

Abstract
PIEZO1 encodes a mechanoreceptor, a cation channel activated by mechanical stimuli. Gain‐of‐function (GoF) variants in PIEZO1 cause dehydrated hereditary stomatocytosis (DHS), or xerocytosis, a pleiotropic syndrome characterized by anemia and iron overload. DHS patients develop hepatic iron overload independent of the degree of anemia and transfusion regimen. PIEZO1‐GoF variants suppress hepcidin expression in both hepatic cellular model and constitutive/macrophage‐specific Piezo1‐GoF mice model. Therefore, PIEZO1‐GoF variants regulate hepcidin expression by a crosstalk between hepatocytes (HCs) and macrophages with a still unknown mechanism. Transcriptomic and proteomics analysis in the human hepatic Hep3B cells engineered for the PIEZO1‐R2456H variant (PIEZO1‐KI) revealed alterations in the actin cytoskeleton regulation, MAPK cascade, and RAS signaling. These changes mainly occur through a novel key regulator, RRAS, whose protein and mRNA levels are regulated by PIEZO1 activation and inhibition. This regulation was further confirmed in C57BL/6 mouse primary HCs treated with Yoda‐1 and/or GsMTx‐4. Indeed, PIEZO1‐KI cells exhibited hyper‐activated RAS‐GTPase activity that is rescued by PIEZO1 inhibition, restoring expression of the hepcidin gene HAMP. A negative correlation between RAS signaling and HAMP regulation was confirmed by inhibiting RAS‐GTPase and MEK1‐2 activity. Conversely, rescued HAMP gene expression requires downregulation of RRAS, confirming negative feedback between RAS‐MAPK and BMP/SMADs pathways in HAMP regulation. We demonstrated that PIEZO1‐GoF variants influence the actin cytoskeleton organization by activating the hepatic RAS signaling system. Understanding the role of RAS signaling in regulating iron metabolism could pave the way for new therapeutic strategies in DHS and other conditions characterized by iron overload.
1. INTRODUCTION
Dehydrated hereditary stomatocytosis (DHS) is an autosomal dominant pleiotropic syndrome characterized by different phenotypes such as hemolytic anemia with altered red blood cell (RBC) membrane permeability, pre‐perinatal edema, pseudohyperkalemia, and hepatic iron overload. 1 , 2 , 3 DHS is mainly caused by mutations in PIEZO1 gene (90% of cases). 4 , 5 PIEZO1 encodes a mechanoreceptor, a cation channel activated by mechanical stimuli that functions as a biological pressure sensor in both vertebrates and invertebrates. 6 , 7 , 8 , 9 , 10 It is widely expressed in various nonsensory cells and can detect mechanical stresses such as static pressure, shear stress, and membrane stretch. 11 , 12 The mutations identified in PIEZO1, mostly missense, 4 , 5 , 6 , 13 , 14 , 15 , 16 , 17 show a gain‐of‐function (GoF) phenotype with a delayed inactivation of the channel that results in cation leak of the monovalent cations Na+ and K+ and, subsequently, altered cell volume. 1 , 4 , 6 , 13 , 18 Moreover, PIEZO1 mutants have been reported to induce changes in responses to osmotic stress and altered membrane protein trafficking. 19
The phenotype of patients with DHS is highly heterogeneous, ranging from asymptomatic to severe. Generally, anemia is well‐compensated with macrocytosis, reticulocytosis, and mild jaundice. 20 , 21 , 22 In most cases, patients with DHS suffer from iron overload, which represents the most harmful complication. 23 , 24 , 25 , 26 Genotype–phenotype correlations revealed that the clinical heterogeneity depends on the mutations affecting PIEZO1. Severely affected patients (mainly with hepatic iron overload) carry mutations in the pore domain of PIEZO1, while mildly affected patients present mutations in the non‐pore domain of the protein. 16 , 23 We recently discovered the involvement of PIEZO1 in the regulation of iron metabolism. PIEZO1 GoF variants suppress the expression of hepcidin in human hepatoma cells. Particularly, PIEZO1 GoF variants in hepatic cell lines, HuH7 and HepG2, induce a hyper‐phosphorylation of the MAP kinases, extracellular‐signal‐regulated kinase 1 and 2 (ERK1/2), that in turn reduce the activation of the R‐SMADs that fail to activate hepcidin. 27 In mice, constitutive or macrophage‐specific expression of GoF Piezo1 allele causes hepcidin suppression and iron overload. 28 Accordingly, DHS patients develop severe hepatic iron overload independent from the degree of anemia and transfusion regimen. Therefore, PIEZO1 plays a role in the regulation of iron metabolism in the interplay between hepatocytes (HCs) and macrophages, though the precise mechanism remains unclear due to gaps in our understanding of mechanotransduction.
Advances in high‐throughput technologies have enabled the generation of vast amounts of data on multiple layers of a biological system, including genomics, transcriptomics, epigenomics, proteomics, metabolomics, and more. Numerous omics approaches identified genes, proteins, and metabolites that are associated with specific diseases or phenotypes of interest. 29 Herein, we used an integrated multi‐omics approach to dissect the intracellular pathways modifications induced by GoF variants in PIEZO1 in a CRISPR‐Cas9 edited—human hepatic cell line, Hep3B, engineered for the variant R2456H. Our results highlighted the importance of cytoskeleton modification in PIEZO1‐mediated signaling 30 and revealed the role of RAS as a new regulator of hepcidin expression in HCs.
2. MATERIALS AND METHODS
2.1. Cell culture and engineered cell line production
Hep3B cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). A CGX™‐HD v2 4 × 180K array chip (Agilent, Agilent Technologies, CA, USA) was used according to the manufacturer's guidelines for the whole genome screening to test for the zygosity of PIEZO1 in Hep3b cell line. The arrays were scanned using Geno‐glyphix1 software (Signature Genomics, Spokane, WA), referring to Hg19 Genome Assembly (GRCh37/hg19) to evaluate the zygosity of PIEZO1 in Hep3B cell line. The result showed the dizygosity of PIEZO1. Hep3B cells enriched for 18% in PIEZO1‐R2456H variant, PIEZO1‐ 4′,6‐diamidino‐2‐phenylindole knock‐in (PIEZO1‐KI) cells by CRISPR/Cas9 technology were produced by Synthego Corporation (Synthego, Menlo Park, CA, USA). Cells were maintained in Eagle's Minimum Essential Medium (EMEM) (Sigma‐Aldrich, Milan, Italy) supplemented with 10% fetal bovine serum (FBS; Life Technologies; California, USA), 100 U/mL penicillin (Life Technologies), and 100 mg/mL streptomycin (Life Technologies) in a humidified 5% CO2 atmosphere at 37°C, according to the manufacturer's instructions. Hep3B cells from the Synthego CRISPR‐edited pool were diluted to 0.5–1 cells per 100 μL and plated on 96‐well plates to obtain the isolation and the expansion of single clones. PIEZO1‐wild type (PIEZO1‐WT) cells were parallelly amplified, and isolation and the expansion of single clones were carried out. Single clones were tested for PIEZO1 gene and protein expression. PIEZO1‐WT #1 and PIEZO1‐KI #1 clones were selected for all subsequent experiments (Figure S1A).
2.2. Isolation of primary murine hepatocytes
Animal studies were reviewed and approved by the Italian Ministry of Health (number D5A89.63/2023). In situ liver perfusion of 8‐week‐old C57BL/6N Wt mice was performed with Liver Perfusion Medium and Liver Digest Medium (Thermo Fisher Scientific; pump flux: 5 mL/min). After liver digestion, debris and membranes were filtered through a 100 μm cell strainer. HCs were separated from nonparenchymal cells through low‐speed centrifugation (50 g for 3 min), resuspended in Williams‐E medium (4% FBS, 1% P/S, Glutamax; Thermo Fisher Scientific) and plated into collagen‐coated 12‐well (2.5–3 × 105 cells/well; Corning‐Thermo Fisher Scientific).
2.3. Drug treatment
Hep3B‐WT single clone, named PIEZO1‐WT, and PIEZO1‐R2456H single clone (PIEZO1‐KI cells) were treated with Yoda‐1 (activator of PIEZO1 at 15 μM; Cat. N° SML1558; Sigma‐Aldrich) for 60 min with and without GsMTx‐4 (inhibitor of PIEZO1 at 5 μM; Cat. N°—ab141871 Abcam) for 30 min, as previously described and harvested for the subsequent analyses. Primary murine HCs were treated with Yoda‐1, at 50 μM for 30 min or Yoda‐1 plus GsMTx‐4 (30 μM for 30 min).
U0126 dual specificity mitogen‐activated protein kinase kinase 1 and 2 (MEK1/2) inhibitor; Cat. N° 1144; Bio‐techne TOCRIS) was added to PIEZO1‐WT and PIEZO1‐KI cells at 10 μM for 30 and 60 min. The RAS inhibitor Salirasib (2‐(((2E,6E)‐3,7,11‐trimethyl‐2,6,10‐dodecatrienyl)sulfanyl)benzoic acid; FTS, farnesylthiosalicylic acid, S‐farnesylthiosalicylic acid; Cat. N° SML1166; Sigma‐Aldrich) was used at 25 and 100 μM for 24 h.
