

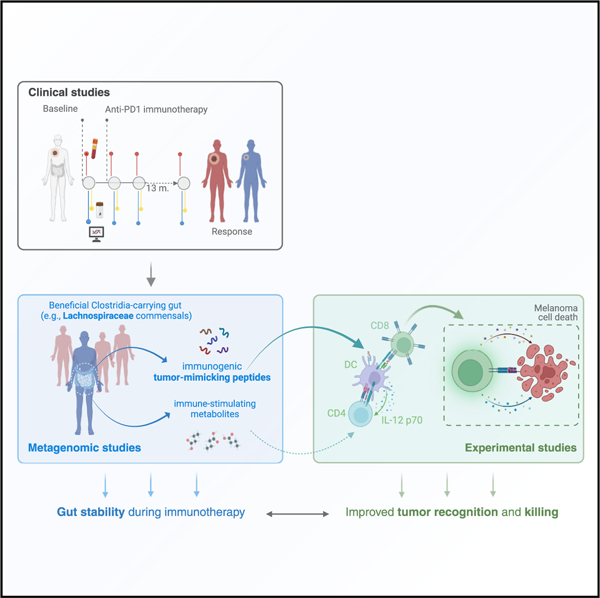
SUMMARY
Immune checkpoint inhibitors (ICIs) improve outcomes in advanced melanoma, but many patients are refractory or experience relapse. The gut microbiota modulates antitumor responses. However, inconsistent baseline predictors point to heterogeneity in responses and inadequacy of cross-sectional data. We followed patients with unresectable melanoma from baseline and during anti-PD-1 therapy, collecting fecal and blood samples that were surveyed for changes in the gut microbiota and immune markers. Varying patient responses were linked to different gut microbiota dynamics during ICI treatment. We select complete responders by their stable microbiota functions and validate them using multiple external cohorts and experimentally. We identify major histocompatibility complex class I (MHC class I)-restricted peptides derived from flagellin-related genes of Lachnospiraceae (FLach) as structural homologs of tumor-associated antigens, detect FLach-reactive CD8+ T cells in complete responders before ICI therapy, and demonstrate that FLach peptides improve antitumor immunity. These findings highlight the prognostic value of microbial functions and therapeutic potential of tumor-mimicking microbial peptides.
Graphical abstract
In brief
Macandog et al. utilized longitudinal data to identify key gut microbial features in immunotherapy-responsive patients with melanoma. Functions stably carried by responders are specific to gut commensals that reportedly evolved mechanisms for human tolerance. These functions could provide advantage during immunotherapy by mimicking tumor antigens that stimulate effective tumor-clearing immunity.
INTRODUCTION
The introduction of immune checkpoint inhibitor (ICI) therapy in the last decade set a precedent for melanoma treatment, significantly improving patient outcome1,2 in an otherwise therapy-resistant cancer type.2 However, a large proportion of advanced melanoma remains non-responsive to ICI (nR). In this regard, the influence of the gut microbiome has been widely reported, with independent studies demonstrating that the gut microbiome of patients with melanoma responding to ICI (R) is compositionally and functionally different from those with nR.3–8 More importantly, recent clinical studies showed that fecal microbiota transplantation (FMT) from either R9,10 or healthy donors11 can broaden the benefit of ICI to more patients, including some with ICI-refractory melanoma, suggesting that targeted modulation of the gut microbiota has still untapped therapeutic potential. However, results of large-scale meta-analyses point to high variability across cohorts as a major limitation,7,8 and considering that microbiota is only one of many factors that impact response, aggregating patients based on response outcomes alone and assuming they will have similar microbial features could be an incorrect strategy that overlooks biologically relevant gut differences. Thus, successfully consolidating these studies could elucidate what constitutes a therapeutic, immune-modulating gut and guide the development of rationally designed microbiome-based therapeutics.
On the other hand, the detection of immunogenic bacterial peptides in melanoma as well as glioblastoma tumors and the evidence that ICI therapy can induce the translocation of gut bacteria12,13 indicate that a deeper mechanistic understanding of the interplay between the gut microbiome and host immunity in the context of immunotherapy is an essential step toward precise clinical interventions. However, current gut microbiome-response associations are mostly founded on pre-ICI baseline data,3,4,6,8 and longitudinal studies that follow gut-host dynamics from pre-ICI through therapy are scant.11,14
Here, we generate fecal 16S and metagenomic data and analyze them with systemic immune features to provide a longitudinal profile of patients with melanoma for up to 13 months of anti-PD-1 therapy. First, we study gut microbiota profiles at baseline and on therapy, where we use longitudinal diversity measures to explore associations between gut stability, response, and the systemic immune state in the ICI setting. More importantly, we demonstrate that longitudinally stable microbial functions have generalizable prognostic value, and we support experimentally the potential of tumor-mimicking peptides in therapy.
RESULTS
Longitudinal profiling of microbiome in melanoma patients undergoing anti-PD-1 therapy
To delineate gut microbiota changes related to host ICI response and identify gut and host factors involved, patients with advanced melanoma (n = 23) were enrolled in two Italian hospitals between January 2018 and October 2022, and, along with clinical information, fecal and blood samples were collected at baseline and before every following injection of single-agent anti-PD-1 treatment (Figure 1A). The objective response rate (ORR) was 56% and the disease control rate (DCR) was 74%. Cohort characteristics are summarized in Table 1 and Figure S1A and detailed in the STAR Methods. Fecal samples were collected from each patient within the window of 0 up to 13 months of therapy, employing the same standardized procedures for harvesting and storage. Processing and sequencing of all samples were centralized to reduce technical bias. In parallel, blood samples were obtained at every time point for whole blood cell (WBC) count and serum from a subgroup of patients (n = 8) prepared for inflammatory soluble factor quantification. The complete list of longitudinal samples included in the study is reported in Table S1.
Figure 1. Longitudinally stable functions are enriched in the gut microbiome of patients with melanoma responsive to anti-PD-1 immunotherapy.
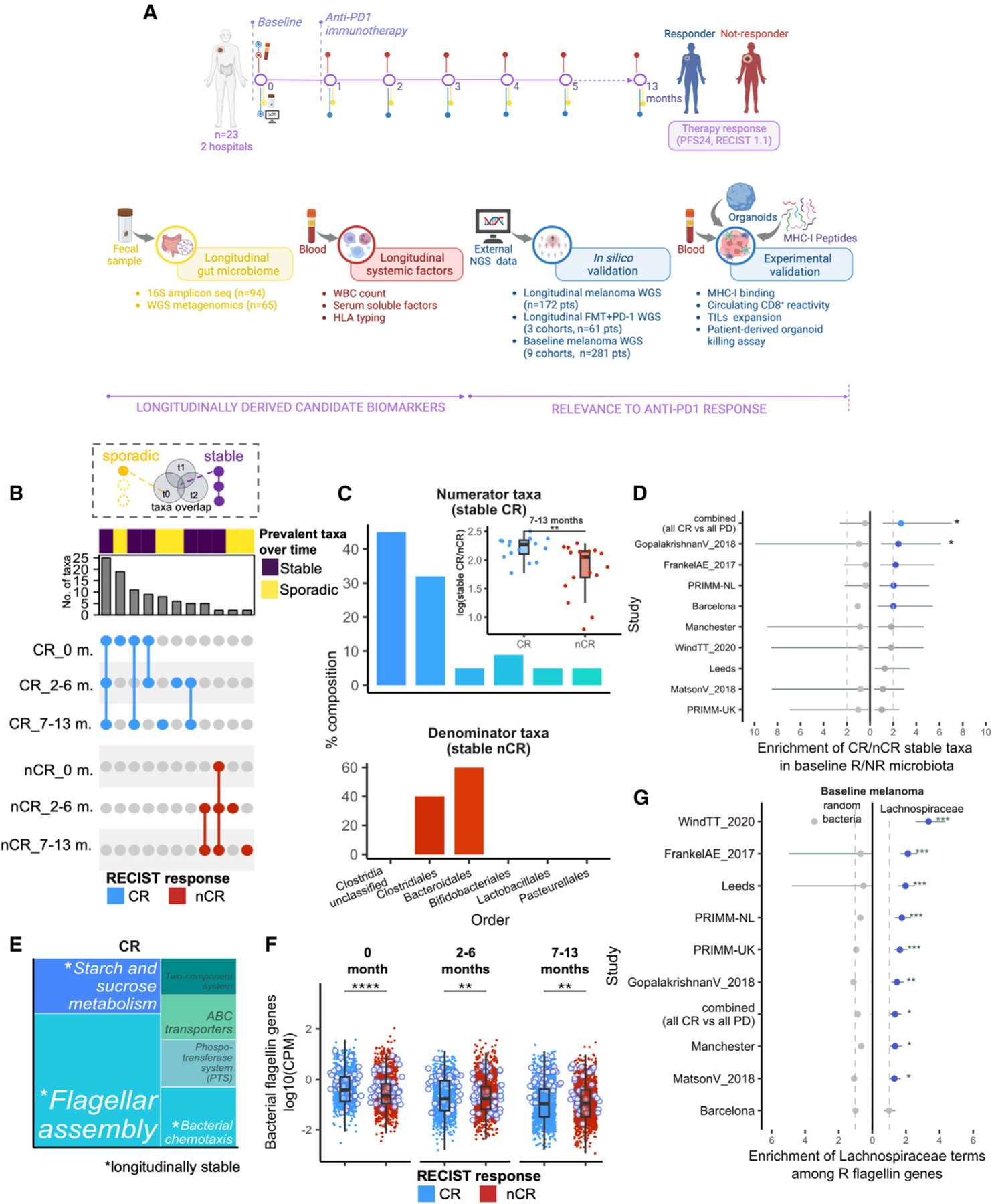
(A) Overview of study design. See also Figure S1A and Table S1.
(B) UpSet plot showing overlaps of prevalent taxa (present in >80% of samples) found in complete responder (CR) and non-CR (nCR) groups at 0, 2–6, and 7–13 months of therapy. Connected dots indicate prevalent taxa shared between time point groups (“stable”), whereas non-connected dots indicate prevalent taxa detected only in one time point group (“sporadic”). Plot is filtered to exclude taxa that have overlaps across CR and nCR (not group-specific). Stable CR is defined as prevalent taxa that are present across 0, 2–6, and 7–13 months, whereas stable nCR is defined as prevalent taxa that are present in at least two time point groups. See also Figures S1B–S1F.
(C) Order-level composition of stable CR (top) and nCR taxa (bottom) and the corresponding stable taxa measure in CR and nCR (i.e., log-ratio of their sum-aggregated relative abundances (inset). Asterisks indicate significance by Wilcoxon rank sum test. See also Figures S2A–S2D. For further biological correlations of stable taxa, see Figures S3 and S4.
(D) Baseline enrichment of stable CR and stable nCR taxa among R- and nR-prevalent taxa, respectively, across the indicated nine external melanoma cohorts. Dashed line indicates arbitrary cutoff for enrichment (Fisher’s exact test |OR|=2), line plots in blue indicate enrichment, line plots in gray indicate no enrichment, asterisks indicate significant enrichment at unadjusted p < 0.05 (Fisher’s exact test), and whiskers depict the interval for 95% confidence.
(E) Treemap of over-represented pathways in CR at p-adj < 0.05, based on KOs associated with CR across all samples (|LM coefficient| > 1.5). Terms in white indicate stable pathways, defined as over-represented pathways appearing at 0, 2–6, and 7–13 months (p-adj < 0.05) within CR, whereas terms in gray indicate non-stable pathways. See also Figures S5A–S5C and Table S3.
(F) Average gene family abundances (log10(CPM)) per patient of bacterial flagellin-related terms in CR and nCR, compared at 0, 2–6, and 7–13 months. Points outlined in blue indicate the top CR-associated flagellin genes by log2(fold-change). See also Figures S5D and S5E and Table S5.
(G) Baseline enrichment of Lachnospiraceae-associated terms among R-associated flagellin gene families across nine melanoma cohorts, dashed lines indicate arbitrary cutoff for enrichment in R (Fisher’s exact test |OR|>1), line plots to the left indicate enrichment of non-specific bacterial flagellin terms subsampled to the same size, line plots in gray indicate no enrichment, whiskers depict the interval for 95% confidence, and asterisks indicate significant enrichment at p-adj<0.05 (Fisher’s exact test). See also Tables S4 and S5.
Legend: with overlap (violet), spurious (yellow). CR (light blue), nCR (red). p-adj<0.001 (***), p-adj<0.01 (**), p-adj<0.05 (*), ns (unannotated).
Table 1.
Breakdown of patient characteristics
| Demographics | n (tot = 23) | % |
|---|---|---|
| Age, years (median) | 54 (33–78) | – |
|
Sex | ||
| Male | 17 | 74% |
| Female | 6 | 26% |
| BMI, kg/m2 (range) | 26.6 (23.2–33.6) | – |
|
RECIST 1.1 (median OS, PFS, months) | ||
| CR | 7 (108, 50) | 30% |
| PR | 6 (104, 36) | 26% |
| SD | 4 (53, 21) | 18% |
| PD | 6 (25, 2) | 26% |
|
ECOG performance score | ||
| 0–1 | 18 | 79% |
| 2 | 3 | 13% |
| NA | 2 | 8% |
|
Tumor stage at study entry (AJCC 8th Edition) | ||
| Unresectable stage III (M0) | 3 | 13% |
| Skin, soft tissue ± nonregional nodes (M1a) | 1 | 4% |
| Distant metastasis to lung (M1b) | 2 | 8% |
| Metastases to visceral non-CNS (M1C) | 12 | 53% |
| Metastases CNS (M1d) | 4 | 18% |
| NA | 1 | 4% |
|
BRAF mutated | ||
| Yes | 9 | 39% |
| No | 12 | 53% |
| NA | 2 | 8% |
|
Previous adjuvant therapy | ||
| Vemurafenib | 1 | 4% |
| Tafinlar | 1 | 4% |
| Ipilumab | 1 | 4% |
| Target therapy | 2 | 8% |
| ORR | – | 56% |
| DCR | – | 74% |
|
Adverse event | ||
| G1-G2 | 10 | 43% |
| G3 | 1 | 4% |
| NA | 12 | 53% |
BMI, body mass index; BOR, best overall response (RECIST 1.1); CR, complete response; PR, partial response; SD, stable disease; PD, disease progression; OS, overall survival; PFS, progression-free survival; DRR, durable response rate (CR/PR); DCR, disease control rate (CR/PR/SD).
Stable taxa are associated with complete response to immunotherapy in melanoma
Leveraging the longitudinality of our dataset, we sought to identify temporally robust features associated with clinical response. We hypothesized that gut features consistently detected across time could help delineate biologically relevant features from confounders. Shotgun metagenomic sequencing was performed on samples collected at baseline (n = 14) and during therapy (n = 51, Figure S1B; Table S1), and based on the number of taxa retrieved (Figure S1C) and enrichment in response-associated features (Figure S1D), we fixed a prevalence cutoff of 80% within each response group. Considering that the majority of complete responders (CRs) achieve excellent long-term outcomes that endure for years after treatment discontinuation,15,16 we focused on this group and compared it with the rest of the patients (non-CR[nCR]), obtaining a well-balanced sample size between the two response groups at each time point (Figure S1B).
