

Abstract
Replicative errors, inefficient repair, and proximity to sites of reactive oxygen species production make mitochondrial DNA (mtDNA) susceptible to damage with time. We explore in vivo allotopic expression (re-engineering mitochondrial genes and expressing them from the nucleus) as an approach to rescue defects arising from mtDNA mutations. We used a mouse strain C57BL/6J(mtFVB) with a natural polymorphism (m.7778 G>T) in the mitochondrial ATP8 gene that encodes a protein subunit of the ATP synthase. We generated a transgenic mouse with an epitope-tagged recoded mitochondrial-targeted ATP8 gene expressed from the ROSA26 locus in the nucleus and used the C57BL/6J(mtFVB) strain to verify successful incorporation. The allotopically expressed ATP8 protein in transgenic mice was constitutively expressed across all tested tissues, successfully transported into the mitochondria, and incorporated into ATP synthase. The ATP synthase with transgene had similar activity to non-transgenic control, suggesting successful integration and function. Exogenous ATP8 protein had no negative impact on measured mitochondrial function, metabolism, or behavior. Successful allotopic expression of a mitochondrially encoded protein in vivo in a mammal is a step toward utilizing allotopic expression as a gene therapy in humans to repair physiological consequences of mtDNA defects that may accumulate in congenital mitochondrial diseases or with age.
Keywords: allotopic expression, mitochondrial DNA mutation, mtDNA, ATP8 gene, transgenic mouse, in vivo gene therapy, safe harbor expression
Graphical abstract
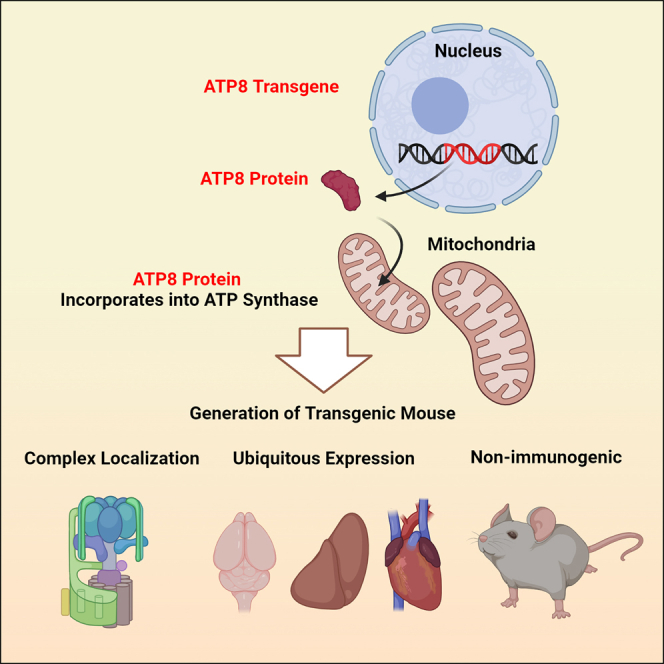
Boominathan and colleagues evaluated allotopic gene therapy for feasibility and safety in vivo. The authors created and characterized a transgenic animal model that expresses a single copy of the mitochondrial ATP8 gene from the nucleus. The model shows constitutive transgene expression across tissues over time with no deleterious functional or inflammatory response.
Introduction
Mitochondrial myopathies are a collective group of diseases predicted to affect nearly 1 in 200 individuals1 that arise from mutations in the nuclear DNA or the mitochondrial DNA (mtDNA) and adversely impact the functions of the organelle. The human mtDNA is 16.5 kb and encodes 13 vital proteins in the respiratory chain. This circular DNA also encodes 22 tRNAs and 2 rRNAs essential to translating the organelle genome. Mutations in the 13 respiratory complex (oxidative phosphorylation [OXPHOS]) genes and their associated non-protein coding genes tend to be associated with severe consequences as they lead to disruption in the production of the core subunits of the electron transport chain (ETC), thus introducing instability into the subunit structure and causing loss of function.
Defects in OXPHOS can cause disease in any organ, but symptoms occur predominantly in neurological, cardiovascular, and skeletal muscle tissue,2 organs whose cells are postmitotic and that rely on high energy turnover for proper function. Leber hereditary optic neuropathy (LHON)3,4 Leigh syndrome,5 mitochondrial encephalopathy,5 chronic progressive external ophthalmoplegia,6 and Kearns-Sayre syndrome6,7 are examples of neurologic diseases linked to mtDNA mutation. Other mtDNA diseases include non-hypoxic lactic acidosis and Pearson marrow-pancreas syndrome.8 Somatic mutations in mtDNA are also implicated in human aging.9
Current therapeutic approaches to address pathologies of mtDNA mutations include using small molecules,10,11 cell and organelle replacement techniques,12,13,14 gene editing, and gene therapy strategies.15,16,17 Most of these approaches in the clinic are still palliative and cannot address the root cause of these diseases. Nonetheless, gene therapy using a recombinant adenovector for the ND4 gene in isolated compartments such as the eye has been implemented with apparent clinical benefit in LHON patients (ClinicalTrials.gov, identifiers NCT03293524 and NCT02064569).
One major challenge in the field is the need for appropriate in vivo models to evaluate the potential of such therapies. Many recent advances in nuclear gene editing strategies are still to be proven applicable in manipulating the mtDNA.18 Organelle DNA inheritance patterns and maintenance impose further challenges in generating in vivo models. The mtDNA is maternally inherited and often exhibits a state of heteroplasmy in disease states. Here, both wild-type and mutant forms of mtDNA co-exist within cells and lead to a disease phenotype when the mutant mtDNA copy number exceeds a critical threshold. Severe mutations that impair OXPHOS function are typically non-viable and are eliminated early during development. For example, transmitochondrial mice carrying two mutations (1) T6589C in COX1 and (2) 13885insC frameshift mutation in ND6, eliminated the more severe ND6 13885insC frameshift mutation within four generations.19 Similarly, efforts to generate in vivo mouse models for the 4.7 kb common deletion mutation were unsuccessful beyond the F3 generation.20 Researchers are therefore limited to studying naturally occurring variants with mild phenotypes, for example, such as those observed due to missense mutations in the ATP821 and ND6 genes.22 Alternatively, researchers have also explored strategies like the co-expression or over expression of mutant allotopic genes in addition to wild-type versions as in the case of ND423 and ATP6 transgenic mice23,24 to mimic disease conditions such as Leber heriditary optic neuropathy (LHON) and Neuropathy, ataxia, and retinitis pigmentosa (NARP). Mouse models describing compromised OXPHOS function in nuclear encoded mitochondrial subunits and other genes are reviewed elsewhere.25,26
Previously, using patient-derived cybrid cells null for the ATP8 protein, we showed that a codon-optimized allotopic gene construct for the human ATP8 gene rescued several biochemical and functional parameters upon stable integration into the nuclear genome.27 Here, we use the ATP8 polymorphism (m.7778 G>T) observed in C57BL/6J(mtFVB) mice to demonstrate gene therapy in vivo. In our hands, some of the reported behavior phenotypes associated with this polymorphism, such as anxiety, could not be recapitulated. We took advantage of the single-nucleotide polymorphism and the resulting amino acid change from aspartic acid to tyrosine at position 5 in the conserved N-terminal region of the protein to demonstrate the utility of a gene therapy product. Using the safe harbor expression approach,28 we placed a single copy of the codon-optimized mouse ATP8 gene in the mouse ROSA26 locus. We show that germline nuclear integration of a single copy of the allotopic mitochondrial ATP8 gene is constitutively expressed across tissues, over time, and is faithfully transmitted to the progeny for up to four generations.
Results
Generation of transgenic mouse expressing optimized ATP8 from a safe harbor locus in the nucleus
To assess the feasibility of using allotopic nuclear expression of ATP8 in vivo, we generated a transgenic mouse model that expresses an epitope-tagged and codon-optimized ATP8 mitochondrial gene (oATP8) under the CAG promoter from the nucleus. The oATP8 construct (ATP5G1MTS-oATP8-FLAG) contains a codon-optimized N-terminal mitochondrial targeting sequence (MTS) from nuclear-encoded ATP synthase subunit ATP5G1 to facilitate transgene localization29 and a C-terminal MYC and FLAG tag for immunodetection (Figure 1A). Using TARGATT homologous recombination technology,30 we inserted a single copy of the oATP8 construct via ΦC31 integrase and attPx3 docking sites into the mouse ROSA26 locus on chromosome 6 in the C57BL/6J(mtC57BL/6J) strain (Figure 1B).
Figure 1.

Generation of transgenic mouse expressing optimized ATP8 from a safe harbor locus in the nucleus
Schematic of the oATP8 construct (A) and diagram portraying the ɸC31-mediated recombination of the oATP8 construct into the mouse ROSA26 locus (NC_000072.7::113052993) (B). An overview of the breeding scheme used to generate the experimental groups (C).
To assess the efficacy of allotopic expression as a gene therapy in vivo, our aim was to test the ability of the exogenous oATP8 protein to compete with an existing endogenous ATP8 mutant protein. The FVB/NJmtFVB/NJ mouse (JAX strain no. 001800) strain is the only available model harboring a spontaneous single-nucleotide polymorphism in the mitochondrial ATP8 gene. The ATP8 gene in this mouse strain contains a transversion (m. 7778G-T) that results in an aspartic acid to tyrosine substitution in the N terminus of the ATP8 protein. This mutation does not produce a null model, as the mutant ATP8 protein is synthesized and incorporated into a fully assembled ATP synthase complex. We utilized a conplastic C57BL/6JmtFVB/NJ/IbraJ (JAX strain no. 010810; from here on C57BL/6J(mtFVB)) strain which contains the mitochondrial FVB/NJ polymorphism with a C57BL/6J nuclear genome, free from potential modifiers that allows for the assessment of the impact of the mitochondrial mutation in the absence of any confounding variation of the FVB/NJmtFVB/NJ nuclear genome.31
As mitochondria are maternally inherited, the conplastic model was developed by crossing male C57BL/6J(mtC57BL/6J) mice onto female FVB/NJmtFVB/NJ mice to produce the C57BL/6J(mtFVB) mouse that contains a C57BL/6J nuclear genome and an FVB/NJ mitochondrial genome.31 We verified that the C57BL/6J(mtFVB) strain harbors the FVB/NJ polymorphism using Sanger sequencing (Figure S1A). To generate the C57BL/6J(mtFVB) carrying the oATP8 nuclear transgene, we crossed female C57BL/6J(mtFVB) with male transgenic C57BL/6J(mtC57BL/6J) (Figure 1C) mice. All animals generated using the breeding scheme in Figure 1C were viable and fertile and born at the expected Mendelian ratio without apparent abnormalities in adult mice. About half the progeny, 48.2% ± 4.3% (SEM) and 49.8% ± 5.7% (SEM) in the C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB), respectively, were born transgenic. For the generation of all experimental groups, non-transgenic and oATP8 transgene littermates were used as controls (Figure 1C).
PCR analysis of tail DNA using specific primer sets corresponding to the regions spanning the ROSA26 locus and oATP8 transgene showed that successful recombination occurred in both the transgenic C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB) strains, but not in the littermate non-transgenic controls (Figures 2A and S1B). Sanger sequencing on purified PCR products confirmed the correct sequences spanning the ROSA26 locus and the oATP8 transgene (data not shown). We conducted next-generation sequencing to verify oATP8 transgene location and assess off-target random insertions. Next-generation sequencing analysis revealed that a single copy of the oATP8 transgene was correctly inserted in the ROSA26 locus (NC_000072.7::113053020) (7 pair-end reads spanning the insertion site were identified with a confidence score of 96). These results suggest the successful recombination of a single copy of the oATP8 transgene into the C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB) mouse strains. The oATP8 transgene is transferred in the C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB) strain offspring for up to four generations (Figure 2B).
Figure 2.

Confirmation of transgene in C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB)
Sequencing products utilizing primers spanning transgene and insertion site for identification of transgene and site-specific insertion into the ROSA26 locus (n = 6 animals) (A). PCR results confirm the presence of the oATP8 construct within the fourth generation of mouse pups born from transgenic C57BL/6J(mtC57BL/6J) or C57BL/6J(mtFVB) parents (B).