Silencing of RAS‐related protein R‐RAS (RRAS) was performed by small interfering RNA (siRNA). Scrambled negative control siRNA (Origene Cat. N° SR30004) and siRNA specific for RRAS (Origene Cat. N° SR304191) were added to PIEZO1‐KI cells at 3 μM for 30 h.
Cellular iron overload was performed by treating PIEZO1‐WT and PIEZO1‐KI cells with 30 μM Holo‐transferrin (Holo‐Tf) (Cat n° T0665, Sigma‐Aldrich) at different time points (2, 4, and 16 h).
2.4. Cell cycle analysis
For cell cycle analysis, PIEZO1‐WT and PIEZO1‐KI cells were harvested by centrifugation, resuspended in PBS containing 3.75% Nonidet P‐40, 100 μg/mL RNase A, and 40 μg/mL propidium iodide, and incubated at room temperature for 3 h in the dark. Samples were analyzed on a FACS flow cytometer (Becton Dickinson Immunocytometry Systems [BDIS]).
2.5. Genotyping of Hep3B PIEZO1‐KI clones
Genomic DNA extraction, polymerase chain reaction (PCR) amplification of PIEZO1 (NM_001142864.4—exon 51), and Sanger sequencing were performed for each single clone from the CRISPR‐edited pool to confirm the editing. Synthego's Inference of CRISPR edits (ICE) tool was used to analyze the sequence data and to obtain the knock‐in score (https://ice.synthego.com). Single clones presenting the R2456H variant in heterozygous state were selected.
2.6. Gene expression analysis
Total RNA was extracted from cell lines using TRIzol reagent (Life Technologies). Synthesis of cDNA was performed using SensiFAST cDNA Synthesis Kit (Bioline). Quantitative RT‐PCR (qRT‐PCR) using Power SYBR Green PCR Master Mix (Bioline) was performed on Applied Biosystems 7900HT Sequence Detection System using standard cycling conditions. β‐actin was used as an internal control. Real‐time‐PCR was performed using a standard TaqMan PCR kit protocol for ID1 and ID3 genes. The 20‐μL PCRs included 3.5 μL reverse transcriptase product, normalized μL TaqMan Universal PCR Master Mix (Applied Biosystems), 1 μL of TaqMan probe mix (20×). The reactions were incubated in a 96‐well plate at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, and 60°C for 1 min. Relative gene expression was calculated by using the 2−ΔCt method, where ΔCt indicates the differences in the mean Ct between the selected genes and the normalization control Glyceraldehyde 3‐ Phosphate dehydrogenase (GAPDH). 31 Oligo sequences are listed in Table S1.
2.7. Protein extraction and Western blotting
Proteins were extracted from cells using RIPA lysis buffer containing protease inhibitor cocktail (Halt™ Protease Inhibitor Cocktail, 100X Cat. N° 78 438; Thermo Fisher Scientific). Equal amounts of protein from each lysate, as determined by a Bradford assay, were subjected to sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and blotted onto polyvinylidene difluoride membranes (Biorad). Detection was performed with rabbit anti‐PIEZO1 (1:1000) (15939‐1‐AP, Proteintech) 4 ; rabbit monoclonal anti‐ferritin (Cat. N° ab75973; 1:1000 dilution; Abcam); rabbit polyclonal anti‐pErk1/pErk2 (Cat. N° ab32538; 1:250 dilution; Abcam); rabbit polyclonal anti‐Erk1/2 (Cat. N° ab17942; 1:1000 dilution; Abcam); rabbit monoclonal anti‐pSMAD 1/5/9 (Cat. N° 13 820; 1:500 dilution; Cell Signaling, Leiden, The Netherlands); rabbit polyclonal anti‐SMAD 1/5/8/9 (Cat. N° ab13723; 1:1000 dilution; Abcam); rabbit polyclonal anti‐RRAS (Cat. N° 8446S; 1:1000 dilution; Cell Signaling); rabbit polyclonal anti‐pMek1 (Cat. N° 9127S; 1:1000; Cell Signaling); mouse monoclonal anti‐Mek1/2 (Cat. N° 4694S; 1:1000; Cell Signaling); anti‐GAPDH (Cat. N° 14C10; 1:1000 dilution; Cell Signaling Technology), was used as the loading control. Immunostained proteins were detected by chemiluminescence (SuperSignal™ West Pico PLUS Chemiluminescent Substrate Cat. N° 34 580, Thermo Fisher Scientific), and densitometric analysis was performed with the BioRad ChemiDoc using Quantity One software (BioRad) to obtain an integrated optical density (OD) value. Ratio between “Adjusted volume” (percentage of correct volume relative to the total volume of the gel or the set of bands analyzed) from analyzed protein(s) and loading control was used to quantify the amount of protein(s).
2.8. RAS activity detection assay
Quantification of the active total RAS was carried out by using the Active RAS Detection Kit (Cell Signaling, #8821) following the manufacturers' instructions. Briefly, untreated PIEZO1‐WT and PIEZO1‐KI cells were treated with Yoda‐1 (15 μM) and Yoda‐1 plus GsMTx‐4 (5 μM), harvested, and lysed to isolate proteins. The GTP‐bound GTPase pull‐down was performed parallelly on 1 μg for each sample and for positive (GTPγS) and negative guanosine diphosphate (GDP) control to ensure the immunoprecipitation procedures are working properly. GST‐Raf1‐RBD fusion protein is used to bind the activated form of GTP‐bound RAS, which can then be immunoprecipitated with glutathione resin. Ras activation levels are then determined by western blotting using a Ras Mouse mAb. The eluted samples (5 μL) were loaded on an SDS‐PAGE, and western blotting was performed as previously described.
2.9. Assays of intracellular Ca2+ and K+ transport
Quantification of total intracellular Ca2+ was carried out in vitro using the Cal‐520 assay (Cat. N° ab171868; AbCam). The assay was performed using Cal‐520 at 2.5 μM and fluorescence was measured as previously described. 27 Briefly, 3 × 104 cells were plated in a tissue culture microplate with black walls and clear bottom the day before. On the day of the experiment, cells were loaded with Cal‐520 AM (2.5 μM) and incubated in a cell incubator for 90 min, and then at room temperature for 30 min. The experiment was run at Ex/Em = 490/525 nm at room temperature. The free calcium concentration was calculated as follows: [[Ca]free = [F − Fmin]/Fmax ─ F], where F is the measured fluorescence, Fmin is the fluorescence in the absence of calcium and Fmax is the fluorescence of the calcium‐saturated probe.
Potassium ion channels and transporter activities were detected with the FluxOR™ Potassium Ion Channel Assay (Cat n. F10016‐F10017, Thermo Fisher Scientific). Cells were incubated in the loading buffer at 18–24°C for 60 min, then replaced with the assay buffer and the stimulus buffer. All buffers were prepared according to the manufacturer's instructions. Buffers composition is detailed in Table S2. Fluorescence was measured every 1–2 s for 1–3 min using a kinetic dispense microplate reader, setting the excitation wavelength to 460–490 nm and the emission wavelength to 520–540 nm. The assay was performed in the absence of ouabain and/or bumetanide.
2.10. Immunofluorescence and flow cytometry analysis
To perform immunofluorescence analysis, 1.5 × 105 PIEZO1‐WT and PIEZO1‐KI cells were seeded on slides placed in 24‐well plates. After 24 h, cells were stained with Phalloidin (Cat. N°sc‐499 438; 1:500; Santa Cruz) for 15 min. Immunofluorescence was performed as previously described. 32 Cells were immunologically stained with rabbit anti‐PIEZO1 (1:100) (15939‐1‐AP, Proteintech) and secondary antibody (1:200) (Alexa Fluor 488 anti‐rabbit, Life Technologies). Nuclei were stained with 1 mg/mL 4',6‐diamidino‐2‐phenylindole (DAPI) (D9542, Sigma). Samples were mounted in Mowiol mounting media and imaged using a LEICA TCS SP8 metaconfocal microscope, equipped with an oil immersion plan Apochromat 63X objective 1.4 NA. The following settings were used: green channel excitation of Alexa488 by the argon laser 488 nm line was detected with the 505–550 nm emission bandpass filter. Red channel excitation of Alexa546 by the Helium/Neon laser 543 nm line was detected with the 560–700 nm emission bandpass filter (using the Meta monochromator). Blue channel excitation of DAPI by the blue diode laser 647 nm and emission bandpass filter. Flow cytometry analysis of membrane PIEZO1 was performed on both PIEZO1‐WT and PIEZO1‐KI cells (3.0 × 105 cells) stained with rabbit anti‐PIEZO1 (1:100), and secondary antibody (1:200) (Alexa Fluor 488), without permeabilization. Unstained PIEZO1‐WT and PIEZO1‐KI cells (nude) were used as negative controls. Threshold for positivity was set at 10. 4 A total of 10 000 events were acquired by FACS Canto II (Beckton Dickinson Biosciences, Sparks Glencoe, MD, USA).
