

Abstract
The pathobiology of IL‐17 in lung fibrogenesis is controversial. Here we examined the role of IL‐17A/F in bleomycin (BLM) and adenoviral TGF‐β1‐induced lung fibrosis in mice. In both experimental models, WT and IL17af−/− mice showed increased collagen contents and remodeled lung architecture as assessed by histopathological examination, suggesting that IL‐17A/F is dispensable for lung fibrogenesis. However, IL17af−/− mice responded to the BLM challenge with perturbed lung leukocyte subset recruitment. More specifically, bleomycin triggered angiocentric neutrophilic infiltrations of the lung accompanied by increased mortality of IL17af −/− but not WT mice. WT bone marrow transplantation failed to correct this phenotype in BLM‐challenged IL17af −/− mice. Conversely, IL17a/f −/− bone marrow transplantation → WT did not perturb lung leukocytic responses upon BLM. At the same time, IL17af−/− mice treated with recombinant IL‐17A/F showed an attenuated lung inflammatory response to BLM. Together, the data show that the degree of BLM‐driven acute lung injury was critically dependent on the presence of IL‐17A/F, while in both models, the fibrotic remodeling process was not.
Keywords: Fibrosis, IL‐17, Injury, Lung, Th17, γδ T cells
Bleomycin‐induced acute lung injury but not fibrosis is regulated in WT mice by the presence of radioresistant innate IL‐17A/F‐producing T cells and is exacerbated in IL17af−/− mice. Recombinant IL‐17A/F therapy but not WT bone marrow transplantation ameliorates bleomycin induced acute lung injury in IL17af−/− mice.
Introduction
Idiopathic pulmonary fibrosis (IPF) is an interstitial lung disease (ILD) characterized by progressive scarring of the lung tissue with a concomitant decline of lung function and fatal outcome within 3–5 years after diagnosis. It has an incidence of 3–9 cases per 100,000 individuals per year [1, 2]. The pathogenesis of IPF is still poorly understood. Increasing evidence indicates a multistep disease process involving a continuously decreasing constructive/regenerative potential of lung progenitor populations. This decline is, at least in part, triggered by recurrent injurious events eventually causing atypical lung tissue remodeling along with increased (myo‐) fibroblast‐dependent deposition of extracellular matrix proteins and subsequent stiffening of the lung [3]. Besides resident lung cells like epithelial cells, fibroblasts, and myofibroblasts, several innate and adaptive immune cell populations such as macrophages, neutrophils, and lymphocyte subsets contribute to the process of lung tissue remodeling [4].
IL‐17, originally named cytotoxic T‐lymphocyte‐associated protein 8 gained attention in several immunopathological processes including mucosal barrier integrity and host defense against bacterial and fungal pathogens [5, 6, 7]. The IL‐17 cytokine family consists of six members named IL‐17A‐IL‐17F, with IL‐17A and IL‐17F representing the most abundant ones sharing 55% homology. IL‐17 is produced by various immune cells including CD4pos T helper (Th) 17 cells, γδ T cells, group 3 innate lymphoid cells, natural killer cells, natural killer T cells, and neutrophils. IL‐17A and IL‐17F are homodimeric proteins that can also form heterodimeric IL‐17A/F complexes. These cytokines appear to signal through a ternary complex with the IL‐17RA and IL‐17RC receptors [8, 9, 10]. More recent studies showed that IL‐17F may also form homodimeric complexes with IL‐17RC independent of IL‐17A [11]. While IL‐17RA is expressed in various tissues and cell types, IL‐17RC expression is confined to non‐hematopoietic tissues [9]. Both IL‐17A and IL‐17F bind as homo‐ or heterodimers to the IL‐17RA/IL‐17RC receptor complex. They are considered to play a central role in neutrophil recruitment through the stimulation of target cells that secrete neutrophil‐attracting chemokines including CXCL1 and CXCL2, which act as functional homologs of human IL‐8 in mice. CXCL1 and CXCL2, along with proinflammatory cytokines and growth factors like G‐CSF, play important roles in the activation and attraction of neutrophils in mice [12, 13].
The role of IL‐17 in lung fibrosis is controversial. Bronchoalveolar lavage (BAL) fluids of IPF patients display increased levels of IL‐17A compared with healthy controls, and IL‐17A‐producing cells were found in active disease areas of IPF lungs [14, 15]. In mice, several studies reported a profibrotic function of IL‐17A, and IL‐17A deficiency (or IL‐17A neutralizing antibodies) attenuated BLM‐induced lung fibrosis [14, 16–18]. In contrast, other reports demonstrated increased lung fibrosis in IL‐17A KO mice after BLM treatment [19].
To the best of our knowledge, the effect of combined IL‐17A/F double knockout (IL17af−/− ) on experimental lung fibrosis has not been explored so far [17, 20]. Therefore, we examined the role of combined IL‐17A/F deficiency in two well‐defined models of BLM and adenoviral TGF‐β1 (AdTGF‐β1) induced experimental lung fibrosis in mice, thereby ruling out mutual compensatory effects that might occur in single KO mice. We found that the acute phase of bleomycin‐induced lung injury but not the ensuing fibrotic remodeling process itself was critically dependent on IL‐17A/F. In contrast, AdTGF‐β1 induced lung fibrosis was completely independent of IL‐17A/F.
Materials and methods
Animals
WT mice (C57Bl/6J) were purchased from Janvier Lab. IL17af −/− mice were generated on a C57Bl/6J background as previously described [21, 22], and were bred at the Central Animal Facility of Hannover Medical School under specific pathogen‐free conditions. Mice were aged 8–14 weeks and were sex‐matched in all experiments. All animal experiments corresponded to the European guideline 2010/63/EU as well as the German Animal Welfare Act and were authorized by the Lower Saxony State Office for Consumer Protection and Food Safety, Germany.
Reagents
Bleomycin sulfate was purchased from Medac. Lipopolysaccharide (LPS) was purchased from Sigma‐Aldrich. Recombinant mouse IL‐17A/F protein (rIL‐17A/F) was purchased from R&D Systems. Antibodies employed for flow cytometry, including anti‐CD3 FITC (clone 145‐2C11), anti‐CD3 V450 (clone 500A2), anti‐CD4 PerCP‐Cy5.5 (clone RM4‐5), anti‐CD4 BV786 (clone RM4‐5), anti‐CD8 BV510 (clone 53–6.7), anti‐CD19 FITC (clone 1D3), anti‐CD19 PE (clone 1D3), anti‐CD45 PECy7 (clone 30‐F11), anti‐Ly6G PE (clone 1A8), anti‐TCRαβ V450 (clone H57‐597), anti‐TCRγδ PE (clone GL‐3), and anti‐TCRγδ BV650 (clone GL‐3) were purchased from BD Biosciences. Anti‐CD25 APC (clone PC61.5), anti‐Foxp3 PE (clone FJK16s, rat IgG2a), anti‐IL‐17A FITC (clone eBio17B7, mouse IgG2aκ), and anti‐IL‐17F PE (clone eBio18F10, rat IgG2aκ) were purchased from Thermo Fisher Scientific.
Treatment of mice
Replication‐deficient adenoviral vectors either carrying the cDNA of biologically active TGF‐β1 (AdTGF‐β1) or empty control vector (AdCL) were prepared as previously detailed [23]. WT or IL17af −/− mice were anesthetized with an intraperitoneal injection of ketamine (75 mg/kg body weight; cp‐pharma) and xylazine (3 mg/kg body weight; Bayer) and were then intubated with an Abbocath‐T 26G catheter (Abbott), which was inserted into the trachea. Mice received either intratracheal installations of 0.03 units/mouse BLM diluted in 50 µL saline, 10 µg LPS diluted in 50 µL PBS, or 1.5 × 108 plaque‐forming units of AdTGF‐β1 diluted in 50 µL PBS. Mice of the control group received instillations of either 0.9% saline, PBS, or empty control vector (AdCL), respectively. Following instillation, mice were set back into their cages with free access to food and water. Mice were monitored twice daily for disease symptoms on the basis of an established scoring system and were euthanized as soon as reaching a previously defined moribund status.
Generation of bone marrow chimeric mice
Bone marrow chimeric mice were generated as outlined previously [24, 25, 26]. Briefly, WT and IL17af−/− mice were subjected to whole‐body irradiation (total dose, 8 Gy) followed by i.v. injection of 107 purified bone marrow cells per mouse collected from either WT or IL17af−/− mice. To monitor the effect of a hematopoietic deletion or reconstitution of IL‐17A/F on BLM‐induced lung injury, four different groups of bone marrow chimeric mice were generated, that is, WT→WT, WT→KO, KO→WT, KO→KO chimeric mice. In selected experiments, IL17af−/− mice expressing the CD45.2 alloantigen received bone marrow cells from CD45.1 expressing WT mice (CD45.1 WT→IL‐17 dKO) and vice versa, CD45.1 expressing WT mice received bone marrow cells from CD45.2 expressing IL17af−/− mice (IL‐17 dKO→CD45.1 WT), allowing us to determine leukocyte subset engraftment/turnover efficiencies based on the detection of CD45.1 versus CD45.2 alloantigens [27]. Reconstitution of the hematopoietic system was achieved after 7 weeks, with engraftment efficacies of >92%. Chimeric mice were kept in individually ventilated cages with free access to autoclaved food and water.
