

Abstract
Pancreatic fibrosis (PF) is primarily distinguished by the stimulation of pancreatic stellate cells (PSCs) and excessive extracellular matrix deposition, which is the main barrier impeding drug delivery and distribution. Recently, nanomedicine, with efficient, targeted, and controllable drug release characteristics, has demonstrated enormous advantages in the regression of pancreas fibrotic diseases. Notably, paracrine signals from parenchymal and immune cells such as pancreatic acinar cells, islet cells, pancreatic cancer cells, and immune cells can directly or indirectly modulate PSC differentiation and activation. The intercellular crosstalk between PSCs and these cells has been a critical event involved in fibrogenesis. However, the connections between PSCs and other pancreatic cells during the progression of diseases have yet to be discussed. Herein, we summarize intercellular crosstalk in the activation of PSCs and its contribution to the development of common pancreatic diseases, including pancreatitis, pancreatic cancer, and diabetes. Then, we also examine the latest treatment strategies of nanomedicine and potential targets for PSCs crosstalk in fibrosis, thereby offering innovative insights for the design of antifibrotic nanomedicine. Ultimately, the enhanced understanding of PF will facilitate the development of more precise intervention strategies and foster individually tailored therapeutic approaches for pancreatic diseases.
Key words: Nanomedicine, Pancreatic fibrosis, Pancreatic stellate cell, Intercellular crosstalk, Pancreatitis, Pancreatic cancer, Diabetes, Therapeutic strategies
Graphical abstract
The latest progress on fibrosis and intercellular crosstalk in the development of common pancreatic diseases was highlighted. It also provides therapeutic strategies of nanomedicine modulating intercellular crosstalk.
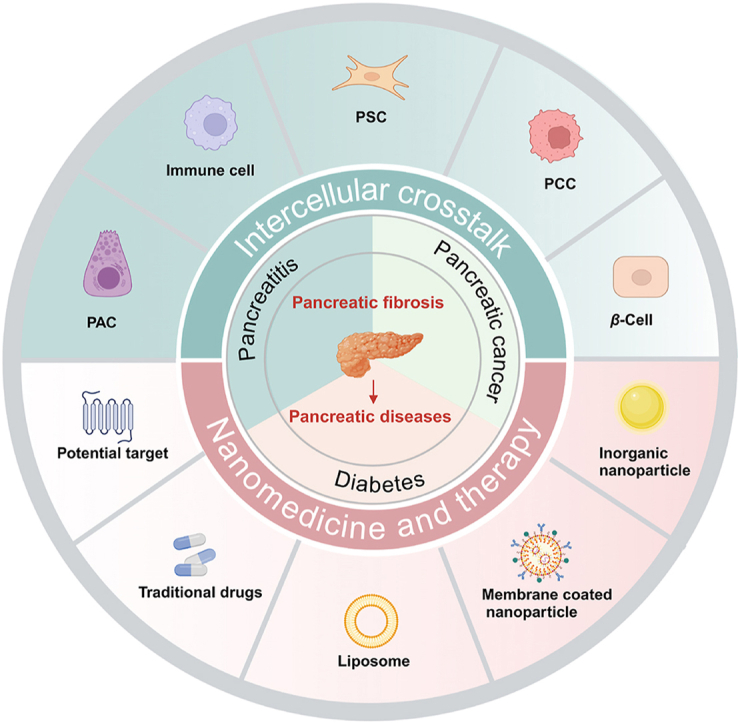
1. Introduction
The pancreas has both exocrine and endocrine functions and is the second largest digestive organ in the human body after the liver. Pancreatic parenchyma is structurally divided into exocrine and endocrine glands1,2. The exocrine pancreas primarily consists of pancreatic acinar cells (PACs), duct cells, and PSCs, which collectively account for over 95% of the pancreatic mass and are responsible for digestion. The endocrine gland refers to the islets, which predominantly consist of five distinct types of islet cells. The endocrine gland, known as the islets, encompasses five distinct islet cell types and plays a crucial role in modulating glucose homeostasis1,2. Both the exocrine and endocrine functions are impaired after the stimulation of pancreatic injury, thereby contributing to the progression of various pancreatic diseases, including pancreatitis, pancreatic cancer (PC), and diabetes3, 4, 5. Given the escalating incidence of pancreatic diseases over time, they pose a significant threat to human life and well-being.
Pancreatic fibrosis (PF) is the result of progressive inflammation and continuous wound healing and is the major pathological feature of pancreatitis, pancreatic cancer, and diabetes4,6,7. It is increasingly recognized as an important determinant of pancreatic disease etiology, treatment response, and progression8. In the absence of effective intervention, PF leads to dysfunction of the pancreas and an increased risk of cancer, failure to therapy, and recurrence9. Fibrosis is a dynamic pathological process under the control of various cells. PSCs are usually considered as the key cells of PF that promote extracellular matrix (ECM) deposition and form a pathological barrier to drug release and accumulation4. In addition to PSCs, multiple cells, including PACs, islet cells, pancreatic cancer cells (PCCs), and immune cells, actively participate in pancreatic fibrosis through modulating PSCs by cytokines, inflammatory mediators, and extracellular matrix components9, 10, 11. Intercellular crosstalk between PSCs and various cells has been an essential event involved in fibrogenesis that constantly persists and may impact the efficacy of treatment. Degradation of the deposited ECM and intervention of intercellular crosstalk will facilitate targeted drug delivery and promote PF reversal.
Recently, nanoparticles have emerged as targeted delivery platforms for therapeutic genes, peptides, and small molecules. These nanoparticles have been extensively studied for the therapy of fibrotic diseases due to their exceptional druggability, remarkable biodegradability, and excellent biocompatibility12, 13, 14. Li et al.15 reported that a peptide nanoparticle anti-fibrosis agent (IGNP-JQ1) inhibited the proliferation of hepatic stellate cells (HSCs) and reduced collagen deposition, an integrated systemic strategy for the therapy of liver fibrosis. Additionally, Bai et al.16 designed an inhalable and mucus-penetrative nanoparticle (NP) system incorporating siRNA against IL-11 (siIL11@PPGC NPs). This system has been shown to increase drug accumulation in the lung, significantly reducing fibrotic development and improving lung function in idiopathic pulmonary fibrosis. Recently, Han et al.17 used a nano-system based on the “nanodrill” strategy (LA-PC) to target PSCs and enhance the accumulation of all-trans retinoic acid (ATRA). The drug delivery efficiency through LA-PC was more than five times higher than free ATRA, and the degree of pancreatic fibrosis was significantly reduced. The use of nanomedicines has revolutionized the field of precision medicine. Compared to conventional drugs, nanomedicines can significantly increase the duration of blood circulation, improve targeting capabilities, and enhance therapeutic effects.
Accumulating evidence suggests that intercellular crosstalk between PSCs and other pancreatic cells significantly contributes to PSC activation and fibrogenesis18,19. However, the interactions occurring during disease progression have yet to be discussed. Hence, this review further summarizes fibrosis in the pathogenesis of three prevalent pancreatic disorders, namely pancreatitis, pancreatic cancer, and diabetes, along with an analysis of the intricate cellular interactions within the fibrotic microenvironment (Fig. 1). Meanwhile, we investigate potential therapeutic strategies utilizing nanomedicine and identify promising targets for mitigating fibrosis associated with pancreatic diseases.
Figure 1.

Intercellular crosstalk of PSCs and therapy in pancreatic diseases. Fibrosis is a dynamic pathological process involving multiple cells. There are different intercellular interactions in different diseases including pancreatitis, pancreatic cancer and diabetes. Paracrine signals from parenchymal and immune cells such as PACs, PCCs, islet cells, and macrophages can directly or indirectly modulate PSCs and participate in fibrosis. Nanomedicine and traditional therapy modulate intercellular crosstalk of PSCs and promote fibrosis regression.
2. The relationship between PSCs and pancreatic diseases
2.1. The role of PSCs in pancreatic fibrosis
In 1998, Bachem et al.20 first isolated pancreatic stromal cells from human and rat pancreatic stroma that specifically express ECM components such as collagen types I (Col I), Col III and fibronectin (FN). These cells are named PSCs for their similarity to HSCs, which are enriched in vitamin A lipid droplets. PSCs are the stromal cells and account for about 4%–7% of the total number of pancreatic cells, mainly around the base of pancreatic acinar cells20. Pancreatic injury and inflammation activate PSCs, exhibiting myofibroblast-like phenotypes, such as expressing α-smooth muscle actin (α-SMA), producing ECM, and increasing proliferation19,21. PSC activation involves various growth factors, cytokines, and environmental stressors. For instance, treatment with platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β) can activate downstream signaling pathways, leading to PSC activation21. PF is caused by the excessive deposits of ECM and collagen fibers during repeated necrosis to repair damaged pancreatic tissue.
2.2. PSC activation and pancreatic diseases
PF is a pathological feature of pancreatitis, pancreatic cancer, and diabetes. Currently, the pathogenesis of PF remains unclear, but it is believed to result from the activation of PSCs. This activation causes excessive ECM production and rapid proliferation, migration, and proliferation of PSCs, ultimately reducing pancreatic parenchymal cells and promoting fibrosis in exocrine glands and islets22. In summary, activated PSCs lead to pancreatic fibrosis, resulting in the loss of pancreatic function and potentially promoting the spread of pancreatic cancer cells, thus accelerating the development of pancreatic disease.
Several studies have demonstrated that pancreatic fibrosis is a characteristic of pancreatitis and plays an active role in the development of CP. Recent evidence suggests that gene-associated pancreatitis or alcohol-induced CP can be characterized by progressive parenchymal fibrosis23. PSC activation is considered the core event in the development of pancreatic fibrosis. The activated PSCs further secrete more inflammatory factors, which regulate immune cells and fibrosis24. During tumorigenesis, stroma cells and pancreatic cancer cells will secret a variety of stimulating factors to activate PSCs. Then, active PSCs can create a suitable microenvironment and facilitate cancer progression25. There has been plenty of evidence that confirms the importance of PSCs in pancreatic cancer development. Jin et al.18 demonstrated that the cell supernatant of activated PSCs induced cancer cell proliferation and migration mediated by PDGF. Pothula et al.6 discovered that PSCs can induce tumor-promoting paracrine effects. Furthermore, CP poses a high risk for PC development, indicating the role of the fibrotic microenvironment in PC progression. Different degrees of fibrosis exist in the islet areas in diabetes patients and animal models26. Changes in the local microenvironment of the islet, high glucose and hyperinsulinemic, oxidative stress and inflammatory factors can induce PSC activation27. Activated PSCs secreted various products, interleukin 1 (IL-1), IL-6 and tumor necrosis factor-α (TNF-α), which lead to local inflammatory response in islets, induce apoptosis of β cells and promote diabetes progression7. Therefore, PSC activation may be a central link between islet fibrosis and islet β-cell dysfunction.
3. Intercellular crosstalk of PSCs and therapeutic strategies in pancreatitis
Pancreatitis is a common gastrointestinal disorder that, depending on its progression, can be categorized as either acute pancreatitis (AP) or chronic pancreatitis (CP). It is generally accepted that there is a continuum of AP, recurrent AP and CP4. About 20% of patients who receive an initial diagnosis of AP later have a relapse, and about 35% of relapsed AP cases progress to CP28. CP is a disorder featured by recurrent inflammation of the pancreas, resulting in the development of fibroinflammatory histology. Pathologic changes include acinar atrophy, chronic inflammation, extensive interstitial fibrosis, and exocrine and endocrine dysfunction29. Irreversible fibrosis, which has a profound effect on the physiological function of the pancreas, is one of the most prominent pathological features of CP.
3.1. Intercellular crosstalk of PSCs in chronic pancreatitis
As the pancreatic-specific mesenchymal stromal cells, PSCs tend to aggregate in the vicinity of blood vessels and conduits within pancreatic tissue and the basal portion of glandular follicles. PSCs constitute only approximately 4% of the total number of pancreatic cells when quiescent, and their cytoplasm is entirely of vitamin A lipid droplets20. The presence of desmin (+), glial fibrillary acidic protein (GFAP) (+), and the lack of α-SMA (‒) serve as indicators for the cells30. In response to pancreatic tissue injury and stress, PSCs undergo a process of activation and transition into a myofibroblast phenotype. This change is marked by the vanishing of lipid droplets in the cytoplasm containing vitamin A and the activation of α-SMA expression. Additionally, activated PSCs exhibit an enhanced capacity for ECM synthesis and secretion of various cytokines31. The development of single-cell RNA sequencing technology has facilitated the discovery of more subtypes of fibroblasts. Yang et al.11 showcased the diversity of PSCs in CP and identified a specific group of PSCs that express the very low-density lipoprotein receptor (VLDLR) and have a heightened lipid metabolism. These cells have the potential to facilitate immunological responses and stimulate PF. Further elucidation is required to determine if distinct subcategories of PSCs are present at various phases of pancreatitis, as well as the governing mechanisms that ensue upon the activation of specific fibroblast subtypes.
The activation of PSCs is essential for developing a pathological microenvironment characterized by excessive fibrous extracellular matrix. Various factors, including cytokines, non-coding RNAs, oxidative stress, hyperglycemia, and ion channel signaling trigger PSC activation32. When the PACs are exposed to harmful stimuli, the macrophages and neutrophils become active and release inflammatory cytokines such as TGF-β, TNF-α, IL-1/6, and PDGF33,34. Inflammatory cytokines can activate PSCs, which in turn secrete additional inflammatory factors such as connective tissue growth factor (CTGF), chemotactic protein 1, IL-1/6, PDGF, C-X3-C Motif Chemokine Ligand 1 (CX3CL1), and endothelin-1 (ET-1)35. It suggests that PSCs can induce their activation or the activation of neighbouring cells, hence facilitating the progression of pancreatic fibrosis. When the pancreas is exposed to endogenous or exogenous stimuli, abnormal activation of trypsinogen in acinar cells leads to acinar cell necrosis, necrotic acinar cells release damage-associated molecular patterns (DAMPs), which promote PSC activation and macrophage infiltration and polarization. Polarized macrophages secrete inflammatory cytokines to activate PSCs further36. Activated PSCs secrete pro-fibrotic cytokines, which eventually induce massive extracellular matrix deposition, contributing to the formation of pancreatic fibrosis (Fig. 2).
