

Abstract
Background
Hereditary angioedema (HAE) in children has specific features and requires multidisciplinary management.
Methods
We performed a literature search and underwent in‐depth discussions to provide practical tools for physicians.
Results
HAE is a rare, life‐threatening genetic disorder. Its epidemiology is poorly documented in children. Clinical manifestations usually appear during childhood or early adolescence. Classical signs, often preceded by prodromal symptoms, include transient, localized, non‐pitting, non‐pruritic swelling of deep dermal/subcutaneous or mucosal/submucosal tissues, leading to oedema of the extremities, face, lips, tongue, trunk and genitals, recurring gastrointestinal symptoms and laryngeal edema possibly causing asphyxiation and death. Diagnosis is often delayed due to low awareness in the medical community, and particularly challenging in case of isolated abdominal crises or atypical presentation and in neonates or infants. It relies on biological tests (measurement of serum/plasma levels of C1INH function, C1INH protein, and C4), genetic testing in selected cases, and imaging for differential diagnosis of acute abdominal crises. Main differential diagnosis for peripheral oedema is mast cell‐mediated oedema that accounts for 95% of angioedema in clinical practice. Quality of life can be significantly impaired. Disease management includes treatment of attacks, short‐term and long‐term prophylaxis, psychological support, avoidance of triggers, patients’ and parents’ education and coordination of all stakeholders, ideally within a specialized healthcare network. New plasma kallikrein inhibitors, namely lanadelumab (subcutaneous route) and berotralstat (oral route) have facilitated long‐term prophylaxis thanks to improved usability.
Conclusion
Diagnostic and treatment of HAE are particularly challenging in children and require specific management by multiple stakeholders.
Keywords: daily diary, iatrogenic, suicide ideation
Abbreviations
- ACEi
Angiotensin‐converting enzyme (ACE) inhibitor
- C1INH
C1 inhibitor
- HAE
hereditary angioedema
- HAE‐I
hereditary angioedema type I
- HAE‐II
hereditary angioedema type II
- HAE‐C1INH
hereditary angioedema due to C1 inhibitor deficiency
- HAE‐nC1INH
hereditary angioedema with normal C1 inhibitor
- HaT
hereditary alpha tryptasemia
- CREAK
French National Reference Center for Angioedema
Key message.
Very few publications on HAE are dedicated to pediatric patients, in particular, exhaustive general reviews are missing. As major changes in the management of HAE children are currently implemented thanks to availability of new drugs, this article aims at presenting an overview on diagnosis and management, to provide pediatricians who are not familiar with this rare disease with essential knowledge and valuable tips for their daily practice.
1. INTRODUCTION
Hereditary angioedema (HAE) is a rare, life‐threatening genetic disorder characterized by acute, recurrent, and unpredictable episodes of cutaneous or submucosal angioedema, mediated by bradykinin, due to C1 inhibitor (C1INH) abnormalities in the vast majority of cases. 1 C1INH is the main primary inhibitor of plasma kallikrein, regulating the release of bradykinin, acting at several steps of the cascade by inhibition of plasmin, coagulation factor XII (Hageman) and kallikrein activity. 2 , 3 Insufficient activity of C1INH results therefore in the overproduction of bradykinin, which increases permeability of blood vessels leading to tissue swelling 4 (Figure 1). The C1INH protein is a member of the serine protease inhibitor (serpin) superfamily. The gene coding for C1INH called SERPING1 is located on chromosome 11. 4
FIGURE 1.
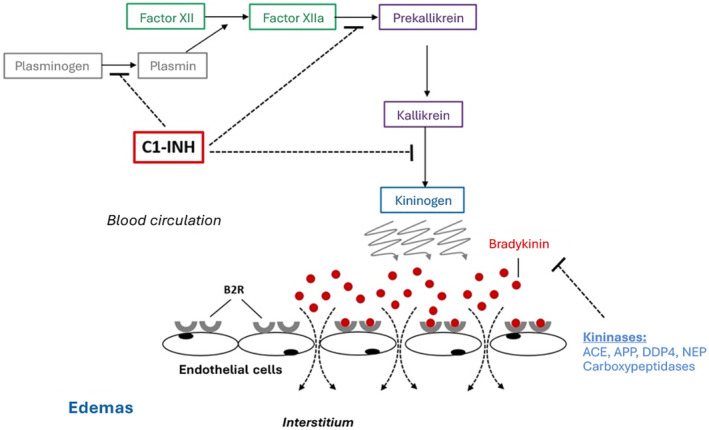
Pathophysiology of C1‐INH HAE. C1‐INH, C1 inhibitor; ACE, Angiotensin converting enzyme; APP, Aminopeptidase P; DPP4, Dipeptidylpeptidase‐4; NEP, Neprilysin; B2R, Bradykinin type 2 constitutive receptor.
HAE can be classified into 3 groups 3 , 5 :
HAE type I (HAE‐I), related to C1INH deficiency, with low antigenic and functional C1INH levels,
HAE type II (HAE‐II) due to C1INH dysfunction, with normal antigenic but low functional C1INH levels.
HAE with normal C1INH (HAE‐nC1INH) characterized by normal C1INH antigenic and functional levels, resulting from mutations in various genes other than SERPING1. Mutations in FXII, ANGPT1, PLG, KNG1, MYOF, or HS3ST6 have been described. Recently, new mutations linked to HAE with normal C1INH have been described, namely a missense variant (p.D239N) in DAB2IP, located in the C2‐domain 6 and 3 variants of the CPN1 gene encoding the catalytic 55‐kDa subunit of CPN: c.533G>A, c.582A>G, and c.734C>T. 7
HAE types I and II are referred to as HAE‐C1INH.
HAE type I accounts for 85% of HAE‐C1INH while HAE‐nC1INH is very rare.
Most SERPING1 mutations consist of missense variants or deletions, duplications, and indels. 8 Although harboring the same mutation, individuals may experience various degrees of disease severity. 8
2. EPIDEMIOLOGY
HAE‐C1INH being an autosomal dominant disease, the risk of being affected is 50% for the offspring. However, 25% of patients exhibit de novo mutations in SERPING1. 4
While the overall prevalence of HAE is estimated between 1/30,000 and 1/80,000, 9 limited data are available on the epidemiology of the disease in children. A French study collected cases of patients who were ageless than 18 years at the date of March 2013, diagnosed with HAE‐C1INH or factor XII mutation before the 31st of August 2014, registered through the French National Reference Center of angioedema (CREAK). 10 In total, 101 patients were identified, among whom 71%, 16%, and 13% were classified HAE‐I, HAE‐II, and factor XII mutated, respectively.
In contrast with HAE, non‐hereditary angioedema is a frequent disease with lifetime prevalence estimated around 5%–7.5% 11 and in the vast majority of cases, angioedema is due to non‐specific mast‐cell activation and degranulation, also referred to as mast cell‐mediated edema. 12 However accurate epidemiological figures are unknown, in particular, the distribution of allergic and non‐allergic cases for mast‐cell related cases, or the percentages of angiotensin‐converting enzyme inhibitors (ACEi)‐related bradykinin‐related cases are not documented in children (Figure 2). Exposure to ACEi is low in this population but their role in angioedema might be underestimated.
FIGURE 2.
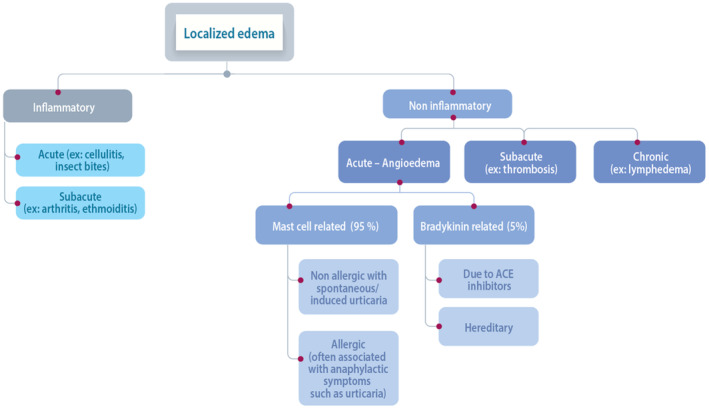
Etiologies of localized edema and distribution of patients.
3. CLINICAL PRESENTATION OF HAE IN CHILDREN
3.1. Clinical signs and symptoms
Several cohorts of children with HAE‐C1INH have been published. Those reporting detailed data or presenting the highest numbers of patients are summarized in Table 1. Other series include a set of 111 children in the United Kingdom, 13 25 in the United States, 14 41 in Denmark, 15 30 in Iran. 16 Publication of data on 269 French pediatric patients with HAE‐C1INH or HAE‐nC1INH is expected soon (submission ongoing). In HAE‐C1INH, clinical manifestations are rare in neonates and infant but usually appear during childhood or early adolescence. 3 Age at symptom onset varies across series. It is commonly admitted that 50%–75% and 90% of patients have experienced first symptoms by the age of 12 and 20 years, respectively. 17 In some series, the mean age at symptom onset is very young (3.3 years) and more than 14% of patients experience first clinical manifestations before the age of 1 year. 18 In other cohorts, mean age at symptom onset is around 5–7 years. 10 , 19 , 20 Of note, attacks appear earlier in females than in males. 3 Most studies show that patients with early symptom onset experience a more severe course of disease than those who have first symptoms later in life. 21 , 22 For instance, in the French cohort, severe episodes occurred in 38.7% and 25% of children who had first symptoms before or after the age of 5 years, respectively. 10 , 19
TABLE 1.