Application of rIL‐17A/F
In selected experiments, IL17af−/− mice received i.v. applications of 10 µg rIL‐17A/F protein diluted in 150 µL of sterile PBS/HSA on days 0, 1, 3, and 5 post‐BLM treatment. Mice were sacrificed on day 6 post‐BLM treatment for immunophenotypic analysis of BAL leukocyte subsets and histopathological examination of lung tissue specimens.
Bronchoalveolar lavage, preparation of lung homogenates, and collection of plasma
BAL of mice was performed as previously described [28]. Briefly, the lungs were removed and homogenized in PBS supplemented with a protease inhibitor cocktail (Roche). For preparation of cell‐free lung homogenate supernatants, lung tissue homogenates were filtered through a 100 µm cell strainer (Greiner) and centrifuged twice at 13,000 rpm for 10 min at 4°C. Whole blood was withdrawn from the vena cava inferior using Lithium‐heparin monovettes (Sarstedt), and plasma was collected by centrifugation of heparinized blood at 4500 rpm at room temperature for 6 min. Cell‐free BAL fluids (BALF), lung homogenate supernatants, and plasma samples were frozen at –20°C until further analysis.
Analysis of leukocyte subsets in bone marrow, peripheral blood, lung tissue, and BALF of mice
Blood smears were prepared on glass slides using anticoagulated blood, followed by Pappenheim staining for differentiation and subsequent enumeration of peripheral blood leukocyte subsets. Leukocyte subsets collected from bone marrow, digested lung tissue, and BALF were processed for flow cytometric gating and immunophenotypic analysis as well as fluorescence‐activated cell sorting, as previously described [29, 30, 31]. Briefly, the femurs and tibias of mice were flushed with RPMI/ 10% FCS for collection of bone marrow cells. The lung lobes of mice were teased into small pieces and digested in RPMI 1640 supplemented with collagenase A (5 mg/mL) and DNase I (1 mg/mL). For fluorescence‐activated cell sorting of TCRαβpos and TCRγδpos T cells, CD3pos T cells were enriched using a magnetic cell separation kit (Miltenyi Biotec). T cells were identified according to their forward scatter area versus side scatter area characteristics and identified as either CD3/TCRαβ or CD3/TCRγδ expressing cells. For immunophenotyping of lung leukocytes, cell suspensions were enriched for CD45pos leukocytes (Magnetic cell separation kit, Miltenyi Biotec). Neutrophils were characterized by their Ly6Gpos cell surface expression. Lymphocytes including regulatory T cells (Tregs) were gated according to their forward scatter area versus side scatter area characteristics and were further distinguished by their CD3posCD4pos, CD3posCD8pos, CD4posCD25posFoxP3pos, CD3posTCRγδpos, or CD19pos immunophenotype. For the determination of intracellular IL‐17A and IL‐17F proteins in respective T‐cell subsets, CD90.2pos T cells were fixed and permeabilized using an intracellular IL‐17A/F staining kit (eBioscience). Specific staining of IL1‐7A and IL‐17F was confirmed by use of respective isotype controls (for anti‐IL‐17A: clone eBio17B7, mouse IgG2aκ; for anti‐IL‐17F: clone eBio18F10, rat IgG2aκ). Immunophenotypic analyses were performed using a BD LSR Fortessa flow cytometer. Postacquisition compensation settings were performed using the BD FACSDiva software.
Flow sorting of lung lymphocyte subsets was carried out under sterile conditions at 4°C with a BD FACSAria III flow cytometer (BD Biosciences) equipped with a 70 µm nozzle. Post‐sort analysis revealed purities of >98% for sorted CD3pos TCRαβpos T cells and >93% for sorted CD3pos TCRγδpos T cells.
Stimulation of T cells for intracellular staining of IL‐17A/F
T cells were isolated from the spleens or lungs of WT and IL17af−/− mice using CD90.2 microbeads, according to the manufacturer's instructions (Miltenyi). Approximately 1.5 × 106 T cells suspended in 150 µL RPMI supplemented with FCS, glutamine, Pen/Strep, and β‐mercaptoethanol were incubated with a cell stimulation cocktail containing PMA (0.05 µg/mL) and ionomycin (1 µg/mL) in the presence of protein transport inhibitors Brefeldin and Monensin at 37°C, 5% CO2. After 3 h of incubation, T cells were washed and stained with anti‐CD3 V450, anti‐CD4 PerCP‐Cy5.5, and anti‐CD8 APC antibodies, followed by fixation and permeabilization (FixPerm, ThermoFisher) for 30 min. Subsequently, cells were stained with FITC‐conjugated anti‐IL‐17A and PE‐conjugated anti‐IL17F antibodies (ThermoFisher) for 30 min at room temperature. FACS analysis was performed on a BD LSR Fortessa flow cytometer equipped with FACSDiva software.
Real‐time PCR
Sorted CD3pos TCRαβpos or TCRγδpos T cells were subjected to isolation of total RNA using the RNAeasy Micro kit (Qiagen) according to the manufacturer's instructions. Synthesis of cDNA and real‐time RT‐PCR were performed as previously described [32]. PCR primers for murine IL‐17A (forward primer 5′‐ACCGCAATGAAGACCCTGAT‐3′; reverse primer 5′‐TCCCTCCGCATTGACACA‐3′) and IL‐17F (forward primer 5′‐CTGAGGCCCAGTGCAGACA‐3′; reverse primer 5′‐GCTGAATGGCGACGGAGTT‐3′) were designed based on gene sequence data retrieved from GenBank (IL‐17A, U43088.1, IL‐17F, AF458064.1). Murine β‐actin (forward primer 5′‐CCACAGCTGAGAGGGAAATC‐3′; reverse primer 5′‐TCTCCAGGGAGGAAGAGGAT‐3′) was used as a housekeeping gene. The 2−ΔΔCt method was used for the calculation of mean fold changes between groups [33].
Mouse lung histopathology
Mice were euthanized at indicated time points and lungs were inflated in situ with 4% formalin fixation medium (ROTI Histofix, Carl Roth), removed en bloc, and immersed in fixation medium for 24 h at room temperature (RT). Individual lung lobes were embedded in paraffin, sectioned at 2.5 µm, and stained with hematoxylin/eosin. Lung tissue sections were examined under blinded conditions using an Olympus BX53 microscope (Olympus).
Determination of lung collagen contents
Lung collagen contents were determined using a hydroxyproline dye binding assay according to previously published protocols [34, 35, 36].
Determination of lung permeability
Mice received an intravenous injection of FITC‐labeled bovine serum albumin (1 mg/mouse in 100 µL saline; Sigma‐Aldrich) 1 h before sacrifice. FITC fluorescence intensities of undiluted BAL supernatants and plasma samples (diluted 1/100 in saline) were measured using a fluorescence spectrometer (FLx800 microplate fluorescence reader; Bio‐Tek) operating at 485 nm absorbance and 528 ± 20 nm emission wavelengths. Lung permeability is defined as the ratio of FITC fluorescence in undiluted BAL supernatants relative to 1:100 diluted plasma samples [37].
Quantification of cytokines
Cytokine levels of IL‐17A and IL‐17F (detection limits 15.6 pg/mL), murine TGF‐β1 (detection limit 31.2 pg/mL), and G‐CSF (detection limit, 14.1 pg/mL) were measured in cell‐free BAL fluids and lung homogenate supernatants using commercially available ELISA kits (R&D Systems). Levels of CXCL‐1 and 2 were measured using Luminex bead arrays (R&D Systems; detection limits for CXCL1, 29.7 pg/mL and for CXCL2, 4.8 pg/mL).
Statistical analysis
All data were analyzed with GraphPad Prism software (version 7). Differences between groups were analyzed by the Mann–Whitney U‐test, and survival data were compared using the log‐rank test. Statistically significant differences between groups were assumed when p‐values were ≤0.05.