Figure 2.

Intercellular crosstalk of PSCs in chronic pancreatitis. Injured PACs release DAMPs, which promote the activation of PSCs and the infiltration and polarisation of macrophages. Figure created with Biorender.com.
3.1.1. The injury of pancreatic acinar cells triggers PSC activation
PACs are the predominant cells found in the pancreas. Damage to these cells is the primary cause of pancreatitis and fibrosis. Research has shown that crucial elements involved in the stimulation of PSCs, such as TGF-β and CTGF, were elevated in acinar cells located in regions of PF in individuals with CP37,38. The necrosis of PACs is ultimately caused by the abnormal activation of trypsinogen, which is induced by either endogenous or external stimuli. The release of DAMPs from PACs enhances the activation of PSCs and the infiltration and polarisation of macrophages, thus enhancing pancreatic fibrosis39,40. PACs modulate the microenvironment and recruit immune cells to influence PSC activation and play a subsidiary role in fibrosis. After co-cultured with PSCs, pancreatic acinar cells exhibited significant morphological changes and became insensitive to post-stimulation secretion. Co-culture promoted the migratory activity and expression of ECM protein in PSCs41. In their study, Wang et al.42 discovered that PACs regulate PSCs by producing exosomal miR-130a-3p and targeting peroxisome proliferator-activated receptor gamma (PPAR-γ). The authors observed that suppressing miR-130a-3p resulted in a substantial amelioration of PF. Meanwhile, The PAC-derived trypsinogen serves as a means of interaction between PACs and PSCs. It can be internalized by PSCs and has an impact on the activation of PSCs and the progression of CP43. PSCs underwent apoptosis after phagocytosis of necrotic acinar cells, whereas surviving PSCs were activated and continued to proliferate6. Recently, Wang et al.44 have elucidated the intricate mechanism through which PACs activate PSCs during the process of pancreatic fibrogenesis in CP. A significant increase of sphingosine kinase 1 (SPHK1) and sphingosine 1-phosphate (S1P) was observed in acinar cells in animal models of CP caused by cerulein injection or pancreatic duct ligation. PAC-derived SPHK1 catalyzes the conversion of sphingosine to S1P, which subsequently binds to S1PR2 receptors located on the surface of PSCs. This interaction promotes the activation of PSCs by inducing autophagic flux via the AMPK-mTOR pathway. The deposition of ECM further contributes to pancreatic fibrosis and local hypoxia, exacerbating PAC injury. Consequently, crosstalk is established between PAC and PSCs mediated by the hypoxia/SPHK1/S1P pathway, playing a pivotal role in both the onset and the progression of CP.
3.1.2. Immune cells promote PSC activation by releasing inflammatory factors
In addition to PACs and PSCs, immune cells such as macrophages, lymphocytes, and neutrophils are also involved in the progression of pancreatic fibrosis. Macrophages have garnered significant attention among these immune cell types due to their pivotal involvement. Macrophages, derived from monocytes, can be stimulated and polarized into specific subtypes to exert targeted effects. These special monocytes are crucial in inflammatory, immunological and oncological processes45,46. Typically, M1 macrophages primarily participate in the immune response, engaging in phagocytosis and promoting inflammation development. Conversely, M2 macrophages are mainly associated with suppressing inflammation while facilitating wound healing and fibrosis47. In physiological conditions, there are relatively few resident macrophages in the pancreas. Following the fragmentation of damaged local PACs, chemokines are produced, attracting monocytes that traverse the vascular endothelium and differentiate into macrophages within the injured tissue18. Various models of chronic pancreatitis have demonstrated an increase in pancreatic macrophages. The initial stages of chronic pancreatitis are characterized by local inflammation, and M1 macrophages activate PSCs via secreting TNF-α and IL-648. Xue et al.49 explored the correlation between macrophages and PF. Co-culturing normal mouse bone marrow-derived macrophages (BMDM) with isolated PSCs from CP mice revealed heightened secretion of IL-4, IL-5, TGF-β, IL-10, IL-13, and other cytokines associated with Th2 cells. The alteration in M1 and M2 markers signifies the ability of activated PSCs to induce polarization of macrophages towards the M2 phenotype. Following the blockade of IL-4/IL-13, there was a notable reduction in the expression levels of α-SMA, which significantly alleviated PF49. These essays emphasize the pivotal function of macrophages in promoting the advancement of PF. The communication between macrophages and PSCs within the local microenvironment potentially drives a vicious cycle of CP and fibrosis.
Neutrophil granulocytes are frequently considered as a reference marker for pancreatic inflammation50. Neutrophils release reactive oxygen species (ROS), proteases, and antimicrobial peptides to eliminate infections. Neutrophils are the primary generator of reactive ROS during pancreatitis51. They can cause oxidative damage to acinar cells and increase trypsinogen activation50,51. The secretion of proteases facilitates acinar cell dissociation and tissue degradation. These findings suggest a clear relationship between neutrophils and disease severity52. It was verified by using anti-neutrophils serum to deplete neutrophils, which decreased pancreatic damage and protease activation51.
3.2. Therapeutic strategies related to crosstalk of PSCs in pancreatitis
Multiple cells interact with PSCs to promote activation and influence the development of CP. Regulating the communication between PSCs and other cells in the pancreas may be an effective therapeutic strategy (Fig. 3).
Figure 3.

Antifibrotic therapy of nanomedicine in chronic pancreatitis. Nanomedicine therapeutic strategies in CP include targeting PSC, macrophage, cytokine and ECM. Figure created with Biorender.com.
3.2.1. Targeting pancreatic acinar cells and PSCs for CP
The precise mechanisms underlying acinar cell injury contributing to CP and pancreatic fibrosis remain unclear. Meanwhile, specific pharmacological interventions for treatment still need to be improved. Dysregulation of various signaling proteins, transcription factors, kinases, and cytokines secreted by acinar cells presents potential targets for intervention in pancreatic fibrosis. Activating the SPHK1/S1P pathway in PACs triggers autophagy and activates PSCs, thereby promoting fibrogenesis of chronic pancreatitis. SPHK1 and S1PR2 represent promising therapeutic targets for clinical treatment and inhibitors targeting SPHK1 and S1PR2 effectively mitigate CP and fibrosis progression44,53. Parathyroid hormone-related protein (PTHrP), released by PACs, influences the function of acinar cells by regulating the production of inflammatory cytokines and stimulating PSCs in paracrine. Several animal models of CP exhibit elevated levels of PTHrP. The disruption of the PTHrP gene in PACs protects by decreasing histological harm and fibrosis54. Vacuole membrane protein 1 (VMP1), an endocytoplasmic reticulum (ER) membrane protein, has been found to have a vital function in autophagosome production. In particular, VMP1 is down-regulated in AP and CP. The absence of pancreatic VMP1 leads to a decrease in the ability to break down cellular components through autophagy, causing stress in the endoplasmic reticulum and triggering the activation of the nuclear factor, erythroid 2 like 2 (NFE2L2/NRF2) pathway. The elimination of VMP1 in the acinar cells of the pancreas induces inflammation, the transformation of acinar cells into ductal cells, and the development of fibrosis in mice55.
The research group has designed and synthesised lipid nanoparticles termed AT-CC, loaded with dual drugs ATRA and ammonium tetrathiomolybdate (TM). These nanoparticles are decorated with collagen-binding peptide (CBP) and collagenase I. This enables effective transport of ATRA and TM into the pancreas. The delivered ATRA efficiently decreases excessive collagen synthesis by preventing the activation of PSCs. TM inhibits lysyl oxidase activation, resulting in a decrease in collagen cross-linking. The synergistic action of ATRA and TM suppresses PSC activation and collagen synthesis and promotes the discovery of PACs function. These results demonstrate that AT-CC is a secure and effective collagen-targeting drug delivery technology for alleviating PF. These innovative nanoparticles represent a novel approach to targeted therapy for pancreatic fibrosis, demonstrating improved precision and efficacy56.
3.2.2. Targeting macrophages and PSCs for CP
Inhibition of the interaction between macrophages and PSCs or reduced macrophage infiltration within the pancreatic tissue has demonstrated improved therapeutic outcomes. Targeting macrophages presents a promising avenue for the treatment of CP and fibrosis. Dasatinib, a second-generation multiple tyrosine kinase receptor inhibitor, was found to inhibit the promotion of M2 macrophage polarization by PSCs in a study investigating their crosstalk. Zeng et al.57 demonstrated that dasatinib led to an amelioration of fibrosis severity in cerulein-induced CP mice models and exerted notable antifibrotic and anti-inflammatory effects through the glycogen synthase kinase-3β/TKs/β-linker pathway. Sulindac, a non-steroidal anti-inflammatory medicine, effectively decreases the levels of pro-inflammatory cytokines, specifically TNF-α and monocyte chemotactic protein-1 (MCP-1), and the production of TGF-α and PDGF-β in chronic pancreatic tissue. Furthermore, sulindac demonstrates notable effectiveness in diminishing the intensity of chronic pancreatitis by decreasing the loss of acinar cells, infiltration of inflammatory cells, and fibrosis of the stroma58. Berberine (BR), as an AMPK activator, inhibits TGF-β signaling and modulates M2 macrophage polarisation. In the animals with CP, berberine significantly reduced PSC activation, inflammatory cell infiltration and collagen deposition59. Isoliquiritigenin (ILG), a chalcone-type flavonoid isolated from liquorice roots, has recently been reported to attenuate inflammation and PF by inhibiting PSC activation and macrophage infiltration into the pancreas60.
In addition to the promising therapeutic agents mentioned above, Khurana et al.61 found that cerium oxide (NC) nanoparticles effectively inhibited the activity of pro-inflammatory cytokines and chemokines via disrupting macrophage signaling. In addition, NC showed a remarkable ability to attenuate fibrogenesis through inhibition of TGF-β signaling, endoplasmic reticulum stress and epithelial-to-mesenchymal transition. A nanotechnology-based CO-bound haemoglobin vesicle (CO-HbV) was developed by Nagao et al.62 Treatment with CO-HbV resulted in polarization of macrophages towards an M2-like phenotype and inhibition of pancreatic neutrophil infiltration. Cell membrane coating technology opened an avenue for nanoparticles. Wang et al.63 developed a biohybrid nanosystem to treat CP. It comprises a polymer nanoparticle loaded with Somatostatin (SST) and a macrophage membrane surface. The nanoparticle reduced PF and showcased the benefits of combining the synthetic polymer with biological membranes in the development of nanoplatforms for sophisticated and efficient peptide delivery. We have summarized more detailed information on examples of nanomedicine treatment of pancreatitis and fibrosis in Table 1 15,56,61, 62, 63, 64.
Table 1.
Therapeutic strategies related to intercellular crosstalk of PSCs in CP.
| Nanomedicine | Targeting therapeutic agent | Mechanisms of action | Pancreatitis model | Preclinical/clinical | Ref. |
|---|---|---|---|---|---|
| LA-PC | All-trans retinoic acid (ATRA) | Inhibiting of PSCs activation by ATRA and reducing the level of PDGF-BB by a down-regulated ERK pathway. | Mice with CP induced by caerulein | Preclinical | 17 |
| AT-CC | ATRA and ammonium tetrathiomolybdate (TM) | Inhibition of PSCs activation and reduction of collagen cross-linking by lipid nanoparticles loaded with ATRA and TM. | Mice with CP induced by caerulein | Preclinical | 56 |
| NC | Cerium oxide | Nanoparticles of cerium oxide (NC) effectively suppressed inflammatory cytokines and chemokines through the disruption of macrophage signaling. | Mice with CP induced by caerulein and rats with CP induced by l-arginine | Preclinical | 61 |
| CO-HbV | Carbon monoxide (CO) | CO-bound haemoglobin vesicles (CO-HbV) polarised macrophages and suppressed neutrophil invasion into the pancreas. | Mice with severe acute pancreatitis induced by CDE diet | Preclinical | 62 |
| MP-SST | Somatostatin (SST) peptide | The nanoparticles loaded with SST and camouflaged with macrophage membrane (MP-SST) resulted in the suppression of factors associated with CP and mitigated the histological damage of the pancreas. | Mice with CP induced by the diethyldithiocarbamate (DDC) | Preclinical | 63 |
| NY | Yttrium oxide | Yttrium oxide nanoparticles (NY) reduced pancreatic oxidative-nitrosative stress and inflammatory cytokines and chemokines resulting in the inhibition of fibrosis signaling. | Rats with CP induced by l-arginine | Preclinical | 64 |
4. Intercellular crosstalk of PSCs and therapeutic strategies in pancreatic cancer
Pancreatic cancer is a common and aggressive malignancy that affects the digestive system, ranking 14th and 4th among all malignant tumors in terms of new cases and deaths worldwide in 2020, respectively, according to GLOBOCAN statistics65. Pancreatic ductal adenocarcinoma (PDAC), arising from glandular epithelium, accounts for approximately 90% of pancreatic cancers. PDAC is one of the most stroma-rich cancers due to its unique interstitial fibrosis feature compared to other tumors66,67. The cancer cells are surrounded by an abundance of dense fibrous stromal components, with the stroma constituting around 80%–90% of the tumor mass. The ECM of pancreatic cancer hinders the adequate transportation of chemotherapeutic medicines to cancer cells and sustains their growth68. Moreover, this stromal mass comprises various cells, such as PSCs, fibroblasts, myofibroblasts, immune cells and vascular endothelial cells. PSCs are the most prominent component, which is up to 50% of its total mass, and perform a crucial role in the tumor microenvironment (TME) of PC68,69. Their continuous activity and active contact with tumor cells in the TME might enhance tumor growth and spread, leading to resistance to chemotherapy in PC through many signaling pathways. Tumor heterogeneity, interstitial fibrosis, and an immunosuppressive microenvironment collectively contribute to the elevated fatality rate observed in pancreatic cancer.
