

Abstract
Prime editors are CRISPR-based genome engineering tools with significant potential for rectifying patient mutations. However, their usage requires experimental optimization of the prime editing guide RNA (PegRNA) to achieve high editing efficiency. This paper introduces the deep transformer-based model for predicting prime editing efficiency (DTMP-Prime), a tool specifically designed to predict PegRNA activity and prime editing (PE) efficiency. DTMP-Prime facilitates the design of appropriate PegRNA and ngRNA. A transformer-based model was constructed to scrutinize a wide-ranging set of PE data, enabling the extraction of effective features of PegRNAs and target DNA sequences. The integration of these features with the proposed encoding strategy and DNABERT-based embedding has notably improved the predictive capabilities of DTMP-Prime for off-target sites. Moreover, DTMP-Prime is a promising tool for precisely predicting off-target sites in CRISPR experiments. The integration of a multi-head attention framework has additionally improved the precision and generalizability of DTMP-Prime across various PE models and cell lines. Evaluation results based on the Pearson and Spearman correlation coefficient demonstrate that DTMP-Prime outperforms other state-of-the-art models in predicting the efficiency and outcomes of PE experiments.
Keywords: MT: Bioinformatics, CRISPR, prime editing, PegRNA, off-target, deep learning, transfer learning, DNABERT
Graphical abstract
Alipanahi and colleagues developed DTMP-Prime, a tool for predicting PegRNA activity and prime editing efficiency. Using a novel encoding algorithm for PegRNA-DNA pairs and the ClinVar dataset, they identified key features affecting PE efficiency and assessed their significance in both the PE2 and PE3 systems.
Introduction
Prime editing (PE)1 is a cutting-edge gene editing technology that represents a significant advancement over the CRISPR-Cas9 system. This innovative tool allows for the precise modification of DNA by facilitating a wide range of base transitions and transversions, as well as enabling the targeted insertion of custom sequences (up to 44 nucleotides [nt]) and deletions (up to 80 nt).1
Multiple PE systems are available, including PE1, PE2, PE3, PE3b, PEmax, and ePPE (engineered plant prime editor) editors.2 The only difference between PE1 and PE2 is the Moloney murine leukemia virus reverse transcriptase fused to the Cas9 enzyme. hyPE2 is a more efficient variant of PE2, which adds the Rad51 DNA-binding domain to PE2. The PE3 system uses nick gRNA (ngRNA), which introduces the non-edited strand to increase the editing efficiency. PE3b is a modification of PE3, in which a single guide RNA (sgRNA) is designed to bind only the edited DNA sequence. To further improve the PE3b system, an ngRNA spacer is designed to match the edited sequences so that it only binds to the edited DNA sequences. Furthermore, protein optimization resulted in PEmax architecture, which enhanced editing efficiency.2 The ePPE is based on PPE (plant prime editor) but removes RT’s RNase H domain and incorporates a viral nucleocapsid protein.
Despite its great potential, PE technology is still in its infancy and requires the overcoming of several limitations to fully realize its capabilities. A significant constraint of PE is its low editing efficiency.3 To address this issue, multiple strategies have been devised to enhance PE efficiency, including utilizing an engineered PE protein, refining the design of the PE guide RNA, manipulating the mismatch repair (MMR) pathway, and optimizing the delivery strategy.2
To improve PE efficiency, researchers have been conducting studies on various aspects of the technique such as:
-
(1)
Optimization of guide RNA design,4 using various strategies that consider factors such as secondary structure, target accessibility, and off-target effects.
-
(2)
Engineering of PE components, especially developing variant forms of the Cas9 enzyme and RT, to improve their activity and efficiency in PE. These novel enzymes could have enhanced processivity, fidelity, or DNA repair capabilities, leading to more efficient and accurate PE outcomes.
-
(3)
Identifying optimal repair templates, several types of repair templates, such as single-stranded oligonucleotides or linear dsDNA fragments, are used in PE experiments; however, the impact of repair template length and delivery methods needs more research.
-
(4)
Modulating DNA repair pathways5 to favor the desired editing outcomes. Researchers are exploring several ways to modulate DNA repair pathways, including manipulating factors involved in homology-directed repair, non-homologous end joining, or base excision repair.6
-
(5)
High-throughput screening approaches, to identify factors that impact PE efficiency.2 This involves testing libraries of gRNAs, repair templates, or other components to identify optimal combinations for efficient PE. These screening approaches enable the identification of key factors that can be further optimized to enhance editing outcomes.2,7
Conducting real-world experiments can be time-consuming, expensive, and resource-intensive. Researchers can find mistakes, risks, limitations, or challenges in the process by guessing how PE will work before they start a real experiment. This lets them improve the designs of their experiments and lower the number of unintended mutations or off-target effects.3
Recently, numerous algorithms and computational approaches such as PrimeDesign,8 PegFinder,9 PnB Designer,10 and PINE-CONE11 have emerged for the purpose of evaluating the efficiency and specificity of PE in silico. These predictive tools play a crucial role in guiding experimental design and aiding in the identification of factors that can enhance editing efficiency, accuracy, and safety.11 Also, several deep learning (DL)-based approaches including DeepPE,12 Easy-Prime,13 PRIDICT,14 and PE-Designer15 have been introduced for predicting PE activity.
Models such as DeepPE,12 Easy-Prime,13 and PRIDICT14 heavily depend on manual feature engineering, which includes calculating various predetermined PegRNA features such as GC count and minimum self-folding free energy. This approach may overlook crucial information, leading to reduced accuracy and applicability. Furthermore, these models lack interpretability and are akin to black boxes.
To address these limitations, transformer-based models such as OPED16,17 aim to automatically learn a comprehensive and interpretable representation of the target DNA and PegRNA pair.17 These advancements will contribute to the further development and widespread application of this novel genome editing technique.18
There is a significant similarity between human language and DNA sequences, particularly the noncoding regions, ranging from alphabets and lexicons to grammar and phonetics. Recently, DL has been employed in high-throughput biology methods,4 leading to profound change in our comprehension of biology.19
Convolutional neural network (CNN) architectures have the potential to extract local signals, but they are limited in their ability to capture sequential information and semantic dependencies within long-range contexts due to the constraints imposed by filter size. In contrast, recurrent neural networks (RNNs), exemplified by LSTM (long short-term memory) and GRU (gated recurrent unit), have been employed to effectively extract sequential information and long-term dependencies.20,21 However, the base RNN models suffer from vanishing gradients and low-efficiency problems.22
To achieve a more precise representation of DNA as a human language, a computational approach should consider all contextual information on a global scale.21 Both CNN and RNN architectures fail to satisfy these requirements. To overcome this limitation, we employed a specialized version of the bidirectional encoder representations from transformers (BERT)23 model known as DNABERT. DNABERT20 leverages an attention-based architecture to comprehensively capture contextual information from the entire input sequence and has demonstrated exceptional performance in extracting potential relationships among various elements of a DNA sequence without the need for human intervention.24
To build a computational model capable of accurately predicting the outcomes of PE, it is imperative to first gain a thorough understanding of the key features that influence experiment efficiency. Subsequently, the construction of a model that can predict the efficiency and other relevant outcomes by analyzing these features and inputs.13 Some of these features pertinent to the design of an optimal PE complex have been investigated by computational models.8,13,15 Li et al.13 have classified the effective features into five categories: spCas9 activity features, oligo features, target mutation features, position features, and RNA folding features. Easy-Prime13 and DeepPE12 demonstrate the importance of the spCas9 activity feature and PBS GC content as more effective features.
The main objective of this paper is to introduce a DNABERT-based model, called DTMP-Prime, which is designed for the prediction of PegRNA activity and PE efficiency. In our work, we have extended the existing framework proposed in Easy-prime13 by introducing a new category to the 5 feature categories. Our network is capable of extracting and analyzing a wide range of effective features (43 in total) in both PE2 and PE3 systems. Using DeepSHap,25 we measured the correlation between them and PE efficiency. DTMP-Prime serves as a valuable tool for assisting in the selection of candidate PegRNAs and facilitating the design of appropriate PegRNA sequences.
The DTMP-Prime model, which is based on BERT22 and utilizes a bi-directional transformer architecture with multi-head attention layers, has been designed to incorporate multi-head attention in the embedding layer. This approach allows us to achieve several key objectives: (1) capturing features related to the identity and position of each nucleotide and k-mer separately in PegRNA or DNA sequences. (2) Understanding the relationship and correlation between each nucleotide and k-mer with other nucleotides and k-mers within the PegRNA or DNA sequences. (3) Examining the relationship and correlation between each nucleotide and k-mer with other nucleotides and k-mers within both the PegRNA and DNA sequences.
The combination of these features with the new encoding strategy has significantly enhanced the efficiency of DTMP-Prime in predicting off-target sites. Furthermore, the utilization of multi-head attention architecture has enabled us to improve the accuracy and generalizability of DTMP-Prime across diverse PE models and cell lines.
