

ABSTRACT
Propylene glycol ethers (PGEs) are organic solvents commonly found as technical grade on the commercial market, as mixtures of secondary (α‐isomer) and primary (β‐isomer, generally < 5%) alcohols. After handling products containing PGEs, they readily enter the human body where they are metabolized. The minor β‐isomer is oxidized by alcohol dehydrogenase (ADH) followed by aldehyde dehydrogenase (ALDH) to a potentially harmful metabolite. Although the enzymatic rate is needed to estimate both parent and metabolite internal exposures, kinetic data for many PGEs are still scarce. Therefore, we generated in vitro hepatic intrinsic clearance data for propylene glycol methyl ether β‐isomer (β‐PGME) and its metabolite methoxy propionic acid (2‐MPA) and integrated these data into an in silico toxicokinetic (TK) model. Hepatic clearance values for the model were generated using an established in vitro 3D culture of the human HepaRG cell line and human S9 liver fraction. Our results showed the presence of ADH and ALDH and consequently, the formation of 2‐MPA in the 3D HepaRG and S9 fraction, which was slow to medium. We integrated the hepatic clearance values into the TK model to predict urinary 2‐MPA concentrations. The simulated urinary 2‐MPA concentrations fitted well (within twofold error from observed experimental data) for both liver systems, showing that they were both able to reliably predict the hepatic clearance of β‐PGME. Although S9 is suitable for short‐term studies, 3D cell culture models maintain metabolic competence over days and weeks. This opens the opportunity for long‐term metabolism studies applying the 3D HepaRG model alone or in multi‐organ systems.
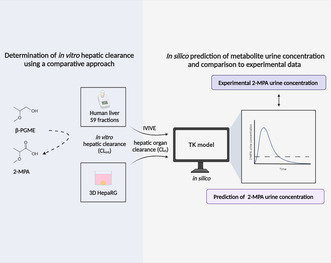
Visual abstract. Overview of the experimental workflow. The in vitro hepatic intrinsic clearance (CLint) for propylene glycol methyl ether β‐isomer (β‐PGME) to its corresponding metabolite methoxy propionic acid (2‐MPA) was determined using human liver S9 fractions and 3D HepaRG. Urinary 2‐MPA concentrations were simulated by integrating the hepatic organ clearance (CLh) into a developed toxicokinetic (TK) model, following in vitro‐in vivo extrapolation (IVIVE). The results were compared with existing human exposure data. Illustration created with BioRender.com.
Abbreviations
- 2‐MAA
methoxy acetic acid
- 2‐MPA
methoxy propionic acid
- ADH
alcohol dehydrogenase
- ALDH
aldehyde dehydrogenase
- CLh
hepatic organ clearance
- CLint
hepatic intrinsic clearance
- CYP
cytochrome P450
- EGE
ethylene glycol ether
- EGME
ethylene glycol methyl ether
- fu,b
plasma binding
- fu,inc
incubational binding
- GC–MS
gas chromatography mass‐spectroscopy
- HPLC‐MS/MS
high‐performance liquid chromatography mass‐spectroscopy
- IVIVE
in vitro‐in vivo extrapolation
- K m
Michaelis–Menten constant
- PGE
propylene glycol ether
- PGME
propylene glycol methyl ether
- pHH
primary human hepatocytes
- S9
human liver subcellular fraction
- TK
toxicokinetic
- V max
maximum reaction rate
1. Introduction
The general population, especially workers, are exposed to a wide range of propylene glycol ethers (PGEs) over a lifetime using cleaning products, surface coatings, and other products [1]. PGEs are the less toxic replacement solvents for ethylene glycol ethers (EGEs) regarding hemolysis, teratogenicity, developmental and reproductive toxicity. Still, occupational exposure to solvents has been associated with numerous neurological disorders [2] and the evaluation of neurotoxicity is conspicuously absent for PGEs [3, 4, 5, 6, 7]. Recently, Hopf et al. defined a novel strategy to assess the neurotoxicity of several PGEs by combining in vitro and in silico methods with human‐controlled exposure studies [8].
Commercial PGEs are commonly sold as technical grade mixtures of secondary (α‐isomer) and primary (β‐isomer, generally < 5%) alcohols. Similar to some banned EGEs, the primary alcohol group of PGEs can be enzymatically oxidized by alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH) to alkoxy propionic acid metabolites [9, 10, 11, 12, 13]. Several studies have proposed that ADH class I (ADH1A/B/C) plays a major role in the metabolism of solvents [10, 11, 14] and the involvement of ALDH2 is well documented [15, 16].
Although alkoxy acetic acid metabolites are reported to be responsible for the adverse effects observed for EGEs, the metabolites and their effects have yet to be assessed for PGEs. Indeed, the metabolism of the widely produced PGEs is poorly described and only a few animal and human studies exist. Based on reported in vivo studies in rats and mice, it is assumed that most propylene glycol methyl ether β‐isomer (β‐PGME) is excreted as methoxy propionic acid (2‐MPA) in urine, a minor fraction exhaled as carbon dioxide (CO2), and marginally eliminated unchanged or as glucuronide and sulfate conjugate in the urine [13, 17, 18, 19] (Figure 1). Concordantly, urinary 2‐MPA concentrations were observed in volunteers exposed to mixtures of α‐PGME (> 99.5%) and β‐PGME (< 0.5%) in controlled exposure studies [20]. However, in vitro metabolism studies, which are faster and safer alternatives to animal or human studies, have not yet been reported for β‐PGME. Yet, knowing the metabolic clearance is important for estimating parent and metabolite internal exposures and predicting toxic effects produced in the body [21].
FIGURE 1.
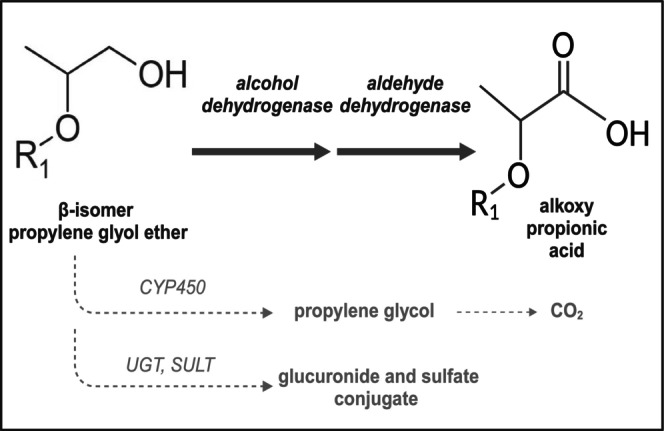
Metabolic pathway proposed for β‐isomer propylene glycol ether. The primary alcohol group of β‐isomer propylene glycol ether is majorly metabolized via the alcohol dehydrogenase and aldehyde dehydrogenase to an alkoxy propionic acid metabolite. Minor pathways are proposed to be the metabolism to propylene glycol via CYP450 enzymes, followed by the exhalation of CO2, and the formation of glucuronide and sulfate conjugates by uridine diphosphate glycosyltransferases (UGTs) and sulfotransferases (SULTs); R1 = alkyl chain. Illustration created with BioRender.com. Data based on literature [13, 17, 18].
Several liver models can be considered when studying the in vitro metabolism of compounds, whereby ease‐of‐use systems like microsomes and cell fractions (S9), primary human hepatocyte suspensions, and 2D cultures are commonly employed for metabolic stability assays. However, their use is limited because of the short enzyme functionality [22]. More complex systems, such as 3D liver cultures, offer a higher degree of in vivo‐like conditions as they retain their hepatic functionality and enzyme performance over multiple weeks. Thus, they can be used to determine the clearance of low turn‐over compounds [23, 24, 25]. Furthermore, the toxicity of compounds and their metabolites can be directly studied in the model or after its integration within in vitro multi‐organ systems [22]. Among liver in vitro models, primary human hepatocytes are considered the gold standard for studying in vitro metabolism, but comparable enzyme functionality has been reported in the HepaRG cell line [26, 27, 28]. In this cell line, the expression of ADH and ALDH has been presented on the gene‐ and protein level, whereas the metabolism of substrates has not yet been described [29, 30, 31].
In this work, our goal was to generate novel in vitro hepatic kinetic data (clearance) for the formation of 2‐MPA from β‐PGME, which were integrated into an in silico model for estimating internal human metabolite exposure, contributing to the approach of Hopf et al. [8]. β‐PGME was the solvent chosen for the study because a calibrated toxicokinetic model for α‐PGME exists from Reale et al. [32] which can be adapted to β‐PGME metabolism. We hypothesized that an established 3D HepaRG model could serve as a tool to determine hepatic kinetic parameters as an alternative to commonly used human liver S9 fractions. Applying the hepatic clearance values derived from both systems (3D HepaRG and S9 fraction), we simulated in vivo urinary 2‐MPA concentrations and compared the predictions to previously reported human data [20]. This allowed the predictive potential of both liver systems to be assessed.
2. Materials and Methods
2.1. Chemicals and S9 Fractions
2‐Methoxyethanol (EGME, > 99.8%), GA11 (SML2028), fomepizole (222569), 1‐Methoxy‐propan‐2‐ol (commercial PGME, > 99.5%), 2‐Methoxyacetic acid (2‐MAA, 97%), and 2‐Methoxypropionic acid (2‐MPA, 98%) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). 2‐Methoxy‐propan‐1‐ol (β‐PGME, 98%) was manufactured by AA Blocks (San Diego, CA, USA). Glycol ethers and their metabolites are listed in Table 1. Ultrapure water was obtained from a Millipore Milli‐Q Plus purification system (Beford, MA, USA). Phosphate buffer was prepared in‐house, adjusted to pH 7.4, and stored at 4°C. All other chemicals and reagents were of analytical grade and purchased from commercial sources. Pooled adult mixed‐gender human liver subcellular fraction (S9) was purchased from ThermoFisherScientific (Reinach, Switzerland; HMS9PL) and stored at −80°C after aliquoting.
TABLE 1.
Overview of the glycol ethers and metabolites and their physicochemical properties.
| CAS number | IUPAC name | Trivial name | Abbreviation | Molecular weight (MW) a | logP a | pKa b | fu,b |
|---|---|---|---|---|---|---|---|
| 109‐86‐4 | 2‐Methoxyethanol | Ethylene glycol methyl ether | EGME | 76.09 | −0.80 | 15.1 (A) | ND |
| 625‐45‐6 | 2‐Methoxyacetic acid | Methoxy acetic acid | 2‐MAA | 90.08 | −0.30 | 3.83 (A) | ND |
|
107‐98‐2 1589‐47‐5 |
1‐Methoxypropan‐2‐ol 2‐Methoxypropan‐1‐ol |
Propylene glycol methyl ether | PGME (commercial) β‐PGME |
90.12 90.12 |
−0.49 −0.49 |
14.9 (A) 14.7 (A) |
0.540 c 0.540 c |
|
4324‐37‐2 |
2‐Methoxypropionic acid | Methoxy propionic acid | 2‐MPA | 104.1 | 0.10* |
3.99 (A) |
ND |
Abbreviation: ND, not determined.
Data derived from Pubchem. PubChem (nih.gov).
Data derived from Chemicalize, Chemicalize‐Instant Cheminformatics Solutions.
Measured data derived from literature [32].
Computed data.
2.2. Cell Culture
The HepaRG cell line was obtained from Biopredic International (Saint Grégoire, France; HPR101) and cultured for 4 weeks in basal medium with growth supplements (Biopredic International; ADD710). Cell passage was performed using Trypsin–EDTA (Sigma, Taufkirchen, Germany; 59417C‐100ML), and cells at passages below 20 were used. Cell differentiation over 2 weeks was induced using medium containing a 50:50 mixture of growth and differentiation supplements (Biopredic International; ADD720) for 4 days, following the use of medium containing solely differentiation supplements for additional 10 days. For cell aggregation, we used William's E Medium + GlutaMAX (ThermoFisherScientific; 32551087), 1 × ITS (Sigma; 11074547001), 100 nM dexamethasone (Sigma; D1756), 1% penicillin and streptomycin (P/S; Sigma; P4333‐100ML), and 20% fetal bovine serum (FBS; ThermoFisherScientific; 10270‐106). Exchange to serum‐free supplemented William's E Medium at Day 5 after aggregation was done for all treatment conditions.
Cryopreserved primary human hepatocytes (pHH) were obtained from Lonza (Basel, Switzerland; HUCPI). Detailed donor information of the two hepatocyte lots is summarized in Table S1. Cells were carefully thawed and resuspended in a thawing medium (Lonza; MCHT50), before being seeded in fresh plating medium (Lonza; MP100/MP250) or aggregation medium for either plated use or 3D culturing, respectively. Plates utilized for 2D cell culture were coated 1 h before seeding with rat tail collagen type I (1.3 μg/cm2, Sigma; 08115) and the medium daily exchanged with maintenance medium (Lonza; CC‐3198). Similar cell aggregation (3D pHH) and treatment media were used as described for the HepaRG before. All cells were cultured at 37°C with 5% CO2 in a humified incubator.