4.1. Intercellular crosstalk of PSCs in microenvironment
PSCs are the main cellular components responsible for developing the desmoplastic reaction in PC. During the development of tumors, these cells experience a gradual process of activation and secrete collagen fibers that surround and protect cancer cells. The presence of collagen in the surrounding tissue plays a vital role in the development of tumors, as cell interactions are essential at all stages of tumor formation, including the beginning, advancement, and spread to other parts of the body. Besides, multiple interactions between PSCs and PCCs, endothelial cell, neurons, other immune cells play different roles in pancreatic cancer (Fig. 4).
Figure 4.

Role of activated PSCs in pancreatic cancer. Activated PSCs produce abundant ECM proteins and remodel the ECM. PDGF, platelet-derived growth factor; CTGF, connective tissue growth factor; IL-4,-6,-8,-13, interleukin-1,-4,-6,-8,-13; bFGF, basal fibroblast growth factor; VEGF, vascular endothelial growth factor; CYR61, cysteine-rich angiogenic inducer 61; MMP-2, matrix metalloproteinase-2; CXCL12, C-X-C motif chemokine ligand 12; BGF, bone growth factor; BNGF, beta-nerve growth factor. Figure created with Biorender.com.
4.1.1. The interactions between PSCs and PCCs mediate the progression of cancer
There is strong evidence of an interaction between PSCs and PCCs, as shown in laboratory experiments involving co-cultures or exposure to conditioned media. PSCs display heightened proliferation, migration, and ECM formation when exposed to PCCs. Conversely, PCCs exhibit heightened cell division but decreased programmed cell death when exposed to secretions from PSCs70,71. Cancer cells stimulate the activation of PSCs by utilizing signaling pathways such as the phosphoinositide 3-kinase (PI3K)/Akt, the Janus kinase (JAK)-signal transducer and activator of transcription (STAT), and Hedgehog. Additionally, they employ inflammatory cytokines and ROS components. Furthermore, PCCs stimulate autophagy in PSCs, resulting in releasing alanine, which serves as a substitute carbon source for mitochondrial oxidative pathway utilization by cancer cells under nutrient-limited conditions71. Knockdown of autophagy proteins in PSC resulted in a decrease in ECM and IL-6 production. Additionally, when these PSCs were combined with PCCs and administered to nude mice, they formed smaller tumors and fewer metastases31. Moreover, PSCs have been shown to stimulate cancer cell migration or epithelial-mesenchymal transition (EMT). The study revealed that the presence of PSC in co-culture with PCC resulted in the suppression of epithelial cell markers (E-cadherin, β-catenin) and the stimulation of mesenchymal cell markers (Vimentin). It indicates that PSC plays a crucial role in the initiation of EMT in PCCs72. Activation of TGF-β, Notch pathway, and Wnt/β-Catenin pathway drives target gene transcription and up-regulates mesenchymal marker expression. The epithelial cells lose polarity and intercellular adhesion and are transformed into a mesenchymal phenotype73,74. By activating PSCs, an optimal tumor microenvironment is established that facilitates the proliferation of PCCs and ultimately contributes to PC progression.
4.1.2. The crosstalk of PSCs and immune cells promotes immunosuppressive TME
Tumor-associated macrophages (TAMs) The TME consists of a wide range of immune cells, such as macrophages, neutrophils, dendritic cells, natural killer cells, effector T cells, regulatory T cells, and B cells75. TAMs infiltrate tumors and are the predominant immune cells in regions densely populated by PSCs76. Multiple studies have shown that PSCs promote the attraction and specialization of monocytes into pro-tumor M2 macrophage subsets using diverse regulatory molecules. This process impairs effector T-cell responses and induces immunosuppression of the TME77,78. Mace et al.79 observed that cancer-associated fibroblast (CAF)-derived macrophage colony-stimulating factor 1 (M-CSF1), IL-6, and C–C motif chemokine ligand 2 (CCL2) play an essential role in attracting monocytes to PC and causing a subsequent rise in the M2/M1 macrophage ratio. The study also showed the importance of fibroblast activation protein (FAP), a marker for PSCs, in facilitating communication between PSCs and TAMs. It revealed that FAP played a role in the interaction between PSCs and TAMs primarily through the cleavage of type I collagen and the subsequent increase in macrophage adhesion. In turn, TAMs also modulate the activation and progression of PSCs. Confirmed that PSCs promote TAMs, M2 macrophages can also enhance EMT progress by secretion of factors including IL-6 and stromal cell-derived factor-1 (SDF-1) to stimulate PSC activation80. Activated M2-like macrophages contribute to ECM remodelling through the secretion of matrix metalloproteinases (MMPs), which negatively impact ECM. Tissue-resident macrophages, the predominant subset in TME, express prolactin receptors, and it has been reported that prolactin contributes to TME fibrosis81. Recent research has examined the impact of PSCs on TAMs, but the reciprocal influence of macrophages on PSCs has yet to be thoroughly explored and elucidated.
Other immune cells The nuclear factor-kappaB (NF-κB) activity in PSCs promotes tumor growth by increasing the expression of C-X-C chemokine ligand 12 (CXCL12), which prevents CD8+ T cells from infiltrating the tumor and killing cancer cells. Blocking CXCL12 in pancreatic tumor cells might promote antitumor immunity82. TGF-β promotes tumor immune evasion through activation of PSCs, and combined inhibition of TGF-β and PDL1 synergistically increases CD8+ T cell infiltration and cytotoxicity to exert antitumor immunity83. PSCs also trigger differentiation in myeloid-derived suppressor cells (MDSCs) by releasing inflammatory cytokines such as IL-16. PSCs stimulate the migration of MDSCs to pancreatic tumors by secreting IL-1679. IL-6 secretion by PSCs promotes immunosuppression, and PSCs treated with a heat shock protein 90 inhibitor reduce IL-6 secretion84. It has been reported that PSCs can activate mast cells, which produce IL-13 and tryptase, thereby stimulating the proliferation of cancer cells and PSCs85. The C-X-C motif chemokine receptor 2 (CXCR2) is expressed in leukocytes, including neutrophils, natural killer cells (NK cells), monocytes, macrophages, and T cells. Its function is to regulate the migration of neutrophils to inflammatory sites by binding to IL-886.
4.1.3. The crosstalk of PSCs and endothelial cells
Activated PSCs express a variety of proangiogenic factors, such as VEGF, bFGF, IL-8, PDGF, and periostin. Additionally, MMP-9 is produced by PSCs to degrade the basement membrane and aids in the development of blood vessels87. VEGF stimulates angiogenesis by enhancing endothelial cell viability, proliferation, and permeability88. Hepatocyte growth factor (HGF) produced by PSCs is the ligand for the c-Met expressed on cancer cells as well as endothelial cells. HGF/cellular mesenchymal-epithelial transition (c-MET) pathway plays a vital role in PSC-induced tube formation of endothelial cells89. Another study has shown that activated PSCs promote tumor cell proliferation and endothelial cell tube formation through the therapeutic miRNAs 199a-3p and 214-3p90.
4.1.4. The crosstalk of PSCs and neurons
PSCs may be involved in neural invasion as pancreatic cancer progresses. The study showed that PSCs can induce neuron outgrowth. It was demonstrated by incubating dorsal root ganglia with the conditioned medium collected from PSCs derived from human pancreatic cancer91. Li et al.92 reported that activated PSCs promote neurite outgrowth towards pancreatic cancer cells and facilitate neural invasion by cancer cells. More recently, it has been demonstrated that PSCs cause pain in pancreatic cancer by stimulating the release of neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and nerve growth factor (BGF), which causes the dorsal root ganglia to secrete pain factors93.
4.2. Therapeutic strategies related to crosstalk of PSCs in pancreatic cancer
The phenomenon of cancer-associated desmoplasia and fibrotic response is a multifaceted and intricate entity that has extensive ramifications. The interconnection of various cells in the TME is crucial for the progression and spread of solid pancreatic cancers. Introducing intercellular crosstalk as an intervention provides a new approach to hinder the advancement of PCs effectively (Fig. 5).
Figure 5.

Nanomedicine therapeutic strategies in pancreatic cancer. Nanomedicine therapeutic strategies in pancreatic cancer including (1) Targeting ECM and promoting ECM remodeling; (2) Targeting fibrotic mechanism; (3) Targeting fibrotic mesenchymal components. Figure created with Biorender.com.
4.2.1. Targeting PCCs and PSCs for PC
Due to the pivotal role of PSCs in the development of PC, medicines that modulate their activation hold great potential as therapeutic candidates. Several anticancer agents have been shown to influence PSC function and ECM production, thereby suppressing tumor growth. In the context of PC, Extensive research has been conducted on medication delivery methods that utilize polymers and nanocarriers. ATRA is the biologically active form of vitamin A. It controls the state of PSC inactivity by controlling the transcription of specific genes when it binds to corresponding nuclear receptors94,95. The nanosystem Au@PP/RA/siHSP47, via AuNP nanocarriers, simultaneously delivered all-trans retinoic acid, which induces the restoration of PSC to the resting state, and siRNA targeting heat shock protein. The drug delivery and penetration were significantly improved in the model. The progression of invasive pancreatic tumors was inhibited considerably96. A composite of magnetic nanoparticles, co-modified with gemcitabine (GEM) and pH (low) insertion peptide (pHLIP), was created and labeled as GEM-MNP-pHLIP. The intelligent nanocarrier consisted of a pH-responsive transmembrane unit and a controlled release unit, in addition to Fe3O4 nanoparticles that enable the tracking of targeted drug delivery using magnetic resonance imaging (MRI). In the animal trials, we administered metformin (MET) through intraperitoneal injection to deplete the thick stromal barrier of PDAC. It was done before injecting the aforementioned nano agents in order to enhance the efficient delivery of GEM97.
Gemcitabine resistance in cancer cells can be induced by PSCs through both direct and indirect coculture systems. The secretion of FN by PSCs within the ECM has been identified as a pivotal contributor to gemcitabine resistance via extracellular signal-related kinases 1 and 2 (ERK1/2) activation98. The arsenic trioxide (ATO)-loaded nanoparticles were synthesized through self-assembly utilizing poly(d,l-lactide) and poly(ethylene glycol) (PEG-PDLLA) and subsequently modified with the single-chain antibody of FAP-α (scAbFAP-α) to form scAb-ATO-NPs. ATO nanoparticles significantly prevent the activation of the PI3K/AKT/galectin-1 pathway, resulting in a decrease in stromal secretion and a reduction in the tumor-promoting potential of PSCs. In addition, this method increases the responsiveness of cancer cells to GEM, thereby offering a new viewpoint on the treatment of pancreatic cancer99. The integration of photothermal therapy (PTT) with the purpose of depleting the stroma locally, increasing the permeability of GEM, and synthesizing C-G NPs has shown significant promise in suppressing the activation of PSCs, deposition of collagen fibers, and production of TGF-β. These findings offer vital insights into the development of resistance to gemcitabine in PC and propose promising therapeutic options to inhibit the progression of PC100. Consequently, these results hold significant promise for future clinical translation. A highly efficient MMP-9-cleavable linker was developed to selectively remove a PEG shield from a polymeric nanocarrier (Magh@PNPs-PEG-RegaCP-PEG) based on PLGA-b-PEG. This linker was evaluated in the mouse model of pancreatic cancer, which is clinically relevant. The heightened tumor-specific presence and functionality of MMP-9 can be utilized to provide an MMP-9-activatable nanoparticle, thereby diminishing liver metastasis and significantly reducing the average size of colonies101. MMPs can be utilised as ECM-based targets or stimulants for the treatment of PDAC in order to optimize therapeutic strategies.
4.2.2. Potential tumor associated macrophage (TAM) targeting strategies
Typically, these therapy strategies focus on addressing different aspects of TAMs, such as their ability to survive, change their features, attract other cells, engulf foreign particles, and promote the growth of new blood vessels. Recently, the utilization of nanotechnology has been evaluated as a strategy for explicitly targeting macrophage polarization. Numerous types of nanoparticles have been assessed for their ability to target TAMs102,103. Iron oxides have shown the ability to transform M2 into the M1 macrophages, while also increasing the production of ROS and triggering death in cancer cells. Zhao et al.104 fabricated tumor-derived antigenic microparticles (T-MPs) incorporating nano-iron oxide. The surface of T-MPs was modified by including adjuvant CPG to develop a Fe3O4/T-MPs-CpG/Lipo vaccine. Following the implementation of combination vaccination in TME, a phenomenon called repolarization was observed, wherein M2-like macrophages underwent a conversion into an M1-like phenotype. Another approach to regulate the polarization of macrophages in the TME of PC is to interfere with the galectin-9/dectin axis. Previous studies have demonstrated that this axis plays a role in promoting the conversion of macrophages into M2. The delivery mechanism comprises exosomes obtained from bone marrow mesenchymal stem cells (BM-MSCs), which are filled with galectin-9 siRNA and coated with an oxaliplatin (OXA) prodrug. The purpose of this alteration is to stimulate immunogenic cell death. Utilizing biomaterials, particularly BM-MSC exosomes, can significantly improve the effectiveness of targeting tumors, leading to a higher concentration of drugs at the tumor location. The siRNA-EXO-OXA (iEXO-OXA) combination therapy elicits anti-tumor immunity by polarizing tumor-suppressive macrophages, attracting cytotoxic T cells, and suppressing Tregs105. It has demonstrated significant therapeutic efficacy in cancer treatment. Finally, we have summarized more detailed information on examples of nanomedicine for the treatment of pancreatic cancer in Table 2 83,96,97,99, 100, 101,104,105.
Table 2.