2.11. Stress fibers analysis to evaluate the cytoskeleton alteration
Analysis of stress fibers was carried out by using the open‐source software FIJI (ImageJ) along with its plugins Analyze Skeleton (2D/3D). 33 Briefly, the selected images (from immunofluorescence described in the previous paragraph) are binarized, and no threshold mask is applied. Images are then segmented (Process › Binary › Skeletonize) and analyzed (Analyze › Skeleton › Analyze Skeleton [2D/3D]). The plugin tags all pixels/voxels in a skeleton image and then counts all its junctions, triple and quadruple points, and branches, and measures their average and maximum length. The tags are shown in a new window displaying every tag in a different color. After classification, a “Results” window is displayed, showing for each skeleton in the image: (i) the number of branches (slab segments, usually connecting end‐points, end‐points and junctions, or junctions and junctions); (ii) the number of voxels of every type: end‐point, slab, and junction voxels; (iii) the number of actual junctions (merging neighbor junction voxels) with an arbitrary number of projecting branches; (iv) the number of triple points (junctions with exactly three branches) and quadruple points (four branches); (v) the average and maximum length of branches, in the corresponding units. A minimum of six different slices have been acquired and processed for each sample.
2.12. Differential proteomics and RNA sequencing analyses
PIEZO1‐WT and PIEZO1‐KI cells were parallelly expanded in triplicates for RNA sequencing and differential proteomics analyses. All acquired raw files were processed using MaxQuant (1.6.2.10) and the implemented Andromeda search engine. For protein assignment, spectra were correlated with the Uniprot Homo sapiens (v. 2019), including a list of common contaminants. Searches were performed with tryptic specifications and default settings for mass tolerances for mass spectrometry (MS) and MS/MS spectra. For further analysis, the Perseus software (1.6.2.3) was used and first filtered for contaminants and reverse entries as well as proteins that were only identified by a modified peptide (First filter). The LFQ ratios were presented as logarithms, grouped, and filtered for min.valid number (min. 3 in at least one group) (second filter). Data resulting from proteomics (4265 total proteins identified) were analyzed considering difference ≥±1 and −log10(p value) ≥ 1.3 statistically significant.
Human lncRNA‐seq and mRNA sequencing (RNAseq) was performed by Novogene—Research Services, using Illumina sequencing PE150. For RNAseq analysis, FASTQs were aligned with hg19 genome and transcriptome with TopHat (Bowtie2). Transcript abundance estimation versus hg19 genome and transcriptome was performed by cufflinks to generate fragments per kilobase of transcript per million mapped fragments (FPKM)data. Cuffdiff was used to evaluate differential expression. Genes with log2(FC) ≥ ±1 and −log10(p value) ≥ 1.3 were considered statistically significant. Pathway analysis for both transcriptome and proteome was performed using DAVID Bioinformatics Resources (https://david.ncifcrf.gov) for Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis.
2.13. Statistical analysis
Statistical significances of the differences in protein and gene expression were determined using Student's t‐tests. Statistical significance of multiple comparisons was calculated using analysis of variance (ANOVA), and post hoc correction was performed using Sidak's multiple comparison tests. The association between variables was assessed using Pearson correlation or Spearman rank correlation, as appropriate. When not differently specified, data are presented as mean ± standard deviation (SD). A two‐sided p < .05 was considered statistically significant.
3. RESULTS
3.1. Hep3B PIEZO1‐KI cells showed iron metabolism alterations
Gene and protein expression analysis of PIEZO1 showed no variations in Hep3B PIEZO1‐R2456H (PIEZO1‐KI) single cell clone compared to PIEZO1‐WT single cell clone (Figure 1A,B). Further, we investigated the cell cycle status of both PIEZO1‐WT and PIEZO1‐KI cells, as PIEZO1 has previously been linked to deregulated cell division mechanisms in various cell types. 34 , 35 , 36 This analysis revealed no alterations in the cell cycle, with PIEZO1‐KI cells showing similar percentages of cells in the S, G1, and G2 phases as the PIEZO1‐WT cells (Figure 1C). Moreover, we quantified the amount of PIEZO1 protein at membrane, cytoplasm, and nucleus through immunofluorescence analysis with PIEZO1 antibody and the cytoskeleton marker, phalloidin, in PIEZO1‐WT and PIEZO1‐KI cells. We found no significant differences in subcellular localization of PIEZO1 between PIEZO1‐WT and KI for this mutant (Figures 1D and S1B,C), as previously demonstrated. 19 Accordingly, we observed no variations in PIEZO1 expression at membrane level by flow cytometry analysis in both PIEZO1‐WT and PIEZO1‐KI cells stained with PIEZO1 on unpermeabilized cells (Figures 1E and S1D).
FIGURE 1.
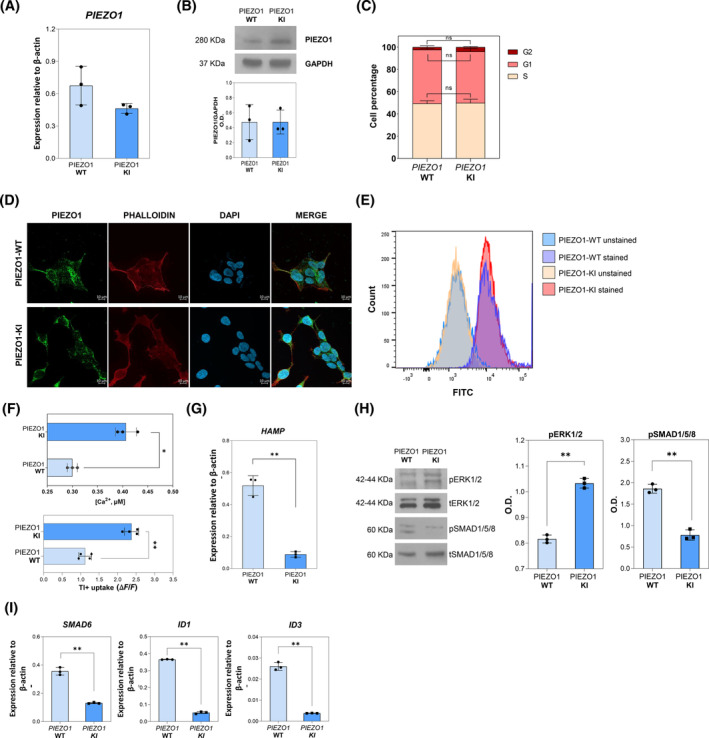
Characterization of Hep3B cell line engineered for PIEZO1 R2456H variant (PIEZO1‐KI). (A) mRNA expression of PIEZO1, relative to β‐actin, in PIEZO1‐WT and PIEZO1‐KI cells. Data are means ± standard deviation (SD) of three independent experiments. (B) Representative immunoblot of PIEZO1 in Hep3B‐WT and PIEZO1‐KI cells. Lower panel: Quantification by densitometric analysis of three separate Western blots with similar results. Data are means ± SD of three independent experiments and are normalized on GAPDH. (C) Histograms showing the number of PIEZO1‐WT and PIEZO1‐KI cells on total events (%) in the G1, S, and G2 phases of the cell cycle. Data derived from three experiments are presented as mean ± SD. (D) Representative confocal imaging by ZEISS LSM 980 Airyscan 2 of PIEZO1‐WT and PIEZO1‐KI cells is shown. Rabbit anti‐PIEZO1 antibody was used to stain the PIEZO1 protein (green). Phalloidin was used as a cytoskeleton marker (red), and DAPI was used as a nuclear marker (blue). Overlapping of both signals (MERGE) is shown on the right (yellow). Scale bar 10 μm. (E) Representative histograms showing fluorescence intensity of PIEZO1 in Hep3b PIEZO1‐WT and PIEZO1‐KI cells stained or not (unstained) with PIEZO1‐Alexa Fluor 488. Data are mean ± SD of three independent acquisitions. Ten thousand events were acquired for each sample. (F) Upper panel: Quantification of total intracellular Ca2+ concentrations in Hep3B cells expressing PIEZO1‐WT and PIEZO1‐KI at steady state. Data are means ± SD of three experiments and are normalized on protein concentrations (*p < .05, PIEZO1‐KI vs. PIEZO1_WT, Student's t‐test). Lower panel: Quantification of Tl + uptake in PIEZO1‐WT and PIEZO1‐KI at steady state. ΔF/F is the difference between the mean of fluorescence (four replicates) and fluorescence at 0 time (**p < .01, PIEZO1‐KI vs. PIEZO1‐WT, Student's t‐test). (G) Quantification of HAMP gene expression normalized to β‐actin in PIEZO1‐WT and PIEZO1‐KI. Data are means ± SD of three independent experiments (**p < .01; Student's t‐test, PIEZO1‐WT vs. PIEZO1‐KI). (H) Representative immunoblotting of pERK1/2 and pSMAD1/5/8 and relative densitometric analysis (right panel) normalized to tERK1/2 and tSMAD1/5/8, in PIEZO1‐WT and PIEZO1‐KI cells. Data are means ± SD of three independent experiments (**p < .01; Student's t‐test, PIEZO1‐WT vs. PIEZO1‐KI). (I) SMAD6, ID1, and ID3 gene expression normalized to β‐actin in PIEZO1‐WT and PIEZO1‐KI. Data are means ± SD of three independent experiments (**p < .01; Student's t‐test, PIEZO1‐WT vs. PIEZO1‐KI). [Color figure can be viewed at wileyonlinelibrary.com]
To confirm the GoF phenotype, we measured the intracellular calcium content and K+ channel activity in both PIEZO1‐WT and PIEZO1‐KI cells. We observed that PIEZO1‐KI cells showed a relative increase in intracellular Ca2+ and enhanced K+ permeability, as measured by Tl+ indicator dye, both consistent with PIEZO1 hyperactivation (Figure 1F).