Patients' characteristics in pediatric cohorts.
| United States Nanda 2015 | Italy Cancian 2019 | United States Tachdjian 2020 | France Dermesropian 2015 | Hungary Andrasi 2022 | Brazil Araujo‐Simoes | |
|---|---|---|---|---|---|---|
| Study type | Retrospective, single center study | Retrospective and prospective, 17 HAE reference centers | Retrospective study (administrative claims database of HAE‐related treatments) | Prospective ongoing study (national database) | Retrospective single center analysis HAE‐C1INH | Retrospective analysis 17 HAE centers |
| Inclusion criteria | HAE‐C1INH, age ≤18 years at diagnosis | HAE, ≤21 years for the pediatric cohort | ≥ pharmacy claim for HAE‐related treatment | HAE‐C1INH or factor XII mutated HAE, age ≤18 years, diagnosis before August 2014 | Diagnosis between 2001 and 2020. Age < 19 years at diagnosis | Age <18 years, confirmed diagnosis of HAE‐C1INH |
| N | 21 | 54 / 983 patients in total | 143/ 1429 patients in total | 101 | 49 | 95 |
| Males/females, % | 71/29 | 59/41 | 41/59 | 52/49 | 47/53 | 54/46 |
| Family history, % | 86 | 90.4 | 73 | 84 | ||
|
Median age (IQR) Mean age ± SD |
13.2 (9.1–18.8) | 11.8 ± 5.4 | 12.3 (4.2) | 7 | ||
| Median age at symptom onset (IQR) | 5.7 (5–9) | 4.9 | 6.5 (0.5–18) | 3.3 | ||
| Diagnosis before age of 10 | 13/15 | |||||
| Median age at diagnosis | 5.0 (4–8) | 5.5 | 6.7 b (0–18) | 7.2 | ||
| Asymptomatic, n (%) | 5 (24) | 17 (32) | 33% | 15 (16) | ||
| Abdominal attacks, % | 93 a | 60 | 57 | |||
| Peripheral attacks, % | 73 | 82 | 73.5 | |||
| Laryngeal attacks, % | 27 | 29 | 10 | |||
| Comorbidities, % | ||||||
| No | 65 | Not provided | ||||
| Atopy/allergy/anaphylaxis | 15 | 57.3 | ||||
| Respiratory infections | 10 | |||||
| Anxiety or depression | 24.5 | |||||
| Fluid and electrolyte disorders | 16.1 | |||||
| Urticaria | 13.3 | |||||
| Mean ER visits per year (IQR) | 1.4 (0–12) |
At least once.
Mean.
Despite the early onset of symptoms, diagnosis is often delayed due to low awareness in the medical community, low specificity of symptoms especially abdominal crises very frequent in children, and absence of family history in 25% of cases. 21 In pediatric cohorts, mean age at symptom onset and mean age at diagnosis are close, although children with family history are diagnosed a few years earlier since systematic presymptomatic screening is recommended although not always implemented. 15 , 19 Diagnosis is also made earlier in reference centers. In contrast, in cohorts of adult patients with HAE, reported diagnostic delay ranges from 8 to 20 years, 21 , 22 , 23 with median ages at diagnosis around 19–23 years. 22 , 24 , 25
In this context, diagnosis is rarely made by pediatricians. The ongoing Icatibant Outcome Survey that includes HAE‐C1INH patients showed that pediatricians and pediatrician‐immunologists diagnosed only 3.0% and 2.5% of patients, respectively, despite a median age at symptom onset of 12 years and a family history in 73% of cases. 25 In this study, most patients were diagnosed by an allergist (29.5%), a clinical immunologist (18.3%), or a dermatologist (16.1%). Delay in diagnosis was however the shortest when made by a pediatrician (median of 1.1 year). Diagnostic delay can be still higher in low‐ and middle‐income countries. 26
Classical signs of disease consist of transient, localized, non‐pitting, non‐pruritic swelling of deep dermal/subcutaneous or mucosal/submucosal tissues due to increased permeability of blood vessels, leading to edema of the extremities, face, lips, tongue, trunk and genitals, recurring gastrointestinal symptoms and laryngeal edema 27 (Figure 3).
FIGURE 3.
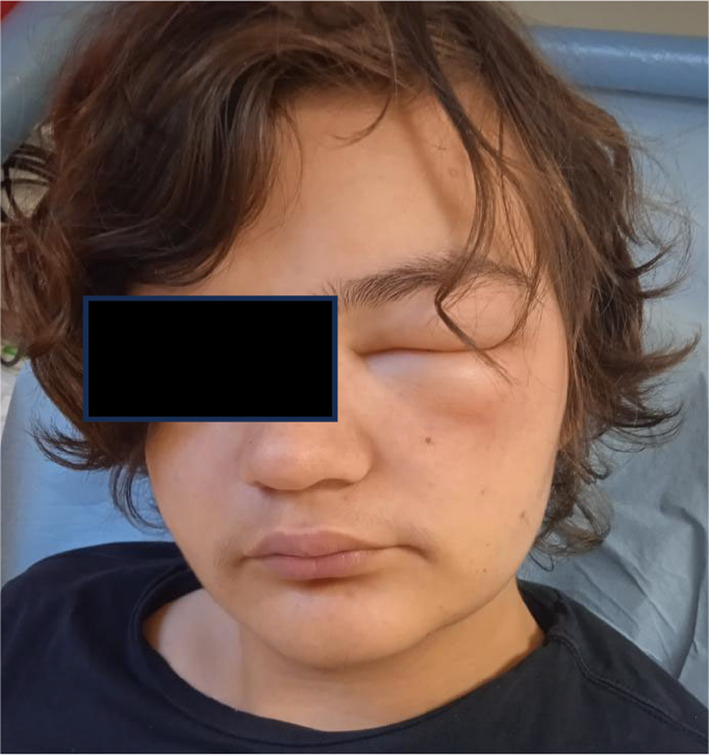
Facial edema.
When occurring in loose tissues, swelling can provoke significant deformation. 12 In boys, edema can provoke paraphimosis. Laryngeal edema is quite rare in young children 15 , 21 but it can lead to asphyxiation and death, especially due to the small diameter of airways. Most frequent presenting symptoms are swelling of upper extremities associated with various degrees of disability, and abdominal pain. 14 Peripheral edema can be very severe, to such an extent that fasciotomy has been described to treat compartment syndrome. 4 Crises can occur spontaneously, but in some cases, they are provoked by various triggers (Figure 4). Of note, if a trauma triggers an attack, a radiograph should be performed to exclude a fracture, which could be incomplete in children (greenstick fracture) and hidden by edema.
FIGURE 4.
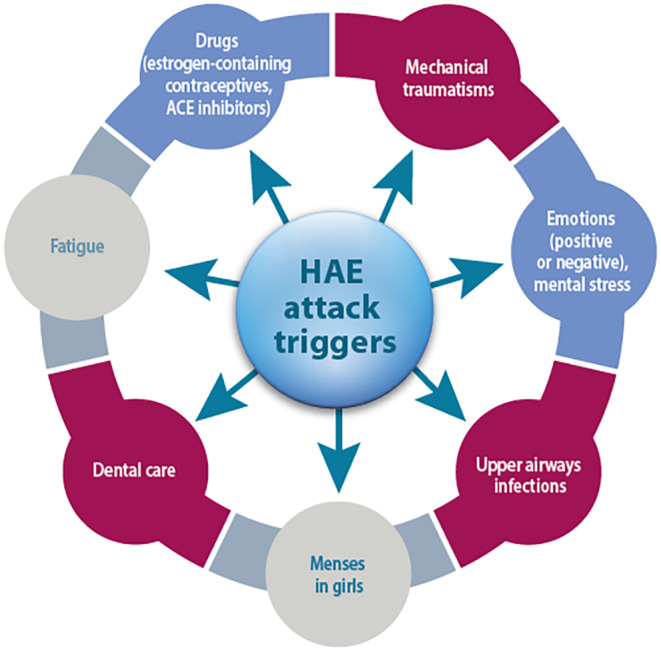
Most frequent triggers provoking HAE attacks.
Abdominal crises manifest as abdominal pain that is usually severe and accompanied by abdominal distension, nausea, vomiting and/or watery diarrhea, prostration, pallor, and dehydration, more rarely ascites and pleural effusion being signs of severity. 21 , 28 A visual analogic scale (VAS) can be used to evaluate acute pain, but iterative utilization by parents or caregivers could trigger a nocebo effect. 29 Moreover, VAS has been developed for adults and its use has been extrapolated in children without strong validation. Faces pain scale can be used by young children who cannot use VAS. In the youngest, who are unable to describe their symptoms and graduate their pain, crying, mood changes and tantrums can be the only manifestations in case of isolated abdominal crises. 30 In this case, specific heteroevaluation tools can be useful to help parents, such as the EVENDOL scale 31 (https://pediadol.org/wp‐content/uploads/2022/03/Evendol‐A4‐05‐2021.pdf). Diagnosis must be considered in rare case of recurrent severe laryngitis otherwise unexplained.
Other sites may be affected by edema in children, including urinary bladder, kidneys, muscles, joints, pericardium, pleura, and central nervous system. 32 These manifestations are however rare in clinical practice.
In patients with HAE‐nC1INH, the disease usually begins later, during adolescence or in young adults, penetrance is incomplete across sexes, and the influence of estrogens is more pronounced in girls. 21 , 27 , 33 Clinical presentation of HAE‐nC1INH due to factor XII mutations has been described in several cohorts of patients from France, Brazil, Germany, or Spain 34 , 35 , 36 , 37 , 38 (Box 1). In Brazilian patients, 37 a high percentage of symptomatic males (53%) and patients with estrogen‐independent FXII‐HAE suggests that searching for FXII mutations is relevant even in males. Due to a disease onset usually delayed in patients with factor XII mutations, occurring during late adolescence or in young adults, the diagnosis might not be made by pediatricians.
BOX 1. Characteristics of HAE‐nC1INH due to factor XII mutations.
Later onset of symptoms (around 20 years of age).
Female predominance (75%–80%).
Asymptomatic forms frequent in males (except in the Brazilian cohort 37 ), infrequent in women.
Influence of estrogens (frequent onset at puberty or at initiation of contraception, exacerbation during estrogen‐based contraception or during pregnancy).
Prodromal symptoms are often reported. The most frequent and most specific in children is erythema marginatum, present in approximately 50% of cases, that can be, however, misdiagnosed as urticaria or livedo. 3 , 21 It consists of a macular erythematous‐serpiginous, fleeting, non‐pruritic, non‐raised rash (Figure 5). Other possible prodromes are less specific and vary across individuals and affected sites. They include fatigue, muscle pain, abdominal pain and nausea, and skin tingling in the affected area 12‐24H before an attack. 21 , 30
FIGURE 5.
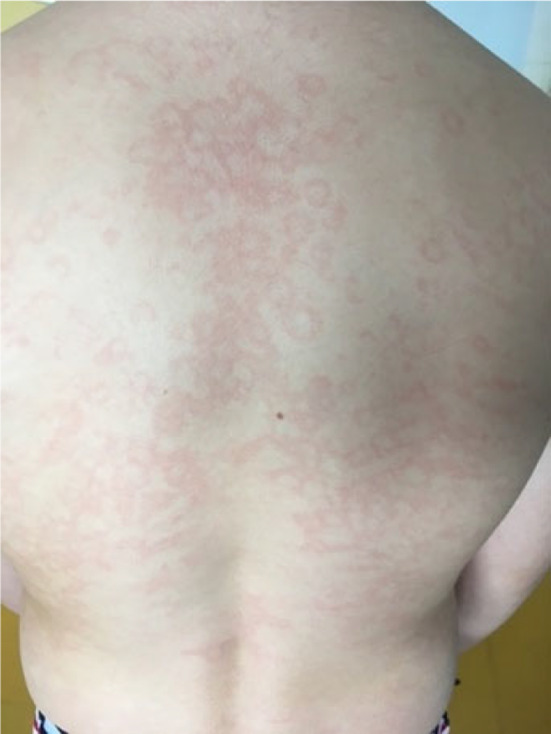
Example of erythema marginatum.