2.3. Cell Viability Assay
For 2D cultures, HepaRG cells were seeded 7 days prior exposure at 2.25 × 105 cells/cm2 in a 96‐well plate (Corning, NY, USA; 353072). For 3D cultures, HepaRG cells were seeded 7 days prior exposure at 3000 cells per well to form 3D‐spheroids in Nunclon Sphera 96‐well U‐shaped‐bottom microplates (ThermoFisherScientific; 174925). The acidic pH condition resulting from metabolites in the highest treatment condition (Figure S1) was neutralized to pH 7.4 (measured with an electrode) using 1 N NaOH and sterilized by filtration before further diluted. For the 3D cultures, half of the medium was removed, and twofold concentrated compound solution was added. To prevent solvent evaporation, we sealed all cell plates with an adhesive gas permeable seal (ThermoFisherScientific; AB0718). Cell viability was measured for β‐PGME and 2‐MPA, and for commercial PGME, ethanol, EGME, and 2‐MAA as comparators using concentration ranges of 10–1000 mM for the glycol ethers, 1–600 mM for ethanol, and 1–25 mM for the metabolites. We assessed cell viability by measuring ATP content using the CellTiter‐Glo Luminescent Cell Viability Assay (Promega, USA; G7570) at 48 h and 7 days after exposure, following the manufacturer's instructions. Luminescence was measured on a Flexstation 3 microplate reader (Molecular Devices) at 1000 ms. Following this, the EC50 was calculated using GraphPad Prism.
2.4. CYP Metabolism Study
HepaRG cells were seeded 7 days prior treatment in a micro‐mold system generating 35 × 3D‐spheroids (Sigma; Z764051‐6EA) in a 24‐well plate (Huberlab, Aesch, Switzerland; 7.662102) at 3000 cells per spheroid. Cytochrome P450 (CYP) functional activity, as a phenotypical characteristic of hepatocytes, was assessed using non‐toxic concentrations of the corresponding CYP substrates 50 μM testosterone (CYP3A; Sigma; 86 500) [33] and 100 μM diclofenac (CYP2C9; Combi‐Blocks, San Diego, CA, USA; OR‐0229). CYPs were induced using a reported concentration of 20 μM rifampicin (Sigma; R3501) [33, 34] at Day 5 and Day 6 of aggregation phase (48 and 24 h before substrate exposure). Supernatant was collected at defined timepoints (10 min, 6 h, 24 h, and 48 h) and stored at –80°C for further analysis by HPLC‐MS/MS.
2.5. Albumin ELISA Assay
Quantification of the albumin concentration in supernatant of 3D HepaRG unexposed or exposed to a reported toxic concentration of 16 mM acetaminophen (APAP; Sigma; A5000) [35] for over 48 h, was determined using the Human Albumin ELISA Kit (Bethyl laboratories, USA; E80‐129) according to the manufacturer's instructions.
2.6. Gene Expression Analysis
HepaRG cells and pHH were seeded at 2.25 × 105 cells/cm2 in a 96‐well plate for 7 days. The total RNA was isolated with the Qiazol Lysis Reagent (Qiagen, Basel, Switzerland; LT‐02241) and mRNA was isolated following the standard TRIzol extraction procedure with glycogen (ThermoFisherScientific; 79306). Reverse transcription was performed using the M‐MLV Reverse transcriptase (Promega, Dübendorf, Switzerland; M1705) and oligo dT (Qiagen; 79237). Real‐time PCR was conducted with specific TaqMan probes for selected genes (see Table 2) and FastStart TaqMan Probe Master (Sigma; 4673417001). The q‐RT‐PCR program was following: 10 min denaturation at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. The Ct values were produced using the LightCycler 480II Systems (Roche Diagnostics, Rotkreuz, Switzerland) and processed on GraphPad Prism using Beta‐2‐Microglobulin (B2M) as the internal standard for normalization. Relative gene expression was calculated as ∆Ct = Ct of the Gene of Interest—Ct B2M.
TABLE 2.
Taqman probes of selected genes obtained from ThermoFisherScientific.
| Gene of Interest | Ref. Nr. |
|---|---|
| Cytochromes P450 3A4 (CYP3A4) | Hs00604506_m1 |
| Cytochromes P450 2B6 (CYP2B6) | Hs04183483_g1 |
| Cytochromes P450 2E1 (CYP2E1) | Hs00559368_m1 |
| Alcohol dehydrogenase isoform 1A (ADH1A) | Hs00605167_g1 |
| Alcohol dehydrogenase isoform 1B (ADH1B) | Hs00605175_m1 |
| Alcohol dehydrogenase isoform 1C (ADH1C) | Hs02383872_s1 |
| Aldehyde dehydrogenase 2 (ALDH2) | Hs01007998_m1 |
| Beta‐2‐Microglobulin (B2M) | Hs00187842_m1 |
2.7. Immunohistochemistry
3D HepaRG were fixed with 4% PFA (Polysciences, Warrington, PA; USA; 18814‐10) for 1 h and washed in 1× PBS (Sigma; 11666789001) + Mg2+ + Ca2+. We permeabilized with 0.5% Triton X‐100 in 1× PBS (ROTH, Arlesheim, Switzerland; 3051.3) for 4 h, washed 3 × 5 min, and blocked with 3% BSA (Sigma; A2153), 0.1% Triton X‐100 in 1× PBS overnight at 4°C. After washing again (3 × 5 min), the primary antibody was incubated overnight (4°C), washed (3 × 1 h), and followed by the incubation of the secondary antibody overnight (4°C). Washed 3D HepaRG (2 × 1 h) were counterstained with DAPI (ThermoFisherScientific; 62248, 1:1000) (1 h, RT) prior to imaging.
Antibodies and the applied dilutions in 1% BSA, and 0.1% Triton X‐100 are listed in Table 3. The images were taken with the Olympus Fluoview FV3000 Confocal Microscope.
TABLE 3.
Antibodies used for immunohistochemistry.
| Protein of interest | Primary antibody | Secondary antibody |
|---|---|---|
| Albumin |
Rabbit monoclonal antibody (Abcam, Cambridge, United Kingdom; ab207327)/1:700 |
Goat anti‐rabbit Alexafluor 488 (ThermoFisherScientific; A‐11070)/1:1000 |
|
Cytochromes P450 3A4 (CYP3A4) |
Rabbit polyclonal antibody (Sigma; AB1254)/1:2000 |
Goat anti‐rabbit Alexafluor 488 (ThermoFisherScientific; A‐11070)/1:1000 |
|
Alcohol dehydrogenase 1 (ADH1A/B/C) |
Goat polyclonal antibody (ThermoFisherScientific; PAB6725)/1:250 |
Rabbit anti‐goat Alexafluor 488 (ThermoFisherScientific; A‐11078)/1:1000 |
|
Aldehyde dehydrogenase 2 (ALDH2) |
Mouse monoclonal antibody (ThermoFisherScientific; MA5‐17029)/1:250 |
Rabbit anti‐mouse Alexafluor 546 (ThermoFisherScientific; A‐11060)/1:1000 |
2.8. Western Blot
Samples of S9 and HepaRG cell lysate were run on a 10% SDS‐Page and transferred to a PVDF membrane (Sigma; GE1060004) using the Pierce Powerplot cassette and power station (ThermoFisherScientific). Protein concentration was measured according to the standard Bradford assay protocol using ROTI Nanoquant (ROTH; K880.1). Membranes were blocked for 1.5 h with 4% milk in TBS‐T (0.1% Tween‐20) and incubated overnight with the primary antibody at 4°C. We washed the membranes with TBS‐T and applied the secondary antibody for 1 h at RT. All antibodies were diluted in 3% milk in TBS‐T and are listed in Table 4. Imaging was performed using the OdysseyCLx imaging system (LI‐COR Biosciences, Bad Homburg, Germany).
TABLE 4.
Antibodies used for Western blot.
| Protein of interest | Primary antibody | Secondary antibody |
|---|---|---|
|
Alcohol dehydrogenase 1 (ADH1A/B/C) |
Goat polyclonal antibody (ThermoFisherScientific; PAB6725)/1:1000 |
Donkey anti‐goat IRDye 800CW (LI‐COR Biosciences; 925–32 214)/1:20000 |
|
Aldehyde dehydrogenase 2 (ALDH2) |
Mouse monoclonal antibody (ThermoFisherScientific; MA5 17 029)/1:2000 |
Donkey anti‐mouse IRDye 800CW (LI‐COR Biosciences; 926–32 212)/1:20000 |
|
Glyceraldehyde‐3‐Phosphate Dehydrogenase (GAPDH) |
Rabbit polyclonal antibody (ThermoFisherScientific; PA1‐987)/1:1000 |
Donkey anti‐rabbit IRDye 680CW (LI‐COR Biosciences; 926–68 073)/1:20000 |
The relative signal mean intensity was calculated using ImageJ by measuring the area of ADH1A/B/C and ALDH2 bands and normalization to the area of GAPDH.
2.9. S9 Fraction Incubation for the Assessment of Michaelis–Menten Kinetics
The incubation mixture (final volume 200 μL) contained S9 (2 mg/mL), 100 mM potassium phosphate buffer (pH 7.4), 3 mM MgCl2, 1 mM NAD + (Sigma; NAD100‐RO), 1 mM NADPH (Sigma; NADPH‐RO), and the respective substrate concentrations (0.1, 0.5, 0.7, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 7, and 8 mM). After 5 min of preincubation, the reaction was initiated with the addition of NAD+ and NADPH, and the incubations were conducted (45 min, 300 rpm, 37°C) on a Thermomixer (Eppendorf, Hamburg, Germany). Incubations without NAD+ and NADPH were included as a control. Reactions were stopped by the addition of 400 μL ice‐cold acetonitrile (ACN; Sigma; 439134). After, the samples were vortexed and centrifuged (15 min, 2500 × g, 4°C) and the supernatant stored at −80°C for further analysis. The reaction rates were linear with respect to time of incubation and the protein concentration (Figure S2). Michaelis–Menten kinetic parameters (V max and K m) were obtained by nonlinear regression using GraphPad Prism applying the Michaelis–Menten equation. Finally, the in vitro intrinsic clearance (CLint) was calculated according to the equations described below. For the inhibition studies, only NAD+ was used as co‐factor and final concentrations of 50 μM fomepizole [36] and 10 μM GA11 [37] were applied. We selected a β‐PGME concentration (5 mM) below expected ADH enzyme saturation, as fomepizole is also described to inhibit CYP2E1 at ADH saturation [38].
2.10. S9 Fraction Incubation for the Evaluation of Solvent Loss and Metabolite Formation
The incubation mixture (final volume 400 μL) contained S9 (2 mg/mL), 100 mM potassium phosphate buffer (pH 7.4), 3 mM MgCl2, 1 mM NAD+, 1 mM NADPH, and the respective substrate concentration (700 μM β‐PGME). The applied substrate concentration was eightfold below the determined K m value. After 5 min of preincubation, the reaction was initiated with the addition of NAD+ and NADPH, and the incubations conducted (45 min, 300 rpm, 37°C) on a Thermomixer (Eppendorf). Incubations without NAD+ and NADPH were included as a control. At defined timepoints (1, 12, 23, 34, and 45 min), the reactions were stopped by the addition of 80 μL ice‐cold ACN to 40 μL collected sample. The samples were vortexed, centrifuged (15 min, 2500 × g, 4°C) and stored at −80°C for further analysis. The reaction rates were linear with respect to time of incubation and the protein concentration (Figure S2). The in vitro CLint was calculated according to the equations described below.
2.11. Assessment of Metabolite Formation in the 3D Liver Models
3D HepaRG and 3D pHH were seeded 7 days before treatment into the 96‐well ultra‐low attachment Elplasia plate (Corning; 4442) with 219 000 cells per well to form 79 × 3D‐spheroids in micro‐arrays (2772 cells/spheroid) per incubation, following centrifugation (2 min, 300 × g). For treatment, 100 μL medium from the well was removed and 50 μL of threefold concentrated β‐PGME solution was added to the remaining 100 μL in the well for the final solvent concentration (5 mM). As the experimental set‐up required a high nominal concentration of β‐PGME to meet the limit of quantification for the formed metabolite 2‐MPA, the applied substrate concentration was higher than for the S9 incubations. Still, the concentration was slightly below the previously determined K m value. At defined time‐points within the first hour (1′, 15′, 30′, 45′, only for HepaRG), and at 1 h, 6 h, and 24 h (HepaRG and pHH), the supernatant was collected, and the samples quenched with ice‐cold ACN. The samples were stored at −80°C for further analysis. All incubations were performed at 37°C, 5% CO2 in a humidified incubator and the plates covered with a gas permeable seal. Before analysis, samples were centrifuged (20 min, 2500 × g, 4°C). The in vitro CLint was calculated according to the equations described below. For comparison of metabolite production within the 3D HepaRG and 3D pHHs, the amount of metabolite (pmol) was normalized for each biological repeat to the protein amount present per incubation (μg) (Table S5). Determination of the protein content per incubation was performed using the Pierce BCA Protein assay kit (ThermoFisherScientific; 23225) according to the manufacturer's instructions.
2.12. Chemical Analytical Methods
(1) 2‐MPA concentrations were quantified with a high‐performance liquid chromatography (HPLC) with quadrupole mass spectrometer detection (MS/MS). The samples were injected into the HPLC (1200 Series Gradient HPLC system, Agilent, Santa Clara, CA, USA) equipped with a C18 column (InfinityLab Poroshell 120 CS‐C18 column (2.1 × 50 mm, 2.7 μm), Agilent; 699775942) connected to the MS/MS in electrospray ionization (ESI) mode (Triple Quadrupole 6475 mass spectrometer, Agilent) with a data acquisition software (MassHunter Quantitative Analysis 10.1, Agilent). (2) β‐PGME concentrations were quantified with gas chromatography (GC) with MS detection. The samples were injected into a GC (Agilent; 6890N) equipped with a capillary column (Rxi‐624Sil, 60 m, 0.25 mm ID, 1.4 μm, Restek, Bad Soden, Germany) coupled to the MS (5973 Network mass selective detector, Agilent) with a data acquisition software (Agilent MassHunter Quantitative Analysis 10.0). (3) Diclofenac, 4‐OH‐diclofenac, 6β‐OH‐testosterone, and testosterone were quantified with a HPLC with MS/MS detection. The samples were injected into the HPLC (Agilent 1260 Infinity II Prime LC system, Agilent) equipped with a C8 column (ZORBAX SB‐C8 column (4.6 × 50 mm, 1.8 μm), Agilent; 822975906) connected to the MS/MS in ESI mode (Triple Quadrupole 6475, Agilent) with a data acquisition software (MassHunter LCMS Acquisition Console and MassHunter Quantitative Analysis 10.1, Agilent). Additional information on the chemical analytical methods is provided in the Supporting Information.