Of note, our experimental conditions induce a basal PIEZO1 activation due to the membrane stretch caused by cell–cell interactions, a known trigger for PIEZO1 activation in epithelial cells. 37 We measured the calcium influx in PIEZO1‐KI cells at different cell densities without other stimuli. A marked increase in intracellular calcium concentration correlated with higher cell densities, confirming that cell–cell interactions and membrane stretching alone are sufficient to activate PIEZO1. The involvement of PIEZO1 in mediating calcium internalization under these conditions was further validated by specifically inhibiting it with the GsMTx‐4 molecule (Figure S1E).
According to our previous data, 27 we observed strong suppression of HAMP gene expression in PIEZO1‐KI cells compared to PIEZO1‐WT (Figure 1G). Additionally, we confirmed alterations in the phosphorylation levels of the MAP kinases ERK1/2 and SMAD1/5/8. We found that pERK1/2 protein levels were upregulated in PIEZO1‐KI cells, while pSMAD1/5/8 levels were downregulated in PIEZO1‐KI cells compared to PIEZO1‐WT (Figure 1H). Finally, we confirmed the downregulation of bone morphogenetic protein/small mother against decapentaplegic (BMP/SMADs) signaling by measuring mRNA levels of BMP target genes (SMAD6, ID1, and ID3) that were all downregulated in PIEZO1‐KI cells compared to PIEZO1‐WT cells (Figure 1I).
3.2. Comparative transcriptomic and proteomic profiling of Hep3B PIEZO1‐KI cells reveals alterations in actin cytoskeleton regulation
To unravel the molecular signaling pathways altered in PIEZO1‐KI cells leading to HAMP gene suppression, we analyzed the transcriptome and proteome of PIEZO1‐KI cells compared to PIEZO1‐WT ones. Briefly, 1546 genes and 213 proteins were found differentially regulated in PIEZO1‐KI cells compared to PIEZO1‐WT ones. Of note, 509 out of 1546 genes (32.9%) were found upregulated, while 1037 genes (66.1%) were downregulated compared to WT cells. On the other hand, we observed 142 out of 213 proteins (66.7%) upregulated versus 71 proteins (33.3%) downregulated in the PIEZO1‐KI model. Further, we observed that 59 of the deregulated genes were paralleled by changes in proteins. (Figure 2A) Particularly, 58 out of 59 genes were concordantly regulated at protein level, while 1 out of 59 presented a discordant regulation (upregulated mRNA and downregulated protein expression). Subsequently, we performed a KEGG pathway analysis on both genes and proteins that were significantly deregulated (Figure 2B). We first focused on the only two pathways commonly deregulated at both RNA and protein levels, that is, metabolic pathway and regulation of actin cytoskeleton. The metabolic pathway is huge and encompasses numerous biological processes resulting not being informative; thus, we further focused on the regulation of the actin cytoskeleton pathway. The analysis of single genes belonging to this latter pathway showed that only 3 out of 34 genes, RRAS (encoding for RAS‐related protein R‐RAS), ITGB4 (encoding for Integrin β4), and F2 (encoding for prothrombin), were commonly deregulated at both RNA and protein levels, suggesting their potential roles as key regulatory candidates (Figure 2B). Additionally, analysis of Protein–Protein Interaction Networks by STRING web tool revealed that the protein encoded by these genes strongly interact with each other, and particularly we observed a separated cluster enriched in genes encoding for integrins (red colored circles, interaction score: 0.700) (Figure 2C).
FIGURE 2.
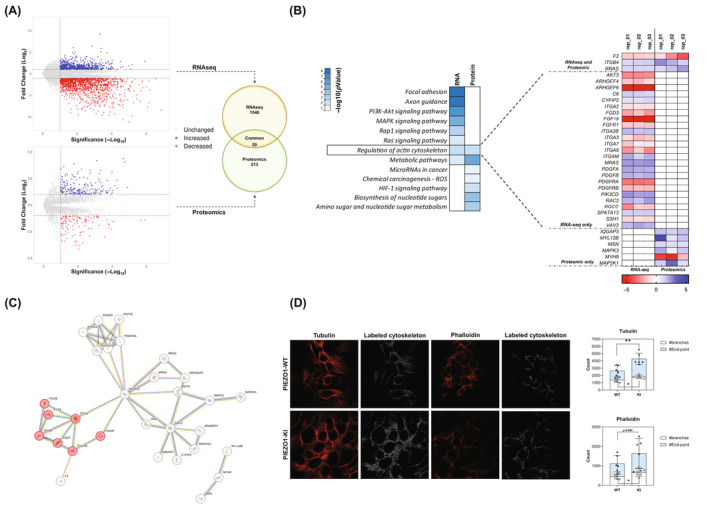
Comparative transcriptomic and proteomic profiling of PIEZO1‐KI cells. (A) Left panel: Volcano plot representing the significantly upregulated (blue), significantly downregulated (red), and unchanged (gray) genes (upper) and protein (lower). Right panel: Venn diagram showing the number of significantly deregulated genes/proteins identified by RNAseq only (n = 1564) and Proteomics only (n = 213). Intersection represents the commonly deregulated genes/proteins (n = 59). (B) KEGG enrichment pathway analysis of deregulated genes and proteins in PIEZO1‐KI cells compared to PIEZO1‐WT. Enrichment is expressed as −log10 of pathway p value. Right panel: Expression heatmap for upregulated (blue) and downregulated (red) genes and protein (three replicates are shown) associated with the pathway named “Regulation of actin cytoskeleton.” The common genes/proteins are shown on the top; RNAseq‐only genes are shown in the middle, and protein‐only genes are shown on the bottom. Undetected expression is shown as a white square (N/D). (C) Interaction analysis (by STRING) of genes/proteins belonging to the “Regulation of actin cytoskeleton” pathway. Integrin cluster indicated as red circles. (D) Representative confocal imaging by ZEISS LSM 980 Airyscan 2 of PIEZO1‐WT and PIEZO1‐KI cells is shown. Tubulin and phalloidin were used as cytoskeleton markers (red). Right panel: Stress fibers were tagged and quantified with ImageJ software. Scale bar 10 μm. Histograms show the number of branches and end‐points in both PIEZO1‐WT and PIEZO1‐KI. Data are means ± standard deviation of six different acquisitions. *p < .05, **p < .01 by unpaired t‐test. [Color figure can be viewed at wileyonlinelibrary.com]
To functionally demonstrate that PIEZO1‐GoF variants are linked to alteration in cytoskeleton organization, we quantified and characterized differences in stress fibers. Immunofluorescence on PIEZO1‐WT and PIEZO1‐KI cells stained with both phalloidin and tubulin antibodies, binding respectively F‐actin and microtubules, was carried out. The analysis was performed on binarized images, and different parameters were evaluated as the number of branches, length, and overall complexity of fibers. Particularly, we observed a significant increase in the number of branches in PIEZO1‐KI compared to PIEZO1‐WT, presenting more junctions and longer paths. The higher complexity of the cytoskeleton of PIEZO1‐KI cells was also observed by correlating the number of branches with their average length and with the end‐point. We observed that PIEZO1‐KI cells presented an increased ratio between the number of end‐points and branches, describing a more complex structure with more ramifications (Figures 2D and S2A,B).
3.3. RRAS is not dispensable in PIEZO1‐mediated HAMP suppression via ERK signaling
To verify the direct correlation of RRAS, ITGB4, and F2 with PIEZO1 GoF variant, PIEZO1‐KI cells were either activated with the PIEZO1 chemical activator, Yoda‐1, or inhibited with GsMTx‐4. RRAS and ITGB4 were upregulated in PIEZO1‐KI cells compared to PIEZO1‐WT, while F2 mRNA expression was reduced in PIEZO1‐KI. Interestingly, only RRAS expression was significantly regulated according to PIEZO1 activation and inhibition. Indeed, Yoda‐1 treatment induced upregulation of RRAS mRNA and protein expression compared to both untreated PIEZO1 WT and PIEZO1‐KI cells, while PIEZO1 inhibition by GsMTx‐4 completely rescued RRAS expression (Figure 3A,B). To confirm that RRAS deregulation was directly dependent on PIEZO1 activity, its expression was also tested in PIEZO1‐WT cells upon PIEZO1 activating or inhibiting conditions (Figure 3C). Yoda‐1 treatment increased RRAS expression and led to HAMP gene downregulation compared to non‐treated cells. Conversely, GsMTx‐4 treatment decreased RRAS expression and upregulated HAMP gene expression. Of note, we confirmed our findings in primary murine HCs isolated from C57BL/6N Wt mice and treated with Yoda‐1 and Yoda‐1 plus GsMTx‐4. We observed an inverse correlation between Hamp and Rras expression upon activating and inhibiting conditions (r = −0.9997; p = .0164) (Figure 3D).
FIGURE 3.