Whereas taxa selected by differential abundance were mostly sporadic across time (Figure S1E), prevalent taxa in both groups were more stable (CR: n = 25, nCR: n = 7, Figure 1B). Among the identified stable CR taxa, 46% ranked highly (i.e., ≥75th percentile) by prevalence and 27% by differential abundance (Figure S1F). Notably, the three most prevalent among the stable CR taxa were also among the top differentially prevalent when comparing CR and nCR (namely Clostridia unclassified SGB6369, Clostridia unclassified SGB14951, and Anaerostipes caccae; Table 2), and CR and nCR can be distinguished at 7–13 months on therapy by the log-ratio of pooled abundance of stable species (Figure 1C inset, but not of higher taxonomy levels, Figure S2A). Taxonomically, stable CR taxa were dominated by Clostridia (Bacillota/Firmicutes phylum, 77%), whereas stable nCR taxa were split between Bacteroidales (Bacteroidetes phylum, 60%) and Clostridiales (40%) (Figure 1C), in agreement with previous reports on baseline3,17 and on therapy7,18 associations. Individually, 20 out of 25 of the stable CR taxa were longitudinally consistent, showing a significant difference in prevalence at baseline as well as during therapy in patients from both hospital sites (Figures S2B and S2C). Furthermore, 13 out of 25 stable CR taxa demonstrated an increase in relative abundance from baseline to therapy in patients with CR (Figure S2D), whereas those with nCR did not exhibit the same trends (Figure S2D).
Table 2.
Stable complete responder/non-CR taxa definition
| Group | Longitudinal overlap |
List of taxa | ||
|---|---|---|---|---|
| 0 month | 2–6 months | 7–13 months | ||
| core CR (n = 25) | x | x | x | Evtepia gabavorous SGB15120 |
| Clostridium SGB6179 | ||||
| Anaerostipes caccae SGB4529 | ||||
| Bacteroides nordii SGB1858 | ||||
| Clostridiales bacterium Choco116 SGB15149 | ||||
| Clostridiales bacterium BX7 SGB72833 | ||||
| Clostridia unclassified SGB14951 | ||||
| Candidatus Metaruminococcus gallistercoris SGB14870 | ||||
| Candidatus Cryptoclostridium obscurum SGB61016 | ||||
| Bifidobacterium pseudolongum SGB17279 | ||||
| Veillonellaceae SGB5809 group | ||||
| Clostridia unclassified SGB6369 | ||||
| Clostridia unclassified SGB14016 | ||||
| Clostridia unclassified SGB6293 | ||||
| Eubacterium maltosivorans SGB4078 | ||||
| Carnobacterium maltaromaticum SGB7902 | ||||
| Candidatus Metalachnospira gallinarum SGB5181 | ||||
| Candidatus Roslinia caecavium SGB4165 | ||||
| Candidatus Gallimonas caecicola SGB14041 | ||||
| Candidatus Heteroscilispira lomanii SGB63278 | ||||
| Bifidobacterium pullorum SGB17264 | ||||
| Firmicutes SGB47515 | ||||
| Bacteroidales unclassified SGB2173 | ||||
| Aggregatibacter sp oral taxon 458 SGB9733 | ||||
| Clostridiales bacterium S5_A14a SGB3977 | ||||
| core nCR (n = 7) | x | x | x | Alistipes sp AF17_16 SGB2326 |
| Clostridiaceae bacterium NSJ_31 SGB14839 | ||||
| – | x | x | Blautia schinkii SGB4825 | |
| Candidatus Saccharibacteria unclassified SGB19893 | ||||
| Bacteroidaceae SGB1808 | ||||
| Bacteroides sp Marseille P3684 SGB1429 | ||||
| Alistipes sp An66 SGB2306 | ||||
Stable CR is defined as prevalent taxa in CR (>80% of samples) across 0, 2–6, and 7–13 months of therapy; stable nCR is defined as prevalent taxa in nCR across at least two time points.
Interactions between the gut microbiome and host are bidirectional: while the host state can induce changes in the gut,12 features of the intestinal ecosystem can directly influence the systemic immunological state.19 We examined patients with paired fecal and blood samples at baseline and on therapy (n = 8, Table S1) and analyzed if the gut microbiota was following the counts of white blood cells over time. We found significantly higher lymphocytes and lower neutrophils and neutrophil-to-lymphocyte ratio (NLR) during therapy in CR compared with nCR (Figure S3A), in line with previous reports.20,21 The systemic state was reflected in the gut microbiota, where these blood cell markers were significantly associated with stable CR taxa, such as Clostridia spp. and Anaerostipes caccae (Figures S3B and S3C). Conversely, low lymphocytes, high neutrophils, and high NLR were associated with a few stable nCR taxa (Figure S3D). These immune cell profiles were complemented by the quantification of 41 soluble inflammatory molecules from serum samples (n = 40, Table S1), taken at baseline and during therapy (Figure S4A). Five cytokines were most important in classifying CR status: interleukin (IL)-12p70, which was associated with CR, and CX3CL1/FRACTALKINE, IL-7, IL-8, and HGF, which were associated with nCR (Figure S4B, upper). These CR and nCR cytokines distinguished between response groups during therapy (Figure S4B, lower), the log-ratio of which associated positively with lymphocyte count and inversely with neutrophils (Figure S4C). With respect to the gut microbiota, CR-associated cytokine environments co-occurred mostly with stable CR taxa (Figure S4D), while nCR-associated cytokine profiles were related with only one stable nCR taxon, namely Blautia schinkii (Figure S4E), in line with the more sporadic nature of nCR microbiota. In all, we show that longitudinally defined gut microbiota features are associated with complete response to ICI and co-occurred with systemic markers of immune response, most notable of which are low NLR and high IL-12p70 levels.
Next, we sought to validate our findings on larger and more diverse external datasets. Because stable CR taxa pertain to bacteria that are present and prevalent in the CR gut from the beginning of therapy, we interrogated nine baseline melanoma cohorts (total n = 281) from Europe, the UK, and the USA.8 While previous analysis found limited reproducibility of microbiome-based signatures across these cohorts,7,8 our results demonstrated enrichment of stable CR taxa in patients with melanoma responsive to ICI in four of the tested studies (enrichment score ≥2), two of which reached statistical significance (Figure 1D). Notably, the highest enrichment of stable taxa was observed Notably, the highest enrichment of stable taxa was observed when subsetting to the two extreme response groups of CR (n = 29) and PD (n = 118), again supporting heterogeneity in immunotherapy responses as a limitation in discerning response-related factors from confounders.
Longitudinally stable functions are enriched in the gut microbiome of patients with melanoma responsive to anti-PD-1 immunotherapy
Having demonstrated compositional differences in the gut microbiota between CR and nCR, we reasoned that microbial functions may better depict response-related mechanisms than individual taxa, which can be more sensitive to geographic distribution7 or dietary habits.17,18 Thus, we analyzed our longitudinal metagenomic data for genes and pathways that are maintained in these communities (see STAR Methods). We re-grouped gene family abundances into KEGG orthologs (KOs) and mapped them to higher-level pathways to determine over-represented functions. Among the main pathways associated with CR were flagellar assembly and bacterial chemotaxis as well as starch and sucrose metabolism (Figure 1E), which were consistently significant from baseline through early and late therapy time points. By contrast, pathways associated with nCR were more variable across time (Figure S5A). The log-ratio of KOs that mapped to these pathways discriminated between CR and nCR specifically at 7–13 months (Figure S5B), which included enzymes involved in fatty acid metabolism (acetyl-CoA carboxylase), butanoate metabolism (4-hydroxybutyrate dehydrogenase), and membrane transport (major facilitator superfamily transporter) (Figure S5C; Table S3). Thus, patients with melanoma achieving CR also carry a subset of stable metagenomic functions that distinguish them from nCR.
Seeing flagellar assembly as a top pathway in CR, we tested its potential involvement in gut microbiota-mediated response to ICI, observing that flagellin-related gene families (n = 1,563 from UniRef90, see STAR Methods) were significantly more abundant in CR from baseline through therapy (Figure 1F). We validated these findings across the nine baseline cohorts, detecting significantly higher abundances of flagellin gene families in R in three datasets3,6,22 (Figure S5D). Furthermore, we interrogated metagenomic functional data from a recently published longitudinal melanoma cohort14 and observed similar associations between flagellin abundance and response during therapy (Figure S5E). Bacterial flagellins are of particular interest because of their well-known modulatory effects on innate as well as adaptive immunity. Top CR-associated flagellin genes retrieved from our dataset mostly annotated to Lachnospiraceae taxa (particularly to butyrate-producer Roseburia inulinivorans, Table S4), with 62% (77 out of 124) of CR-associated flagellin terms (log2(fold-change) > 0) annotated to known Lachnospiraceae genera (Table S5). Flagellin has been shown to play immunomodulatory roles that can influence antitumor response23 and those belonging to Lachnospiraceae specifically have been implicated in host tolerance24 We thus restricted our investigation to Lachnospiraceae-associated flagellin terms (n = 391, UniRef90), with which we obtained significant gene set enrichment in patients with melanoma responsive to ICI in eight out of the nine external cohorts used for validation (Figure 1G). Overall, these results demonstrate the clinical relevance of longitudinally delineated gut microbiota functions, such that they stratify patients by response across baseline cohorts in a way that was not demonstrated with baseline-derived microbiome taxonomic signatures.8
Gut microbiome stability characterizes response to anti-PD-1 immunotherapy in patients with melanoma
We have shown that peculiar microbial functions in the gut of CRs distinguish them from other patients experiencing more heterogeneous responses to ICI. Because such functions are defined by their longitudinal stability rather than taxonomy alone, we investigated whether they reflect differences in gut microbiota dynamics from baseline through ICI treatment. We addressed this point leveraging our single-batch metagenomic dataset (n = 65, Table S1) and stratified patients based on progression-free survival at 24 months (PFS24) (Figure S6A). Significant differences in beta diversity (Aitchison) were observed between patients with long survival (PFS ≥24 months, PFS-L) and those with short survival (PFS <24 months, PFS-S) starting from 5 months of therapy up to 13 months (Figure 2A), but not at baseline (Figure 2B and Figure S6B) or earlier therapy time points. 16S sequencing data from the same patients aligned with this increasing trend (see STAR Methods, Figures S5C and S5D), even when patients were stratified using RECIST 1.1 measures (Figures S5E–S5G).
Figure 2. Patients with melanoma responding differently to anti-PD-1 therapy have progressively different gut microbiome diversity.
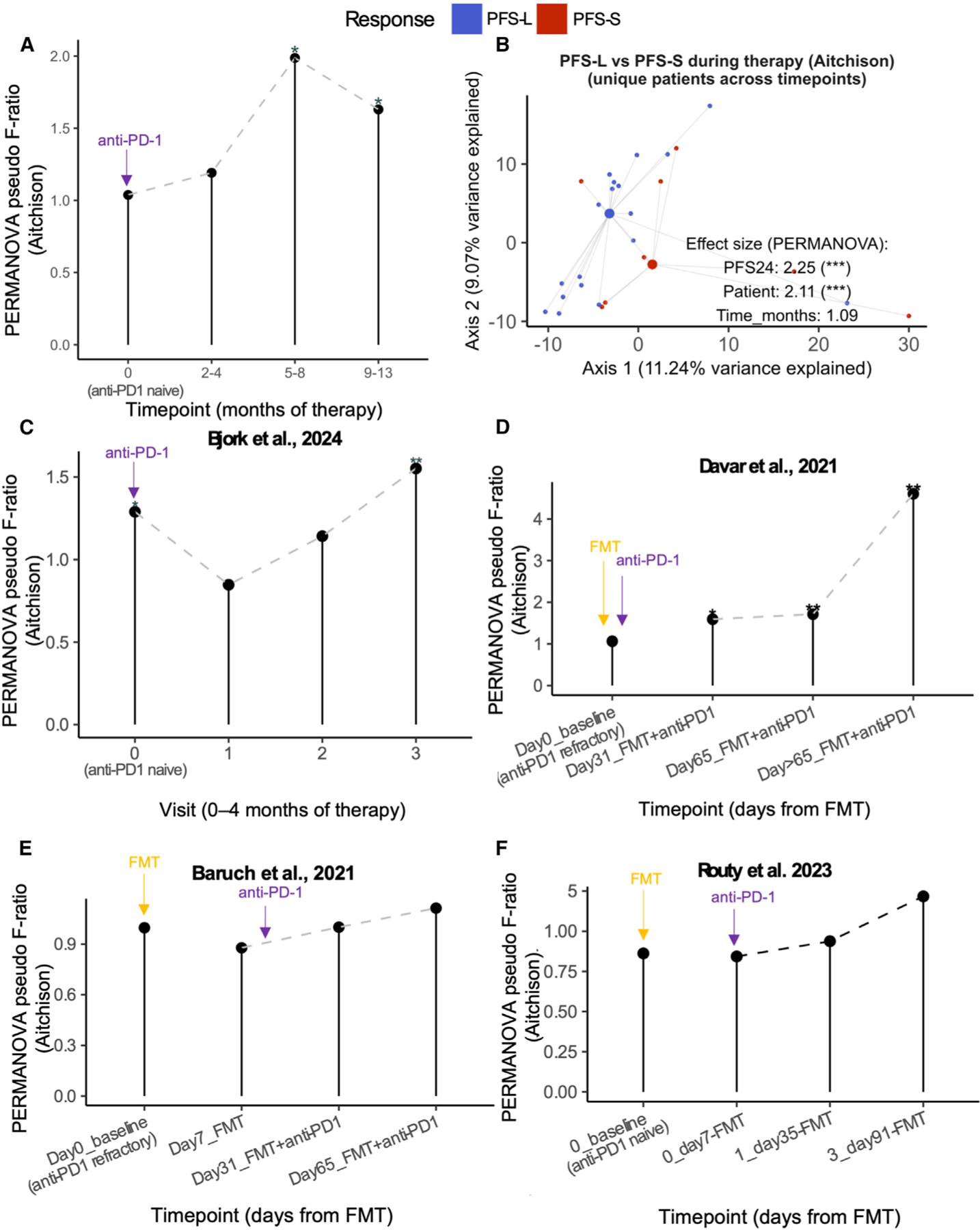
(A) Aitchison gut composition distance (PERMANOVA pseudo F-ratio) between PFS-L (PFS > 24 months) and PFS-S (PFS < 24 months) groups compared across time (0, 2–4, 5–8, and 9–13 months of therapy). Asterisk indicates PERMANOVA p-adj (distance~Response) at the given time point group. See also Table S1 and Figure S6A.