Exogenous optimized ATP8 is ubiquitously expressed, localizes to mitochondria, and incorporates into the ATP synthase complex
The expression of FLAG-tagged oATP8 protein was readily detected in whole-cell lysates of the brain, liver, skeletal muscle, kidney, and spleen in both male and female 12-week-old C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB) mice, but not in age-matched littermate non-transgenic animals (Figures 3A and 3C). Using denaturing western blots probed for FLAG-tagged oATP8, we detected a single product at ∼14 kDa corresponding to the processed exogenous oATP8 protein (Figure S2A). Normalizing the protein band intensities to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) indicated that the relative transgene expression levels are comparable in both C57BL/6J(mtC57BL/6J) (Figure 3B) and C57BL/6J(mtFVB) (Figure 3D) mice, with minor variations at the organ level and between replicates. To determine the extent of the exogenous oATP8 protein fully localizing to the mitochondria, we isolated the cytoplasmic and mitochondrial fractions from 50-week-old mouse livers. Using denaturing western blot, we determined that the FLAG-tagged oATP8 protein is fully compartmentalized to the mitochondrial fraction in both male and female C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB) strains (Figure 4A). The transgenic oATP8 protein intensities were normalized to either the cytosolic marker phosphoglycerate kinase 1 (PGK1) or the mitochondrial marker, aconitase (ACO2) in respective samples (Figure 4B).
Figure 3.

Exogenous optimized ATP8 is ubiquitously expressed
Denaturing PAGE western blots from processed liver, brain, heart, skeletal muscle, kidney, and spleen of 12-week-old non-transgenic and transgenic C57BL/6J(mtC57BL/6J) (A) and C57BL/6J(mtFVB) (C) mice expressing the oATP8 protein. The C-terminal FLAG epitope was immunodetected with mouse anti-FLAG antibody against the oATP8 protein. GAPDH was used as a loading control. Approximately twenty-five micrograms of protein was loaded per lane (n = 5 biological replicates). Values are expressed as means ± standard error of the mean. The immunoblot bands of oATP8 were quantified by densitometry analysis (using ImageJ), normalized to GAPDH (B and D).
Figure 4.

Transgenic optimized ATP8 localizes to mitochondria and incorporates into ATP synthase complex
Cytoplasmic (Cyto.) and mitochondrial (Mito.) fractions were isolated from liver from 50-week-old transgenic C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB) mice. Denaturing PAGE western blots were used to assess the compartmentalization of oATP8 to the mitochondria by probing for oATP8 using mouse anti-FLAG, cytosolic contents using rabbit anti-PGK1, and mitochondrial protein using mouse anti-ACO2. GAPDH was used as a loading control. The animal ID alongside the gender (“M” for male and “F” for female) is listed at the top of the lanes. Cytoplasmic fractions were run with ∼50 μg protein and mitochondrial fractions with ∼35 μg protein per lane (n = 4 biological replicates; 2 males and 2 females) (A). The immunoblot bands of oATP8 were quantified by densitometry analysis (using ImageJ), normalized to aconitase for mitochondrial fractions, and to PGK1 for cytoplasmic fractions (B). Values are expressed as means ± standard error of the mean. Blue Native PAGE western blots using 25 μg protein from purified liver mitochondrial fractions of 12-week-old non-transgenic and transgenic C57BL/6J(mtC57BL/6J) and C57BL/6J (mtFVB mice). Integration of oATP8 into ATP synthase complex was detected by ATP synthase complex monomer (∗) and dimer (∗∗) formation using mouse anti-FLAG antibody (Ci). ATP5O/OSCP (Cii), and ATP6 (Ciii) were probed as controls for ATP synthase complex proteins (n = 3 animals).
We then asked if the exogenous oATP8 protein was incorporated into the ATP synthase by analyzing isolated mitochondria from a 12-week-old mouse liver using blue native (BN)-polyacrylamide gel electrophoresis (PAGE). The expressed FLAG-tagged oATP8 protein integrated into monomeric and dimeric ATP synthase in both C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB) strains (Figure 4Ci), as the FLAG-tagged oATP8 bands co-migrate with endogenous ATP synthase subunits ATP5O/OSCP (ATP synthase peripheral stalk subunit OSCP) (Figure 4Cii) and ATP6 (mitochondrially encoded ATP6) (Figure 4Ciii). The expression of oATP8 does not affect ETC subunit levels or assembly, specifically of GRIM-19 (complex I subunit), SDHB (succinate dehydrogenase complex iron sulfur subunit B; complex II subunit), CORE-2 (ubiquinol-cytochrome c reductase core protein 2; complex III subunit), and CO2 (mitochondrially encoded cytochrome c oxidase II; complex IV subunit) (Figures S2Bi–S2Biv). The following data demonstrate that the exogenous oATP8 gene is constitutively expressed, translated, and targeted in vivo into the correct mitochondrial complex in all the tissues we checked.
Stable expression of exogenous oATP8 with time
We proceeded to ask whether exogenous oATP8 has prolonged expression with time. Using RT-qPCR, we quantitatively assessed the RNA expression levels of exogenous oATP8 with time in brain. We found that exogenous oATP8 RNA expression did not significantly change between 6- and 50-week-old animals in both strains assessed (Figures 5A and 5B). We evaluated whether stable mRNA expression with time was reflected in stable exogenous oATP8 protein abundance. Whole-cell liver lysates from 6-, 12-, 30-, and 50-week-old animals were probed for FLAG-tagged oATP8 via denaturing western blot. Both male and female C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB) strains expressed the exogenous oATP8 protein up to age 50 weeks (Figure 6A). Overexpression of an exogenous protein may negatively regulate endogenous protein expression. Using RT-qPCR, we tested the impact of exogenous oATP8 expression on endogenous ATP8 and ATP6 levels. Nuclear oATP8 expression did not significantly alter the levels of endogenous ATP8 (Figure 5C) or ATP6 mRNA (Figure S3). At the protein level, probing for endogenous and exogenous ATP8 revealed that the exogenous FLAG-tagged ATP8 was consistently expressed at higher levels in C57BL/6J(mtFVB) transgenic mice compared with wild-type C57BL/6J(mtC57BL/6J) transgenic mice (Figure 6A). For ratiometric analysis, we normalized the endogenous and exogenous ATP8 protein intensities to GAPDH (Figure 6B). The apparent ratios of oATP8/ATP8 (Figure 6C) indicated that the proportion of transgenic ATP8 was significantly higher in the C57BL/6J(mtFVB) transgenic mice, with statistical significance observed at later time points (30 and 50 weeks). However, we observed considerable variation in the transgene intensities between replicates, particularly in the C57BL/6J(mtC57BL/6J) control samples. To mitigate variations in the efficiencies of tissue lysis, we performed a similar experiment with purified mitochondria. Mitochondria from non-transgenic controls and transgenic C57BL/6J(mtC57BL/6J) or C57BL/6J(mtFVB) liver samples (n = 3) were purified using Dounce homogenization as described in the materials and methods. Using denaturing PAGE we estimated the apparent ratio of oATP8/ATP8 in the mitochondrial lysates (Figure 6D). The intensities were normalized to the mitochondrial marker aconitase (Figure 6E). It is evident that the transgenic oATP8 is approximately twice the amount of the endogenous ATP8 in the C57BL/6J(mtFVB) transgenic mice relative to C57BL/6J(mtC57BL/6J) transgenic mice, suggesting better incorporation of oATP8 in the FVB mitochondria (Figures 6E and 6F). We did not observe statistical significance in the wild-type transgenic mitochondria despite appreciable transgene expression across samples. It is important to emphasize that densitometry analysis of western blots is a semiquantitative technique and determining the exact ratios of oATP8/ATP8 warrants further analysis using alternative methods.
Figure 5.

Stable expression of transgenic oATP8 with time
Quantitative RT-PCR detection of mRNA levels for transgenic oATP8 and endogenous ATP8 from brain of 50-week-old mice. Transgenic oATP8 was normalized to ACTIN (A) and COX10, a nuclear-encoded mitochondrial gene (B). Endogenous ATP8 mRNA was normalized to COX3, a mitochondria-encoded gene (C) (n = 3–4 animals, performed in triplicate). Transgenic oATP8 levels were not detected in non-transgenic animals (data not shown). Error bars show SEM. two-way ANOVA was performed.
Figure 6.

Distribution of transgenic versus endogenous ATP8
Denaturing PAGE western blots of mouse liver tissue from 6-, 12-, 30-, and 50-week-old transgenic C57BL/6J(mtC57BL/6J) mice (A left panel) and C57BL/6J(mtFVB) mice (A, right panel). The immunoblot bands of oATP8 and endogenous ATP8 were quantified by densitometry analysis (using ImageJ), normalized to GAPDH (B) and the ratio of oATP8 and endogenous ATP8 were calculated (C). Denaturing PAGE western blots of mouse liver mitochondria from non-transgenic and transgenic C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB) mice (D). The immunoblot bands of oATP8 and endogenous ATP8 were quantified by densitometry analysis (using ImageJ), normalized to aconitase (E) and the ratio of oATP8 and endogenous ATP8 were calculated (F). Approximately fifty micrograms of protein was run per lane (n = 3 animals per group). The animal ID alongside the gender (“M” for male and “F” for female) is listed at the top of the lanes. FLAG-tagged oATP8 protein was immunodetected with mouse anti-FLAG antibody and endogenous ATP8 was immunodetected with rabbit anti-ATP8 antibody. GAPDH and ACO2 were used as loading controls for nuclear and mitochondrial fractions. Error bars show SEM. (B) Two-way ANOVA with Šídák’s multiple comparisons: p > 0.05, ∗∗p = 0.0018, ∗∗∗∗p ≤ 0.0001. (C) Two-way ANOVA with Šídák’s multiple comparisons: p > 0.05, ∗p = 0.0205, ∗∗p = 0.00015. (E) Two-way ANOVA with Šídák’s multiple comparisons: p > 0.05, ∗p = 0.0114. (F) Unpaired t test with Welch’s correction. p > 0.05; NS, not significant.
Preliminary mass spectrometry analysis using HPLC LC-MS/MS strategies indicated that peptides spanning V28SSQTFPLAPSPK40 and I58YLPHSLPAQQ67 in the ATP8 protein were readily detected and could be used to distinguish transgenic and endogenous ATP8 proteins. Particularly, the sequence I58YLPHSLPAQQ67 is converted to I58YLPHSLPAQQLEQK71 upon the addition of the FLAG tag. The values for the peptides obtained with and without the FLAG tag compared with an internal control such as V28SSQTFPLAPSPK40 could report the levels of endogenous ATP8 and exogenous oATP8 quantitatively. We generated heavy atom stable isotope-labeled I58YLPHSLˆˆPAQQ67 (Lˆˆ with 13C6) and I58YLPHSLPAQQLEQKˆˆ71 (Kˆˆ with 13C6, 15N2) peptides to use as synthetic peptides in enhancing the signal. Unfortunately, the variants in the endogenous peptides were not detected by mass spectrometry, presumably because of inadequate ionization in the assay. Therefore, their relative quantitation in the ATP synthase complex was not possible. In addition, we also generated stable isotope-labeled MPQLDTSTWFITIISSMITLFILFQLKˆˆ and MPQLTTSTWFITIISSMITLFILFQLKˆˆ peptides (Kˆˆ with 13C6, 15N2) to enhance the signal for the N-terminal region of the ATP8 protein reflecting the aspartic acid to tyrosine mutation at the fifth position; however, these peptides were not detected in our mass spectroscopy assays.
