtype. d, Cold-competition of WT hCNT3 or WT bCNT3 mediated [3H]-ribavirin uptake by cold NHC or GS4. (ChoCl – sodium-free negative control condition with 96 mM choline chloride; 15-minute uptake with 0.1 μM [3H]-ribavirin; n = 3 biological replicates with individual replicates and mean ± s.e.m. shown). e, Representative size-exclusion chromatography profile and corresponding SDS-PAGE analysis of purified nanodisc reconstituted bCNT3. Fractions pooled for cryo-EM analysis indicated with an asterisk (*). Purification and nanodisc reconstitution of bCNT3 was repeated routinely with similar results for each cryo-EM structure reported in this study. f, Representative cryo-EM micrograph (top) and 2D class averages (bottom) of nanodisc reconstituted bCNT3. Micrographs of similar quality were recorded for each cryo-EM dataset.
Extended Data Fig. 2. Cryo-EM image processing of drug-bound and drug-free IFS states of bCNT3.

a, Cryo-EM image processing procedures. b, Fourier-shell correlation plots (top) and angular orientation distributions (bottom) from the final reconstructions. c, Local resolution of the final reconstructions (local resolution determined in cryoSPARC).
Extended Data Fig. 3. Local cryo-EM map quality. Local cryo-EM maps for secondary structure segments.
From top to bottom are NHC, PSI-6206, Ribavirin, GS441524 (subset), GS441524 (consensus) and apo, respectively. Cryo-EM densities are shown at map threshold values of 6σ for NHC, PSI-6206, ribavirin, GS4 sub., apo, or 7σ for GS4 cons.
Extended Data Fig. 4. Sodium ion coordination in the inward-facing state.

a, Location of the two sodium ion binding sites relative to the nucleoside binding site within the NHC bound IFS bCNT3 structure. b, Architecture and coordination geometries of the two sodium ion binding sites in the 2.31Å cryo-EM map (map thresholds used denoted in panel insets).
Extended Data Fig. 5. TEVC studies of molnupiravir interaction with CNT3.

a, TEVC recordings of inward currents elicited by application of 100 μM uridine, or current block by co-application of 100 μM uridine + 1 mM MPV in WT hCNT3 expressing oocytes (representative trace at left, summary of peak currents from n = 6 biological replicates at right with individual measurements and mean ± s.e.m. shown; values are baseline corrected to leak current per oocyte). b, TEVC recordings of inward currents elicited by application of 100 μM uridine, or current block by co-application of 100 μM uridine + 1 mM MPV in WT bCNT3 expressing oocytes (representative trace at left, summary of peak currents from n = 4 biological replicates at right with individual measurements and mean ± s.e.m. shown; values are baseline corrected to leak current per oocyte).
Extended Data Fig. 6. Cryo-EM image processing of molnupiravir-bound OFS, INT1, INT2, INT3 conformational states of bCNT3.

a, Initial cryo-EM image processing procedure for the ‘MPV condition 1’ dataset. b, Cryo-EM ensemble analysis of ‘MPV condition 1’. Final Fourier shell correlation plots and angular orientation distribution plots shown next to every reconstruction obtained.
Extended Data Fig. 7. Cryo-EM image processing of molnupiravir-bound INT1 and INT2 conformational states of bCNT3.

a, Initial cryo-EM image processing procedure for the ‘MPV condition 2’ dataset. b, Cryo-EM ensemble analysis of ‘MPV condition 2’. Final Fourier shell correlation plots and angular orientation distribution plots shown next to every reconstruction obtained.
Extended Data Fig. 8. MPV ligand densities in representative OFS, INT1, INT2, and INT3 cryo-EM reconstructions.

Cryo-EM densities around the nucleoside binding site from representative reconstructions containing OFS, INT1, INT2, or INT3 protomers (reconstructions used: OFS-INT1-INT1 highest res; INT1-INT1- INT1 ‘condition 1’ ensemble analysis; INT2-INT2-INT2 ‘condition 2’ ensemble analysis; INT1-INT1-INT3 ‘condition 1’ ensemble analysis). Sharpened maps shown (threshold = 6σ).