The number and severity of attacks peak at 3–6 years of age and around puberty. 39 In the French cohort, 22% of children had a history of severe crises (abdominal, laryngeal, or face). 10 In a Brazilian study, clinical presentation was low, moderate, and severe in 37.5%, 30%, and 27.5%, respectively. 18 Main factors that should make clinicians evoke HAE diagnosis in children are listed in Box 2.
BOX 2. Situations in which HAE diagnosis should be considered in children.
Familial history.
Recurrent angioedema (face, extremities, genitals, and upper airways).
Recurrent unexplained abdominal crises.
Erythema marginatum.
Absence of urticarial.
No response to antihistamines, adrenaline, corticosteroids and omalizumab.
3.2. Impact on quality of life
The impact of symptoms on quality of life (QoL) is very important but difficult to assess since there are no HAE‐specific and validated tools for the youngest. Nevertheless, QoL has been described in a few pediatric studies.
A Danish study concluded that QoL scores (PedsQL) in HAE‐C1INH children (median age 12 years) were comparable on average with normal scores for healthy children. 40 However, the sample size was very small (n = 14) and the study had no control group. Of note, children with frequent recent attacks exhibited lower QoL scores but scores were independent of whether the disease was familial or sporadic. In contrast, a study that analyzed QoL in 34 HAE‐C1INH children and 64 healthy controls showed that symptomatic children experienced impaired QoL compared to controls, across total score, school, and psychosocial dimensions of the PedQL™ questionnaire. 41 QoL impairment increased with the number of attacks and was more pronounced in children with multisite attacks compared with those who had peripheral attacks only. QoL scores were not significantly different in asymptomatic HAE‐C1INH children (n = 11) and controls. In the same study, the authors evaluated anxiety in 33 HAE‐C1INH patients and 52 healthy controls, using the State‐Trait Anxiety Inventory (STAI). 42 Both state (transient, situational anxiety) and trait (proneness to anxious behavior) scores were significantly higher in HAE‐C1INH children, reflecting higher levels of anxiety, with an inverse correlation between anxiety and QoL. Even children with no attacks had higher anxiety state scores than controls. Among HAE‐C1INH children, anxiety traits were significantly higher in those with attacks than in asymptomatic ones. Finally, children with laryngeal attacks appeared more anxious than those with abdominal and/or peripheral attacks only. Another study that compared HAE children with children suffering from mast‐cell‐related angioedema showed that QoL was more impaired in the former. 43 QoL evaluation must be improved by developing adapted and validated tools to better capture QoL impairment in children.
It has also been shown that children with HAE‐C1INH suffer from alexithymia (difficulties in identifying and describing emotions), similar to children with other chronic diseases, but reaching more frequently a level considered as critical. 44 Alexithymia levels correlated with disease severity and stress. Inability to identify and describe feelings may result in reduced ability to cope with stress, that in turn may trigger attacks and increase disease severity, resulting in a vicious circle that needs to be broken.
In a US cohort, 44.8% of pediatric patients had claims for medication related to anxiety or depression. 45 Another deleterious consequence of HAE is the impact on education for children who experience frequent attacks, due to recurrent school absenteeism. 30 It has been estimated that children miss an average of 20 school days per year. 9
3.3. Diagnostic workup in symptomatic children
Early diagnosis is key to appropriate management of the disease and improvement of patients' lives, including survival: indeed, mortality in severe laryngeal attacks with asphyxiation is significantly higher in undiagnosed than in diagnosed patients. 46 However, diagnosis can be challenging in children, as shown by the low percentage of patients diagnosed during childhood. Diagnosis starts with medical history including family history, examination, and thorough patient's and family interview on previous episodes. Biochemical tests must be performed even in children without family history because 25% of cases of HAE‐C1INH appear de novo. 3
3.3.1. Biological tests
A detailed diagnostic algorithm has been established in recent WAO/EAACI guidelines. 3 Biochemical tests include measurement of serum/plasma levels of C1INH function, C1INH protein, and C4 (Table 2). The combination of these three tests is associated with high diagnostic accuracy. 3 C4 levels are usually low even between attacks in HAE‐C1INH 39 and tests should be performed during attack‐free periods, or repeated after attacks if done at the time of attack, and in the absence of inflammatory process. 47 Tests should be repeated for diagnostic confirmation.
TABLE 2.
Biochemical and genetic profiles of various types of hereditary or acquired angioedema. 12
| Hereditary AE | Acquired AE | ||||
|---|---|---|---|---|---|
| HAE type I | HAE type II | HAE‐nC1INH | Acquired HAE‐C1INH | ACE‐induced AE | |
| C1‐INH level | <50% | Normal | Normal | <50% | Normal |
| C1‐INH function | <50% | <50% | Normal | <50% | Normal |
| C4 | Low | Low | Normal | Low | Normal |
| C1q | Normal | Normal | Normal | Low | Normal |
| Anti C1‐INH antibody | Absent | Absent | Normal | Positive in 50% of cases | Normal |
| Mutation | SERPING1 gene | SERPING1 gene | FXII, PLG, ANGPT1, KNG1, MYOF, HS3ST6 | None | None |
3.3.2. Genetic testing
The place of genetic testing is controversial. Most authors and guidelines consider that it is not necessary in most cases when patients are symptomatic, being only useful when biochemical tests are inconclusive or in children less than 1 year, or in patients who have evocative symptoms but normal C1INH. 27 In those cases, analysis of F12, PLG, ANGPT1, and KNG1 should be performed on top of SERPING1, although in 5% to 10% of cases, no mutation can be identified. 21
3.3.3. Imaging
Gastrointestinal involvement sometimes mimics an acute abdomen.
Ultrasonography (US) is a very useful non‐invasive and rapidly available diagnostic tool for pediatric acute abdomen. In children, it can show typical thickening of the intestinal wall and sometimes the presence of ascites. 21 , 47 , 48 , 49 , 50 , 51 High‐frequency ultrasound can detect small bowel thickening with uniform prominent hypoechoic folds, sometimes associated with intraperitoneal free fluid. The distribution of the disease may be either segmental or diffuse. The uniform hypoechoic folds are better appreciated on the longitudinal view (Figure 6A). On axial view, a non‐stratified thickening with luminal narrowing is observed (Figure 6B). Thickening of the digestive wall may be responsible for secondary intussusception but it is a rare complication. In children who have not been previously diagnosed, HAE must be considered when typical sonographic features are present. However, US findings are transient and bowel wall edema and ascites may quickly resolve and appear normal if examination is delayed. Most frequent differential diagnosis in children with small bowel thickening is Henoch–Schönlein purpura. Importantly, mild edema can be unseen, therefore normal US does not always rule out the diagnosis. Computed tomography scan is useful in overweight or obese children, or in teenagers reaching adult size.
FIGURE 6.
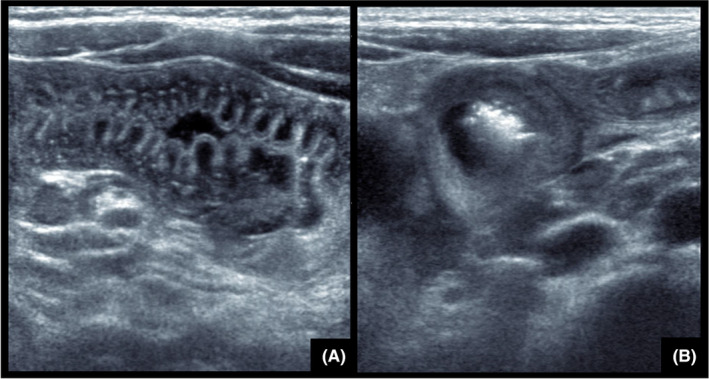
Abdominal ultrasonography. (A) Uniform prominent hypoechoic folds better appreciated on longitudinal view. (B) Non‐stratified thickening of small bowel with luminal narrowing on axial view.
3.4. Differential diagnosis
The classical association of peripheral edema, abdominal crises, and laryngeal edema is quite rare in children, leading to frequent misdiagnoses of isolated attacks. 28 Initial diagnostic errors are reported in up to 50% of patients. 23 In peripheral edema clinical characteristics, must be carefully checked, are helpful. However, some localizations are more challenging (Box 3).
BOX 3. Key messages for diagnosis.
Differentiate bradykinin and mast cell‐related angioedema (presence of urticaria).
Evoke HAE in children with erythema marginatum preceding the crisis.
Search for peripheral and laryngeal edema in case of recurrent abdominal crises.
Perform ultrasonography in case of recurrent abdominal pain during the crisis (or if negative tomography).
A second measurement of C1‐INH protein, function, and C4 must be performed during attack‐free periods or repeated after attacks, in the absence of any inflammatory process.
Test all family members when an index case has been identified by measurement of C1‐INH protein, function, and C4; for infants, repeat tests after 1 year of age.
Genetic testing in selected cases.
Once diagnosis is made, refer to an expert center for appropriate management.
Laryngeal edema can be mistaken for anaphylaxis or epiglottal inflammation. 39 Abdominal pain is one of the most frequent complaints and reason for visits to emergency departments in the pediatric population, reported in 13% and 17% of children and adolescents at median ages of 12.6 and 15.6 years, respectively. 28 HAE abdominal crises can also mimic inflammatory bowel diseases with long diagnostic delay. 52
Main differential diagnosis for peripheral edema is mast cell‐mediated edema that accounts for 95% of angioedema in clinical practice. 2 , 12 Measurement of serum tryptase levels can be used to differentiate non‐specific mast cell activation (normal tryptase), which is very frequent among mast cell‐mediated edema, from mast cell activation syndrome, although diagnostic criteria established for adults 53 (tryptase increase during crisis followed by normal levels when the crisis is terminated) are missing in children. Tryptase elevation also allows detection of hereditary alpha tryptasemia (HaT), a common autosomal dominant genetic trait, defined by increased TPSAB1 gene copy number. While the majority of individuals with HaT are asymptomatic, the presence of HaT sometimes increases the frequency and severity of immediate hypersensitivity reactions. 54 Systemic mastocytosis is rare in children.