Exogenous expression of the oATP8 protein does not negatively impact mitochondrial function, physiology, or behavior
To assess whether the exogenous oATP8 protein impacts mitochondrial function, we used an in-gel ATPase activity assay to determine whether ATP hydrolysis occurred in monomeric and dimeric forms of the ATP synthase (Figure 7A). In-gel ATP hydrolysis activity was comparable among non-transgenic and transgenic samples tested. Using the Agilent XFe Seahorse, we quantitatively assessed the impact of the exogenous oATP8 protein on maximal ATP hydrolysis activity (Vmax), substrate concentration giving reaction rate of 1/2 Vmax of the ATP synthase to ATP (Km), ATP-linked respiration, maximal respiration, and oligomycin sensitivity. Maximal ATP hydrolysis activity (Vmax) and Km (ATP) of the ATP synthase were not significantly changed between the transgenic and non-transgenic groups in isolated liver mitochondria (Figure 7B; Table S1). ATP-linked respiration (state 3) and maximal respiration (state 3u) were not affected in isolated liver and skeletal muscle mitochondria (Figures 7D and 7E). Lastly, the IC50 of oligomycin in isolated liver mitochondria was not significantly changed when comparing transgenic C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB) animals to the relevant non-transgenic controls (Figure 7C; Table S2).
Figure 7.

Exogenous expression of oATP8 protein does not negatively impact mitochondrial function
In gel activity analysis in 12-week-old non-transgenic and transgenic C57BL/6J(mtC57BL/6J) and C57BL/6J(mtFVB) mice to detect ATPase activity. Mouse liver mitochondria were purified and ∼25 μg mitochondrial protein was used for clear native-PAGE. ATPase activity was documented and stained with Coomassie to detect ATP synthase monomer and dimer formation (Ai and Aii). Quantitative assessment of ATP hydrolysis activity was performed using isolated liver mitochondria from 12-week-old male mice (B). The Vmax and Km of ATP synthase to ATP substrate was calculated with the 95% confidence interval (CI). Lines were fit using “Non-linear fit: Michaelis-Menten” in GraphPad Prism. Values are means ± SEM (n = 5 animals) (Table S1). ATP synthesis and maximal respiratory capacity were monitored in isolated liver and skeletal muscle mitochondria from 9- to 12-week-old male mice. Error bars show SEM. Two-way ANOVA was performed with no significant differences between groups (n = 5 for liver mitochondria, n = 3 for skeletal muscle mitochondria) (D and E). The sensitivity of the ATP synthase to oligomycin was assessed by monitoring ATP hydrolysis activity. Values are means ± SEM (n = 3 animals) (C). Lines were fit using “Non-linear fit: [Inhibitor] vs. normalized response – Variable slope” in GraphPad Prism and IC50 values and 95% CI were calculated (Table S2).
To further assess the safety of the allotopic gene therapy, we sought to determine whether exogenous oATP8 construct or protein negatively impacted mouse physiology and behavior. We conducted a series of behavioral assays in animals between age 6 and 9 weeks (Figure 8A). We utilized the open-field and elevated plus maze to assess changes in anxiety levels, exploratory behavior, or locomotion. To ensure there were no systemic metabolic defects with this gene therapy approach, we conducted the Promethion metabolic cage assay to monitor for systemic metabolic changes, feeding and drinking behavior, and general locomotion. We also tested whether exercise capacity and tolerance were altered using the treadmill exhaustion test. Our results show that the presence of the transgene did not significantly change any of the aforementioned parameters (Figure 8A; Tables S3–S9). The weight of the transgenic animals was not significantly altered from their non-transgenic controls up to age 50 weeks (Figure 8B). Since gene therapy can elicit host immune responses, we assessed mouse plasma samples via inflammatory cytokine assay as an additional means to characterize the safety of our approach. The transgenic animals did not show significant changes in plasma inflammatory cytokine levels compared with their relevant non-transgenic controls (Figure 8C). Furthermore, global proteome analysis of liver mitochondria purified from gender-controlled, non-transgenic, and transgenic mice did not reveal any major changes (Figure S4). The proteins Plbd1 and Glb1 were upregulated in male C57BL/6J(mtC57BL/6J) transgenic mice compared with their non-transgenic counterparts (Figure S4i) and Actb was upregulated in female C57BL/6J(mtC57BL/6J) non-transgenic mice compared with female transgenic mice (Figure S4ii). Elevated levels of proteins Maoa, Acss3, and Dhodh were observed in male C57BL/6J(mtFVB) non-transgenic mice compared with their transgenic cohorts. Plbd1 and Hba were upregulated in male C57BL/6J(mtFVB) transgenic mice when compared with control C57BL/6J(mtFVB) male mice (Figure S4iii). In addition, the proteins Actb, Coro1a, Lcp1, Oxct1, and Pkm were all upregulated in female C57BL/6J(mtFVB) non-transgenic mice compared with female mutant transgenic mice (Figure S4iv).
Figure 8.

Influence of exogenous oATP8 on physiology and behavior
Schematic representing the timeline of behavioral assays conducted (A). Animal weight was monitored at 9 weeks (n = 8 animals), 18 weeks (n = 7–14 animals), 30 weeks (n = 4 animals), and 50 weeks (n = 6–8 animals) (B). Heatmap portraying the ELISA results of a pro-inflammatory cytokine assay conducted on plasma from 12-week-old mice (C).
Discussion
The allotopic expression of mitochondrial genes is defined as the re-engineering of functional mitochondrial genes and their expression from the nucleus. Challenges to successful allotopic expression have been described previously, including (1) the engineering and nuclear integration of an optimized construct to express from the nucleus, (2) the targeting of an exogenous cytosolic protein to the mitochondria, and (3) the functional assembly and rescue of a mutant mitochondrial phenotype.32 Our previous work has addressed several of these challenges as we have shown that mitochondrial genes require codon optimization for robust expression from the nucleus33 and demonstrated how simultaneous allotopic expression of two mitochondrial genes, the minimally recoded ATP6 and the codon-optimized ATP8, can rescue a patient-derived cybrid model null for the ATP8 protein.27
The concept of allotopic expression was initially tested in yeasts34,35,36 and plants37 to target soluble proteins into the mitochondrion, and was later adapted for membrane proteins.38 Nagley et al. were the first to implement allotopic expression for mtDNA proteins by recoding the yeast ATP8 gene for the nuclear DNA code. Their work demonstrated successful restoration of respiratory growth in ATP8-null cells.39 Subsequently, several studies describing the transition of mtDNA genes to the nuclear-cytosolic machinery have been reported in unicellular eukaryotes and mammalian systems with varying success. The therapeutic potential of this strategy is actively being explored for more common mtDNA mutations, such as those in the ATP6 and ND4 genes. Mutations in the ATP6 gene result in clinically diverse symptoms including NARP and Leigh’s syndrome due to compromised ATP synthase activity. Allotopic expression of ATP6 in patient cybrids with specific mutations such as the mt.8993T>G or (mt.8529G>A) shows promise and can restore partial function.27,29,40 Bilateral injection of lenadogene nolparvovec (an AAV-engineered allotopic ND4 gene) in the eyes of LHON patients carrying the m.11778G>A MT-ND4 mutation improved visual acuity and has shown excellent safety profile.41
Successful allotopic expression of mtDNA-encoded genes requires optimization of important coding and non-coding elements such as (1) mandatory changes to the gene sequence to synchronize nuclear-cytosolic translation, (2) appending a suitable MTS to target the protein to the mitochondria, and (3) appropriate 5′ and 3′ UTR sequences in regulating transcription and translation. We and others have used the mitochondrial targeting sequence from the ATP5G127,29,33 COX VIII42 and COX 1043,44 nuclear mitochondrial genes to target mammalian ATP8, ATP6, ND1, ND4, or ND6 proteins with varying efficiencies. A recent study by Chin et al. screened 31 combinations of MTSs reflecting specific properties such as species origin, length, charge, and hydrophobicity of the MTSs or by attaching two or more MTSs in tandem to achieve proper targeting. Particularly, attaching a long MTS such as the one from Zea mays mitochondrial inner membrane protein 4-hydroxybenzoate polyprenyltransferase (116 amino acids) seemed beneficial in targeting the human allotopic ATP6 construct and showed OXPHOS function recovery in Leigh’s model ATP6 8993T>G mutant cybrids.43 Similarly, appending a triple chimeric MTS from Z. mays and human mitochondrial genes to the codon-optimized ND3 gene was able to rescue complex I function in patient fibroblasts with two different mutations, the m.10197G>C or the m.10191T>C missense mutation, in the MT-ND3 gene.45 Our previous studies indicate that nuclear codon optimization for all the mtDNA genes significantly improves translation and association with mitochondria; however, the appended MTS, in this case that of ATP5G1, was not always efficiently processed.33 Only 8 of the 13 codon-optimized genes showed stable expression. In general, proteins targeted to the inner membrane of mitochondria have been identified to employ both N-terminal cleavable and internal non-cleavable sequences for their efficient import into mitochondria. Further studies examining the hydrophobicity of cargo proteins, their topology after import into the inner membrane, and their assembly into OXPHOS complexes are crucial for determining the optimal conditions for successful allotopic expression.
More than 1,100 genes of the mitochondrial proteome are nuclear-encoded, translated on cytosolic ribosomes, and subsequently imported into mitochondria. Increasing evidence suggests that eukaryotic cells use multiple mechanisms—co-translational, pre-translational, and post-translational—to target cargo proteins to mitochondria. Sequences in the 3′ UTR have been particularly identified to play an important role in localizing a subset of mRNAs to the outer mitochondrial membrane for a co-translational import.46 More recently, using proximity ligation studies Fazal et al. further delineate a ribosome-dependent or -independent mechanism for the localization of such mRNAs to the outer mitochondrial membrane.47 The ribosome-independent mechanism likely utilizes specific RNA binding proteins such as PUM (pumilio RNA binding family member), CLUH (clustered mitochondria homolog), and SYNJ2BP (synaptojanin 2 binding protein) that bind to specific regions in the 3′ UTR preventing premature translation of such RNAs until they reach their destination. Appending the SOD2 3′ UTR to allotopic ATP648 or the Cox 10 3′ UTR to complex I (ND1, ND4) and ATP synthase (ATP6) genes improved the expression and targeting of their respective proteins.44,49 However, appending several other putative 3′ UTRs to allotopic COX2 or the truncated version of ND4 mRNAs similarly did not increase mitochondrial colocalization in other studies.43
The potential for using allotopic expression in treating mtDNA disorders requires the demonstration of its feasibility and safety in an animal model. In this study, we generated and characterized a mouse model in the C57BL/6J(mtC57BL/6J) and the C57BL/6J(mtFVB) strains that allotopically express a single copy of the transgenic oATP8 mitochondrial gene from the mouse ROSA26 safe harbor locus. This is the first time that whole-body expression of a mitochondrial gene from a defined locus in the nucleus has been implemented. Previous approaches have predominantly used recombinant adenoviruses to target specific organs, such as the eye, expressing allotopic ND4,23 ATP6,24 or ND150 transgenes. In these studies, the allotopic ATP6 gene was randomly incorporated into the nuclear genome of C57BL/6 or B6(B6SJLF1) backgrounds.24 We utilized the safe harbor integration system to precisely control the transgene’s nuclear location, copy number, and the surrounding 5′ and 3' UTR elements. Despite these controls, we observed variability in transgene levels across different organs and samples. The organ-level variation in transgene expression could be attributed to varying demands for mitochondrial biogenesis and function. Tissues such as the heart and brain rely more on oxidative phosphorylation for their energy demands, whereas the spleen is less so. The sample level variations could be due to selective pressures such as transcriptomic changes surrounding the transgene, although we observed appreciable transgene expression in all samples tested. The mROSA26 locus was originally identified in mouse ESCs for stable expression of promoter-less transgenes using lentivirus knockin screens51; however, this locus is in the third intron region of the THUMPD3 gene that is yet to be characterized. In humans, this region is also surrounded by proto-oncogenes that could influence transgene expression.52 Recent advances in genomics and epigenetics guided identification of genomic safe harbors accounting for human population variations53,54 could uncover neutral regions in the human genome more suited for stable transgene expression.