Models such as DeepPE,12 in addition to using sequence-based features and structural features such as melting temperature, use other features such as 4 nt after RT-PBS or 4 nt before RTT. These features are not included in DTMP-Prime. However, with the integration of a novel encoding algorithm in DTMP-Prime, our model can effectively extract relationships between all combinations of 6 nt at any position within both the PegRNA and DNA sequences. This enhancement allows for a more comprehensive analysis of sequence characteristics and significantly improves the model’s capacity to predict PegRNA activity and PE efficiency.
Results
Our proposed DTMP-Prime model has three new ideas. It uses a special type of network called a multi-head attention-based transformer for selecting and analyzing features that influence PE efficiency and predict PE efficiency according to these features; a newly developed encoding algorithm designed for encoding PegRNA-DNA pairs; and the integration of DNABERT20 as a PegRNA activity classifier.
To show the superiority and impact of the three aforementioned innovations, we conducted three sets of evaluations. These evaluations involved analyzing the proposed model for predicting PE efficiency and detecting effective features, followed by a comparison of the results with those from prior studies. In addition, we implemented the encoding layer based on the proposed encoding algorithm and one-hot encoding algorithm separately. A detailed comparison of the outcomes generated by these two algorithms is presented in encoding algorithm below. Furthermore, we incorporated the deep layer of DTMP-Prime, employing DNABERT, transformer + attention, RNN, and other deep models separately. We then compared these models with each other and with previous research findings in DNABERT model.
Our performance comparison with other state-of-the-art models has demonstrated that DTMP-Prime shows a better result in predicting PegRNA activity and PE efficiency. Consequently, it is anticipated to emerge as a valuable tool in the field of CRISPR and PE research. Moreover, DTMP-Prime exhibited superior performance compared with other off-target prediction models in CRISPR systems.
Our model integrates various features categorized into six groups to predict editing efficiency and prioritize candidate PegRNA sequences. Beside sequence and activity features recognized for their association with PE efficiency, our model facilitates the exploration of latent features, including RNA secondary structure, RNA folding, gene interaction information, and PegRNA-DNA sequence interaction. This comprehensive approach allows for a more thorough analysis and prediction of PegRNA activity and PE efficiency.
With more than 170,000 data records of PE experiments conducted across numerous research studies,4,8,9,10 we have developed an extensive database that captures the specific characteristics of each PegRNA and their efficiency. The dataset used to build and test our model is provided in Tables S1 and S2, respectively. To characterize features that influence PE efficiency, we investigated various PegRNA sequences with up to 63 nt lengths in 5 genomic sites in human cell lines. We analyzed their activity and efficiency using transformer-based deep network and discovered that Cas activity, sequence length, nucleotide composition, secondary structure, RNA folding, MMR proficiency, and cell type all affect PE efficiency. Combining all the above features into a multi-head attention-based transformer model, we can predict the efficiency of new PE experiments with high precision.
DTMP-Prime
In this section, we evaluate the performance of the DTMP-Prime model in predicting PE efficiency. To overcome the problem of lack of data and improve the accuracy of DTMP-Prime, we adopted pre-training and fine-tuning strategies through transfer learning. This approach initially establishes a general-purpose learning from massive amounts of unlabeled data and subsequently addresses various applications with task-specific data with minimal adjustments.
Initially, DTMP-Prime was pre-trained using data obtained from ClinVar. The variant summary from ClinVar was accessed on September 29, 2021, and we specifically focused on the Homo sapiens genome assembly GRCh38/hg38.26 To ensure data integrity, we filtered the variants by allele ID to eliminate duplicates and by clinical significance to prioritize pathogenic variants. Subsequently, we categorized the variants into three groups based on their variant type: single-nucleotide variants (SNVs), insertions, and deletions.
To facilitate the installation and correction of these pathogenic variants, we enumerated all candidate PegRNAs and sgRNAs for each variant using specific criteria. These criteria included: (1) a maximum distance of 50 nt from the editing site to the PegRNA nicking site, (2) the presence of NGG PAM or NG PAM, (3) a minimum homology of 5 nt downstream of the edit, (4) a minimum PBS length of 8 nt and maximum PBS length of 18 nt, (5) a minimum RTT length of 8 nt and maximum RTT length of 68 nt, and (6) a minimum sgRNA-nick-to-PegRNA-nick distance of 0 nt and maximum nick-to-nick distance of 100 nt.
Our pre-train dataset consists of 77,738 records for correcting ClinVar variants (containing 51,473 SNVs, 1,833 insertions, and 24,432 deletions) and 229,035,543 records for installing of ClinVar variants (containing 152,685,709 SNVs, 4,399,142 insertions, and 71,950,692 deletions).
We design all candidate PegRNAs (229,035,543 PegRNAs to install and 213,459,730 PegRNAs to correct these pathogenic variants) and use them for pre-training DTMP-Prime. More details are provided in Table S3.
After pre-training the model, we fine-tuned over 72,261 PE experiment records gathered from DeepPE,13 PRIDICT,12 DeepPrime,27 Easy-Prime,13 and PrimeDesign8 projects (Table S1). To prevent overfitting, the model underwent validation through a 5-fold cross-validation procedure. In essence, the complete dataset was randomly partitioned into five equivalent segments. Each of these segments was sequentially excluded to create an external set for validating the model constructed using the remaining four segments. This iterative process was reiterated five times, ensuring that every sequence in the dataset was predicted.
We randomly selected 1,013 records of the DeepPE dataset to assess the performance of DTMP-Prime (Table S2). As a widely used method for evaluating model performance, we analyzed the correlation between the measured PE efficiency in a real experiment and the predicted PE efficiency by DTMP-Prime in an external dataset. The results, depicted in Figure 1A, indicate that DTMP-Prime achieved an R value (Pearson correlation coefficient) of 0.8 and an R value (Spearman correlation coefficient) of 0.77 on the selected test dataset.
Figure 1.

Comparison between DTMP-Prime and state-of- art models predicting PE efficiency
(A) Scatterplot with correlation between measured PE efficiency in a real experiment and predicted PE efficiency by DTMP-Prime in an external dataset. (B–E) Comparison of DTMP-Prime with other machine learning models including RNN + Attention, BiLSTM + Attention, GRU + Attention, support vector regression, multi-layer perceptron K-nearest neighbors, DT, RF, AdaBoost, gradient-boosted DTs, over various critics: (B) Pearson correlation coefficient, (C) Spearman correlation coefficient, (D) mean absolute error (MAE), and (E) root mean-squared error (RMSE). (F–I) Comparison between DTMP-Prime and other models predicting PE efficiency such as OPED and PRIDICT over critics such as Pearson correlation coefficient, Spearman correlation coefficient, MAE, and RMSE, respectively. (J) Comparison between DTMP-Prime, Easy-prime, and PrimeDesign based on PE efficiency of designed PegRNAs in five loci.
To showcase the effectiveness of DTMP-Prime, which leverages deep transformer layers and a multi-head attention mechanism, we initially compared its results with those of other machine learning models such as RNN + Attention, BiLSTM + Attention, GRU + Attention, support vector regression, multi-layer perceptron, K-nearest neighbors, decision tree (DT), random forest (RF), AdaBoost, and gradient-boosted DTs. Figures 1B–1E present a comparison between DTMP-Prime and these models based on criteria including Pearson and Spearman correlation coefficient, mean absolute error and root mean-squared error. As mentioned before, for the final test of DTMP-Prime, 1,013 records of the DeepPE project were selected (Table S2). We compare DTMP-Prime with other models according to achieved results on these 1,000 selected records. Also, Figures 1F–1I present a comparison between DTMP-Prime and DL-based models such as OPED, DeepPE, and PRIDICT based on the above criteria.
In subsequent analyses, we evaluated the performance of DTMP-Prime against state-of-the-art models, Easy-Prime13 and PrimeDesign.27 To ensure a fair comparison, we employed the same loci used in Li et al.'s study.13 DTMP-Prime was tested on five variants associated with blood traits. We designed our own PegRNAs and compared them with the same PegRNAs designed in Easy-Prime and PrimeDesign. Detailed sequences for each model are provided in Table S4. For loci rs3785098, rs9386791, and rs2251964, DTMP-Prime generated PegRNAs with distinct RTT and PBS sequences. Conversely, for rs55935819 and rs760369, DTMP-Prime produced identical sequences to Easy-Prime and PrimeDesign, respectively. For each of these two cases, we have different approaches.
In an effort to save time and cost, we utilized the reported observed efficiencies for rs55935819 and rs760369 from Li et al. (Table S2 of associated paper13). For rs3785098 and rs9386791, we supplemented their comparison with the results of DTMP-Prime (designed PegRNA and predicted efficiency). For the remaining loci (rs3785098, rs9386791, and rs2251964), we provide comprehensive data, including designed PegRNAs, predicted efficiencies, and observed efficiencies.