2.13. Calculation of the In Vitro Clearance and Processing to Hepatic Organ Clearance
The in vitro CLint for the solvent loss and the metabolite formation was determined according to Equation (1) for the S9 incubations and Equation (2) for the cell incubations [39]:
| (1) |
| (2) |
where the elimination constant (−k) and the formation constant (k) were derived from the slope of the linear regression from the ln‐transformed % of solvent remaining versus the incubation time and the ln‐transformed metabolite concentration versus the incubation time, respectively.
The in vitro CLint for the S9 incubations was calculated as following:
| (3) |
where V max is the maximum velocity and K m the Michaelis–Menten constant, both predicted from GraphPad Prism.
The in vivo hepatic intrinsic clearance (CLint, in vivo) was calculated using physiological scaling factors for hepatocellularity [40, 41], for the protein [42], and liver weight [43] according to Equations (4 and 5):
| (4) |
| (5) |
The hepatic organ clearance (CLh) was predicted using the well‐stirred liver model according to Equation (6) [44]:
| (6) |
where Q H is the hepatic blood flow (20.7 mL/min/kg) [45]. Incubational binding for the test system (f u,inc) was predicted using the IQVIVE platform (qivivetools.wur.nl), taking into account the physicochemical properties of the compounds. For the S9 incubations, the S9 protein concentration (2 mg/mL) was considered and the Hallifax and Houston method applied [46]. For the 3D HepaRG experiments, the cell concentration (1.46 × 106 cells/mL) was considered and the Kilford method applied [47]. A measured plasma binding value (f u,b) for PGME was used [32].
2.14. β‐PGME Toxicokinetic Model Development and Calibration
The compartmental toxicokinetic (TK) model developed for α‐PGME from Reale et al. [32], included the excretion of parent compound and the metabolism of α‐PGME to propylene glycol (PG) and conjugation products. This TK model was adapted for the present study to incorporate metabolism of β‐PGME to 2‐MPA, with equal physiological parameters used for both isomers. Due to the low excretion of conjugated β‐PGME (3%) and the minimal amounts (below the limit of quantification) of the parent compound (β‐PGME) [48], these metabolic routes were excluded and only the formation of PG and the main metabolite 2‐MPA were included. Table 5 summarizes the 2‐MPA specific parameters derived from literature and computer tools, and the generated in vitro predicted hepatic organ clearance (CLh). The CLh value obtained from solvent loss measurement was used under the assumption that the clearance of β‐PGME loss was expected to be equal to the clearance of 2‐MPA formation, with 2‐MPA being the main metabolite. The kinetic constant for the metabolism of β‐PGME to PG was not available from literature, therefore, we used a literature kinetic constant of EGME to ethylene glycol (EG). As β‐PGME is structurally and chemically similar to EGME, we assumed that their metabolism to PG and EG, respectively, were comparable.
TABLE 5.
Parameters used in the β‐PGME toxicokinetic model, in addition to the ones already reported in Reale et al. [32].
| Parameters | Symbol | Units | Values | References |
|---|---|---|---|---|
| 2‐MPA‐specific parameters | ||||
| Molecular weight | MWMPA | [g/mol] | 104.105 | Comptox Chemicals Dashboard (CCD) a |
| Blood‐air partition coefficient | PbaMPA | — | 200 357 | Kramer et al. [49] |
| Central‐air partition coefficient | PcaMPA | — | 243 185 | Tibaldi et al. [50] |
| Brain–blood partition coefficient | PBRMPA | — | 0.71 | Rodgers & Rowland [51] |
| Unbound fraction in plasma | Fu,pMPA | — | 0.46 | Sipes et al. [52] |
| Blood‐to‐plasma ratio | RbMPA | — | 0.98 | ADMET predictor Software |
| Urinary excretion rate of 2‐MPA | KMPA | [1/h] | 0.09 | Devanthéry et al. [20] |
| Unbound fraction in brain | Fu,BMPA | — | 0.53 | Watanabe et al. [53] |
| Permeability coefficient (BBB) | PeMPA | [cm/min] | 0.006 | Kerns et al. [54] |
| Kinetic constant EGME → EG (ethylene glycol) | CLmetPG | [L/h/Kg] | 0.3 | Gargas et al. [55] |
| In vitro predicted hepatic organ clearance (CL h ) | ||||
| S9, metabolite formation, calculated (V max/K m) | CLmetMPA | [L/h/Kg] | 0.0216 | Predicted b |
| S9, solvent loss, measured | CLmetMPA | [L/h/Kg] | 0.3000 | Predicted b |
| S9, metabolite formation, measured | CLmetMPA | [L/h/Kg] | 0.9840 | Predicted b |
| 3D HepaRG, metabolite formation, measured | CLmetMPA | [L/h/Kg] | 0.5442 | Predicted b |
CompTox Chemicals Dashboard (epa.gov).
Predicted values based on the measured in vitro CLint applying in vitro‐in vivo extrapolation as described in the methods section.
The adapted TK model included a central compartment representing the body fluids, a brain compartment to compute β‐PGME brain concentrations, and a blood–brain barrier to assess the β‐PGME rate for entering the brain (Figure 2). We assumed that β‐PGME is mainly metabolized in the liver (disregarding other tissues that may metabolize β‐PGME) and that the isomer distributions are equal in the central compartment; 2‐MPA distributes in the same central compartment as the parent compound; and lastly, the central and brain TK compartment diffusions for β‐PGME and 2‐MPA are the same as for the α‐isomer. Finally, the model was run with a fraction of 0.3% β‐PGME in Berkeley Madonna software (version 8.3.18, University of California, USA) based on reported literature data [20].
FIGURE 2.
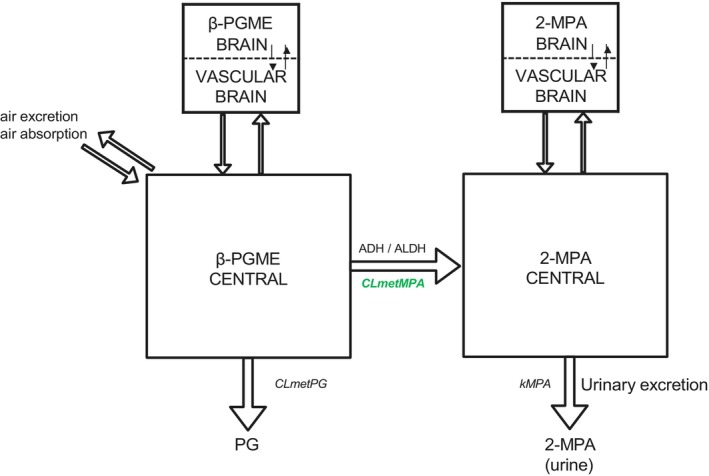
Developed β‐PGME toxicokinetic model in the present study. The simulation model for β‐isomer propylene glycol methyl ether (β‐PGME) and methoxy propionic acid (2‐MPA) was based on the model of Reale et al. [32]. Arrows represent flow rates, expressed as mg/h, and rectangles represent model compartments. ADH, alcohol dehydrogenase; ALDH, aldehyde dehydrogenase; CLmetMPA, clearance of the formation of 2‐MPA; CLmetPG, clearance of the formation of propylene glycol; kMPA, urinary excretion rate of 2‐MPA.
2.15. TK Model Evaluation
We used published data [20] from a human study (exposure to 99.7% α‐PGME, 0.3% β‐PGME) to evaluate the developed β‐isomer TK model. Key information on the pharmacokinetic dataset used for model evaluation is summarized in Table 6. We could not validate the TK model due to the lack of experimental data; however, we used three criteria to evaluate the TK model: (1) Model performance according to the WHO criteria stating that a model is acceptable if its predicted values match with the experimental kinetic profiles within a twofold difference [56]; (2) goodness‐of‐fit assessed with linear regressions with an acceptable fit defined as a coefficient of determination (R 2) equal or higher than 0.75 [57]; and (3) mean absolute percentage error (MAPE) was acceptable if MAPE < 50%, good if MAPE < 20%, and > 10%, and excellent if MAPE < 10% [58]. Microsoft Excel 2016 was used to calculate linear regression and MAPE.
TABLE 6.
Pharmacokinetic study on PGME in humans used for β‐TK model development and evaluation.
| Exposure concentration | Duration | Measured concentration | Matrix | Number of time points | References |
|---|---|---|---|---|---|
| 95 ppm | 6 h with 30′ break after 3 h | CurMPA | Urine | 9 | Devanthéry et al. [20] |
| 50 ppm | 6 h with 30′ break after 3 h | CurMPA | Urine | 7 | Devanthéry et al. [20] |
Note: CurMPA: 2‐MPA concentration in urine. PGME consisted of 99.7% α‐PGME and 0.3% β‐PGME.
2.16. Sensitivity Analysis and Uncertainty Analysis
A sensitivity analysis was performed to identify model parameters (p) that had the greatest influence on the model output, for example the 48 h area under the curves (AUC) of 2‐MPA in urine and blood. MS Excel 2016 was used to perform sensitivity analysis. Normalized sensitivity coefficients (NSC) of the TK model output to any selected parameter of the model were calculated as follows:
| (7) |
where Δoutput and Δp are the differences between output and p values, respectively, before and after increasing p by 1%. Parameters were categorized depending on the influence they had on the output parameters following these criteria: low impact if |NSC| < 0.2, medium impact if 0.2 ≤ |NSC| < 0.5, high impact if |NSC| ≥ 0.5 [56]. The uncertainty of parameters with high impact on model output (|NSC| ≥ 0.5) was qualitatively assessed depending on the parameter source following these criteria [58, 59]: low uncertainty, if a parameter value was obtained from human data parameters or verified through successful use in physiologically based pharmacokinetic models; medium uncertainty, if a parameter value was obtained from a different species with a high probability that scaling holds across species; high uncertainty, if a value was not available for a parameter and assumptions had to be made.
2.17. Statistical Analysis
Data representation and statistical analysis were performed using Microsoft Excel (Microsoft, Redmond, WA) and GraphPad Prism (GraphPad Software, San Diego, CA, USA; Version 10.0.2). Data are expressed as mean values ± SD. For the statistical analysis of two groups, we used the Student's t‐test. One‐way ANOVA was used for statistical analysis of multiple concentrations of the same treatment. For multiple concentrations and different treatments, we used two‐way ANOVA. p < 0.05 was considered as significant (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001, ****p ≤ 0.0001).
2.18. Nomenclature of Targets and Ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY [60, 61, 62], and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 [63].
3. Results
3.1. The 3D HepaRG Model Displays Key Hepatocellular and Metabolic Functions
Immunostainings (Figure 3A) showed the presence of albumin in 3D HepaRG cultured over 7 days. Albumin was observed intracellularly in untreated HepaRG cells (CTR) and released into the medium (78.4 ± 22.9 ng/mL) (Figure 3B). The albumin secretion was significantly reduced (1.78 ± 0.68 ng/mL) by treating the cells with the hepatotoxicant acetaminophen (APAP), proving the ability of the cells to respond to hepatotoxic effects. The 3D cultures were able to metabolize two known reference drugs, namely testosterone and diclofenac, which are substrates for CYP3A and CYP2C9, respectively. As illustrated in Figure 3C, the HepaRG successfully converted the test compounds to their hydroxylated metabolites over 48 h, indicating active phase I metabolism. We observed higher metabolite formation for HepaRG cells preincubated with 20 μM rifampicin, mostly for testosterone metabolism. The in vitro intrinsic hepatic clearance (CLint) of testosterone was higher than for diclofenac. Concordantly, immunostainings of 3D HepaRG showed significant induction of CYP3A4 by rifampicin: As depicted in Figure 3D/E, the integrated density/area DAPI for the induced cells was higher than for the uninduced cells.
FIGURE 3.