Genetic testing should be performed in cases of mast cell‐mediated edema refractory to second‐line treatments (omalizumab or dupilumab), regardless of IgE value. Indeed, there is no correlation between IgE values and mast cell activation. Of note, this applies to total IgE, but not to specific IgE.
A retrospective study analyzed differences between children with HAE (n = 57, median age 8.9 years) or recurrent mast‐cell angioedema (HA) (n = 42, median age 11.5 years). 11 Median age at symptom onset was 6.0 and 7.8 years, respectively, frequency of episodes within last year was 3 and 5, respectively, duration of symptoms was 48 and 24 h, respectively. Involvement of lips and eyelids was more frequent in the HA group as well as itching as a prodromal symptom, while gastrointestinal involvement and trauma as a triggering factor were more frequent in HAE. Slow onset to peak swelling and duration of 2–5 days must evoke bradykinin edema rather than other etiologies. 30 Characteristics of bradykinin‐ and mast cell‐related edemas are summarized in Table 3.
TABLE 3.
Characteristics of bradykinin‐related and mast cell‐related edemas.
| Bradykinin edema | Mast cell‐related edema | |
|---|---|---|
| Prodrome | Erythema marginatum | Itching |
| Type of onset | Gradual onset | Rapid onset |
| Associated urticaria | Absent/Infrequent | Frequent |
| Abdominal localization | Recurrent gastrointestinal attacks | Short duration, less intense |
| Duration | 2–5 days | 12–24 hours |
| Response to treatment | Refractory to treatment with corticosteroids, antihistamines, and adrenaline |
Sensitive to treatment with corticosteroids, antihistamines, and adrenaline Fast response to adrenaline |
| Family context | Frequent family history (75%) | Infrequent family history |
| Food triggers | Absent | Occasional |
Normal C1q values must be confirmed to exclude acquired angioedema diagnosis (rare in children but possible, especially in autoimmune diseases like lupus).
Interestingly, several publications reported cases of mast‐cell‐related urticaria and angioedema in the context of COVID‐19. 55 , 56 , 57
Misdiagnosis can lead to unnecessary surgery or hospitalization in case of abdominal crises. 21 Over time, delay in diagnosis increases anxiety and impairs QoL, prevents the administration of appropriate treatment, and increases the risk of death from laryngeal edema. 30
4. SCREENING AND GENETIC COUNSELING IN ASYMPTOMATIC CHILDREN
Prenatal diagnosis is not generally recommended due to the risk of miscarriage and because pregnancy interruption is not justified because of the variability of disease severity and the existence of efficient therapies. However, in case of in vitro fertilization, preimplantation genetic diagnosis can be used to select embryos devoid of HAE‐C1INH. 21 The whole family must be screened as soon as an index case has been identified, by measurement of plasma C4, C1INH antigen levels and C1INH functional activity. 30
In families with HAE history, newborns and infants must be considered affected until the diagnosis has been reliably ruled out by appropriate tests. 3 , 21 But caution is required for the interpretation of biochemical measurements since the complement system is immature during the first months/years of life. Reference values in children are lacking and adult levels are only achieved between 6 and 36 months. 58 Thus, measurement of serum C4 and C1INH should be repeated after the age of 12 months. 3 , 21 Nevertheless, cases of false negative diagnoses based on serum measurement have been described in children actually carrying SERPING1 mutations. 20 Recommendations might therefore evolve towards systematic genetic screening to avoid such errors.
Sensitivity of low complement C4 in cord blood for the diagnosis of HAE was only 75% in a Danish cohort study. 15 Antigenic and functional C1INH levels are estimated at 70% and 62% of adult values, respectively. 3 Therefore, neonates can be erroneously diagnosed with HAE while they are not affected. Therefore, cord blood testing is not recommended. 47
5. ANNOUNCEMENT OF DIAGNOSIS AND INITIAL MANAGEMENT
The first consultation after diagnostic confirmation must be long enough to give time to the patient and caregiver to ask questions and fully understand the most important messages, in particular recognition of crises and appropriate management (administration of treatment, emergency call, identification of at‐risk situations and requirement for short‐term prophylaxis, proposition for being trained for self‐administration). A patient card is provided, and family screening must be planned rapidly. 59 Families cannot retain all information at first consultation, so that priority must be given to secure the organization of emergency management while other aspects will be emphasized during following visits, and above all, during therapeutic education sessions. Information should be provided on life expectancy, which is similar for HAE and non‐HAE patients, and on the availability of treatment, including prophylaxis, at all ages and during pregnancy for women.
In the specific case of presymptomatic diagnosis, caution is required to avoid anxiety and distress in children who, after announcement, will live in constant fear of attacks, with a sword of Damocles hanging over their head while it is not possible to predict the severity of their upcoming disease, if any.
6. MANAGEMENT OF THE DISEASE IN CHILDREN
The ultimate objectives of treatment are to obtain disease control and improve quality of life. 1 The Angioedema Control Test (AECT), designed to evaluate disease control has been validated in HAE. 60 , 61 However, it has been developed and validated in adults.
Although there are currently no criteria to define disease control in pediatrics, it is common sense that it should translate into reducing the number of attacks as much as possible. Improving quality of life is a more complex issue that integrates a wide range of criteria, including constraints and tolerability of treatment, and can vary across patients according to their profile and situation. However, it can be approached by their desire to live a normal life whatever their definition of normal life. 30 For a child, a key objective is also to allow normal education and social life. Therefore, the clinician should make every effort to reduce the burden of disease without increasing the burden of therapy.
In particular, the administration route is very important to consider in children who are often reluctant to undergo injections, all the more that they must be repeated for long‐term therapy. While not all young children are needle‐phobic, most of them dread injection‐related pain and feel uncomfortable with needles. Recurrent intravenous injections are challenging in children due to the small diameter of veins while subcutaneous injections can provoke site reactions. Reluctance to treatment administration can lead to poor compliance. On the other hand, injections performed by a nurse ensure full compliance to treatment but add other constraints. Overall, these therapies are burdensome to pediatric patients and their caregivers alike. Adolescence is also a period known to be tricky for long‐term therapy. 62 , 63 , 64 , 65 While no studies have been performed in HAE adolescents receiving long‐term prophylaxis, experience from other chronic diseases with long‐term curative or prophylactic treatment, show the frequent refusal of therapy at this period. A survey performed in adults showed that treatment burden was perceived higher in case of IV injections compared to SC injections. 66 Time spent for preparation and administration as well as inconvenience for storage were other subjects of complaint. Of note, although frequent IV injections are not only burdensome but also challenging because of progressive loss of venous capital, placement of a port has never been recommended, due to the risk of infections. However, this issue became less crucial thanks to the availability of non‐IV treatments.
Management of HAE includes treatment of attacks, short‐term and long‐term prophylaxis. Access to treatment can differ across countries, thus management should be adapted to local available options. Other key requirements are summarized in Box 4.
BOX 4. Key messages for pediatric HAE management.
Create a treatment action plan.
Make sure that patients have on‐demand treatment available, even before the first attack if diagnosis is made by systematic screening.
Avoid estrogen‐based contraceptives in female adolescents.
Identify triggers and consider actions to avoid/reduce their frequency.
Review the need for long‐term prophylaxis at least once a year.
Detect and consider counseling for anxiety and depression.
Encourage participation in a therapeutic education program.
Propose participation in a patient association.
Consider non‐pharmacological supportive care.
Provide education for other adults on the child's care team (teachers, coaches, etc.).
6.1. Treatment of attacks
All attacks should be considered for on‐demand treatment, which becomes mandatory if upper airways are affected or could be affected by diffusion of edema. 3 Laryngeal attacks must be considered as emergencies since clinical course is unpredictable and death is possible. 3 Therefore, patients must be provided with appropriate medication with at least 2 doses, that should be available whether at home or out of home. Home therapy should be preferred as far as possible, allowing a dramatic decrease in hospitalization rate.
In Europe, the preferred option for the treatment of attacks is icatibant, a bradykinin B2 receptor specific and selective antagonist, administered via subcutaneous route. The dose is determined according to body weight (from 10 mg in children between 12 and 25 kg to 30 mg in those above 65 kg). Only newborns, pregnant women and individuals in whom icatibant proved inefficient or poorly tolerated are treated with plasma‐derived C1 inhibitors that aim at compensating quantitative or qualitative deficiency in endogenous C1‐INH. Indeed, C1 inhibitors which are also recommended as first‐line treatment in guidelines, are administered by intravenous (IV) route, which is clearly less convenient for home therapy. According to the marketing authorizations, there is no lower limit of age for Berinert® (20 IU/kg) while Cynrize® (1000 IU for children aged 2–11 years weighing more than 25 kg or 500 IU for those weighing less than 25 kg) is indicated for children ≥2 years, however both are used indiscriminately in clinical practice.
Tranexamic acid (TXA) (10 mg/kg every 4 h during 12–24 h, with a maximum dose of 3 g/day), although not approved in this indication, can be used to treat non‐severe abdominal and peripheral attacks in a child who has no access to other treatments, or in whom IV injections are not justified, but only if efficacy has been undoubtedly observed in this child. Being orally available, TXA can be easily administered in school or during other activities out of home, which can have a positive impact on children's comfort and quality of life. However, no studies evaluated its efficacy with a high level of evidence and its utilization only relies on clinical experience and expert opinion. Pending the results of ongoing studies in children aged 2–12 using subcutaneous or oral route, TXA is a convenient alternative. Tranexamic acid is mainly used for on‐demand and prophylactic treatment in a developing country. 18
6.2. Short‐term prophylaxis
Few good‐quality data are available regarding short‐term prophylaxis (STP) in children and adolescents and studies are controversial since some of them show a low incidence of attacks (6%–30%) while others indicate an 80% incidence of attacks in patients who underwent procedures without prophylaxis. 67 However, it has been shown that STP decreases the risk of attacks, and the benefit/risk ratio of STP is therefore favorable since therapies used in this indication are generally well tolerated.
In high‐risk procedures (medical, surgical, and dental procedures associated with mechanical impact to the upper aerodigestive tract), plasma derived C1 inhibitors are recommended (20 units/kg 6 h before the procedure). 3 , 67 , 68 Icatibant is not approved for short‐term prophylaxis.
Importantly, as no study demonstrated that long‐term prophylaxis makes STP unnecessary, physicians should be aware of the risk of attacks and on‐demand treatment must always be available. 3
6.3. Long‐term prophylaxis
Disappearance of attacks, that allow normal quality of life, 41 is the ultimate goal of long‐term prophylaxis (LTP) that should be systematic in all children who had at least one life‐threatening episode, whatever their age. In other cases, since it can be very burdensome to children and adolescents, the decision must be made on a case‐by‐case basis, considering disease activity, benefits and constraints, quality of life, child's environment, and healthcare resources. 3 It must be re‐evaluated regularly (at least once a year) and adapted to the disease evolution.