Results
BLM triggers expression of IL‐17A and IL‐17F in mouse lungs
We initially examined gene expression levels of IL‐17A and IL‐17F in sorted TCRαβpos and TCRγδpos T cells of mice challenged with BLM. TCRαβpos T cells showed a strongly increased IL‐17A and IL‐17F gene expression peaking at 24 h posttreatment (Fig. 1A, Supporting Information Fig. S1). Similarly, TCRγδpos T cells responded to BLM with increased IL‐17A and IL‐17F gene expression, particularly at 72 h posttreatment (Fig. 1B). Analysis of intracellular IL‐17A and IL‐17F protein in lung CD4pos, CD8pos, and TCRγδpos T cells of WT mice revealed IL‐17A and IL‐17F positive CD4pos T cells but not CD8pos T cells (data not shown), and IL‐17A and F protein expressing TCRγδpos T cells at 72 h post‐BLM, relative to saline treatment. As expected, T‐cell subsets of IL17af−/− mice used as negative controls lacked any detectable IL‐17A/F protein (Fig. 1C and D). In selected experiments, we confirmed the principal inducibility of IL‐17A and IL‐17F protein, particularly in CD4+ T cells but also TCRγδpos T cells of untreated and BLM‐challenged mice after stimulation with PMA/ionomycin in vitro (Supporting Information Fig. S2). IL‐17A levels increased in BALF of mice as early as 24 h post‐BLM challenge, whereas in lung homogenates, peak IL‐17A levels were observed at day 3 posttreatment (Fig. 1E and F). In contrast, IL‐17F protein was not detectable in BALF or lung homogenates of BLM‐challenged WT mice (data not shown). Collectively, the BLM challenge triggers both IL‐17A/F RNA transcription particularly in CD4pos T cells and TCRγδpos T cells, leading to a predominant IL‐17A protein release in the lungs of mice.
Figure 1.
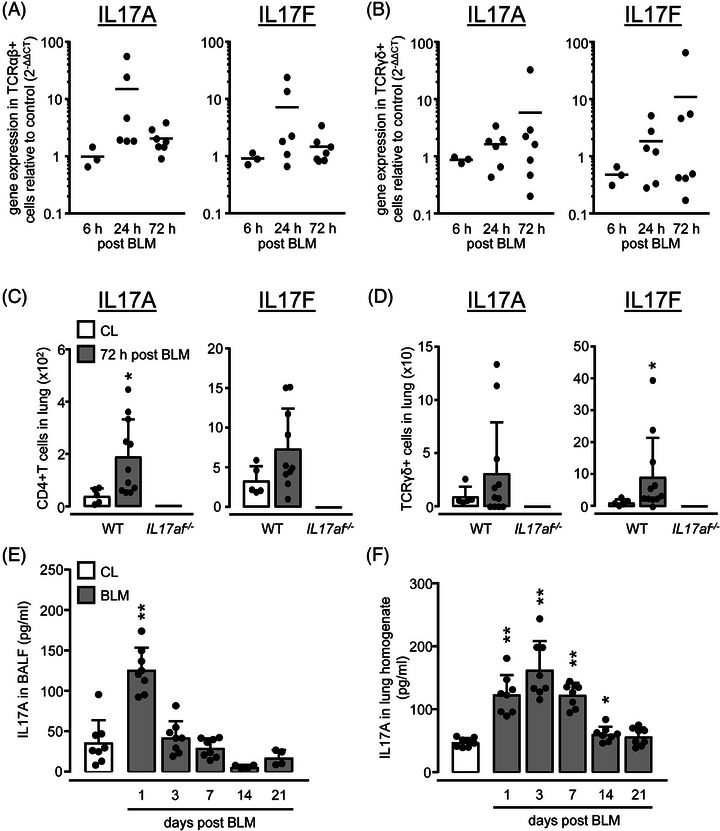
IL‐17A and IL‐17F gene and protein expression in lungs of BLM challenged mice. WT mice were treated with saline (CL, white bars) or BLM (grey bars) for 6 h, 24 h, 72 h, 7 days, 14 days, or 21 days, as indicated. (A, B) Gene expression levels of IL‐17A and IL‐17F in sorted TCRαβpos (A) and TCRγδpos T cells collected from lungs of BLM‐exposed WT mice. (C, D) Intracellular IL‐17A and IL‐17F protein expression of CD4pos T cells (C) and TCRγδpos T cells (D) of WT and IL17af −/− mice was measured 72 h post‐BLM by flow cytometry. (E, F) IL‐17A protein levels in BALF (E) and lung (F) of saline vs. BLM‐treated mice quantified by ELISA. Data are shown as scatter plots with mean values indicated as horizontal lines (A, B) or as mean ± SD of n = 9–12 (A, B), or n = 5–11 (C, D), or n = 4–8 (E, F) mice per group and time point. Note that in A and B, one data point represents a pool of T cells sorted from n = 2–3 mice per experimental group and time point. Data in (C–F) are representative of two independent experiments. *p ≤ 0.05, **p ≤ 0.01 compared with mice from the control group (Mann–Whitney U‐test).
IL‐17af−/− mice are highly susceptible to BLM‐induced lung injury
Next, we examined what effect IL‐17A/F double deficiency would have on fibrotic lung remodeling in BLM‐challenged mice. Unexpectedly, IL17af −/− mice had to be euthanized according to our previously defined scoring system by days 6–8 post‐BLM treatment, with an overall mortality of > 75% relative to WT mice (Fig. 2A). Importantly, this increased mortality was not due to aggravated lung fibrosis in the double mutant mice, as hydroxyproline levels were similarly increased in lungs of WT and IL17af −/− mice in response to BLM without significant differences between groups (Fig. 2B). BAL fluid levels of TGF‐β1 were similar in WT and IL17af −/− mice post‐BLM (Fig. 2C).
Figure 2.
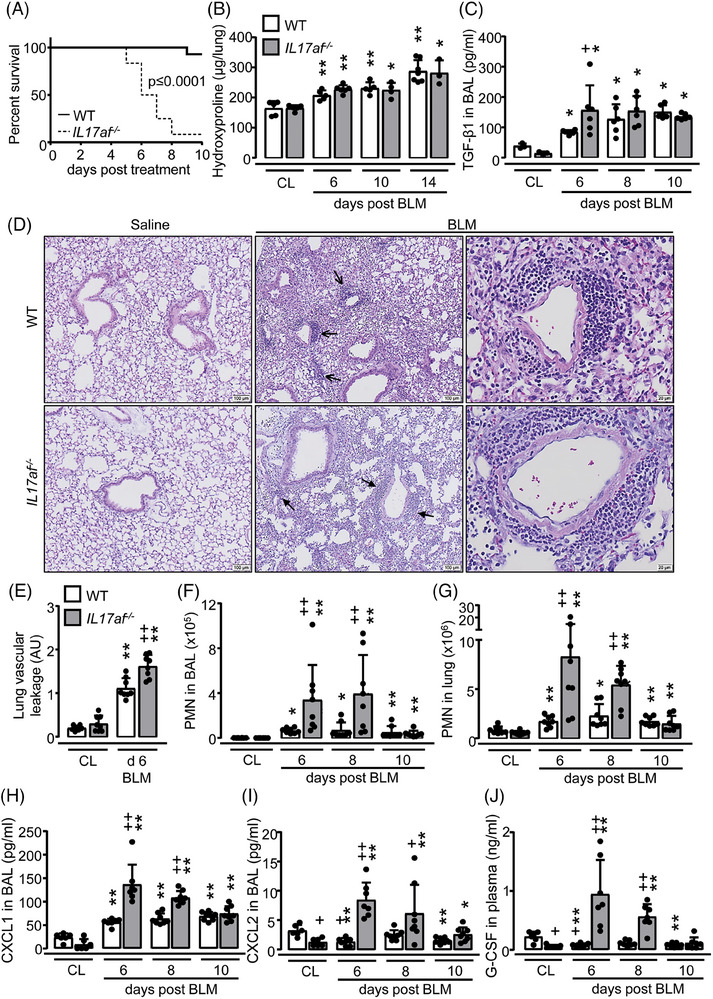
Neutrophilic angiocentric inflammation in lungs of BLM‐treated IL17af −/− mice. WT (white bars) and IL17af −/− mice (grey bars) were treated with either saline (CL) or BLM for up to 14 days. (A) Outcome of WT and IL17af −/− mice after treatment with BLM (n = 8 mice per group) (log‐rank test). (B, C) Hydroxyproline levels in lung tissue (B) and murine TGF‐β1 protein levels in BALF (C) of saline vs. BLM‐treated WT and IL17af −/− mice at days 6, 8, 10, and 14 post application, as indicated. (D) Histopathology of hematoxylin/eosin‐stained lung tissue sections of saline vs. BLM‐treated WT and IL17af −/− mice at day 6 posttreatment. Open arrows indicate lymphoplasmacellular infiltrates and closed arrows indicate angiocentric neutrophilic infiltration. Representative images from a total of n = 4 mice per experimental group are shown at original magnifications of ×10 and ×40 (scale bars = 100 and 20 µm). (E) Lung vascular leakage in saline vs. BLM‐treated WT and IL17af −/− mice at day 6 posttreatment. (F, G) Immunophenotypic analysis of neutrophils in BALF (F) and lung tissue (G) of BLM‐treated WT and IL17af −/− mice at days 6, 8, and 10 post application. (H–J) CXCL1 (H) and CXCL2 (I) protein levels in BALF and G‐CSF (J) protein levels in the plasma of saline vs. BLM‐treated WT and IL17af −/− mice at days 6, 8, and 10 post application. Data are shown as mean ± SD of n = 3–7 (B, C) or n = 6–8 (E–J) mice per group and time point. *p ≤ 0.05, **p ≤ 0.01 compared with mice from the control group. + p ≤ 0.05, ++ p ≤ 0.01 IL17af −/− relative to WT mice (Mann–Whitney U‐test).