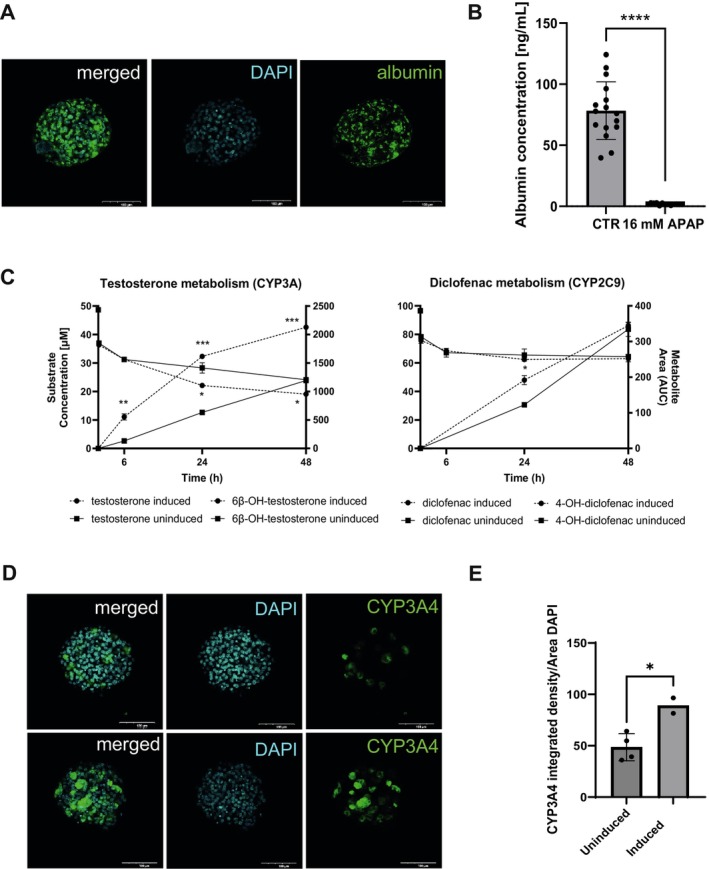
Proof for hepatic functionality of 3D HepaRG. (A) 3D HepaRG were stained for albumin (green) and counterstained with DAPI (blue). Scale bar representing 100 μm. (B) 3D HepaRG were exposed to 16 mM APAP (acetaminophen, positive control) for 48 h. Albumin was measured using the Bethyl Laboratories ELISA kit. Data are expressed as albumin concentration (ng/mL) per 3D HepaRG spheroid. N = 2 independent biological experiments with six to eight technical replicates. Bar graphs represent single measurements with mean ± SD; statistical analysis based on an unpaired t‐test: ****p < 0.0001. (C) Uninduced and induced (20 μM rifampicin) 3D HepaRG were exposed to 50 μM testosterone and 100 μM diclofenac for over 48 h. Conversion of testosterone and diclofenac into 6β‐hydroxy testosterone and 4‐hydroxy diclofenac, respectively, was determined at 6, 24, and 48 h by HPLC‐MS/MS. Data are expressed as concentrations (mM) calculated using calibration curves. Area under the curve in arbitrary units (AUC) was used for calculating 6β‐hydroxy testosterone and 4‐hydroxy diclofenac; N = 1 biological experiment with two technical replicates. Statistical analysis based on an unpaired t‐test: ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05. (D) Uninduced and induced (20 μM rifampicin) 3D HepaRG were stained for CYP3A4 (green) and counterstained with DAPI (blue). Scale bar representing 100 μm. (E) Immunostaining images (n = 2–4) were quantified for CYP3A4 using ImageJ. Bar graphs represent mean ± SD; statistical analysis based on an unpaired t‐test: *p ≤ 0.05.
More importantly, HepaRG cells also expressed the enzymes directly involved in the metabolism of PGME. Figure 4A shows the results for q‐RT‐PCR calculated as ∆Ct (in comparison with B2M expression) for the isoforms ADH1A, ADH1B, ADH1C, and ALDH2. ADH1B showed the highest relative gene expression across all models; HepaRG, primary human hepatocytes (pHH) and human total liver RNA as a comparator. ADH1A expression was lower, but similar in all tested models. Expression of ADH1C in HepaRG was similar to that in pHH and slightly lower than in RNA isolated from fresh liver tissue. ALDH2 expression in HepaRG was lower than in pHH and fresh tissue. Furthermore, immunostainings of the 3D HepaRG confirmed the protein expression of ADH1A/B/C and ALDH2 in the cells (Figure 4B). In addition, Western blot results showed distinctive bands for ADH1A/B/C and ALDH2 for HepaRG and pHH cell lysates (Figure 4C), whereby the signal mean intensity relative to GAPDH was marginally higher for pHH compared to HepaRG (Figure 4D). For both cell culture models, ALDH2 was considered abundant, with a higher expression level than GAPDH.
FIGURE 4.
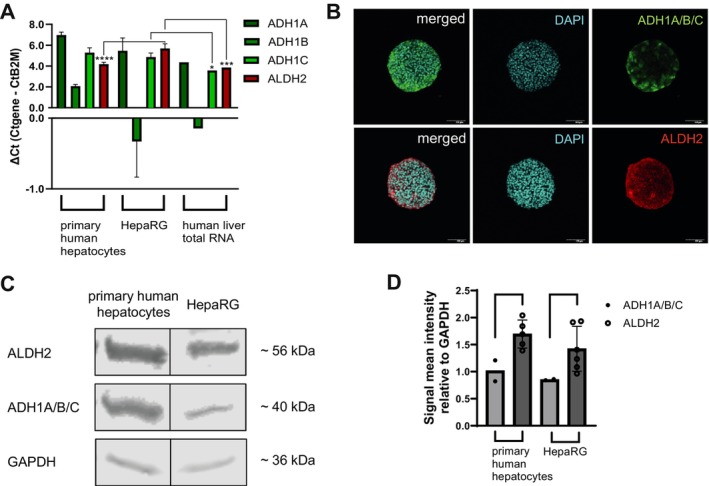
Confirmation of ADH1A/B/C and ALDH2 in 3D HepaRG and comparison to other liver systems. (A) Gene expression of ADH1A, ADH1B, ADH1C and ALDH2 in primary human hepatocytes (pHH), HepaRG, and human liver total RNA was detected using q‐RT‐PCR; N = 3 independent biological repeats with two to four technical replicates for each pHH donor, four to six technical replicates for HepaRG and N = 1 biological experiment with two technical replicates for human liver total RNA. Bar graphs represent mean ± SD; statistical analysis based on one‐way ANOVA followed by Dunnett's pairwise comparison: *p ≤ 0.05; ***p ≤ 0.001; ****p ≤ 0.0001. (B) 3D HepaRG were stained for ADH1A/B/C (green), ALDH2 (red), and counterstained with DAPI (blue). Scale bar representing 100 μm. (C) Western blot of ALDH2, ADH1A/B/C, and GAPDH (loading control) in pHH and HepaRG cell lysate. (D) Signal mean intensity relative to GAPDH for the target proteins was determined using ImageJ. Cell lysate from both pHH donors was each used in total N = 2 independent Western blots for ADH1A/B/C, and two to three times in total N = 5 independent Western blots for ALDH2. HepaRG cell lysate was used in N = 2 independent Western blots for ADH1A/B/C, and N = 6 independent Western blots for ALDH2. Bar graphs represent mean ± SD; statistical analysis based on an unpaired t‐test.
3.2. Effects of the Solvents and Their Metabolites on Liver Cells
Relative cell viability (cellular ATP content) measurements showed that 3D HepaRG cultures were fairly resistant to all treatments with EC50 values in the millimolar range (Figure 5A and Table 7). For all substances, EC50 values were lower after 7 days of treatment than after 48 h exposure. A comparison of the calculated EC50 values shows that EGME appeared to be less toxic than commercial PGME and β‐PGME. Notably, we observed initially higher ATP content (100%–130%) for all compounds after exposure over 7 days. Compared to the solvents β‐PGME and EGME, the metabolites showed approximately fourfold (2‐MPA) and 50‐fold (2‐MAA) higher toxicities, increasing with longer exposure time. Interestingly, 2‐MAA (EGME metabolite) was more toxic than 2‐MPA (β‐PGME metabolite), although β‐PGME and commercial PGME seemed to be more toxic than EGME. Compared to 2D cultures, the 3D HepaRG were more sensitive as indicated by the lower EC50 values.
FIGURE 5.
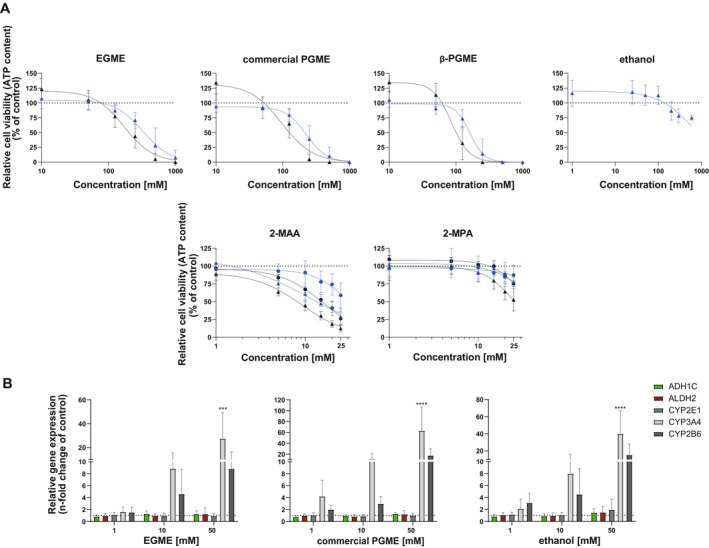
Effects of glycol ethers, metabolites, and ethanol on HepaRG. (A) Cells were exposed to EGME, commercial PGME, β‐PGME, ethanol, 2‐MAA, and 2‐MPA for 48 h (2D: ●, 3D: ▲) and 7 days (2D: ●, 3D: ▲). Viability was assessed using the CellTiter‐Glo Luminescent Cell Viability Assay and expressed as relative ATP content (% of untreated control), N = 3–4 independent biological repeats with three technical replicates. Graphs represent mean ± SD. (B) Gene expression of ADH1C, ALDH2, CYP3A4, CYP2E1, and CYP2B6 after treatment with EGME, commercial PGME, and ethanol for 7 days was detected using q‐RT‐PCR; N = 3 independent biological repeats with two to three technical replicates. Bar graphs represent mean ± SD; statistical analysis based on two‐way ANOVA followed by Dunnett's pairwise comparison: ***p ≤ 0.001; ****p ≤ 0.0001.
TABLE 7.
EC50 values and the corresponding 95% confidence interval (CI) for EGME, commercial PGME, β‐PGME, ethanol, 2‐MAA, and 2‐MPA assessed on 2D and 3D HepaRG cultures upon 48 h or 7 days of exposure.
| 2D HepaRG (48 h) | 2D HepaRG (7 days) | 3D HepaRG (48 h) | 3D HepaRG (7 days) | |||||
|---|---|---|---|---|---|---|---|---|
| EC50 [mM] | 95% CI | EC50 [mM] | 95% CI | EC50 [mM] | 95% CI | EC50 [mM] | 95% CI | |
| EGME | NA | NA | NA | NA | 326 | 261–397 | 171 | 139–205 |
| PGME | NA | NA | NA | NA | 226 | 194–262 | 96 | 73–126 |
| β‐PGME | NA | NA | NA | NA | 162 | 146–183 | 86 | 75–99 |
| ethanol | NA | NA | NA | NA | 610 | 464–898 | NA | NA |
| 2‐MAA | 31 | 27–45 | 16 | 15–17 | 11 | 5–15 | 9 | 8–10 |
| 2‐MPA | 81 | ND | 34 | 31–40 | 46 | ND | 26 | 23–30 |
Abbreviations: NA, not available; ND, not determinable.
Regarding metabolic inductions, q‐RT‐PCR results showed a significant concentration‐dependent increase (30‐ to 60‐fold) of the relative gene expression of the phase I CYP450 enzymes CYP3A4 and CYP2B6 after 7‐day exposure to EGME, commercial PGME, and ethanol in HepaRG (Figure 5B). On the contrary, the expression of CYP2E1, ADH1C, and ALDH2 was not induced by the exposure to the solvents.
3.3. Determination of Hepatic Kinetic Parameters in Human Liver S9 Incubations
We confirmed ADH1A/B/C and ALDH2 protein presence in human liver S9 fraction by Western blot (Figure 6A). Results showed distinctive bands for both enzymes with signal mean intensities relative to GAPDH of 1.27 ± 0.60 for ADH1A/B/C and 1.73 ± 0.47 for ALDH2 (Figure 6B).
FIGURE 6.
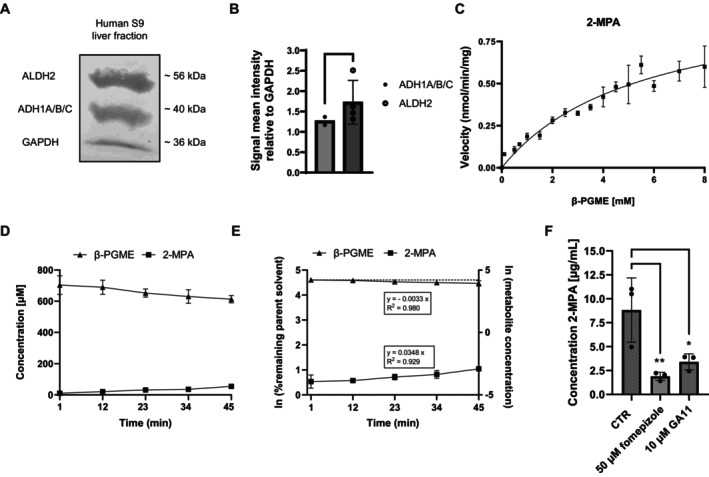
Hepatic kinetic of β‐PGME conversion to 2‐MPA using human liver S9 fractions. (A) Western blot of ALDH2, ADH1A/B/C, and GAPDH (loading control) in S9 cell lysate. (B) Signal mean intensity relative to GAPDH for the target proteins was determined using ImageJ (N = 2–4 independent biological repeats). Bar graphs represent mean ± SD; statistical analysis based on an unpaired t‐test. (C) Representative velocity against substrate concentration plot for Michaelis–Menten kinetics in S9 incubations. 2‐MPA was quantified using HPLC‐MS/MS. The Michaelis–Menten curve was fitted to the data using Graphpad Prism and K m and V max predicted. Data points represent mean ± SD of N = 3 independent incubations with two technical replicates. (D) Solvent (β‐PGME) loss‐time profile (▲) and 2‐MPA metabolite formation (■) in S9 incubations. Data points represent mean ± SD of N = 3 independent incubations with two technical replicates. (E) Ln of % remaining parent (▲) and ln of metabolite concentration (■) versus time profile in S9 incubations. The in vitro CLint was calculated from the slope as described in the methods section. Data points represent mean ± SD of N = 3 independent incubations summarizing calculated slopes derived from each single graph. The dotted line represents the parent value at time‐point 1 min. (F) Inhibition of 2‐MPA formation. 2‐MPA was quantified after sampling at time‐point 45 min for control incubations (CTR) and incubations applying 50 μM fomepizole and 10 μM GA11. Data points represent N = 3 incubations with two technical replicates. Bar graphs represent mean ± SD; statistical analysis based on one‐way ANOVA followed by Dunnett's pairwise comparison; *p ≤ 0.05, **p ≤ 0.01.
