

SUMMARY
The BonnMu resource is a transposon tagged mutant collection designed for functional genomics studies in maize. To expand this resource, we crossed an active Mutator (Mu) stock with dent (B73, Co125) and flint (DK105, EP1, and F7) germplasm, resulting in the generation of 8064 mutagenized BonnMu F2‐families. Sequencing of these Mu‐tagged families revealed 425 924 presumptive heritable Mu insertions affecting 36 612 (83%) of the 44 303 high‐confidence gene models of maize (B73v5). On average, we observed 12 Mu insertions per gene (425 924 total insertions/36 612 affected genes) and 53 insertions per BonnMu F2‐family (425 924 total insertions/8064 families). Mu insertions and photos of seedling phenotypes from segregating BonnMu F2‐families can be accessed through the Maize Genetics and Genomics Database (MaizeGDB). Downstream examination via the automated Mutant‐seq Workflow Utility (MuWU) identified 94% of the presumptive germinal insertion sites in genic regions and only a small fraction of 6% inserting in non‐coding intergenic sequences of the genome. Consistently, Mu insertions aligned with gene‐dense chromosomal arms. In total, 42% of all BonnMu insertions were located in the 5′ untranslated region of genes, corresponding to accessible chromatin. Furthermore, for 38% of the insertions (163 843 of 425 924 total insertions) Mu1, Mu8 and MuDR were confirmed to be the causal Mu elements. Our publicly accessible European BonnMu resource has archived insertions covering two major germplasm groups, thus facilitating both forward and reverse genetics studies.
Keywords: genetic resource, mutagenesis, Zea mays, Mutator, transposon, Mu‐seq
Significance Statement
Our resource provides a wide array of sequence‐indexed mutants covering diverse genetic backgrounds, thereby facilitating functional genomics studies in maize.
INTRODUCTION
Maize (Zea mays L.) has a long history of genetic investigation. Since the early 1900s geneticists have been collecting and describing maize mutations affecting a broad range of biological processes. However, to date, only a few hundred genes have been functionally characterized based on visible morphological mutant phenotypes (Schnable & Freeling, 2011; https://www.maizegdb.org). Forward genetic experiments represent a classical method to unravel gene functions by cloning a gene based on a mutant phenotype. In contrast, reverse genetic screens enable the identification of mutant phenotypes by utilizing disrupted gene sequences (Candela & Hake, 2008). Both approaches have been successfully applied in the past as exemplified by their application in identifying and characterizing multiple genes underlying maize root development (Li et al., 2016; Nestler et al., 2014; Xu et al., 2015).
Genome‐wide insertional mutagenesis represents a powerful reverse genetics tool to generate loss‐of‐function mutations for virtually all genes within a genome. Meanwhile, several maize genomes of two major germplasm, such as B73 and W22 of the dent pool (Jiao et al., 2017; Schnable et al., 2009; Springer et al., 2018) and F7, DK105, and EP1 of the European flint pool (Haberer et al., 2020) were sequenced. Dent and flint pools are genetically divergent with respect to several traits including early vigor and cold tolerance, which is due to their historic geographical separation and adaptation to different environmental conditions. The progenitors of the European flint pool reached higher latitudes, which required selection for cold tolerance and early maturation (Haberer et al., 2020). Therefore, the flint lines are important genetic resources for central European maize research due to their stable growth properties under temperate climatic conditions.
To facilitate genome‐wide insertional mutagenesis screens in maize, Mutator (Mu) transposons are used as biological mutagens, because they can move from one location in the genome to another, thereby disrupting genes (Lisch, 2015). Transposons, also known as transposable elements, are mobile DNA sequences first discovered in maize (McClintock, 1951). They are classified into two major classes: class I retrotransposons, which necessitate an RNA intermediate for transposition and class II DNA transposons, which directly transpose via DNA transposase. Mu transposons, the most active class II transposon family in maize, consist of an autonomous element (MuDR; Robertson, 1978) and multiple non‐autonomous elements (Lisch, 2002, 2015; Tan et al., 2011). All identified Mu transposons contain highly similar 215 bp terminal inverted repeats (TIRs) at both ends of the elements and create 9 bp target site duplications directly flanking the Mu transposon sequences upon insertion. Mu elements randomly target genes throughout the maize genome (Lisch, 2015). As such, Mu insertion site frequencies were observed to strongly correlate with gene density (Schnable et al., 2009). To date, three public sequence‐indexed mutant libraries have been established by Mu transposon insertional mutagenesis as invaluable resources for conducting functional genetics studies in maize (UniformMu: McCarty et al., 2013; ChinaMu: Liang et al., 2019; BonnMu: Marcon et al., 2020). These mutant collections are ideal starting points for forward and reverse genetic screens (i) to functionally characterize novel mutants regulating various developmental processes (Dai et al., 2021; Hunter et al., 2014) and (ii) to validate candidate genes by additional allelic mutations. The BonnMu resource utilizes random Mu insertions to disrupt genes and employs the Mutant‐seq (Mu‐seq; McCarty et al., 2013) method to identify these disruptions. Mu‐seq enables the identification of maize F2‐families carrying transposon insertions in a sequence‐indexed (i.e., transposon tagged) population, by high‐throughput next‐generation sequencing. The analysis of this European‐based sequence‐indexed resource has been optimized and accelerated by the MuWU bioinformatic pipeline (Stöcker et al., 2022), which facilitates unbiased and high‐throughput analysis of sequence data from mutagenized BonnMu F2‐families.