(B) PCA plot of PFS-L and PFS-S Aitchison beta diversity using randomly sampled unique patient samples at each time point group during therapy (2–4, 5–8, and 9–13 months). Statistics are from multi-variate PERMANOVA (distance~Response+Patient+Time point). See also Figure S6B. For similar analyses on 16S data, see Figures S5C–S5G. For comparison against tumor-free, see Figures S7A–S7C and Table S2.
(C–F) Aitchison gut composition difference between R and nR from Shotgun sequencing of (C) longitudinal melanoma-ICI study by Bjork et al.16, compared at visits 0, 1, 2, and 3, spanning 0 to 1–4 months of therapy, depending on patient. Cutoff of < 4 months of therapy was set for this analysis based on sample size distribution across time points of this study, and FMT melanoma-ICI studies by (D) Davar et al., 2021,10 (E) Baruch et al.,24 and (F) Routy et al.13 and compared at different time points as indicated. Asterisk indicates PERMANOVA p-adj (distance~group) at the given time point group. Arrows indicate time point at which fecal microbiota transplant (FMT, yellow) and/or anti-PD1 immunotherapy (violet) commenced for that study. Differences among these studies are outlined in detail in Table S8.
Legend: PFS-L (blue), PFS-S (red). p-adj<0.001 (***), p-adj<0.01 (**), p-adj<0.05 (*), ns (unannotated).
Similarly, we found increasing differences between response groups during ICI treatment when re-analyzing publicly available longitudinal metagenomic data from a longitudinal melanoma study averaging two months of ICI therapy per patient14 (Figure 2C) as well as three independent studies whose recipients received anti-PD-1 treatment in combination with FMT9–11 (Figures 2D–2F). These differences may be particularly contributed by an increasingly diverging microbiota in patients with melanoma non-responsive to ICI, as resulting from the comparison of the distance-among-samples between each of the two response groups and a reference group (n = 14) of tumor-free subjects (see STAR Methods, Table S2; Figures S7A–S7C), suggesting that the microbiota of PFS-L and PFS-S patients may have different dynamics on therapy.
MHC class I-restricted peptides derived from FLach genes show structural homology with human TAAs
Next, we sought to precisely define the mechanism underlying the modulation of the response to ICI by longitudinally defined gut microbiota functions. Preclinical evidence supports the contribution of immunomodulatory bacterial metabolites, direct stimulation of antitumor T cell responses, and molecular mimicry between shared bacterial and tumor epitopes.12,13,25 In this regard, gut-residing Lachnospiraceae can be a rich source of tumor-mimicking epitopes26; thus, we searched our shortlist of CR-associated flagellin-related Lachnospiraceae (FLach) proteins for peptides predicted to bind to HLA class I receptors (HLA-I), then matched them against a public database of experimentally validated tumor-associated antigens (TAAs, n = 271, see STAR Methods). Out of 14 FLach proteins tested, 13 encoded for 9-mers predicted to bind to HLA-I (Table S6). Among those showing some extent of sequence homology to TAAs (Table S6, see STAR Methods for details), 3 FLach-TAA epitope pairs were predicted to share strong binding affinity (SB) to their respective HLA-I (<100 nM), while in the other 10 pairs only the FLach was an SB. The 3 SB TAAs were all associated with melanoma, namely: preferentially expressed antigen in melanoma (PRAME), a melanoma-associated antigen expressed in 87% of metastatic and 83.2% of primary melanomas27; melanoma antigen recognized by T cells 1 (MART-1/Melan-A), one of the oldest identified tumor antigens found in most melanomas28,29; and secernin 1 (SCRN1), a protein involved in MMP2/9 exocytosis,30,31 and overexpressed in amelanotic melanoma32
We confirmed by immunohistochemistry the expression of Melan-A (p = 0.033, Fisher’s exact test) and PRAME (p = 0.018, Fisher’s exact test) on available primary tumors from CR (n = 2) as compared with nCR (PR n = 7 and PD n = 2), with CR expressing both antigens while none of them was detected on PD samples (Figure S8A). On the other hand, in silico modeling revealed high structural homology between the 3 SB FLach peptides and their respective SB TAAs in terms of 3D conformation and points of contact to HLA (cyan Figure 3A) and T cell receptor (TCR, green Figure 3A). We consolidated these predictions by directly measuring the binding affinity of FLach to purified major histocompatibility complex class I (MHC class I) in vitro (Figure 3B), demonstrating that some of the bacterial-derived 9-mers bind their predicted MHC class I with a higher affinity than prototypic control peptides (Table 3). Overall, these data demonstrate that analysis of stable gut metagenomes associated with complete response to ICI leads to the identification of MHC class I-restricted bacterial antigens with previously unexplored tumor-antigen mimicry potential.
Figure 3. MHC class I-restricted peptides derived from flagellin-related Lachnospiraceae gene families show structural homology with human tumor-associated antigens.
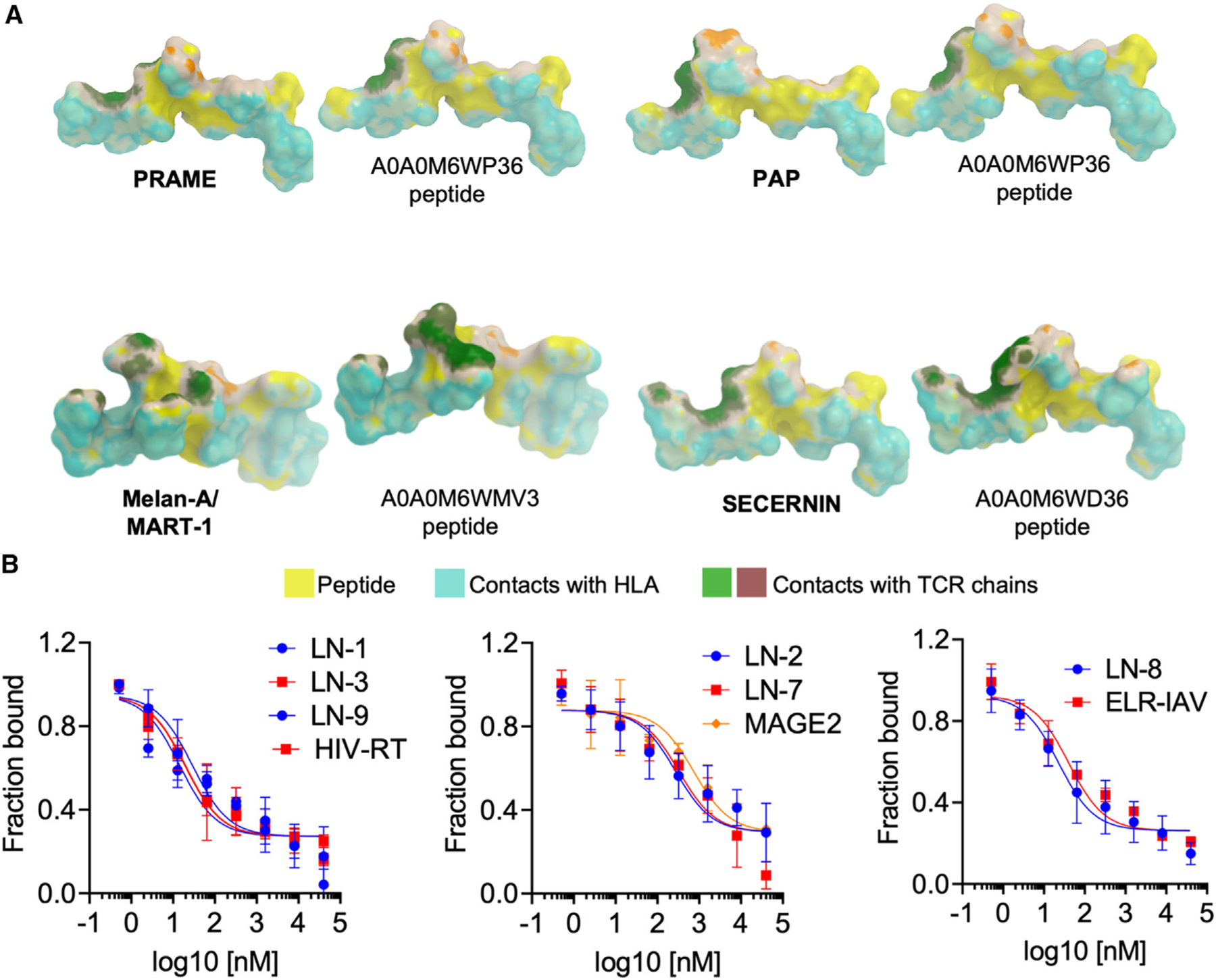
(A) Structural simulation of three candidate epitopes (right of pair) predicted from CR-associated flagellin gene families, determined to have sequence and structural homology to a tumor-associated antigen (TAA) (left of pair) and strong affinity to the same HLA allele as that of the TAA (<100 nM). See also Table S6. For immunohistochemistry results, also see Figure S8A.
(B) Experimental binding affinities of the selected flagellin peptides toward A02:01 (left), B07:02 (center), and B08:01 (right), calculated by titrating different concentrations of the test peptide in the presence of 1 μM of protein and fixed concentration (25 nM) of probe peptide HIV-RT (left), MAGE2 (center), and ELR-IAV (right), respectively. See Table 3.
Table 3.
Binding affinity of FLach peptides to MHC class I
| Peptide | Sequence | Allele | Predicted Affinity (nM) | Experimental Affinity (nM) |
|---|---|---|---|---|
| LN-1 | KMSYEDIEL | A02:01 | 71.69 | 110.7 |
| LN-3 | ALNETSAIL | A02:01 | 67.47 | 64.02 |
| LN-9 | GLDALNNLL | A02:01 | 66.44 | 111.9 |
| HIV-RT | ILKEPVHGV | A02:01 | 40.85 | 63.81 |
| LN-2 | MPKDGAAFI | B07:02 | 93.13 | 271.7 |
| LN-7 | SVRGRLGAF | B07:02 | 66.44 | 325.5 |
| MAGE2 | VPISHLYIL | B07:02 | 46.13 | 730.3 |
| LN-8 | FPELKHFTM | B08:01 | 41.60 | 64 |
| ELR-IAV | ELRSRYWAI | B08:01 | 6.53 | 143 |
NetMHCpan4.1 predicted and experimentally calculated affinity of indicated peptides to MHC class I.
Complete response is associated with a higher reactivity against FLach peptides in circulating and tumor-infiltrating CD8+ T cells in melanoma patients
The data above indicate that bacteria-derived FLach peptides are constitutively present in CRs and may be involved in anti-tumoral immune responses elicited by ICI therapy. We tested this hypothesis experimentally, first by assessing whether we can detect T cells with reactivity against FLach peptides in the peripheral immune compartment of melanoma patients before any ICI treatment. Specifically, we pulsed peripheral blood mononuclear cells (PBMCs) isolated from patients with melanoma undergoing ICI therapy (baselines, n = 11) with two pools of purified FLach peptides, namely the FLach-G pool (including the 3 SB bacterial-derived peptides that share structural homology with melanoma SB TAAs, Figure 3A; Table S7 in green) and the FLach-R pool (a mix of the 7 SB bacteria-derived peptides matching low-affinity TAAs, Table S7 in red), and measured CD8+ T cell-specific reactivity by flow cytometry. Compared with patients with non-responding melanoma (n = 4), CD8+ T cells of CR (n = 7) showed greater activation following peptide stimulation, as measured by an increased proportion of IFNγ -secreting CD8+ T cells (peptide mix/control ratio 1.3 ± 0.2 vs. 0.6 ± 0.1 for FLach-G and 1.4 ± 0.2 vs. 0.3 ± 0.2 for FLach-R in CR vs. nCR, respectively) in the large majority of CR tested (5/7 and 6/7 with FLach-G and FLach-R pools, respectively, Figures 4A–4B), especially with the FLach-G pool, which resulted in upregulation of the IL-2 receptor subunit CD25 (Figure S8B). No differences in terms of CD4/CD8 ratio nor naive T cells were observed (Figures S8C and S8D). Importantly, neither mix showed TLR5 agonist activity (Figure S8E), suggesting that the immune modulatory effects of FLach peptides are completely distinct from those of flagellins.24,33,34
Figure 4. Complete response associates with a higher FLach-directed reactivity and antitumor immunity in melanoma patients.
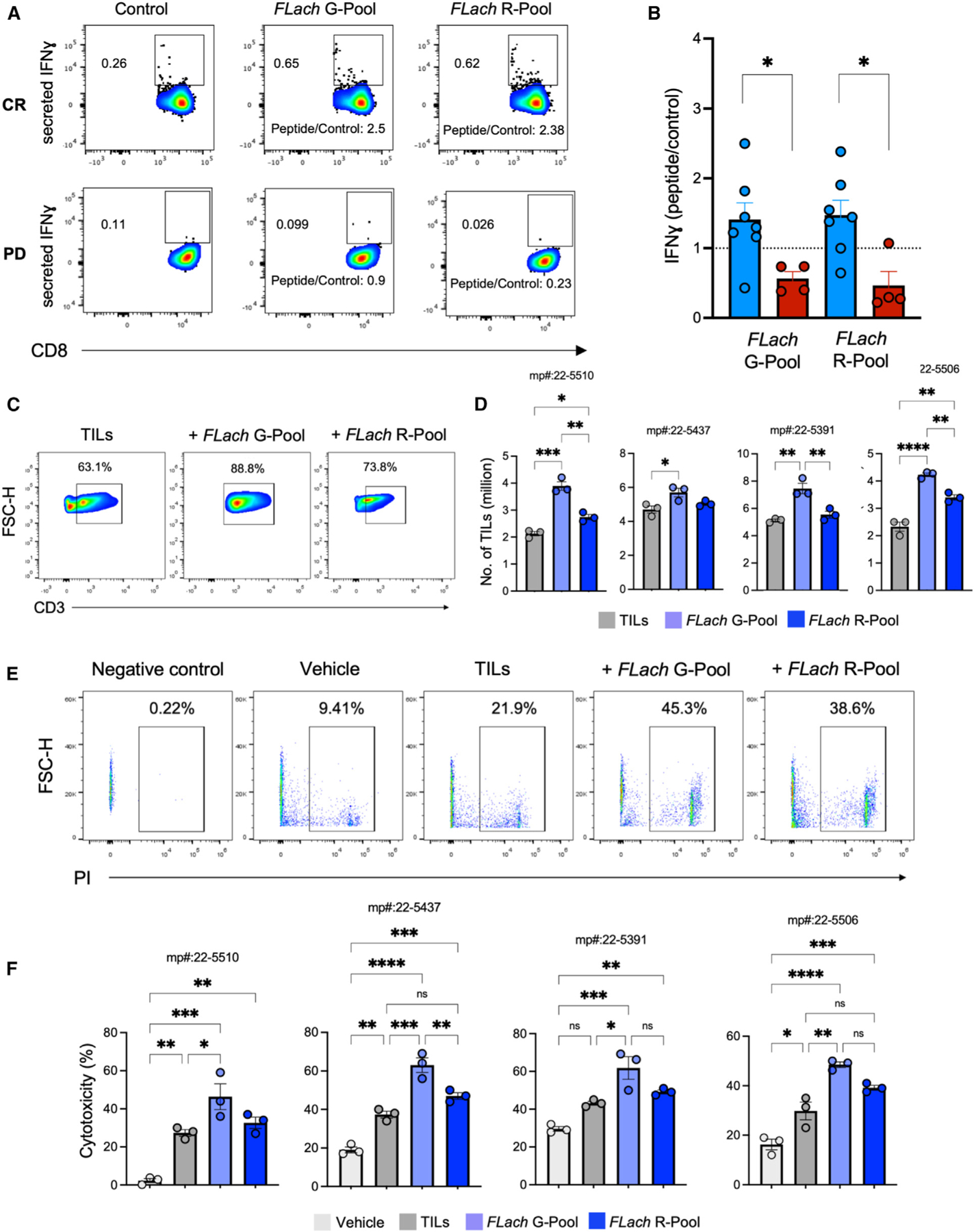
(A and B) (A) Representative flow cytometry contour plots and (B) MFI measurements of IFN-γ (normalized on untreated controls) on CD8+ T cells in peripheral blood monocytic cells (PBMCs) from patients with melanoma grouped by response to ICI (responsive = 7, non-responsive = 4). See also Figures S8B–S8E and Table S7.