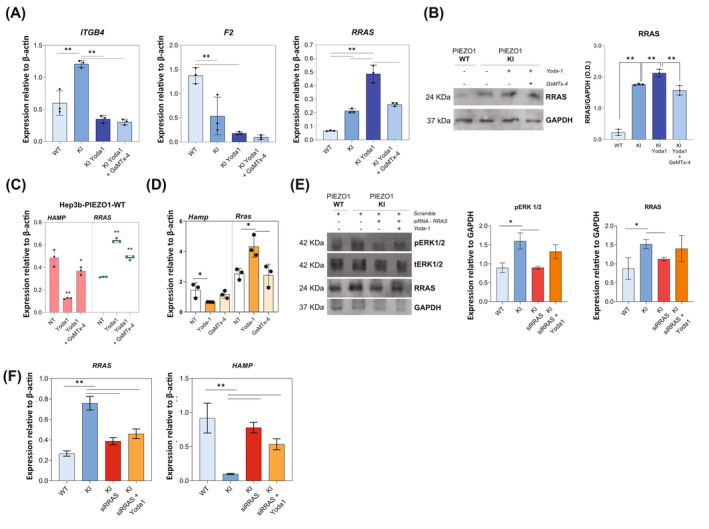
Crosstalk between RRAS deregulation and HAMP expression. (A) mRNA expression relative to β‐actin of ITGB4, F2, and RRAS, in PIEZO1‐WT and PIEZO1‐KI cells at steady state and PIEZO1‐KI treated with Yoda‐1 (15 μM for 60 min) or Yoda‐1 plus GsMTx‐4 (5 μM for 30 min) cells. (B) Left panel: Representative immunoblot of RRAS in PIEZO1‐WT and PIEZO1‐KI cells at steady state and PIEZO1‐KI treated with Yoda‐1 (15 μM for 60 min) or Yoda‐1 plus GsMTx‐4 (5 μM for 30 min) cells. Right panel: Quantification by densitometric analysis of three separate Western blots with similar results. Data are means ± standard deviation (SD) (**p < .01, ANOVA adjusted for Sidak's multiple comparisons test) and are normalized on GAPDH. (C) mRNA expression of HAMP (red histograms, left) and RRAS (blue scatter dot plot, right) in PIEZO1‐WT cells at steady state (NT) and treated with Yoda‐1 and GsMTx‐4. Data are means ± SD of three independent experiments. *p < .05, **p < .01 by ANOVA test and post hoc correction by Sidak's multiple comparison tests. (D) mRNA expression of Hamp and Rras in primary murine hepatocytes at steady state (NT), treated with Yoda‐1 (50 μM for 30 min) and Yoda‐1 plus GsMTx‐4 (30 μM for 30 min). Data are means ± SD of three independent experiments. *p < .05, by ANOVA test and post hoc correction by Sidak's multiple comparison tests. r‐value by Pearson correlation analysis of Hamp and Rras gene expression. (E) Left panel: Representative immunoblots of pERK1/2 and RRAS in total cells lysate of PIEZO1‐WT and PIEZO1‐KI cells treated with siRRAS and siRRAS + Yoda‐1 (15 μM for 60 min); tERK1/2 and GAPDH were used as loading control. Right panel: Histograms showing quantification by densitometric analysis of three separate western blots with similar results. Data are means ± SD (*p < .05, **p < .01, ANOVA test and post hoc correction by Sidak's multiple comparison tests). (F) Histograms showing HAMP and RRAS gene expression in PIEZO1‐WT and PIEZO1‐KI cells treated with siRRAS and siRRAS + Yoda‐1 (15 μM for 60 min). Data are means ± SD of three independent experiments (*p < .05, **p < .01, ANOVA test and post hoc correction by Sidak's multiple comparison tests). [Color figure can be viewed at wileyonlinelibrary.com]
To further confirm the role of RRAS as a key player in the molecular signaling alteration mediated by PIEZO1, we silenced RRAS by siRNA in Hep3B PIEZO1‐KI cells, both treated and untreated with Yoda‐1. Our results revealed a direct correlation between RRAS downregulation and a reduction in the phosphorylation levels of ERK1/2 (r = 0.9590, p = .04), as well as an inverse correlation between RRAS downregulation and an increase in HAMP gene expression (r = −0.9902, p = .01) (Figure 3E,F).
3.4. MAPK cascade and RAS‐mediated signaling are key regulatory pathways of HAMP gene expression in Hep3b PIEZO1‐KI cells
Along the deregulated intracellular pathways detected in the PIEZO1‐KI cellular model, we observed that RRAS takes part also in the mitogen‐activated protein kinase (MAPK) signaling pathway, Rap1, and RAS signaling pathway (Figure S3A). Therefore, we investigated the involvement of this signaling in PIEZO1‐mediated regulation of hepcidin by MAPK and RAS signaling inhibition. At steady state, PIEZO1‐KI cells treated with U0126, a potent MEK kinase inhibitor, showed decreased phosphorylation of ERK1/2 and restored HAMP expression to levels comparable to those in PIEZO1‐WT cells (Figure 4A,B).
FIGURE 4.
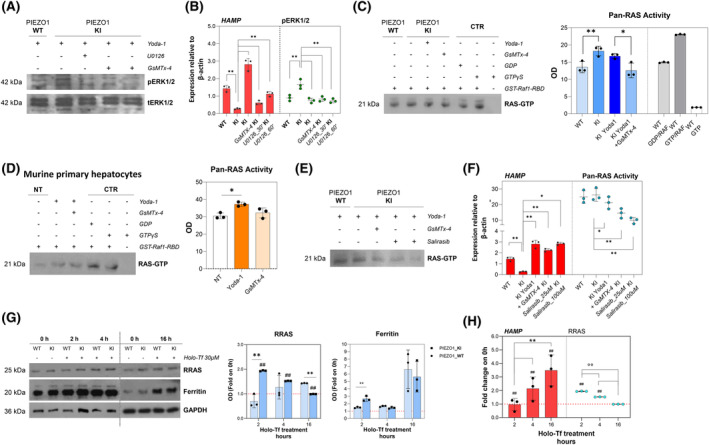
RRAS is a key regulator of PIEZO1‐mediated HAMP gene regulation, modulating MAPK cascade and RAS‐mediated signaling. (A) Representative immunoblots of pERK1/2 in total cells lysate of PIEZO1‐WT and PIEZO1‐KI cells treated with Yoda‐1 (15 μM for 60 min), PIEZO1‐KI treated with Yoda‐1 plus GsMTx‐4 (5 μM for 30 min) and PIEZO1‐KI treated with U0126 for 30 and 60 min. tERK1/2 was used as the loading control. (B) Histograms showing HAMP gene expression in PIEZO1‐WT and PIEZO1‐KI treated with Yoda‐1 (15 μM for 60 min), PIEZO1‐KI treated with Yoda‐1 plus GsMTx‐4 (5 μM for 30 min), and PIEZO1‐KI treated with U0126 for 30 and 60 min. Scatter dot plot showing quantification by densitometric analysis of three separate western blots with similar results. Data are means ± standard deviation (SD) of three independent experiments (*p < .05, **p < .01, ANOVA test and post hoc correction by Sidak's multiple comparison tests). (C) Left panel: Representative immunoblots of RAS GTP proteins purified by affinity precipitation with the RAS binding domain (RBD) of RAF1 from total cell lysate of PIEZO1‐WT and PIEZO1‐KI cells at steady state and PIEZO1‐KI treated with Yoda‐1 (15 μM for 60 min) or Yoda‐1 plus GsMTx‐4 (5 μM for 30 min). Lanes 5 and 6 are the positive controls (cell lysate treated with GST‐Raf1‐RBD plus GDP or GTP); lane 7 is the negative control (without GST‐Raf1‐RBD). Right panel: Quantification by densitometric analysis of three separate western blots with similar results. Data are means ± SD (*p < .05, PIEZO1‐WT vs. PIEZO1‐KI; **p < .01, PIEZO1‐KI vs. PIEZO1‐KI + GsMTx‐4; ANOVA test and post hoc correction by Sidak's multiple comparison tests). (D) Left panel: Representative immunoblots of RAS GTP proteins purified by affinity precipitation with the RBD of RAF1 from total cell lysate of primary murine hepatocytes (HCs) treated with vehicle, Yoda‐1 (50 μM for 30 min) or Yoda‐1 plus GsMTx‐4 (30 μM for 30 min). Lanes 5 and 6 are the positive controls (cell lysate treated with GST‐Raf1‐RBD plus GDP or GTP); lane 7 is the negative control (without GST‐Raf1‐RBD). Right panel: Histograms showing quantification by densitometric analysis of three separate western blots with similar results. Data are means ± SD (*p < .05, hepatocytes + Yoda‐1 vs. hepatocytes vehicle‐treated by ANOVA test and post hoc correction by Sidak's multiple comparison tests). (E) Left panel: Representative immunoblots of RAS GTP proteins purified by affinity precipitation with the RBD of RAF1 from total cell lysate of PIEZO1‐WT and PIEZO1‐KI cells at steady state and PIEZO1‐KI treated with Yoda‐1, Yoda‐1 plus GsMTx‐4 or Salirasib 25 and 100 μM. Right panel: Histograms showing HAMP gene expression in PIEZO1‐WT and PIEZO1‐KI treated with Yoda‐1 (15 μM for 60 min), PIEZO1‐KI treated with Yoda‐1 plus GsMTx‐4 and PIEZO1‐KI treated with Salirasib 25 and 100 μM. Boxes showing quantification by densitometric analysis of three separate Western blots with similar results. Data are means ± SD of three independent experiments. (*p < .05, PIEZO1‐WT vs. PIEZO1‐KI; **p < .01, PIEZO1‐KI vs. PIEZO1‐KI + GsMTx‐4; ANOVA test and post hoc correction by Sidak's multiple comparison tests). (G) Left panel: Representative immunoblots of RRAS, and ferritin in total cells lysate of PIEZO1‐WT and PIEZO1‐KI cells treated with Holo‐transferrin (Holo‐Tf) (30 μM) for 0, 2, 4, or 16 h. GAPDH is the loading control. Right panel: Quantification by densitometric analysis of three separate Western blots with similar results. Data are means ± SD (*p < .05, **p < .01 PIEZO1‐WT vs. PIEZO1‐KI; ##p < .01 PIEZO1‐KI 2, 4, or 6 vs. PIEZO1‐KI NT [0 h]; ##p < .01 PIEZO1‐KI vs. PIEZO1‐WT. ANOVA test and post hoc correction by Sidak's multiple comparison tests). (H) Histograms showing HAMP gene expression normalized to β‐actin (red), and scatter dot plot showing quantification of and RRAS protein expression in PIEZO1‐KI cells treated with Holo‐Tf (30 μM) for 2, 4, or 16 h. Data are means ± SD of three independent experiments and are represented as fold on NT (##p < .01 vs. NT (0 h);°°p < .01, PIEZO1‐KI 2 h vs. PIEZO1‐KI 4 h, PIEZO1‐KI 4 h vs. PIEZO1‐KI 16 h ANOVA and PIEZO1‐KI 2 h vs. PIEZO1‐KI 16 h by ANOVA test and post hoc correction by Sidak's for multiple comparisons). OD, optical density. [Color figure can be viewed at wileyonlinelibrary.com]
Then, we evaluated whether PIEZO1 GoF mutation also affects the RAS activation, in addition to gene/protein expression. We quantified the overall RAS activity (pan‐RAS activity), in terms of GTP‐bound RAS, in PIEZO1‐WT and PIEZO1‐KI cells at steady state and after activation or inhibition of PIEZO1. We found that PIEZO1‐KI cells, including those treated with Yoda‐1, exhibited a significant increase in RAS activity compared to PIEZO1‐WT cells (Figure 4C). The inhibition of PIEZO1 with GsMTx‐4 was able to rescue the hyperactivation of RAS signaling to PIEZO1‐WT levels, confirming the direct regulation of PIEZO1 on RAS‐GTPase activity (Figure 4C). The data were confirmed also in primary murine HCs upon activation and inhibition of Piezo1 (Figure 4D).