Histopathological examination of lung tissue of saline‐treated WT and IL17a/f − /−mice showed a normal bronchial and alveolar architecture. BLM‐treated IL17af −/− mice displayed pneumonia along with an angiocentric accentuated neutrophilic infiltration of the lung parenchyma. In line with this finding, IL17af −/− mice exhibited significantly increased lung vascular leakage at day 6 post‐BLM when compared with WT mice showing a predominant lymphoplasmacellular infiltration of the peribronchovascular space (Fig. 2D and E, Supporting Information Fig. S3).
We next analyzed the neutrophilic response in WT and IL17af −/− mice exposed to BLM. At baseline, mice of both groups exhibited low neutrophil counts in BALF and lung tissue. However, a significant neutrophilia was observed in IL17af −/− mice upon BLM compared with WT, and neutrophil counts concomitantly increased in BALF and lung tissue of IL17af −/− mice on days 6 and 8 post‐BLM (Fig. 2F and G, Supporting Information Fig. S4). This neutrophil‐dominated phenotype was accompanied by increased levels of neutrophil‐attracting chemokines CXCL1 and CXCL2 in BALF as well as G‐CSF cytokine levels in the plasma of the dKO mice relative to WT post‐BLM (Fig. 2H–J). Collectively, these data show that IL‐17A/F particularly plays a regulatory role in BLM‐induced neutrophil‐dependent acute lung injury, but not in the later developing phase of lung fibrotic remodeling.
Lack of IL‐17A/F attenuates lymphocyte accumulations in response to BLM
IL17af −/− mice showed strongly decreased CD4pos and CD8pos T‐ and B‐cell accumulations in BAL and lungs post‐BLM when compared with WT (Fig. 3A–D, Supporting Information Fig. S1). Numbers of CD4/CD25pos regulatory T cells (Tregs) were also reduced in lungs of IL17af −/− mice post‐BLM (Fig. 3E). In contrast, numbers of γδ T cells were increased in lungs of IL17af −/− mice post‐BLM, relative to WT (Fig. 3F).
Figure 3.
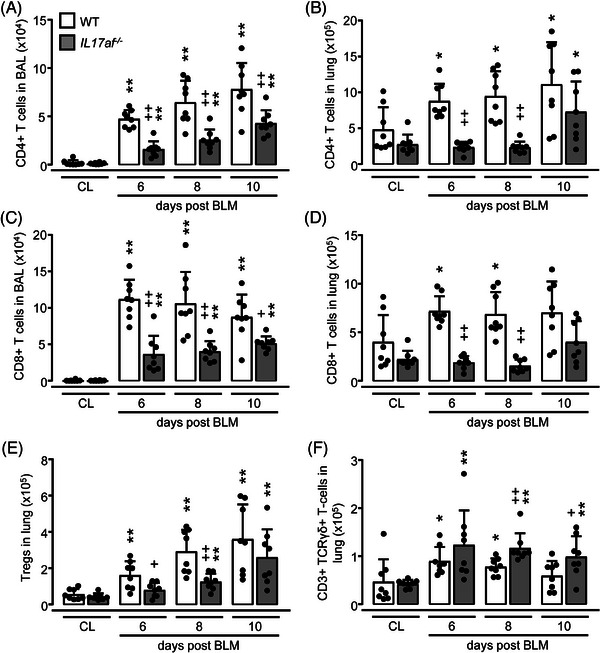
Immunophenotypic analysis of T‐cell subsets in BALF and lung tissue of BLM‐treated WT and IL17af−/− mice. WT (white bars) and IL17af−/− mice (grey bars) were treated with either saline (CL) or BLM for 6, 8, or 10 days. (A–D) Immunophenotypic analysis of CD4pos and CD8pos T cells in BALF (A, C) and lung tissue (B, D) of WT and IL17af −/− mice exposed to saline vs. BLM. (E, F) Numbers of CD4pos/CD25pos/FoxP3pos Tregs (E) and TCRγδpos T cells (F) in lung tissue of saline vs. BLM‐treated WT and IL17af −/− mice. Data are shown as mean ± SD of n = 8 mice per group and time point and representative of two independent experiments. *p ≤ 0.05, **p ≤ 0.01 compared with mice from the control group. + p ≤ 0.05, ++ p ≤ 0.01 IL17af −/− relative to WT mice (Mann–Whitney U‐test).
Effect of LPS on leukocyte subset recruitment in IL‐17A/F deficient mice
Based on the finding that BLM‐challenged IL17a/f −/− mice had a disparate lymphocytic response compared with WT, we questioned whether this would be an intrinsic feature of the knockout. However, treatment with intratracheal LPS triggered similar neutrophilic alveolitis in IL17a/f −/− mice as observed in WT mice (Fig. S5). Both WT and IL17af −/− mice displayed similar numbers of CD4pos and CD8pos T cells as well as Tregs in BALF and lungs upon LPS, while numbers of TCRγδpos T cells were significantly increased in IL17af −/− mice at days 6 to 10 post‐LPS compared with WT (Fig. S5). Together, these data show that the observed accentuated neutrophilic response of IL17af −/− mice to BLM challenge is a stimulus‐ rather than a strain‐specific feature of the double‐mutant mice.
Protection from BLM injury is mediated by radioresistant γδ T17 cells and cannot be compensated by Th17 cells
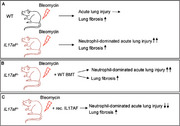
γδ T cells are the main producers of IL‐17 in the mouse lung. These cells are fetal‐derived and relatively radioresistant [38]. To investigate the contribution of other IL‐17‐producing lymphocytes, such as CD4+ Th17 cells to the inflammatory response to BLM, we next questioned whether the neutrophil‐dominated phenotype in BLM‐challenged IL17af −/− mice could be corrected by transplantation of bone marrow from WT mice. Irradiated IL17af −/− mice reconstituted with the hematopoietic system of IL17af −/− mice (KO→KO) had increased neutrophil counts in BAL and lung upon BLM challenge as opposed to WT→WT transplanted mice (Fig. 4A and B). Transplant of WT bone marrow into IL17af −/− mice (WT→KO) did not attenuate lung neutrophilia in response to BLM when compared with KO→KO mice (Fig. 4A and B). In contrast, WT mice reconstituted with the bone marrow of IL17af −/− mice (KO→WT) did not show accentuated neutrophil counts in BAL and lung after BLM challenge (Fig. 4A and B). At the same time, we found a nearly inverse relationship of lung lymphocyte counts in chimeric mice challenged with BLM: low numbers of CD4pos and CD8pos T cells, as well as Tregs, were found in lungs of KO→KO and WT→KO mice, while increased lung lymphocyte counts were found in chimeric WT→WT and KO→WT mice post‐BLM (Fig. 4C–G). Collectively, these data support that fetal‐derived radioresistant IL‐17A/F producing γδ T cells are sufficient to attenuate the acute lung injury developing in IL17af−/− mice upon BLM. In contrast, reconstituted peripheral IL‐17A/F‐producing T cells such as CD4+ Th17 cells appear to be inefficient regulators of BLM‐driven lung injury.
Figure 4.
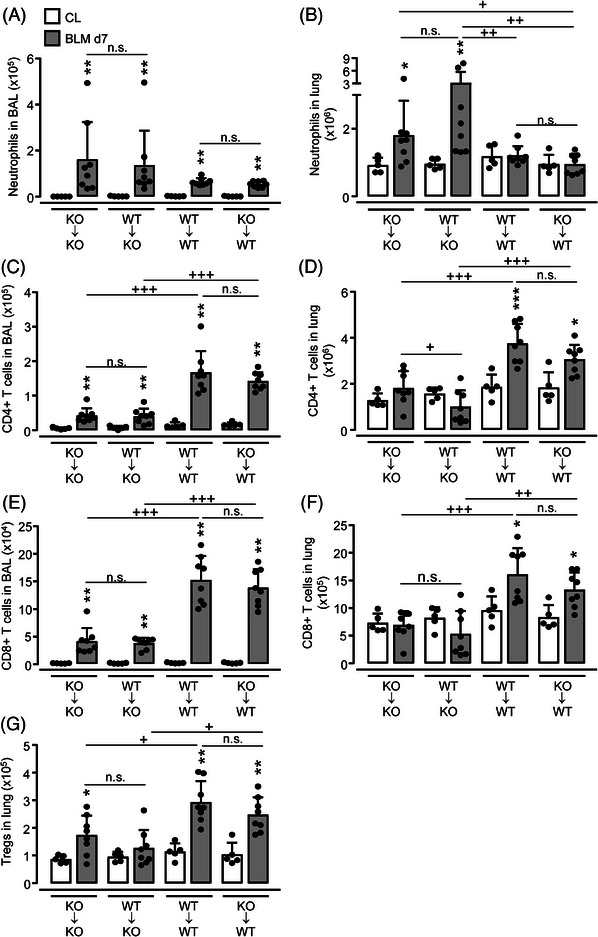
Lung leukocyte recruitment in BLM‐treated bone marrow chimeric mice. WT and IL17af −/− mice were subjected to whole‐body irradiation followed by intravenous application of bone marrow cells collected from either WT or IL17af −/− mice to generate KO→KO, WT→KO, WT→WT, and KO→WT mice, as indicated. Eight weeks posttransplantation chimeric mice were exposed to either saline (CL, white bars) or BLM (grey bars) for 7 days. (A–F) Immunophenotypic analysis of neutrophils, CD4pos, and CD8pos T cells in BALF (A, C, E) and lung tissue (B, D, F) of WT and IL17af−/− mice exposed to saline vs. BLM. (G) Numbers of CD4pos/CD25pos/FoxP3pos Tregs in lung tissue of saline vs. BLM‐treated WT and IL17af−/− mice. Data are shown as mean ± SD of n = 5–8 mice per group and time point and are representative of two independent experiments. *p ≤ 0.05, **p ≤ 0.01 compared with mice from the control group. + p ≤ 0.05, ++ p ≤ 0.01, +++ p ≤ 0.001 as indicated (Mann–Whitney U‐test). n.s. not significant.