(C and D) (C) Representative flow cytometry contour plots and (D) number of TILs from four human melanoma tumors expanded with or without adding the indicated peptide pools. See also Figure S8F.
(E and F) (E) Flow cytometry and (F) bar plots showing cytotoxicity (measured as % of PI-positive melanoma cells) of TILs expanded with or without adding the indicated peptide pools and tested on four matching melanoma patient-derived organoids.
While providing evidence of FLach peptide immunogenicity, the fact that PBMC reactivity is differentially correlated with response suggests that CRs bear the potential to mount a FLach-directed CD8+ T cell-mediated response, targeting structurally matching TAAs expressed on their tumors. To evaluate this hypothesis, we next analyzed tumor-infiltrating lymphocytes (TILs) isolated from fresh melanoma tumors (n = 4) and expanded in vitro in the presence of IL-2, alone or in combination with either FLach-G or FLach-R pools. After 14 days of culture, TIL expansion was significantly higher in the presence of FLach compared with control, especially with the FLach-G pool (which contained SB FLach SB matching SB TAAs), indicating a specific reactivity of TILs against those FLach antigens mimicking known melanoma-associated TAAs (Figures 4C and 4D).
Finally, we assessed direct TIL-killing ability on melanoma patient-derived organoids (MPDOs), co-cultured by semi-immersion on Matrigel (Figure S8F, see STAR Methods). Similar to parental tumors, MPDO retained diverse immune cell populations, including antigen-presenting cells (APCs), enabling testing the effect of FLach peptides on the cytotoxicity of expanded autochthonous TILs. Indeed, live/dead cell staining revealed that TILs expanded by IL-2 plus FLach-G pool exhibited significantly higher antitumor activity than FLach-R pool or bulk TILs expanded with IL-2 alone in all four patients (Figures 4E and 4F), demonstrating that FLach peptides can improve antitumor immune responses. In all, these data show that (1) T cells with reactivity against HLA-I-restricted FLach peptides exist in the peripheral blood and correlate with ICI response, (2) a fraction of FLach-directed T cells cross-recognizing TAAs infiltrates the tumor, and (3) they can mount an antigen-specific antitumor response.
DISCUSSION
Amidst the body of work relating gut microbiota to ICI response in melanoma patients, precisely identifying response-modulating microbial features remains a challenge. Hurdles in consolidating such studies include biological heterogeneity inherent to clinical responses and scarce data on the gut microbiota during therapy when response modulation takes place. We address these problems by leveraging a unique longitudinal dataset of patients with melanoma who, unlike other longitudinal studies,11,14,35 were followed from pre-ICI up to 13 months of ICI treatment in an adjuvant setting. Despite its limited size, this unique cohort enabled the identification of enduringly stable features that were fundamental in capturing nuances between response groups, as opposed to differential features identified at single time points. In addition, we validated insights about the long-term ICI-exposed gut and host immune state using multiple external cohorts and, experimentally, on patient-derived samples.
Parallel evaluation of the gut microbiota and the systemic immune state of CR patients helped us reduce the overwhelming influence of heterogeneity when matching clinical and biological features throughout the study. Indeed, this approach identified a group of Clostridia-dominated taxa that stably inhabits the gut and associates with distinctly low NLR and high levels of IL-12p70. Although only correlative, these immunological differences prompt questions on how the gut and systemic immunity reciprocally influence each other during ICI therapy. While Clostridia have been previously associated with response to ICI3,7,18,36 and successful FMT engraftment,37,38 IL-12p70 is produced by dendritic cells either upon antigenic stimulation (where it is implicated in helper T cell differentiation)39 or in response to the IFNγ released by anti-PD-1 activated T cells40 As such, IL-12p70 has been associated with severe immune-related adverse events (irAEs)41 and enhancement of anti-PD-1 response40,42 respectively. Future experiments should be designed to disentangle any mechanistic link between stable gut microbes and immune markers.
Fitting the gut-immune narrative, ICI-induced translocation of specific enteric bacteria from the gut to secondary lymphoid organs has been recently demonstrated in preclinical models, eliciting antitumor T cell activity systemically.12 However, our results suggest that defined shared functions rather than individual taxa may be key to immune modulation. Among them, genes for starch and sucrose metabolism are stably carried in CR gut metagenomes. Starch granules are abundant in many natural foods (i.e., potatoes, rice, and cereal grains) and include polymers of glucose linked to an α-glucan by linkages either soluble (amylopectin) or resistant to enzymatic degradation (amylose). While the soluble portion is processed by the host, resistant starch is metabolized by gut microorganisms via the collective metabolic activities of primary and secondary degraders, the latter scavenging partially digested polymers and completing their fermentation to end-products such as short-chain fatty acids (SCFAs), including butyrate.43 Beneficial effects of SCFAs on intestinal epithelial homeostasis44,45 and immune functions46–50 have been widely reported, but their role in host immunity during immunotherapy response appears ambiguous51–53 with concurrent immune-activating (driven by IFN-γ and IL-12)54,55 and suppressive (mediated by Treg)55 effects described. However, a higher intake of dietary fibers, which are metabolic precursors of SCFAs, has been shown to favorably impact immunotherapy outcome,18 prompting dietary intervention trials (e.g., NCT04645680). Effects on antitumor immunity18 and intestinal inflammation17 likely contribute to the beneficial impact of dietary fiber on cancer immunotherapy outcome. However, more studies are needed to tease apart systemic versus local intestinal immunomodulatory interactions and to mechanistically define how the type and source of dietary fiber drive changes in the gut community. Current methods suffer from a limited ability to predict enzymatic degradation activities from metagenomic data and ascribe them to specific microbial groups, especially non-bacterial microbiota (i.e., fungi and protists) whose metabolic contributions are largely unknown.
Based on our longitudinal analyses of the microbiota, we also propose that patients with better or worse outcomes may exhibit different gut dynamics during therapy. A previous cross-sectional study from McCulloch et al. reported the strongest association between baseline microbiota and ICI response in melanoma patients at around one year of treatment,7 suggesting a more pronounced role of microbiota in response at a specific window during therapy. Here, following gut diversity on individual patients, we add that, while the microbiota is more baseline-like over time in patients with a better survival (PFS-L), it is increasingly more variable in those experiencing earlier disease progression (PFS-S), demonstrating different gut dynamics over long-term ICI in patients with different outcomes. Notably, we cannot rule out that changes observed in the gut during ICI may be due either to an overall improvement in health among responders or to other sources of variation (i.e., diet, psychological, and lifestyle factors), and disentangling causative from confounding factors is untenable with observational data alone. Nevertheless, different predictive abilities of gut microbiota for response in monotherapy (i.e., anti-PD-1) and combination (i.e., anti-PD-1 and CTLA-4) settings across cancer types support that the ICI regimen itself exerts a specific influence on the microbiota.56 In any case, measuring the gut dynamic could be developed as a tool for decision-making, either in the neoadjuvant (e.g., determining optimal surgery time) or adjuvant (e.g., guiding therapy completion) therapies.
Our functional data reveal a previously unappreciated role for the flagellum in the microbiota-host crosstalk during ICI. We detected significantly increased flagellin-related gene families in the CR metagenomes, both in our patients as well as in responders of various international cohorts, and their presence at baseline suggests that they were not incidentally emerging from lifestyle and diet changes during therapy. However, comprehensive longitudinal lifestyle and diet surveys provided by each patient would be needed to rule this out unequivocally. Interestingly, a large portion of flagellin-related genes we retrieved in the CR gut were carried by Lachnospiraceae, which reportedly exhibit a peculiar weak-agonist activation of Toll-like receptor (TLR)5 immunity,24 allowing the carriage of a “silent,” host-tolerated repertoire of antigens with broad tumor-mimicry potential. A study demonstrated the capacity of commensals in mediating a host-tolerated immunogenic response in a colonized mouse model57 and, here, we provide proof that a distinct pre-ICI immunity directed against FLach peptides exists in patients with CR, in the peripheral immune compartment, as well as in the tumor. The latter is important, as the role of tumor antigens in ICI response is a re-emerging paradigm. Recent studies have shown the success of ICI on MMR-deficient (dMMR) patients, which is linked, at least in part, to a higher immune “visibility” of their highly mutated and presumably more neoantigenic tumors.58–61 Extending this concept, trials combining immunotherapy with personalized tumor-specific neoantigen vaccines are showing early promise (NCT03897881).62 The immunomodulatory (and TLR-5 mediated) effect of one flagellin has also been demonstrated,33 and its safety and efficacy as part of a live biotherapy is being clinically tested on patients with cancer, alone (NCT03934827) or in combination with immunotherapy (NCT03637803). While suggesting that a subgroup of patients is predisposed to complete response to ICI treatment and that FLach reactivity can be used as a non-invasive biomarker to stratify patients, our results demonstrate that FLach peptides can also be used therapeutically to either improve the expansion of autochthonous TILs for adoptive T cell therapy or, potentially, as a pre-conditioning treatment on selected patients undergoing ICI therapy. Mechanistic and preclinical studies are underway to address these outstanding points.
In conclusion, we propose that the immune response elicited by ICI therapy shapes a host niche favorable for gut taxa that synergistically support immune cell function and tumor recognition while being well-adapted to host conditions, such as flagellin-carrying gut commensals under the Clostridia clade. Patients that carry such a beneficial gut are thus disposed to respond well to ICI and manifest gut stability as a direct or indirect consequence of this healthy response (Figure S9). Addressing how to harness or induce such an ICI-conducive gut microbiota or its host immune reactivity will pave the way to more efficient treatment of melanoma and potentially other solid tumors.
Limitations of the study
The longitudinality of our study was instrumental, first, to establish “homogeneity” among CRs based on gut and systemic profiling and, second, to utilize stability as a marker of response by gut dynamic and persistence of gut features during ICI. Nevertheless, the limited size of the initial cohort remains a grounding caveat, and despite having our longitudinally derived gut markers corroborated by multiple baseline studies, the overarching hypothesis of gut stability among ICI responders would require validation in larger, long-term longitudinal studies.
Furthermore, we cannot rule out that gut and systemic features found in CRs were the result of an overall improvement in health or other confounding factors rather than linked to immune response—although their presence since baseline would support the latter. In this regard, complementary in vivo studies on preclinical animal models would help deconvolute causative factors, elucidate underlying gut-immune mechanisms related to response, and conclusively demonstrate the therapeutic effect of FLach peptides.
RESOURCE AVAILABILITY
Lead contact
Inquiries and requests related to the study should be made out to the lead contact, Luigi Nezi (luigi.nezi@ieo.it).
Materials availability
All reagents are available from the corresponding authors upon reasonable request. Patient-derived biological samples were limited and may not be further available. Peptides were designed in-house and purchased from GenScript Biotech (NJ, USA).
STAR★METHODS
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Study design and enrollment
Fecal samples were obtained from patients with melanoma treated with anti-PD-1 immunotherapy enrolled at Istituto Europeo di Oncologia (IEO, Milan) and at Istituto Nazionale Tumori IRCCS “Fondazione G. Pascale” (INT-FGP, Naples) between January 2018 and October 2022. All subjects gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Declaration of Helsinki and approved by the IEO and INT-FGP Ethical Committee (studies registered as R845/18-IEO 889 and 37/22 oss, respectively). A total of 23 patients were assessed for eligibility and enrolled. Inclusion criteria encompassed patients with a confirmed diagnosis of unresectable or metastatic cutaneous melanoma (BRAF wild-type or mutant) that did not meet any of the exclusion criteria (i.e systemic use of antibiotics during the last month before the start of therapy, concomitant use of immunosuppressive medications, known autoimmune conditions). The median age of patients at study entry was 54 years (33–78 years) and 17 (74%) were male. Participants underwent anti-PD1 therapy (480mg every four weeks). Eighteen (79%) patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1, with 9 patients (39%) had stage IV disease, including 3 (15%) with brain metastases and 4 (20%) with an elevated baseline serum lactate dehydrogenase (LDH). Patient baseline characteristics are summarized in Table 1. At the time of data cutoff (15 December 2022), the median follow-up time was 20.7 (7.5–42.3) months, with six patients (30%) still on anti-PD-1 therapy. In parallel, fourteen donors (median age of 42) were enrolled as tumor-free group.
METHOD DETAILS
Sample collection
Melanoma patients were followed at baseline and over the course of anti-PD1 immunotherapy and collected fecal (Canvax Biotech, ES) and blood samples for at least one timepoint. Select fecal samples were processed for 16S amplicon (n=94) and Shotgun metagenomic sequencing (n=65), whereas blood samples were processed as whole blood for white blood cell count (n=58) and as serum for soluble factor quantification (n=40).
For the 16S sequencing, 19 patients have baseline samples, 17 have therapy samples, and 13 have baseline and at least 1 therapy sample. Of the 13, 8 had samples both at 2–7 months (early therapy) and 8–13 months (late therapy); these 8 patients were used for the 16S longitudinal analysis.
For the Shotgun sequencing, 14 patients have baseline samples, 11 have samples at 2–7 months (early therapy) and 7 have samples at 8–13 months (late therapy). These samples were used for stable taxa and functional analyses.
16S amplicon sequencing
Fecal samples were processed for 16S sequencing to compare the microbiota structure and composition at baseline (t0) (i.e., before therapy) and at each cycle of therapy (t1, t2, t3… tn). Briefly, DNA was extracted from feces of melanoma patients and healthy donors using the DNeasy PowerSoil Pro kit (Qiagen), after which the V3-V4 region of 16S was amplified. Libraries were prepared following the 16S sequencing library preparation protocol (Illumina), and sequenced by a 2×250 bp paired end chemistry on a MiSeq platform.