Using this approach, we found sustained allotopic expression of oATP8 in animal models up to 50 weeks. This exogenous oATP8 protein is ubiquitously expressed, localizes to the ATP synthase in the mitochondria, and does not disrupt endogenous ATP synthase assembly, mitochondrial function, or the measured aspects of systemic behavior and physiology. We also observed that the transgene and the exogenous oATP8 protein do not elicit an inflammatory host immune response. The absence of detectable differences between transgenic and non-transgenic animals highlights that allotopic gene therapy for the ATP8 gene is feasible and safe to use in vivo. However, ATP8 is a small protein (∼8 kDa) in the mitochondrial genome with a single transmembrane domain. The applicability of this approach to other mtDNA genes may require a case-by-case validation, considering factors such as protein length, number of transmembrane domains, and the efficiency of import and targeting to their respective OXPHOS complexes. Mitochondrial proteome analysis for liver mitochondria purified from transgenic and non-transgenic mice did not show any discernible changes indicating that the expression of the allotopic ATP8 protein is well tolerated and does not result in adverse proteostasis.
The capacity of the exogenous protein to integrate and compete with an existing endogenous mutant protein, the likely scenario in many mitochondrial diseases, warrants further investigation in vivo. We addressed this question using western blotting. By comparing the apparent ratios of transgenic ATP8 with endogenous ATP8, normalized to a mitochondrial marker such as ACO2, our data suggest that transgenic ATP8 integrates as well as, or better than, in the C57BL/6J(mtFVB) strain compared with the C57BL/6J(mtC57BL/6J) strain. This might be attributed to enhanced protein stability of transgenic ATP8 in the FVB strain due to differences in the aspartic acid substitution at the fifth position. In contrast, in the C57BL/6J(mtC57BL/6J) strain, the apparent levels of transgenic and endogenous ATP8 were comparable and did not show significant differences.
Previously, we have shown how only a fraction of allotopically expressed mitochondrial protein integrates into the proper mitochondrial complex.27,33 In our mouse models, we observe that the majority of the exogenous oATP8 protein is processed and localized into the mitochondria. In addition, we have found that the expression of the oATP8 transgene does not alter transcription levels of endogenous ATP8 in 6- and 50-week-old animals.
We recognize that a limitation of the generated models is the overexpression of the exogenous oATP8 protein compared with endogenous levels of ATP8. To incorporate transcriptional regulation of the transgenic construct, future studies on transcriptional regulation of the mitochondrial genome by the mitochondria and nucleus must be completed. In addition, our model does not demonstrate functional rescue of a dysfunctional phenotype due to a mitochondrial mutation. There is a lack in animal models with specific mtDNA mutations that systemically elicit behavioral or functional alterations without being deleterious, it is difficult therefore to functionally assess for systemic rescue using allotopic expression in vivo. In our study, we utilized the conplastic C57BL/6J(mtFVB) mouse strain with a naturally occurring ATP8 missense mutation that exhibits mild phenotypes not significantly different from C57BL/6J(mtC57BL/6J) animals.31 Previously, behavior studies comparing the conplastic C57BL/6J(mtFVB) mice with the wild-type mitochondrial variant indicated an elevated anxiety phenotype in the C57BL/6J(mtFVB) mice.31 Other pathophysiological observations, such as increased reactive oxygen species in the β cells of the pancreas, have also been reported in these mice. This polymorphism was also observed to confer resistance to acute models of liver failure55 and experimental autoantibody-induced skin inflammations,56 indicating altered cellular metabolism without affecting the OXPHOS activity. In our hands, we could not recapitulate the anxiety phenotype in the C57BL/6J(mtFVB) mice. There were no differences in exploratory behavior, locomotion, or anxiety using the open-field and the elevated maze tests between the C57BL/6J and the mtFVB mitochondrial background strains.
The pathological effects of mtDNA mutations that arise with age extend beyond localized regions, highlighting the necessity for more systemic and global approaches. The development of this gene therapy technology in vivo provides the opportunity to demonstrate the rescue of whole-body function in models that exhibit pathological mtDNA mutations.
Materials and methods
Antibodies
The following antibodies were used in this study: anti-FLAG (cat. no. F1804, Millipore Sigma, Burlington, MA), anti-ATP8 polyclonal (mitochondrially encoded ATP8; cat. no. PA5-109987, Thermo Fisher Scientific, Waltham, MA), anti-SDHB (succinate dehydrogenase complex iron sulfur subunit B; cat. no. AP19974b, Abcepta, San Diego, CA), anti-ATP6 (mitochondrially encoded ATP6; clone no. 1G7-1G2, mAbdx, Eugene, OR), anti-CO2 (mitochondrially encoded cytochrome c oxidase II; cat. no. PA5-102933, Thermo Fisher Scientific), anti-ATP5O/OSCP (ATP synthase peripheral stalk subunit OSCP; cat. no. TA804572, Origene Technologies, Rockville, MD), anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase; cat. no. G9545, Sigma-Aldrich, St. Louis, MO), anti-PGK1 (phosphoglycerate kinase 1; cat. no. A14039, Abclonal, Woburn, MA), anti-GRIM19 (cat. no. ab110240, Abcam, Fremont, CA), anti-CORE-2 (ubiquinol-cytochrome c reductase core protein 2; cat. no. ab14745, Abcam), anti-ACO2 (aconitase 2; cat. no. TA500824, Origene Technologies). Horseradish peroxidase-conjugated secondary antibodies to mouse (cat. no. G21040), rabbit (cat. no. A16023), and goat (cat. no. G21040) was obtained from Life Technologies (Carlsbad, CA).
Mice
Four-week-old male and female C57BL/6J (strain no. 000664) and female C57BL/6J-mtFVB/NJ/IbraJ [C57BL/6J(mtFVB)] (strain no. 010810) were purchased from The Jackson Laboratory (Bar Harbor, ME). The C57BL/6J nuclear background was maintained on the C57BL/6J(mtFVB) strain by backcrossing female C57BL/6J(mtFVB) animals to C57BL/6J(mtC57BL/6J) males.31 All lines used in the studies were in the C57BL/6J nuclear background strain.
All mice in the colony were housed 2–5 per cage at 22°C ± 0.5°C under specific pathogen-free conditions in a 12-h light/12-h dark cycle and had free access to food and water. Cages and bedding were changed once per week. Standard mouse chow diet was obtained from Envigo (2018 Tekland Global 18% protein diet, cat no. 2918). In all the studies, efforts were made to use equal numbers of male and female mice within each experimental group. The following groups were used in this study: (1) C57BL/6J(mtC57BL/6J), (2) C57BL/6J(mtFVB), (3) C57BL/6J(mtC57BL/6J) transgenic, and (4) C57BL/6J(mtFVB) transgenic.
All experiments involving the use of mice were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the Buck Institute and conducted with strict adherence to the National Institutes of Health Animal Care and Guidelines under protocol A10226.
Generation of ATP8-expressing transgenic mice
We generated a transgenic mouse model expressing an optimized ATP8 (oATP8) sequence in the mouse ROSA26 locus, allowing stable ubiquitous expression of the transgene while mitigating potential insertion site side effects that may occur with random transgene integration. We used the TARGATT technology developed by Applied Stem Cell (ASC, Milpitas, CA) to integrate a large transgene into the ROSA26 safe harbor locus by pronuclear injection.30 In brief, the codon-optimized transgene sequence ATP5G1MTS-oATP8-FLAG (oATP8 construct) under the CAG promoter was subcloned into the TARGATT system vector designed to facilitate ɸC31 integrase-mediated recombination at the ROSA26 locus. ASC has developed a C57BL/6J ROSA26-3attP mouse line by knocking in three tandem attP sites via CRISPR-Cas9 into the ROSA26 locus. The oATP8 construct contains attB docking sites for ɸC31 integrase-mediated recombination. The ɸC31 mRNA (50 ng/μL) along with the oATP8 construct (5 ng/μL) was microinjected into the pronucleus of 153 zygotes in the C57BL/6J ROSA26-3attP mouse line, and the injected zygotes were implanted into six CD1 foster mice. Successful oATP8 construct integration in the founder mice was identified with PCR analyses of genomic DNA using primers targeting optimized ATP8 and the TARGATT insertion site. After PCR screening, five males and one female positive founders (transgenic oATP8 mice) were identified. All mice produced by ASC had a C57BL/6J genetic background. Male transgenic oATP8 mice were crossed with female conplastic mice C57BL/6J(mtFVB) at the Buck Institute to produce mice with an optimized ATP8 transgene expression in animals with the FVB/NJ mitochondrial background. The transgenic and non-transgenic animals used were littermates. All the mice produced were viable, fertile, and born at the expected Mendelian ratio without apparent abnormalities in adult mice.
Design of mouse ATP8 transgene
The sequence for the mouse ATP8 gene was recoded for nuclear-cytosolic translation. The mitochondrial targeting sequence, including the first 66 amino acids for the mouse ATP5G1 gene, was appended at the 5′ end. This chimeric construct was codon optimized (GenScript Biotech, Piscataway, NJ) and cloned under a CAG promoter and the SV40 poly(A) 3′ UTR.
Preparation of mouse genomic DNA for genotyping and PCR
Two millimeters of mouse tail tissue was cut and placed into PCR Eppendorf tubes at 4°C. DNA was extracted using the KAPA Biosystems Mouse DNA Extraction Kit (KR0385) according to the manufacturer’s protocol. For genotyping by PCR, serial primer pairs were used as listed in (Table S10). KAPA2G Fast (HotStart) Genotyping Mix (KR0385) was used according to the manufacturer’s protocol. The following PCR conditions were used: 95°C for 5 min; 95°C for 15 s, 60°C for 15 s, 72°C for 15 s (35 cycles); and 72°C for 5 min. A 1% agarose gel was used for electrophoresis.
RNA and cDNA preparation
Approximately 50 mg of frozen mouse brain tissue (n = 3–4) from each experimental group was individually added to 600 μL of TRIzol (Invitrogen). Brain tissues were disrupted using a handheld electric homogenizer (Model H100, Waverly) until the tissue was fully homogenized. The homogenized tissue was vortexed, then left to incubate for 2–3 min, then centrifuged at 12,000 × g for 15 min.
RNA from mouse brains was processed using a Quick-RNA MiniPrep Kit (Zymo Research, Irvine, CA) according to the manufacturer’s guidelines. RNA from the Zymo-spin IIICG column was eluted using 25 μL of DNAse/RNAse-free water by centrifugation for 1 min at 12,000 × g. The quantity and quality of mouse RNA samples were determined using a NanoDrop 1000 spectrophotometer (Thermo Scientific, Waltham, MA). Total RNA (1 μg) from mouse brain was used in a total reaction volume of 20 μL containing 4 μL of iScript Reverse Transcription Supermix (Bio-Rad, Hercules, CA) to synthesize cDNA according to the manufacturer’s guidelines. cDNA was stored at −20°C before carrying out quantitative RT-PCR.
qRT-PCR
A 10 μL reaction volume containing 50 ng of cDNA, 500 nM of forward and reverse primer, and 5 μL of SensiFAST SYBR No-ROX Kit (BIOLINE, Meridian Biosciences, Cincinnati, OH) was used in a qPCR reaction performed using a LightCycler 480 Real-Time PCR System (Roche Applied Science). The primer pairs used for RT-qPCR are listed in Table S11. For the mouse liver samples, fold changes in gene expression were determined using the 2(–ΔΔCt) method. Gene expression for nuclear encoding genes was normalized to β-actin and COX10. p values were calculated using a two-way ANOVA with Sidak’s multiple comparisons test using the GraphPad Prism 10.2 software. Mitochondrially encoded genes were normalized to COX3. A no-reverse transcriptase control was included in each reaction set to rule out non-specific priming.
Preparation of purified mitochondrial DNA and Sanger sequencing
We prepared purified mtDNA from conplastic animals to verify for homoplasmy of the mitochondrial 7778 G-T mutation in the ATP8 gene. Mitochondrial DNA was isolated from approximately 50 mg of mouse brain or liver tissue using the DNeasy Blood and Tissue kit from QIAGEN (Hilden, Germany). The region spanning the mutation was PCR amplified using mouse-mtDNA-7611-forward (5′GTGGATCTAACCATAGCTTTATGCCC3′) and mouse-mtDNA-8052-reverse (5′GAGACGGTTGTTGATTAGGCG3ʹ). The resulting PCR product (441 bp) was gel purified and subjected to Sanger sequencing (Sequetech, Mountain View, CA).
Preparation of genomic DNA for Sanger sequencing and next-generation sequencing
To verify for construct insertion in the ROSA26 locus, we conducted next-generation sequencing.