To prove whether or not bone marrow‐derived (donor‐type) as compared with lung resident leukocytes (recipient‐type) contributed to the increased neutrophil (versus lymphocyte) accumulations in chimeric IL17af −/− or WT mice post‐BLM, we transplanted bone marrow cells from CD45.1 alloantigen‐expressing WT mice into irradiated CD45.2 alloantigen‐expressing IL17af −/− mice (CD45.1 WT→IL‐17a/f −/−) and vice versa IL‐17a/f −/−→CD45.1 WT mice. As expected, the vast majority of neutrophils in the lungs of chimeric mice of either experimental group were bone marrow‐derived donor cells (>97%), both in response to saline or BLM treatment (Fig. S6A, C, E, and H). Similarly, donor cell engraftment of blood CD4pos and CD8pos T cells in recipient chimeric mice was above 80% (Supporting Information Fig. S6B, D, F, and G). However, the ratio of donor‐ versus recipient‐type lung T cells was approximately 60:40, meaning that ∼60% of lung T‐cell subsets of chimeric mice were of donor genotype (i.e. from the hematopoietic system), whereas ∼40% of lung CD4pos and CD8pos T cells were of recipient genotype (Fig. S6I and J). These data support the view that the fraction of ∼40% of nondepleted recipient‐type CD45.1pos T cells observed in lungs of chimeric IL‐17af −/−→CD45.1 WT mice were sufficient to contribute to normalized lung neutrophil and lymphocyte counts upon BLM challenge.
Therapeutic application of rIL‐17AF attenuates BLM‐induced lung inflammation in IL17af −/− mice
We further examined whether treatment of IL17af −/− mice with mouse recombinant IL‐17A/F protein (rIL‐17A/F) would ameliorate the severe phenotype in response to BLM (Fig. 5A). Opposed to the profound neutrophil‐dominated angiocentric infiltrations of the lung in the presence of vascular coagulation in vehicle‐treated, BLM challenged IL17af −/‐ mice, BLM challenged IL17af −/− mice treated with rIL‐17A/F developed angiocentric lymphocytic infiltrations of the lung in the absence of vascular coagulation (Fig. 5B). Consistent with these results, numbers of neutrophils were decreased, while lymphocyte subset counts were increased in BALF of rIL‐17A/F treated, BLM challenged IL17af −/− mice compared with vehicle treatment and no mortality was observed in this group (Fig. 5C–E). These data demonstrate that therapy with soluble recombinant IL‐17A/F protein is sufficient to attenuate BLM‐induced acute lung injury in IL17af −/− mice.
Figure 5.
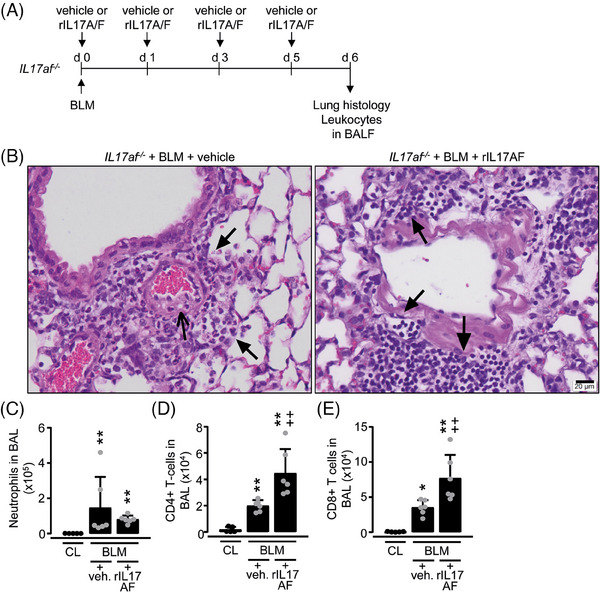
Therapy of BLM‐treated IL17af −/− mice with recombinant IL‐17A/F protein. (A) Experimental design. BLM‐exposed IL17af −/− mice were treated i.v. with rIL‐17A/F protein or vehicle, as indicated. (B) Histopathology of hematoxylin/eosin‐stained lung tissue section of IL17af −/− mice with or without rIL‐17A/F protein therapy at day 6 post‐BLM treatment. Closed arrows in (B, left histology) indicate angiocentric neutrophilic infiltrations, while open arrows in (B, left histology) denote intravascular coagulation in IL17af −/− mice treated with BLM for 6 days. Closed arrows in (B, right histology) indicate lymphoplasmacellular infiltrates in BLM‐treated IL17af −/− mice with rIL‐17A/F protein therapy. Representative histology images from a total of n = 4 mice per experimental group are shown at original magnifications of ×40 (Scale bar 20 µm). (C–E) Immunophenotypic analysis of neutrophils (C), CD4pos T cells (D), and CD8pos T cells (E) in BALF of BLM‐treated IL17af −/− mice in the absence or presence of rIL‐17A/F protein therapy. Data are shown as mean±SD of n = 5–6 mice per group and time point and are representative of two independent experiments. *p ≤ 0.05, **p ≤ 0.01 compared with mice from the control group (CL). ++ p ≤ 0.01 rIL‐17A/F application relative to vehicle (Mann–Whitney U‐test).
IL‐17A/F is dispensable in AdTGF‐β1‐induced experimental lung fibrosis
We also assessed the role of IL‐17A/F in a well‐established model of AdTGF‐β1 induced lung fibrosis in mice [29]. Compared with empty control vector‐treated mice, AdTGF‐β1 treatment did not increase IL17A protein release in BALF and lung homogenates of mice (Fig. 6A and B), and only a very low IL‐17F protein release was noted during the observation period of 21 days (data not shown). Hydroxyproline contents were similarly increased in lung tissue lysates of WT and IL17af −/− mice upon AdTGF‐β1 exposure starting by day 10 up until days 14 and 21 posttreatment without significant differences between groups (Fig. 6C). Finally, histopathological examination of lung tissue sections revealed interstitial lung collagen deposition along with widened alveolar septa in AdTGF‐β1 exposed IL17af −/− mice on days 14 and 21 posttreatment, with no overt differences compared with controls (Fig. 6D). Notably, both WT and IL17af −/− mice did not develop angiocentric neutrophilic lung inflammation upon AdTGF‐β1, and no mortality was observed in either group over time (data not shown). Together, the data show that IL‐17A/F deficiency does not appear to play a role in AdTGF‐β1‐induced lung fibrosis in mice.
Figure 6.
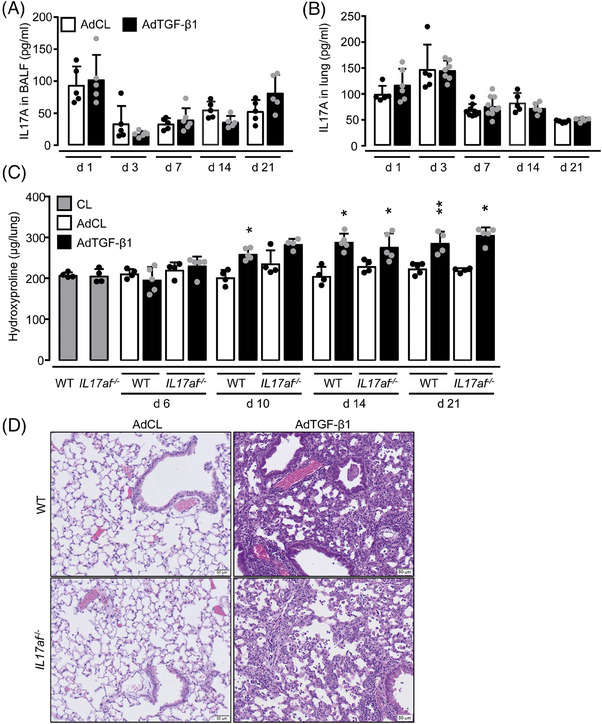
AdTGF‐β1 induced lung fibrosis in WT and IL17af −/− mice. WT and IL17af −/− mice were left untreated (grey bars) or were treated with either AdCL (white bars) or AdTGF‐β1 (black bars) for up to 21 days. (A, B) IL‐17A protein levels in BALF (A) and lung (B) of AdCL vs. AdTGF‐β1‐treated mice. (C) Hydroxyproline levels in lung tissue of AdCL‐ vs. AdTGF‐β1‐treated WT and IL17af −/− mice. (D) Histopathology of hematoxylin/eosin‐stained lung tissue sections of WT and IL17af−/− mice at day 14 post‐AdCL vs. AdTGF‐β1 application. Data are shown as mean ± SD of at least n = 5–8 mice per experimental group and time point and are representative of two independent experiments. *p ≤ 0.05, **p ≤ 0.01 compared with AdCL.