16S sequence processing
For each 16S sequencing run, data were filtered and denoised using the DADA2 plug-in in qiime2 (qiime2-2018.11)68 where parameters were set to trim sequences at the 5′ region by the length of the primer, to generate a counts table of amplicon sequence variants (ASVs) for that dataset.
For the rest of the core 16S analyses, qiime2-2019.7 was used. Phylogenetic tree reconstruction for downstream diversity analyses was done with the q2-fragment-insertion plug-in,70 using SILVA 128 database64 as reference sequence. For taxonomy assignment the q2-feature-classifier plug-in was used. Briefly, full-length reference sequences from the SILVA 132 database were downloaded from the SILVA resources page for qiime (https://www.arb-silva.de/download/archive/qiime), after which the sequences were used to train a Naïve-Bayes classifier using the fit-classifier-naïve-bayes function.71 The trained classifier was run on the representative sequences output of DADA2 using the classify-sklearn function to generate taxonomic assignments for each ASV.
All downstream analyses were performed in R, after exporting the taxonomy table, ASV counts table, phylogenetic tree, and metadata in R and converting into a phyloseq object.74
16S-related analyses are further described under ‘quantification and statistical analysis’ section.
Shotgun metagenomic sequencing
DNA extracts from fecal samples of select melanoma samples from our study were subjected to metagenomic sequencing, where libraries were prepared using the Illumina DNA Prep Kit according to manufacturer’s protocol. Libraries were multiplexed using dual indexing and sequencing was performed with a 300-bp paired-end chemistry, using the Illumina NovaSeq6000 platform according to manufacturer’s protocol.
Shotgun sequence pre-processing
Shotgun metagenomic sequencing was performed at the NGS Core Facility at University of Trento. The quality of all sequenced metagenomes was controlled using the preprocessing pipeline implemented in https://github.com/SegataLab/preprocessing, which consists of three main stages: (1) initial quality control by removing low-quality reads (quality score <Q20), fragmented short reads (<75 bp) and reads with more than two ambiguous nucleotides; (2) contaminant DNA removal using Bowtie 272 and the sensitive local parameter, removing both the phiX174 Illumina spike-in and human-associated reads (hg19); and (3) sorting and splitting for the creation of standard forward, reverse and unpaired reads output files for each metagenome.
Core diversity from Shotgun data
Beta diversity distances were computed from the relative abundances using the calculate_diversity.R function from MetaPhlAn, setting the method to ‘aitchison’. The distances were ordinated by PCoA using the ape::pcoa function. Beta-diversity group differences were computed from the distance matrices by PERMANOVA,88 checking for balance in dispersion by PERMDISP.75 All reported significant p-values by PERMANOVA were checked to have non-significant dispersion. The overall composition of the samples was projected into a PCA after computing the Aitchison distance on the transformed abundance, testing group differences on the abundance table by PERMANOVA while checking for balance in dispersion by PERMDISP.
Metagenomic analyses
Metagenomic sequence data from our study as well as from previously published baseline melanoma cohorts8 were run through the biobakery 3 pipeline,63 which leverages a set of 99,200 high-quality and fully annotated reference microbial genomes spanning 16,800 species and the 87.3 million UniRef9066 functional annotations available in UniProt as of January 2019. Taxonomic profiling of taxa composition of all metagenomic samples was performed with MetaPhlAn v4.0.365 using default parameters and CHOCOPhlAnSGB v202103 as database. Functional potential analysis of the metagenomic samples was performed with HUMAnN v3.663 using default parameters.
Shotgun-related analyses are further described under ‘quantification and statistical analysis’ section.
Epitope prediction and tumor antigen matching
Candidate flagellin proteins determined from metagenomic data were tested for tumor mimicking potential following the methods described in Ragone et al.26 Briefly, protein sequences of our candidate flagellins were queried, NetMHCstabpan 4.173 was used to shortlist 9-mer peptides with predicting strong-binding (SB) affinity (<100 nM) to MHC class I HLA alleles. BLAST homology search was performed on these SB peptides against a pre-determined list of tumor-associated antigens (n=271), after which homologous sequences were queried for epitope prediction using NetMHCstabpan 4.1. Among the homologous sequences, those peptides with matching HLA allele affinity to any of the TAAs were selected for structural prediction.
Molsoft Mol Browser (version 3.8-7d) (Molsoft LLC, San Diego, CA) was used for epitope modeling and molecular docking and conformation calculations for the shortlisted peptides and matching TAAs.
Assembly of peptide-filled MHC class I molecules
HLA-B*0801 and HLA-B*0702 molecules were reconstituted in vitro as described previously.89 Briefly, urea-solubilized inclusion bodies of class I heavy chain (HC) (1 mM) and b2m (2 mM) were combined with a synthetic peptide (10 mM) (FLRGRAYGL for HLA-B*0801; APRTVALTA for HLA-B*0702) in an oxidative refolding buffer. The refolding mixtures containing MHC I/peptide complexes were concentrated and purified on a Superdex 200 Increase 10/300 gel filtration column at 4°C. Stock solutions of purified MHC I/peptide complexes (10–30 mg/mL) in 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, were kept at −80°C.
Assembly of peptide-deficient MHC class I molecules
Peptide-deficient HLA-B*0801 and HLA-B*0702 molecules were reconstituted in vitro from the denaturation of peptide-filled molecules.90 In brief, peptide-filled molecules were incubated in buffered 6M guanidine hydrochloride for 5 hours at room temperature. Denatured HC and b2m subunits were separated from the peptide ligand by extensive washes in centrifugal filters (MWCO 10K). The retentate fraction containing HC and b2m were diluted to 0.1 mg/mL and dialyzed overnight in buffered 8M urea, after which an excess of b2m (to 0.1 mg/mL) was added to the dialysis bag. Renaturation and assembly of the two subunits was initiated by extensive dialysis in 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, at 4°C. Glycerol (to 15%) was added to the crude mixture of peptide-deficient molecules prior to concentration. Peptide-deficient molecules were purified on a Superdex 200 Increase 10/300 gel filtration column in 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, at 4°C. Desired fractions of peptide-deficient molecules were concentrated in the presence of 15% glycerol and analyzed by MALDI mass spectrometry to ascertain that the molecules are devoid of peptides. Stock solutions of purified peptide-deficient molecules were kept at −80°C.
HLA-A2:01, B07:02 and B08:01 peptide binding exchange assay
A fluorescence polarization competition binding assay was used to calculate the binding affinity of the proposed peptides to HLA-A02:01 or B07:02, or B08:01. HIV-RT (ILKEPVCGV), MAGE2 (VPICHLYIL) and ELR-IAV (ELRSRCWAI) were labeled with Alexa Fluor 488 C5-maleimide and used as the probe peptide for A02:01, B07:02 and B08:01 respectively. Alexa488 labeled and unlabelled probe peptides for these studies were obtained from 21st Century Biochemicals (Marlborough, MA). The binding reactions were carried in buffer conditions of 1X PBS, 0.1% β-octylglucoside, 1X protease cocktail inhibitor. The A02:01, B07:02 and B08:01 concentration used was selected by titrating the proteins against fixed labeled peptide concentration (25 nM) of HIV-RT peptide, MAGE2 peptide and ELR-IAV peptide respectively. We chose the concentration of the proteins for the assay that showed ~50% of maximum binding. IC50 values were calculated by incubating 1μM of protein with their respective Alexa488 labeled peptides in presence of 5-fold dilution of the test peptides starting from 40μM to 0.5μM. The reactions were incubated at 4°C for 16hrs. The capacity of each test peptide to compete for binding of probe peptide was measured by FP after 16 hrs at 4°C. The assay was read using a SpectraMax plate reader (Molecular Devices). FP values were converted to fraction bound by calculating [(FP_sample - FP_free)/(FP_no_comp - FP_free)], where FP_sample represents the FP value in the presence of test peptide; FP_free represents the value for free Alexa488-conjugated peptide; and FP_no_comp represents values in the absence of competitor peptide. We plotted fraction bound versus concentration of test peptide and fit the curve to the equation y = 1/ (1 +[pep]/IC50), where [pep] is the concentration of test peptide, y is the fraction of probe peptide bound at that concentration of test peptide, and IC50 is the 50% inhibitory concentration of the test peptide.
Immunohistochemistry
Slides (5 mm) from formalin-fixed paraffin embedded (FFPE) samples were processed for deparaffinization and rehydration. Heat-induced antigen retrieval (citrate buffer pH 6 for MEL-A or 9 for PRAME, Thermofisher) was performed using the microwave and incubation in 3% H2O2 was used to inactivate endogenous peroxidase. Slides were incubated ON at 4 °C with rabbit anti-human MEL-A (1:400) and rabbit anti-human PRAME (Abcam, Ab219650; 1:400), in a blocking solution composed of 3% BSA, 5% goat serum (Invitrogen, 10000 C) and 0.1% Triton in PBS. After incubation with HRP-secondary antibody (30 min) and Aminoethyl Carbazole (AEC) + High Sensitivity Substrate Chromogen (Dako) slides were counterstained with hematoxylin and mounted and acquired by using Aperio (Leica).
In vitro PBMC reactivity assay
Cryopreserved PBMCs of patients with melanoma R and NR were used for the in vitro stimulation after challenge with FLA peptides. After thawing, washing, and resting PBMCs in pre-warmed RPMI-1640 (Euroclone, Cat. No. ECM2001L) with 10% FBS (Euroclone, Cat. No. ECS0165L), 10mM Hepes (Sigmna, Cat. No. H0087), 1X Sodium Pyruvate (Eurocloene, Cat. No. ECM0742D), 1X Pen/Strep (Euroclone, Cat. No. ECB3001D) and 100 U/ml IL2 (PROLEUKIN), cells were plated at 1×106/ml (200,000 cells/well), pulsed with peptides (1ug/ml), and incubated for 5 days, after which the media was changed. At day 8 cells were restimulated with 2ug/ml of peptide and 1ug/ml of anti-CD28/49d (BD, Cat. No. 347690) for 6h. After 4h, Golgi plug (BD, Cat. No. 555029) was added. Staining was performed using Fixable Viability Stain BV510 (BD, Cat. No. 564406), CCR7 BV421 (BD, Cat. No. 150503), CD8 BV605 (BD, Cat. No. 564116), CD25 BV786 (BD, Cat. No. 741035), CD45RA PE (BD, Cat. No. 561883), CD69 APC (BD, Cat. No. 555533), CD3 APCR700 (BD, Cat. No. 565119) and CXCR3 PerCP-Cy5.5 (BD, Cat. No. 560832); then, cells were fixed and permeabilized using Fixation/Permeabilization Solution Kit (BD, Cat # 554714), and stained intracellularly with an antibody cocktail (IL17A FITC (Invitrogen, Cat. No. 11-7179-42), IFNG PE-CF594 (BD, Cat. No. 562392), TNFA APC-CY7 (BioLegend, Cat. No. 502944) in the permeabilization solution. Samples were acquired using a FACSCelesta BVYG equipped with FACSDiva software version 8.0.1 (all from BD Biosciences).
Analysis of TLR5 activation pathway
To determine the effects of the peptide pools on TLR5 signaling, a HEK reporter cell line, purchased from Invivogen (San Diego, USA), was used. This reporter cell line expresses Secreted Embryonic Alkaline Phosphatase (SEAP), which is coupled to the nuclear factor κB/Activating protein-1 (NF-κB/AP-1) promotor. Upon activation of the TLRs by a specific agonist, high levels of intracellular NF-κB will lead to the secretion of SEAP which can be quantified by spectrophotometer at 620–655nm. The HEK-Blue-hTLR5 cell line expressing TLR5 was cultured in DMEM medium, containing 10% heat-inactivated FBS, 2 mM l-glutamine, 4.5 g L−1 glucose, 50 U mL−1 and 50 mg mL−1 penicillin/streptomycin and 100 mg mL−1 Normocin. All reporter cell lines were cultured for 3 passages before they were maintained in a cell medium containing selective antibiotics (blasticidin 10 ug/mL, zeocin 300 ug/mL). HEK cells were seeded into a flat-bottom 96-well plate at a cell density following the manufacturer’s protocol (25000 cells in 180 ul per well) directly into Hek-Blue Detection medium. To determine the activation of TLR5 by the pool peptides, cells were stimulated 16h (37 °C, 5% CO2) with 20 uL of 1ug/mL of the pools G and R as well as increasing concentrations of the Ultrapure flagellin from Salmonella Typhimurium from InvivoGen (San Diego, USA). After 16h, the plate was read at 643 nm by Tecan spectrophotometer.
The expression of TLR5 was also measured in PBMCs from R and NR patients by means of flow cytometry. After thawing PBMCs as previously described, the staining was performed using Fixable Viability Stain BV510 (BD, Cat. No. 564406), HLADR BV605 (BD, Cat. No. 562845) and TLR5 APCR700 (Bio-techne, Cat. No. FAB6704N). Then, cells were fixed (BD, Cat. No. 554722) and acquired using a FACSCelesta BVYG equipped with FACSDiva software version 8.0.1.
Analysis of blood markers
White blood cell counts (% of total WBC) and soluble factor data (mean-centered and scaled by SD of pg/mL) were analyzed, making group comparison on the quantities by Wilcoxon rank sum test. For the soluble factor data, important features were determined by the mean decrease in accuracy computed from random forest regression using randomForest::randomForest91 with mtry=6 and ntree=1000. Regression was performed on data that was controlled for even patient and response representation across timepoints, selecting for unique patients in timepoints of 0, 1–4, 5–8, 9–11, and >11 months.
Bead-based multiplexed ELISA
Multiplexed ELISA on patients sera were performed on a Luminex 200 platform (Luminex Inc.,) using custom kits of pre-mixed antibody-coated beads (R&D System Inc., MN), which included the following analytes: CCL11_eotaxin, CCL13_MCP4, CCL2_MCP1, CCL22_MDC, CCL26_EOTAXIN3, CCL5_RANTES, CD25_IL2Ra, CX3CL1_FRACTALKINE, CXCL1_GROa, CXCL10_IP10, CXCL2, CXCL6_GCP2, CXCL9, EGF, IFNγ, GMCSF, HGF, IL10, IL1ra_IL1F3, IL7, IL8_CXCL8, TRAIL, VEGFA, IL1b_IL1F2, IL5, IL6, IL-17F, IL23, TNFα, PDGFBB, CCL3_MIP1a, CCL4_MIP1b, FGFbasic_FGF2, GCSF, IL1a_IL1F1, IL2, IL21, IL-12 p70, IL13, IL15, IL-17/IL17A. The assay was performed based on manufacturer recommendations. Briefly, 50 μl of samples together with kit standards were added to each well in duplicate and incubated with the diluted Microparticle Cocktail at 4 °C, ON, on a shaker at 850 rpm. Unbound soluble molecules were removed by washing the plate. The Biotin-Antibody Cocktail specific to the analytes of interest was added to each well for 1 h at RT. After washing again, the Streptavidin-Phycoerythrin conjugated was added for 30 min at RT. After the final washing steps, the microparticles are resuspended in kit buffer and read on a Luminex 200 platform. The outputs (pg/mL) were visualized and statistically analyzed in R upon centering and scaling using the scale function in R (SD from mean pg/mL). Data were visualized together with sample annotations using the ComplexHeatmap package.84 Group comparisons were visualized as boxplots using the ggpubr package and statistically analyzed by applying the non-parametric Wilcoxon signed-rank test on the values, where significance is set at p-value <0.05. Soluble molecule data were associated with other experimental data by Spearman correlation and visualized using the corrplot package.