Genomic DNA from ∼50 mg of brain tissue from the transgenic mice was fragmented to an average size of ∼350 bp and subjected to DNA library creation using established Illumina paired-end protocols. The Illumina Novaseq 6000 platform (Illumina, San Diego, CA) was utilized for genomic DNA sequencing in Novogene Bioinformatics Technology (Beijing, China) to generate 150-bp paired-end reads with a minimum coverage of 10× for ∼99% of the genome (mean coverage of 30×).
The original image data generated were converted into sequence data via base calling (Illumina pipeline CASAVA v.1.8.2) and then subjected to a quality control procedure to remove unusable reads by fastp with the parameters as “-g -q 5 -u 50 -n 15 -L 150 --min_trim_length 10 --overlap_diff_limit 1 --overlap_diff_percent_limit 10”: (1) discard the adapter-containing reads and force poly(G) tail trimming, (2) discard the paired reads when uncertain nucleotides (N) constitute more than 10% of either read, (3) discard the paired reads when low-quality nucleotides (base quality less than 5, Q ≤ 5) constitute more than 50% of either read.
Sequencing reads were aligned to the reference genome using the Burrows-Wheeler Aligner with default parameters.57 Subsequent processing, including duplicate removal was performed using samtools and PICARD (http://picard.sourceforge.net).
The raw SNP/InDel sets are called by GATK with the parameters as ”--gcpHMM 10 -stand_emit_conf 10 -stand_call_conf 30”. Then we filtered these sets using the following criteria: SNP: QD < 2.0, FS > 60.0, MQ < 30.0, HaplotypeScore >13.0, MappingQualityRankSum < −12.5, ReadPosRankSum < −8.0 INDEL: QD < 2.0, FS > 200.0, ReadPosRankSum < −20.0. Breakdancer was used for SV detections with the parameters as “-q 20 -c 4”.58,59 ANNOVAR was used for functional annotation of variants. The UCSC known genes was used for gene and region annotations.60
To assess the read distribution along the construct insertion site and the construct, we extracted the contiguous sequences that span 5 kb before and 5 kb after the construct insertion site (NC_000072.7:113053020), the reads that mapped to the construct, and the reads that mapped to the TARGATT insertion site using SOAPdenovo software.61 We further confirmed the presence of the construct in the ROSA26 locus using the program blastn62 to align the contiguous sequences mapped to the construct and insertion site to the ROSA26 locus on the mouse reference genome (GRCm3963).
Construct insertion into the ROSA26 locus was further ascertained by Sanger sequencing using the proprietary primers specific to the ROSA26 locus provided by Applied Stem Cell (Milipitas, CA). The following PCR conditions were used: 95°C for 5 min; 95°C for 30 s, 60°C for 30 s, 72°C for 50 s (35 cycles); and 72°C for 5 min. The PCR QIAquick Gel Extraction Kit (QIAGEN, cat. no. 28704) was used to purify and concentrate the PCR product excised from the 1% agarose gel.
Cardiac puncture, plasma sample collection, and mouse tissue harvesting
Upon euthanasia with CO2, a terminal cardiac puncture was performed by inserting a 23–27 gauge needle and a 1 cc syringe between the left ventricle and septum of the heart. Blood was collected into 1.3 mL microcentrifuge tubes with EDTA (1.6 mg EDTA/mL) and stored at 4°C. About 200–500 μL of blood was collected per mouse. Blood was centrifuged at 13,000 rpm using a fixed-angle rotor centrifuge (Eppendorf) for 5 min at 4°C. The resulting supernatant is designated plasma. Plasma was immediately transferred into a clean 1.5 mL tube and stored at −80°C until use. After cardiac puncture and blood sample collection, the liver, spleen, brain, skeletal muscle (gastrocnemius and soleus), heart, and kidney were isolated and immediately flash frozen in liquid nitrogen and then stored at −80°C until use.
Isolation of mitochondria from liver tissue
Liver mitochondria were prepared from mice using established methods with few amendments.64 In brief, mice were euthanized using CO2 and the liver was rapidly removed and placed in an isolation medium composed of 250 mM sucrose, 5 mM Tris-HCl, 2 mM EGTA (pH 7.4) at 4°C. Liver tissue (0.2–0.3 g) was finely chopped into 2- to 3-mm pieces and homogenized using a 7 mL Dounce tissue grinder with loose pestle clearance (0.089–0.14 mm) using five consecutive strokes. Cellular debris was removed by centrifuging homogenized tissue at 700 × g for 10 min at 4°C using a fixed-angle rotor (Beckman Centrifuge J250). Following centrifugation, fat/lipid was carefully aspirated, and the remaining supernatant was carefully transferred through two layers of cheesecloth to a new tube and centrifuged at 8,000 × g for 10 min at 4°C to pellet the mitochondria. The supernatant following this spin was decanted into a separate tube, centrifuged again at 8,000 × g for 10 min, and saved separately as the “cytoplasmic fraction” at −80°C until use. The light-brown edge (composed of dead mitochondria, microsomes) of the mitochondrial pellet was removed using the edge of a pestle and the darker pellet was resuspended in isolation medium and centrifuged again at 8,000 × g for 10 min at 4°C. The final mitochondrial pellet was resuspended in isolation medium, stored at 4°C, and used within 1–3 h for subsequent Seahorse experiments or stored at −80°C until use in other assays. Protein was assessed using Biuret assay.65 Typically, ∼6.0 mg of mitochondrial protein (100 μL volume) was obtained from a single mouse liver.
Isolation of mitochondria from skeletal muscle tissue
Skeletal muscle mitochondria were isolated from the mouse hindlimbs in 4°C Chappell-Perry medium 1 (CP-1; 100 mM KCl, 50 mM Tris, 2 mM EGTA [pH 7.4] at 4°C) and Chappell-Perry medium 2 (CP-2; CP1 supplemented with 0.5% [w/v] fatty acid-free BSA, 5 mM MgCl2, 1 mM ATP, and 250 units/100 mL protease type VIII [pH 7.4]) following established methods.66
Mitochondrial respiration measurement and analysis
We measured rates of oxygen consumption using the XFe96 Agilent Seahorse Analyzer (Seahorse Bioscience) in mitochondrial assay solution (MAS) composed of 70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, and 0.2% (w/v) fatty acid-free BSA (pH 7.2) at 37°C. Stocks of succinate, malate, glutamate, and ADP (1 M) were made in RO water and adjusted to pH 7.2 with potassium hydroxide. Stocks of 4 mM FCCP (carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone), 4 mM rotenone, 10 mg/mL oligomycin, and 2 mM myxothiazol were made in 95% ethanol.
We conducted a mitochondrial stress test following established protocols.67 Mitochondrial respiratory control was measured by sequentially exposing mitochondria to a final concentration of 2 mM ADP (state 3), 2 μg/mL oligomycin (state 3o), 4 μM FCCP (state 3u), and 2.5 μM myxothiazol (non-mitochondrial respiration). To measure maximal respiratory capacity, we simultaneously exposed the mitochondria to 2 μg/mL oligomycin and 4 μM FCCP in the first injection. We quantified the respiratory control ratio using these metrics after subtracting rates with myxothiazol in each well (RCR, state 3/state 4o, or state 3u/state 4o). ADP, oligomycin, FCCP, and myxothiazol were loaded into the injection ports in a volume of 20 μL. We adhered 1 μg of isolated liver mitochondria or 1 μg of isolated skeletal muscle mitochondria per well in a volume of 20 μL of MAS buffer by centrifuging the plate in a swinging bucket rotor at 2,000 × g for 20 min at 4°C. After centrifugation, 100 μL of MAS buffer with substrate, 10 mM succinate, and 2 μM rotenone, warmed to 37°C, was added to each well. Prior to running the assay, we titrated the total amount of mitochondrial protein plated to ensure that the linear range of mitochondrial response to stimuli was within the dynamic range of the instrument. We also carefully titrated the FCCP exposure to the mitochondria to ensure maximal respiratory capacity was captured.
The Wave software (Agilent) was used to extract rates of oxygen consumption. Protein values (μg/well) were used to normalize the rates of oxygen consumption in each well. Mix and measurement cycles were completed twice per segment after calibration and equilibration. Each segment consisted of a mix (1 min), wait (1 min), and measurement (3 min) period repeated for each injection port.
ATP hydrolysis activity
We monitored hydrolase activity of the F1FO complex by measuring pH changes in the XFe96 Agilent Seahorse Analyzer (Seahorse Bioscience). The hydrolysis of ATP to ADP generates H+ and acidifies the experimental medium, therefore oligomycin-sensitive changes in the acidification rate of the medium can be attributed to the activity of the F1FO catalyzing hydrolysis. To access matrix enzymes without the need for facilitated transport across the inner membrane, we used alamethicin to form non-selective pores permeable to solutes up to 3 kDa (∼10–20 Å) for unrestricted delivery of substrates.68 We purchased alamethicin from Sigma (A4665) and kept it as a 25 mg/mL stock solution in ethanol at −20°C. To determine the optimal alamethicin concentration, we titrated alamethicin in an NADH-driven respiration experiment in the XFe96 Agilent Seahorse Analyzer, as described previously.68
Liver mitochondria (4 μg/well) were assayed in an amended MAS medium (220 mM mannitol, 70 mM sucrose, 10 mM MgCl2, 2 mM HEPES, 1 mM EGTA, 0.2% [w/v] fatty-acid-free BSA [pH 7.2] at 37°C) supplemented with 1 μM FCCP (item no. 15218; Cayman Chemical), 1 μM rotenone (R8875, Sigma), 1 μM myxothiazol (T5580, Sigma), and 10 μg/mL of alamethicin in a total volume of 120 μL. Where indicated, mitochondria were initially exposed to 20 mM Mg-ATP (A9187, Sigma). Mg-ATP was also initially titrated to determine the Km of the F1FO complex. Oligomycin (2 μg/mL) (Cayman Chemical) in a volume of 20 μL was added acutely via the injector port of the Agilent Seahorse Analyzer at 7× the final concentration. The IC50 of oligomycin at the F1FO complex was calculated by titrating the oligomycin concentration. Proton production rates were calculated by fitting to a pH standard curve generated by serial injections of HCl from the injection ports, as described previously.69 The Wave software (Agilent) was used to extract acidification rates. Protein values (μg/well) were used to normalize the rates in each well.
Open-field assay
We used the open-field (TruScan 2) assay to assess for locomotor and anxiety behavior. The open-field apparatus is 7 inches (17.78 cm) by 7 inches (17.78 cm) by 12 inches (30.48 cm) in size. Six-week-old male and female mice were used from each of the experimental groups. Animals were acclimated to the testing room for 30–60 min prior to testing. Room lighting was kept consistent during acclimation and testing. Each mouse was left undisturbed in the apparatus for a 10 min period. The observer would sit behind a tall nylon curtain panel to avoid being identified as an external cue. Each mouse was tested three times over three consecutive days. All behavior experiments were conducted at the same time between 8 a.m. and 1 p.m. during the light phase in a dimly lit room. TruScan 2 software was used to record and analyze animal behavior. Open-field assays were analyzed by assessing locomotive activity within various areas in the open-field arena. Locomotive activity can be quantified via speed, time spent, or total distance. Analysis of the open-field data was completed blind to the experimental groups. To quantify anxiety behavior, the following metrics were calculated: (1) total distance moved, or time spent in the center field, and (2) total distance moved, or time spent in the marginal/outer field. Differences in locomotion were assessed by calculating (1) total distance moved in the entire arena and (2) total number of moves in the entire arena.