Figure 1J illustrates the predicted and observed efficiencies of DTMP-Prime, PrimeDesign, and Easy-Prime for rs3785098, rs9386791, and rs2251964. Figure 1K compares the predicted efficiencies of DTMP-Prime and Easy-Prime for rs55935819, where both models generated identical PegRNAs. Figure 1L presents a similar comparison between DTMP-Prime and PrimeDesign for rs3785098. As shown in Figures 1J–1L, in all loci, DTMP-Prime achieved higher efficiency than PrimeDesign and in three loci (rs760369, rs9386791, and rs2251964) more than Easy-Prime.
It is worth mentioning that, to predict the PE efficiency, models used different features. Table 1 provides valuable insights into the utilization of various features in models such as DeepPE,12 Easy-prime,13 PrimeDesign,8 PegFinder,9 and DTMP-Prime.
Table 1.
Categories of features used to predict PE efficiency in different tools
| Categories | Used in |
|---|---|
| Cas9 activity | DeepPE, Easy-Prime, DTMP-Prime |
| Sequence features | DeepPE, PrimeDesign, PegFinder, Easy-Prime, DTMP-Prime, etc. |
| RNA folding features | Easy-Prime, DTMP-Prime |
| Mutation features | DeepPE, PrimeDesign, PegFinder, Easy-Prime, DTMP-Prime, etc. |
| Position features | DeepPE, PrimeDesign, PegFinder, Easy-Prime, DTMP-Prime, etc. |
| Structural feature | DTMP-Prime |
Features analysis
One of the major focuses of this study is to derive the contribution and importance of effective features in PE experiments and predict the final outcome. Analyzing the importance of the various components of the input sequences will allow new insights into PE efficiency and preferences. In this study, we systematically characterize how the length, composition, and secondary structure of PegRNA, as well as its interaction with cell line and target site, affect PE efficiency.
As stated, Li et al.'s research13 classified the effective features on PE efficiency into 5 categories. In this project, we have introduced an additional category. Based on our investigation, we have identified 43 features that are categorized into 6 categories that significantly affect the efficiency of PE experiments. The feature categories include: (1) sequence features, (2) positional features, (3) mutation features, (4) structural features, (5) RNA folding features, and (6) Cas9 activity features. Subsequently, we expound upon each category with greater elaboration. Table 2 outlines the features of each category and an abbreviation for each feature to be used as a reference in the rest of the paper. These features are employed in our proposed solution to rapidly design all candidate PegRNAs and other essential components for a desired edit (step 2 in Figure 2), subsequently scoring, ranking, and finally selecting the optimal PegRNA.
Table 2.
Six categories and 43 effective features influencing the efficiency of PE experiments
| Category (number of features) | Features | Abbreviation | Effects on PE efficiency |
|---|---|---|---|
| Sequence features (16) | index of genomic position | IGP | – |
| strand (+/−) | Strand | – | |
| index of edited position | IEP | – | |
| target sequence chromosome ID | Target_Cho_ID_Strands | – | |
| length of PBS | PBS_Len | strong impact with length (6–16 nt) | |
| length of RTT | RTT_Len | strong impact with length (7–23 nt) | |
| GC Content in PBS | GC_Content_PBS | increase | |
| GC Content in RTT | GC_Content_RTT | increase | |
| GC Content in PBS + RTT | GC_Content_PBS_RTT | increase | |
| GC frequency in PBS | GC_Freq_PBS | increase | |
| GC frequency in RTT | GC_Freq_RTT | increase | |
| GC frequency in PBS + RTT | GC_Freq_PBS_RTT | increase | |
| CC at position 18 of PBS+RTT | CC_18_ PBS_RTT | no significant effect | |
| GA at position 40 of wide sequence | GA_40 | no significant effect | |
| G at position 14 of RTT+PBS | G_14_ PBS_RTT | no significant effect | |
| occurrence of consecutive T and A | Poly_T | decreases | |
| Positional features (3) | number of nucleotides from target mutation to the end of RTT sequence | Target_end_flank | no significant effect |
| distance between target mutation and sgRNA nick site | Target_pos | no significant effect | |
| distance between ngRNA nick site and sgRNA nick site | ngRNA_pos | no significant effect | |
| Mutation features (3) | type (substitution, deletion, insertion) | Mutation_Sub, Mutation_Ins, Mutation_Del | no significant effect |
| length of mutation | Mutation_Len | no significant effect | |
| the number of mismatches | Mismatch_Num | no significant effect | |
| Structural features (8) | melting temperature of PBS | PBS_Melt_Temp | direct correlation |
| melting temperature of RTT | RTT_Melt_Temp | direct correlation | |
| melting temperature of PBS+RTT | PBS_RTT_Melt_Temp | direct correlation | |
| reverse-transcribed cDNA and PAM-opposite DNA strand | Tm_3 | direct correlation | |
| RT template region and reverse-transcribed cDNA | Tm_4 | direct correlation | |
| minimum free energy of PegRNA | MFE_PegRNA | reverse correlation | |
| minimum free energy of spacer | MFE_ spacer | reverse correlation | |
| RNA-DNA hybridization energies | RNA_DNA_HE | significantly anti-correlated | |
| RNA folding features (11) | the RNA folding disruption score for the first 10 positions in the RTT | RTT_Disrup_Score | significant reverse correlation (first 5 positions of RTT) |
| PAM disruption feature | PAM_Disrup_Feature | increase | |
| Cas9 activity features (2) | SpCas9 activity | SpCas9_Activity | increase |
| chromatin accessibility | Chromatin_Accessibility | increase |
Figure 2.

The main steps of DTMP-Prime
Category 1 encompasses various features of the DNA sequence both before and after the desired edit, including the number and frequency of G and C nucleotides in each of the PBS and RTT sequences, as well as their overall count. In addition, it involves the length of PBS and RTT sequences. In category 1, the chromosome number, type of strand (+/−), and the beginning position of the edit are replaced.
Category 2 comprises positional features, which refer to the relative distances between the PegRNA nick site and the target mutation (Target_pos), distances between the PegRNA nick site and the ngRNA nick site (ngRNA_pos or nick position), and distances between the target mutation and the end of the RTT (Target_end_flank), or minimal homology downstream of the edit. The downstream homology target (Target_end_flank) represents the number of nucleotides following the target up to the RTT end position.13
Features such as the type of edit (including insertion, deletion, and mutation) and the length of edit belong to category 3. The other feature in this category is whether a target mutation disrupts the PAM sequence or the protospacer of the ngRNA.
Category 4 includes features such as temperature of melting (TM) and minimum free energy, which are related to the second structure of PegRNA. We can never calculate the exact number of these features, but there are some useful tools and applications that estimate these two features according to the second structure of PegRNA. The secondary structure of the PegRNA consists of the sgRNA, the scaffold, the RTT, and the PBS. Furthermore, we calculate the energy of RNA-DNA hybridization by computing the difference in length-normalized Gibbs free energy at a temperature of 37°C between a paired RNA-DNA oligomer and two unpaired oligonucleotides.16
Category 5 refers to the features related to RNA folding. We calculate the probability of different positions on the RTT sequence disrupting the secondary structure of the RNA scaffold. For example, the C-base at the first position in the RTT can pair with G81 in the RNA scaffold,13,17 which affects the proper gRNA structure required for the interaction between Cas9 and gRNA,28 leading to lower PE efficiency.
Category 6 consists of only two traits, namely Cas9 activity and chromatin accessibility.14 Cas9 activity pertains to the capacity of the Cas9 enzyme to cleave DNA strands.29,30 Cas9 exhibits varying cleavage activity in different editing scenarios. Fortunately, the predictability of Cas9 activity allows for the employment of helpful tools such as DeepCpf31 and DeepSpCas9.32 As improved in DeepCpf1, the performance of Cas9 activity prediction models obviously improved when chromatin accessibility information was considered. So, we fine-tune DeepSpCas9 using a data subset such as Endo_Cas9_1A and binary chromatin accessibility information, leading to the development of a fine-tuned model predicting Cas9 activity based on both target sequence information and chromatin accessibility.
The RNA-folding disruption score is defined as the maximal pairing probability between a position in the RTT and the whole scaffold sequence. A higher score indicates a stronger interaction between the RTT and the RNA scaffold, which can potentially disrupt the RNA secondary structure. To calculate RNA-folding disruption, firstly, the second structure of RNA is estimated. Secondly, the binding score is estimated. As is known, the binding score is a good measure to estimate the degree of connection between PegRNA and DNA.
To estimate the binding score, DeepBind33 is deployed in our work. The RNA_folding score is estimated by features of categories 2, 4, and 5 from Table 2.