Cross-data feature association
To make associations between metagenomic features and systemic data, Fisher’s exact test was used. Binary states of metagenomic features (present: >0, absent: 0 abundance) and systemic features (high: >median; low: ≤median) were transformed into a contingency table, with the odds ratio computed by Fisher’s exact test.
For cross-data correlations, Spearman r coefficients were computed with rstatix::cor_test.77
Cross-cohort feature set enrichment analysis
Enrichment analysis was performed by Fisher’s exact test.92 Briefly, contingency tables were computed; binary categories for feature states were generated, by prevalence (prevalent: |OR|>1; not-prevalent: |OR|<1) for the taxonomic data and abundance (abundant: | fold-change|>1.1; not-abundant: |fold-change|<1) for the flagellin abundance data, and presence of the term in the query feature set (present: TRUE; absent: FALSE). Enrichment magnitude was determined by Fisher’s exact test OR; unadjusted p-values were reported (*p<0.05).
QUANTIFICATION AND STATISTICAL ANALYSIS
Core diversity from rarefied 16S data
All downstream analyses were performed in R, after exporting the taxonomy table, ASV counts table, phylogenetic tree, and metadata in R and converting into a phyloseq object. Alpha and beta diversity analyses were performed using the vegan package,75 using counts rarefied by the lowest sequence depth within a given dataset. For alpha-diversity analysis by standardized Faith’s phylogenetic diversity, the ses.pd function from the picante package was used.76 For computing higher level taxa ratios, raw counts were first aggregated to that level before rarefaction. Group differences based on alpha-diversity and counts ratio were computed by Wilcoxon rank sum test93; beta-diversity differences were computed from the distance matrices by PERMANOVA,88 checking for balance in dispersion by PERMDISP.75 All reported significant p-values by PERMANOVA were checked to have non-significant dispersion.
Batch correction
For batch correction, raw counts tables were first transformed by centered-log-ratio (CLR) following the mixOmics workflow for pre-processing microbiota data.94 Briefly, an offset of 1 was added to the raw counts, low-frequency OTUs with an overall abundance of <0.01% were filtered out, and outlier samples with library size greater than 2×105 were removed. Batch effect correction was applied using COMBAT with non-parametric setting.78 After computing the euclidean distance on the transformed abundance (i.e., Aitchison) using the stats::dist function, the distances were ordinated by PCoA using the ape::pcoa function. Group differences were tested on the distances by PERMANOVA while checking for balance in dispersion by PERMDISP.
Distance-based comparisons
For handling distance data the usedist package79 was used: dist_subset for subsetting distances, dist_get for retrieving specific group-wise distances, dist_groups for sorting within- and between-group distances.
Within-patient gut distance
To normalize for patient variability in the longitudinal analysis of 16S data, within-patient gut distance was used as measure, computing the Euclidean distance between a baseline and a timepoint sample from taxonomic abundance data (CLR-transformed and batch-corrected), and doing boxplot group comparisons on this distance, using non-parametric Wilcoxon rank sum as statistical test.
Between-patient gut distance
As measure of gut similarity, patients were ranked by between-patient gut distance: first, the Euclidean distance from taxonomic abundance data (CLR-transformed and batch-corrected) was computed, then, distances between-patient distances was retrieved, extracting between-group distances after using patient ID metadata in the dist_groups function from usedist.
Stable taxa analysis from Shotgun data
Within CR and nCR groups, prevalent taxa were identified as taxa having a non-zero abundance in > 80% of samples. This was determined within each timepoint group, 0, 2–7, and 8–13 months. Cut-off at 80% was determined as the minimum prevalence at which the median differential prevalence score is significantly greater than that at the starting cut-off of 40% prevalence (i.e., not highly prevalent) (Figure S4C). To account for having two studies in the analysis, prevalent taxa identification was performed as follows: first, putting together both studies, and second, subsetting only for INT–FGP samples. The union of taxa identified within-group with each dataset was taken as the prevalent taxa set for that group.
To utilize longitudinality of our data, prevalent taxa were further filtered for longitudinal consistency. “Stable” taxa were thus determined as prevalent taxa detected in all three timepoint groups for CR (0, 2–7, and 8–13 months), and in at least two timepoint groups nCR (0 and 2–7 or 0 and 8–13 months).
Taxonomic differential ranking
To determine significantly differentially abundant taxa, Wilcoxon rank sum test was performed on taxonomic abundance data from MetaPhlAn (relative abundance), with p-value adjustment by FDR. To determine the group with which a feature was associated, the corresponding log2 fold-change was also computed, by adding an offset of 0.01 to the abundances, getting the mean relative abundance of each feature across samples within a group, and, using the foldchange package, getting the fold-change (gtools)80 of the group means and converting it to log-ratio (foldchange2logratio).
Differential prevalence was performed as follows. First, from the taxonomy counts table, abundances > 0 were all converted to 1 to signify presence. A contingency table was built by counting the presences and absences at a binary category being tested, after which Fisher’s exact test92 was performed on the table. P-adj were used to determine significantly differentially prevalent taxa, and the calculated odds ratio (OR) for each feature was used to determine direction of association.
Differential ranking was performed on the hits by sorting the features by the −log10(p-adj) of each feature, determining the group of association by the sign of either the log2 fold-change (for differential abundance) or Fisher’s OR (for differential prevalence). The hits within a group were percentile-ranked using dplyr::ntile81 with n=100, and the hits that fall in the 75th percentile or more were considered as top ranking taxa. Differential abundance and prevalence indices of stable taxa were measured by taking the size of stable taxa and dividing against the size of top-ranking hits.
Additive log-ratio
To account for compositionality of taxonomic data, group comparisons on metagenomic taxonomy abundance data were performed by first subsetting the abundance table (relative abundance) to the features of interest (e.g., features that were determined to define each group of comparison), sum-aggregating relative abundances across features, taking the ratio of the values between the two groups, and log-transforming the sum.68 For response group comparisons, values pertaining to the CR group were always used as numerators. An additive log-ratio approach was similarly applied to soluble factor quantities (pg/mL) for testing differential ability of top CR and nCR cytokines.
Differential abundance on Shotgun functional data
To determine significant terms associated with a group, differential abundance was performed using Maaslin2.82 First, gene family abundances from Humann were re-grouped from gene family terms (Uniprot90 IDs) to KEGG orthologs (KOs). Group-associated KOs were determined by converting abundances in RPK to CPM, linearly modeling log-transformed relative abundances against response status of samples at 0, 2–7, and 8–13 months. Significance for differential abundance was set at p-adj < 0.05.
Over-representation analysis of pathways
Next, over-representation analysis was performed to examine higher level pathways within the CR and nCR groups and pathways. For each group of comparison, KOs with |LM coefficient| > 1.5 were taken as input, filtering for those taxa associated with the given group by the sign of LM coefficient (eg, +LM values only for comparison group; –LM values only for reference group). Over-representation of pathways was tested using the enricher function from clusterProfiler in R67, using the pathway–KO mapping file from https://github.com/picrust/picrust2/blob/master/picrust2/default_files/pathway_mapfiles/KEGG_pathways_to_KO.tsv and KO–pathway mapping file from https://github.com/biobakery/humann/blob/master/humann/data/misc/map_kegg-pwy_name.txt.gz. Significantly over-represented pathways present across timepoints were defined as “stable”.
Flagellin-related analyses
To determine flagellin-related terms from the gene family abundance table, a list was generated from the UniProt website (https://www.uniprot.org/), searching for “(taxonomy_id:2) AND flagellin” within the UniRef database and matching for UniRef90 hits as was used for the gene family data. With this list, we retrieve 1,563 features present in our dataset and 4,241 features in the multi-cohort baseline melanoma data.
To determine Lachnospiraceae-related terms the bacterial flagellin list was subset to those whose Genus designation in the ‘Organism’ column falls in Lachnospiraceae (n=59). With this list, we retrieve 391 features present in the multi-cohort baseline melanoma data.
In our dataset, group comparisons for abundance were performed by Wilcoxon rank sum test, applying p-value adjustment with FDR and visualizing as boxplots by log10-transforming the abundances. To perform flagellin differential testing on the multi-cohort and our dataset, abundances were sum-aggregated before performing group comparisons by Wilcoxon rank sum test. Log2 fold-change was computed by getting the median of the sum-aggregate abundance across samples within a group, and, using the gtools package,80 getting the fold-change (foldchange) of the group medians and converting it to log-ratio (foldchange2logratio).
Candidate complete response flagellin markers
To shortlist response-related flagellin markers from our longitudinal data, log2 fold-change was computed by getting the median abundance of each flagellin across samples within CR and nCR, and, using the gtools package,80 getting the fold-change (fold-change) of the group medians and converting it to log-ratio (foldchange2logratio). The top 14 flagellin markers with the highest log2 fold-change were identified as candidate CR flagellin markers.
Statistical and analytical parameters
Statistical tests were set at a significance level of p-adj<0.05 unless otherwise stated. Multiple comparisons adjustment was performed by FDR. Statistics were computed using the rstatix package77; p-values were adjusted for multiple comparisons by FDR using stats::p.adjust; where random sampling was performed, a seed of 1 was used (base::set.seed).67
Plot visualizations
Plots were visualized with ggplot281 except for heatmaps and UpSet plots, which were generated using ComplexHeatmap,84and ridgeplots, which were generated with ggridges::geom_ridgeline.85 Statistics were annotated to figures with ggpubr::stat_pvalue_ manual.86 Figures were assembled in R using grid.arrange and arrangeGrob functions from gridExtra.87
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| MEL-A | Dako | M7196 | |
| PRAME | Abcam | Cat#Ab219650; RRID:AB_219650 | |
| CCR7 BV421 | BD Biosciences | Cat#150503 | |
| CD8 BV605 | BD Biosciences | Cat#564116; RRID:AB_2869551 | |
| CD25 BV786 | BD Biosciences | Cat#741035; RRID:AB_2740625 | |
| CD45RA PE | BD Biosciences | Cat#561883; RRID:AB_10895572 | |
| CD69 APC | BD Biosciences | Cat#555533; RRID: AB_398602 | |
| CD3 APCR700 | BD Biosciences | Cat#565119; RRID:AB_2744385 | |
| CXCR3 PerCP-Cy5.5 | BD Biosciences | Cat#560832; RRID:AB_2033945 | |
| IL17A FITC | Invitrogen | Cat#11-7179-42 | |
| IFNG PE-CF594 | BD Biosciences | Cat#562392; RRID:AB_11153859 | |
| TNFA APC-CY7 | BioLegend | Cat#502944; RRID:AB_2562870 | |
| HLADR BV605 | BD Biosciences | Cat#562845 | |
| TLR5 APCR700 | Bio-techne R&D Systems | Cat#FAB6704N; RRID:AB_3651878 | |
|
Biological Samples | |||
| Fecal and Blood Samples from Patients with Melanoma | Istituto Europeo di Oncologia; Istituto Nazionale Tumori IRCCS Fondazione G. Pascale | R845/18-IEO 889; 37/22 oss | |
|
Chemicals, Peptides, and Recombinant Proteins | |||
| Stool Sample Collection & Stabilization Kit | Canvax | Cat#SC0012 | |
| Bovine Serum Albumin | Seqens | Cas#1005-70 | |
| Goat Serum | Invitrogen | Cat#10000 C | |
| HRP-Secondary Antibody (Anti-Rabbit) | Agilent Dako | Cat#K4003; RRID:AB_2630375 | |
| Aminoethyl Carbazole (AEC) + High Sensitivity Substrate Chromogen | Agilent Dako | Cat#K3461 | |
| Hematoxylin | Sigma | Cat#MHS 16-500ML | |
| RPMI-1640 | Euroclone | Cat#ECM2001L | |
| DMEM | Euroclone | Cat#ECM0103L | |
| FBS | Euroclone | Cat#ECS0165L | |
| Hepes | Sigma | Cat#H0087 | |
| Sodium Pyruvate | Euroclone | Cat#ECM0742D | |
| Pen/Strep | Euroclone | Cat#ECB3001D | |
| Glutamine | Euroclone | Cat#LOBE17605E | |
| Normocin | InvivoGen | Cat#ant-nr | |
| Recombinant Human IL-2 | Novartis | Cat#CLB-P-476-800-13980 IT | |
| Anti-CD28/49d | BD Biosciences | Cat#347690; RRID:AB_647457 | |
| Golgi Plug | BD Biosciences | Cat#555029 | |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat#554714 | |
| Cytofix/Cytoperm | BD Biosciences | Cat#554722 | |
| Fixable Viability Stain BV510 | BD Biosciences | Cat#564406 | |
| Purified Custom Peptides | GenScript (See Table S7) | NA | |
| UltraPure Flagellin from Salmonella typhimurium | InvivoGen | Cat#TLR-STFLA | |
|
Critical Commercial Assays | |||
| DNeasy PowerSoil Pro Kit | Qiagen | Cat#47016 | |
| MiSeq Reagent Kit V2 | Illumina | Cat#MS-102-2003 | |
| Nextera XT Index Kit V2 Set A | Illumina | Cat#FC-131-2001 | |
| Qubit dsDNA HS Assay Kit | Thermofisher | Cat#Q32851 | |
| Kapa HiFi HotStart ReadyMix | Fisher Scientific | Cat#50-196-5299 | |
| Human Magnetic Luminex Screening Assay | Bio-techne | Cat#LXSAHM-23 | |
|
Deposited Data | |||
| Raw 16S and/or Shotgun data | This paper; Baruch et al.9; Davar et al.10; Routy et al.5; Bjork et al.14 | PRJEB61942; PRJNA678737; PRJNA672867; PRJNA928744; PRJEB70966, PRJEB43119 | |
| Analyzed Shotgun data | Lee et al.8; Bjork et al.14 | https://bioconductor.org/packages/curatedMetagenomicData/ | |
| Source code/scripts to analyse the data | This paper; Biobakery63 |
https://github.com/ADGM/melanoma-longitudinal-paper
https://github.com/SegataLab/preprocessing |
|
| SILVA 128 database; SILVA 132 database | Quast et al.64 |