Elevated plus maze
The elevated plus maze was used to assess anxiety behavior. The elevated plus maze apparatus consists of four arms of 15 inches (38.1 cm) in length and 2 inches (5.08 cm) width each. Two opposing arms are enclosed in opaque 6.5 inch (16.51 cm) high side and end walls. The center of the maze is 2 by 2 inches (5.08 × 5.08 cm). The elevation of the structure is 30.5 inches above the ground. The structure was surrounded by tall nylon curtain panels to avoid having the observer being identified as an external cue. EthoVision XT 15 Software was used to record and assess animal behavior. Seven-week-old male and female mice were used from each of the experimental groups. Animals were acclimated to the testing room for 30–60 min prior to testing. Room lighting was kept consistent during acclimation and testing. Mice were put into the central position of the maze facing an open arm and left undisturbed for 10 min. Their behavior was recorded using a video camera and later analyzed by a blinded observer. For all mice, testing was performed between 8 a.m. and 1 p.m. to minimize the influence of the time of day. Two animals that slipped off the platform were excluded from the analysis. We tested anxiety based on the following parameters: (1) percentage of entries into open arms, (2) percentage of time spent in open arms, and (3) distance traveled in open arms. Locomotor activity was determined as (1) total entries into open and closed arms, (2) entries into closed arms, and (3) distance traveled in closed arms.
Treadmill
We used the treadmill (E8700TS series Panlab Harvard Apparatus) to assess for non-voluntary fatigue-like behavior in mice. Nine-week-old female and male animals from all experimental groups were used in this study by following previously published protocols70 with several amendments. The treadmill apparatus consists of five lanes, inclined at 10°, with an electric shock administered at 2 mA as an external motivator. We defined the criterion for fatigue as spending five continuous seconds in the designated fatigue zone on the treadmill apparatus (i.e., one body length region ranging from the rear of the treadmill including the shock grid). Prior to the assay, animals were trained on utilizing the platform. During the training phase, animals were allowed to explore the platform for 3 min. Afterward, the treadmill speed was gradually increased to 12 m/min by 1.2 m/min increments every minute for 10 min. This training phase was repeated on the following day. The animals were returned to their cages for 24 h before the testing phase of the assay. During the testing phase, the treadmill speed was initiated at 10 m/min and brought to 15 m/min after 5 min. Afterward, the treadmill speed was gradually increased by 3 m/min increments every 2 min until no mice remained on the platform. Animals were weighed before and after the exercise assay. The time spent and distance run on the platform were recorded and normalized to the animal’s respective weight.
Metabolic cage
We monitored changes in metabolic function of mice in standard (22°C) conditions using the Promethion Metabolic Cage system. Metabolic function was monitored using the following parameters: (1) food and water consumption, (2) volumes of O2 consumed and CO2 produced, (3) total distance moved, and (4) total movement in the activity wheel. Eight-week-old mice were individually housed under a 12/12-h light/dark cycle with free access to food and water for 3 days. Animals were acclimated to being singly housed in their standard housing room for 3 days prior to beginning the metabolic cage experiment. We excluded the first 24 h as the animals were acclimating to the metabolic cage. Metabolic data were subset for analysis by light and dark cycle and statistically analyzed using the CalR Analysis Tool.71
Plasma cytokine assay
Plasma levels of the cytokines GM-CSF, IFN-γ, IL-1β, IL-2, IL-4, IL-6, IL-10, IL-12p70, MCP-1, and TNF-α were determined using a Multiplexing LASER Bead Assay (Mouse Cytokine Array, Chemokine Array 10-Plex [MDF10], Eve Technologies, Canada).
Homogenization of mouse tissues using RIPA lysis buffer
Mouse tissues were dissected and processed at The Buck Institute. Stainless steel beads (250 mg) (Next Advance, 0.9 2MM BLND RNASE SS BEAD 4ML. SKU SSB14BRNA) were weighed and added to tubes (Axygen; SKU: ST-150-C-S) filled with 200 μL of lysis buffer (1× RIPA buffer [100 mL]: 5 mL of 1 M Tris [pH 7.4], 1 mL Triton X-100, 1 g Na-deoxycholate [0.25%], 3 mL of 5 M NaCl, 200 μL of 0.5M EDTA [pH 8]). Frozen tissues were thawed on ice and 25–30 mg of tissues was weighed and added to fresh 1.5 mL tubes. Tissues were homogenized at speed 8–10 for 5–7 min (speed and time is dependent on tissue type) using a Next Advance Bullet Blender Storm 24 Place Bead Homogenizer (SKU: BBY24M). Tissue lysate was transferred to new 1.5 mL tubes, diluted 1:3 using lysis buffer, and stored at −80°C until future use. One Complete Mini Protease Inhibitor Cocktail Tablet (Sigma-Aldrich, 11836153001) and 100 μL of 100 mM PMSF was added to 10 mL of lysis buffer before starting the experiment.
Protein estimation and SDS-PAGE for mouse tissue lysates
A Pierce BCA Protein Assay Kit (Pierce, cat. no. 23227) was used for the estimation of protein content. Tissues were diluted 1:3 (10 μL tissue + 20 μL of RO water) and 10 μL of sample was added to each well of a microplate in triplicates. Absorbance at 562 nm was measured to estimate protein concentration using a plate reader. Equivalent amounts (25–30 μg/well) of tissue lysates were analyzed on 12% SDS NuPage PAGE gels. Immunoblotting for the epitope tags (anti-FLAG), mitochondrial proteins (anti-ATP5O/OSCP and anti-aconitase), and cytosolic marker/loading control (anti-PGK1 and anti-GAPDH) were achieved using specific antibodies as described above.
BN-PAGE for mouse mitochondria
BN-PAGE was performed on 3%–12% Native PAGE gels (Invitrogen, Carlsbad, CA). Purified mouse liver mitochondria were washed once by diluting into Liver Mito Isolation Medium (250 mM sucrose, 10 mM Tris, 1 mM EGTA) followed by centrifugation at 10,000 × g for 5 min at 4°C. Pellets were lysed by incubating in 2× BN lysis buffer (40 mM HEPES KOH [pH 7.4], 100 mM NaCl, 2 mM EDTA, 20% glycerol, 2 mM PMSF, 2× PIM) and 2% digitonin (v/v) for 30 min on ice. The lysates were centrifuged at 17,000 × g for 30 min. Supernatants were transferred into new tubes and Serva CBG 250 (5 mg/mL) (10× solution: 50 mg/mL Serva CBG 250 in 0.5 M ϵ-aminocaproic acid and 0.1 M Bis-Tris [pH 7.0]) was added to supernatants. The samples were further cleared by centrifugation at 10,000 rpm for 5 min at 4°C prior to loading on gels (25 μg protein/lane). Immunoblotting was performed by transferring the proteins onto PVDF membranes by tank transfer.
In-gel ATPase activity
In-gel ATPase activity was prepared as described previously.27 Purified mitochondria from mouse liver tissues were prepared similarly to that for BN-PAGE. Following lysis and centrifugation, the supernatant was transferred to fresh 1.5 mL Eppendorf tubes and glycerol was added to supernatants for a final concentration of 5%. The samples were then cleared by centrifugation at 10,000 rpm for 5 min at 4°C prior to loading on gels. Clear native-PAGE (CN-PAGE) was performed on 3%–12% native PAGE gels (Invitrogen). After 1 h incubation in equilibration buffer (35 mM Tris-base, 70 mM glycine [pH 8.3]), the gel was incubated for 2 h in ATP synthase complex assay buffer (35 mM Tris-base, 70 mM glycine, 14 mM MgSO4, 8 mM ATP, 0.2% Pb(NO3)2) with gentle shaking (60 rpm) at room temperature (RT). To visualize total protein load in the gel, Coomassie stain (GelCode Blue Stain Reagent; Thermo Fisher Scientific, cat. no. 24590) was added and developed over 1 h.
Proteomic analysis
Protein digestion and desalting
Aliquots of 200 μg protein lysates for each sample were brought to the same overall volume of 100 μL with water, reduced using 20 mM dithiothreitol in 50 mM TEAB at 50°C for 10 min, cooled to RT and held at RT for 10 min, and alkylated using 40 mM iodoacetamide in 50 mM TEAB at RT in the dark for 30 min. Samples were acidified with 12% phosphoric acid to obtain a final concentration of 1.2% phosphoric acid. S-Trap buffer consisting of 90% methanol in 100 mM TEAB at pH ∼7.1, was added and samples were loaded onto the S-Trap micro spin columns. The entire sample volume was spun through the S-Trap micro spin columns at 4,000 × g and RT, binding the proteins to the micro spin columns. Subsequently, S-Trap micro spin columns were washed twice with S-Trap buffer at 4,000 × g and RT and placed into clean elution tubes. Samples were incubated for 1 h at 47°C with sequencing-grade trypsin (Promega, San Luis Obispo, CA) dissolved in 50 mM TEAB at a 1:25 (w/w) enzyme:protein ratio, and then digested overnight at 37°C.
Peptides were sequentially eluted from micro S-Trap spin columns with 50 mM TEAB, 0.5% formic acid (FA) in water, and 50% acetonitrile (ACN) in 0.5% FA. After centrifugal evaporation, samples were resuspended in 0.2% FA in water and desalted with Oasis 10-mg Sorbent Cartridges (Waters, Milford, MA). The desalted elutions were then subjected to an additional round of centrifugal evaporation and re-suspended in 0.1% FA in water at a final concentration of 1 μg/μL. Four microliters of each sample were diluted with 2% ACN in 0.1% FA to obtain a concentration of 200 ng/μL. Two microliters of synthetic peptides (equimolar addition, 1,000-fold dilution) were added to each sample, thus bringing up the total volume to 20 μL.72
Synthetic ATP8 peptides
Details for synthetic ATP8 peptides are provided below. All synthetic peptides were obtained from Vivitide (Louisville, KY) with >99% isotopic purity. The synthetic peptide (1) IYLPHSLˆˆPQQ contains a stable isotope-labeled leucine residue (Lˆˆ with 13C6) with >98% chemical purity. The synthetic peptide (2) IYLPHSLPQQLEQKˆˆ contains a stable isotope-labeled lysine residue (Kˆˆ with 13C6, 15N2) with >98% chemical purity. The remaining synthetic peptides (3) VSSQTFPLAPSPKˆˆ, (4) MPQLDTSTWFITIISSMITLFILFQLKˆˆ, and (5) MPQLTTSTWFITIISSMITLFILFQLKˆˆ each contain a stable isotope-labeled lysine residue (Kˆˆ with 13C6, 15N2) with 95% chemical purity. Synthetic peptides were diluted in 2% ACN and 0.1% FA in H2O for MS analysis. Peptides were dissolved to obtain a concentration of 1 μg/μL and diluted 1,000-fold before being added to prepared digests. Peptides 1–3 were combined as one aliquot and peptides 4 and 5 were combined as another aliquot. We aimed to use these synthetic peptides to quantify the relative expression of allotopically expressed ATP8 protein and endogenous ATP8 protein in the intact ATP synthase of transgenic mice using mass spectrometry.
Mass spectrometric analysis
Reverse-phase HPLC-MS/MS analyses were performed on a Dionex UltiMate 3000 system coupled online to an Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). The solvent system consisted of 2% ACN, 0.1% FA in water (solvent A) and 80% ACN, 0.1% FA in ACN (solvent B). Digested peptides (400 ng) were loaded onto an Acclaim PepMap 100 C18 trap column (0.1 × 20 mm, 5 μm particle size; Thermo Fisher Scientific) over 5 min at 5 μL/min with 100% solvent A. Peptides (400 ng) were eluted on an Acclaim PepMap 100 C18 analytical column (75 μm × 50 cm, 3 μm particle size; Thermo Fisher Scientific) at 300 nL/min using the following gradient: linear from 2.5% to 24.5% of solvent B in 125 min, linear from 24.5% to 39.2% of solvent B in 40 min, up to 98% of solvent B in 1 min, and back to 2.5% of solvent B in 1 min. The column was re-equilibrated for 30 min with 2.5% of solvent B, and the total gradient length was 210 min. Each sample was acquired in data-independent acquisition (DIA) mode.73,74 Full MS spectra were collected at 120,000 resolution (Automatic Gain Control [AGC] target: 3e6 ions, maximum injection time: 60 ms, 350–1,650 m/z), and MS2 spectra at 30,000 resolution (AGC target: 3e6 ions, maximum injection time: auto, normalized collision energy: 30, fixed first mass 200 m/z). The isolation scheme consisted of 26 variable windows covering the 350–1,650 m/z range with an overlap of 1 m/z.75
DIA data processing and statistical analysis
DIA data were processed in Spectronaut (version 17.6.230428.55965) using directDIA. Data extraction parameters were set as dynamic and non-linear item response theory (iRT) calibration with precision iRT was selected. Data were searched against the Mus musculus reference proteome with 58,430 entries accessed on 01/31/2018 (UniProtKB-TrEMBL). DIA data were also searched against sequences for endogenous ATP8, transgenic ATP-8, and ATP-8 with FLAG tag. Trypsin/P was set as the digestion enzyme and two missed cleavages were allowed. Cysteine carbamidomethylation was set as a fixed modification while methionine oxidation and protein N terminus acetylation were set as dynamic modifications. Identification was performed using 1% precursor and protein q value. Quantification was based on the peak areas of extracted ion chromatograms of 3–6 MS2 fragment ions, specifically b and y ions, with local normalization and q value sparse data filtering applied (Table S12). Differential protein expression analysis comparing (1) male control transgenic to male control non-transgenic, (2) female control transgenic to female control non-transgenic, (3) male mutant transgenic to male mutant non-transgenic, or (4) female mutant transgenic to female mutant non-transgenic was performed using a paired t test, and p values were corrected for multiple testing using the Storey method.76 Specifically, group-wise testing corrections were applied to obtain q values. Protein groups with at least two unique peptides, q < 0.01, and absolute Log2(fold change) > 0.58 are significantly altered (Tables S13 and S14).