Therapeutic strategies related to intercellular crosstalk of PSCs in PC.
| Nanomedicine | Targeting therapeutic agent | Mechanism of action | Combination therapy | Cancer model | Preclinical/clinical | Ref. |
|---|---|---|---|---|---|---|
| LYiClustersiPD-L1 | TGF-β receptor inhibitors (LY2157299) and siRNA targeting PD-L1 (siPD-L1) | LY2157299 can effectively inhibit the activation of PSCs and result in a reduction in type I collagen. SiPD-L1 silenced PD-L1 gene expression in tumor cells, increased tumor infiltrating CD8+ T cells and provoked antitumor immunity. | Immunotherapy | A subcutaneous pancreatic tumor model induced by Panc02 cells | Preclinical | 83 |
| Au@PP/RA/siHSP47 | ATRA and siHSP | By utilizing AuNP nanocarriers, we can transport both ATRA, which triggers the restoration of PSC to its resting state, and siRNA that specifically targets heat shock protein. | N/A | Panc-1/PSC co-inoculated subcutaneous xenograft tumor model | Preclinical | 96 |
| GEM-MNP-pHLIP | GEM | The composite magnetic nanoparticles, consisting of pH-responsive transmembrane unit and controlled release unit, were formed by combining GEM and pH (low) insertion peptide (pHLIP) with Fe3O4 nanoparticles (MNPs). | N/A | A tumor subcutaneous xenograft tumor model | Preclinical | 97 |
| scAb-ATO-NPs | Arsenic trioxide (ATO) | Targeting the AP4/AKT/PI3K galectin-1 pathway using ATO nanoparticles effectively inhibits its activation, leading to a reduction in stromal secretion and attenuation of the tumor-promoting capability of PSCs. | N/A | MIA Panc-2/PSC co-inoculated subcutaneous xenograft tumor model | Preclinical | 99 |
| C-G NPs | Gemcitabine | C-G nanoparticles (NPs) are created by incorporating acid-responsive photothermal molecules with gemcitabine. These NPs are used to study the combined effects of chemophotothermal therapy and the interaction between photothermal therapy (PTT) and the TGF-β pathway in pancreatic ductal adenocarcinoma. | Photothermal therapy (PTT) | Metastatic xenograft PDAC tumor mouse model | Preclinical | 100 |
| Magh@PNPs-PEG-RegaCP-PEG | MMP-9 | A highly efficient MMP-9-cleavable linker was developed to selectively remove a PEG shield from a polymeric nanocarrier (Magh@PNPs-PEG-RegaCP-PEG) based on PLGA-b-PEG. This linker was tested in a mouse model of pancreatic cancer, which is therapeutically relevant. | N/A | Liver metastasis induced by injection of the well-established L-CI.5s experimental liver metastasis model | Preclinical | 101 |
| Fe3O4/T-MPs-CpG/Lipo | Tumor-derived antigenic microparticles (T-MPs) | Reversal of infiltrating TAMs to a tumor-suppressive M1 phenotype and modulating the tumor immunosuppressive network. | Immunotherapy | Tumor mouse model induced by injection of cells | Preclinical | 104 |
| iEXO-OXA | OXA | The delivery mechanism comprises exosomes obtained from bone marrow mesenchymal stem cells (BM-MSCs), which are filled with siRNA that specifically targets galectin-9 and are externally modified with oxaliplatin (OXA). The nanoparticles induce the formation of anti-tumor immunity by promoting the polarization of tumor-suppressive macrophages. | Immunotherapy | Orthotopic PANC-02 pancreatic tumor models | Preclinical | 105 |
5. Intercellular crosstalk of PSCs and therapeutic strategies in diabetes
Diabetes is a metabolic disorder characterized by chronic hyperglycaemia and glucose intolerance due to impaired insulin action or deficiency. Worldwide, 537 million adults (aged 20–79 years) are living with diabetes, according to the IDF Diabetes Atlas 2021. These figures are projected to rise to 643 million by 2030 and 783 million by 2045. Diabetes causes life-threatening, disabling and costly complications and reduces life expectancy, making it one of the most severe and common chronic diseases of our time106,107. In related studies, fibrosis is another essential pathogenic factor in pancreatic islet dysfunction. β-Cell dysfunction plays a critical role in the pathogenesis of diabetes, with multiple factors contributing to the loss of β-cell mass and function. These factors include glucose lipotoxicity, chronic inflammation, endoplasmic reticulum stress and epigenetic modifications108. Pathological studies have revealed varying degrees of fibrosis in pancreatic islets and peri-islet regions in both diabetes patients and animal models, alongside exocrine pancreatic fibrosis. Immunohistochemical staining has detected ECM deposits, primarily composed of Col I, Col III, and FN, and have been detected in the fibrotic areas of DM pancreatic islets. In contrast to fibrosis of pancreatic exocrine tissue, pancreatic fibrosis in diabetes is mainly confined to the pancreatic islets, which suggests that islet fibrosis in diabetes is associated with different activation pathways in pancreatic exocrine disease109,110. However, the specific pathogenesis needs to be further clarified.
5.1. Intercellular crosstalk between PSCs and β-cells
The association between PSCs and diabetes seems to be reciprocal. Within the realm of diabetes, elevated blood sugar levels, known as hyperglycemia, can initiate the activation of PSCs through the renin-angiotensin system (RAS)111. Matrix deposition led to the development of islet-specific fibrosis in mice models. In pancreatic islets, the cells responsible for insulin secretion experience higher levels of glucose and insulin in cases of diabetes, unlike the rest of the pancreatic tissue. The simultaneous occurrence of ERK1/2 phosphorylation activation and proliferation of PSCs can result in a decrease in insulin secretion and an elevation in β-cell mortality caused by TNF-α, INF-γ, and IL-1β112 (Fig. 6). Significantly, directing efforts towards the RAS system shows potential as a viable treatment strategy for treating diabetes.
Figure 6.

Intercellular crosstalk between PSCs and β-cells in islet fibrosis. High-glucose levels, insulin and angiotensin II (Ang II), and a release of pro-inflammatory cytokines induce PSC activation. Activated PSCs cause decrease of insulin secretion and increase of β-cell death by TNFα、INFγ、IL1β. Pancreatic fibrosis arises with extracellular matrix production. Figure created with Biorender.com.
5.1.1. Hyperglycemia contributes to the activation of PSCs and islet fibrosis
Diabetes is a condition characterized by persistent high levels of blood glucose. The presence of hyperglycemia and hyperinsulinemia triggers the RAS, which is renowned for its conventional role in regulating blood pressure, fluid balance, and electrolyte homeostasis through its effects on vascular smooth muscle cells and aldosterone secretion. Angiotensin II (Ang II) acts as the main active peptide in the signaling pathway of the RAS, mainly exerting its effects through Ang II receptor type 1 (AT1R). Furthermore, Ang II functions as a growth factor that promotes the multiplication of cells and the inflammation of tissues, which is essential in the development of fibrosis and impaired function in different organs associated with diseases112,113. In individuals with diabetes, increased glucose levels trigger the synthesis of Ang II and the activation of AT1R, resulting in an increase in the production of TGF-β through the interaction of Ang II with AT1R. TGF-β is a pivotal mediator in the activation of PSCs. Islet fibrosis is further exacerbated by the activation and proliferation of PSCs and the increased production of ECM proteins, which is produced by the signaling of ATII and TGF114,115. Remarkably, in the OLETF rat model, the activation of PSCs and the development of fibrosis are mainly confined to the islets, even though hyperglycemic circumstances impact the entire pancreas. Glucose and insulin can stimulate the proliferation of PSCs and the phosphorylation of ERK 1/2. In turn, it leads to the production of the CTGF gene and the activation of PSCs116. Consequently, hyperglycemia and hyperinsulinemia are critical mitogenic factors that drive PSC activation and proliferation, potentially elucidating islet-specific fibrosis in diabetes.
Advanced glycation end products (AGEs), metabolites involved in diabetes, also activate PSCs. AGEs are formed through non-enzymatic glycation and oxidation of proteins, lipids, and glucose117,118. The production of AGEs is significantly augmented in diabetic patients due to elevated blood glucose levels, playing crucial roles in the development and progression of vascular complications related to diabetes119,120. Studies have demonstrated that AGEs can induce endothelial-to-mesenchymal transition (EndMT) in islet endothelial cells and contribute to islet fibrosis in diabetic mice. It suggests that EndMT induced by AGEs may contribute to the pathogenesis of islet fibrosis associated with diabetes121. However, the comprehensive molecular mechanisms underlying this phenomenon during the diabetic condition remain unclear.
5.1.2. Activated PSCs induced β-cells injury and diabetes progression
It has been suggested that activated PSCs induce apoptosis of pancreatic islet β-cells. Activated PSCs secrete various products, including IL-1, IL-6, TNF-α, MCP-1, and galectin-1 (Gal-1), leading to local inflammatory reactions in pancreatic islets and promoting the generation of ROS. Consequently, this process induces apoptosis of islet β-cells and accelerates the progression of diabetes7. Among these factors, IL-1β plays a critical role in inhibiting β-cell apoptosis. It can upregulate Fas expression in β-cells along with interferon (IFN) γ and TNF-α; however, IL-1β exhibits the most significant impact. Furthermore, IL-1β binds to the IL-1 receptor on β-cell membranes to activate the NF-κB signaling pathway, mitogen-activated protein kinases (MAP)/stress-activated protein kinases (SAPK) pathway, and protein kinase C (PKC) pathway. In contrast, IFN-γ binds to the IFN-γ receptor on β-cell membranes, triggering signaling and activation of transcription factor-1 (STAT-1), which regulates caspase expression. TNF-α induces apoptosis in a time- and dose-dependent manner in β-cells122, 123, 124. Additionally, islets co-cultured with PSCs in a hypoxia-conditioned medium exhibited a reduction in cellular viability and an elevation in apoptotic events. PSCs are activated in hypoxia and promote β-cell apoptosis and death through exosomal miR-23a-3p. The addition of N-acetyl-l-cysteine, an antioxidant, mitigated the activation of PSCs in islets125,126. In conclusion, these studies support that PSC activation within the islets increases β-cell death and contributes to the progression of diabetes.
5.2. Therapeutic strategies related to fibrosis in diabetes
The therapies of diabetes-associated fibrosis include traditional drugs and nanomedicine. Some antidiabetic agents on the market have been reported to have anti-fibrotic effects, but more explicit evidence of efficacy is still needed. Meanwhile, the discovery of new targets provides another way for anti-fibrotic therapy in diabetes (Fig. 7).
Figure 7.

Diabetes-associated fibrosis and therapeutic strategies. In addition to islet, fibrosis has a role in the pathophysiology of diabetic nephropathy, neuropathy, cardiomyopathy, and retinopathy. The therapies of diabetes-associated fibrosis include traditional drugs and nanomedicine. Figure created with Biorender.com.
5.2.1. Antidiabetic therapy
Some antidiabetic agents have demonstrated efficacy in lowering blood sugar levels and exhibiting therapeutic potential as antifibrotic agents in diabetes. (1) Administration of the glucagon like peptide-1 (GLP-1) analogue exendin-4 has demonstrated the ability to inhibit the onset of diabetes in a rat model of type 2 diabetes following partial pancreas removal. This treatment has led to a significant 40% increase in the mass of β-cells127. In addition, exendin-4 demonstrates anti-inflammatory properties and functions as an antifibrotic agent in mesangial cells. Exendin-4 efficiently decreases the synthesis of Ang II and TGF-β1 by decreasing the creation of ROS while not influencing the phosphorylation of ERK128,129. (2) Sodium-glucose cotransporter 2 (SGLT2) inhibitors are a new oral medication for lowering blood sugar levels. They work independently of insulin and successfully reduce blood glucose levels by increasing the amount of glucose excreted in urine130. The precise mechanisms behind the protective effects of SGLT2 inhibitors luseogliflozin on islet β-cells remain incompletely comprehended. Mice treated with luseogliflozin showed a notable decrease in the expression levels of genes associated with fibrosis, such as TGF-β, FN, Col I, and Col III131. These compelling findings suggest that SGLT2 inhibitors are promising therapeutic agents for safeguarding β-cells against islet fibrosis.
Long-term diabetes causes several significant organ damage and dysfunctions, as seen in both diabetic individuals and animal models. The development of fibrosis, which primarily involves the deposition of ECM, is one pathological reaction to tissue injury. It has been proposed that fibrosis has a role in the pathophysiology of diabetic nephropathy, neuropathy, cardiomyopathy, and retinopathy. Individuals who have diabetes for an extended period frequently have large fibrotic lesions in their liver, kidney, heart, etc. Currently, there are no approved antifibrotic treatments available. Insulin replacement therapy is an important antiglycemic agent approach to diabetes132. Due to gastrointestinal effects, insulin and other macromolecular agents must be injected subcutaneously, which can be inconvenient and painful, leading to poor patient compliance133. Nanotechnology is being applied to improve insulin replacement therapy's simplicity, efficacy and safety. Hunt et al.134 recently applied a silver sulfide quantum dots platform to insulin with a pH- and enzyme-sensitive encapsulating polymer. The formulation is insoluble in acidic environments and sensitive to glucosidase enzymes that trigger insulin release. As a responsive oral insulin nanoformulation, it shows the potential to control blood glucose without hypoglycemia orally. Cheng et al.135 developed insulin-loaded nanoparticles decorated with folic acid to target intestinal cells. Folate is transported through a proton-coupled folate transporter (PCFT), which was upregulated in the intestinal epithelial cells of diabetic rats. Folate-modified nanoparticles loaded with insulin have a higher oral insulin bioavailability than subcutaneous insulin injection.