Next, we determined the Michaelis–Menten kinetics K m and maximum reaction rate (V max) using the S9 incubations. Figure 6C illustrates the resulting β‐PGME concentration versus velocity plot. Results revealed a K m value of 5572 ± 2171 μM and V max of 1.04 ± 0.28 nmol/min/mg for the reaction of β‐PGME to 2‐MPA (Table 8). The in vitro hepatic intrinsic (CLint) for V max/K m was calculated as 0.19 ± 0.03 μL/min/mg.
TABLE 8.
Obtained Michaelis–Menten kinetic parameters (V max, K m) from human liver subcellular fraction (S9) incubations, in vitro hepatic intrinsic clearance (CLint, in vitro) calculated on V max/K m and measured from S9 and 3D HepaRG metabolism studies, predicted in vivo hepatic intrinsic clearance (CLint, in vivo) and in vitro predicted hepatic organ clearance (CLh). Data represent mean values ± SD.
| β‐PGME → 2‐MPA | S9 incubations | 3D HepaRG |
|---|---|---|
| V max [nmol/min/mg] | 1.04 ± 0.28 | |
| K m [μM] | 5572 ± 2171 |
| Calculated | Measured | Measured | Measured | |
|---|---|---|---|---|
| (Vmax/Km) | Solvent loss | Metabolite formation | Metabolite formation | |
| f u,inc a | 0.87 | 0.87 | 0.87 | 0.94 |
| CLint, in vitro [μL/min/mg] | 0.19 ± 0.03 | 3.80 ± 2.14 | 40.9 ± 5.19 | 9.62 ± 0.85 b |
| CLint, in vitro [μL/min/10 6 cells] | 9.34 ± 0.83 | |||
| Predicted CL int, in vivo [mL/min/kg] | 0.60 ± 0.09 | 11.8 ± 6.67 | 127 ± 16.1 | 28.2 ± 2.50 |
| Predicted CL h [mL/min/kg] | 0.36 ± 0.05 | 5.03 ± 2.52 | 16.4 ± 0.47 | 9.07 ± 0.44 |
Data derived from IQVIVE, https://www.qivivetools.wur.nl .
Converted from 9.34 ± 0.83 μL/min/106 cells, using 117.5 × 106 cells/g liver and 121 mg S9 protein/g liver.
We illustrated the results for the 2‐MPA metabolite formation and β‐PGME depletion experiments in Figure 6D, following a linear manner over 45 min. We calculated the measured in vitro CLint for the solvent loss from the ln of % remaining parent solvent as 3.80 ± 2.14 μL/min/mg (Figure 6E and Table 8). The measured in vitro CLint calculated from the ln of % 2‐MPA metabolite concentration was 40.9 ± 5.19 μL/min/mg. Both values appeared to be higher than the in vitro CLint calculated from V max/K m.
Further, studies applying a competitive inhibitor for ADH (fomepizole = 4‐methyl pyrazole) and for ALDH (GA11 = 2,6‐diphenylimidazo[1,2‐a]pyridine) showed a significantly reduced 2‐MPA formation compared to control incubations (Figure 6F).
Finally, we performed hepatic organ clearance (CLh) predictions through in vitro‐in vivo extrapolation (IVIVE) based on the in vitro CLint obtained from the performed measurements as well as the in vitro CLint calculated from K m and V max. Although the determined in vitro CLint based on the metabolite formation (40.9 ± 5.19 μL/min/mg) was much higher than for the solvent loss (3.80 ± 2.14 μL/min/mg), the in vitro predicted CLh values showed to be nearly within threefold deviation (metabolite formation: 16.4 ± 0.47 mL/min/kg; solvent loss: 5.00 ± 2.52 mL/min/kg). Besides, the calculated in vitro predicted CLh (K m/V max), 0.36 ± 0.05 mL/min/kg, was much lower than the measured values. All values are summarized in Table 8.
3.4. Assessment of Hepatic Kinetic Parameters in the 3D HepaRG Model
Given linear 2‐MPA formation within the first hour, we calculated the measured in vitro CLint in the 3D HepaRG from the ln of % 2‐MPA metabolite concentration within 60 min (Figure 7A,B). The in vitro CLint for the β‐PGME loss was not determined, as only a small quantity of the 5 mM applied β‐PGME was metabolized, and the obtained depletion curves were not reliably measurable with the analytical methods. The in vitro CLint for the metabolite formation was 9.34 ± 0.83 μL/min/106 cells or 9.62 ± 0.85 μL/min/mg after applying the previously described scaling factors (summarized in Table 8). Similar to the S9 incubations, clearance was greater than the in vitro CLint calculated from V max/K m. In comparison to the measured in vitro CLint in the S9 incubations, the in vitro CLint determined in the 3D HepaRG appeared to be smaller. Nevertheless, the in vitro predicted CLh values following IVIVE were close within twofold deviation.
FIGURE 7.
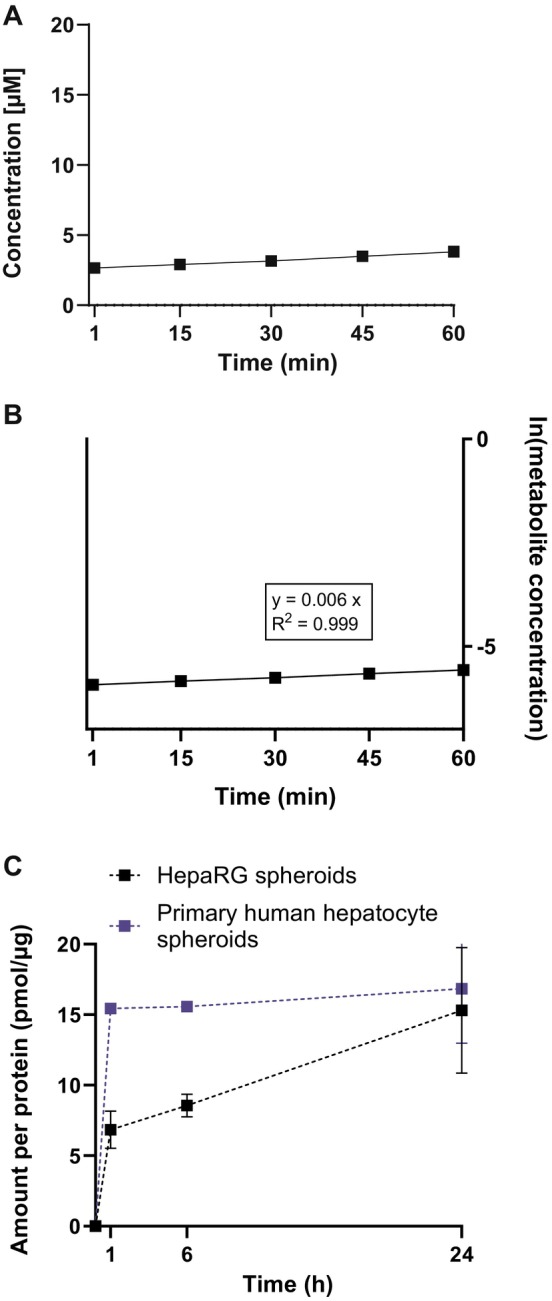
Long‐term metabolism profile in 3D HepaRG and 3D primary human hepatocytes and hepatic kinetic for β‐PGME conversion to 2‐MPA using 3D HepaRG. (A) 2‐MPA metabolite formation within the first hour (■) in 3D HepaRG. Data points represent mean ± SD of N = 3 independent incubations with two technical replicates. (B) Ln of 2‐MPA concentration (■) versus time profile in 3D HepaRG. The in vitro CLint was calculated from the slope as described in the methods section. Data points represent mean ± SD of N = 3 independent incubations summarizing calculated slopes derived from each single graph. The dotted line represents the parent value at time‐point 1 min. (C) 2‐MPA formation‐time profile in 3D HepaRG and 3D primary human hepatocytes (pHH). 3D‐spheroids were exposed to 5 mM β‐PGME and the conversion into 2‐MPA was determined at 1, 6, and 24 h by HPLC‐MS/MS. Data are expressed as amount of metabolite formed per incubational protein content (pmol/μg) calculated using calibration curves; N = 3 independent biological repeats with two technical replicates (HepaRG) and N = 2 independent biological repeats using each pHH donor once with one to two technical replicates. Graphs represent mean ± SD.
Simultaneous incubations with 3D HepaRG and 3D primary human hepatocytes (pHH) showed higher concentrations of 2‐MPA at 1 and 6 h for the 3D pHH than for the 3D HepaRG, yet the amount of formed metabolite was similar at the end of the incubation (Figure 7C). Artifacts due to stability, adsorption, or evaporation of β‐PGME and 2‐MPA over time are unlikely based on our data (Figures S3 and S4).
3.5. Implementation of In Vitro Data Into the Simulation Model
We generated simulations applying the β‐isomer PGME toxicokinetic (TK) model integrating the in vitro predicted hepatic organ clearance (CLh) values (Table 8). No calibration was carried out and no parameters were fitted, enabling both sets of experimental data [20] (PGME exposure at 95 ppm and 50 ppm) to be used for model evaluation. Figure 8 illustrates the comparisons of predicted urinary 2‐MPA concentrations versus experimental data. Our results show a low fit for the simulations based on the in vitro predicted CLh calculated for S9 (V max/K m) with R2 = 0.16 (below the acceptable threshold of 0.75) and a MAPE of 64.1% (unacceptable). Most data points were out of twofold range from observed data (Figure 9A). In contrast, the model predictions using in vitro predicted CLh from S9 (solvent loss) correlated well with the observed data with R 2 = 0.87 and a MAPE of 23.9% (acceptable). Similar fits presented the simulations based on the in vitro predicted CLh from S9 (metabolite formation) with R 2 = 0.93 and a MAPE of 21.5% (acceptable) and 3D HepaRG (metabolite formation) with R 2 = 0.92 and a MAPE of 22.4% (acceptable). Those predictions were all within twofold error from observed data (Figure 9B–D).
FIGURE 8.
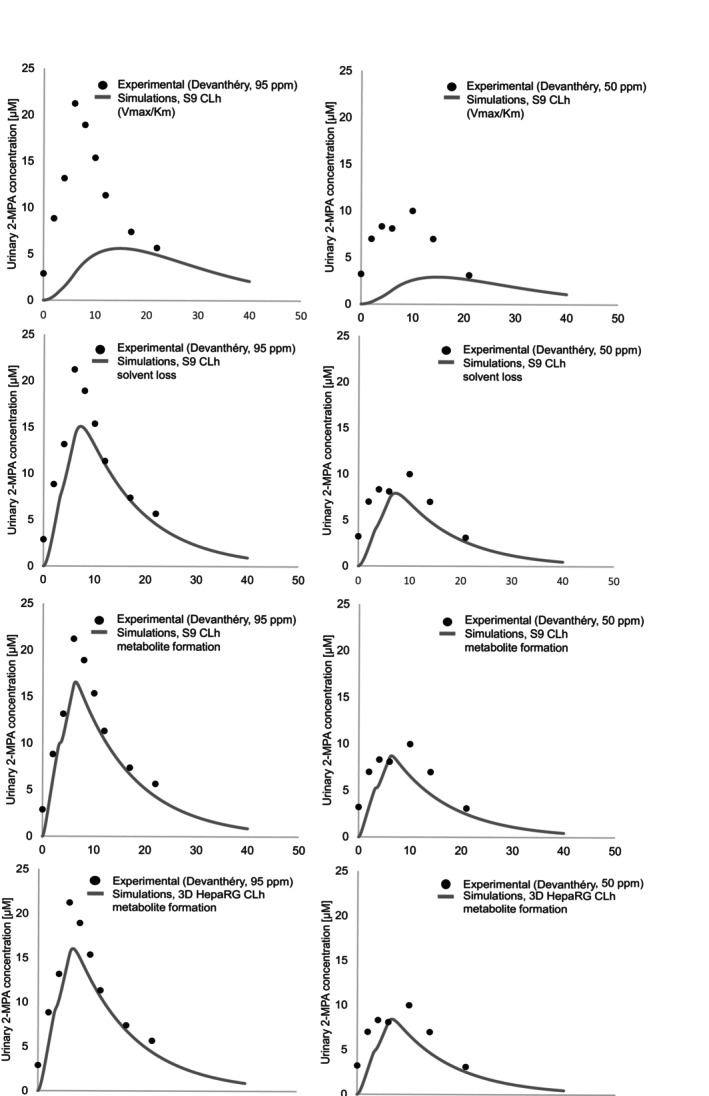
Toxicokinetic model evaluation: Comparison of predicted versus experimental data. Predicted urinary 2‐MPA concentrations (lines) based on in vitro predicted CLh from S9 fraction (V max/K m), S9 fraction (solvent loss), S9 (metabolite formation), and 3D HepaRG (metabolite formation) were compared to experimental data (dots) derived from Devanthéry et al. [20]. The dip in concentration at 3 h observed in the graphs is due to a 30‐min break from exposure as described in Table 6.
FIGURE 9.