Statistical analysis
The overall statistical analyses for each test were performed with the GraphPad Prism 10.0 software for Windows, Software (San Diego, CA, www.graphpad.com) assuming a confidence interval (CI) of 95%. Data are presented as means ± SEM and analyzed using two-Way ANOVA with Sidak’s multiple comparisons post-test or one-way ANOVA with Tukey’s post-test for inter-group differences. The significance level was set at α = 0.05. Vmax and Km values were calculated with 95% CI using “Non-linear fit: Michaelis-Menten.” IC50 values with 95% CI were determined through curve fitting using GraphPad Prism 10 (version 10.2) with “Non-linear fit: [Inhibitor] vs. normalized response – Variable slope” (Table S15).
Data and code availability
Raw data and complete MS datasets have been uploaded to the Mass Spectrometry Interactive Virtual Environment (MassIVE) repository, developed by the Center for Computational Mass Spectrometry at the University of California San Diego, and can be downloaded using the following link: https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=368e5a7a28e84af5b0133da7ec9df5df (MassIVE ID no. MSV000093685; ProteomeXchange ID: PXD047911).
Acknowledgments
We would like to thank Dr. Alexandra Stolzing, Dr. Matthew O’Connor, and Ms. Caitlin Lewis for constructive discussions during the early stages of this work. We would also like to thank Dr. Elena Magay and Ms. Anne Corwin for excellent technical support, and Dr. Kathlene Joyce and Mr. Michael Rae for critical feedback. We acknowledge financial support from SENS Research Foundation, LifeSpan.io, Longecity Foundation, and Foster Foundation.
Author contributions
A.B. and M.D.B. conceptualized the study. D.V.B., B.D., C.T., C.D.K., and M.A.W. developed the methodology and performed the experiments. D.V.B., B.D., C.T., M.A.W., C.D.K., and A.B. curated and analyzed the data. A.B. procured the funding. D.V.B., B.D., C.D.K., and A.B. wrote the original draft. M.A.W., B.S., M.D.B., and A.B. supervised the study. All authors contributed to the review of the manuscript and provided final approval.
Declaration of interests
A patent application has been filed on the "Allotopic expression of mtDNA genes" in 2023 in the USA (PCT/US23/76302) (B.D. and A.B.). The authors declare no other competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101372.
Contributor Information
Martin D. Brand, Email: mbrand@buckinstitute.org.
Amutha Boominathan, Email: amutha.boominathan@sens.org.
Supplemental information
References
- 1.Elliott H.R., Samuels D.C., Eden J.A., Relton C.L., Chinnery P.F. Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet. 2008;83:254–260. doi: 10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cwerman-Thibault H., Sahel J.A., Corral-Debrinski M. Mitochondrial medicine: to a new era of gene therapy for mitochondrial DNA mutations. J. Inherit. Metab. Dis. 2011;34:327–344. doi: 10.1007/s10545-010-9131-5. [DOI] [PubMed] [Google Scholar]
- 3.Howell N., Bindoff L.A., McCullough D.A., Kubacka I., Poulton J., Mackey D., Taylor L., Turnbull D.M. Leber hereditary optic neuropathy: identification of the same mitochondrial ND1 mutation in six pedigrees. Am. J. Hum. Genet. 1991;49:939–950. [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace D.C., Singh G., Lott M.T., Hodge J.A., Schurr T.G., Lezza A.M., Elsas L.J., 2nd, Nikoskelainen E.K. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science (New York, N.Y.) 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 5.Gerbitz K.D., Obermaier-Kusser B., Lestienne P., Zierz S., Müller-Höcker J., Pongratz D., Paetzke-Brunner I., Deufel T. Mutations of the mitochondrial DNA: the contribution of DNA techniques to the diagnosis of mitochondrial encephalomyopathies. J. Clin. Chem. Clin. Biochem. 1990;28:241–250. doi: 10.1515/cclm.1990.28.4.241. [DOI] [PubMed] [Google Scholar]
- 6.Shoffner J.M., Lott M.T., Voljavec A.S., Soueidan S.A., Costigan D.A., Wallace D.C. Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: a slip-replication model and metabolic therapy. Proc. Natl. Acad. Sci. USA. 1989;86:7952–7956. doi: 10.1073/pnas.86.20.7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Comte C., Tonin Y., Heckel-Mager A.M., Boucheham A., Smirnov A., Auré K., Lombès A., Martin R.P., Entelis N., Tarassov I. Mitochondrial targeting of recombinant RNAs modulates the level of a heteroplasmic mutation in human mitochondrial DNA associated with Kearns Sayre Syndrome. Nucleic Acids Res. 2013;41:418–433. doi: 10.1093/nar/gks965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van den Ouweland J.M., de Klerk J.B., van de Corput M.P., Dirks R.W., Raap A.K., Scholte H.R., Huijmans J.G., Hart L.M., Bruining G.J., Maassen J.A. Characterization of a novel mitochondrial DNA deletion in a patient with a variant of the Pearson marrow-pancreas syndrome. Eur. J. Hum. Genet. 2000;8:195–203. doi: 10.1038/sj.ejhg.5200444. [DOI] [PubMed] [Google Scholar]
- 9.Larsson N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010;79:683–706. doi: 10.1146/annurev-biochem-060408-093701. [DOI] [PubMed] [Google Scholar]
- 10.Zhao X., Zhang Y., Lu L., Yang H. Therapeutic Effects of Idebenone on Leber Hereditary Optic Neuropathy. Curr. Eye Res. 2020;45:1315–1323. doi: 10.1080/02713683.2020.1736307. [DOI] [PubMed] [Google Scholar]
- 11.Enns G.M., Cohen B.H. Clinical Trials in Mitochondrial Disease: An Update on EPI-743 and RP103. J. Inborn Errors Metab. Screen. 2017;5 doi: 10.1177/2326409817733013. [DOI] [Google Scholar]
- 12.Bax B.E., Levene M., Bain M.D., Fairbanks L.D., Filosto M., Kalkan Uçar S., Klopstock T., Kornblum C., Mandel H., Rahman S., et al. Erythrocyte Encapsulated Thymidine Phosphorylase for the Treatment of Patients with Mitochondrial Neurogastrointestinal Encephalomyopathy: Study Protocol for a Multi-Centre, Multiple Dose, Open Label Trial. J. Clin. Med. 2019;8:1096. doi: 10.3390/jcm8081096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirano M., Carelli V., De Giorgio R., Pironi L., Accarino A., Cenacchi G., D'Alessandro R., Filosto M., Martí R., Nonino F., et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): Position paper on diagnosis, prognosis, and treatment by the MNGIE International Network. J. Inherit. Metab. Dis. 2021;44:376–387. doi: 10.1002/jimd.12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sendra L., García-Mares A., Herrero M.J., Aliño S.F. Mitochondrial DNA Replacement Techniques to Prevent Human Mitochondrial Diseases. Int. J. Mol. Sci. 2021;22:551. doi: 10.3390/ijms22020551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang J.X., Pyle A., Taylor R.W., Oláhová M. Interrogating Mitochondrial Biology and Disease Using CRISPR/Cas9 Gene Editing. Genes. 2021;12:1604. doi: 10.3390/genes12101604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang X., Jiang J., Li Z., Liang J., Xiang Y. Strategies for mitochondrial gene editing. Comput. Struct. Biotechnol. J. 2021;19:3319–3329. doi: 10.1016/j.csbj.2021.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nissanka N., Moraes C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020;21 doi: 10.15252/embr.201949612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin T., Luo J., Huang D., Li H. Current Progress of Mitochondrial Genome Editing by CRISPR. Front. Physiol. 2022;13 doi: 10.3389/fphys.2022.883459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan W., Waymire K.G., Narula N., Li P., Rocher C., Coskun P.E., Vannan M.A., Narula J., Macgregor G.R., Wallace D.C. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science (New York, N.Y.) 2008;319:958–962. doi: 10.1126/science.1147786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoue K., Nakada K., Ogura A., Isobe K., Goto Y., Nonaka I., Hayashi J.I. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat. Genet. 2000;26:176–181. doi: 10.1038/82826. [DOI] [PubMed] [Google Scholar]
- 21.Weiss H., Wester-Rosenloef L., Koch C., Koch F., Baltrusch S., Tiedge M., Ibrahim S. The mitochondrial Atp8 mutation induces mitochondrial ROS generation, secretory dysfunction, and β-cell mass adaptation in conplastic B6-mtFVB mice. Endocrinology. 2012;153:4666–4676. doi: 10.1210/en.2012-1296. [DOI] [PubMed] [Google Scholar]
- 22.Lin C.S., Sharpley M.S., Fan W., Waymire K.G., Sadun A.A., Carelli V., Ross-Cisneros F.N., Baciu P., Sung E., McManus M.J., et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl. Acad. Sci. USA. 2012;109:20065–20070. doi: 10.1073/pnas.1217113109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu H., Ozdemir S.S., Koilkonda R.D., Chou T.H., Porciatti V., Chiodo V., Boye S.L., Hauswirth W.W., Lewin A.S., Guy J. Mutant NADH dehydrogenase subunit 4 gene delivery to mitochondria by targeting sequence-modified adeno-associated virus induces visual loss and optic atrophy in mice. Mol. Vis. 2012;18:1668–1683. [PMC free article] [PubMed] [Google Scholar]
- 24.Dunn D.A., Pinkert C.A. Nuclear expression of a mitochondrial DNA gene: mitochondrial targeting of allotopically expressed mutant ATP6 in transgenic mice. J. Biomed. Biotechnol. 2012;2012 doi: 10.1155/2012/541245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vempati U.D., Torraco A., Moraes C.T. Mouse models of oxidative phosphorylation dysfunction and disease. Methods (San Diego, CA, U. S.) 2008;46:241–247. doi: 10.1016/j.ymeth.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruzzenente B., Rötig A., Metodiev M.D. Mouse models for mitochondrial diseases. Hum. Mol. Genet. 2016;25:R115–R122. doi: 10.1093/hmg/ddw176. [DOI] [PubMed] [Google Scholar]
- 27.Boominathan A., Vanhoozer S., Basisty N., Powers K., Crampton A.L., Wang X., Friedricks N., Schilling B., Brand M.D., O'Connor M.S. Stable nuclear expression of ATP8 and ATP6 genes rescues a mtDNA Complex V null mutant. Nucleic Acids Res. 2016;44:9342–9357. doi: 10.1093/nar/gkw756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao C., Farruggio A.P., Bjornson C.R.R., Chavez C.L., Geisinger J.M., Neal T.L., Karow M., Calos M.P. Recombinase-mediated reprogramming and dystrophin gene addition in mdx mouse induced pluripotent stem cells. PLoS One. 2014;9 doi: 10.1371/journal.pone.0096279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manfredi G., Fu J., Ojaimi J., Sadlock J.E., Kwong J.Q., Guy J., Schon E.A. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat. Genet. 2002;30:394–399. doi: 10.1038/ng851. [DOI] [PubMed] [Google Scholar]