The increased RAS activity in PIEZO1‐KI cells was further validated by using salirasib, a drug blocking all forms of RAS by dislodging RAS from its membrane anchoring sites, facilitating its degradation. 38 We observed that HAMP gene expression in PIEZO1‐KI cells was restored to PIEZO1‐WT levels after 24 h of treatment with two different concentrations of salirasib (25 and 100 μM), confirming the role of RAS signaling in HAMP regulation (Figure 4E,F). Since salirasib has been reported to inhibit the growth of multiple hepatocarcinoma cell lines, we tested cell viability in both PIEZO1‐WT and KI cells, observing no differences in either viability or cell growth (Figure S3B,C). Moreover, since pan‐RAS activity is increased in PIEZO1‐KI cells, we analyzed gene and protein expression of other RAS superfamily members in our transcriptomic and proteomics data. We found that only RRAS was significantly deregulated at both gene and protein levels (Figure S3D). Additionally, we analyzed some of these genes (HRAS, NRAS, and RRAS2) by qRT‐PCR analysis, demonstrating no alterations in PIEZO1‐KI cells compared to PIEZO1‐WT ones (Figure S3E).
As final proof of concept, we examined the regulation of RRAS in PIEZO1‐KI cells under iron‐loading conditions. PIEZO1‐KI cells were iron overloaded with Holo‐Tf for 2, 4, and 16 h to mimic the transferrin‐mediated hepatic iron overload. Firstly, we tested for ferritin protein expression to assess the efficacy of treatment. Of note, at steady state, PIEZO1‐KI cells showed higher ferritin expression compared to PIEZO1‐WT cells (Figure 4G), confirming the direct correlation between hepatic iron overload and PIEZO1‐GoF variant. Interestingly, an inverse correlation was observed between HAMP gene expression (which was strongly upregulated in Holo‐Tf‐treated cells compared to untreated ones) and the RRAS protein expression (r = −1.000; p = .0023) (Figure 4H).
4. DISCUSSION
In recent years, the growing interest in the pathogenic mechanisms of iron overload in patients affected by DHS has shed light on the important role of mechanotransduction in iron metabolism. Indeed, genotype–phenotype correlation studies have revealed that patients suffering from DHS with mutations in PIEZO1 gene show a more significant tendency toward iron overload compared to those with KCNN4 mutations. 22 , 23 To unravel the molecular basis of iron overload due to PIEZO1 mutations, both cellular 27 and mouse models 28 have been generated. Our previous findings reported that PIEZO1 mutations are associated with altered MAPK pathway and BMP/SMAD signaling, leading to HAMP gene suppression in a calcium‐mediated mechanism. 27 Additionally, both constitutive and macrophagic‐specific Piezo1 GoF mice developed age‐dependent iron overload. 28 Mechanotransduction has a pivotal role in liver physiology and disease, affecting different cell types. 39 , 40 In HCs, mechanotransduction has been found to be essential not only in proliferation and cell fate maintenance but also in rewire metabolism, although molecular mechanisms are poorly understood. 41 , 42 The conversion of mechanical forces into biological signals in the HCs can implicate static pressure, shear stress, and membrane stretch. 43
To dissect the role of PIEZO1 GoF variants in the HCs, we developed an integrated multi‐omics approach composed of proteomic and transcriptomic profiles of human hepatic cell line engineered for PIEZO1 R2456H variant (PIEZO1‐KI cells). The hepatic PIEZO1‐KI model presented a GoF phenotype with an increased intracellular calcium concentration and Tl+ uptake as a K+ surrogate, without alteration in gene and protein expression of PIEZO1, as demonstrated in erythrocytes. 4 The pathway analysis performed on both genes and proteins revealed that regulation of the actin cytoskeleton was a commonly deregulated pathway in both the proteome and transcriptome. Mechanotransduction is a well‐known mechanism inducing cytoskeleton remodeling and several studies demonstrated that PIEZO1 transduces mechanical signals into biochemical cues through integrins, G protein‐coupled receptors, transient receptor potential ion channels, and other intracellular pathways. 43 , 44 To demonstrate a deregulation in the actin cytoskeleton, we characterized stress fibers. The formation, stability, dynamics, and morphology of the stress fibers are highly regulated by mechanical and biochemical cues. For instance, upregulation of contractility, actin polymerization, and matrix adhesion promote the formation and thickening of stress fibers, whereas cell relaxation, the inhibition of contractility, and actin cytoskeletal disruption lead to their disassembly and disintegration. 45 We found that PIEZO1‐KI cells presented an increased number of stress fibers that appear longer than ones of PIEZO1‐WT cells. Moreover, the cytoskeleton of PIEZO1‐KI cells appears more complex, presenting an increased number of junctions compared to WT cells and, more interestingly, an increased number of end‐points/branches, describing a branched‐out structure that mirrors the alteration of the cytoskeleton in response to the PIEZO1‐GoF mutation.
The analysis of individual gene/protein expressions within the actin cytoskeleton regulatory pathway revealed that most genes were significantly deregulated in RNAseq data (25 genes vs. 6 proteins out of 34 members of the pathway). Interestingly, we observed that only three genes/proteins were concomitantly deregulated in both analyses, RRAS, ITGB4, and F2. Among them, only RRAS expression was regulated in response to PIEZO1 activation and inhibition, suggesting its potential role as a key regulator in PIEZO1‐dependent intracellular signaling deregulation. RRAS is an atypical member of the RAS subfamily of small GTPases that enhances integrin‐mediated adhesion and signaling. 46 RRAS function is related to the integrin function. It regulates cell adhesion through integrins and downstream signaling events, in a bi‐directional mechanism, as RRAS is activated downstream of integrin‐mediated adhesion. 47 Interestingly, PIEZO1 and RRAS have already been described as functionally associated with a Ca2+‐mediated integrin activation mechanism. Particularly, a PIEZO1‐mediated spatial regulation of RRAS has been hypothesized. 48 We observed that RRAS protein is included in other enriched pathways, such as MAPK, RAP1, and RAS signaling pathways, besides a role in axon guidance. PIEZO1‐KI cells exhibited an alteration in both MAPK and RAS signaling cascades. Firstly, we confirmed the tight relationship between PIEZO1 activation and phosphorylation levels of ERK1/2 and the inverse correlation with HAMP gene expression. Indeed, suppressing HAMP gene expression is rescued not only by PIEZO1 inhibition but also by the inhibition of ERK phosphorylation that is sufficient to restore HAMP expression to WT levels. The role of ERK1/2 kinases in HAMP regulation has been proven so far. It has been demonstrated that in iron‐loading conditions, a Tfr2‐mediated HAMP regulation occurs by inducing phosphorylation of ERK1/2. 49 , 50 Of note, the inhibition of ERK1/2 is less effective in restoring HAMP expression to WT levels than the PIEZO1 inhibition alone.