For estimating the sequence score, features from categories 1, 2, and 3 are used. As is known, features such as the number and frequency of G and C nucleotides in the two PBS and RTT sequences, as well as factors such as the presence or absence of specific nucleotides in the defined position of PegRNA, affect PE efficiency.
Finally, for the off-target score, we try to predict and estimate the number of off-target sites. For the assessment of off-targets generated by the mismatch, we use Cas-OFFinder.34 DTMP-Prime can incorporate off-target scoring predictions29,35,36,37 into its ranking system, and nominate PegRNAs for increasing editing efficiency.
As mentioned previously, the final score of PegRNAs is calculated based on the four scores described above. All PegRNAs are sorted according to their final score (step 4 in Figure 2). Then, the 10 PegRNAs with the highest score are selected (step 5 in Figure 2) and aligned with the whole DNA to find off-targets (step 6 in Figure 2). In the final step, for aligned positions with less than three mismatches, we predict the activity of PegRNA38 and associated PE efficiency by using a multi-head attention-based deep transformer model (step 6 in Figure 2). It is worth mentioning that DTMP-Prime supports three types of edits including insertion, deletion, and substitutions, but mutation length is limited to 3 bp because we predict the activity of PegRNAs and associated PE efficiencies with less than three mismatches in step 6.
Further details regarding this model are provided in the materials and methods.
The results of the analysis of the six categories of features are shown in Table 2. The relative importance of each feature (Figures 3A and 3B) and the overall importance of the six categories in PE2 or PE3 systems are shown in Figure 3C.
Figure 3.

Analyze of effective features
(A and B) The relative importance based on mean absolute SHAP value (y axis) of effective features (x axis) in PE2 and PE3 systems, respectively. (C) The overall importance based on mean absolute SHAP value (y axis) of the six feature categories in PE2 and PE3 systems. (D) Relationship between the PegRNA sequence length (x axis) and PE efficiency (y axis). (E and F) The effect of the lengths of the RT template and PBS (x axis) on the efficiency of off-target prediction (y axis) in HEK293T cells, respectively. (G) Correlation between PE efficiency and Gibbs free energy (ΔG). PE efficiency (y axis) of 200 sequences with 30 nt length in the HEK3 locus in HEK293T cells were predicted and correlated with their ΔG (x axis). (H) The effect of the number of consecutive “T”/“A” in spacer sequence or PegRNA extension (x axis) on editing efficiency (y axis). (I) Distribution diagram of high-efficiency edits in different temperatures (y axis). (J) Correlation between the RNA-folding disruption score and the PE efficiency (y axis) for each of the first 16 positions in the RTT (x axis).
There is a complex relationship between the PegRNA sequence length and efficiency. As shown in Figure 3D, the efficiency of the short PegRNAs with 1–4 nt RTT sequences was 1.2- to 3.7-fold lower than that for longer ones. One possible explanation is the proficiency MMR. The MMR pathway recognizes and excises short mismatches of less than 13 nt and could therefore remove short insertions before the nicked strand is relegated6 across the target sites. Sequences between 15 and 21 nt are 1.3–1.6 times more efficient than 10–14 nt ones. Sequences between 15 and 21 nt have the highest efficiency, while longer sequences are activated less frequently, but still at moderate efficiency even for sequences larger than 60 nt. This disparity is potentially due to steric issues for reverse transcription and base pairing of the unedited strand.6
Our investigation shows that the length of PBS (6–16 nt), and the length of RT template (7–23 nt), had a strong impact on the editing efficiency. We investigated different PBS or RT-template lengths and calculated the off-target score. Overall, changing the RT template length did not affect PE2 specificity. Indeed, fewer off-target sites were associated with a relatively short PBS template (11–13 nt) than a long RT template (14–17 nt). Figures 3E and 3F show the effect of the lengths of the RT template and PBS on the efficiency of off-target editing in HEK293T cells. The dependence of the occurrence of off-target edits on the lengths of the PBS or RT template was investigated using PegRNAs with variable PBS lengths (10–17 bp) or RT lengths (10–17 bp), respectively.
We examined secondary structures of varying strength, including some sequences with perfect hairpins. Two hundred sequences of 30 nt in length in the HEK3 locus in HEK293T cells were selected randomly and PE efficiency was predicted. Their secondary structure free energy is quantified by the Vienna fold ΔG. It is important to mention that more negative ΔG values indicate stronger secondary structures.39 As shown in Figure 3G, ΔG is correlated with PE efficiency and the average Pearson’s is R = −0.42. It is considered that the structure of both PegRNA 3′ extension (comprising the primer binding site and the reverse transcription template) and the target site are important. Also, as noticed in Table 2, for all combinations of PBS length and RTT length, the PBS RNA-DNA hybridization energy was significantly anti-correlated with predicted PE efficiency. This dependency increases for PegRNAs with shorter PBS length.
Selecting sequences with high GC, and especially cytosine content, prone to forming strong secondary structures, enhances the efficiency. Indeed, the stronger secondary structure of the PegRNA 3′ extension led to higher edit efficiency. One potential explanation for this observation is that structured PegRNAs are more protected from digestion by 3′-exonucleases.40 Also, we noticed that incorporating structured motifs at the 3′ end of PegRNAs and preventing degradation of the 3′ extension12 improved PE efficiency by 3- to 4-fold. Alternatively, structured PegRNAs could ease the pairing of the edited strand with the non-edited strand due to being sterically smaller via folding onto themselves.
To make the secondary structure of PegRNA more stable, a non-C/G pair can be changed to a C/G pair in the small hairpin and achieve higher frequencies of targeted insertions and deletions. Also, the incorporation of structured RNA motifs, such as an exoribonuclease-resistant RNA motif, can enhance PegRNA stability and prevent degradation.
The presence of TTTT sequence acts as a transcription terminator for RNA polymerase III and strongly impairs PegRNA expression. The final efficiency of these sequences is 5- to 11.6-fold lower compared with sequences without TTTT. Similarly, stretches of AAAA have a 1.5- to 1.8-fold reduction in efficiency. Figure 3H shows the effect of consecutive “T”/“A” in spacer sequence or PegRNA extension on editing efficiency.
Since the PBS and RTT are important for the initial reverse transcription process, the TM becomes a key factor for the stability of DNA, RNA, and DNA/RNA double-stranded hybrid.40 We assessed the efficiency of about 41,000 different sequences at different temperatures and monitored the TM of PBS, TM of target DNA region corresponding to RT template, TM of reverse-transcribed cDNA and PAM-opposite, TM of RT template region and reverse-transcribed cDNA, and DeltaTM (mentioned in our database as TM1, TM2, TM3, TM4, TM3–TM2, respectively). For edits with high-efficiency (more than 50%), we investigated the distribution diagram of different temperatures. As shown in Figure 3I, more than 90% of high-efficiency edits occurred at temperatures between 30° and 50°.
As mentioned, for the characterization of the RNA-folding features, we defined the RNA-folding disruption score as the maximal pairing probability between a position in the RTT and the whole scaffold sequence. A higher score indicates a stronger interaction between the nucleotide and the RNA scaffold. Nucleotides with high scores can potentially disrupt the RNA secondary structure. Nucleotides at multiple positions in the RTT have variable importance in predicting PE efficiency. For instance, the appearance of the C-base at the first position in the RTT dramatically decreases the editing efficiency of PEs. One possible explanation for this effect is that the appearance of the C-base at the first position in the RTT can pair with G81 in the RNA scaffold and disrupt the interaction between G81 and Y1356 in Cas9.13,41 In fact, pairing between the RTT and scaffold changes the correct gRNA structure needed for Cas9 and gRNA to interact, which makes PE less effective. We calculated the correlation between the RNA-folding disruption score and the PE efficiency for each of the first 16 positions in the RTT. As shown in Figure 3J, the first five positions have a significant reverse correlation with PE efficiency, but the overall significance is low for all positions. This correlation declines from the sixth position and is no longer significant beyond the tenth position.
As mentioned, pairing between the RTT and scaffold could destabilize the RNA secondary structure and decrease the activity of PEs.42,43 This correlation declines from the 6th to the 10th position and is no longer significant beyond the 11th. As seen, the first five positions are more important for overall PE efficiency. Indeed, the probability of interaction of specific nucleotides with scaffold sequences decreases as the distance of nucleotides from the start of RTT increases.
By examining the impact of all the effective features described in Table 2 and comparing all the features and categories with each other (see Figures 3A and 3B), we conclude that the spCas9 activity, RNA folding, and PBS GC content are the top 3 most important features in the PE2 system. This demonstrates the importance of spCas9 activity and second structure features more than pure sequence features in the PE2 system. The PAM disruption feature, the target mutation position (Target_pos), and RNA folding are three important features in the PE3 system. In contrast, the numbers of mutations are lower-ranked features in both models, which demonstrates that mutation type does not affect PE efficiency significantly. This issue confirms that PE is a versatile tool for different kinds of genome editing.