6.3.1. Before age of 12 years
Fortunately, LTP is rarely mandatory in very young children because attacks are usually infrequent and rarely severe. 69 However, if a life‐threatening episode has occurred, LTP should be initiated. Plasma‐derived C1 inhibitors can be used at any age and until recently, they were also the only treatment available in children aged between 2 and 12 years. However, lanadelumab (Takhzyro®), a human monoclonal IgG1 antibody administered by subcutaneous route, is now approved from 2 years of age. It has been evaluated in children aged 2–12 years in the SPRING study for the treatment of attacks as well as long‐term prophylaxis over 1 year, administered every 4 weeks or every 2 weeks (NCT04070326). 70 Twenty‐one patients (4 aged 2–6 years) were enrolled and treated for 52 weeks with lanadelumab 150 mg Q2W for those aged 6–12 years and Q4W for those aged 2–6 years. Median age at symptom onset was 2.0 years and mean baseline attack rate was 1.84/month. The attack rate decreased by 94.8% from baseline (from 1.84 to 0.08). Overall, 76.2% of children remained attack‐free during the full study period. Efficacy was not affected by patients' age or weight. No serious adverse event or treatment‐related discontinuation was reported. Improvement in quality of life occurred as early 4 weeks after start of treatment and was maintained over time.
In the United States, treatment with subcutaneous C1INH is available. Used in long‐term prophylaxis, it has shown efficacy and safety similar to those observed in adults in the extension phase of the pivotal COMPACT study. 71
TXA remains an alternative if other options are not available or not tolerated (25–75 mg/kg/day in 2 or 3 uptakes with a maximal dose of 3 g/day). 72 TXA is usually well tolerated, side effects being mainly myalgia, creatine kinase increase, vascular thrombosis, and postural hypotension. 69 Contraindications include history of thromboembolism or documented thrombophilia defect. 32 Long‐term administration of danazol must be avoided in children at least before puberty (Tanner stage V) and even after puberty in girls since it can provoke growth retardation and several endocrine adverse effects occurring not only during childhood but also all life long. 32 , 69 , 73 , 74 It should be used only in last resort, in countries where no other options are available, at a low dose (2.5 mg/kg/day initially, increased to 5 mg/kg/day with a maximum of 200 mg), using an intermittent regimen (every other day or 3‐day intervals).
6.3.2. From 12 years
Recommendations for long‐term prophylaxis are similar in children after the age of 12 and in adults. First‐line treatment consists of kallikrein inhibitors. These drugs act by binding to plasma kallikrein, therefore blocking their binding site and preventing cleavage of high‐molecular‐weight kininogen which results in decreased bradykinin production. 75 Two drugs are currently available: lanadelumab and berotralstat (Orladeyo®).
Lanadelumab has been investigated in a randomized phase 3 study (HELP) that included 125 patients aged at least 12 years, presenting with confirmed HAE I or II who experienced at least one attack within 4 weeks prior to inclusion. 76 Patients received lanadelumab 150 mg every 4 weeks (Q4W, n = 28), or 300 mg Q4W (n = 29), or 300 mg Q2W (n = 27), or placebo (n = 21); they were treated for 26 weeks. Only 10 patients were <18 years old. In the overall population, the mean monthly number of attacks in lanadelumab arms was significantly lower than in the placebo arms, ranging from 0.26 to 0.53 compared to 1.97 at baseline. In patients who received 300 mg Q2W (dosing regimen recommended in the drug SmPC), the mean number of attacks during the treatment period was 0.26 per month, representing an 87% reduction compared to preinclusion rate, and 44% of patients had no attacks. The most frequent adverse events in experimental arms were injection site reactions and dizziness. An open‐label extension phase was implemented, allowing inclusion of new patients, where all participants received lanadelumab 300 mg Q2W. 77 Twenty‐one patients aged less than 18 years participated. After a mean follow‐up of 29 weeks, the mean HAE attack rate was reduced by 87.4% overall, 75% of patients achieved a ≥90% reduction and 37% were attack‐free throughout the study period. Analysis of quality‐of‐life data showed clinically significant improvements with mean changes in AE‐QoL total scores compared to baseline of −10.2 points for patients who switched from the initial phase and −19.5 points for new patients. In clinical practice, the dosing interval can be extended to 4 weeks in patients without attacks.
Berotralstat is a small molecule orally available with once‐a‐day administration, that selectively inhibits kallikrein activity and therefore prevents cleavage of high molecular kininogen and overproduction of bradykinin. In the phase 3, double‐blind APeX‐2 study, it was compared to placebo in 120 adults and children of 12 years diagnosed with HAE‐C1INH who experienced at least 2 attacks within 56 days prior to inclusion. 78 Two doses, namely 110 mg/day (n = 41) and 150 mg/day (n = 40) were compared to placebo (n = 39). Both demonstrated a statistically significant reduction in the rate of attacks during the 24‐week treatment period; the effect was rapid as the number of attacks began to decrease during the first month and sustained. At the recommended dose of 150 mg/day, the monthly rate of attacks was 1.31 versus 2.35 in the placebo arm, p < .001. The mean attack rate decreased by more than 70% in one‐half of patients who received berotralstat 150 mg/day. The overall AE‐QoL mean change from baseline was clinically significant at 24 weeks, namely −14.6 points Most frequent adverse events were digestive disorders and back pain, grade 1 or 2 in most cases. At the end of this 6‐month phase, patients from the experimental arms continued berotralstat at the same dose and those from the placebo arm were randomized to berotralstat 110 or 150 mg for another 24 weeks of treatment. In the phase 2 randomized APEX‐S safety study, 14 adolescents who received 150 mg/day during at least 48 weeks (mean age 13.9 years, mean time since diagnosis of HAE 7.5 years). 79 After a mean exposure of 515 days, the monthly mean and median attack rates were 0.4 and 0.0 through week 48, and more than 70% of patients remaining attack‐free during this period. Global satisfaction average scores increased from 78.6 at baseline to 91.8 at week 48, the largest improvement being observed in convenience with a 29.4‐point increase. Berostralstat was generally well tolerated in this age group.
However, it should be kept in mind that, similar to short‐term prophylaxis, LTP is not 100% efficient and on‐demand therapy should always be available to treat possible breakthrough attacks. 27
Post‐pubertal female patients should receive counseling on contraception as appropriate during treatment with berostralstat and lanadelumab.
6.4. Organization of children's management
Several healthcare professionals are involved in the child's management. Attacks are treated in hospitals by emergency specialists, long‐term therapy is overseen by an HAE expert and everyday health concerns are managed by a general practitioner or a non‐specialized pediatrician. The challenge is to coordinate all these stakeholders and to avoid errors in any medical decisions.
The child should be registered in a local hospital for emergency treatment. The mobile and on‐site emergency teams must have received all necessary information on the child's medical condition, and they must have been provided with clear therapeutic instructions in order to save time in case of severe attack. The patient must also have a patient card with information on his disease, emergency number to call, and specific treatment to administer if an attack occurs at any place.
For optimal coordination, the child should be followed in a care network. Healthcare networks are of utmost importance in the management of rare diseases because only networks are able to gather sufficient knowledge and expertise in all aspects of these complex and poorly investigated entities, to conduct clinical research, to involve as many specialists as required by the disease characteristics, to organize communication and to obtain sufficient resources to develop new skills. 80 Collaboration between networks and expert centers are key to reduce time to diagnosis and optimize management.
Patient associations are also useful to help children and parents share their experience, acquiring practical knowledge and coping with the disease.
7. ONGOING CLINICAL DEVELOPMENT
Children from 2 to 12 years;
An ongoing study is assessing berotralstat in children aged 2–12 years (NCT05453968). The main objective of the study is to evaluate safety and pharmacokinetics in this population while frequency and severity of attacks will also be assessed in the long‐term extension phase.
An ongoing study is investigating pharmacokinetics and safety of garadacimab in children aged 2–12 years (NCT05819775).
Children from 12 to 18 years.
Sebetralstat (KVD900), an oral small molecule, selective inhibitor of kallikrein is being developed for on‐demand treatment. 75 A phase 3 double‐blind, three‐way crossover study (KONFIDENT) evaluated the drug for treatment of HAE attacks in adults and adolescents (≥12 years of age). 81 Sebetralstat provided faster symptom relief, more frequent complete resolution and decreased severity of attacks compared to placebo. A PK substudy in adolescents aged 12–17 years is ongoing (NCT06467084).
Donidalorsen is a ligand‐conjugated investigational antisense oligonucleotide targeted at prekallikrein in the liver. 75 It is delivered subcutaneously every 8 weeks. It has been investigated as LTP in the double‐blind, randomized, placebo‐controlled 3‐arm, OASIS‐HAE study that included adults and children from 12 years. 82 Out of 90 patients included, 22 were aged 12–17 years, all in donidalorsen arms. The mean attack rate from week 1 to week 25 was 81% lower in the 4‐week group than in the placebo group (p < .001) and 55% lower in the 8‐week group than in the placebo group (p = .004); the median reduction in the number of attacks per month from baseline was 90% in the 4‐week group (from 3.61 to 0.44), 83% in the 8‐week group (from 3.18 to 1.02), and 16% in the placebo group (from 2.90 to 2.26).
Garadacimab is a fully human monoclonal antibody that inhibits factor XII. It has been evaluated for prophylaxis of HAE attacks in adults and adolescents (≥12 years of age) suffering from HAE I/II in the double‐blind, randomized, placebo‐controlled phase III VANGUARD study. 83 In total, 65 patients were recruited, including 30 children aged 12–17 years (47%). During the 6‐month treatment period, the mean number of monthly attacks was significantly lower in the experimental arm (0.27) than in the control arm (2.01), p < .0001. The median number of attacks per month was 0 (IQR 0.00–0.31) and 1.35 (IQR 1.00–3.20), respectively. Compared with baseline, the mean reduction of attacks was 91% and 20%, respectively. The drug was not associated with an increased risk of bleeding or thromboembolic events.
These new drugs, added to the therapeutic armentarium, will offer a large panel of options, allowing better adaptation of treatment to each patient.