5.2.2. Antifibrotic therapy
RAS blockers, including angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs), have shown promise in efficiently avoiding or reducing islet fibrosis in persons with high blood sugar levels by directly inhibiting PSCs. Ramipril has demonstrated efficacy in mitigating pancreatic islet fibrosis over an extended period of treatment136. Candesartan and ramipril have proven effective in attenuating the increased expression of TGF-β following elevated levels of Ang II. The significant elevation observed in CTGF and type IV collagen proteins was effectively reduced by candesartan and ramipril111. Furthermore, troglitazone, a PPAR-γ agonist, has demonstrated inhibitory effects on the profibrogenic activity of PSCs137. Taurine and tempol are both potent antioxidants with notable antifibrotic properties. They reduced the activation of PSCs and the development of fibrosis in the islets of OLETF rats in response to high glucose138,139. Prior studies have shown that mitogen-activated protein kinase inhibitors can effectively decrease the phosphorylation of ERK 1/2 and the proliferation of pancreatic stellate cells triggered by both glucose and insulin140. However, additional inquiries are required to ascertain their precise impacts. Furthermore, a recent study on the antifibrotic drug pirfenidone showed a decrease in the expression of α-SMA and a restriction of islet fibrosis in the pancreas of OLETF rats. However, this therapy did not increase glucose tolerance or insulin secretion116. Despite these limitations, targeting PSCs directly could be considered a focused strategy, either alone or in combination with other drugs.
Ahangarpour et al.141 have prepared solid lipid nanoparticle (SLN) of myricitrin. In a mouse model of diabetic nephropathy (DN), moderate and low doses of SLN were found to be more effective in reducing TGF-β gene expression and ameliorate fibrosis of kidney compared to a diabetes group. Hussein et al.142 designed a type of Zinc oxide nanocrystals (ZnO-NPs) using hydroxyethylcellulose as a stabiliser and directing agent. It was showed that levels of C-reactive and cytokines (IL-1 and IL-6) decreased and NO level increased in the diabetic group. ZnO-NPs can effectively attenuate vascular diseases as a promising agent for diabetic vascular diseases and hyperglycaemia. More detailed information of nanomedicine for the treatment of diabetes and fibrosis can be found in Table 3134,135,141,142.
Table 3.
Antifibrotic treatment strategies in diabetes mellitus.
| Nanomedicine | Targeting therapeutic agent | Mechanism of action | Diabetes model | Preclinical/clinical | Ref. |
|---|---|---|---|---|---|
| QD-INS-CS/GS | Insulin | Silver sulfide (Ag2S) quantum dots (QDs) are orally bioavailable and increase the rate of absorption. Insulin release is mediated by enzymatic cleavage of polymerization chitosan and glucose copolymer. | NOD/ShiLtJ mice | Preclinical | 134 |
| Ins/PBCA NPs | Insulin | Folate is actively transported through proton-coupled folate transporter (PCFT). Due to increasing intracellular trafficking, folate-modified nanoparticles loaded with insulin have a high oral bioavailability. | Diabetic rats induced by alloxan | Preclinical | 135 |
| SLN | Myricitrin | Myricitrin targeted into kidney cell with solid lipid nanoparticle (SLN) to inhibit the expression of TGF-β and reduce oxidative stress and cytotoxicity. | Diabetic mice induced by streptozotocin-nicotinamide (STZ-NA) | Preclinical | 141 |
| ZnO-NPs | ZnO | Zinc oxide nanocrystals (ZnO-NPs) decrease levels of C-reactive and cytokines (IL-1 and IL-6) and increase NO level. | Diabetic rats induced by STZ | Preclinical | 142 |
6. Conclusions and future perspectives
Pancreatic fibrosis is a long-term and ongoing process that ultimately results in pancreatic endocrine and exocrine insufficiency. This dynamic pathological process exhibits varying compositions and roles across different diseases, necessitating a comprehensive understanding of fibrosis within the unique microenvironment associated with each disease. Firstly, CP, PC and diabetes are interrelated and may act as risk factors for one another. CP is characterized by persistent and permanent damage in pancreatic tissue. The injury results in the gradual loss of the exocrine to endocrine compartments of the pancreas, which are replaced by atrophy or fibrosis78. The development of CP leads to organ dysfunction and increases the risk of developing PC. Pancreatic fibrosis is also a typical feature of PC, which promotes the activation and recruitment of CAFs8. Pancreatic fibrosis can be used as a diagnostic marker of PC. Type 2 diabetes, particularly when accompanied by obesity, is a known risk factor for the development of PC. Meanwhile, CP and PC are the most common cause of pancreatogenic diabetes, also known as type 3c diabetes145. In addition, fibrosis is found in the endocrine pancreas55. However, the mechanism underlying PSC activation in the islet still needs to be discovered, and the relationship between fibrosis and diabetes warrants further attention.
In a normal pancreas, the gland is divided into many indistinct lobules by the connective tissue of the capsule. The pancreatic tissue exhibits few fibrous components. However, the tissue of CP is characterized by complete atrophy of the acinar cells and progressive fibrotic destruction of the pancreatic secretory parenchyma143. The fibrotic component of pancreatitis is primarily located in the interlobular or perilobular area. The incomplete obstruction of the pancreatic duct and the presence of cells expressing α-SMA, a marker of myofibroblasts, are significant factors in interlobular fibrosis143. The most common type of PC is ductal adenocarcinoma. During tumor progression, fibrosis progresses due to duct obstruction and the proliferation of interlobular myofibroblasts. The tissue of PC has a dense fibrous stroma with excessive connective tissue hyperplasia and fibrosis144. Different from pancreatitis and pancreatic cancer, where fibrosis occurs in the exocrine division, the fibrosis of diabetes appears in the capillaries, intra-islet and peri-islet of the endocrine gland110 as shown in Table 4110,143,144. Although the characteristics of fibrotic tissue and the triggers for PSC activation vary in different disease contexts, PSCs are vital mediators of fibrosis. They are activated to produce large amounts of ECM deposited between tissues8. Hence, targeted inhibition of PSCs is a universal therapeutic strategy for antifibrotic treatment of different diseases. Interventions targeting PSCs include changing cells from activated to resting states and promoting apoptosis, with the ultimate goal of reducing the production of extracellular matrix proteins.
Table 4.
Differences of PSCs activation in CP, PC and diabetes.
| Comparison | CP | PC | Diabete |
|---|---|---|---|
| Fibrosis mechanism | PSCs are key mediators of fibrosis. PSCs are activated to produce large amounts of ECM deposited between tissues. | ||
| The triggers for PSCs activation | Pancreatic acinar cells | Pancreatic cancer cells | β-Cells |
| Location of fibrosis | Exocrine gland Interlobular or perilobular area |
Exocrine gland Periductal and interlobular |
Endocrine gland Islet |
| Pathological features | The tissue is characterized by complete atrophy of the acinar cells and progressive fibrotic destruction of the pancreatic secretory parenchyma143. | The tissue is characterized by a dense fibrous stroma with excessive connective tissue hyperplasia and highly fibrotic144. | Capillaries, intra-islet and peri-islet wrapped in fibrous tissue110. |
| Masson of tissue |  |
 |
 |
PF is a dynamic, cell-mediated pathological process in which activation of PSCs is generally recognized as the central event. In response to intracellular and extracellular microenvironmental stimuli, activated PSCs interact with other pancreatic cells, including PACs, islet cells, PCCs and immune cells. Notably, these cells play essential roles in activating PSCs in various diseases through different molecular and cellular signaling mechanisms. Firstly, PSCs are activated by paracrine profibrogenic chemo and molecular signals. Besides, pancreatic cells inhibit the activation of PSCs and eliminate activated PSCs through the induction of death and senescence. In order to gain new insights into therapeutic interventions for PF, it is essential to unravel the specific cellular mechanisms underlying cellular interactions in PSC activation. Further investigations should prioritize identifying of the pivotal elements that govern the activation of PSCs by these specific pancreatic cells. It will aid in the determination of precise approaches for intervening in PF. Additionally, the intercellular crosstalk in the regression of PF still needs to be better understood. In particular, which factor plays a leading role in regulating PSC activation during this process has yet to be discovered. A better understanding of this critical process would facilitate the development of more effective strategies to promote the regression of fibrosis and delay the progression of pancreatic disease.
Currently, numerous studies have provided the limitations of conventional drug delivery systems. To date, numerous studies have provided evidence of the limitations of conventional clinical drug delivery systems. They are mostly related to lack of potency and reduced efficacy or altered effects for several reasons: (1) restricted route of administration, (2) inappropriate or ineffective dosage, (3) low biological half-life, (4) lack of target specificity, and (5) side effects of the agents. Therefore, developing novel nanocarrier formations to improve bioavailability, efficacy and safety is expected to improve traditional drug delivery systems in fibrosis therapy. Recent research in the development of various nanocarriers such as polymeric NPs, liposomes and micelles has progressed phenomenally and shown promising advances in the treatment of antifibrosis and pancreatic diseases. In nanomedicine design, the aspects of material selection shall be considered including better biocompatibility and biodegradability, low immunogenicity, and accessibility. Due to interaction with organs, tissues and cells, nanoparticles are exposed in humans and induce side effects and toxicity. We shall further emphasize the long-term safety of nanomedicine.
As mentioned above, the crosstalk between PSCs and other pancreatic cells plays a crucial role in pathological conditions of PF. Some nanomedicines have been developed to treat PF and pancreatic diseases by targeting the crosstalk between PSCs and other cells. There still needs to be more understanding of the complex mechanisms that cause PSCs to become active and the molecular processes that lead to pancreatic fibrosis. Additional inquiry is necessary to understand and solve these intricacies and create inventive treatment approaches. Continued endeavors in researching PSCs and their interactions with other cellular constituents have immense promise for substantial advancements in the management of pancreatic disorders. In addition, our team has examined the significant function of various cells in other fibrotic conditions and conducted therapeutic experiments with nanomedicine intervention. Mesenchymal stem cells (MSCs) are self-renewing pluripotent stem cells with multidirectional differentiation potential, and their application in organ transplantation and other aspects has been extensively explored146. We developed a platform (MSCs-Lip@NCAF) using MSCs, and liposomes modified with type I collagenase loaded with nintedanib. MSCs-Lip@NCAF can be migrated to fibrotic lungs because of the homing characteristic of MSCs and then Lip@NCAF was sensitively released. Subsequently, Lip@NCAF ablated collagen fibers, delivered nintedanib into fibroblasts, and inhibited fibroblast overactivation. MSCs differentiated into AECIIs to repair the alveolar structure and ultimately promote the regeneration of damaged lungs in aged mice. MSCs-Lip@NCAF was designed for the treatment of severe fibrosis and exhibited exceptional efficacy in treating fibrosis in elderly mice147 (Fig. 8). MSCs have unique anti-inflammatory and immunomodulatory effects, which will be a potential therapeutic to reduce the inflammatory response, inhibit fibrosis, and rescue pancreatic function. Ultimately, it is crucial to understand, regulate, or take advantage of the abnormal microenvironment to maximize drug effectiveness. This task presents numerous obstacles and possibilities that require urgent consideration. There will be increased research attention on regulating the intercellular crosstalk in the future. Progress in this area will significantly contribute to the field of precision medicine.
Figure 8.

Illustration of lung-targeting nanoengineered MSCs-Lip@NCAF designed to reverse PF in young and aged mice. (A) Preparation of MSCs-Lip@NCAF. PC, phosphatidylcholine. (B) MSCs-Lip@NCAF migrated to injured lungs because of their homing ability, and Lip@NCAF was released in response to MMP-2. Then, Lip@NCAF degraded collagen fibers and inhibited fibroblast overactivation. Moreover, MSCs repaired injured AEC IIs in young mice and differentiated into AEC IIs to participate in alveolar reestablishment in aged mice. Reprinted with the permission from Ref. 147. Copyright © 2023 American Association for the Advancement of Science.
Author contributions
Hui Wang: Writing – original draft, Conceptualization. Liang Qi: Writing – original draft. Han Han: Writing – original draft. Xuena Li: Data curation, Investigation. Mengmeng Han: Data curation, Resources. Lei Xing: Supervision. Ling Li: Writing – review & editing. Hulin Jiang: Conceptualization, Writing – review & editing.
Conflicts of interest
The authors have no conflicts of interest to declare.
Acknowledgments
This work was financially supported by the National Key R&D Program of China (2022YFE0198400) and the National Natural Science Foundation Major International (Regional) Joint Research Program (Nos. 82020108029, 82320108003, China), National Natural Science Foundation (Nos. 82170845, 82073398, 82003677, 82000740, 81970717, China). This work was also supported by the Project of State Key Laboratory of Natural Medicines, China Pharmaceutical University (SKLNMZZ202021, China), the “111” Project from the Ministry of Education of China, the State Administration of Foreign Experts Affairs of China (B16046), and Double First-Rate construction plan of China Pharmaceutical University (CPU2018GY06, CPU2022QZ18, China) and the Key Research & Development Program (BE2022853, China) and Medical Key Discipline (ZDXK202203, China) of Jiangsu Province.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Ling Li, Email: lingli@seu.edu.cn.
Hulin Jiang, Email: jianghulin3@163.com.