As a comprehensive computational model, we aimed to capture all effective features in DTMP-Prime. Some of these features are selected in the hand-crafted feature selection layer but most of them are analyzed through our powerful transformer-based deep layer. We present the results of our comprehensive feature analysis in Table 2. We believe this table will be a useful guide for developing other machine learning and DL-based models for optimal PegRNA design or predicting the PegRNA’s activity and efficiency.
Encoding algorithm
This section provides the evaluation outcomes of the performance analysis carried out on our encoding algorithm (see encoding layer for more information). To achieve this objective, our model’s encoding layer is constructed utilizing two distinct encoding schemes: one-hot encoding and our novel encoding algorithm. The classic one-hot method of encoding PegRNA and DNA sequence involves using two vectors with a dimension of 4 × L as input, where L is the length of the PegRNA. In contrast, our new encoding strategy utilizes an 8 × L vector as input.
To evaluate the performance of the two encoding methods, various traditional deep architectures including CNN and RNN with diverse topologies are tested. It is widely recognized that certain hyperparameters, including the number of layers and parameters, significantly influence the performance of the deep network models. Hence, it is imperative to meticulously devise the model architecture to ensure its suitability for our intended objective. In this section, one of our main objectives was to evaluate the individual impacts of three key factors: kernel sizes, the number of feature mappings per layer, and the number of layers.
Initially, we assessed the efficacy of the model at various kernel sizes. We have determined and set the specific number of layers and feature maps. The kernel size of 3 × 3 showed better performance in comparison with the other kernel sizes. Subsequently, we maintained constant kernel sizes, and the optimal outcome was attained by employing two convolutional layers and one pooling layer. Rectified linear units were selected as the activation function for each convolutional layer, and average pooling was applied after the convolution layer. The CNN classifier was trained using the base rate of learning (0.005), momentum (0.9), and batch size of 2,000 examples, all of which were conventional settings.
As mentioned before, the DTMP-Prime model is applicable for predicting off-target sites in CRISPR experiments. In addition, the likelihood of an off-target site in PE experiments is significantly lower compared with such sites in CRISPR experiments. Moreover, there has been limited research conducted on the prediction of off-target sites in the PE area.
As a common dataset based on Cas9 nuclease, and to ensure a fair comparison with other models that are utilized for off-target prediction, we used the CRISPOR dataset to train and test our model. We employed leave-one-gRNA-out cross-validation on the CRISPOR dataset to evaluate the prediction performance of the two encoding schemes. Also, it is worth mentioning that the CRISPOR database contains valuable information pertaining to CRISPR experiments, while our main dataset, derived from the ClinVar database, comprises records specifically related to PE experiments.
Identifying a site as off- or on-target is a classification issue. Hence, accuracy, F1 score, and precision were used for this evaluation. Due to the imbalanced nature of the CRISPOR dataset, models can easily achieve a high accuracy value. In addition, the precision, recall, and F1 score measures are challenging to accurately represent the overall performance, because the elevation method tends to label many samples as negative. This results in lower values for precision, recall, and F1 score metrics. The precision/recall area under the curve (AUC) provides a more comprehensive representation of the overall situation. Figure 4 compares two encoding methods using different deep models based on metrics such as AUC values of (1) the true positive rate against the false positive rate and (2) the precision/recall curve.
Figure 4.

Comparison of true positive/false positive rate and AUC score of the two encoding schemes through different deep models
(A) The top 4 diagrams represent one-hot encoding results, and (B) the last four diagrams represent the proposed encoding results.
As noted, that DTMP-Prime has the capability to predict off-target sites in CRISPR editing systems. There are several tools for off-target prediction in these systems38 such as CFD, CNN_std, AttnToMismatch_CNN, and CRISPR-OFF.44,45 In the rest of this section, we wanted to compare DTMP-Prime with other models commonly used for off-target prediction in CRISPR experiments. We compared DTMP-Prime with other off-target prediction models in Figure 5A.
Figure 5.

Performance of DTMP-Prime
(A) Comparison of DTMP-Prime with other advanced off-target prediction models based on critics including accuracy, F1 score, and precision (y axis). (B) Comparison of the PegRNA activity prediction performance (x axis) of DTMP-Prime utilizing two encoding mechanism, the proposed encoding and one-hot encoding. (C and D) Comparison of DTMP-Prime utilizing three different deep models including DNABERT, DistilBERT and a transformer model with three encoders and decoders plus multi-head attention layers over (C) MAE and RMSE criteria (y axis). (D) Pearson and Spearman correlation coefficient (y axis).
As shown in Figure 5B, the PegRNA activity prediction performance of our new encoding algorithm is significantly better than the classic one-hot encoding scheme. In other words, our encoding algorithm can express the information of sequence pairs more effectively, leading to better results.
Furthermore, we evaluated the performance of our proposed encoding model in conjunction with the multi-head attention transformer layer and other architectures such as the DL model based on the gated recurrent unit and attention mechanism, support vector regression, multi-layer perceptron, K-nearest neighbors, DT, and RF. The results demonstrate that our proposed encoding model, along with the DNABERT and CNN5 (refer to model with CNN architecture consisting of three convolutional layers) architectures, yields the most favorable outcomes. See more detail in Figures 1B–1I and 5B–5C.
DNABERT model
In this section, we evaluate the performance of our fine-tuned DNABERT20 model for embedding effective features of input sequence to predict PE efficiency; as for the deep layer in our model, we adopted pre-training and fine-tuning strategies in the embedding layer of DTMP-Prime too.
DNABERT is a DNA sequence model trained on sequences from the human reference genome Hg38. We used pre-trained DNABERT-6 (a pre-trained model with k-mer size = 6) in the embedding layer and fine-tuned it with 43,149 records of the PE-associated dataset (Table S5) to learn more about structure of DNA and PegRNA sequences. For more information see the DTMP-Prime repository on GitHub.
To validate the effectiveness of our DNABERT-based embedding layer, we compared it with other embedding models such as DNABERT, -DistilBERT, and a transformer model with three encoders and decoder plus multi-head attention layers. All these three models were fine-tuned on the same data (extracted from the DeepPE project12). Figures 5C and 5D show the results of this comparison.
Discussion
PE facilitates protein tagging, the correction of pathogenic deletions, and many other exciting applications. An ideal tool to enable these applications would integrate the edits efficiently, accurately, and safely, avoiding unintended outcomes or double-strand break stress, which has hampered Cas9-based therapies. In response to this need, we introduce DTMP-Prime, a DNABERT-based model, to predict PE efficiency with high accuracy. DTMP-Prime assists in the design and ranks all candidate PegRNAs for a desired experiment and selects the optimal one to do an efficient edit; thus, reducing off-target edits.
Our transformer-based model contains the analysis of all effective features in PE efficiency. Some of these features are selected manually, but most of them are analyzed through our powerful transformer-based deep layer and new encoding algorithm. We believe that the results of our analysis will be a useful guide for developing other machine learning and DL-based models for optimal PegRNA design or predicting the PegRNA’s activity or PE efficiency.
As proved, in addition to the described category of features, cell type, repair pathways, epigenetic modification, and PE systems have a significant influence on PE efficiency. Fortunately, models trained specifically on one target site still outperformed predictions on other sites. Here, we just used data from four genomic sites in human cell lines to train our model, but more cell type-specific features, such as chromatin openness and epigenetic modification, will be investigated in the feature development. Another important feature not considered in this study is the repair pathway. As noticed, the insertion efficiency of short RTT sequences (<10 nt) was variable, with high rates in MMR-deficient cell lines but not in MMR-proficient ones. Indeed, MMR antagonizes PE. So, another major effort in future development will be the analysis of the effect of the repair pathway on PE efficiency. Hence, the use of engineered PegRNAs will likely reduce the need for exhaustive screening and substantially advance the application scope of PE.
We investigated the effective features of PegRNAs in PE2 or PE3 systems and built two separate models for each one. We expect that we will be able to add the implementation of PE4 and PE5 systems as soon as more PE data become available.
In the next step, we will integrate extra biological features in our model—associated with the prime editing mechanism in all PE systems and cell lines—and develop accurate models with precise and resistant outcomes.
Materials and methods
In this section, we present DTMP-Prime, a novel multi-head attention-based deep transformer model for predicting PE efficiency and PegRNA activity. DTMP-Prime enables the proper design of PegRNA and ngRNA as urgent components for a desired PE experiment. Using a multi-head attention-based transformer layer leads to the capture of any relationship and correlation between each nucleotide and k-mer with other nucleotides and k-mers within the PegRNA and DNA sequences. The combination of a DNABERT-based20 embedding layer with the new encoding strategy has significantly enhanced the efficiency of DTMP-Prime in predicting off-target sites. Furthermore, the utilization of multi-head attention architecture has enabled us to improve the accuracy and generalizability of DTMP-Prime across diverse PE models and cell lines. In addition, the proposed feature selection mechanism, encoding algorithm, and embedding mechanism are covered in this section.