- 30.Tasic B., Hippenmeyer S., Wang C., Gamboa M., Zong H., Chen-Tsai Y., Luo L. Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proc. Natl. Acad. Sci. USA. 2011;108:7902–7907. doi: 10.1073/pnas.1019507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X., Gimsa U., Wester-Rosenlöf L., Kanitz E., Otten W., Kunz M., Ibrahim S.M. Dissecting the effects of mtDNA variations on complex traits using mouse conplastic strains. Genome Res. 2009;19:159–165. doi: 10.1101/gr.078865.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saravanan S., Lewis C.J., Dixit B., O'Connor M.S., Stolzing A., Boominathan A. The Mitochondrial Genome in Aging and Disease and the Future of Mitochondrial Therapeutics. Biomedicines. 2022;10:490. doi: 10.3390/biomedicines10020490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewis C.J., Dixit B., Batiuk E., Hall C.J., O'Connor M.S., Boominathan A. Codon optimization is an essential parameter for the efficient allotopic expression of mtDNA genes. Redox Biol. 2020;30 doi: 10.1016/j.redox.2020.101429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banroques J., Delahodde A., Jacq C. A mitochondrial RNA maturase gene transferred to the yeast nucleus can control mitochondrial mRNA splicing. Cell. 1986;46:837–844. doi: 10.1016/0092-8674(86)90065-6. [DOI] [PubMed] [Google Scholar]
- 35.Banroques J., Perea J., Jacq C. Efficient splicing of two yeast mitochondrial introns controlled by a nuclear-encoded maturase. EMBO J. 1987;6:1085–1091. doi: 10.1002/j.1460-2075.1987.tb04862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchirico M., Tzellas A., Fox T.D., Conrad-Webb H., Periman P.S., Mason T.L. Relocation of the unusual VAR1 gene from the mitochondrion to the nucleus. Biochem. Cell Biol. 1995;73:987–995. doi: 10.1139/o95-106. [DOI] [PubMed] [Google Scholar]
- 37.Pineau B., Mathieu C., Gérard-Hirne C., De Paepe R., Chétrit P. Targeting the NAD7 subunit to mitochondria restores a functional complex I and a wild type phenotype in the Nicotiana sylvestris CMS II mutant lacking nad7. J. Biol. Chem. 2005;280:25994–26001. doi: 10.1074/jbc.M500508200. [DOI] [PubMed] [Google Scholar]
- 38.Hoffmann A., Hildebrandt V., Heberle J., Büldt G. Photoactive mitochondria: in vivo transfer of a light-driven proton pump into the inner mitochondrial membrane of Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. USA. 1994;91:9367–9371. doi: 10.1073/pnas.91.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagley P., Farrell L.B., Gearing D.P., Nero D., Meltzer S., Devenish R.J. Assembly of functional proton-translocating ATPase complex in yeast mitochondria with cytoplasmically synthesized subunit 8, a polypeptide normally encoded within the organelle. Proc. Natl. Acad. Sci. USA. 1988;85:2091–2095. doi: 10.1073/pnas.85.7.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zullo S.J., Parks W.T., Chloupkova M., Wei B., Weiner H., Fenton W.A., Eisenstadt J.M., Merril C.R. Stable transformation of CHO Cells and human NARP cybrids confers oligomycin resistance (oli(r)) following transfer of a mitochondrial DNA-encoded oli(r) ATPase6 gene to the nuclear genome: a model system for mtDNA gene therapy. Rejuvenation Res. 2005;8:18–28. doi: 10.1089/rej.2005.8.18. [DOI] [PubMed] [Google Scholar]
- 41.Newman N.J., Yu-Wai-Man P., Subramanian P.S., Moster M.L., Wang A.G., Donahue S.P., Leroy B.P., Carelli V., Biousse V., Vignal-Clermont C., et al. Randomized trial of bilateral gene therapy injection for m.11778G>A MT-ND4 Leber optic neuropathy. Brain. 2023;146:1328–1341. doi: 10.1093/brain/awac421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perales-Clemente E., Fernández-Silva P., Acín-Pérez R., Pérez-Martos A., Enríquez J.A. Allotopic expression of mitochondrial-encoded genes in mammals: achieved goal, undemonstrated mechanism or impossible task? Nucleic Acids Res. 2011;39:225–234. doi: 10.1093/nar/gkq769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chin R.M., Panavas T., Brown J.M., Johnson K.K. Optimized Mitochondrial Targeting of Proteins Encoded by Modified mRNAs Rescues Cells Harboring Mutations in mtATP6. Cell Rep. 2018;22:2818–2826. doi: 10.1016/j.celrep.2018.02.059. [DOI] [PubMed] [Google Scholar]
- 44.Bonnet C., Augustin S., Ellouze S., Bénit P., Bouaita A., Rustin P., Sahel J.A., Corral-Debrinski M. genes restores respiratory chain complex I activity in fibroblasts harboring mutations in these genes. Biochim. Biophys. Acta. 2008;1783:1707–1717. doi: 10.1016/j.bbamcr.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 45.Borna N.N., Kishita Y., Shimura M., Murayama K., Ohtake A., Okazaki Y. Identification of a novel MT-ND3 variant and restoring mitochondrial function by allotopic expression of MT-ND3 gene. Mitochondrion. 2024;76 doi: 10.1016/j.mito.2024.101858. [DOI] [PubMed] [Google Scholar]
- 46.Golani-Armon A., Arava Y. Localization of Nuclear-Encoded mRNAs to Mitochondria Outer Surface. Biochemistry. 2016;81:1038–1043. doi: 10.1134/S0006297916100023. [DOI] [PubMed] [Google Scholar]
- 47.Fazal F.M., Han S., Parker K.R., Kaewsapsak P., Xu J., Boettiger A.N., Chang H.Y., Ting A.Y. Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell. 2019;178:473–490.e26. doi: 10.1016/j.cell.2019.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaltimbacher V., Bonnet C., Lecoeuvre G., Forster V., Sahel J.A., Corral-Debrinski M. mRNA localization to the mitochondrial surface allows the efficient translocation inside the organelle of a nuclear recoded ATP6 protein. RNA (N. Y.) 2006;12:1408–1417. doi: 10.1261/rna.18206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonnet C., Kaltimbacher V., Ellouze S., Augustin S., Bénit P., Forster V., Rustin P., Sahel J.A., Corral-Debrinski M. Allotopic mRNA localization to the mitochondrial surface rescues respiratory chain defects in fibroblasts harboring mitochondrial DNA mutations affecting complex I or v subunits. Rejuvenation Res. 2007;10:127–144. doi: 10.1089/rej.2006.0526. [DOI] [PubMed] [Google Scholar]
- 50.Liu Y., Eastwood J.D., Alba D.E., Velmurugan S., Sun N., Porciatti V., Lee R.K., Hauswirth W.W., Guy J., Yu H. Gene therapy restores mitochondrial function and protects retinal ganglion cells in optic neuropathy induced by a mito-targeted mutant ND1 gene. Gene Ther. 2022;29:368–378. doi: 10.1038/s41434-022-00333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friedrich G., Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- 52.Sadelain M., Papapetrou E.P., Bushman F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer. 2011;12:51–58. doi: 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
- 53.Shrestha D., Bag A., Wu R., Zhang Y., Tang X., Qi Q., Xing J., Cheng Y. Genomics and epigenetics guided identification of tissue-specific genomic safe harbors. Genome Biol. 2022;23:199. doi: 10.1186/s13059-022-02770-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aznauryan E., Yermanos A., Kinzina E., Devaux A., Kapetanovic E., Milanova D., Church G.M., Reddy S.T. Discovery and validation of human genomic safe harbor sites for gene and cell therapies. Cell Rep. Methods. 2022;2 doi: 10.1016/j.crmeth.2021.100154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eipel C., Hildebrandt A., Scholz B., Schyschka L., Minor T., Kreikemeyer B., Ibrahim S.M., Vollmar B. Mutation of mitochondrial ATP8 gene improves hepatic energy status in a murine model of acute endotoxemic liver failure. Life Sci. 2011;88:343–349. doi: 10.1016/j.lfs.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 56.Schilf P., Künstner A., Olbrich M., Waschina S., Fuchs B., Galuska C.E., Braun A., Neuschütz K., Seutter M., Bieber K., et al. A Mitochondrial Polymorphism Alters Immune Cell Metabolism and Protects Mice from Skin Inflammation. Int. J. Mol. Sci. 2021;22:1006. doi: 10.3390/ijms22031006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen K., Wallis J.W., McLellan M.D., Larson D.E., Kalicki J.M., Pohl C.S., McGrath S.D., Wendl M.C., Zhang Q., Locke D.P., et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li R., Zhu H., Ruan J., Qian W., Fang X., Shi Z., Li Y., Li S., Shan G., Kristiansen K., et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 63.Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2016;44:D67–D72. doi: 10.1093/nar/gkv1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frezza C., Cipolat S., Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 65.Cammarata R.J., Rodnan G.P., Fennell R.H. Serum anti-gamma-globulin and antinuclear factors in the aged. JAMA. 1967;199:455–458. [PubMed] [Google Scholar]
- 66.Mookerjee S.A., Quinlan C.L., Wong H.S., Dighe P., Brand M.D. Plate-Based Measurement of Respiration by Isolated Mitochondria. Methods Mol. Biol. 2018;1782:301–313. doi: 10.1007/978-1-4939-7831-1_17. [DOI] [PubMed] [Google Scholar]
- 67.Rogers G.W., Brand M.D., Petrosyan S., Ashok D., Elorza A.A., Ferrick D.A., Murphy A.N. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Divakaruni A.S., Andreyev A.Y., Rogers G.W., Murphy A.N. In situ measurements of mitochondrial matrix enzyme activities using plasma and mitochondrial membrane permeabilization agents. Anal. Biochem. 2018;552:60–65. doi: 10.1016/j.ab.2017.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mookerjee S.A., Brand M.D. Measurement and Analysis of Extracellular Acid Production to Determine Glycolytic Rate. J. Vis. Exp. 2015;106 doi: 10.3791/53464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dougherty J.P., Springer D.A., Gershengorn M.C. The Treadmill Fatigue Test: A Simple, High-throughput Assay of Fatigue-like Behavior for the Mouse. J. Vis. Exp. 2016;111 doi: 10.3791/54052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mina A.I., LeClair R.A., LeClair K.B., Cohen D.E., Lantier L., Banks A.S. CalR: A Web-Based Analysis Tool for Indirect Calorimetry Experiments. Cell Metabol. 2018;28:656–666.e1. doi: 10.1016/j.cmet.2018.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bruderer R., Bernhardt O.M., Gandhi T., Xuan Y., Sondermann J., Schmidt M., Gomez-Varela D., Reiter L. Optimization of Experimental Parameters in Data-Independent Mass Spectrometry Significantly Increases Depth and Reproducibility of Results. Mol. Cell. Proteomics. 2017;16:2296–2309. doi: 10.1074/mcp.RA117.000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Burger T. Gentle Introduction to the Statistical Foundations of False Discovery Rate in Quantitative Proteomics. J. Proteome Res. 2018;17:12–22. doi: 10.1021/acs.jproteome.7b00170. [DOI] [PubMed] [Google Scholar]
- 74.Escher C., Reiter L., MacLean B., Ossola R., Herzog F., Chilton J., MacCoss M.J., Rinner O. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. 2012;12:1111–1121. doi: 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Collins B.C., Hunter C.L., Liu Y., Schilling B., Rosenberger G., Bader S.L., Chan D.W., Gibson B.W., Gingras A.C., Held J.M., et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 2017;8:291. doi: 10.1038/s41467-017-00249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gillet L.C., Navarro P., Tate S., Röst H., Selevsek N., Reiter L., Bonner R., Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data and complete MS datasets have been uploaded to the Mass Spectrometry Interactive Virtual Environment (MassIVE) repository, developed by the Center for Computational Mass Spectrometry at the University of California San Diego, and can be downloaded using the following link: https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=368e5a7a28e84af5b0133da7ec9df5df (MassIVE ID no. MSV000093685; ProteomeXchange ID: PXD047911).