References
- 1.Zhou Q., Melton D.A. Pancreas regeneration. Nature. 2018;557:351–358. doi: 10.1038/s41586-018-0088-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beheiry R., Abdel-Raheem W., Balah A., Salem H., Karkit M. Morphological, histological and ultrastructural studies on the exocrine pancreas of goose. Beni Suef Univ J Basic Appl Sci. 2018;7:353–358. [Google Scholar]
- 3.Wood L.D., Canto M.I., Jaffee E.M., Simeone D.M. Pancreatic cancer: pathogenesis, screening, diagnosis, and treatment. Gastroenterology. 2022;163:386–402. doi: 10.1053/j.gastro.2022.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beyer G., Habtezion A., Werner J., Lerch M.M., Mayerle J. Chronic pancreatitis. Lancet. 2020;396:499–512. doi: 10.1016/S0140-6736(20)31318-0. [DOI] [PubMed] [Google Scholar]
- 5.Kautzky-Willer A., Harreiter J., Pacini G. Sex and gender differences in risk, pathophysiology and complications of type 2 diabetes mellitus. Endocr Rev. 2016;37:278–316. doi: 10.1210/er.2015-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pothula S.P., Pirola R.C., Wilson J.S., Apte M.V. Pancreatic stellate cells: aiding and abetting pancreatic cancer progression. Pancreatology. 2020;20:409–418. doi: 10.1016/j.pan.2020.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Cnop M., Welsh N., Jonas J.C., Jorns A., Lenzen S., Eizirik D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–S107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 8.Kleeff J., Whitcomb D.C., Shimosegawa T., Esposito I., Lerch M.M., Gress T., et al. Chronic pancreatitis. Nat Rev Dis Primers. 2017;3 doi: 10.1038/nrdp.2017.60. [DOI] [PubMed] [Google Scholar]
- 9.Mateus G.L., Pereira E., Werneck D.C.J., Bernal-Mizrachi E., Almaca J. Islet pericytes convert into profibrotic myofibroblasts in a mouse model of islet vascular fibrosis. Diabetologia. 2020;63:1564–1575. doi: 10.1007/s00125-020-05168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarkar R., Xu Z., Perera C.J., Apte M.V. Emerging role of pancreatic stellate cell-derived extracellular vesicles in pancreatic cancer. Semin Cancer Biol. 2023;93:114–122. doi: 10.1016/j.semcancer.2023.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Yang X., Chen J., Wang J., Ma S., Feng W., Wu Z., et al. Very-low-density lipoprotein receptor-enhanced lipid metabolism in pancreatic stellate cells promotes pancreatic fibrosis. Immunity. 2022;55:1185–1199. doi: 10.1016/j.immuni.2022.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Puri S., Mazza M., Roy G., England R.M., Zhou L., Nourian S., et al. Evolution of nanomedicine formulations for targeted delivery and controlled release. Adv Drug Deliv Rev. 2023;200 doi: 10.1016/j.addr.2023.114962. [DOI] [PubMed] [Google Scholar]
- 13.Wicki A., Witzigmann D., Balasubramanian V., Huwyler J. Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Control Release. 2015;200:138–157. doi: 10.1016/j.jconrel.2014.12.030. [DOI] [PubMed] [Google Scholar]
- 14.Zoulikha M., Xiao Q., Boafo G.F., Sallam M.A., Chen Z., He W. Pulmonary delivery of sirna against acute lung injury/acute respiratory distress syndrome. Acta Pharm Sin B. 2022;12:600–620. doi: 10.1016/j.apsb.2021.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li F., Zhao Y., Cheng Z., Wang Y., Yue Y., Cheng X., et al. Restoration of sinusoid fenestrae followed by targeted nanoassembly delivery of an anti-fibrotic agent improves treatment efficacy in liver fibrosis. Adv Mater. 2023;35 doi: 10.1002/adma.202212206. [DOI] [PubMed] [Google Scholar]
- 16.Bai X., Zhao G., Chen Q., Li Z., Gao M., Ho W., et al. Inhaled sirna nanoparticles targeting IL11 inhibit lung fibrosis and improve pulmonary function post-bleomycin challenge. Sci Adv. 2022;8 doi: 10.1126/sciadv.abn7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han H., Chen B., Ding J., Si J., Zhou T., Wang Y., et al. A PDGFRβ-targeting nanodrill system for pancreatic fibrosis therapy. Chin Chem Lett. 2024;35 [Google Scholar]
- 18.Jin G., Hong W., Guo Y., Bai Y., Chen B. Molecular mechanism of pancreatic stellate cells activation in chronic pancreatitis and pancreatic cancer. J Cancer. 2020;11:1505–1515. doi: 10.7150/jca.38616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherman M.H. Stellate cells in tissue repair, inflammation, and cancer. Annu Rev Cel Dev Biol. 2018;34:333–355. doi: 10.1146/annurev-cellbio-100617-062855. [DOI] [PubMed] [Google Scholar]
- 20.Bachem M.G., Schneider E., Gross H., Weidenbach H., Schmid R.M., Menke A., et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115:421–432. doi: 10.1016/s0016-5085(98)70209-4. [DOI] [PubMed] [Google Scholar]
- 21.Duner S., Lopatko L.J., Ansari D., Gundewar C., Andersson R. Pancreatic cancer: the role of pancreatic stellate cells in tumor progression. Pancreatology. 2010;10:673–681. doi: 10.1159/000320711. [DOI] [PubMed] [Google Scholar]
- 22.Wynn T.A., Ramalingam T.R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones T.E., Bellin M.D., Yadav D., Freeman M.L., Schwarzenberg S.J., Slivka A., et al. The histopathology of SPINK1-associated chronic pancreatitis. Pancreatology. 2020;20:1648–1655. doi: 10.1016/j.pan.2020.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng L., Xue J., Jaffee E.M., Habtezion A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology. 2013;144:1230–1240. doi: 10.1053/j.gastro.2012.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diazzi S., Tartare-Deckert S., Deckert M. Bad neighborhood: fibrotic stroma as a new player in melanoma resistance to targeted therapies. Cancers (Basel) 2020;12:1364. doi: 10.3390/cancers12061364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Homo-Delarche F., Calderari S., Irminger J.C., Gangnerau M.N., Coulaud J., Rickenbach K., et al. Islet inflammation and fibrosis in a spontaneous model of type 2 diabetes, the GK rat. Diabetes. 2006;55:1625–1633. doi: 10.2337/db05-1526. [DOI] [PubMed] [Google Scholar]
- 27.Hayden M.R. Islet amyloid and fibrosis in the cardiometabolic syndrome and type 2 diabetes mellitus. J Cardiometab Syndr. 2007;2:70–75. doi: 10.1111/j.1559-4564.2007.06159.x. [DOI] [PubMed] [Google Scholar]
- 28.Ahmed A.U., Issa Y., Hagenaars J.C., Bakker O.J., van Goor H., Nieuwenhuijs V.B., et al. Risk of recurrent pancreatitis and progression to chronic pancreatitis after a first episode of acute pancreatitis. Clin Gastroenterol Hepatol. 2016;14:738–746. doi: 10.1016/j.cgh.2015.12.040. [DOI] [PubMed] [Google Scholar]
- 29.Bynigeri R.R., Jakkampudi A., Jangala R., Subramanyam C., Sasikala M., Rao G.V., et al. Pancreatic stellate cell: pandora's box for pancreatic disease biology. World J Gastroenterol. 2017;23:382–405. doi: 10.3748/wjg.v23.i3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mccarroll J.A., Phillips P.A., Santucci N., Pirola R.C., Wilson J.S., Apte M.V. Vitamin a inhibits pancreatic stellate cell activation: implications for treatment of pancreatic fibrosis. Gut. 2006;55:79–89. doi: 10.1136/gut.2005.064543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sousa C.M., Biancur D.E., Wang X., Halbrook C.J., Sherman M.H., Zhang L., et al. Pancreatic stellate cells support tumor metabolism through autophagic alanine secretion. Nature. 2016;536:479–483. doi: 10.1038/nature19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sherman M.H., Yu R.T., Engle D.D., Ding N., Atkins A.R., Tiriac H., et al. Vitamin d receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramakrishnan P., Loh W.M., Gopinath S., Bonam S.R., Fareez I.M., Mac G.R., et al. Selective phytochemicals targeting pancreatic stellate cells as new anti-fibrotic agents for chronic pancreatitis and pancreatic cancer. Acta Pharm Sin B. 2020;10:399–413. doi: 10.1016/j.apsb.2019.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 35.Carapuca E.F., Gemenetzidis E., Feig C., Bapiro T.E., Williams M.D., Wilson A.S., et al. Anti-stromal treatment together with chemotherapy targets multiple signaling pathways in pancreatic adenocarcinoma. J Pathol. 2016;239:286–296. doi: 10.1002/path.4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang R., Lotze M.T., Zeh H.J., Billiar T.R., Tang D. Cell death and damps in acute pancreatitis. Mol Med. 2014;20:466–477. doi: 10.2119/molmed.2014.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J., Gao M., Nipper M., Deng J., Sharkey F.E., Johnson R.L., et al. Activation of the intrinsic fibroinflammatory program in adult pancreatic acinar cells triggered by hippo signaling disruption. PLoS Biol. 2019;17 doi: 10.1371/journal.pbio.3000418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang W., Jin L., Ju D., Lu Z., Wang C., Guo X., et al. The pancreatic clock is a key determinant of pancreatic fibrosis progression and exocrine dysfunction. Sci Transl Med. 2022;14 doi: 10.1126/scitranslmed.abn3586. [DOI] [PubMed] [Google Scholar]
- 39.Wang Z., Liu J., Wang Y., Guo H., Li F., Cao Y., et al. Identification of key biomarkers associated with immunogenic cell death and their regulatory mechanisms in severe acute pancreatitis based on WGCNA and machine learning. Int J Mol Sci. 2023;24:3033. doi: 10.3390/ijms24033033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Habtezion A., Gukovskaya A.S., Pandol S.J. Acute pancreatitis: a multifaceted set of organelle and cellular interactions. Gastroenterology. 2019;156:1941–1950. doi: 10.1053/j.gastro.2018.11.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blauer M., Laaninen M., Sand J., Laukkarinen J. Reciprocal stimulation of pancreatic acinar and stellate cells in a novel long-term in vitro co-culture model. Pancreatology. 2016;16:570–577. doi: 10.1016/j.pan.2016.03.012. [DOI] [PubMed] [Google Scholar]
- 42.Wang Q., Wang H., Jing Q., Yang Y., Xue D., Hao C., et al. Regulation of pancreatic fibrosis by acinar cell-derived exosomal miR-130a-3p via targeting of stellate cell PPAR-γ. J Inflamm Res. 2021;14:461–477. doi: 10.2147/JIR.S299298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao W., Luo D., Lv Z., Yang Y., Wang L., Ma B., et al. The RABEP1-mediated endocytosis and activation of trypsinogen to promote pancreatic stellate cell activation. Biomolecules. 2022;12:1063. doi: 10.3390/biom12081063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang D., Han S., Lv G., Hu Y., Zhuo W., Zeng Z., et al. Pancreatic acinar cells-derived sphingosine-1-phosphate contributes to fibrosis of chronic pancreatitis via inducing autophagy and activation of pancreatic stellate cells. Gastroenterology. 2023;165:1488–1504. doi: 10.1053/j.gastro.2023.08.029. [DOI] [PubMed] [Google Scholar]
- 45.Shapouri-Moghaddam A., Mohammadian S., Vazini H., Taghadosi M., Esmaeili S.A., Mardani F., et al. Macrophage plasticity, polarization, and function in health and disease. J Cel Physiol. 2018;233:6425–6440. doi: 10.1002/jcp.26429. [DOI] [PubMed] [Google Scholar]
- 46.Xia Y., Rao L., Yao H., Wang Z., Ning P., Chen X. Engineering macrophages for cancer immunotherapy and drug delivery. Adv Mater. 2020;32 doi: 10.1002/adma.202002054. [DOI] [PubMed] [Google Scholar]
- 47.Hu Q., Lyon C.J., Fletcher J.K., Tang W., Wan M., Hu T.Y. Extracellular vesicle activities regulating macrophage- and tissue-mediated injury and repair responses. Acta Pharm Sin B. 2021;11:1493–1512. doi: 10.1016/j.apsb.2020.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baer J.M., Zuo C., Kang L.I., de la Lastra A.A., Borcherding N.C., Knolhoff B.L., et al. Fibrosis induced by resident macrophages has divergent roles in pancreas inflammatory injury and PDAC. Nat Immunol. 2023;24:1443–1457. doi: 10.1038/s41590-023-01579-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xue J., Sharma V., Hsieh M.H., Chawla A., Murali R., Pandol S.J., et al. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun. 2015;6:7158. doi: 10.1038/ncomms8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gukovskaya A.S., Vaquero E., Zaninovic V., Gorelick F.S., Lusis A.J., Brennan M.L., et al. Neutrophils and nadph oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology. 2002;122:974–984. doi: 10.1053/gast.2002.32409. [DOI] [PubMed] [Google Scholar]
- 51.Sendler M., Dummer A., Weiss F.U., Kruger B., Wartmann T., Scharffetter-Kochanek K., et al. Tumor necrosis factor alpha secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut. 2013;62:430–439. doi: 10.1136/gutjnl-2011-300771. [DOI] [PubMed] [Google Scholar]
- 52.Mayerle J., Schnekenburger J., Kruger B., Kellermann J., Ruthenburger M., Weiss F.U., et al. Extracellular cleavage of E-cadherin by leukocyte elastase during acute experimental pancreatitis in rats. Gastroenterology. 2005;129:1251–1267. doi: 10.1053/j.gastro.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 53.Cui L., Li C., Zhang G., Zhang L., Yao G., Zhuo Y., et al. S1P/S1PR2 promote pancreatic stellate cell activation and pancreatic fibrosis in chronic pancreatitis by regulating autophagy and the NLRP3 inflammasome. Chem Biol Interact. 2023;380 doi: 10.1016/j.cbi.2023.110541. [DOI] [PubMed] [Google Scholar]