Discussion
The role of IL‐17A/F in lung fibrosis is still unclear [39]. In this study, IL‐17A/F deficiency caused profound angiocentric neutrophilic infiltration of the lung, vasculopathy, and increased lung leakage leading to early mortality by day 6 post‐BLM. Importantly, the effect of IL‐17A/F deficiency in mice was (1) limited to the early phase of BLM‐induced acute lung injury, (2) was attenuated in IL‐17a/f −/− mice receiving recombinant IL‐17A/F protein therapy, and (3) was not observed in a model of LPS induced acute lung injury or a model of AdTGF‐β1 induced lung fibrosis, thus pointing to a BLM‐specific effect. Moreover, although BLM‐induced acute lung injury was fatal in IL‐17a/f −/− mice, the ensuing fibrogenic response was similar between groups, demonstrating that BLM‐induced lung fibrogenesis was uncoupled from the phase of increased acute lung injury.
The role of IL‐17A/F in fibrotic lung tissue remodeling is controversially discussed (reviewed in [40]). Previous studies reported a profibrotic function of IL‐17 in experimental lung fibrosis, though most of those studies focused on IL‐17A only while leaving IL‐17F unaddressed [14, 16, 18]. Both IL‐17A and IL‐17F as well as the IL‐17A/F heterodimer bind to the same IL‐17RA receptor, and IL‐17RA KO mice exhibited significantly less neutrophilic lung inflammation in the BLM or silica model of lung injury [17, 41, 42]. Other studies found that lack of IL‐17RA increased lung collagen deposition in BLM‐treated mice [43]. In our models of AdTGF‐β1 or BLM‐induced lung fibrosis, lack of IL‐17A/F did not affect lung collagen deposition but rather caused substantially increased acute lung injury and early mortality in the BLM model only. As such, the current study does not support a role for IL‐17A/F in the process of lung fibrogenesis.
IL‐17A KO mice were previously found to exhibit significantly reduced lung neutrophil recruitment and less airway inflammation upon either BLM or LPS challenge, suggesting a proinflammatory role for IL‐17A in models of acute lung injury [20, 44, 45]. According to more recent work, IL‐17A and IL‐17F are no longer believed to have identical biological functions [46, 47, 48, 49]. Although both IL‐17A and IL‐17F are considered neutrophil‐inducing factors, lack of IL‐17A/F caused profound angiocentric neutrophilic infiltration of the lung, resulting in endothelial activation, intravascular coagulation, and lung leakage, most likely mediating the increased early mortality upon BLM challenge, although organ systems other than the lung were not examined in the current study. Of note, this scenario was specific for BLM, as lipopolysaccharide‐treated IL17af −/− mice showed a similar lung neutrophilic response as in WT mice, and these inflammatory sequelae were also not observed in AdTGF‐β1 exposed IL17af −/− mice. Therefore, the data support the notion that IL‐17 A/F deficiency particularly aggravates BLM‐induced acute lung injury, but not the ensuing fibrotic remodeling phase of this model.
Previous studies reported that neutralization of IL‐17A protein attenuated the neutrophilic response, while IL‐17F neutralizing antibodies had no effect on neutrophil counts in BALF of BLM‐challenged mice [17]. In the current study, we found that deletion of IL‐17A/F led to a profound lung neutrophilic response and increased mortality to BLM challenge, and therapeutic application of recombinant IL‐17A/F protein significantly improved the lung inflammatory phenotype of BLM‐challenged IL17af −/− mice. These data demonstrate the importance of secreted IL‐17A/F protein in the regulation of BLM‐induced acute lung injury. However, differences between our study and the previous report make a direct comparison difficult. The previous study employed single antibody neutralization rather than double KO mice to approach the role of individual IL‐17 family members in lung injury/fibrosis, which may have been confounded by compensatory effects. Moreover, administration routes and dosages of applied BLM were different between our study and the previous report. Finally, neutralizing antibodies are less effective compared with stable genetic models as employed in the current study.
IL17af −/− mice demonstrated strongly increased lung neutrophil counts along with significantly reduced lymphocyte counts in response to the BLM challenge. These data suggest that IL1‐7A/F has a regulatory function in both lung neutrophil and lymphocyte mobilization upon BLM challenge. We found that TCRγδpos T cells and perhaps also CD4pos T cells represented a major source for IL‐17A and IL‐17F in BLM‐challenged mice. In preliminary studies, diphtheria toxin‐induced depletion of γδpos T cells diminished CD4+ and CD8+ T lymphocytes but not neutrophil counts in BLM‐challenged mice, suggesting that γδ T‐cell‐derived IL‐17A/F particularly affects lymphocyte rather than neutrophil recruitment in response to BLM (data not shown), thereby adding to previous studies reporting a similar reduction of CD4pos and CD8pos T cells in BALF of BLM‐treated γδ T‐cell KO mice [50]. Along this line, CD4pos/ CD25pos/Foxp3pos regulatory T cells (Tregs) were also significantly reduced in the lungs of BLM‐challenged IL17a/f −/− mice.
The interplay between IL‐17‐secreting T cells and Tregs is only partially understood (reviewed in [51]). Th17 cells and Tregs appear to have at least in part an interdependent relationship while exerting partially antagonistic functions, with Th17 cells believed to exert a profibrotic function in BLM‐driven lung fibrosis, while Tregs mostly appear to exert antifibrotic functions in later stages of BLM induced lung fibrosis. In the current study, we found diminished numbers of lung Tregs to coincide with neutrophil‐dominated lung injury in BLM‐challenged IL17af−/− mice. Moreover, we recently found an important role for Tregs in limiting the exacerbation of experimental lung fibrosis in mice [31]. In line with this, several studies reported that an imbalance of the Th17/Treg axis may affect pulmonary fibrogenesis [52, 53, 54]. However, differences in BLM dosage, route of administration, age, gender, and genetic background of mice make it rather difficult to directly compare the results between these studies.
Hematopoietic reconstitution of IL17af −/− mice with bone marrow from WT mice did not abrogate the angiocentric neutrophilic accumulates observed in these mice, but treatment with recombinant IL‐17A/F protein was sufficient to attenuate the fatal inflammatory response to BLM. Mechanistically, it appears that radioresistant hematopoietic lung resident recipient‐type IL‐17A/F producing cells, including γδ T cells play an important regulatory role in lung lymphocytic/neutrophilic responses to BLM. Radioresistance of immune cells including lung macrophages, lymphocytes, and natural killer cells (but not neutrophils) is a well‐described phenomenon [55, 56, 57]. Of note, our engraftment experiments enrolling irradiated CD45.1/CD45.2 alloantigen‐expressing mice confirmed an incomplete depletion of lymphocytes in whole‐body irradiated chimeric mice, just opposed to the high turnover rates observed for radiosensitive neutrophils. As a consequence, IL‐17‐producing recipient‐type T cells surviving in the lungs of irradiated WT mice transplanted with IL17af −/− bone marrow were apparently sufficient to regulate lymphocytic and neutrophilic responses to BLM similar to the extent observed in irradiated WT→WT mice challenged with BLM.
In summary, we found that IL‐17A/F deficiency aggravated BLM‐induced acute lung injury in mice without affecting lung fibrogenesis, and also did not play a role in LPS‐induced acute lung injury or AdTGF‐β1 induced lung fibrosis in mice, supporting the view that IL‐17A/F is dispensable in lung fibrogenesis. The disturbed leukocytic response observed in the lungs of IL17af −/− mice was amenable to recombinant IL‐17A/F protein therapy but not to WT bone marrow transplantation, suggesting that IL‐17A/F secreting lung resident rather than circulating lymphocyte subsets contribute to the regulation of BLM induced acute lung injury in mice. As a consequence, it appears to be imperative for the BLM model of lung injury to evaluate whether a given mediator under study directly affects the acute lung injury phase of the model with effects on the ensuing phase of lung fibrogenesis, or indeed directly exerts effects on the fibrogenesis phase of this model.