8. OVERALL DISEASE MANAGEMENT
Management of chronic diseases carries specific features in the pediatric population, with additional particularities in rare diseases. Patients suffering from HAE must be followed in a reference center comprising at least pediatricians and HAE experts, which can be different from the local site dedicated to emergency situations. Visits in the reference center should be performed at least once a year. 59
Therapeutic education is the cornerstone of disease management. It involves both children and parents, the participation of each of them evolving over time. Therapeutic education comprises theoretical training to explain the basics of the disease, pathophysiology, triggers, and alert signals, as well as practical training to develop operational skills such as injections or other life‐saving emergency actions. It can be delivered in various settings (individual or group sessions, actual or virtual), using different dedicated tools that should always be adapted to the patient or caregiver. 59 , 84 Meeting other children and families facing the same disease is often helpful as counseling can be more efficient when it comes from non‐healthcare professionals. In particular, motivation can be low when symptoms are infrequent and mild. When a parent is affected by HAE, it is usually easier to rely on the other parent to manage injections since it can be traumatizing for the affected one to provoke in his child the negative experience that he endured when he was a child. Simulation in healthcare was shown to be feasible and relevant for therapeutic education of patients. 85 It consists of acting in various situations mimicking real life, after appropriate training (recognition of symptoms, call to emergency departments, injection).
It is crucial to teach the child when he must raise alerts to adults. Self‐injections become possible at various ages depending on the child, but the earliest is always the best to develop autonomy as soon as possible. Self‐administration is usually feasible in adolescents as shown in a Danish cohort where the age of training for self‐administration ranged between 10.4 and 16.4 years. 15 The children must also be involved in the decision‐making process as soon as possible and children must be regularly screened for anxiety, depression or any other psychological disorder. 30 Parents are sometimes reluctant to accept the child's autonomy for injections. Whenever this attitude is identified, discussions must be held, including psychologists if necessary.
A new challenge recently emerged with optimization of disease management: thanks to early and appropriate treatment, many adolescents and young adults have never experienced any severe crisis. They do not know how to recognize it and they are not familiar with injections, which put them paradoxically in high‐risk situation. Therapeutic education programs must insist on training even in asymptomatic and well‐controlled patients.
Once triggers have been identified, they must be avoided as far as possible. However, a balance must be found to allow children living a normal life and avoid excessive protection that could also increase stress and be counterproductive. In particular, children must not feel different from their classmates, to prevent any segregation, a very frequent phenomenon at this period of life. Since infections are frequent triggers of attacks, vaccination is recommended when available for most frequent infectious diseases. Drugs known to increase bradykinin levels must be avoided, such as estrogen‐containing contraceptives in girls who start contraception or angiotensin‐converting enzyme inhibitors (ACEi). Of note, racécadotril (Tiorfan®) 86 and methylphenidate (Ritaline®) 87 interact with ACEi and can therefore increase the risk associated with ACEi administration. However, these interactions are rarely seen in clinical practice as few children are treated by ACEi. Nevertheless, all drug‐induced cases of angioedema should be reported in everyday clinical practice.
Beyond the child's family, teachers or any other individuals involved in frequent interactions with the child must be informed and trained to recognize symptoms and act appropriately. Personalized care project can be implemented in school and for extracurricular activities, to allow the child having a life as normal as possible 28
8.1. Transitioning
Transitioning from pediatric to adult healthcare has been well defined in frequent chronic diseases such as diabetes or asthma, but there are few recommendations adapted to rare diseases. Overall, preparation for transition should start as early as possible to empower adolescents and develop skills to manage their condition. 88 The overall process can be divided into 3 phases (to be adapted to each individual): preparation from 12 to 16, transfer from 16 to 22 and engagement from 20 to 24. 88 Multidisciplinary care and specific therapeutic education programs must be implemented to optimize the process. In France, the healthcare network on rare immune and hematological diseases (MARIH) https://marih.fr/documentation/transition_d_un_service_pediatrique_a_adulte/ and the health autoimmune and autoinflammatory diseases pathway (FAI2R) (https://www.fai2r.org/transition/) developed recommendations and practical tools to help medical teams.
8.2. Psychological management
Regular screening of psychological distress, anxiety and depression is mandatory and appropriate psychological support should be provided as needed. Management of pediatric HAE should also rely on an integrative holistic approach with non‐pharmacological interventions to relieve psychological symptoms, 46 including but not limited to hypnotherapy, music or art therapy. Taking into account the multiple aspects of disease management, from emergency care to highly specialized long‐term treatment or supportive care, a multidisciplinary approach is mandatory to optimize the children health and well‐being.
FUNDING INFORMATION
Financial support was provided by Biocryst.
CONFLICT OF INTEREST STATEMENT
Anne Pagnier: Consultant/advisor for Takeda, Biocryst, Behring. Investigator in clinical trials for Takeda, Biocryst, Kalvista. Angelina Dermesropian: None. Charlotte Kevorkian‐Verguet: speaker, advisor, and/or received research and consultancy grants from the following companies: GSK, Takeda, Novartis, Sanofi, Shire, Biocryst, Fresenius Kabi. Mélisande Bourgoin‐Heck: Consultant/advisor activities for Biocryst, Takeda, ALK; Presenting fees from DBV, Takeda; Research support from Thermofischer, Novartis, Astra‐Zeneca; Itravel grants from ALK, Stallergènes, Takeda, Bioprojet. Cyrille Hoarau: Investigator in clinical trials for ALK, Biocryst, CSL Behring, LFB, Stallergenes; consultant/advisor for ALK, Biocryst, Blueprint, Pilège, Octapharma, Sanofi, Stallergenes, Takeda; research support from ALK and Stallergenes. Héloïse Reumaux: None. Frédérique Nugues: None. Christine Audouin‐Pajot: None. Sibylle Blanc: None. Aurélia Carbasse: None. Anne‐Laure Jurquet: None. Mélanie Voidey: None. Mona Villedieu: Employee of Biocryst. Laurence Bouillet: Speaker for/Engaged in research and educational projects with/Travel grants from the following companies: BioCryst, CSL Behring, Takeda, Novartis, GSK, Blueprint, Kalvista, pharvaris, Intellia. Isabelle Boccon‐Gibod: Speaker, advisor, engaged in research and educational project and/or received research and consultancy grants from the following companies: Takeda, CSL Behring, Pharming, Kalvista, Pharvaris, Novartis, BioCryst.
PEER REVIEW
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer‐review/10.1111/pai.14268.
ACKNOWLEDGMENTS
We thank Dr. Anne Visbecq for medical writing assistance.
Pagnier A, Dermesropian A, Kevorkian‐Verguet C, et al. Hereditary angioedema in children: Review and practical perspective for clinical management. Pediatr Allergy Immunol. 2024;35:e14268. doi: 10.1111/pai.14268
Editor: Marina Atanaskovic‐Markovic
REFERENCES
- 1. Maurer M, Aygoren‐Pursun E, Banerji A, et al. Consensus on treatment goals in hereditary angioedema: a global Delphi initiative. J Allergy Clin Immunol. 2021;148(6):1526‐1532. doi: 10.1016/j.jaci.2021.05.016 [DOI] [PubMed] [Google Scholar]
- 2. Launay D, Bouillet L, Boccon‐Gibod I, et al. Hereditary angioedema and its new treatments: an update. Rev Med Interne. 2023;44:344‐353. doi: 10.1016/j.revmed.2023.01.020 [DOI] [PubMed] [Google Scholar]
- 3. Maurer M, Magerl M, Betschel S, et al. The international WAO/EAACI guideline for the management of hereditary angioedema‐the 2021 revision and update. Allergy. 2022;77(7):1961‐1990. doi: 10.1111/all.15214 [DOI] [PubMed] [Google Scholar]
- 4. Zafra H. Hereditary angioedema: a review. WMJ. 2022;121(1):48‐53. https://www.ncbi.nlm.nih.gov/pubmed/35442579 [PubMed] [Google Scholar]
- 5. Reshef A, Buttgereit T, Betschel SD, et al. Definition, acronyms, nomenclature, and classification of angioedema (DANCE): AAAAI, ACAAI, ACARE, and APAAACI DANCE consensus. J Allergy Clin Immunol. 2024;154:398‐411.e1. doi: 10.1016/j.jaci.2024.03.024 [DOI] [PubMed] [Google Scholar]
- 6. D'Apolito M, Santacroce R, Vazquez DO, et al. DAB2IP associates with hereditary angioedema: insights into the role of VEGF signaling in HAE pathophysiology. J Allergy Clin Immunol. 2024;154:698‐706. doi: 10.1016/j.jaci.2024.05.017 [DOI] [PubMed] [Google Scholar]
- 7. Vincent D, Parsopoulou F, Martin L, et al. Hereditary angioedema with normal C1 inhibitor associated with carboxypeptidase N deficiency. J Allergy Clin Immunol Glob. 2024;3(2):100223. doi: 10.1016/j.jacig.2024.100223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Santacroce R, D'Andrea G, Maffione AB, Margaglione M, d'Apolito M. The genetics of hereditary angioedema: a review. J Clin Med. 2021;10(9):2023. doi: 10.3390/jcm10092023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Norris M, Ashoor Z, Craig T. Management of pediatric hereditary angioedema types 1 and 2: a search for international consensus. Allergy Asthma Proc. 2022;43(5):388‐396. doi: 10.2500/aap.2022.43.220052 [DOI] [PubMed] [Google Scholar]
- 10. Dermesropian A. Epidémiologie descriptive de l'angioedème à bradykinines chez l'enfant sur le territoire français. Joseph Fourier–Faculté de médecine de Grenoble 2015.