- 54.Bhatia V., Rastellini C., Han S., Aronson J.F., Greeley G.J., Falzon M. Acinar cell-specific knockout of the PTHrP gene decreases the proinflammatory and profibrotic responses in pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2014;307:G533–G549. doi: 10.1152/ajpgi.00428.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang S., Chao X., Jiang X., Wang T., Rodriguez Y., Yang L., et al. Loss of acinar cell VMP1 triggers spontaneous pancreatitis in mice. Autophagy. 2022;18:1572–1582. doi: 10.1080/15548627.2021.1990672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qi L., Han H., Han M.M., Sun Y., Xing L., Jiang H.L., et al. Remodeling of imbalanced extracellular matrix homeostasis for reversal of pancreatic fibrosis. Biomaterials. 2023;292 doi: 10.1016/j.biomaterials.2022.121945. [DOI] [PubMed] [Google Scholar]
- 57.Zeng X.P., Wang L.J., Guo H.L., He L., Bi Y.W., Xu Z.L., et al. Dasatinib ameliorates chronic pancreatitis induced by caerulein via anti-fibrotic and anti-inflammatory mechanism. Pharmacol Res. 2019;147 doi: 10.1016/j.phrs.2019.104357. [DOI] [PubMed] [Google Scholar]
- 58.Bai H., Chen X., Zhang L., Dou X. The effect of sulindac, a non-steroidal anti-inflammatory drug, attenuates inflammation and fibrosis in a mouse model of chronic pancreatitis. BMC Gastroenterol. 2012;12:115. doi: 10.1186/1471-230X-12-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bansod S., Doijad N., Godugu C. Berberine attenuates severity of chronic pancreatitis and fibrosis via AMPK-mediated inhibition of TGF-β1/Smad signaling and M2 polarization. Toxicol Appl Pharmacol. 2020;403 doi: 10.1016/j.taap.2020.115162. [DOI] [PubMed] [Google Scholar]
- 60.Wang L.J., He L., Hao L., Guo H.L., Zeng X.P., Bi Y.W., et al. Isoliquiritigenin ameliorates caerulein-induced chronic pancreatitis by inhibiting the activation of PSCs and pancreatic infiltration of macrophages. J Cel Mol Med. 2020;24:9667–9681. doi: 10.1111/jcmm.15498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khurana A., Saifi M.A., Godugu C. Nanoceria ameliorates fibrosis, inflammation, and cellular stress in experimental chronic pancreatitis. ACS Biomater Sci Eng. 2023;9:1030–1042. doi: 10.1021/acsbiomaterials.2c00933. [DOI] [PubMed] [Google Scholar]
- 62.Nagao S., Taguchi K., Sakai H., Yamasaki K., Watanabe H., Otagiri M., et al. Carbon monoxide-bound hemoglobin vesicles ameliorate multiorgan injuries induced by severe acute pancreatitis in mice by their anti-inflammatory and antioxidant properties. Int J Nanomedicine. 2016;11:5611–5620. doi: 10.2147/IJN.S118185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang F., Deng Y., Yu L., Zhou A., Wang J., Jia J., et al. A macrophage membrane-polymer hybrid biomimetic nanoplatform for therapeutic delivery of somatostatin peptide to chronic pancreatitis. Pharmaceutics. 2022;14:2341. doi: 10.3390/pharmaceutics14112341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khurana A., Saifi M.A., Godugu C. Yttrium oxide nanoparticles attenuate l-arginine induced chronic pancreatitis. Biol Trace Elem Res. 2023;201:3404–3417. doi: 10.1007/s12011-022-03446-6. [DOI] [PubMed] [Google Scholar]
- 65.Miller K.D., Nogueira L., Devasia T., Mariotto A.B., Yabroff K.R., Jemal A., et al. Cancer treatment and survivorship statistics, 2022. CA Cancer J Clin. 2022;72:409–436. doi: 10.3322/caac.21731. [DOI] [PubMed] [Google Scholar]
- 66.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., et al. Global cancer statistics 2020: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 67.Huang J., Lok V., Ngai C.H., Zhang L., Yuan J., Lao X.Q., et al. Worldwide burden of, risk factors for, and trends in pancreatic cancer. Gastroenterology. 2021;160:744–754. doi: 10.1053/j.gastro.2020.10.007. [DOI] [PubMed] [Google Scholar]
- 68.Sherman M.H., Beatty G.L. Tumor microenvironment in pancreatic cancer pathogenesis and therapeutic resistance. Annu Rev Pathol. 2023;18:123–148. doi: 10.1146/annurev-pathmechdis-031621-024600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bear A.S., Vonderheide R.H., O'Hara M.H. Challenges and opportunities for pancreatic cancer immunotherapy. Cancer Cell. 2020;38:788–802. doi: 10.1016/j.ccell.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murray E.R., Menezes S., Henry J.C., Williams J.L., Alba-Castellon L., Baskaran P., et al. Disruption of pancreatic stellate cell myofibroblast phenotype promotes pancreatic tumor invasion. Cell Rep. 2022;38 doi: 10.1016/j.celrep.2021.110227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yan Z., Ohuchida K., Fei S., Zheng B., Guan W., Feng H., et al. Inhibition of ERK1/2 in cancer-associated pancreatic stellate cells suppresses cancer-stromal interaction and metastasis. J Exp Clin Cancer Res. 2019;38:221. doi: 10.1186/s13046-019-1226-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karnevi E., Rosendahl A.H., Hilmersson K.S., Saleem M.A., Andersson R. Impact by pancreatic stellate cells on epithelial-mesenchymal transition and pancreatic cancer cell invasion: adding a third dimension in vitro. Exp Cel Res. 2016;346:206–215. doi: 10.1016/j.yexcr.2016.07.017. [DOI] [PubMed] [Google Scholar]
- 73.Dongre A., Weinberg R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cel Biol. 2019;20:69–84. doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 74.Cao X., Hu Y., Luo S., Wang Y., Gong T., Sun X., et al. Neutrophil-mimicking therapeutic nanoparticles for targeted chemotherapy of pancreatic carcinoma. Acta Pharm Sin B. 2019;9:575–589. doi: 10.1016/j.apsb.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schalck A., Sakellariou-Thompson D., Forget M.A., Sei E., Hughes T.G., Reuben A., et al. Single-cell sequencing reveals trajectory of tumor-infiltrating lymphocyte states in pancreatic cancer. Cancer Discov. 2022;12:2330–2349. doi: 10.1158/2159-8290.CD-21-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu Y., Xiong J., Sun X., Gao H. Targeted nanomedicines remodeling immunosuppressive tumor microenvironment for enhanced cancer immunotherapy. Acta Pharm Sin B. 2022;12:4327–4347. doi: 10.1016/j.apsb.2022.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tan B., Shi X., Zhang J., Qin J., Zhang N., Ren H., et al. Inhibition of RSPO-LGR4 facilitates checkpoint blockade therapy by switching macrophage polarization. Cancer Res. 2018;78:4929–4942. doi: 10.1158/0008-5472.CAN-18-0152. [DOI] [PubMed] [Google Scholar]
- 78.Hu B., Wang Z., Zeng H., Qi Y., Chen Y., Wang T., et al. Blockade of DC-SIGN(+) tumor-associated macrophages reactivates antitumor immunity and improves immunotherapy in muscle-invasive bladder cancer. Cancer Res. 2020;80:1707–1719. doi: 10.1158/0008-5472.CAN-19-2254. [DOI] [PubMed] [Google Scholar]
- 79.Mace T.A., Ameen Z., Collins A., Wojcik S., Mair M., Young G.S., et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73:3007–3018. doi: 10.1158/0008-5472.CAN-12-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hashimoto O., Yoshida M., Koma Y., Yanai T., Hasegawa D., Kosaka Y., et al. Collaboration of cancer-associated fibroblasts and tumor-associated macrophages for neuroblastoma development. J Pathol. 2016;240:211–223. doi: 10.1002/path.4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tang P.C., Chung J.Y., Xue V.W., Xiao J., Meng X.M., Huang X.R., et al. Smad3 promotes cancer-associated fibroblasts generation via macrophage-myofibroblast transition. Adv Sci. 2022;9 doi: 10.1002/advs.202101235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garg B., Giri B., Modi S., Sethi V., Castro I., Umland O., et al. NFκB in pancreatic stellate cells reduces infiltration of tumors by cytotoxic T cells and killing of cancer cells, via up-regulation of CXCL12. Gastroenterology. 2018;155:880–891. doi: 10.1053/j.gastro.2018.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y., Gao Z., Du X., Chen S., Zhang W., Wang J., et al. Co-inhibition of the TGF-β pathway and the PD-L1 checkpoint by pH-responsive clustered nanoparticles for pancreatic cancer microenvironment regulation and anti-tumor immunotherapy. Biomater Sci. 2020;8:5121–5132. doi: 10.1039/d0bm00916d. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Y., Ware M.B., Zaidi M.Y., Ruggieri A.N., Olson B.M., Komar H., et al. Heat shock protein-90 inhibition alters activation of pancreatic stellate cells and enhances the efficacy of PD-1 blockade in pancreatic cancer. Mol Cancer Ther. 2021;20:150–160. doi: 10.1158/1535-7163.MCT-19-0911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ma Y., Hwang R.F., Logsdon C.D., Ullrich S.E. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res. 2013;73:3927–3937. doi: 10.1158/0008-5472.CAN-12-4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Admyre C., Axelsson L.G., von Stein O., Zargari A. Immunomodulatory oligonucleotides inhibit neutrophil migration by decreasing the surface expression of interleukin-8 and leukotriene B4 receptors. Immunology. 2015;144:206–217. doi: 10.1111/imm.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tang D., Wang D., Yuan Z., Xue X., Zhang Y., An Y., et al. Persistent activation of pancreatic stellate cells creates a microenvironment favorable for the malignant behavior of pancreatic ductal adenocarcinoma. Int J Cancer. 2013;132:993–1003. doi: 10.1002/ijc.27715. [DOI] [PubMed] [Google Scholar]
- 88.Xu Z., Vonlaufen A., Phillips P.A., Fiala-Beer E., Zhang X., Yang L., et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am J Pathol. 2010;177:2585–2596. doi: 10.2353/ajpath.2010.090899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Patel M.B., Pothula S.P., Xu Z., Lee A.K., Goldstein D., Pirola R.C., et al. The role of the hepatocyte growth factor/c-MET pathway in pancreatic stellate cell-endothelial cell interactions: antiangiogenic implications in pancreatic cancer. Carcinogenesis. 2014;35:1891–1900. doi: 10.1093/carcin/bgu122. [DOI] [PubMed] [Google Scholar]
- 90.Kuninty P.R., Bojmar L., Tjomsland V., Larsson M., Storm G., Ostman A., et al. MicroRNA-199a and -214 as potential therapeutic targets in pancreatic stellate cells in pancreatic tumor. Oncotarget. 2016;7:16396–16408. doi: 10.18632/oncotarget.7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Demir I.E., Ceyhan G.O., Rauch U., Altintas B., Klotz M., Muller M.W., et al. The microenvironment in chronic pancreatitis and pancreatic cancer induces neuronal plasticity. Neurogastroenterol Motil. 2010;22(480–90):e112–e113. doi: 10.1111/j.1365-2982.2009.01428.x. [DOI] [PubMed] [Google Scholar]
- 92.Li X., Wang Z., Ma Q., Xu Q., Liu H., Duan W., et al. Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer. Clin Cancer Res. 2014;20:4326–4338. doi: 10.1158/1078-0432.CCR-13-3426. [DOI] [PubMed] [Google Scholar]
- 93.Han L., Ma J., Duan W., Zhang L., Yu S., Xu Q., et al. Pancreatic stellate cells contribute pancreatic cancer pain via activation of sHH signaling pathway. Oncotarget. 2016;7:18146–18158. doi: 10.18632/oncotarget.7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li M., Wang Y., Li M., Wu X., Setrerrahmane S., Xu H. Integrins as attractive targets for cancer therapeutics. Acta Pharm Sin B. 2021;11:2726–2737. doi: 10.1016/j.apsb.2021.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Charbe N.B., Amnerkar N.D., Ramesh B., Tambuwala M.M., Bakshi H.A., Aljabali A., et al. Small interfering RNA for cancer treatment: overcoming hurdles in delivery. Acta Pharm Sin B. 2020;10:2075–2109. doi: 10.1016/j.apsb.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Han X., Li Y., Xu Y., Zhao X., Zhang Y., Yang X., et al. Reversal of pancreatic desmoplasia by re-educating stellate cells with a tumor microenvironment-activated nanosystem. Nat Commun. 2018;9:3390. doi: 10.1038/s41467-018-05906-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Han H., Hou Y., Chen X., Zhang P., Kang M., Jin Q., et al. Metformin-induced stromal depletion to enhance the penetration of gemcitabine-loaded magnetic nanoparticles for pancreatic cancer targeted therapy. J Am Chem Soc. 2020;142:4944–4954. doi: 10.1021/jacs.0c00650. [DOI] [PubMed] [Google Scholar]
- 98.Chen C., Zhao S., Zhao X., Cao L., Karnad A., Kumar A.P., et al. Gemcitabine resistance of pancreatic cancer cells is mediated by IGF1R dependent upregulation of CD44 expression and isoform switching. Cell Death Dis. 2022;13:682. doi: 10.1038/s41419-022-05103-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao Y., Yao H., Yang K., Han S., Chen S., Li Y., et al. Arsenic trioxide-loaded nanoparticles enhance the chemosensitivity of gemcitabine in pancreatic cancer via the reversal of pancreatic stellate cell desmoplasia by targeting the AP4/galectin-1 pathway. Biomater Sci. 2022;10:5989–6002. doi: 10.1039/d2bm01039a. [DOI] [PubMed] [Google Scholar]