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Author contributions
Marie T. Moog performed the experiments and wrote the manuscript. Melina Baltes, Tina Röpke, Franziska Aschenbrenner, Regina Maus, Jennifer Stolper, and Danny Jonigk performed the experiments. Immo Prinz, Martin Kolb, and Ulrich A. Maus designed the study and wrote the manuscript.
Abbreviations
- AdTGF‐β1
adenoviral TGF‐β1
- BAL
bronchoalveolar lavage
- BALF
BAL fluids
- BLM
bleomycin
- ILD
interstitial lung disease
- IPF
idiopathic pulmonary fibrosis
- LPS
lipopolysaccharide
Supporting information
Supporting Information
Acknowledgements
The study has been funded by the Federal Ministry of Education and Research by supporting the German Center for Lung Research. All experiments involving animals have been approved by the local governmental authorities (permission numbers 18/3056, 19/3086, and 20/3349). All authors concur with the submission of the article.
Open access funding enabled and organized by Projekt DEAL.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Raghu, G. , Collard, H. R. , Egan, J. J. , Martinez, F. J. , Behr, J. , Brown, K. K. , Colby, T. V. et al., An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence‐based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011. 183: 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. King, T. E., Jr. , Pardo, A. and Selman, M. , Idiopathic pulmonary fibrosis. Lancet 2011. 378: 1949–1961. [DOI] [PubMed] [Google Scholar]
- 3. Rogliani, P. , Mura, M. , Assunta Porretta, M. and Saltini, C. , New perspectives in the treatment of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2008. 2: 75–93. [DOI] [PubMed] [Google Scholar]
- 4. Desai, O. , Winkler, J. , Minasyan, M. and Herzog, E. L. , The role of immune and inflammatory cells in idiopathic pulmonary fibrosis. Front. Med. (Lausanne) 2018. 5: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ross, P. J. , Sutton, C. E. , Higgins, S. , Allen, A. C. , Walsh, K. , Misiak, A. , Lavelle, E. C. et al., Relative contribution of Th1 and Th17 cells in adaptive immunity to Bordetella pertussis: towards the rational design of an improved acellular pertussis vaccine. PLoS Pathog. 2013. 9: e1003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Murakami, T. , Hatano, S. , Yamada, H. , Iwakura, Y. and Yoshikai, Y. , Two types of interleukin 17A‐producing gammadelta T cells in protection against pulmonary infection with Klebsiella pneumoniae . J. Infect. Dis. 2016. 214: 1752–1761. [DOI] [PubMed] [Google Scholar]
- 7. Kudva, A. , Scheller, E. V. , Robinson, K. M. , Crowe, C. R. , Choi, S. M. , Slight, S. R. , Khader, S. A. et al., Influenza A inhibits Th17‐mediated host defense against bacterial pneumonia in mice. J. Immunol. 2011. 186: 1666–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wright, J. F. , Guo, Y. , Quazi, A. , Luxenberg, D. P. , Bennett, F. , Ross, J. F. , Qiu, Y. et al., Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J. Biol. Chem. 2007. 282: 13447–13455. [DOI] [PubMed] [Google Scholar]
- 9. Kuestner, R. E. , Taft, D. W. , Haran, A. , Brandt, C. S. , Brender, T. , Lum, K. , Harder, B. et al., Identification of the IL‐17 receptor related molecule IL‐17RC as the receptor for IL‐17F. J. Immunol. 2007. 179: 5462–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang, S. H. and Dong, C. , A novel heterodimeric cytokine consisting of IL‐17 and IL‐17F regulates inflammatory responses. Cell Res. 2007. 17: 435–440. [DOI] [PubMed] [Google Scholar]
- 11. Goepfert, A. , Lehmann, S. , Blank, J. , Kolbinger, F. and Rondeau, J. M. , Structural analysis reveals that the cytokine IL‐17F forms a homodimeric complex with receptor IL‐17RC to drive IL‐17RA‐independent signaling. Immunity 2020. 52: 499–512.e495. [DOI] [PubMed] [Google Scholar]
- 12. McGeachy, M. J. , Cua, D. J. and Gaffen, S. L. , The IL‐17 family of cytokines in health and disease. Immunity 2019. 50: 892–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang, X. , Angkasekwinai, P. , Dong, C. and Tang, H. , Structure and function of interleukin‐17 family cytokines. Protein Cell 2011. 2: 26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilson, M. S. , Madala, S. K. , Ramalingam, T. R. , Gochuico, B. R. , Rosas, I. O. , Cheever, A. W. and Wynn, T. A. , Bleomycin and IL‐1beta‐mediated pulmonary fibrosis is IL‐17A dependent. J. Exp. Med. 2010. 207: 535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nuovo, G. J. , Hagood, J. S. , Magro, C. M. , Chin, N. , Kapil, R. , Davis, L. , Marsh, C. B. et al., The distribution of immunomodulatory cells in the lungs of patients with idiopathic pulmonary fibrosis. Mod. Pathol. 2012. 25: 416–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Segawa, S. , Goto, D. , Iizuka, A. , Kaneko, S. , Yokosawa, M. , Kondo, Y. , Matsumoto, I. et al., The regulatory role of interferon‐γ producing gamma delta T cells via the suppression of T helper 17 cell activity in bleomycin‐induced pulmonary fibrosis. Clin. Exp. Immunol. 2016. 185: 348–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gasse, P. , Riteau, N. , Vacher, R. , Michel, M. L. , Fautrel, A. , di Padova, F. , Fick, L. et al., IL‐1 and IL‐23 mediate early IL‐17A production in pulmonary inflammation leading to late fibrosis. PLoS One 2011. 6: e23185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cipolla, E. , Fisher, A. J. , Gu, H. , Mickler, E. A. , Agarwal, M. , Wilke, C. A. , Kim, K. K. et al., IL‐17A deficiency mitigates bleomycin‐induced complement activation during lung fibrosis. FASEB J. 2017. 31: 5543–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim, H. S. , Go, H. , Akira, S. and Chung, D. H. , TLR2‐mediated production of IL‐27 and chemokines by respiratory epithelial cells promotes bleomycin‐induced pulmonary fibrosis in mice. J. Immunol. 2011. 187: 4007–4017. [DOI] [PubMed] [Google Scholar]
- 20. Sonnenberg, G. F. , Nair, M. G. , Kirn, T. J. , Zaph, C. , Fouser, L. A. and Artis, D. , Pathological versus protective functions of IL‐22 in airway inflammation are regulated by IL‐17A. J. Exp. Med. 2010. 207: 1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haas, J. D. , Ravens, S. , Düber, S. , Sandrock, I. , Oberdörfer, L. , Kashani, E. , Chennupati, V. et al., Development of interleukin‐17‐producing γδ T cells is restricted to a functional embryonic wave. Immunity 2012. 37: 48–59. [DOI] [PubMed] [Google Scholar]
- 22. Sandrock, I. , Reinhardt, A. , Ravens, S. , Binz, C. , Wilharm, A. , Martins, J. , Oberdörfer, L. et al., Genetic models reveal origin, persistence and non‐redundant functions of IL‐17‐producing γδ T cells. J. Exp. Med. 2018. 215: 3006–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sime, P. J. , Xing, Z. , Graham, F. L. , Csaky, K. G. and Gauldie, J. , Adenovector‐mediated gene transfer of active transforming growth factor‐beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 1997. 100: 768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maus, U. A. , Wellmann, S. , Hampl, C. , Kuziel, W. A. , Srivastava, M. , Mack, M. , Everhart, M. B. et al., CCR2‐positive monocytes recruited to inflamed lungs downregulate local CCL2 chemokine levels. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005. 288: L350–358. [DOI] [PubMed] [Google Scholar]
- 25. Maus, U. A. , Waelsch, K. , Kuziel, W. A. , Delbeck, T. , Mack, M. , Blackwell, T. S. , Christman, J. W. et al., Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2‐CCR2 axis. J. Immunol. 2003. 170: 3273–3278. [DOI] [PubMed] [Google Scholar]
- 26. Ostermann, L. , Seeliger, B. , David, S. , Flasche, C. , Maus, R. , Reinboth, M. S. , Christmann, M. et al., S100A9 is indispensable for survival of pneumococcal pneumonia in mice. PLoS Pathog. 2023. 19: e1011493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maus, U. A. , Janzen, S. , Wall, G. , Srivastava, M. , Blackwell, T. S. , Christman, J. W. , Seeger, W. et al., Resident alveolar macrophages are replaced by recruited monocytes in response to endotoxin‐induced lung inflammation. Am. J. Respir. Cell Mol. Biol. 2006. 35: 227–235. [DOI] [PubMed] [Google Scholar]