Extended Data Fig. 9. Comparison of conformational trajectories in MPVloaded bCNT3 and apo nwCNT.

a, Structural superposition of bCNT3 (top left) and nwCNT (bottom left) conformers. Alignment based on the trimerization interface, TM3 and TM6. Relative positions of HP1 (middle) and HP2 (right), with lines denoting a marker residue for each conformational state. b, Transport domain movement quantified by its tilt angle relative to a representative IFS structure (X-axis) and distance displacement of nucleoside binding site center of mass (Y-axis). For bCNT3, coordinates corresponding to the highest resolution instance of each conformer used for the analysis (OFS-INT1-INT1 highest res; INT1-INT1-INT1 ‘condition 1’ ensemble analysis; INT2-INT2-INT2 ‘condition 2’ ensemble analysis; INT1-INT1-INT3 ‘condition 1’ ensemble analysis; all relative to the GS4 consensus IFS structure). c, Structural superposition of the transport domain for OFS, INT1, INT3, INT3 and IFS (NHC) structures. d, [3H]-ribavirin uptake (1.0 μM) in 10 minutes into oocytes expressing WT or mutant bCNT3 (n = 3; mean ± s.e.m. and individual replicates shown).
Extended Data Fig. 10. Map-to-model analysis for reconstructions obtained from the cryo-EM ensemble analysis.

a, Map-to-model of every trimer reconstruction obtained from MPV ‘condition 1’ with the ensemble analysis. Map-to-model correlation analysis shown below each reconstruction (for details see Methods). b, Map to model of every trimer reconstruction obtained from MPV ‘condition 2’ with the ensemble analysis. Map-model correlation analysis shown below each reconstruction (for details see Methods).
Supplementary Material
Acknowledgements:
Cryo-EM data were screened and collected at the Duke University Shared Materials Instrumentation Facility (SMIF), the National Cancer Institute’s National Cryo-EM Microscopy facility (NCEF) at the Frederick National Laboratory for Cancer Research, the Pacific Northwest Center for Cryo-EM (PNCC) at OHSU, and the National Institute of Environmental Health Sciences (NIEHS). We thank Janette Myers at PNCC, Adam Wier, Tara Fox, and Ulrich Baxa at NCEF, and Nilakshee Bhattacharya at SMIF for assistance with the microscope operation. We thank M. Hirschi for initial biochemistry of bCNT3 and K. Tsolova for help with part of the radioactive tracer uptake assay, as well as manuscript reading. This research was supported by a National Institutes of Health grant R21AI166134 (S.-Y.L.) and by the National Institute of Health Intramural Research Program; US National Institutes of Environmental Health Science (ZIC ES103326 to M.J.B.); National Science Foundation MCB-2111728 (W.I.). A portion of this research was supported by NIH grant U24GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research. DUKE SMIF is affiliated with the North Carolina Research Triangle Nanotechnology Network, which is in part supported by the NSF (ECCS-2025064).
Footnotes
Competing Interests: The authors declare no competing interests.