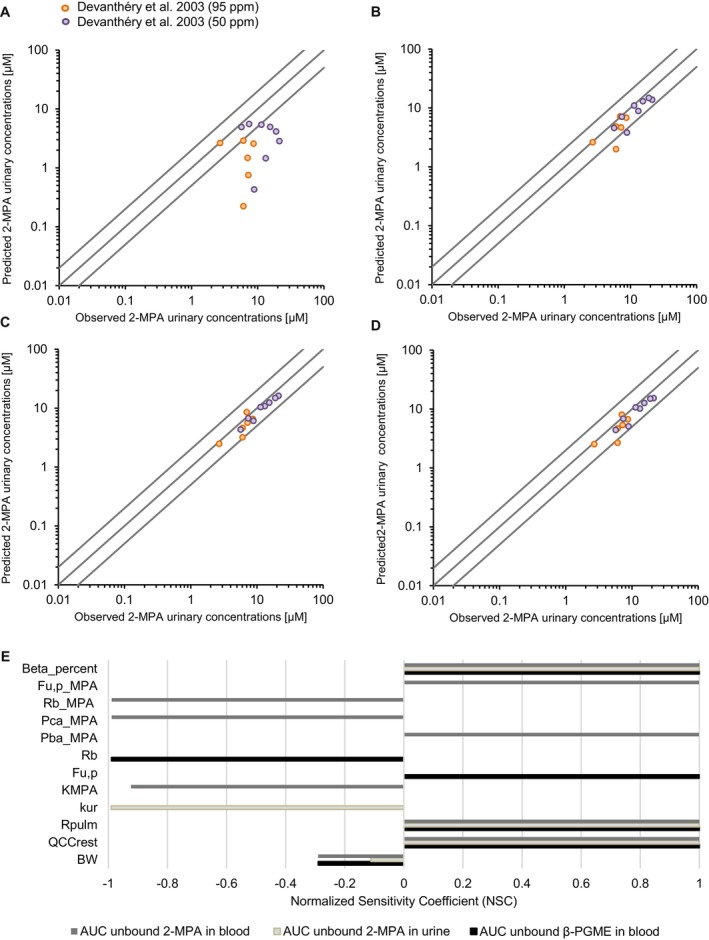
Plot of predicted versus experimental data and results of sensitivity analysis. Observed urinary 2‐MPA concentrations measured by Devanthéry et al. [20] were plotted versus concentrations predicted by our toxicokinetic model using the clearance value determined with (A) S9 fraction (V max/K m), (B) S9 fraction (solvent loss), (C) S9 fraction (metabolite formation), and (D) 3D HepaRG model (metabolite formation). The middle line is the line of unity, and the outer lines represent twofold deviations. (E) Normalized sensitivity coefficients (NSC) for the area under the concentration curves (AUC) of unbound 2‐MPA in blood and urine and unbound β‐PGME in blood. Only parameters with medium or high impact on the output (|NSC| ≥ 0.2) are shown in the graph. Abbreviations for the parameters can be found in Table 5.
Results from the sensitivity analysis are shown in Figure 9E, whereby the parameters with medium or high impact (|NSC| ≥ 0.2) on the selected output are illustrated. We observed that all three selected area under the curves (AUC) outputs (2‐MPA urine concentration, 2‐MPA blood concentration, and β‐PGME blood concentration) were highly sensitive to cardiac output (QCCrest), β‐PGME pulmonary retention (R pulm), and the percentage of β‐isomer PGME present in the mixture used for the exposure experiment (Beta_percent). Additionally, AUC of unbound β‐PGME in blood was highly sensitive to β‐PGME unbound fraction in plasma (Fu,p) and to β‐PGME blood‐toplasma ratio (R b). The AUC of 2‐MPA in blood was highly sensitive to 2‐MPA unbound fraction in plasma (Fu,p_MPA), 2‐MPA blood‐to‐plasma ratio (Rb_MPA), blood‐air partition coefficient (PC) of 2‐MPA (Pba_MPA), central‐air PC of 2‐MPA (Pca_MPA), and to the urinary excretion rate of 2‐MPA (K MPA). Among the parameters with high impact, those with high uncertainty were Pba_MPA, Pca_MPA Rb_MPA, and Fu,p_MPA (Table S6).
4. Discussion
In this study, we used a 3D HepaRG liver model and human liver S9 fraction to assess the hepatic clearance of β‐isomer propylene glycol methyl ether (β‐PGME) as in vitro input data for a newly developed toxicokinetic (TK) model.
Our results show that HepaRG cultured in 3D‐spheroids successfully metabolized the solvent β‐PGME. This stable 3D‐cellular model could satisfy the increasing demand for determining the clearance of compounds with high metabolic stability and/or with prolonged exposure [25]. In this context, short‐term in vitro assays such as S9 incubations are of limited use due to their short‐lived enzyme activity [64]. Active metabolism of 3D HepaRG was expected based on the gene and protein expression levels of the metabolizing enzymes derived from 2D HepaRG, which were comparable to those detected in 2D primary human hepatocytes. From a quantitative point of view, the amount of 2‐MPA initially formed by the 3D HepaRG was approximately two‐ to threefold lower than that detected with 3D primary human hepatocytes. This discrepancy may be attributed to a slightly lower enzyme activity in the cell line. Alternatively, inter‐individual variation on hepatocyte donors may have played a role. Both primary hepatocyte donors used in this study were African Americans, and may have been carriers of the alcohol dehydrogenase (ADH1B*3) gene variant exclusive to populations of African ancestry [65]. While we did not genotype the cells, we specifically observed high gene expression of ADH1B in HepaRG and human liver total reference RNA, but not in these primary hepatocytes lots. Although we showed ADH and aldehyde dehydrogenase (ALDH) enzyme presence in 3D HepaRG, these data cannot conclusively demonstrate that these enzymes were responsible for the metabolism of PGME, as HepaRGs are known for expressing a wide range of enzymes [29]. However, we demonstrated the specific conversion of β‐PGME by inhibiting this pathway in S9 incubations. Using the ADH inhibitor fomepizole significantly reduced 2‐MPA formation, which was expected because of its wide application in the treatment of patients suffering from methanol or ethylene glycol poisoning [36, 66]. Furthermore, we confirmed the in vitro inhibitory potential of GA11, previously described only through computational approaches as an inhibitor of ALDH2 [37].
In comparison to published rat studies that reported more than 60% of β‐PGME being excreted as 2‐MPA [19, 48], we observed low metabolism of β‐PGME in vitro. This could have resulted from species differences, extra‐hepatic metabolism of β‐PGME, or possibly analytical sensitivity and capacity issues [67]. As our experiments using 3D HepaRG and S9 exhibited the formation of 2‐MPA inversely parallel to the depletion of β‐PGME, we do not expect that other metabolic reactions have occurred that would significantly exceed 2‐MPA generation. We would like to highlight here, that the greater clearance values measured for the 2‐MPA formation than the β‐PGME loss derived from the S9 incubations could be due to the use of different analytical techniques for compound quantification, introducing potential interdependent errors.
To this date, no in vivo or in vitro metabolic kinetic studies reporting the Michaelis–Menten parameters (K m, V max) for the conversion of β‐PGME to 2‐MPA are known. Aligning our results for Michaelis–Menten kinetics with an existing study performed on EGME using human hepatocytes, we find values in a similar range (millimolar) with a reported K m of 1700 μM and V max of 61.3 nmol/h/106 cells [4, 5]. This results in a calculated in vitro CLint of 0.62 μL/min/mg applying the scaling factors from our study, which can be classified as a low clearance rate, [68] resulting in a slow metabolite generation. Similarly, the calculated in vitro CLint (V max/K m) for β‐PGME obtained in our study was low. Despite the low reported hepatic clearance, EGME is a known toxicant that has been described for adverse effects on the testis, bone marrow, the central nervous system, and many more, because of its metabolite methoxy acetic acid (2‐MAA) [4, 6]. Therefore, we conclude that we cannot exclude adverse effects by 2‐MPA, especially as 2‐MPA showed an approximately four times lower EC50 value in 3D HepaRG cultures than β‐PGME. Moreover, based on the overall generated in vitro predicted CLh values, we would propose that β‐PGME is a low to medium clearance compound, considering reported clearance rankings [68, 69, 70]. In translation to the human body, this suggests that β‐PGME would exhibit moderate persistence before being metabolized into the potentially harmful metabolite 2‐MPA.
Although the calculated clearance (V max/K m) for β‐PGME was close to the reported value for EGME, the performed TK simulations resulted in a low fit compared to the experimental data.
Generally, we observed underprediction of the experimental urinary 2‐MPA concentrations for all simulated curves, though not as significant as for those using the calculated clearance (V max/K m). It has become recognized that in vitro data tend to underestimate the observed metabolic clearance in humans [71]. Reasons represent the use of standard biological scaling factors, extrinsic factors such as storage conditions and/or the preparation process, the absence of appropriate correction for non‐specific binding, as well as extra‐hepatic metabolism in vivo [67, 72, 73]. However, we cannot explain whether the notable underestimation of the calculated in vitro CLint (V max/K m) derived from an underpredicted V max value or an overpredicted K m value.
Next, we argue that 3D HepaRG represents a more physiological system compared to the S9 fraction. It requires that compounds permeate into the cells to be metabolized and takes into consideration the full range of enzymes and cofactors, which allows for evaluation of the metabolism via ADH/ALDH in the presence of other physiological metabolic routes [29, 74]. In our study, we showed the active performance of Phase I metabolism in an inducible manner and the secretion of albumin comparable to reported levels, two phenotypical characteristics for hepatocytes [33, 75]. Moreover, the similar simulation outcomes for the 3D HepaRG and the S9 incubations confirmed effective ADH and ALDH enzyme activities in HepaRG and we expect a high (not clearance‐limiting) permeability of β‐PGME into the cells.
In the context of β‐PGME, the linear formation of 2‐MPA within the first hour did not necessitate a complex, long‐lasting in vitro system for the determination of the hepatic kinetic parameter. Besides, the S9 incubations showed to deliver reliable input data as well. Nevertheless, the characterized 3D HepaRG model can serve as a tool to study the metabolism of other compounds converted via the ADH/ALDH pathway, especially low‐turnover compounds that require a longer incubation time. Moreover, these more complex cell cultures can be maintained for days and weeks and are hence suitable for implementation into more complex, multiorgan systems such as Organ on Chip (OOC) or microphysiological systems (MPS) [76, 77]. This would greatly help to better understand adverse effects of β‐PGME and its metabolite in the body and under chronic exposure.
In comparison to simpler liver systems like the S9 cell fraction, the advanced 3D HepaRG model moreover offers the possibility for studying direct hepatocellular toxicity caused by the formed metabolite over extended incubation periods [78]. Given the low observed metabolite formation in the 3D HepaRG, we propose that the evaluated cytotoxicity of the solvents is primarily due to the induction of cell permeability rather than caused by the metabolites [79]. Furthermore, we observed viability curves initially exceeding 100%, which could be explained by an adaptive metabolic cell response after first compound exposures. Previous chronic toxicity studies in animals using EGEs have shown the induction of liver enzymes and changes in liver weights [17, 80, 81]. In line with this, we observed increased gene expression of CYP3A4 and CYP2B6 after treatment of HepaRG with non‐cytotoxic concentrations of EGME, commercial PGME, and ethanol, but not for CYP2E1, ADH1A/B/C, and ALDH2. This was unexpected given the reported induction effects described after alcohol exposure [38]. So far, the significance of these findings for humans is not clear, as high in vitro concentrations were applied and a prediction of human liver exposure concentrations would be needed. However, our results may be relevant since occupational exposures take place over a lifetime and cumulative mixture effects cannot be excluded. In addition, as many chemicals are substrates, inhibitors, or inducers of CYP3A4 and CYP2C9, drug interactions after long‐term exposure to the solvents should be considered [82]. Moreover, polymorphism of the ALDH2 enzyme is well known and should be taken into account [16].
In summary, using the established 3D HepaRG model, we were able to generate in vitro hepatic kinetic data for the conversion of β‐PGME to 2‐MPA that reliably predicted the in vivo situation in a similar magnitude (twofold) to S9 fraction. We conclude that the 3D HepaRG model can be used to generate in vitro hepatic metabolism and clearance data for compounds metabolized via ADH and ALDH. Moreover, it offers a promising tool to simultaneously study enzyme induction and metabolite‐induced hepatotoxicity and to be integrated into multiorgan systems. Future studies should provide further liver input data on other PGEs for the TK model, which will help fill the gap of knowledge on metabolism and systemic toxicity of PGEs.
Author Contributions
Sophie Werner, Lucie Hegg, Nancy B. Hopf, Laura Suter‐Dick participated in research design. Sophie Werner conducted experiments. Sophie Werner contributed new reagents or analytic tools. Sophie Werner, Lucie Hegg performed data analysis. Sophie Werner, Lucie Hegg, Nancy B. Hopf, Myriam Borgatta, Laura Suter‐Dick wrote or contributed to the writing of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Data S1.
Acknowledgments
We would like to thank Timm Hettich and André Büttler at the FHNW for their valuable assistance in the development of the analytical methods. We thank the Swiss Centre for Applied Human Toxicity (SCAHT), as well as our home institutions (FHNW, Unisanté) for funding the research.
Funding: This work was supported by the Swiss Centre for Applied Human Toxicology (SCAHT‐GL 21‐10).
Data Availability Statement
The authors declare that all the data supporting the findings of this study are available within the paper and its Supporting Information.