Figure 2 illustrates the main process flow of DTMP-Prime. As shown in Figure 2, firstly, all possible PegRNAs for a desired edit are designed. Secondly, a PE component is formed, and scored according to four scores that are calculated based on selected features and predicted outcomes. In the next steps, the PE components are ranked according to their acquired efficiency score from the previous step and the top-k (e.g., k = 10) PegRNAs are selected. Finally, the activity of PegRNA score and final efficiency of desired edit are predicted.
The internal mechanism of PE experiments is currently not well understood or clearly defined. So, the feature selection process and score calculation may be challenging. Therefore, we present an innovative computational method to predict PE efficiency, utilizing a DL-based model and multiple PE datasets to train and test the model. DTMP-Prime takes the original and desired DNA sequences as inputs and encodes them as a matrix from which all effective features are extracted. After training the base model according to the available data and the defined effective features, the DeepSHAP (deep shapley additive explanations) tool25 is deployed to analyze the impact of effective features on the PE efficiency. DeepSHAP explains the results of the developed DL-based models and helps researchers understand and interpret the models. In this research, DeepSHAP is utilized to determine the regions of the DNA sequence that contribute to the activity prediction of PegRNAs. The calculation of feature importance is determined by taking the mean absolute value of the DeepSHAP value. This proposed model not only predicts PegRNA activity but also provides a systematic way for developers to assess the impact of different features on PE efficiency and activity.
The following sub-sections address the encoding algorithm, the DNABERT-based embedding mechanism, hand-crafted feature selection layer, and, finally, the multi-head attention-based deep transformer prediction model, respectively.
The architecture of DTMP-Prime
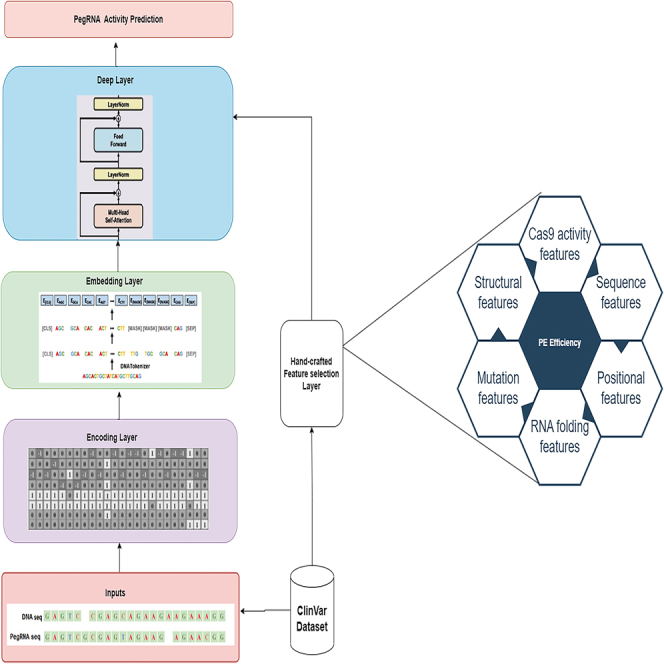
The proposed model is composed of four main components: the encoding layer, the embedding layer, the hand-crafted feature layer, and the neural networks as the deep layer. In the encoding layer, we propose a new encoding method based on PegRNA-DNA sequence pairs, which represents sequence pair information effectively and helps improve the model’s prediction performance. In addition, to better utilize the sequential information of PegRNA and DNA sequence, we added a new embedding layer to our model, which is based on the DNABERT20 model. In the final component, we introduce a new deep transformer layer that learns the effective features of base pairs through transformers. The primary components of the proposed model are shown in Figure 6A. In the following, we provide a detailed description of each layer.
Figure 6.

Architecture of DTMP-Prime
(A) General architecture of DTMP-Prime. (B) Encoding of two sample DNA-PegRNA pairs using the proposed encoding approach into an 8 L matrix. (C) Embedding layer in DTMP-Prime. (D) Deep layer in DTMP-Prime.
Encoding layer
Most computational models for gRNA or PegRNA design use a one-hot encoding scheme to convert the input sequences of nucleotides into numerical vectors of 1 or 0. For instance, adenine is converted to (1,0,0,0), and so on. One-hot encoding increases dimensionality, resulting in a slower and more complex training process. Moreover, one-hot encoding takes more memory space while it adds no new information, since it only changes data representation. Also, most models just encode PegRNA and use only sequence information of PegRNAs. So, PegRNA-DNA pairing information was ignored. To overcome these limitations, we propose a new encoding scheme based on both PegRNA and DNA sequences. In this approach, we initially encode the PegRNA and DNA sequences into a four-dimensional one-hot vector matrix and then encode sequence pairs based on our proposed set of rules. As known, our scheme supports all types of bulges (bulges in both PegRNA and DNA) and mismatches, addressing the encoding of three distinct types of off-target sites, resulting in the capability of DTMP-Prime in handling three edit types including insertion, deletion, and substitutions.
With PegRNA and DNA sequence lengths equal to 73, we design an 8 × 73 matrix in which one column is devoted to each position in the PegRNA/DNA sequence and the first four rows of it are related to the four nucleotides adenine, thymine, cytosine, and guanine, respectively. Eight rules are defined in our encoding scheme for filling out the rows of this matrix as follows but, before using these rules, the encoding mechanism initializes all of the elements of this matrix to 0. Rules 1 to 3 update the first four rows based on the existence of nucleotides in each of the DNA and RNA sequences. Rule 4 encodes the matching of the two sequences in one position. Rules 5 and 6 fill the next two rows, which encode the existence of a gap in any of the two sequences. Finally, rules 7 and 8 fill the last two rows of the matrix, with the former representing the mismatches between the two sequences and the latter indicating the PAM sequence. The rules are as follows:
-
(1)
If the corresponding nucleotide at position x is the same for both PegRNA and DNA sequences, we assign −1 to the corresponding row for each nucleotide.
-
(2)
If the corresponding nucleotides at position x are different in two sequences, we assign the number 1 to the corresponding rows of both nucleotides.
-
(3)
If there is a gap in one of the PegRNA and DNA sequences and the other sequence contains a nucleotide in the same position, we assign 1 to the corresponding row of that nucleotide (rows 1–4).
-
(4)
If the corresponding nucleotide at position x is the same for both PegRNA and DNA sequences, we assign 1 to rows 5 and 6.
-
(5)
If the nucleotide at position x is only in the DNA sequence and a gap has occurred in the PegRNA sequence, we assign 1 to the 5th row.
-
(6)
If the nucleotide at position x is only in the PegRNA sequence, and a gap has occurred in the DNA sequence, we assign 1 to the 6th row.
-
(7)
Row 7 of the matrix represents the mismatches between the two sequences. If the nucleotide at position x is different in the PegRNA and DNA sequences, indicating a mismatch, we assign 1 to the 7th row.
-
(8)
If the nucleotide at position x is part of the PAM string (the last three positions of the PegRNA sequence), we assign 1 to the 8th row.
Figure 6B shows the results of applying our proposed encoding mechanism based on the above rules to two sample DNA and PegRNA sequences.
Embedding layer
By creating a co-occurrence matrix of PegRNA and its corresponding target DNA sequence, we employ an embedding layer to get the global and statistical information of the input sequences. As mentioned, we added a new embedding layer to our model, which is based on DNABERT.20 Similar to the use of BERT22,23 methods to transform alphabetic sequences into numerical compact vectors, we use DNABERT in the embedding layer to transform PegRNA and DNA sequences into compact vectors and extract the effective features of two sequences. Because BERT takes advantage of using three different embedding mechanisms called token, segment, and position embedding, the use of this structure in DTMP-Prime helps us capture contextual and positional information. Therefore, DTMP-Prime can extract sequence, positional, and structural features to use them to predict PE efficiency.
DNABERT is a pre-trained bidirectional encoder representation to capture the global and transferrable understanding of genomic DNA sequences. A pre-trained DNABERT model can be fine-tuned for various sequence analysis tasks,23,24 such as key feature extraction. As mentioned previously, one of the main goals of our model is to extract the important features that affect the efficiency of PE concerning the PegRNA and DNA sequences.
DNABERT is a specialized version of the BERT algorithm that is used for embedding nucleotide sequences. As discussed in Devlin et al.,22 the BERT model is a deep neural network with 110 million parameters. Although transfer learning from large-scale pre-trained models has become prevalent in natural language processing (NLP), operating these large models under constrained computational resources or inference budgets is challenging. In recent years, with the expansion of BERT-based NLP applications,23 developers have tried to change its structure and built simpler versions, including RoBERTa,46 Albert,47 and fastBERT.48 Similar solutions have also been proposed to solve the complexity problem of DNABERT, and simpler models such as DNABERT-DistilBERT49 have been developed. DNABERT-DistilBERT is not usable for sequences longer than 512 nt. Considering that the maximum length of our input sequences is 73 nt, we used the DNABERT-DistilBERT model; as shown in Figures 5C and 5D, its performance is the same as the original DNABERT.