- 100.Hu Q., Xu M., Feng J., Xie H., Li J., He Y., et al. Hyperthermia-induced stellate cell deactivation to enhance dual chemo and pH-responsive photothermal therapy for pancreatic cancers. Nanoscale. 2022;14:15735–15748. doi: 10.1039/d2nr04235e. [DOI] [PubMed] [Google Scholar]
- 101.Grunwald B., Vandooren J., Locatelli E., Fiten P., Opdenakker G., Proost P., et al. Matrix metalloproteinase-9 (MMP-9) as an activator of nanosystems for targeted drug delivery in pancreatic cancer. J Control Release. 2016;239:39–48. doi: 10.1016/j.jconrel.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 102.Halbrook C.J., Pontious C., Kovalenko I., Lapienyte L., Dreyer S., Lee H.J., et al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 2019;29:1390–1399. doi: 10.1016/j.cmet.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhu Y., Yu X., Thamphiwatana S.D., Zheng Y., Pang Z. Nanomedicines modulating tumor immunosuppressive cells to enhance cancer immunotherapy. Acta Pharm Sin B. 2020;10:2054–2074. doi: 10.1016/j.apsb.2020.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhao H., Zhao B., Wu L., Xiao H., Ding K., Zheng C., et al. Amplified cancer immunotherapy of a surface-engineered antigenic microparticle vaccine by synergistically modulating tumor microenvironment. ACS Nano. 2019;13:12553–12566. doi: 10.1021/acsnano.9b03288. [DOI] [PubMed] [Google Scholar]
- 105.Zhou W., Zhou Y., Chen X., Ning T., Chen H., Guo Q., et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials. 2021;268 doi: 10.1016/j.biomaterials.2020.120546. [DOI] [PubMed] [Google Scholar]
- 106.Sun H., Saeedi P., Karuranga S., Pinkepank M., Ogurtsova K., Duncan B.B., et al. IDF diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183 doi: 10.1016/j.diabres.2021.109119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gregory G.A., Robinson T., Linklater S.E., Wang F., Colagiuri S., de Beaufort C., et al. Global incidence, prevalence, and mortality of type 1 diabetes in 2021 with projection to 2040: a modelling study. Lancet Diabetes Endocrinol. 2022;10:741–760. doi: 10.1016/S2213-8587(22)00218-2. [DOI] [PubMed] [Google Scholar]
- 108.Weir G.C., Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. 2004;53(Suppl 3):S16–S21. doi: 10.2337/diabetes.53.suppl_3.s16. [DOI] [PubMed] [Google Scholar]
- 109.Kim J.W., Ko S.H., Cho J.H., Sun C., Hong O.K., Lee S.H., et al. Loss of beta-cells with fibrotic islet destruction in type 2 diabetes mellitus. Front Biosci. 2008;13:6022–6033. doi: 10.2741/3133. [DOI] [PubMed] [Google Scholar]
- 110.Wang J., Li T., Zhou Y., Wang X., Carvalho V., Ni C., et al. Genetic lineage tracing reveals stellate cells as contributors to myofibroblasts in pancreas and islet fibrosis. iScience. 2023;26 doi: 10.1016/j.isci.2023.106988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yang Y., Kim J.W., Park H.S., Lee E.Y., Yoon K.H. Pancreatic stellate cells in the islets as a novel target to preserve the pancreatic β-cell mass and function. J Diabetes Investig. 2020;11:268–280. doi: 10.1111/jdi.13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Forrester S.J., Booz G.W., Sigmund C.D., Coffman T.M., Kawai T., Rizzo V., et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. 2018;98:1627–1738. doi: 10.1152/physrev.00038.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Feng N., Yu H., Wang Y., Zhang Y., Xiao H., Gao W. Exercise training attenuates angiotensin II-induced cardiac fibrosis by reducing POU2F1 expression. J Sport Health Sci. 2023;12:464–476. doi: 10.1016/j.jshs.2022.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ko S.H., Hong O.K., Kim J.W., Ahn Y.B., Song K.H., Cha B.Y., et al. High glucose increases extracellular matrix production in pancreatic stellate cells by activating the renin-angiotensin system. J Cel Biochem. 2006;98:343–355. doi: 10.1002/jcb.20797. [DOI] [PubMed] [Google Scholar]
- 115.Hong O.K., Lee S.H., Rhee M., Ko S.H., Cho J.H., Choi Y.H., et al. Hyperglycemia and hyperinsulinemia have additive effects on activation and proliferation of pancreatic stellate cells: possible explanation of islet-specific fibrosis in type 2 diabetes mellitus. J Cel Biochem. 2007;101:665–675. doi: 10.1002/jcb.21222. [DOI] [PubMed] [Google Scholar]
- 116.Lee E., Ryu G.R., Ko S.H., Ahn Y.B., Song K.H. A role of pancreatic stellate cells in islet fibrosis and β-cell dysfunction in type 2 diabetes mellitus. Biochem Biophys Res Commun. 2017;485:328–334. doi: 10.1016/j.bbrc.2017.02.082. [DOI] [PubMed] [Google Scholar]
- 117.Twarda-Clapa A., Olczak A., Bialkowska A.M., Koziolkiewicz M. Advanced glycation end-products (ages): formation, chemistry, classification, receptors, and diseases related to ages. Cells. 2022;11:1312. doi: 10.3390/cells11081312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Singh R., Barden A., Mori T., Beilin L. Advanced glycation end-products: a review. Diabetologia. 2001;44:129–146. doi: 10.1007/s001250051591. [DOI] [PubMed] [Google Scholar]
- 119.Jud P., Sourij H. Therapeutic options to reduce advanced glycation end products in patients with diabetes mellitus: a review. Diabetes Res Clin Pract. 2019;148:54–63. doi: 10.1016/j.diabres.2018.11.016. [DOI] [PubMed] [Google Scholar]
- 120.Salazar J., Navarro C., Ortega A., Nava M., Morillo D., Torres W., et al. Advanced glycation end products: new clinical and molecular perspectives. Int J Environ Res Public Health. 2021;18:7236. doi: 10.3390/ijerph18147236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tsai P.S., Chiu C.Y., Sheu M.L., Yang C.Y., Lan K.C., Liu S.H. Advanced glycation end products activated endothelial-to-mesenchymal transition in pancreatic islet endothelial cells and triggered islet fibrosis in diabetic mice. Chem Biol Interact. 2021;345 doi: 10.1016/j.cbi.2021.109562. [DOI] [PubMed] [Google Scholar]
- 122.Park S.K., Kwon K., Ryu D., Han J., Choi H.G. Protective effect of neorhodomela aculeata methanolic extract through the suppressive action on NF-κB and stat pathway in IL-1β and IFN-γ induced β-cell damage. Genes Genomics. 2010;32:239–246. [Google Scholar]
- 123.Dinarello C.A. A clinical perspective of IL-1β as the gatekeeper of inflammation. Eur J Immunol. 2011;41:1203–1217. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- 124.Carvalho V., Wang Q., Xu X., Liu L., Jiang W., Wang X., et al. Long-term exercise preserves pancreatic islet structure and β-cell mass through attenuation of islet inflammation and fibrosis. FASEB J. 2023;37 doi: 10.1096/fj.202201879R. [DOI] [PubMed] [Google Scholar]
- 125.Kim J.J., Lee E., Ryu G.R., Ko S.H., Ahn Y.B., Song K.H. Hypoxia increases β-cell death by activating pancreatic stellate cells within the islet. Diabetes Metab J. 2020;44:919–927. doi: 10.4093/dmj.2019.0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee E., Ryu G.R., Ko S.H., Ahn Y.B., Song K.H. Pancreatic stellate cells promote pancreatic β-cell death through exosomal microRNA transfer in hypoxia. Mol Cel Endocrinol. 2023;572 doi: 10.1016/j.mce.2023.111947. [DOI] [PubMed] [Google Scholar]
- 127.Kim C.L.T., Hosaka T., Yoshida M., Harada N., Sakaue H., Sakai T., et al. Exendin-4, a GLP-1 receptor agonist, directly induces adiponectin expression through protein kinase a pathway and prevents inflammatory adipokine expression. Biochem Biophys Res Commun. 2009;390:613–618. doi: 10.1016/j.bbrc.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 128.Li W., Cui M., Wei Y., Kong X., Tang L., Xu D. Inhibition of the expression of TGF-β1 and CTGF in human mesangial cells by exendin-4, a glucagon-like peptide-1 receptor agonist. Cell Physiol Biochem. 2012;30:749–757. doi: 10.1159/000341454. [DOI] [PubMed] [Google Scholar]
- 129.Kim J.W., Park S.Y., You Y.H., Ham D.S., Lee S.H., Yang H.K., et al. Suppression of ROS production by exendin-4 in PSC attenuates the high glucose-induced islet fibrosis. PLoS One. 2016;11 doi: 10.1371/journal.pone.0163187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Macdonald F.R., Peel J.E., Jones H.B., Mayers R.M., Westgate L., Whaley J.M., et al. The novel sodium glucose transporter 2 inhibitor dapagliflozin sustains pancreatic function and preserves islet morphology in obese, diabetic rats. Diabetes Obes Metab. 2010;12:1004–1012. doi: 10.1111/j.1463-1326.2010.01291.x. [DOI] [PubMed] [Google Scholar]
- 131.Okauchi S., Shimoda M., Obata A., Kimura T., Hirukawa H., Kohara K., et al. Protective effects of SGLT2 inhibitor luseogliflozin on pancreatic β-cells in obese type 2 diabetic db/db mice. Biochem Biophys Res Commun. 2016;470:772–782. doi: 10.1016/j.bbrc.2015.10.109. [DOI] [PubMed] [Google Scholar]
- 132.Pasquel F.J., Lansang M.C., Dhatariya K., Umpierrez G.E. Management of diabetes and hyperglycaemia in the hospital. Lancet Diabetes Endocrinol. 2021;9:174–188. doi: 10.1016/S2213-8587(20)30381-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sims E.K., Carr A., Oram R.A., Dimeglio L.A., Evans-Molina C. 100 years of insulin: celebrating the past, present and future of diabetes therapy. Nat Med. 2021;27:1154–1164. doi: 10.1038/s41591-021-01418-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hunt N.J., Lockwood G.P., Heffernan S.J., Daymond J., Ngu M., Narayanan R.K., et al. Oral nanotherapeutic formulation of insulin with reduced episodes of hypoglycaemia. Nat Nanotechnol. 2024;4:534–544. doi: 10.1038/s41565-023-01565-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cheng H., Cui Z., Guo S., Zhang X., Huo Y., Mao S. Mucoadhesive versus mucopenetrating nanoparticles for oral delivery of insulin. Acta Biomater. 2021;135:506–519. doi: 10.1016/j.actbio.2021.08.046. [DOI] [PubMed] [Google Scholar]
- 136.Ko S.H., Kwon H.S., Kim S.R., Moon S.D., Ahn Y.B., Song K.H., et al. Ramipril treatment suppresses islet fibrosis in Otsuka Long-Evans Tokushima fatty rats. Biochem Biophys Res Commun. 2004;316:114–122. doi: 10.1016/j.bbrc.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 137.Tham D.M., Martin-Mcnulty B., Wang Y.X., Wilson D.W., Vergona R., Sullivan M.E., et al. Angiotensin II is associated with activation of NF-kappaB-mediated genes and downregulation of PPARs. Physiol Genomics. 2002;11:21–30. doi: 10.1152/physiolgenomics.00062.2002. [DOI] [PubMed] [Google Scholar]
- 138.Lee E., Ryu G.R., Ko S.H., Ahn Y.B., Yoon K.H., Ha H., et al. Antioxidant treatment may protect pancreatic beta cells through the attenuation of islet fibrosis in an animal model of type 2 diabetes. Biochem Biophys Res Commun. 2011;414:397–402. doi: 10.1016/j.bbrc.2011.09.087. [DOI] [PubMed] [Google Scholar]
- 139.Ryu G.R., Lee E., Chun H.J., Yoon K.H., Ko S.H., Ahn Y.B., et al. Oxidative stress plays a role in high glucose-induced activation of pancreatic stellate cells. Biochem Biophys Res Commun. 2013;439:258–263. doi: 10.1016/j.bbrc.2013.08.046. [DOI] [PubMed] [Google Scholar]
- 140.Xu X.F., Liu F., Xin J.Q., Fan J.W., Wu N., Zhu L.J., et al. Respective roles of the mitogen-activated protein kinase (MAPK) family members in pancreatic stellate cell activation induced by transforming growth factor-beta1 (TGF-β1) Biochem Biophys Res Commun. 2018;501:365–373. doi: 10.1016/j.bbrc.2018.04.176. [DOI] [PubMed] [Google Scholar]
- 141.Ahangarpour A., Oroojan A.A., Khorsandi L., Kouchak M., Badavi M. Antioxidant, anti-apoptotic, and protective effects of myricitrin and its solid lipid nanoparticle on streptozotocin-nicotinamide-induced diabetic nephropathy in type 2 diabetic male mice. Iran J Basic Med Sci. 2019;22:1424–1431. doi: 10.22038/IJBMS.2019.13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hussein J., El-Banna M., Razik T.A., El-Naggar M.E. Biocompatible zinc oxide nanocrystals stabilized via hydroxyethyl cellulose for mitigation of diabetic complications. Int J Biol Macromol. 2018;107:748–754. doi: 10.1016/j.ijbiomac.2017.09.056. [DOI] [PubMed] [Google Scholar]
- 143.Detlefsen S., Sipos B., Feyerabend B., Kloppel G. Fibrogenesis in alcoholic chronic pancreatitis: the role of tissue necrosis, macrophages, myofibroblasts and cytokines. Mod Pathol. 2006;19:1019–1026. doi: 10.1038/modpathol.3800613. [DOI] [PubMed] [Google Scholar]
- 144.Masamune A., Watanabe T., Kikuta K., Shimosegawa T. Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin Gastroenterol Hepatol. 2009;7:S48–S54. doi: 10.1016/j.cgh.2009.07.038. [DOI] [PubMed] [Google Scholar]
- 145.Andersen D.K., Korc M., Petersen G.M., Eibl G., Li D., Rickels M.R., et al. Diabetes, pancreatogenic diabetes, and pancreatic cancer. Diabetes. 2017;66:1103–1110. doi: 10.2337/db16-1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rad F., Ghorbani M., Mohammadi R.A., Habibi R.M. Mesenchymal stem cell-based therapy for autoimmune diseases: emerging roles of extracellular vesicles. Mol Biol Rep. 2019;46:1533–1549. doi: 10.1007/s11033-019-04588-y. [DOI] [PubMed] [Google Scholar]
- 147.Han M.M., He X.Y., Tang L., Qi L., Yang M.Y., Wang Y., et al. Nanoengineered mesenchymal stem cell therapy for pulmonary fibrosis in young and aged mice. Sci Adv. 2023;9 doi: 10.1126/sciadv.adg5358. [DOI] [PMC free article] [PubMed] [Google Scholar]