References
- 1. Borgatta M., Hechon J., Wild P., and Hopf N. B., “Influence of Collection and Storage Materials on Glycol Ether Concentrations in Urine and Blood,” Science of the Total Environment 792 (2021): 148196, 10.1016/j.scitotenv.2021.148196. [DOI] [PubMed] [Google Scholar]
- 2. Brown R. C., Lockwood A. H., and Sonawane B. R., “Neurodegenerative Diseases: An Overview of Environmental Risk Factors,” Environmental Health Perspectives 113, no. 9 (2005): 1250–1256, 10.1289/ehp.7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cicolella A., “Glycol Ethers: A Ubiquitous Family of Toxic Chemicals: A Plea for REACH Regulation,” Annals of the New York Academy of Sciences 1076 (2006): 784–789, 10.1196/annals.1371.049. [DOI] [PubMed] [Google Scholar]
- 4. ECETOC , The Toxicology of Glycol Ethers and Its Relevance to Man, vol. I, 4th ed. (Brussels, Belgium: Technical Report No. 95, Technical Report , 2005), 159–162. I(95), http://www.ecetoc.org/wp‐content/uploads/2014/08/ECETOC‐TR‐095‐Vol‐I.pdf%0Ahttp://www.ecetoc.org/wp‐content/uploads/2014/08/ECETOC‐TR‐095‐Vol‐II.pdf. [Google Scholar]
- 5. ECETOC , The Toxicology of Glycol Ethers and Its Relevance to Man, vol. II, 4th ed. (Brussels, Belgium: Substance Profiles. Technical Report , 2005), 159–162. II(95), http://www.ecetoc.org/wpcontent/uploads/2014/08/ECETOC‐TR‐095‐Vol‐I.pdf%0Ahttp://www.ecetoc.org/wpcontent/uploads/2014/08/ECETOC‐TR‐095‐Vol‐II.pdf. [Google Scholar]
- 6. Bagchi G. and Waxman D. J., “Toxicity of Ethylene Glycol Monomethyl Ether: Impact on Testicular Gene Expression,” International Journal of Andrology 31, no. 2 (2008): 269–274, 10.1111/j.1365-2605.2007.00846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Multigner L., Catala M., Cordier S., et al., “The INSERM Expert Review on Glycol Ethers: Findings and Recommendations,” Toxicology Letters 156, no. 1 SPEC. ISS (2005): 29–37, 10.1016/j.toxlet.2003.12.077. [DOI] [PubMed] [Google Scholar]
- 8. Hopf N. B., Suter‐Dick L., Huwyler J., et al., “Novel Strategy to Assess the Neurotoxicity of Organic Solvents Such as Glycol Ethers: Protocol for Combining in Vitro and in Silico Methods With Human‐Controlled Exposure Experiments,” JMIR Research Protocols 13 (2024): e50300, 10.2196/50300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carney E. W., Pottenger L. H., Johnson K. A., et al., “Significance of 2‐Methoxypropionic Acid Formed From β‐Propylene Glycol Monomethyl Ether: Integration of Pharmacokinetic and Developmental Toxicity Assessments in Rabbits,” Toxicological Sciences 71, no. 2 (2003): 217–228, 10.1093/toxsci/71.2.217. [DOI] [PubMed] [Google Scholar]
- 10. Aasmoe L., Winberg J. O., and Aarbakke J., “The Role of Liver Alcohol Dehydrogenase Isoenzymes in the Oxidation of Glycolethers in Male and Female Rats,” Toxicology and Applied Pharmacology 150, no. 1 (1998): 86–90, 10.1006/taap.1998.8410. [DOI] [PubMed] [Google Scholar]
- 11. Aasmoe L. and Aarbakke J., “Sex‐Dependent Induction of Alcohol Dehydrogenase Activity in Rats,” Biochemical Pharmacology 57, no. 9 (1999): 1067–1072, 10.1016/S0006-2952(99)00003-9. [DOI] [PubMed] [Google Scholar]
- 12. Moslen M. T., Kaphalia L., Balasubramanian H., Yina Y. M., and Au W. W., “Species Differences in Testicular and Hepatic Biotransformation of 2‐Methoxyethanol,” Toxicology 96, no. 3 (1995): 217–224, 10.1016/0300-483X(94)02921-G. [DOI] [PubMed] [Google Scholar]
- 13. Miller R. R., Hermann E. A., Langvardt P. W., McKenna M. J., and Schwetz B. A., “Comparative Metabolism and Disposition of Ethylene Glycol Monomethyl Ether and Propylene Glycol Monomethyl Ether in Male Rats,” Toxicology and Applied Pharmacology 67, no. 2 (1983): 229–237, 10.1016/0041-008X(83)90229-6. [DOI] [PubMed] [Google Scholar]
- 14. Crabb D. W., Matsumoto M., Chang D., and You M., “Overview of the Role of Alcohol Dehydrogenase and Aldehyde Dehydrogenase and Their Variants in the Genesis of Alcohol‐Related Pathology,” Proceedings of the Nutrition Society 63, no. 1 (2004): 49–63, 10.1079/pns2003327. [DOI] [PubMed] [Google Scholar]
- 15. Wang Y. and He W., Endogenous Mitochondrial Aldehyde Dehydrogenase‐2 as an Antioxidant in Liver (Wuhan, China: Elsevier Inc., 2018), 247–259, 10.1016/B978-0-12-803951-9.00021-5. [DOI] [Google Scholar]
- 16. Wang W., Wang C., Xu H., and Gao Y., “Aldehyde Dehydrogenase, Liver Disease and Cancer,” International Journal of Biological Sciences 16, no. 6 (2020): 921–934, 10.7150/ijbs.42300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Corley R. A., Gies R. A., Wu H., and Weitz K. K., “Development of a Physiologically Based Pharmacokinetic Model for Propylene Glycol Monomethyl Ether and Its Acetate in Rats and Humans,” Toxicology Letters 156, no. 1 SPEC. ISS (2005): 193–213, 10.1016/j.toxlet.2003.12.078. [DOI] [PubMed] [Google Scholar]
- 18. Miller R. R., Hermann E. A., and Young J. T., “Ethylene Glycol Monomethyl Ether and Propylene Glycol Monomethyl Ether: Metabolism, Disposition, and Subchronic Inhalation Toxicity Studies,” Environmental Health Perspectives 57 (1984): 233–239, 10.1289/ehp.8457233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scandinavian S., Sato A., and Nakajima T., “Pharmacokinetics of Organic Solvent Vapors in Relation to Their Toxicity Author(s): Akio Sato and Tamie Nakajima Published by : The Scandinavian Journal of Work,” Environment & Health, the Finnish Institute of Occupational Health, the Danish National 13, no. 2 (1987): 81–93. [DOI] [PubMed] [Google Scholar]
- 20. Devanthéry A., Berode M., Droz P. O., and Pulkkinen J., “Propylene Glycol Monomethyl Ether Occupational Exposure (PGME). 4. Analysis of 2‐Methoxypropionic Acid in Urine,” International Archives of Occupational and Environmental Health 76, no. 2 (2003): 151–155, 10.1007/s00420-002-0401-x. [DOI] [PubMed] [Google Scholar]
- 21. Benet L. Z. and Zia‐Amirhosseini P., “Basic Principles of Pharmacokinetics,” Toxicologic Pathology 23, no. 2 (1995): 115–123, 10.1177/019262339502300203. [DOI] [PubMed] [Google Scholar]
- 22. Ingelman‐Sundberg M. and Lauschke V. M., “3D Human Liver Spheroids for Translational Pharmacology and Toxicology,” Basic & Clinical Pharmacology & Toxicology 130, no. 1 (2021): 5–15, 10.1111/bcpt.13587. [DOI] [PubMed] [Google Scholar]
- 23. Bonn B., Svanberg P., Janefeldt A., Hultman I., and Grime K., “Determination of Human Hepatocyte Intrinsic Clearance for Slowly Metabolized Compounds: Comparison of a Primary Hepatocyte/Stromal Cell Co‐Culture With Plated Primary Hepatocytes and hepaRG,” Drug Metabolism and Disposition 44, no. 4 (2016): 527–533, 10.1124/dmd.115.067769. [DOI] [PubMed] [Google Scholar]
- 24. Smith C. M., Nolan C. K., Edwards M. A., et al., “A Comprehensive Evaluation of Metabolic Activity and Intrinsic Clearance in Suspensions and Monolayer Cultures of Cryopreserved Primary Human Hepatocytes,” Journal of Pharmaceutical Sciences 101, no. 10 (2012): 3989–4002, 10.1002/jps.23262. [DOI] [PubMed] [Google Scholar]
- 25. Di L. and Obach R. S., “Addressing the Challenges of Low Clearance in Drug Research,” AAPS Journal 17, no. 2 (2015): 352–357, 10.1208/s12248-014-9691-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guillouzo A., Corlu A., Aninat C., Glaise D., Morel F., and Guguen‐Guillouzo C., “The Human Hepatoma HepaRG Cells: A Highly Differentiated Model for Studies of Liver Metabolism and Toxicity of Xenobiotics,” Chemico‐Biological Interactions 168, no. 1 (2007): 66–73, 10.1016/j.cbi.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 27. Zanelli U., Caradonna N. P., Hallifax D., Turlizzi E., and Houston J. B., “Comparison of Cryopreserved HepaRG Cells With Cryopreserved Human Hepatocytes for Prediction of Clearance for 26 Drugs,” Drug Metabolism and Disposition 40, no. 1 (2012): 104–110, 10.1124/dmd.111.042309. [DOI] [PubMed] [Google Scholar]
- 28. Aninat C., Piton A., Glaise D., et al., “Expression of Cytochromes P450, Conjugating Enzymes and Nuclear Receptors in Human Hepatoma HepaRG Cells,” Drug Metabolism and Disposition 34, no. 1 (2006): 75–83, 10.1124/dmd.105.006759. [DOI] [PubMed] [Google Scholar]
- 29. Hart S. N., Li Y., Nakamoto K., Subileau E. A., Steen D., and Zhong X. B., “A Comparison of Whole Genome Gene Expression Profiles of HepaRG Cells and HepG2 Cells to Primary Human Hepatocytes and Human Liver Tissues,” Drug Metabolism and Disposition 38, no. 6 (2010): 988–994, 10.1124/dmd.109.031831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Attignon E. A., Distel E., Le‐Grand B., et al., “Down‐Regulation of the Expression of Alcohol Dehydrogenase 4 and CYP2E1 by the Combination of α‐Endosulfan and Dioxin in HepaRG Human Cells,” Toxicology in Vitro: An International Journal Published in Association With BIBRA 45, no. June (2017): 309–317, 10.1016/j.tiv.2017.06.029. [DOI] [PubMed] [Google Scholar]
- 31. Ishida K., Kaji K., Sato S., et al., “Sulforaphane Ameliorates Ethanol Plus Carbon Tetrachlorideinduced Liver Fibrosis in Mice Through the Nrf2‐Mediated Antioxidant Response and Acetaldehyde Metabolization With Inhibition of the LPS/TLR4 Signaling Pathway,” Journal of Nutritional Biochemistry 89 (2021): 108573, 10.1016/j.jnutbio.2020.108573. [DOI] [PubMed] [Google Scholar]
- 32. Reale E., Sandstrom J., Culot M., et al., “Predicting Human Neurotoxicity of Propylene Glycol Methyl Ether (PGME) by Implementing In Vitro Neurotoxicity Results Into Toxicokinetic Modelling,” Science of the Total Environment 886, no. February (2023): 163767, 10.1016/j.scitotenv.2023.163767. [DOI] [PubMed] [Google Scholar]
- 33. Messner C. J., Premand C., Gaiser C., Kluser T., Kubler E., and Suter‐Dick L., “Exosomal microRNAs Release as a Sensitive Marker for Drug‐Induced Liver Injury In Vitro,” Applied In Vitro Toxicology 6, no. 3 (2020): 77–89, 10.1089/aivt.2020.0008. [DOI] [Google Scholar]
- 34. Berger B., Donzelli M., Maseneni S., et al., “Comparison of Liver Cell Models Using the Basel Phenotyping Cocktail,” Frontiers in Pharmacology 7, no. NOV (2016): 1–12, 10.3389/fphar.2016.00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Messner C. J., Schmidt S., Özkul D., et al., “Identification of miR‐199a‐5p, miR‐214‐3p and miR‐99b‐5p as Fibrosis‐Specific Extracellular Biomarkers and Promoters of HSC Activation,” International Journal of Molecular Sciences 22 (2021): 9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mégarbane B., “Treatment of Patients With Ethylene Glycol or Methanol Poisoning: Focus on Fomepizole,” Open Access Emergency Medicine 2 (2010): 67–75, 10.2147/OAEM.S5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quattrini L., Gelardi E. L. M., Coviello V., et al., “Imidazo[1,2‐ a]Pyridine Derivatives as Aldehyde Dehydrogenase Inhibitors: Novel Chemotypes to Target Glioblastoma Stem Cells,” Journal of Medicinal Chemistry 63, no. 9 (2020): 4603–4616, 10.1021/acs.jmedchem.9b01910. [DOI] [PubMed] [Google Scholar]
- 38. Zakhari S., “Overview: How Is Alcohol Metabolized by the Body?,” Alcohol Research & Health 29, no. 4 (2006): 245–254. [PMC free article] [PubMed] [Google Scholar]
- 39. Obach R. S., Baxter J. G., Liston T. E., et al., “The Prediction of Human Pharmacokinetic Parameters From Preclinical and In Vitro Metabolism Data,” Journal of Pharmacology and Experimental Therapeutics 283, no. 1 (1997): 46–58. [PubMed] [Google Scholar]
- 40. Barter Z., Bayliss M., Beaune P., et al., “Scaling Factors for the Extrapolation of in Vivo Metabolic Drug Clearance From in Vitro Data: Reaching a Consensus on Values of Human Micro‐Somal Protein and Hepatocellularity per Gram of Liver,” Current Drug Metabolism 8, no. 1 (2006): 33–45, 10.2174/138920007779315053. [DOI] [PubMed] [Google Scholar]