Similar to breaking language texts into words, before applying the DNABERT model to two sequences, we first convert our input sequences into 6-mer using a special tokenization algorithm called DNATokenizer. Thus, for an M nt sequence, a k-mer sequence of length M − k + 1 (M – 5) was obtained. Then, we used learned word embeddings to convert each k-mer to a vector of dimension dl. More details of this process are shown in Figure 6C.
To learn general high-level features, we used pre-trained DNABERT, but six transformer encoders were employed to extract the features outlined in Table 2 and two transformer decoders were specifically designed to extract other hidden features of PegRNA and target sequence, respectively. Multi-head attention in the transformer was calculated as follows:
| (Equation 1) |
| (Equation 2) |
while the letter h represents the number of parallel attention heads, and Q, K, and V matrices symbolize the repositories of queries, keys, and values associated with the input. The matrix W represents the pertinent parameter matrix. The superscript O indicates the output, and T indicates matrix transposition. The variables dk refer to the dimensions of keys. Layer normalization in the transformer was applied as:
| (Equation 3) |
where γ and β are learnable parameters and ε is a very small constant. The variable x represents the input of the layer normalization, while E[x] denotes the expected value or mean of the variable x.
After giving PegRNA and desired DNA as input and preprocessing them, we apply Distillation-DNABERT to transfer sequences to numerical vectors. Distillation-DNABERT is available at https://github.com/joanaapa/Distillation-DNABERT-Promoter.
As shown in Figure 6C, Distillation-DNABERT takes as input a set of sequences represented as k-mer tokens. Each sequence is represented as a matrix M, in which each token is embedded as a numerical vector. Formally, DNABERT captures contextual information by performing the multi-head self-attention mechanism on matrix M, which is subsequently used as input to the deep layer.
Hand-crafted feature selection layer
As one of the main goals of our model, we aim to extract all the important features that affect the efficiency of PE concerning the PegRNA and DNA sequences. According to the details described in effective feature analysis, to calculate the final score of PegRNAs, four scores, including (1) SpCas9_activity_score, (2) RNA_folding score, (3) sequence score, and finally (4) off-target score, are needed based on the features described in Table 2. Although we used DNABERT-DistilBERT40 and fine-tuned it to automatically key feature extraction, some features needed to be selected and calculated manually. To calculate certain features such as spCas9 activity, minimum free energy, and binding degree, we used available computational models or executed defined processes or formulas. Also, some pre-processing needs to be done on sequences to extract complex features. All these processes are performed in this layer.
Deep layer
As shown in Figure 6D, at the core of the deep layer, we employed multi-head attention-based transformers as a deep network to predict PE efficiency. The main faction of multi-head attention-based transformers in this layer is to predict PE efficiency. Also, using off-target score, DTMP-Prime can perform a binary classification of PegRNA activity. Indeed, the output of the last hidden states will be used for activity classification.
Similar to the original DNABERT model, DTMP-Prime consists of 12 transformer layers. The complete architecture of our model is shown in Figure 6D. As shown in Figure 6A, the deep layer takes the output of the hand-crafted feature selection, encoding, and embedding layers as input to determine a sequence activity. To unify all information and consolidate them in one frame, we formed a data frame consisting of three parts: (1) the 43 features (as described in Table 2), (2) the matrix resulting from the encoding of the DNA and PegRNA, and (3) the matrix resulting from the embedded wide DNA and desired DNA. After forming this data frame, the deep layer takes that as input. The input is then fed to 12 transformer blocks.
Data and code availability
The Python code and all of the relevant data supporting the findings of this study are presented in the article or in the supplemental information are deposited in: https://github.com/alipanahiCRISPR/DTMP-Prime. Additional information and other raw data are available from the corresponding author upon reasonable request.
Acknowledgments
We would like to express my gratitude to Mr. Kian Sahafi for his support in creating our website and managing the GitHub page of DTMP-Prime.
Author contributions
Conceptualization, L.S. and A.K.; methodology, R.A.; implementation, R.A.; analysis, R.A., L.S., and A.K.; visualization, R.A.; writing – original draft, R.A.; writing – review & editing, R.A., L.S., and A.K.; supervision, L.S. and A.K.; investigation, L.S. and A.K.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102370.
Supplemental information
References
- 1.Anzalone A.V., Gao X.D., Podracky C.J., Nelson A.T., Koblan L.W., Raguram A., Levy J.M., Mercer J.A.M., Liu D.R. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 2022;40:731–740. doi: 10.1038/s41587-021-01133-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen P.J., Hussmann J.A., Yan J., Knipping F., Ravisankar P., Chen P.F., Chen C., Nelson J.W., Newby G.A., Sahin M., et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell. 2021;184:5635–5652.e29. doi: 10.1016/j.cell.2021.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hillary V.E., Ceasar S.A. Prime editing in plants and mammalian cells: Mechanism, achievements, limitations, and future prospects. Bioessays. 2022;44 doi: 10.1002/bies.202200032. [DOI] [PubMed] [Google Scholar]
- 4.Mathis N., Allam A., Kissling L., Marquart K.F., Schmidheini L., Solari C., Balázs Z., Krauthammer M., Schwank G. Predicting prime editing efficiency and product purity by deep learning. Nat. Biotechnol. 2023;41:1151–1159. doi: 10.1038/s41587-022-01613-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koeppel J., Weller J., Peets E.M., Pallaseni A., Kuzmin I., Raudvere U., Peterson H., Liberante F.G., Parts L. Prediction of prime editing insertion efficiencies using sequence features and DNA repair determinants. Nat. Biotechnol. 2023;41:1446–1456. doi: 10.1038/s41587-023-01678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peterka M., Akrap N., Li S., Wimberger S., Hsieh P.P., Degtev D., Bestas B., Barr J., van de Plassche S., Mendoza-Garcia P., et al. Harnessing DSB repair to promote efficient homology-dependent and independent prime editing. Nat. Commun. 2022;13:1240. doi: 10.1038/s41467-022-28771-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin Q., Jin S., Zong Y., Yu H., Zhu Z., Liu G., Kou L., Wang Y., Qiu J.L., Li J., Gao C. High-efficiency prime editing with optimized, paired pegRNAs in plants. Nat. Biotechnol. 2021;39:923–927. doi: 10.1038/s41587-021-00868-w. [DOI] [PubMed] [Google Scholar]
- 8.Hsu J.Y., Grünewald J., Szalay R., Shih J., Anzalone A.V., Lam K.C., Shen M.W., Petri K., Liu D.R., Joung J.K., Pinello L. PrimeDesign software for rapid and simplified design of prime editing guide RNAs. Nat. Commun. 2021;12:1034. doi: 10.1038/s41467-021-21337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chow R.D., Chen J.S., Shen J., Chen S. pegFinder: A pegRNA designer for CRISPR prime editing. bioRxiv. 2020 doi: 10.1101/2020.05.06.081612. Preprint at. [DOI] [Google Scholar]
- 10.Siegner S.M., Karasu M.E., Schröder M.S., Kontarakis Z., Corn J.E. PnB Designer: a web application to design prime and base editor guide RNAs for animals and plants. BMC Bioinf. 2021;22:1–12. doi: 10.1186/s12859-021-04034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Standage-Beier K., Tekel S.J., Brafman D.A., Wang X. Prime editing guide RNA design automation using PINE-CONE. ACS Synth. Biol. 2021;10:422–427. doi: 10.1021/acssynbio.0c00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim H.K., Yu G., Park J., Min S., Lee S., Yoon S., Kim H.H. Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol. 2021;39:198–206. doi: 10.1038/s41587-020-0677-y. [DOI] [PubMed] [Google Scholar]
- 13.Li Y., Chen J., Tsai S.Q., Cheng Y. Easy-Prime: a machine learning–based prime editor design tool. Genome Biol. 2021;22 doi: 10.1186/s13059-021-02458-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathis N., Allam A., Tálas A., Benvenuto E., Schep R., Damodharan T., Balázs Z., Janjuha S., Schmidheini L., Steensel B.V., et al. Predicting prime editing efficiency across diverse edit types and chromatin contexts with machine learning. bioRxiv. 2023 doi: 10.1101/2023.10.09.561414. Preprint at. [DOI] [Google Scholar]