Data Availability
Atomic coordinates have been deposited in the Protein Data Bank with the PDB IDs: 8TZ2 (apo), 8TZ5 (NHC), 8TZ6 (PSI-6206), 8TZ1 (ribavirin), 8TZ3 (GS4 cons.), 8TZ4 (GS4 subset), 8TZD (INT1-INT1-OFS;MPV cond. 1), 8TZ7 (INT1 homotrimer; MPV cond. 1 ensemble), 8TZ8 (INT1-INT1-INT3;MPV cond. 1 ensemble), 8TZA (INT1-INT1-INT2;MPV cond. 2 ensemble), 8TZ9 (INT2 homotrimer; MPV cond. 2 ensemble). The reconstructed cryo-EM maps have been deposited in the Electron Microscopy Data Bank with the IDs: EMD-41731 (apo), EMD-41734 (NHC), EMD-41735 (PSI-6206), EMD-41730 (ribavirin), EMD-41732 (GS4 cons.), EMD-41733 (GS4 subset), EMD-41755 (INT1-INT1-OFS;MPV cond. 1), EMD-41736 (INT1 homotrimer; MPV cond. 1 ensemble), EMD-41737 (INT1-INT1-INT3;MPV cond. 1 ensemble), EMD-41738 (INT2 homotrimer; MPV cond. 2 ensemble), EMD-41739 (INT1-INT1-INT2;MPV cond. 2 ensemble), EMD-41740 (consensus; MPV cond. 1), EMD-41752 (consensus; MPV cond. 2), EMD-41747 (INT3 homotrimer; MPV cond. 1 ensemble), EMD-41746 (INT3-INT3-INT1;MPV cond. 1 ensemble), EMD-41745 (INT3-INT3-OFS;MPV cond. 1 ensemble), EMD-41744 (INT1-INT1-OFS;MPV cond. 1 ensemble), EMD-41741 (OFS-OFS-OFS;MPV cond. 1 ensemble), EMD-41742 (OFS-OFS-INT1;MPV cond. 1 ensemble), EMD-41743 (OFS-OFS-INT3;MPV cond. 1 ensemble), EMD-41748 (OFS-INT1-INT3 clockwise; MPV cond. 1 ensemble), EMD-41749 (OFS-INT1-INT3 counterclockwise; MPV cond. 1 ensemble), EMD-41751 (INT1 homotrimer; MPV cond. 2 ensemble), EMD-41750 (INT1-INT2-INT2;MPV cond. 2 ensemble). Source data will be provided with the paper online, and additional data relevant to this paper are available upon reasonable request to S-Y. L.
References
- 1.Yates MK & Seley-Radtke KL The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antiviral Res 162, 5–21 (2019). 10.1016/j.antiviral.2018.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seley-Radtke KL & Yates MK The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antiviral Res 154, 66–86 (2018). 10.1016/j.antiviral.2018.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright NJ & Lee SY Toward a Molecular Basis of Cellular Nucleoside Transport in Humans. Chem Rev 121, 5336–5358 (2021). 10.1021/acs.chemrev.0c00644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pruijssers AJ & Denison MR Nucleoside analogues for the treatment of coronavirus infections. Current opinion in virology 35, 57–62 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graci JD & Cameron CE Mechanisms of action of ribavirin against distinct viruses. Rev Med Virol 16, 37–48 (2006). 10.1002/rmv.483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beigel JH et al. Remdesivir for the Treatment of Covid-19 - Final Report. N Engl J Med 383, 1813–1826 (2020). 10.1056/NEJMoa2007764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lawitz E. et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 368, 1878–1887 (2013). 10.1056/NEJMoa1214853 [DOI] [PubMed] [Google Scholar]
- 8.Jayk Bernal A. et al. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N Engl J Med 386, 509–520 (2022). 10.1056/NEJMoa2116044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehellou Y, Rattan HS & Balzarini J. The ProTide Prodrug Technology: From the Concept to the Clinic. J Med Chem 61, 2211–2226 (2018). 10.1021/acs.jmedchem.7b00734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warren TK et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 531, 381–385 (2016). 10.1038/nature17180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheahan TP et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Science translational medicine 12, eabb5883 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan VC & Muller FL Advantages of the Parent Nucleoside GS-441524 over Remdesivir for Covid-19 Treatment. ACS Med Chem Lett 11, 1361–1366 (2020). 