- 11. Ocak M, Nain E, Sahiner UM, et al. Recurrent angioedema in childhood: hereditary angioedema or histaminergic angioedema? Pediatr Dermatol. 2021;38(1):143‐148. doi: 10.1111/pde.14467 [DOI] [PubMed] [Google Scholar]
- 12. Bouillet L. Angioedema. In: Zuberbier T, Grattan C, Maurer M, eds. Urticaria and Angioedema. Springer International Publishing; 2021:133‐147. [Google Scholar]
- 13. Read N, Lim E, Tarzi MD, Hildick‐Smith P, Burns S, Fidler KJ. Paediatric hereditary angioedema: a survey of UK service provision and patient experience. Clin Exp Immunol. 2014;178(3):483‐488. doi: 10.1111/cei.12433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bennett G, Craig T. Hereditary angioedema with a focus on the child. Allergy Asthma Proc. 2015;36(1):70. doi: 10.2500/aap.2015.36.3806 [DOI] [PubMed] [Google Scholar]
- 15. Aabom A, Andersen KE, Fagerberg C, Fisker N, Jakobsen MA, Bygum A. Clinical characteristics and real‐life diagnostic approaches in all Danish children with hereditary angioedema. Orphanet J Rare Dis. 2017;12(1):55. doi: 10.1186/s13023-017-0604-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ayazi M, Fazlollahi MR, Mohammadzadeh I, et al. Delayed diagnosis of hereditary angioedema with C1‐inhibitor deficiency in Iranian children and adolescents. Pediatr Allergy Immunol. 2019;30(3):395‐398. doi: 10.1111/pai.13028 [DOI] [PubMed] [Google Scholar]
- 17. Pancholy N, Craig T. Hereditary angioedema in children: a review and update. Curr Opin Pediatr. 2019;31(6):863‐868. doi: 10.1097/MOP.0000000000000832 [DOI] [PubMed] [Google Scholar]
- 18. Araujo‐Simoes J, Boanova AGP, Constantino‐Silva RN, et al. The challenges in the follow‐up and treatment of Brazilian children with hereditary angioedema. Int Arch Allergy Immunol. 2021;182(7):585‐591. doi: 10.1159/000512944 [DOI] [PubMed] [Google Scholar]
- 19. Nanda MK, Elenburg S, Bernstein JA, Assa'ad AH. Clinical features of pediatric hereditary angioedema. J Allergy Clin Immunol Pract. 2015;3(3):392‐395. doi: 10.1016/j.jaip.2014.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andrasi N, Balla Z, Visy B, et al. Diagnosing pediatric patients with hereditary C1‐inhibitor deficiency‐experience from the Hungarian angioedema Center of Reference and Excellence. Front Allergy. 2022;3:860355. doi: 10.3389/falgy.2022.860355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Campos RA, Valle SOR, Toledo EC. Hereditary angioedema: a disease seldom diagnosed by pediatricians. J Pediatr. 2021;97(Suppl 1):S10‐S16. doi: 10.1016/j.jped.2020.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Christiansen SC, Davis DK, Castaldo AJ, Zuraw BL. Pediatric hereditary angioedema: onset, diagnostic delay, and disease severity. Clin Pediatr (Phila). 2016;55(10):935‐942. doi: 10.1177/0009922815616886 [DOI] [PubMed] [Google Scholar]
- 23. Magerl M, Gothe H, Krupka S, Lachmann A, Ohlmeier C. A Germany‐wide survey study on the patient journey of patients with hereditary angioedema. Orphanet J Rare Dis. 2020;15(1):221. doi: 10.1186/s13023-020-01506-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zanichelli A, Farkas H, Bouillet L, et al. The global registry for hereditary angioedema due to C1‐inhibitor deficiency. Clin Rev Allergy Immunol. 2021;61(1):77‐83. doi: 10.1007/s12016-021-08855-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grumach AS, Longhurst HJ, Aberer W, et al. Pediatricians diagnosed few patients with childhood‐presented hereditary angioedema: Icatibant outcome survey findings. J Allergy Clin Immunol Pract. 2019;7(3):1078‐1080. doi: 10.1016/j.jaip.2018.07.047 [DOI] [PubMed] [Google Scholar]
- 26. Guryanova I, Suffritti C, Parolin D, et al. Hereditary angioedema due to C1 inhibitor deficiency in Belarus: epidemiology, access to diagnosis and seven novel mutations in SERPING1 gene. Clin Mol Allergy. 2021;19(1):3. doi: 10.1186/s12948-021-00141-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mansour E, Veronez CL, Craig T, Grumach AS. Hereditary angioedema in children and adolescents. Allergol Immunopathol (Madr). 2022;50(S Pt 1):1‐6. doi: 10.15586/aei.v50iSP1.535 [DOI] [PubMed] [Google Scholar]
- 28. Pagnier A. L'angioedème héréditaire en pédiatrie: enjeux dagnostique et thérapeutique. Presse Med. 2015;44:89‐95. [DOI] [PubMed] [Google Scholar]
- 29. Brascher AK, Witthoft M, Becker S. The underestimated significance of conditioning in placebo hypoalgesia and nocebo hyperalgesia. Pain Res Manag. 2018;2018:6841985. doi: 10.1155/2018/6841985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johnston DT, Smith RC. Hereditary angioedema: special considerations in children. Allergy Asthma Proc. 2020;41(Suppl 1):S43‐S46. doi: 10.2500/aap.2020.41.200042 [DOI] [PubMed] [Google Scholar]
- 31. Beltramini A, Galinski M, Chabernaud JL, et al. Pain assessment in children younger than 8 years in out‐of‐hospital emergency medicine: reliability and validity of EVENDOL score. Pediatr Emerg Care. 2019;35(2):125‐131. doi: 10.1097/PEC.0000000000000953 [DOI] [PubMed] [Google Scholar]
- 32. Farkas H, Martinez‐Saguer I, Bork K, et al. International consensus on the diagnosis and management of pediatric patients with hereditary angioedema with C1 inhibitor deficiency. Allergy. 2017;72(2):300‐313. doi: 10.1111/all.13001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Banerji A, Li Y, Busse P, et al. Hereditary angioedema from the patient's perspective: a follow‐up patient survey. Allergy Asthma Proc. 2018;39(3):212‐223. doi: 10.2500/aap.2018.39.4123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bork K, Wulff K, Witzke G, Hardt J. Hereditary angioedema with normal C1‐INH with versus without specific F12 gene mutations. Allergy. 2015;70(8):1004‐1012. doi: 10.1111/all.12648 [DOI] [PubMed] [Google Scholar]
- 35. Deroux A, Boccon‐Gibod I, Fain O, et al. Hereditary angioedema with normal C1 inhibitor and factor XII mutation: a series of 57 patients from the French National Center of reference for angioedema. Clin Exp Immunol. 2016;185(3):332. doi: 10.1111/cei.12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marcos C, López Lera A, Varela S, Liñares T, Alvarez‐Eire MG, López‐Trascasa M. Clinical, biochemical, and genetic characterization of type III hereditary angioedema in 13 northwest Spanish families. Ann Allergy Asthma Immunol. 2012;109(3):195‐200.e2. doi: 10.1016/j.anai.2012.05.022 [DOI] [PubMed] [Google Scholar]
- 37. Veronez CL, Moreno AS, Constantino‐Silva RN, et al. Hereditary angioedema with NORMAL C1 inhibitor and F12 mutations in 42 Brazilian families. J Allergy Clin Immunol Pract. 2018;6(4):1209‐1216 e8. doi: 10.1016/j.jaip.2017.09.025 [DOI] [PubMed] [Google Scholar]
- 38. Piñero‐Saavedra M, González‐Quevedo T, de San S, Pedro B, et al. Hereditary angioedema with F12 mutation: clinical features and enzyme polymorphisms in 9 southwestern Spanish families. Ann Allergy Asthma Immunol. 2016;117(5):520‐526. doi: 10.1016/j.anai.2016.09.001 [DOI] [PubMed] [Google Scholar]
- 39. Caballero T. Angio‐oedema due to hereditary C1 inhibitor deficiency in children. Allergol Immunopathol (Madr). 2013;41(1):45‐53. doi: 10.1016/j.aller.2012.01.002 [DOI] [PubMed] [Google Scholar]
- 40. Aabom A, Nguyen D, Fisker N, Bygum A. Health‐related quality of life in Danish children with hereditary angioedema. Allergy Asthma Proc. 2017;38(6):440‐446. doi: 10.2500/aap.2017.38.4093 [DOI] [PubMed] [Google Scholar]
- 41. Engel‐Yeger B, Farkas H, Kivity S, Veszeli N, Kohalmi KV, Kessel A. Health‐related quality of life among children with hereditary angioedema. Pediatr Allergy Immunol. 2017;28(4):370‐376. doi: 10.1111/pai.12712 [DOI] [PubMed] [Google Scholar]
- 42. Kessel A, Farkas H, Kivity S, Veszeli N, Kohalmi KV, Engel‐Yeger B. The relationship between anxiety and quality of life in children with hereditary angioedema. Pediatr Allergy Immunol. 2017;28(7):692‐698. doi: 10.1111/pai.12758 [DOI] [PubMed] [Google Scholar]
- 43. Ocak M, Nain E, Akarsu A, Sahiner UM, Sekerel BE, Soyer O. Health‐related quality of life in children with hereditary angioedema compared with patients with histaminergic angioedema. Allergy Asthma Proc. 2021;42(4):325‐332. doi: 10.2500/aap.2021.42.210019 [DOI] [PubMed] [Google Scholar]
- 44. Savarese L, Bova M, De Falco R, et al. Emotional processes and stress in children affected by hereditary angioedema with C1‐inhibitor deficiency: a multicenter, prospective study. Orphanet J Rare Dis. 2018;13(1):115. doi: 10.1186/s13023-018-0871-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tachdjian R, Johnson KE, Casso D, Oliveria SA, Devercelli G, Jain G. Real‐world cohort study of adult and pediatric patients treated for hereditary angioedema in the United States. Allergy Asthma Proc. 2020;41(3):172‐182. doi: 10.2500/aap.2020.41.200011 [DOI] [PubMed] [Google Scholar]
- 46. Tachdjian R, Kaplan AP. A comprehensive management approach in pediatric and adolescent patients with hereditary angioedema. Clin Pediatr (Phila). 2023;62:973‐980. doi: 10.1177/00099228231155703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wahn V, Aberer W, Aygoren‐Pursun E, et al. Hereditary angioedema in children and adolescents–a consensus update on therapeutic strategies for German‐speaking countries. Pediatr Allergy Immunol. 2020;31(8):974‐989. doi: 10.1111/pai.13309 [DOI] [PubMed] [Google Scholar]
- 48. Dirks K, Deuerling J, Lutz H. Sonography in hereditary angioedema: typical findings demonstrated by the example of 3 cases. Ultraschall Med. 2001;22(4):186‐190. doi: 10.1055/s-2001-16815 [DOI] [PubMed] [Google Scholar]