- 28. Steinwede, K. , Henken, S. , Bohling, J. , Maus, R. , Ueberberg, B. , Brumshagen, C. , Brincks, E. L. et al., TNF‐related apoptosis‐inducing ligand (TRAIL) exerts therapeutic efficacy for the treatment of pneumococcal pneumonia in mice. J. Exp. Med. 2012. 209: 1937–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Knippenberg, S. , Ueberberg, B. , Maus, R. , Bohling, J. , Ding, N. , Tort Tarres, M. , Hoymann, H. G. et al., Streptococcus pneumoniae triggers progression of pulmonary fibrosis through pneumolysin. Thorax 2015. 70: 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moog, M. T. , Hinze, C. , Bormann, T. , Aschenbrenner, F. , Knudsen, L. , DeLuca, D. S. , Jonigk, D. et al., B cells are not involved in the regulation of adenoviral TGF‐beta1‐ or bleomycin‐induced lung fibrosis in mice. J. Immunol. 2022. 208: 1259–1271. [DOI] [PubMed] [Google Scholar]
- 31. Moye, S. , Bormann, T. , Maus, R. , Sparwasser, T. , Sandrock, I. , Prinz, I. , Warnecke, G. et al., Regulatory T cells limit pneumococcus‐induced exacerbation of lung fibrosis in mice. J. Immunol. 2020. 204: 2429–2438. [DOI] [PubMed] [Google Scholar]
- 32. Bormann, T. , Maus, R. , Stolper, J. , Jonigk, D. , Welte, T. , Gauldie, J. , Kolb, M. et al., Role of the COX2‐PGE(2) axis in S. pneumoniae‐induced exacerbation of experimental fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021. 320: L377–L392. [DOI] [PubMed] [Google Scholar]
- 33. Livak, K. J. and Schmittgen, T. D. , Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐delta delta C(T)) method. Methods 2001. 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 34. Sisson, T. H. , Mendez, M. , Choi, K. , Subbotina, N. , Courey, A. , Cunningham, A. , Dave, A. et al., Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010. 181: 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bormann, T. , Maus, R. , Stolper, J. , Tort Tarres, M. , Brandenberger, C. , Wedekind, D. , Jonigk, D. et al., Role of matrix metalloprotease‐2 and MMP‐9 in experimental lung fibrosis in mice. Respir. Res. 2022. 23: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tort Tarres, M. , Aschenbrenner, F. , Maus, R. , Stolper, J. , Schuette, L. , Knudsen, L. , Lopez Rodriguez, E. et al., The FMS‐like tyrosine kinase‐3 ligand/lung dendritic cell axis contributes to regulation of pulmonary fibrosis. Thorax 2019. 74: 947–957. [DOI] [PubMed] [Google Scholar]
- 37. Maus, U. A. , Srivastava, M. , Paton, J. C. , Mack, M. , Everhart, M. B. , Blackwell, T. S. , Christman, J. W. et al., Pneumolysin‐induced lung injury is independent of leukocyte trafficking into the alveolar space. J. Immunol. 2004. 173: 1307–1312. [DOI] [PubMed] [Google Scholar]
- 38. Haas, J. D. , Ravens, S. , Duber, S. , Sandrock, I. , Oberdorfer, L. , Kashani, E. , Chennupati, V. et al., Development of interleukin‐17‐producing gammadelta T cells is restricted to a functional embryonic wave. Immunity 2012. 37: 48–59. [DOI] [PubMed] [Google Scholar]
- 39. Albrecht, M. , Halle, O. , Gaedcke, S. , Pallenberg, S. T. , Camargo Neumann, J. , Witt, M. , Roediger, J. et al., Interleukin‐17A and interleukin‐22 production by conventional and non‐conventional lymphocytes in three different end‐stage lung diseases. Clin. Transl. Immunol. 2022. 11: e1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gurczynski, S. J. and Moore, B. B. , IL‐17 in the lung: the good, the bad, and the ugly. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018. 314: L6–L16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lo Re, S. , Dumoutier, L. , Couillin, I. , Van Vyve, C. , Yakoub, Y. , Uwambayinema, F. , Marien, B. et al., IL‐17A‐producing gammadelta T and Th17 lymphocytes mediate lung inflammation but not fibrosis in experimental silicosis. J. Immunol. 2010. 184: 6367–6377. [DOI] [PubMed] [Google Scholar]
- 42. François, A. , Gombault, A. , Villeret, B. , Alsaleh, G. , Fanny, M. , Gasse, P. , Adam, S. M. et al., B cell activating factor is central to bleomycin‐ and IL‐17‐mediated experimental pulmonary fibrosis. J. Autoimmun. 2015. 56: 1–11. [DOI] [PubMed] [Google Scholar]
- 43. Fabro, A. T. , da Silva, P. H. , Zocolaro, W. S. , de Almeida, M. S. , Rangel, M. P. , de Oliveira, C. C. , Minatel, I. O. et al., The Th17 pathway in the peripheral lung microenvironment interacts with expression of collagen V in the late state of experimental pulmonary fibrosis. Immunobiology 2015. 220: 124–135. [DOI] [PubMed] [Google Scholar]
- 44. Muir, R. , Osbourn, M. , Dubois, A. V. , Doran, E. , Small, D. M. , Monahan, A. , O'Kane, C. M. et al., Innate lymphoid cells are the predominant source of IL‐17A during the early pathogenesis of acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2016. 193: 407–416. [DOI] [PubMed] [Google Scholar]
- 45. Yan, Z. , Xiaoyu, Z. , Zhixin, S. , Di, Q. , Xinyu, D. , Jing, X. , Jing, H. et al., Rapamycin attenuates acute lung injury induced by LPS through inhibition of Th17 cell proliferation in mice. Sci. Rep. 2016. 6: 20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen, R. , Zhang, Q. , Chen, S. , Tang, H. , Huang, P. , Wei, S. , Liang, Z. et al., IL‐17F, rather than IL‐17A, underlies airway inflammation in a steroid‐insensitive toluene diisocyanate‐induced asthma model. Eur. Respir. J. 2019. 53. [DOI] [PubMed] [Google Scholar]
- 47. De Luca, A. , Pariano, M. , Cellini, B. , Costantini, C. , Villella, V. R. , Jose, S. S. , Palmieri, M. et al., The IL‐17F/IL‐17RC axis promotes respiratory allergy in the proximal airways. Cell Rep. 2017. 20: 1667–1680. [DOI] [PubMed] [Google Scholar]
- 48. Ishigame, H. , Kakuta, S. , Nagai, T. , Kadoki, M. , Nambu, A. , Komiyama, Y. , Fujikado, N. et al., Differential roles of interleukin‐17A and ‐17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 2009. 30: 108–119. [DOI] [PubMed] [Google Scholar]
- 49. Yang, X. O. , Nurieva, R. , Martinez, G. J. , Kang, H. S. , Chung, Y. , Pappu, B. P. , Shah, B. et al., Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008. 29: 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Braun, R. K. , Ferrick, C. , Neubauer, P. , Sjoding, M. , Sterner‐Kock, A. , Kock, M. , Putney, L. et al., IL‐17 producing gammadelta T cells are required for a controlled inflammatory response after bleomycin‐induced lung injury. Inflammation 2008. 31: 167–179. [DOI] [PubMed] [Google Scholar]
- 51. Seyran, M. , Melanie, S. , Philip, S. , Amiq, G. and Fabian, B. , Allies or enemies? The effect of regulatory T cells and related T lymphocytes on the profibrotic environment in bleomycin‐injured lung mouse models. Clin. Exp. Med. 2023. 23: 1075–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Galati, D. , De Martino, M. , Trotta, A. , Rea, G. , Bruzzese, D. , Cicchitto, G. , Stanziola, A. A. et al., Peripheral depletion of NK cells and imbalance of the Treg/Th17 axis in idiopathic pulmonary fibrosis patients. Cytokine 2014. 66: 119–126. [DOI] [PubMed] [Google Scholar]
- 53. Chen, Y. , Li, C. , Weng, D. , Song, L. , Tang, W. , Dai, W. , Yu, Y. et al., Neutralization of interleukin‐17A delays progression of silica‐induced lung inflammation and fibrosis in C57BL/6 mice. Toxicol. Appl. Pharmacol. 2014. 275: 62–72. [DOI] [PubMed] [Google Scholar]
- 54. Wang, L. , Wang, X. , Tong, L. , Wang, J. , Dou, M. , Ji, S. , Bi, J. et al., Recovery from acute lung injury can be regulated via modulation of regulatory T cells and Th17 cells. Scand. J. Immunol. 2018. 88: e12715. [DOI] [PubMed] [Google Scholar]
- 55. Hahn, I. , Klaus, A. , Maus, R. , Christman, J. W. , Welte, T. and Maus, U. A. , Dendritic cell depletion and repopulation in the lung after irradiation and bone marrow transplantation in mice. Am. J. Respir. Cell Mol. Biol. 2011. 45: 534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Prosser, J. S. , Survival of human T and B lymphocytes after X‐irradiation. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1976. 30: 459–465. [DOI] [PubMed] [Google Scholar]
- 57. Park, H. R. and Jung, U. , Depletion of NK cells resistant to ionizing radiation increases mutations in mice after whole‐body irradiation. In Vivo 2021. 35: 1507–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.