- 15.Hwang G.H., Jeong Y.K., Habib O., Hong S.A., Lim K., Kim J.S., Bae S. PE-Designer and PE-Analyzer: web-based design and analysis tools for CRISPR prime editing. Nucleic Acids Res. 2021;49:W499–W504. doi: 10.1093/nar/gkab319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu F., Huang S., Hu J., Chen X., Song Z., Dong J., Liu Y., Huang X., Wang S., Wang X., Shu W. Design of prime-editing guide RNAs with deep transfer learning. Nat. Mach. Intell. 2023;5:1261–1274. [Google Scholar]
- 17.Nelson J.W., Randolph P.B., Shen S.P., Everette K.A., Chen P.J., Anzalone A.V., An M., Newby G.A., Chen J.C., Hsu A., Liu D.R. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022;40:402–410. doi: 10.1038/s41587-021-01039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris J.A., Rahman J.A., Guo X., Sanjana N.E. Automated design of CRISPR prime editors for thousands of human pathogenic variants. bioRxiv. 2020 doi: 10.1016/j.isci.2021.103380. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sapoval N., Aghazadeh A., Nute M.G., Antunes D.A., Balaji A., Baraniuk R., Barberan C.J., Dannenfelser R., Dun C., Edrisi M., et al. Current progress and open challenges for applying deep learning across the biosciences. Nat. Commun. 2022;13:1728. doi: 10.1038/s41467-022-29268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji Y., Zhou Z., Liu H., Davuluri R.V. DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics. 2021;37:2112–2120. doi: 10.1093/bioinformatics/btab083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leksono M.A., Purwarianti A. Sequential Labelling and DNABERT For Splice Site Prediction in Homo Sapiens DNA. arXiv. 2022 doi: 10.48550/arXiv.2212.07638. Preprint at. [DOI] [Google Scholar]
- 22.Devlin J., Chang M.W., Lee K., Toutanova K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv. 2018 doi: 10.48550/arXiv.1810.04805. Preprint at. [DOI] [Google Scholar]
- 23.Koroteev M.V. BERT: a review of applications in natural language processing and understanding. arXiv. 2021 doi: 10.48550/arXiv.2103.11943. Preprint at. [DOI] [Google Scholar]
- 24.Xu S., Zhang C., Hong D. BERT-based NLP techniques for classification and severity modeling in basic warranty data study. Insur. Math. Econ. 2022;107:57–67. [Google Scholar]
- 25.Fernando Z.T., Singh J., Anand A. Proceedings of the 42nd International ACM SIGIR Conference on Research and Development in Information Retrieval. 2019. A study on the Interpretability of Neural Retrieval Models using DeepSHAP; pp. 1005–1008. [Google Scholar]
- 26.Landrum M.J., Chitipiralla S., Brown G.R., Chen C., Gu B., Hart J., Hoffman D., Jang W., Kaur K., Liu C., et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48:D835–D844. doi: 10.1093/nar/gkz972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu G., Kim H.K., Park J., Kwak H., Cheong Y., Kim D., Kim J., Kim J., Kim H.H. Prediction of efficiencies for diverse prime editing systems in multiple cell types. Cell. 2023;186:2256–2272.e23. doi: 10.1016/j.cell.2023.03.034. [DOI] [PubMed] [Google Scholar]
- 28.Xue L., Tang B., Chen W., Luo J. Prediction of CRISPR sgRNA activity using a deep convolutional neural network. J. Chem. Inf. Model. 2019;59:615–624. doi: 10.1021/acs.jcim.8b00368. [DOI] [PubMed] [Google Scholar]
- 29.Imani A., Valiant J., Gunawan A.A.S. 2023 International Conference on Computer Science, Information Technology and Engineering (ICCoSITE) IEEE; 2023. Deep Learning-based Approach on sgRNA off-target Prediction in CRISPR/Cas9; pp. 440–444. [Google Scholar]
- 30.Bortesi L., Zhu C., Zischewski J., Perez L., Bassié L., Nadi R., Forni G., Lade S.B., Soto E., Jin X., et al. Patterns of CRISPR/Cas9 activity in plants, animals and microbes. Plant Biotechnol. J. 2016;14:2203–2216. doi: 10.1111/pbi.12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwon K.H., Seonwoo M., Myungjae S., Soobin J., Woo C.J., Younggwang K., Sangeun L., Sungroh Y., Henry K.H. In in the 92nd Keynote Collection of the Japanese Nursery School Annual Conference. 2019. DeepCpf1: Deep learning-based prediction of CRISPR-Cpf1 activity atendogenous sites. [Google Scholar]
- 32.Kim H.K., Kim Y., Lee S., Min S., Bae J.Y., Choi J.W., Park J., Jung D., Yoon S., Kim H.H. SpCas9 activity prediction by DeepSpCas9, a deep learning–based model with high generalization performance. Sci. Adv. 2019;5 doi: 10.1126/sciadv.aax9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alipanahi B., Delong A., Weirauch M.T., Frey B.J. Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015;33:831–838. doi: 10.1038/nbt.3300. [DOI] [PubMed] [Google Scholar]
- 34.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Q., Cheng X., Liu G., Li B., Liu X. Deep learning improves the ability of sgRNA off-target propensity prediction. BMC Bioinf. 2020;21:51. doi: 10.1186/s12859-020-3395-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guan Z., Jiang Z. Transformer-based anti-noise models for CRISPR-Cas9 off-target activities prediction. Brief. Bioinform. 2023;24 doi: 10.1093/bib/bbad127. [DOI] [PubMed] [Google Scholar]
- 37.Liu Q., He D., Xie L. Prediction of off-target specificity and cell-specific fitness of CRISPR-Cas System using attention boosted deep learning and network-based gene feature. PLoS Comput. Biol. 2019;15 doi: 10.1371/journal.pcbi.1007480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alipanahi R., Safari L., Khanteymoori A. CRISPR genome editing using computational approaches: A survey. Front. Bioinform. 2022;2 doi: 10.3389/fbinf.2022.1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corsi G.I., Qu K., Alkan F., Pan X., Luo Y., Gorodkin J. CRISPR/Cas9 gRNA activity depends on free energy changes and on the target PAM context. Nat. Commun. 2022;13:3006. doi: 10.1038/s41467-022-30515-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Z., Shang P., Mohanraju P., Geijsen N. Prime editing: advances and therapeutic applications. Trends Biotechnol. 2023;41:1000–1012. doi: 10.1016/j.tibtech.2023.03.004. [DOI] [PubMed] [Google Scholar]
- 41.Hillary V.E., Ceasar S.A. A review on the mechanism and applications of CRISPR/Cas9/Cas12/Cas13/Cas14 proteins utilized for genome engineering. Mol. Biotechnol. 2023;65:311–325. doi: 10.1007/s12033-022-00567-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X., Zhou L., Gao B.Q., Li G., Wang X., Wang Y., Wei J., Han W., Wang Z., Li J., et al. Highly efficient prime editing by introducing same-sense mutations in pegRNA or stabilizing its structure. Nat. Commun. 2022;13:1669. doi: 10.1038/s41467-022-29339-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang G., Liu Y., Huang S., Qu S., Cheng D., Yao Y., Ji Q., Wang X., Huang X., Liu J. Enhancement of prime editing via xrRNA motif-joined pegRNA. Nat. Commun. 2022;13:1856. doi: 10.1038/s41467-022-29507-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin J., Wong K.C. Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics. 2018;34:i656–i663. doi: 10.1093/bioinformatics/bty554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verma P., Kethar J., Appavu R. Predictability of Off-Targets in CRISPR-Cas9 Gene Editing Systems using Convolutional Neural Networks. J. Stud. Res. 2023;12:10. doi: 10.47611/jsrhs.v12i3.5052. [DOI] [Google Scholar]
- 46.Liu Y., Ott M., Goyal N., Du J., Joshi M., Chen D., Levy O., Lewis M., Zettlemoyer L., Stoyanov V. Roberta: A Robustly Optimized Be Rt Pretraining Approach. arXiv. 2019 doi: 10.48550/arXiv.1907.11692. Preprint at. [DOI] [Google Scholar]
- 47.Lan Z., Chen M., Goodman S., Gimpel K., Sharma P., Soricut R. Albert: A lite bert for self-supervised learning of language representations. arXiv. 2019 doi: 10.48550/arXiv.1909.11942. Preprint at. [DOI] [Google Scholar]
- 48.Liu W., Zhou P., Zhao Z., Wang Z., Deng H., Ju Q. Fastbert: a self-distilling bert with adaptive inference time. arXiv. 2020 doi: 10.48550/arXiv.2004.02178. Preprint at. [DOI] [Google Scholar]
- 49.Sanh V., Debut L., Chaumond J., Wolf T. DistilBERT, a Distilled Version of BERT: Smaller, Faster, Cheaper and Lighter. arXiv. 2019 doi: 10.48550/arXiv.1910.01108. Preprint at. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Python code and all of the relevant data supporting the findings of this study are presented in the article or in the supplemental information are deposited in: https://github.com/alipanahiCRISPR/DTMP-Prime. Additional information and other raw data are available from the corresponding author upon reasonable request.