10.1021/acsmedchemlett.0c00316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williamson BN et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 585, 273–276 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feld JJ & Hoofnagle JH Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436, 967–972 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Murphy BG et al. The nucleoside analog GS-441524 strongly inhibits feline infectious peritonitis (FIP) virus in tissue culture and experimental cat infection studies. Vet Microbiol 219, 226–233 (2018). 10.1016/j.vetmic.2018.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agostini ML et al. Small-Molecule Antiviral beta-d-N(4)-Hydroxycytidine Inhibits a Proofreading-Intact Coronavirus with a High Genetic Barrier to Resistance. J Virol 93 (2019). 10.1128/JVI.01348-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toots M. et al. Characterization of orally efficacious influenza drug with high resistance barrier in ferrets and human airway epithelia. Sci Transl Med 11 (2019). 10.1126/scitranslmed.aax5866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoon JJ et al. Orally Efficacious Broad-Spectrum Ribonucleoside Analog Inhibitor of Influenza and Respiratory Syncytial Viruses. Antimicrob Agents Chemother 62 (2018). 10.1128/AAC.00766-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirschi M, Johnson ZL & Lee SY Visualizing multistep elevator-like transitions of a nucleoside transporter. Nature 545, 66–70 (2017). 10.1038/nature22057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson ZL et al. Structural basis of nucleoside and nucleoside drug selectivity by concentrative nucleoside transporters. Elife 3, e03604 (2014). 10.7554/eLife.03604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson ZL, Cheong CG & Lee SY Crystal structure of a concentrative nucleoside transporter from Vibrio cholerae at 2.4 A. Nature 483, 489–493 (2012). 10.1038/nature10882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Y. et al. Cryo-EM structure of the human concentrative nucleoside transporter CNT3. PLoS Biol 18, e3000790 (2020). 10.1371/journal.pbio.3000790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young JD, Yao SY, Baldwin JM, Cass CE & Baldwin SA The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med 34, 529–547 (2013). 10.1016/j.mam.2012.05.007 [DOI] [PubMed] [Google Scholar]
- 24.Wright NJ & Lee SY Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat Struct Mol Biol 26, 599–606 (2019). 10.1038/s41594-019-0245-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C. et al. Structural basis of the substrate recognition and inhibition mechanism of Plasmodium falciparum nucleoside transporter PfENT1. Nat Commun 14, 1727 (2023). 10.1038/s41467-023-37411-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsubota A. et al. Contribution of ribavirin transporter gene polymorphism to treatment response in peginterferon plus ribavirin therapy for HCV genotype 1b patients. Liver Int 32, 826–836 (2012). 10.1111/j.1478-3231.2011.02727.x [DOI] [PubMed] [Google Scholar]
- 27.Doehring A. et al. Role of nucleoside transporters SLC28A2/3 and SLC29A1/2 genetics in ribavirin therapy: protection against anemia in patients with chronic hepatitis C. Pharmacogenet Genomics 21, 289–296 (2011). 10.1097/FPC.0b013e32834412e7 [DOI] [PubMed] [Google Scholar]
- 28.Rau M. et al. Impact of genetic SLC28 transporter and ITPA variants on ribavirin serum level, hemoglobin drop and therapeutic response in patients with HCV infection. J Hepatol 58, 669–675 (2013). 10.1016/j.jhep.2012.11.027 [DOI] [PubMed] [Google Scholar]