Parallelly, we observed the PIEZO1‐KI cells exhibited enhanced pan‐RAS‐GTPase activity compared to WT cells. This increase in activity was completely rescued following the inhibition of PIEZO1 by GsMTx‐4 treatment, confirming that PIEZO1 directly affects the GTPase activity, thereby modulating downstream signaling pathways. The inhibition of RAS activation by salirasib indeed fully restored HAMP gene expression, confirming a tight correlation between RAS signaling and iron status. This process requires the RRAS protein, as its inhibition by siRNA restores HAMP expression to WT levels via ERK signaling, whose phosphorylation levels are suppressed in RRAS‐silenced conditions. Interestingly, PIEZO1 hyperactivation alone is insufficient to counteract the effect of RRAS silencing, confirming its pivotal role in this regulative axis. This was finally proved by inducing HAMP expression through Holo‐Tf treatment. We observed that increased HAMP gene levels inversely correlate with the expression of RRAS, thus confirming that RAS small GTPase activity correlates with HAMP gene regulation.
Taking together these data established the key role of RRAS protein in regulating HAMP gene expression in response to PIEZO1 GoF via the RAS/MAPK pathway. Herein, we demonstrated that PIEZO1 GoF at the hepatic level alters the organization of the actin cytoskeleton, thereby activating the intracellular RAS signaling pathway. This activation involves RAS‐related GTPases, such as RRAS, linking the regulation of membrane integrity to the intracellular activation of the MAPK pathway. A potential additive role of integrin‐mediated signaling may explain why RAS and ERK inhibitors can only partially rescue the phenotype, unlike the more effective GsMTx‐4 treatment (Figure 5). This suggests that additional mechanisms are involved in this complex and finely‐tuned regulation.
FIGURE 5.
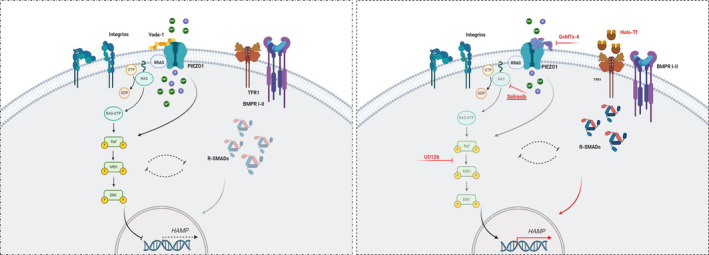
Schematic representation of intracellular deregulation in PIEZO1‐KI model. Schematic models of the intracellular pathways' alteration leading to HAMP deregulation in PIEZO1‐KI hepatic cells here described. Left panel: PIEZO1 activation, due to GoF variants or Yoda‐1 treatment, induced an over‐activation of small GTPase and RAS‐mediated signaling that, in turn, activates MAPK cascade. In this condition, HAMP gene expression is suppressed also because of R‐SMAD downregulation. Right panel: The inhibition of the channel by GsMTx‐4 molecule, RAS‐inhibition and MEK activity inhibition are able to rescue HAMP gene expression to PIEZO1‐WT levels (in red). Finally, the HAMP gene over‐expression induced by Holo‐transferrin (Holo‐Tf) treatment inversely correlates with the protein expression of the small GTPase RRAS, suggesting negative feedback between RAS‐MAPK axis and BMP/SMAD pathway (black dotted lines). [Color figure can be viewed at wileyonlinelibrary.com]
To the best of our knowledge, this is the first proposal of a PIEZO1‐RAS/MAPK‐hepcidin axis. Understanding the role of the RAS signaling pathway in regulating iron metabolism could be crucial for developing new therapeutic strategies for DHS and other conditions characterized by iron overload.
AUTHOR CONTRIBUTIONS
IA and RR designed the study. IA conducted the study. IA and BER wrote the manuscript; VDO and AN performed all of the cell line assays and drug treatments. FME and AnI performed qRT‐PCR and Western blotting; BER and RM performed the establishment of the cellular model and the sequencing analysis of the clones; VAL and MC performed the raw data analysis of RNA sequencing analysis. AcI and RR revised the manuscript.
FUNDING INFORMATION
Cariplo and Telethon Alliance—2021, number 2022‐0583 to Immacolata Andolfo.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests.
PATIENT CONSENT STATEMENT
The authors have nothing to report.
Supporting information
Data S1. Supporting Information.
ACKNOWLEDGMENTS
The authors thank the Mass Spectrometry core facility of the TIGEM Institute (https://www.tigem.it/research/facilities) for the differential proteomics analysis, the Bioinformatic Service for NGS of CEINGE—Biotecnologie Avanzate Franco Salvatore (https://www.ceinge.unina.it/index.php/facility) for the raw data analysis of RNA sequencing analysis; the microscopy facility CEINGE—Biotecnologie Avanzate Franco Salvatore (https://www.ceinge.unina.it/index.php/microscopia); the flow cytometry laboratory of CEINGE—Biotecnologie Avanzate Franco Salvatore. The authors further thank NextGenerationEU and Italian Ministry of Research PNRR—National Center for gene therapy and Drugs based on RNA technology—Spoke 1‐CN3 ‘Genetic Diseases.’
Rosato BE, D'Onofrio V, Marra R, et al. RAS signaling pathway is essential in regulating PIEZO1‐mediated hepatic iron overload in dehydrated hereditary stomatocytosis. Am J Hematol. 2025;100(1):52‐65. doi: 10.1002/ajh.27523
DATA AVAILABILITY STATEMENT
Data available on request from the authors.
REFERENCES
- 1. Andolfo I, Russo R, Gambale A, Iolascon A. New insights on hereditary erythrocyte membrane defects. Haematologica. 2016;101(11):1284‐1294. doi: 10.3324/haematol.2016.142463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andolfo I, Russo R, Gambale A, Iolascon A. Hereditary stomatocytosis: an underdiagnosed condition. Am J Hematol. 2018;93(1):107‐121. doi: 10.1002/ajh.24929 [DOI] [PubMed] [Google Scholar]
- 3. Gallagher PG. Diagnosis and management of rare congenital nonimmune hemolytic disease. Hematology Am Soc Hematol Educ Program. 2015;2015:392‐399. doi: 10.1182/asheducation-2015.1.392 [DOI] [PubMed] [Google Scholar]
- 4. Andolfo I, Alper SL, De Franceschi L, et al. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood. 2013;121(19):3925‐3935. doi: 10.1182/blood-2013-02-482489 [DOI] [PubMed] [Google Scholar]
- 5. Zarychanski R, Schulz VP, Houston BL, et al. Mutations in the mechanotransduction protein PIEZO1 are associated with hereditary xerocytosis. Blood. 2012;120(9):1908‐1915. doi: 10.1182/blood-2012-04-422253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bae C, Gnanasambandam R, Nicolai C, Sachs F, Gottlieb PA. Xerocytosis is caused by mutations that alter the kinetics of the mechanosensitive channel PIEZO1. Proc Natl Acad Sci U S A. 2013;110(12):E1162‐E1168. doi: 10.1073/pnas.1219777110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coste B, Mathur J, Schmidt M, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330(6000):55‐60. doi: 10.1126/science.1193270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ge J, Li W, Zhao Q, et al. Architecture of the mammalian mechanosensitive Piezo1 channel. Nature. 2015;527(7576):64‐69. doi: 10.1038/nature15247 [DOI] [PubMed] [Google Scholar]
- 9. Syeda R, Xu J, Dubin AE, et al. Chemical activation of the mechanotransduction channel Piezo1. Elife. 2015;4:e07369. doi: 10.7554/eLife.07369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao Q, Zhou H, Chi S, et al. Structure and mechanogating mechanism of the Piezo1 channel. Nature. 2018;554(7693):487‐492. doi: 10.1038/nature25743 [DOI] [PubMed] [Google Scholar]
- 11. Tang H, Zeng R, He E, Zhang I, Ding C, Zhang A. Piezo‐type mechanosensitive ion channel component 1 (Piezo1): a promising therapeutic target and its modulators. J Med Chem. 2022;65(9):6441‐6453. doi: 10.1021/acs.jmedchem.2c00085 [DOI] [PubMed] [Google Scholar]
- 12. Xiang Z, Zhang P, Jia C, et al. Piezo1 channel exaggerates ferroptosis of nucleus pulposus cells by mediating mechanical stress‐induced iron influx. Bone Res. 2024;12(1):20. doi: 10.1038/s41413-024-00317-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Albuisson J, Murthy SE, Bandell M, et al. Dehydrated hereditary stomatocytosis linked to gain‐of‐function mutations in mechanically activated PIEZO1 ion channels. Nat Commun. 2013;4:1884. doi: 10.1038/ncomms2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Del Orbe BR, Arrizabalaga B, De la Hoz Rastrollo AB, et al. Hereditary xerocytosis, a misleading anemia. Ann Hematol. 2016;95(9):1545‐1546. doi: 10.1007/s00277-016-2716-9 [DOI] [PubMed] [Google Scholar]
- 15. Shmukler BE, Vandorpe DH, Rivera A, Auerbach M, Brugnara C, Alper SL. Dehydrated stomatocytic anemia due to the heterozygous mutation R2456H in the mechanosensitive cation channel PIEZO1: a case report. Blood Cells Mol Dis. 2014;52(1):53‐54. doi: 10.1016/j.bcmd.2013.07.015 [DOI] [PubMed] [Google Scholar]