- 49. Gonzalez Sanchez FJ, Galante MJ, Gonzalez‐Carrero Sixto C, et al. Small bowel angioedema. An unusual condition with an interesting differential diagnosis. Rev Esp Enferm Dig. 2022;114(3):178‐179. doi: 10.17235/reed.2021.8407/2021 [DOI] [PubMed] [Google Scholar]
- 50. Jamil B, Naeem MS, Anachebe T, Majeed MH. Hereditary angio‐oedema as a rare cause of small‐bowel obstruction. BMJ Case Rep. 2019;12(10):e231186. doi: 10.1136/bcr-2019-231186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Patel N, Suarez LD, Kapur S, Bielory L. Hereditary angioedema and gastrointestinal complications: an extensive review of the literature. Case Rep Immunol. 2015;2015:925861. doi: 10.1155/2015/925861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krogulska A, Lewandowska D, Ludwig H, Dabrowska A, Kowalczyk A. Hereditary angioedema as a disease of different clinical courses and difficult diagnosis, particularly in children–a case report and literature review. Postepy Dermatol Alergol. 2021;38(6):1118‐1121. doi: 10.5114/ada.2021.106249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Valent P, Akin C, Arock M, et al. Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. Int Arch Allergy Immunol. 2012;157(3):215‐225. doi: 10.1159/000328760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lyons JJ. Hereditary alpha Tryptasemia: genotyping and associated clinical features. Immunol Allergy Clin N Am. 2018;38(3):483‐495. (In eng). doi: 10.1016/j.iac.2018.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Abasaeed Elhag SA, Ibrahim H, Abdelhadi S. Angioedema and urticaria in a COVID‐19 patient: a case report and review of the literature. JAAD Case Rep. 2020;6(10):1091‐1094. doi: 10.1016/j.jdcr.2020.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Adelino R, Andres‐Cordon JF, De La Cruz A, Martinez C. Acute urticaria with angioedema in the setting of coronavirus disease 2019. J Allergy Clin Immunol Pract. 2020;8(7):2386‐2387. doi: 10.1016/j.jaip.2020.04.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Muntean IA, Pintea I, Bocsan IC, Dobrican CT, Deleanu D. COVID‐19 disease leading to chronic spontaneous urticaria exacerbation: a Romanian retrospective study. Healthcare (Basel). 2021;9(9):1144. doi: 10.3390/healthcare9091144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. HAS . Angioedème héréditaire–Diagnostic et prise en charge chez l'adulte et chez l'enfant. https://www.has‐sante.fr/jcms/p_3297816/fr/angioedemes‐hereditaires‐diagnostic‐et‐prise‐en‐charge‐de‐l‐adulte‐et‐de‐l‐enfant
- 59. Boccon‐Gibod I. Angioedèmes héréditaires: traitements et éducation thérapeutique. Presse Med. 2015;44:78‐88. [DOI] [PubMed] [Google Scholar]
- 60. Weller K, Donoso T, Magerl M, et al. Validation of the angioedema control test (AECT)‐a patient‐reported outcome instrument for assessing angioedema control. J Allergy Clin Immunol Pract. 2020;8(6):2050‐2057 e4. doi: 10.1016/j.jaip.2020.02.038 [DOI] [PubMed] [Google Scholar]
- 61. Weller K, Donoso T, Magerl M, et al. Development of the angioedema control test‐a patient‐reported outcome measure that assesses disease control in patients with recurrent angioedema. Allergy. 2020;75(5):1165‐1177. doi: 10.1111/all.14144 [DOI] [PubMed] [Google Scholar]
- 62. Friedman IM, Litt IF. Adolescents' compliance with therapeutic regimens. Psychological and social aspects and intervention. J Adolesc Health Care. 1987;8(1):52‐67. doi: 10.1016/0197-0070(87)90246-4 [DOI] [PubMed] [Google Scholar]
- 63. Friedman IM, Litt IF, King DR, et al. Compliance with anticonvulsant therapy by epileptic youth. Relationships to psychosocial aspects of adolescent development. J Adolesc Health Care. 1986;7(1):12‐17. doi: 10.1016/s0197-0070(86)80088-2 [DOI] [PubMed] [Google Scholar]
- 64. Michaud PA, Suris JC, Viner R. The adolescent with a chronic condition. Part II: healthcare provision. Arch Dis Child. 2004;89(10):943‐949. doi: 10.1136/adc.2003.045377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Song M, Omar HA. Discovering the complexities of adolescent compliance to treatment. Int J Adolesc Med Health. 2009;21(1):3‐8. doi: 10.1515/ijamh.2009.21.1.3 [DOI] [PubMed] [Google Scholar]
- 66. Radojicic C, Riedl MA, Craig TJ, et al. Patient perspectives on the treatment burden of injectable medication for hereditary angioedema. Allergy Asthma Proc. 2021;42(3):S4‐S10. doi: 10.2500/aap.2021.42.210025 [DOI] [PubMed] [Google Scholar]
- 67. Ajewole O, Lanlokun M, Dimanche S, Craig T. Short‐term prophylaxis for children and adolescents with hereditary angioedema. Allergy Asthma Proc. 2021;42(3):205‐213. doi: 10.2500/aap.2021.42.210006 [DOI] [PubMed] [Google Scholar]
- 68. Angioedème héréditaire–Diagnostic et prise en charge chez l'adulte et chez l'enfant. Protocole National de Diagnostic et de Soins. 2024. [Google Scholar]
- 69. Farkas H, Varga L, Szeplaki G, Visy B, Harmat G, Bowen T. Management of hereditary angioedema in pediatric patients. Pediatrics. 2007;120(3):e713‐e722. doi: 10.1542/peds.2006-3303 [DOI] [PubMed] [Google Scholar]
- 70. Maurer M, Lumry WR, Li HH, et al. Lanadelumab in patients 2 to less than 12 years old with hereditary angioedema: results from the phase 3 SPRING study. J Allergy Clin Immunol Pract. 2024;12(1):201‐211 e6. doi: 10.1016/j.jaip.2023.09.009 [DOI] [PubMed] [Google Scholar]
- 71. Levy D, Caballero T, Hussain I, et al. Long‐term efficacy of subcutaneous C1 inhibitor in pediatric patients with hereditary angioedema. Pediatr Allergy Immunol Pulmonol. 2020;33(3):136‐141. doi: 10.1089/ped.2020.1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Protocole National de Diagnostic et de soins ‐ Diagnostic et prise en charge chez l'adulte et chez l'enfant. https://www.has‐sante.fr/upload/docs/application/pdf/2021‐11/pnds‐ibg‐5xi2021.pdf
- 73. Guo Y, Zhang H, Lai H, et al. Long‐term prophylaxis with androgens in the management of hereditary angioedema (HAE) in emerging countries. Orphanet J Rare Dis. 2022;17(1):399. doi: 10.1186/s13023-022-02536-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zanichelli A, Senter R, Merlo A, et al. Comorbidities in angioedema due to C1‐inhibitor deficiency: an Italian survey. J Allergy Clin Immunol Pract. 2024;12(4):1029‐1036. doi: 10.1016/j.jaip.2023.12.046 [DOI] [PubMed] [Google Scholar]
- 75. Busse P, Kaplan A. Specific targeting of plasma kallikrein for treatment of hereditary angioedema: a revolutionary decade. J Allergy Clin Immunol Pract. 2022;10(3):716‐722. doi: 10.1016/j.jaip.2021.11.011 [DOI] [PubMed] [Google Scholar]
- 76. Banerji A, Riedl MA, Bernstein JA, et al. Effect of Lanadelumab compared with placebo on prevention of hereditary angioedema attacks: a randomized clinical trial. JAMA. 2018;320(20):2108‐2121. doi: 10.1001/jama.2018.16773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Banerji A, Bernstein JA, Johnston DT, et al. Long‐term prevention of hereditary angioedema attacks with lanadelumab: the HELP OLE study. Allergy. 2022;77(3):979‐990. doi: 10.1111/all.15011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zuraw B, Lumry WR, Johnston DT, et al. Oral once‐daily berotralstat for the prevention of hereditary angioedema attacks: a randomized, double‐blind, placebo‐controlled phase 3 trial. J Allergy Clin Immunol. 2021;148(1):164‐172 e9. doi: 10.1016/j.jaci.2020.10.015 [DOI] [PubMed] [Google Scholar]
- 79. Desai BB, Johnston J, Aggarwal D, Tomita K, Iocca D, Reshef H. A. Berotralstat demonstrates consistently low attack rates in adolescent patients: subgroup analysis from APeX‐S European Academy of Allergy and Clinical Immunology (EAACI). Poster 3362021.
- 80. Bolz‐Johnson M, Clément L, Gahl W, et al. Enhancing the value of clinical networks for rare diseases. Rare Dis Orphan Drugs J. 2022;1(2):9. doi: 10.20517/rdodj.2022.01 [DOI] [Google Scholar]
- 81. Riedl MA, Farkas H, Aygoren‐Pursun E, et al. Oral sebetralstat for on‐demand treatment of hereditary angioedema attacks. N Engl J Med. 2024;391(1):32‐43. doi: 10.1056/NEJMoa2314192 [DOI] [PubMed] [Google Scholar]
- 82. Riedl MA, Tachdjian R, Lumry WR, et al. Efficacy and safety of Donidalorsen for hereditary angioedema. N Engl J Med. 2024;391(1):21‐31. doi: 10.1056/NEJMoa2402478 [DOI] [PubMed] [Google Scholar]
- 83. Craig TJ, Reshef A, Li HH, et al. Efficacy and safety of garadacimab, a factor XIIa inhibitor for hereditary angioedema prevention (VANGUARD): a global, multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2023;401(10382):1079‐1090. doi: 10.1016/S0140-6736(23)00350-1 [DOI] [PubMed] [Google Scholar]
- 84. Boccon‐Gibod I, Pagnier A, Bouillet L. Le programme d'éducation thérapeutique « EDUCREAK »: évaluation quadriennale. Rev Fr Allergol. 2017;57(3):284. doi: 10.1016/j.reval.2017.02.222 [DOI] [Google Scholar]
- 85. Mechineaud M, Berton J, Barbarot S, et al. Use of simulation in healthcare for therapeutic training of the parents of children with hereditary angioedema. Ann Dermatol Venereol. 2020;147(5):340‐349. doi: 10.1016/j.annder.2020.02.001 [DOI] [PubMed] [Google Scholar]
- 86. Lochbaum R, Hoffmann TK, Greve J, Hahn J. Concomitant medication in patients with bradykinin‐mediated angioedema–there's more than ACE inhibitors. J Dtsch Dermatol Ges. 2023;21(11):1283‐1289. doi: 10.1111/ddg.15154 [DOI] [PubMed] [Google Scholar]
- 87. Goyal R, Arroyave A, Malik S. Urticaria and angioedema secondary to methylphenidate exposure in a young child. J Child Adolesc Psychopharmacol. 2015;25(9):731‐733. doi: 10.1089/cap.2014.0040 [DOI] [PubMed] [Google Scholar]
- 88. Georgin‐Lavialle S, Hentgen V, Truchetet ME, et al. Transition from pediatric to adult care: recommendations of the French network for autoimmune and autoinflammatory diseases (FAI(2)R). Rev Med Interne. 2021;42(9):633‐638. doi: 10.1016/j.revmed.2021.02.003 [DOI] [PubMed] [Google Scholar]