- 29.Iikura M. et al. ENT1, a ribavirin transporter, plays a pivotal role in antiviral efficacy of ribavirin in a hepatitis C virus replication cell system. Antimicrob Agents Chemother 56, 1407–1413 (2012). 10.1128/AAC.05762-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morello J. et al. Influence of a single nucleotide polymorphism at the main ribavirin transporter gene on the rapid virological response to pegylated interferon-ribavirin therapy in patients with chronic hepatitis C virus infection. J Infect Dis 202, 1185–1191 (2010). 10.1086/656334 [DOI] [PubMed] [Google Scholar]
- 31.Rasmussen HB et al. Cellular Uptake and Intracellular Phosphorylation of GS-441524: Implications for Its Effectiveness against COVID-19. Viruses 13 (2021). 10.3390/v13071369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang AQ et al. Preclinical Pharmacokinetics and In Vitro Properties of GS-441524, a Potential Oral Drug Candidate for COVID-19 Treatment. Front Pharmacol 13, 918083 (2022). 10.3389/fphar.2022.918083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith KM et al. Cation coupling properties of human concentrative nucleoside transporters hCNT1, hCNT2 and hCNT3. Mol Membr Biol 24, 53–64 (2007). 10.1080/09687860600942534 [DOI] [PubMed] [Google Scholar]
- 34.Hamilton SR et al. Subcellular distribution and membrane topology of the mammalian concentrative Na+-nucleoside cotransporter rCNT1. J Biol Chem 276, 27981–27988 (2001). 10.1074/jbc.M100518200 [DOI] [PubMed] [Google Scholar]
- 35.Fernandez-Calotti P, Casulleras O, Antolin M, Guarner F. & Pastor-Anglada M. Galectin-4 interacts with the drug transporter human concentrative nucleoside transporter 3 to regulate its function. FASEB J 30, 544–554 (2016). 10.1096/fj.15-272773 [DOI] [PubMed] [Google Scholar]
- 36.Damaraju VL et al. Influence of sugar ring conformation on the transportability of nucleosides by human nucleoside transporters. Chembiochem 12, 2774–2778 (2011). 10.1002/cbic.201100567 [DOI] [PubMed] [Google Scholar]
- 37.Arimany-Nardi C. et al. Identification and Characterization of a Secondary Sodium-Binding Site and the Main Selectivity Determinants in the Human Concentrative Nucleoside Transporter 3. Mol Pharm 14, 1980–1987 (2017). 10.1021/acs.molpharmaceut.7b00085 [DOI] [PubMed] [Google Scholar]
- 38.Gucwa M. et al. CMM-An enhanced platform for interactive validation of metal binding sites. Protein Sci 32, e4525 (2023). 10.1002/pro.4525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Errasti-Murugarren E, Molina-Arcas M, Casado FJ & Pastor-Anglada M. The human concentrative nucleoside transporter-3 C602R variant shows impaired sorting to lipid rafts and altered specificity for nucleoside-derived drugs. Mol Pharmacol 78, 157–165 (2010). 10.1124/mol.110.063552 [DOI] [PubMed] [Google Scholar]
- 40.Zhang J. et al. Uridine binding and transportability determinants of human concentrative nucleoside transporters. Mol Pharmacol 68, 830–839 (2005). 10.1124/mol.105.012187 [DOI] [PubMed] [Google Scholar]
- 41.Cao L. et al. The adenosine analog prodrug ATV006 is orally bioavailable and has preclinical efficacy against parental SARS-CoV-2 and variants. Sci Transl Med 14, eabm7621 (2022). 10.1126/scitranslmed.abm7621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schafer A. et al. Therapeutic treatment with an oral prodrug of the remdesivir parental nucleoside is protective against SARS-CoV-2 pathogenesis in mice. Sci Transl Med 14, eabm3410 (2022). 10.1126/scitranslmed.abm3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sciences, G. (https://ClinicalTrials.gov/show/NCT05603143, 2022).