https://www.arb-silva.de/fileadmin/silva_databases/qiime/Silva_128_release.tgz; https://www.arb-silva.de/fileadmin/silva_databases/qiime/Silva_132_release.zip |
|
| CHOCOPhlAnSGB v202103 database | Blanco-Míguez et al.65 | =https://github.com/biobakery/humann?tab=readme-ov-file#download-the-chocophlan-database | |
| UniRef90 database | Suzek et al.66 | https://www.uniprot.org/ | |
| Pathway–KO mapping file | PICRUSt2 | https://github.com/picrust/picrust2/blob/master/picrust2/default_files/pathway_mapfiles/KEGG_pathways_to_KO.tsv | |
| KO–pathway mapping file | Beghini et al.63 | https://github.com/biobakery/humann/blob/master/humann/data/misc/map_kegg-pwy_name.txt.gz | |
|
Experimental models: Cell lines | |||
| HEK-Blue hTLR5 | Invivogen | Cat#hkb-htlr5; RRID:CVCL_IM83 | |
|
Oligonucleotides | |||
| 16S Amplicon PCR Forward Primer TCGTCGGCAGCGTCAGATGTGTATA AGAGACAGCCTACGGGNGGCWGCAG |
Illumina | NA | |
| 16S Amplicon PCR Reverse Primer GTCTCGTGGGCTCGGAGATGTGTA TAAGAGACAGGACTACHVGGGTATCTAATCC |
Illumina | NA | |
|
Software and algorithms | |||
| R version v4.1.1 (2021-08-10) | R Core Team67 | https://www.r-project.org | |
| RStudio v2022.07.2+576 | RStudio | https://posit.co | |
| Inkscape v1.2.2 | Inkscape Developers | https://inkscape.org/ | |
| BioRender | BioRender | https://www.biorender.com/ | |
| qiime2-2018.11; qiime2-2019.7 | Bolyen et al.68 | https://qiime2.org/ | |
| qiime2-2018.11 DADA2 plug-in | Callahan et al.69 | https://docs.qiime2.org/2018.11/plugins/available/dada2/index.html | |
| qiime2-2019.7 SEPP fragment insertion plug-in; qiime2-2019.7 naive Bayes feature classifier plug-in |
Janssen et al.70; Bokulich et al.71 | https://docs.qiime2.org/2019.7/plugins/available/fragment-insertion/sepp/; https://docs.qiime2.org/2019.7/plugins/available/feature-classifier/fit-classifier-naive-bayes/ | |
| Bowtie 2 | Langmead and Salzberg72 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml | |
| MetaPhlAn v4.0.3 | Blanco-Míguez et al.65 | https://github.com/biobakery/MetaPhlAn | |
| HUMAnN v3.6 | Beghini et al.63 | https://github.com/biobakery/humann | |
| NetMHCstabpan v4.1 | Rasmussen et al.73 | https://services.healthtech.dtu.dk/services/NetMHCpan-4.1/ | |
| Molsoft Mol Browser v3.8-7d | Molsoft | https://www.molsoft.com/icm_browser.html | |
| phyloseq v1.36.0 | McMurdie et al.74 | https://joey711.github.io/phyloseq/ | |
| vegan v2.6-8 | Oksanen et al.75 | https://github.com/vegandevs/vegan | |
| picante v1.8.2 | Kembel76 | https://github.com/skembel/picante | |
| rstatix v0.7.2 | Kassambara77 | https://github.com/kassambara/rstatix | |
| sva v3.40.0 | Johnson et al.78 | https://bioconductor.org/packages/release/bioc/html/sva.html | |
| usedist v0.4.0 | Bittinger79 | https://github.com/kylebittinger/usedist | |
| gtools v3.9.5 | Warnes et al.80 | https://cran.r-project.org/web/packages/gtools/index.html | |
| dplyr v1.1.4 | Wickham et al.81 | https://dplyr.tidyverse.org/ | |
| Maaslin2 v1.7.3 | Mallick et al.82 | https://github.com/biobakery/Maaslin2 | |
| clusterProfiler v4.0.5 | Wu et al.83 | https://guangchuangyu.github.io/software/clusterProfiler/ | |
| ggplot2 v3.5.1 | Wickham et al. 81 | https://ggplot2.tidyverse.org/ | |
| ComplexHeatmap v2.8.0 | Gu84 | https://github.com/jokergoo/ComplexHeatmap | |
| ggridges v0.5.6 | Aldahmani and Zoubeidi85 | https://wilkelab.org/ggridges/ | |
| ggpubr v0.6.0 | Kassambara86 | https://github.com/kassambara/ggpubr/releases | |
| gridExtra v2.3 | Auguie87 | https://CRAN.R-project.org/package=gridExtra | |
Highlights.
Microbiome dynamic during immunotherapy correlates with clinical response in melanoma
Stable microbial functions carried by responders hold cross-cohort prognostic value
MHC class I peptides from stable flagellin-related genes mimic tumor-associated antigens
They induce reactivity on PBMC and TIL from responders and improve antitumor immunity
ACKNOWLEDGMENTS
We thank patients involved in the study and their families and nurses, the IEO biobank (B4Med), the Genomic Unit, Cell culture Unit, and the Flow Cytometry Unit at IEO for their technical assistance. L.N. would like to thank TL2 for their unconditional support and dedicates this work to his parents. C.C. is a fellow of Fondazione Umberto Veronesi. L.N. is supported by Worldwide Cancer Research (WWCR 22-0402) and Associazione Italiana per la Ricerca contro il Cancro (AIRC IG 26406). T.M. is supported by AIRC StartUp grant 21474. X.X. is supported by NIH grants CA261608 and 258113. M.C.A. is supported by a Monash Partners-Equity Trustees Cancer Program grant. This work was supported in part by the Italian Ministry of Health with Ricerca Corrente and 5×1000 funds.
Footnotes
DECLARATION OF INTERESTS
L.N., T.M., P.A.A., and A.D.G.M. are named co-inventors on a patent application relating to this work (EP n. 24164184.4 “Flagellin-related peptides and uses”). M.C.A. reports advisory board participation, honoraria, or research funds to institutions from MSD Australia, BMS Australia, and Pierre Fabre Australia and is a named co-inventor on a patent application relating to Methods and compositions for treating cancer (WO2020106983A1), all unrelated to this work. P.A.A. reports grants or contracts from Bristol Myers Squibb, Roche-Genentech, Pfizer, and Sanofi; consulting fees from Bristol Myers Squibb, Roche-Genentech, Merck Sharp & Dohme, Novartis, Merck Serono, Pierre Fabre, Sun Pharma, Sanofi, Sandoz, Italfarmaco, Nektar, Pfizer, Lunaphore, Medicenna, Bio-Al Health, ValoTx, Replimmune, and Bayer; support for attending meetings and/or travel from Pfizer, Bio-Al Health, and Replimmune; and participating on a data safety monitoring board or advisory board for Bristol Myers Squibb, Roche-Genentech, Merck Sharp & Dohme, Novartis, AstraZeneca, Boehringer Ingelheim, Eisai, Regeneron, Daiichi Sankyo, Oncosec, Nouscom, Seagen, iTeos, and Erasca. L.N. reports research funds from BMS Europe unrelated to this work.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.chom.2024.10.006.
Data and code availability
All sequencing data have been deposited on the ENA archive and are accessible under the accession number ENA Project: PRJEB61942. Metadata will be available as well upon request, subject to approval by the Ethical Committees of IEO and INT-FGP.
All in-house computer codes are available on https://github.com/ADGM/melanoma-longitudinal-paper. Remaining data are available within the article, STAR Methods, supplemental information, or source data file.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
REFERENCES
- 1.Luke JJ, Flaherty KT, Ribas A, and Long GV (2017). Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol 14, 463–482. 10.1038/nrclinonc.2017.43. [DOI] [PubMed] [Google Scholar]
- 2.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, et al. (2015). Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med 372, 320–330. 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 3.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. (2018). Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 359, 97–103. 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, Gajewski TF, and Gajewski TF (2018). The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108. 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, et al. (2018). Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 359, 91–97. 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 6.Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, and Koh AY (2017). Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia 19, 848–855. 10.1016/j.neo.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCulloch JA, Davar D, Rodrigues RR, Badger JH, Fang JR, Cole AM, Balaji AK, Vetizou M, Prescott SM, Fernandes MR, et al. (2022). Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat. Med 28, 545–556. 10.1038/s41591-022-01698-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee KA, Thomas AM, Bolte LA, Björk JR, de Ruijter LK, Armanini F, Asnicar F, Blanco-Miguez A, Board R, Calbet-Llopart N, et al. (2022). Cross-cohort gut microbiome associations with immune check-point inhibitor response in advanced melanoma. Nat. Med 28, 535–544. 10.1038/s41591-022-01695-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, Adler K, Dick-Necula D, Raskin S, Bloch N, et al. (2021). Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 371, 602–609. 10.1126/science.abb5920. [DOI] [PubMed] [Google Scholar]
- 10.Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, Deblasio RN, Menna C, Ding Q, Pagliano O, et al. (2021). Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science 371, 595–602. 10.1126/science.abf3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Routy B, Lenehan JG, Miller WH Jr., Jamal R, Messaoudene M, Daisley BA, Hes C, Al KF, Martinez-Gili L, Punčochář M, et al. (2023). Fecal microbiota transplantation plus anti-PD-1 immunotherapy in advanced melanoma: a phase I trial. Nat. Med 29, 2121–2132. 10.1038/s41591-023-02453-x. [DOI] [PubMed] [Google Scholar]
- 12.Choi Y, Lichterman JN, Coughlin LA, Poulides N, Li W, Del Valle P, Palmer SN, Gan S, Kim J, Zhan X, et al. (2023). Immune checkpoint blockade induces gut microbiota translocation that augments extraintestinal antitumor immunity. Sci. Immunol 8, eabo2003. 10.1126/sciimmunol.abo2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bender MJ, McPherson AC, Phelps CM, Pandey SP, Laughlin CR, Shapira JH, Medina Sanchez L, Rana M, Richie TG, Mims TS, et al. (2023). Dietary tryptophan metabolite released by intratumoral Lactobacillus reuteri facilitates immune checkpoint inhibitor treatment. Cell 186, 1846–1862.e26. 10.1016/j.cell.2023.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Björk JR, Bolte LA, Maltez Thomas A, Lee KA, Rossi N, Wind T, Smit LM, Armanini F, Asnicar F, Blanco-Miguez A, et al. (2024). Longitudinal gut microbiome changes in immune checkpoint blockade-treated advanced melanoma. Nat. Med 30, 785–796. 10.1038/s41591-024-02803-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies MA (2020). Is it safe to stop anti–PD-1 immunotherapy in patients with metastatic melanoma who achieve a complete response? J. Clin. Oncol 38, 1645–1647. 10.1200/JCO.20.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Betof Warner A, Palmer JS, Shoushtari AN, Goldman DA, Panageas KS, Hayes SA, Bajwa R, Momtaz P, Callahan MK, Wolchok JD, et al. (2020). Long-term outcomes and responses to retreatment in patients with melanoma treated with PD-1 blockade. J. Clin. Oncol 38, 1655–1663. 10.1200/JCO.19.01464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simpson RC, Shanahan ER, Batten M, Reijers ILM, Read M, Silva IP, Versluis JM, Ribeiro R, Angelatos AS, Tan J, et al. (2022). Diet-driven microbial ecology underpins associations between cancer immunotherapy outcomes and the gut microbiome. Nat. Med 28, 2344–2352. 10.1038/s41591-022-01965-2. [DOI] [PubMed] [Google Scholar]
- 18.Spencer CN, McQuade JL, Gopalakrishnan V, McCulloch JA, Vetizou M, Cogdill AP, Khan MAW, Zhang X, White MG, Peterson CB, et al. (2021). Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science 374, 1632–1640. 10.1126/science.aaz7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrews MC, Reuben A, Gopalakrishnan V, and Wargo JA (2018). Concepts collide: genomic, immune, and microbial influences on the tumor microenvironment and response to cancer therapy. Front. Immunol 9, 946. 10.3389/fimmu.2018.00946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrucci PF, Ascierto PA, Pigozzo J, Del Vecchio M, Maio M, Antonini Cappellini GC, Guidoboni M, Queirolo P, Savoia P, Mandalà M, et al. (2016). Baseline neutrophils and derived neutrophil-to-lymphocyte ratio: prognostic relevance in metastatic melanoma patients receiving ipilimumab. Ann. Oncol 27, 732–738. 10.1093/annonc/mdw016. [DOI] [PubMed] [Google Scholar]
- 21.Capone M, Giannarelli D, Mallardo D, Madonna G, Festino L, Grimaldi AM, Vanella V, Simeone E, Paone M, Palmieri G, et al. (2018). Baseline neutrophil-to-lymphocyte ratio (NLR) and derived NLR could predict overall survival in patients with advanced melanoma treated with nivolumab. J. Immunother. Cancer 6, 74. 10.1186/s40425-018-0383-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wind TT, Gacesa R, Vich Vila A, de Haan JJ, Jalving M, Weersma RK, and Hospers GAP (2020). Gut microbial species and metabolic pathways associated with response to treatment with immune checkpoint inhibitors in metastatic melanoma. Melanoma Res 30, 235–246. 10.1097/CMR.0000000000000656. [DOI] [PubMed] [Google Scholar]
- 23.Hajam IA, Dar PA, Shahnawaz I, Jaume JC, and Lee JH (2017). Bacterial flagellin—a potent immunomodulatory agent. Exp. Mol. Med 49, e373. 10.1038/emm.2017.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clasen SJ, Bell MEW, Borbón A, Lee DH, Henseler ZM, de la Cuesta-Zuluaga J, Parys K, Zou J, Wang Y, Altmannova V, et al. (2023). Silent recognition of flagellins from human gut commensal bacteria by Toll-like receptor 5. Sci. Immunol 8, eabq7001. 10.1126/sciimmunol.abq7001. [DOI] [PubMed] [Google Scholar]
- 25.Davar D, and Zarour HM (2022). Facts and hopes for gut microbiota interventions in cancer immunotherapy. Clin. Cancer Res 28, 4370–4384. 10.1158/1078-0432.CCR-21-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ragone C, Manolio C, Mauriello A, Cavalluzzo B, Buonaguro FM, Tornesello ML, Tagliamonte M, Buonaguro L, and Buonaguro L (2022). Molecular mimicry between tumor associated antigens and microbiota-derived epitopes. J. Transl. Med 20, 316. 10.1186/s12967-022-03512-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lezcano C, Jungbluth AA, Nehal KS, Hollmann TJ, and Busam KJ (2018). PRAME expression in melanocytic tumors. Am. J. Surg. Pathol 42, 1456–1465. 10.1097/PAS.0000000000001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, Miki T, and Rosenberg SA (1994). Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc. Natl. Acad. Sci. USA 91, 3515–3519. 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coulie PG, Brichard V, Van Pel A, Wölfel T, Schneider J, Traversari C, Mattei S, De Plaen E, Lurquin C, Szikora JP, et al. (1994). A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J. Exp. Med 180, 35–42. 10.1084/jem.180.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin S, Jiang T, Yu Y, Tang H, Lu S, Peng Z, and Fan J (2015). Secernin-1 contributes to colon cancer progression through enhancing matrix metalloproteinase-2/9 exocytosis. Dis. Markers 2015, 230703. 10.1155/2015/230703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schnaeker EM, Ossig R, Ludwig T, Dreier R, Oberleithner H, Wilhelmi M, and Schneider SW (2004). Microtubule-dependent matrix metalloproteinase-2/matrix metalloproteinase-9 exocytosis: prerequisite in human melanoma cell invasion. Cancer Res 64, 8924–8931. 10.1158/0008-5472.CAN-04-0324. [DOI] [PubMed] [Google Scholar]