- 41. Musther H., Harwood M. D., Yang J., Turner D. B., Rostami‐Hodjegan A., and Jamei M., “The Constraints, Construction, and Verification of a Strain‐Specific Physiologically Based Pharmacokinetic Rat Model,” Journal of Pharmaceutical Sciences 106, no. 9 (2017): 2826–2838, 10.1016/j.xphs.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 42. Houston J. and Galetin A., “Methods for Predicting in Vivo Pharmacokinetics Using Data From in Vitro Assays,” Current Drug Metabolism 9, no. 9 (2008): 940–951, 10.2174/138920008786485164. [DOI] [PubMed] [Google Scholar]
- 43. Davies B. and Morris T., “Physiological Parameters in Laboratory Animals and Humans,” Pharmaceutical Research 10, no. 7 (1993): 1093–1095, 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 44. Pang K. S. and Rowland M., “Hepatic Clearance of Drugs. I. Theoretical Considerations of a “Wellstirred” Model and a “Parallel Tube” Model. Influence of Hepatic Blood Flow, Plasma and Blood Cell Binding, and the Hepatocellular Enzymatic Activity on Hepatic Drug Clearance,” Journal of Pharmacokinetics and Biopharmaceutics 5, no. 6 (1977): 625–653, 10.1007/BF01059688. [DOI] [PubMed] [Google Scholar]
- 45. Riley R. J., McGinnity D. F., and Austin R. P., “A Unified Model for Predicting Human Hepatic, Metabolic Clearance From In Vitro Intrinsic Clearance Data in Hepatocytes and Microsomes,” Drug Metabolism and Disposition 33, no. 9 (2005): 1304–1311, 10.1124/dmd.105.004259. [DOI] [PubMed] [Google Scholar]
- 46. Austin R. P., Barton P., and Riley R. J., “Response to “Binding of Drugs to Hepatic Microsomes: Comment and Assessment of Current Prediction Methodology With Recommendation for Improvement”,” Drug Metabolism and Disposition 34, no. 4 (2006): 727, 10.1124/dmd.105.009142. [DOI] [PubMed] [Google Scholar]
- 47. Kilford P. J., Gertz M., Houston J. B., and Galetin A., “Hepatocellular Binding of Drugs: Correction for Unbound Fraction in Hepatocyte Incubations Using Microsomal Binding or Drug Lipophilicity Data,” Drug Metabolism and Disposition 36, no. 7 (2008): 1194–1197, 10.1124/dmd.108.020834. [DOI] [PubMed] [Google Scholar]
- 48. Miller R. R., Langvardt P. W., Calhoun L. L., and Yahrmarkt M. A., “Metabolism and Disposition of Propylene Glycol Monomethyl Ether (PGME) Beta Isomer in Male Rats,” Toxicology and Applied Pharmacology 83, no. 1 (1986): 170–177, 10.1016/0041-008X(86)90334-0. [DOI] [PubMed] [Google Scholar]
- 49. Kramer C., Mochalski P., Unterkofler K., Agapiou A., Ruzsanyi V., and Liedl K. R., “Prediction of Blood:Air and Fat:Air Partition Coefficients of Volatile Organic Compounds for the Interpretation of Data in Breath Gas Analysis,” Journal of Breath Research 10, no. 1 (2016): 1–10, 10.1088/1752-7155/10/1/017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tibaldi R., Ten Berge W., and Drolet D., “Dermal Absorption of Chemicals: Estimation by IH SkinPerm,” Journal of Occupational and Environmental Hygiene 11, no. 1 (2014): 19–31, 10.1080/15459624.2013.831983. [DOI] [PubMed] [Google Scholar]
- 51. Rodgers T. and Rowland M., “Physiologically Based Pharmacokinetic Modelling 2: Predicting the Tissue Distribution of Acids, Very Weak Bases, Neutrals and Zwitterions,” Journal of Pharmaceutical Sciences 95, no. 6 (2006): 1238–1257, 10.1002/jps.20502. [DOI] [PubMed] [Google Scholar]
- 52. Sipes N. S., Wambaugh J. F., Pearce R., et al., “An Intuitive Approach for Predicting Potential Human Health Risk With the Tox21 10k Library,” Environmental Science & Technology 51, no. 18 (2017): 1078610796, 10.1021/acs.est.7b00650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Watanabe R., Esaki T., Ohashi R., et al., “Development of an In Silico Prediction Model for Pglycoprotein Efflux Potential in Brain Capillary Endothelial Cells Toward the Prediction of Brain Penetration,” Journal of Medicinal Chemistry 64, no. 5 (2021): 2725–2738, 10.1021/acs.jmedchem.0c02011. [DOI] [PubMed] [Google Scholar]
- 54. Kerns E. H., Blood‐Brain Barrier in Drug Discovery: Optimizing Brain Exposure of CNS Drugs and Minimizing Brain Side Effects for Peripheral Drugs (Maryland, USA: Wiley, 2015). [Google Scholar]
- 55. Gargas M. L., Tyler T. R., Sweeney L. M., et al., “A Toxicokinetic Study of Inhaled Ethylene Glycol Monomethyl Ether (2‐ME) and Validation of a Physiologically Based Pharmacokinetic Model for the Pregnant Rat and Human,” Toxicology and Applied Pharmacology 165, no. 1 (2000): 53–62, 10.1006/taap.2000.8928856. [DOI] [PubMed] [Google Scholar]
- 56. WHO, IPCS , Characterization and Application of Physiologically Based Pharmacokinetic Models in Risk Assessment, Harmonization Project Document (Geneva: World Health Organization, 2010). [Google Scholar]
- 57. Yuan L., Chou W. C., Richards E. D., et al., “A Web‐Based Interactive Physiologically Based Pharmacokinetic (iPBPK) Model for Meloxicam in Broiler Chickens and Laying Hens,” Food and Chemical Toxicology 168, no. March (2022): 113332, 10.1016/j.fct.2022.113332. [DOI] [PubMed] [Google Scholar]
- 58. Lin Z., Jaberi‐Douraki M., He C., et al., “Performance Assessment and Translation of Physiologically Based Pharmacokinetic Models From Acslx to Berkeley Madonna, Matlab, and r Language: Oxytetracycline and Gold Nanoparticles as Case Examples,” Toxicological Sciences 158, no. 1 (2017): 23–35, 10.1093/toxsci/kfx070. [DOI] [PubMed] [Google Scholar]
- 59. Teeguarden J. G., Deisinger P. J., Poet T. S., et al., “Derivation of a Human Equivalent Concentration for n‐Butanol Using a Physiologically Based Pharmacokinetic Model for Nbutyl Acetate and Metabolites n‐Butanol and n‐Butyric Acid,” Toxicological Sciences 85, no. 1 (2005): 429–446, 10.1093/toxsci/kfi103. [DOI] [PubMed] [Google Scholar]
- 60. Harding S. D., Sharman J. L., Faccenda E., et al., “The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and Expansion to Encompass the New Guide to IMMUNOPHARMACOLOGY,” Nucleic Acids Research 46, no. D1 (2018): D1091–D1106, 10.1093/nar/gkx1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alexander S. P. H., Fabbro D., Kelly E., et al., “The Concise Guide to PHARMACOLOGY 2023/24: Enzymes,” British Journal of Pharmacology 180, no. S2 (2023): S289–S373, 10.1111/bph.16181. [DOI] [PubMed] [Google Scholar]
- 62. Alexander S. P. H., Kelly E., Mathie A. A., et al., “The Concise Guide to PHARMACOLOGY 2023/24: Introduction and Other Protein Targets,” British Journal of Pharmacology 180, no. S2 (2023): S1–S22, 10.1111/bph.16176. [DOI] [PubMed] [Google Scholar]
- 63. Alexander S. P. H., Mathie A. A., Peters J. A., et al., “The Concise Guide to PHARMACOLOGY 2023/24: Ion Channels,” British Journal of Pharmacology 180, no. Suppl 2 (2023): S145–S222, 10.1111/bph.16178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hutzler J. M., Ring B. J., and Anderson S. R., “Low‐Turnover Drug Molecules: A Current Challenge for Drug Metabolism Scientists,” Drug Metabolism and Disposition 43, no. 12 (2015): 1917–1928, 10.1124/dmd.115.066431. [DOI] [PubMed] [Google Scholar]
- 65. Scott D. M. and Taylor R. E., “Health‐Related Effects of Genetic,” Alcohol Research & Health 30 (2007): 18–22. [PMC free article] [PubMed] [Google Scholar]
- 66. Venn R. M., “Fomepizole for the Treatment of Ethylene Glycol Poisoning,” Critical Care 1, no. 1 (1999): 1–4, 10.1186/ccf-1999-42. [DOI] [Google Scholar]
- 67. Chiba M., Ishii Y., and Sugiyama Y., “Prediction of Hepatic Clearance in Human From In Vitro Data for Successful Drug Development,” AAPS Journal 11, no. 2 (2009): 262–276, 10.1208/s12248-009-9103-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kratochwil N. A., Meille C., Fowler S., et al., “Metabolic Profiling of Human Long‐Term Liver Models and Hepatic Clearance Predictions From in Vitro Data Using Nonlinear Mixed‐Effects Modeling,” AAPS Journal 19, no. 2 (2017): 534–550, 10.1208/s12248-016-0019-7. [DOI] [PubMed] [Google Scholar]
- 69. Hultman I., Vedin C., Abrahamsson A., Winiwarter S., and Darnell M., “Use of HμREL Human Coculture System for Prediction of Intrinsic Clearance and Metabolite Formation for Slowly Metabolized Compounds,” Molecular Pharmaceutics 13, no. 8 (2016): 2796–2807, 10.1021/acs.molpharmaceut.6b00396. [DOI] [PubMed] [Google Scholar]
- 70. Słoczyńska K., Gunia‐Krzyzak A., Koczurkiewicz P., et al., “Metabolic Stability and Its Role in the Discovery of New Chemical Entities,” Acta Pharmaceutica 69, no. 3 (2019): 345–361, 10.2478/acph-2019-0024. [DOI] [PubMed] [Google Scholar]
- 71. Donato M. T., Hallifax D., Picazo L., et al., “Metabolite Formation Kinetics and Intrinsic Clearance of Phenacetin, Tolbutamide, Alprazolam, and Midazolam in Adenoviral Cytochrome P450transfected HepG2 Cells and Comparison With Hepatocytes and In Vivo,” Drug Metabolism and Disposition 38, no. 9 (2010): 1449–1455, 10.1124/dmd.110.033605. [DOI] [PubMed] [Google Scholar]
- 72. Preiss L. C., Lauschke V. M., Georgi K., and Petersson C., “Multi‐Well Array Culture of Primary Human Hepatocyte Spheroids for Clearance Extrapolation of Slowly Metabolized Compounds,” AAPS Journal 24, no. 2 (2022): 1–12, 10.1208/s12248-022-00689-y. [DOI] [PubMed] [Google Scholar]
- 73. Chan T. S., Yu H., Moore A., Khetani S. R., and Tweedie D., “Meeting the Challenge of Predicting Hepatic Clearance of Compounds Slowly Metabolized by Cytochrome P450 Using a Novel Hepatocyte Model, HepatoPac,” Drug Metabolism and Disposition 47, no. 1 (2019): 58–66, 10.1124/DMD.113.053397FULLARTICLECORRECTION. [DOI] [PubMed] [Google Scholar]
- 74. Mandon M., Huet S., Dubreil E., Fessard V., and Le Hégarat L., “Three‐Dimensional HepaRG Spheroids as a Liver Model to Study Human Genotoxicity In Vitro With the Single Cell Gel Electrophoresis Assay,” Scientific Reports 9, no. 1 (2019): 1–9, 10.1038/s41598-019-47114-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lübberstedt M., Müller‐Vieira U., Mayer M., et al., “HepaRG Human Hepatic Cell Line Utility as a Surrogate for Primary Human Hepatocytes in Drug Metabolism Assessment In Vitro,” Journal of Pharmacological and Toxicological Methods 63, no. 1 (2011): 59–68, 10.1016/j.vascn.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 76. Beckwitt C. H., Clark A. M., Wheeler S., et al., “Liver ‘Organ on a Chip’,” Experimental Cell Research 363, no. 1 (2018): 15–25, 10.1016/j.yexcr.2017.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cox B., Barton P., Class R., et al., “Setup of Human Liver‐Chips Integrating 3D Models, Microwells and a Standardized Microfluidic Platform as Proof‐Of‐Concept Study to Support Drug Evaluation,” Biomaterials and Biosystems 7, no. February (2022): 100054, 10.1016/j.bbiosy.2022.100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gunness P., Mueller D., Shevchenko V., Heinzle E., Ingelman‐Sundberg M., and Noor F., “3D Organotypic Cultures of Human Heparg Cells: A Tool for In Vitro Toxicity Studies,” Toxicological Sciences 133, no. 1 (2013): 67–78, 10.1093/toxsci/kft021. [DOI] [PubMed] [Google Scholar]
- 79. Flores M. V., Voget C. E., and Ertola R. J. J., “Permeabilization of Yeast Cells (Kluyveromyces Lactis) With Organic Solvents,” Enzyme and Microbial Technology 16, no. 4 (1994): 340–346, 10.1016/0141-0229(94)90177-5. [DOI] [Google Scholar]
- 80. Spencer P. J., Crissman J. W., Stott W. T., et al., “Propylene Glycol Monomethyl Ether (PGME): Inhalation Toxicity and Carcinogenicity in Fischer 344 Rats and B6C3F1 Mice,” Toxicologic Pathology 30, no. 5 (2002): 570–579, 10.1080/01926230290105848. [DOI] [PubMed] [Google Scholar]
- 81. Spencer P. J., “New Toxicity Data for the Propylene Glycol Ethers ‐ A Commitment to Public Health and Safety,” Toxicology Letters 156, no. 1 (2005): 181–188, 10.1016/j.toxlet.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 82. Zanger U. M. and Schwab M., “Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation,” Pharmacology & Therapeutics 138, no. 1 (2013): 103–141, 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
The authors declare that all the data supporting the findings of this study are available within the paper and its Supporting Information.