- 44.Qiu B, Matthies D, Fortea E, Yu Z. & Boudker O. Cryo-EM structures of excitatory amino acid transporter 3 visualize coupled substrate, sodium, and proton binding and transport. Sci Adv 7 (2021). 10.1126/sciadv.abf5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erkens GB, Hänelt I, Goudsmits JM, Slotboom DJ & van Oijen AM Unsynchronised subunit motion in single trimeric sodium-coupled aspartate transporters. Nature 502, 119–123 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Georgieva ER, Borbat PP, Ginter C, Freed JH & Boudker O. Conformational ensemble of the sodium-coupled aspartate transporter. Nat Struct Mol Biol 20, 215–221 (2013). 10.1038/nsmb.2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arkhipova V, Guskov A. & Slotboom DJ Structural ensemble of a glutamate transporter homologue in lipid nanodisc environment. Nat Commun 11, 998 (2020). 10.1038/s41467-020-14834-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garaeva AA & Slotboom DJ Elevator-type mechanisms of membrane transport. Biochem Soc Trans 48, 1227–1241 (2020). 10.1042/BST20200290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drew D, North RA, Nagarathinam K. & Tanabe M. Structures and general transport mechanisms by the major facilitator superfamily (MFS). Chemical reviews 121, 5289–5335 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qureshi AA et al. The molecular basis for sugar import in malaria parasites. Nature 578, 321–325 (2020). [DOI] [PubMed] [Google Scholar]
Methods-only references
- 51.Wright NJ et al. Methotrexate recognition by the human reduced folate carrier SLC19A1. Nature 609, 1056–1062 (2022). 10.1038/s41586-022-05168-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimanius D, Dong L, Sharov G, Nakane T. & Scheres SHW New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem J 478, 4169–4185 (2021). 10.1042/BCJ20210708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296 (2017). 10.1038/nmeth.4169 [DOI] [PubMed] [Google Scholar]
- 54.Liebschner D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877 (2019). 10.1107/S2059798319011471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pettersen EF et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Science 30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu EL et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. Journal of Computational Chemistry 35, 1997–2004 (2014). 10.1002/jcc.23702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suh D. et al. CHARMM‐GUI Enhanced Sampler for various collective variables and enhanced sampling methods. Protein Science 31, e4446 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nature methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jo S, Kim T. & Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PloS one 2, e880 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eastman P. et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS computational biology 13, e1005659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates have been deposited in the Protein Data Bank with the PDB IDs: 8TZ2 (apo), 8TZ5 (NHC), 8TZ6 (PSI-6206), 8TZ1 (ribavirin), 8TZ3 (GS4 cons.), 8TZ4 (GS4 subset), 8TZD (INT1-INT1-OFS;MPV cond. 1), 8TZ7 (INT1 homotrimer; MPV cond. 1 ensemble), 8TZ8 (INT1-INT1-INT3;MPV cond. 1 ensemble), 8TZA (INT1-INT1-INT2;MPV cond. 2 ensemble), 8TZ9 (INT2 homotrimer; MPV cond. 2 ensemble). The reconstructed cryo-EM maps have been deposited in the Electron Microscopy Data Bank with the IDs: EMD-41731 (apo), EMD-41734 (NHC), EMD-41735 (PSI-6206), EMD-41730 (ribavirin), EMD-41732 (GS4 cons.), EMD-41733 (GS4 subset), EMD-41755 (INT1-INT1-OFS;MPV cond. 1), EMD-41736 (INT1 homotrimer; MPV cond. 1 ensemble), EMD-41737 (INT1-INT1-INT3;MPV cond. 1 ensemble), EMD-41738 (INT2 homotrimer; MPV cond. 2 ensemble), EMD-41739 (INT1-INT1-INT2;MPV cond. 2 ensemble), EMD-41740 (consensus; MPV cond. 1), EMD-41752 (consensus; MPV cond. 2), EMD-41747 (INT3 homotrimer; MPV cond. 1 ensemble), EMD-41746 (INT3-INT3-INT1;MPV cond. 1 ensemble), EMD-41745 (INT3-INT3-OFS;MPV cond. 1 ensemble), EMD-41744 (INT1-INT1-OFS;MPV cond. 1 ensemble), EMD-41741 (OFS-OFS-OFS;MPV cond. 1 ensemble), EMD-41742 (OFS-OFS-INT1;MPV cond. 1 ensemble), EMD-41743 (OFS-OFS-INT3;MPV cond. 1 ensemble), EMD-41748 (OFS-INT1-INT3 clockwise; MPV cond. 1 ensemble), EMD-41749 (OFS-INT1-INT3 counterclockwise; MPV cond. 1 ensemble), EMD-41751 (INT1 homotrimer; MPV cond. 2 ensemble), EMD-41750 (INT1-INT2-INT2;MPV cond. 2 ensemble). Source data will be provided with the paper online, and additional data relevant to this paper are available upon reasonable request to S-Y. L.