- 16. Andolfo I, Martone S, Rosato BE, et al. Complex modes of inheritance in hereditary red blood cell disorders: a case series study of 155 patients. Genes. 2021;12(7):958. doi: 10.3390/genes12070958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zama D, Giulietti G, Muratore E, et al. A novel PIEZO1 mutation in a patient with dehydrated hereditary stomatocytosis: a case report and a brief review of literature. Ital J Pediatr. 2020;46(1):102. doi: 10.1186/s13052-020-00864-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alper SL. Genetic diseases of PIEZO1 and PIEZO2 dysfunction. Curr Top Membr. 2017;79:97‐134. doi: 10.1016/bs.ctm.2017.01.001 [DOI] [PubMed] [Google Scholar]
- 19. Glogowska E, Schneider ER, Maksimova Y, et al. Novel mechanisms of PIEZO1 dysfunction in hereditary xerocytosis. Blood. 2017;130(16):1845‐1856. doi: 10.1182/blood-2017-05-786004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bruce LJ. Hereditary stomatocytosis and cation‐leaky red cells – recent developments. Blood Cells Mol Dis. 2009;42(3):216‐222. doi: 10.1016/j.bcmd.2009.01.014 [DOI] [PubMed] [Google Scholar]
- 21. Iolascon A, Andolfo I, Russo R. Advances in understanding the pathogenesis of red cell membrane disorders. Br J Haematol. 2019;187(1):13‐24. doi: 10.1111/bjh.16126 [DOI] [PubMed] [Google Scholar]
- 22. Picard V, Guitton C, Thuret I, et al. Clinical and biological features in PIEZO1‐hereditary xerocytosis and Gardos channelopathy: a retrospective series of 126 patients. Haematologica. 2019;104(8):1554‐1564. doi: 10.3324/haematol.2018.205328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andolfo I, Russo R, Rosato BE, et al. Genotype‐phenotype correlation and risk stratification in a cohort of 123 hereditary stomatocytosis patients. Am J Hematol. 2018;93(12):1509‐1517. doi: 10.1002/ajh.25276 [DOI] [PubMed] [Google Scholar]
- 24. Orvain C, Da Costa L, Van Wijk R, et al. Inherited or acquired modifiers of iron status may dramatically affect the phenotype in dehydrated hereditary stomatocytosis. Eur J Haematol. 2018;101(4):566‐569. doi: 10.1111/ejh.13135 [DOI] [PubMed] [Google Scholar]
- 25. Syfuss PY, Ciupea A, Brahimi S, et al. Mild dehydrated hereditary stomatocytosis revealed by marked hepatosiderosis. Clin Lab Haematol. 2006;28(4):270‐274. doi: 10.1111/j.1365-2257.2006.00774.x [DOI] [PubMed] [Google Scholar]
- 26. Rosato BE, Alper SL, Tomaiuolo G, Russo R, Iolascon A, Andolfo I. Hereditary anemia caused by multilocus inheritance of PIEZO1, SLC4A1 and ABCB6 mutations: a diagnostic and therapeutic challenge. Haematologica. 2022;107:2280‐2284. doi: 10.3324/haematol.2022.280799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Andolfo I, Rosato BE, Manna F, et al. Gain‐of‐function mutations in PIEZO1 directly impair hepatic iron metabolism via the inhibition of the BMP/SMADs pathway. Am J Hematol. 2020;95(2):188‐197. doi: 10.1002/ajh.25683 [DOI] [PubMed] [Google Scholar]
- 28. Ma S, Dubin AE, Zhang Y, et al. A role of PIEZO1 in iron metabolism in mice and humans. Cell. 2021;184(4):969‐982.e13. doi: 10.1016/j.cell.2021.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Worheide MA, Krumsiek J, Kastenmuller G, Arnold M. Multi‐omics integration in biomedical research – a metabolomics‐centric review. Anal Chim Acta. 2021;1141:144‐162. doi: 10.1016/j.aca.2020.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aglialoro F, Hofsink N, Hofman M, Brandhorst N, van den Akker E. Inside out integrin activation mediated by PIEZO1 signaling in erythroblasts. Front Physiol. 2020;11:958. doi: 10.3389/fphys.2020.00958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Russo R, Langella C, Esposito MR, et al. Hypomorphic mutations of SEC23B gene account for mild phenotypes of congenital dyserythropoietic anemia type II. Blood Cells Mol Dis. 2013;51(1):17‐21. doi: 10.1016/j.bcmd.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Russo R, Marra R, Andolfo I, et al. Characterization of two cases of congenital dyserythropoietic anemia type I shed light on the uncharacterized C15orf41 protein. Front Physiol. 2019;10:621. doi: 10.3389/fphys.2019.00621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods. 2012;9(7):676‐682. doi: 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wan Y, Wang H, Fan X, et al. Mechanosensitive channel Piezo1 is an essential regulator in cell cycle progression of optic nerve head astrocytes. Glia. 2023;71(5):1233‐1246. doi: 10.1002/glia.24334 [DOI] [PubMed] [Google Scholar]
- 35. Piddini E. Epithelial homeostasis: a Piezo of the puzzle. Curr Biol. 2017;27(6):R232‐R234. doi: 10.1016/j.cub.2017.02.002 [DOI] [PubMed] [Google Scholar]
- 36. Liu S, Xu X, Fang Z, et al. Piezo1 impairs hepatocellular tumor growth via deregulation of the MAPK‐mediated YAP signaling pathway. Cell Calcium. 2021;95:102367. doi: 10.1016/j.ceca.2021.102367 [DOI] [PubMed] [Google Scholar]
- 37. Gudipaty SA, Lindblom J, Loftus PD, et al. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature. 2017;543(7643):118‐121. doi: 10.1038/nature21407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu B, Yuan Y, Liu J, et al. Single‐cell RNA sequencing reveals the mechanism of sonodynamic therapy combined with a RAS inhibitor in the setting of hepatocellular carcinoma. J Nanobiotechnol. 2021;19(1):177. doi: 10.1186/s12951-021-00923-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kang N. Mechanotransduction in liver diseases. Semin Liver Dis. 2020;40(1):84‐90. doi: 10.1055/s-0039-3399502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guo T, Wantono C, Tan Y, Deng F, Duan T, Liu D. Regulators, functions, and mechanotransduction pathways of matrix stiffness in hepatic disease. Front Physiol. 2023;14:1098129. doi: 10.3389/fphys.2023.1098129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pocaterra A, Santinon G, Romani P, et al. F‐actin dynamics regulates mammalian organ growth and cell fate maintenance. J Hepatol. 2019;71(1):130‐142. doi: 10.1016/j.jhep.2019.02.022 [DOI] [PubMed] [Google Scholar]
- 42. Mitten EK, Baffy G. Mechanotransduction in the pathogenesis of non‐alcoholic fatty liver disease. J Hepatol. 2022;77(6):1642‐1656. doi: 10.1016/j.jhep.2022.08.028 [DOI] [PubMed] [Google Scholar]
- 43. Di X, Gao X, Peng L, et al. Cellular mechanotransduction in health and diseases: from molecular mechanism to therapeutic targets. Signal Transduct Target Ther. 2023;8(1):282. doi: 10.1038/s41392-023-01501-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Karska J, Kowalski S, Saczko J, Moisescu MG, Kulbacka J. Mechanosensitive ion channels and their role in cancer cells. Membranes. 2023;13(2):167. doi: 10.3390/membranes13020167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Z, Xia S, Kanchanawong P. An integrated enhancement and reconstruction strategy for the quantitative extraction of actin stress fibers from fluorescence micrographs. BMC Bioinform. 2017;18(1):268. doi: 10.1186/s12859-017-1684-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ada‐Nguema AS, Xenias H, Hofman JM, Wiggins CH, Sheetz MP, Keely PJ. The small GTPase R‐Ras regulates organization of actin and drives membrane protrusions through the activity of PLCepsilon. J Cell Sci. 2006;119(Pt 7):1307‐1319. doi: 10.1242/jcs.02835 [DOI] [PubMed] [Google Scholar]
- 47. Conklin MW, Ada‐Nguema A, Parsons M, Riching KM, Keely PJ. R‐Ras regulates beta1‐integrin trafficking via effects on membrane ruffling and endocytosis. BMC Cell Biol. 2010;11:14. doi: 10.1186/1471-2121-11-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McHugh BJ, Buttery R, Lad Y, Banks S, Haslett C, Sethi T. Integrin activation by Fam38A uses a novel mechanism of R‐Ras targeting to the endoplasmic reticulum. J Cell Sci. 2010;123(Pt 1):51‐61. doi: 10.1242/jcs.056424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Poli M, Luscieti S, Gandini V, et al. Transferrin receptor 2 and HFE regulate furin expression via mitogen‐activated protein kinase/extracellular signal‐regulated kinase (MAPK/Erk) signaling. Implications for transferrin‐dependent hepcidin regulation. Haematologica. 2010;95(11):1832‐1840. doi: 10.3324/haematol.2010.027003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ramey G, Deschemin JC, Vaulont S. Cross‐talk between the mitogen activated protein kinase and bone morphogenetic protein/hemojuvelin pathways is required for the induction of hepcidin by holotransferrin in primary mouse hepatocytes. Haematologica. 2009;94(6):765‐772. doi: 10.3324/haematol.2008.003541 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.
Data Availability Statement
Data available on request from the authors.