- 32.Militaru IV, Rus AA, Munteanu CVA, Manica G, and Petrescu SM (2022). New panel of biomarkers to discriminate between amelanotic and melanotic metastatic melanoma. Front. Oncol 12, 1061832. 10.3389/fonc.2022.1061832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lauté-Caly DL, Raftis EJ, Cowie P, Hennessy E, Holt A, Panzica DA, Sparre C, Minter B, Stroobach E, and Mulder IE (2019). The flagellin of candidate live biotherapeutic Enterococcus gallinarum MRx0518 is a potent immunostimulant. Sci. Rep 9, 801. 10.1038/s41598-018-36926-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng JH, Nguyen VH, Jiang SN, Park SH, Tan W, Hong SH, Shin MG, Chung IJ, Hong Y, Bom HS, et al. (2017). Two-step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Sci. Transl. Med 9, eaak9537. 10.1126/scitranslmed.aak9537. [DOI] [PubMed] [Google Scholar]
- 35.Zeng Y, Shi Q, Liu X, Tang H, Lu B, Zhou Q, Xu Y, Chen M, Zhao J, Li Y, et al. (2023). Dynamic gut microbiota changes in patients with advanced Malignancies experiencing secondary resistance to immune checkpoint inhibitors and immune-related adverse events. Front. Oncol 13, 1144534. 10.3389/fonc.2023.1144534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montalban-Arques A, Katkeviciute E, Busenhart P, Bircher A, Wirbel J, Zeller G, Morsy Y, Borsig L, Glaus Garzon JF, Müller A, et al. (2021). Commensal Clostridiales strains mediate effective anti-cancer immune response against solid tumors. Cell Host Microbe 29, 1573–1588.e7. 10.1016/j.chom.2021.08.001. [DOI] [PubMed] [Google Scholar]
- 37.Ianiro G, Punčochář M, Karcher N, Porcari S, Armanini F, Asnicar F, Beghini F, Blanco-Míguez A, Cumbo F, Manghi P, et al. (2022). Variability of strain engraftment and predictability of microbiome composition after fecal microbiota transplantation across different diseases. Nat. Med 28, 1913–1923. 10.1038/s41591-022-01964-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kazemian N, Ramezankhani M, Sehgal A, Khalid FM, Kalkhoran AHZ, Narayan A, Wong GKS, Kao D, and Pakpour S (2020). The trans-kingdom battle between donor and recipient gut microbiome influences fecal microbiota transplantation outcome. Sci. Rep 10, 18349. 10.1038/s41598-020-75162-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trinchieri G (2003). Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol 3, 133–146. 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 40.Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, et al. (2018). Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity 49, 1148–1161.e7. 10.1016/j.immuni.2018.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lim SY, Lee JH, Gide TN, Menzies AM, Guminski A, Carlino MS, Breen EJ, Yang JYH, Ghazanfar S, Kefford RF, et al. (2019). Circulating cytokines predict immune-related toxicity in melanoma patients receiving anti-PD-1–based immunotherapy. Clin. Cancer Res 25, 1557–1563. 10.1158/1078-0432.CCR-18-2795. [DOI] [PubMed] [Google Scholar]
- 42.Yin P, Liu X, Mansfield AS, Harrington SM, Li Y, Yan Y, and Dong H (2016). CpG-induced antitumor immunity requires IL-12 in expansion of effector cells and down-regulation of PD-1. Oncotarget 7, 70223–70231. 10.18632/oncotarget.11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cerqueira FM, Photenhauer AL, Pollet RM, Brown HA, and Koropatkin NM (2020). Starch digestion by gut bacteria: crowdsourcing for carbs. Trends Microbiol 28, 95–108. 10.1016/j.tim.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 44.Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJ, Pearce EL, Oltz EM, and Stappenbeck TS (2016). The colonic crypt protects stem cells from microbiota-derived metabolites. Cell 165, 1708–1720. 10.1016/j.cell.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson K, Glover L, Kominsky D, Magnuson A, et al. (2015). Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17, 662–671. 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zitvogel L, Daillère R, Roberti MP, Routy B, and Kroemer G (2017). Anticancer effects of the microbiome and its products. Nat. Rev. Microbiol 15, 465–478. 10.1038/nrmicro.2017.44. [DOI] [PubMed] [Google Scholar]
- 47.Rooks MG, and Garrett WS (2016). Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol 16, 341–352. 10.1038/nri.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He Y, Fu L, Li Y, Wang W, Gong M, Zhang J, Dong X, Huang J, Wang Q, Mackay CR, et al. (2021). Gut microbial metabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8+ T cell immunity. Cell Metab 33, 988–1000.e7. 10.1016/j.cmet.2021.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Luu M, Weigand K, Wedi F, Breidenbend C, Leister H, Pautz S, Adhikary T, Visekruna A, and Visekruna A (2018). Regulation of the effector function of CD8+ T cells by gut microbiota-derived metabolite butyrate. Sci. Rep 8, 14430. 10.1038/s41598-018-32860-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luu M, Riester Z, Baldrich A, Reichardt N, Yuille S, Busetti A, Klein M, Wempe A, Leister H, Raifer H, et al. (2021). Microbial short-chain fatty acids modulate CD8+ T cell responses and improve adoptive immunotherapy for cancer. Nat. Commun 12, 4077. 10.1038/s41467-021-24331-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coutzac C, Jouniaux JM, Paci A, Schmidt J, Mallardo D, Seck A, Asvatourian V, Cassard L, Saulnier P, Lacroix L, et al. (2020). Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat. Commun 11, 2168. 10.1038/s41467-020-16079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nomura M, Nagatomo R, Doi K, Shimizu J, Baba K, Saito T, Matsumoto S, Inoue K, Muto M, Inoue K, and Muto M (2020). Association of short-chain fatty acids in the gut microbiome with clinical response to treatment with nivolumab or pembrolizumab in patients with solid cancer tumors. JAMA Netw. Open 3, e202895. 10.1001/jamanetworkopen.2020.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hayase E, and Jenq RR (2021). Role of the intestinal microbiome and microbial-derived metabolites in immune checkpoint blockade immunotherapy of cancer. Genome Med 13, 107. 10.1186/s13073-021-00923-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Danne C, and Sokol H (2021). Butyrate, a new microbiota-dependent player in CD8+ T cells immunity and cancer therapy? Cell Rep. Med 2, 100328. 10.1016/j.xcrm.2021.100328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 56.Gunjur A, Shao Y, Rozday T, Klein O, Mu A, Haak BW, Markman B, Kee D, Carlino MS, Underhill C, et al. (2024). A gut microbial signature for combination immune checkpoint blockade across cancer types. Nat. Med 30, 797–809. 10.1038/s41591-024-02823-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen YE, Bousbaine D, Veinbachs A, Atabakhsh K, Dimas A, Yu VK, Zhao A, Enright NJ, Nagashima K, Belkaid Y, et al. (2023). Engineered skin bacteria induce antitumor T cell responses against melanoma. Science 380, 203–210. 10.1126/science.abp9563. [DOI] [PubMed] [Google Scholar]
- 58.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413. 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, El Dika IH, Segal N, Shcherba M, Sugarman R, et al. (2022). PD-1 blockade in mismatch repair–deficient, locally advanced rectal cancer. N. Engl. J. Med 386, 2363–2376. 10.1056/NEJMoa2201445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Diehl A, Yarchoan M, Hopkins A, Jaffee E, and Grossman SA (2017). Relationships between lymphocyte counts and treatment-related toxicities and clinical responses in patients with solid tumors treated with PD-1 checkpoint inhibitors. Oncotarget 8, 114268–114280. 10.18632/oncotarget.23217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee YJ, Park YS, Lee HW, Park TY, Lee JK, and Heo EY (2022). Peripheral lymphocyte count as a surrogate marker of immune checkpoint inhibitor therapy outcomes in patients with non-small-cell lung cancer. Sci. Rep 12, 626. 10.1038/s41598-021-04630-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weber JS, Carlino MS, Khattak A, Meniawy T, Ansstas G, Taylor MH, Kim KB, McKean M, Long GV, Sullivan RJ, et al. (2024). Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): a randomised, phase 2b study. Lancet 403, 632–644. 10.1016/S0140-6736(23)02268-7. [DOI] [PubMed] [Google Scholar]
- 63.Beghini F, McIver LJ, Blanco-Míguez A, Dubois L, Asnicar F, Maharjan S, Mailyan A, Manghi P, Scholz M, Thomas AM, et al. (2021). Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 10, e65088. 10.7554/eLife.65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glöckner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–D596. 10.1093/NAR/GKS1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blanco-Míguez A, Beghini F, Cumbo F, McIver LJ, Thompson KN, Zolfo M, Manghi P, Dubois L, Huang KD, Thomas AM, et al. (2023). Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol 41, 1633–1644. 10.1038/s41587-023-01688-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suzek BE, Wang Y, Huang H, McGarvey PB, and Wu CH; UniProt Consortium (2015). UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932. 10.1093/BIOINFORMATICS/BTU739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.R Core Team (2016). R A language and environment for statistical computing (R Foundation for Statistical Computing). https://www.scirp.org/reference/ReferencesPapers?ReferenceID=2010931. [Google Scholar]
- 68.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, AlGhalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol 37, 852–857. 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, and Holmes SP (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Janssen S, McDonald D, Gonzalez A, Navas-Molina JA, Jiang L, Xu ZZ, Winker K, Kado DM, Orwoll E, Manary M, et al. (2018). Phylogenetic placement of exact amplicon sequences improves associations with clinical information. mSystems 3, e00021–18. 10.1128/mSystems.00021-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bokulich NA, Dillon MR, Zhang Y, Rideout JR, Bolyen E, Li H, Albert PS, and Caporaso JG (2018). q2-longitudinal: longitudinal and paired-sample analyses of microbiome data. mSystems 3, e00219–18. 10.1128/mSystems.00219-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rasmussen M, Fenoy E, Harndahl M, Kristensen AB, Nielsen IK, Nielsen M, and Buus S (2016). Pan-specific prediction of peptide-MHC Class I complex stability; a correlate of T cell immunogenicity. J. Immunol 197, 1517–1524. 10.4049/JIMMUNOL.1600582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McMurdie PJ, and Holmes S (2013). phyloseq: an R package for Reproducible Interactive Analysis and Graphics of microbiome Census Data. PLoS One 8, e61217. 10.1371/JOURNAL.PONE.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oksanen J, Kindt R, Legendre P, O’Hara B, Simpson GL, Solymos P, Henry M, Stevens H, and Wagner H (2009). The vegan Package Title Community Ecology Package. https://github.com/vegandevs/vegan. [Google Scholar]
- 76.Kembel S (2010). An Introduction to the Picante Package. [Google Scholar]
- 77.Kassambara A (2023). Pipe-Friendly Framework for Basic Statistical Tests [R package rstatix version 0.7.2]. [Google Scholar]
- 78.Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. 10.1093/BIOSTATISTICS/KXJ037. [DOI] [PubMed] [Google Scholar]
- 79.Bittinger K (2020). Distance Matrix Utilities [R package usedist version 0.4.0].. [Google Scholar]
- 80.Warnes GR, Bolker B, Lumley T, and Warnes MGR (2015). Package ‘gtools’. R Package version, 3. [Google Scholar]
- 81.Wickham H, François R, Henry L, and Müller K (2020). Dplyr A grammar of data manipulation. R package version 0.8.4. https://www.scirp.org/reference/referencespapers?referenceid=2761682. [Google Scholar]
- 82.Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Nguyen LH, Tickle TL, Weingart G, Ren B, Schwager EH, et al. (2021). Multivariable association discovery in population-scale meta-omics studies. PLoS Comput Biol 17, e1009442. 10.1371/JOURNAL.PCBI.1009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Cambridge (Mass.)) 2, 100141. 10.1016/J.XINN.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gu Z (2022). Complex heatmap visualization. Imeta 1, e43. 10.1002/IMT2.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aldahmani S, and Zoubeidi T (2020). Graphical group ridge. J. Stat. Comput. Simul 90, 3422–3432. 10.1080/00949655.2020.1803320. [DOI] [Google Scholar]
- 86.Kassambara A (2023). “ggplot2” Based Publication Ready Plots [R package ggpubr version 0.6.0]. [Google Scholar]
- 87.Auguie B (2017). Miscellaneous Functions for “Grid” Graphics [R package gridExtra version 2.3]. [Google Scholar]
- 88.Anderson MJ (2017). Permutational Multivariate Analysis of Variance (PERMANOVA). Wiley StatsRef: Statistics Reference Online, 1–15. 10.1002/9781118445112.STAT07841. [DOI] [Google Scholar]
- 89.Garboczi DN, Hung DT, and Wiley DC (1992). HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc. Natl. Acad. Sci. USA 89, 3429–3433. 10.1073/PNAS.89.8.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bouvier M, and Wiley DC (1998). Structural characterization of a soluble and partially folded class I major histocompatibility heavy chain/b2m heterodimer. Nat. Struct. Biol 5, 377–384. 10.1038/NSB0598-377. [DOI] [PubMed] [Google Scholar]
- 91.Breiman L (2001). Random forests. Mach. Learn 45, 5–32. 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 92.Fisher RA (1992). Statistical Methods for Research Workers. In Breakthroughs in statistics: Methodology and distribution (Springer; New York: ), pp. 66–70. 10.1007/978-1-4612-4380-9_6. [DOI] [Google Scholar]
- 93.Wilcoxon F (1992). Individual Comparisons by Ranking Methods, pp. 196–202. 10.1007/978-1-4612-4380-9_16. [DOI] [PubMed] [Google Scholar]
- 94.Rohart F, Gautier B, Singh A, and Lě Cao KA (2017). mixOmics: an R package for ‘omics feature selection and multiple data integration. PLoS Comput Biol 13, e1005752. 10.1371/JOURNAL.PCBI.1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data have been deposited on the ENA archive and are accessible under the accession number ENA Project: PRJEB61942. Metadata will be available as well upon request, subject to approval by the Ethical Committees of IEO and INT-FGP.
All in-house computer codes are available on https://github.com/ADGM/melanoma-longitudinal-paper. Remaining data are available within the article, STAR Methods, supplemental information, or source data file.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.