In this study, we extend the original BonnMu reverse genetics resource introduced by Marcon et al. (2020). We achieved this expansion by sequencing an additional 6912 mutagenized F2‐families across various genetic backgrounds, including B73, Co125, DK105, EP1, and F7. Utilizing a consistent downstream analysis of all Mu‐seq reads through the MuWU bioinformatic pipeline, we identified transposon‐induced mutations in 83% of all maize genes. The enhanced BonnMu resource is now available for maize geneticists, providing an invaluable tool for molecular and genetic analyses.
RESULTS
BonnMu insertions cover 83% of all annotated B73v5 gene models
The BonnMu F2‐families were generated in different genetic germplasm backgrounds, i.e., B73 and Co125 from the dent pool and F7, EP1, and DK105 from the flint pool (Table S1). In a previous study, we generated two Mu‐seq libraries comprising 1152 BonnMu F2‐families in B73 background (Marcon et al., 2020). For the downstream bioinformatic analysis, we employed the Mu‐seq method described by McCarty et al. (2013) and Liu et al. (2016). In the present study, we complement the two previous Mu‐seq libraries by 12 additional libraries comprising 6912 BonnMu F2‐families. To ensure equal analysis of all 14 datasets, we applied the bioinformatics method MuWU (Stöcker et al., 2022) and integrated the first two libraries (Marcon et al., 2020) into the analysis. Sequencing of 14 Mu‐seq libraries yielded 1 589 204 597 raw read pairs (Table 1). After automated trimming of U‐adapter and TIR sequences, 74% (1 173 003 813 read pairs; Table 1) of the read pairs remained. Among the remaining read pairs 90% were aligned to the B73 genome containing 44 303 high‐confidence gene models (Zm‐B73‐REFERENCE‐NAM‐5.0). After duplicate read removal, more than 235 million reads, i.e., read pairs and unpaired reads, remained and were used to identify presumptive germinal insertion sites at intersections of one row and one column pool. Subsequently, reads were counted in each of the 48 pools per library for insertion site identification. The MuWU analysis exclusively considered insertion sites supported by a minimum of four reads for subsequent analyses. Transposon insertions can occur in germinal or somatic cells. As a consequence, germinal insertions are heritable and transmitted via the gametes to all cells of the offspring. In contrast somatic insertions are not heritable and confined to sectors of the plant which are derived from mutated somatic cells. To exclude most somatic mutations from further analyses we sampled two distinct leaves per seedling for independent row and column pools. By this approach, somatic insertions generally appeared only in row or column pools and were excluded from downstream analyses (Marcon et al., 2020; McCarty et al., 2013). In total, 425 924 distinct presumptive germinal insertion sites were detected in the 14 Mu‐seq libraries tagging 36 612 (83%) of the 44 303 B73v5 genes (Table 1; Data S1). In detail, we counted insertions affecting genic regions, defined as from the start of the 5′ UTR to the end of 3′ UTR of genes, including exons and introns. Additionally, Mu insertions in promoter regions – up to 2100 bp upstream of the start of the 5′ UTR – and close downstream regions of genes – up to 2100 bp downstream of the end of the 3′ UTR – were considered. Based on the number of 425 924 insertion sites, each of the 36 612 tagged genes carried on average 12 insertional alleles (425 924 insertion sites/36 612 affected genes). The majority of 60% of the affected genes (21 812 of 36 612 B73v5 genes) harbored insertions in their coding sequence (Data S1). Among the 8064 BonnMu F2‐families under analysis, 98% (7908 of 8064 F2‐families) carried at least one presumptive germinal insertion. Only for a minority of 2% of the BonnMu F2‐families (156 of 8064 F2‐families), no insertion was detected. Hence on average, each BonnMu F2‐family carried 53 presumptive heritable Mu insertions (425 924 insertion sites/8064 F2‐families). An extreme example is the BonnMu F2‐family F7‐4‐F‐1766 hosting 338 distinct Mu insertions in 457 different genes. In this family, some insertions affected multiple genes, either due to their close proximity or overlapping regions, resulting in a greater number of impacted genes than the total number of insertions.
Table 1.
Alignment statistics for Mu‐seq libraries
| Description | 14 Mu‐seq libraries a |
|---|---|
| Raw read pairs | 1 589 204 597 |
| Read pairs after trimming | 1 173 003 813 |
| Average alignment rate | 90% |
| Read pairs and unique unpaired reads after removing duplicates | 235 240 526 |
| Number of presumptive germinal Mu insertions (unique insertions) | 425 924 |
| Number of Mu‐tagged genes (unique genes) | 36 612 |
Two previously published Mu‐seq libraries (Marcon et al., 2020) were included in the analysis.
The genomes of the European flint lines DK105, EP1, and F7 were recently sequenced (Haberer et al., 2020). In light of this, we explored the number of presumptive germinal insertion sites by mapping the Mu‐seq reads of the 3456 sequenced BonnMu F2‐libraries in DK105, EP1, and F7 genetic background (Table S1) to their respective genomes. Considering only genes that can be assigned to chromosomes, there are 46 726, 43 375, and 44 043 genes in the DK105, EP1, and F7 genomes, respectively. Among the 462 BonnMu F2‐families in DK105 genetic background (Table S1) the MuWU analysis identified 29 986 unique insertions affecting 36% of the DK105 genes (16 704 of 46 726 genes; Data S2). Furthermore, we detected 57 806 distinct insertions in 53% of the EP1 genes (22 830 of 43 375 genes), when the Mu‐seq reads of 690 BonnMu F2‐families in EP1 genetic background were mapped to the EP1 genome (Data S3). Finally, the 2304 analyzed BonnMu F2‐families in F7 genetic background (Table S1) carried 64 839 unique insertions affecting 42% of the F7 genes (18 548 genes of 44 043 genes of the F7 genome; Data S4).
BonnMu affected genes in flint germplasm complement the set of tagged genes identified in dent lines
Based on the analysis of 14 Mu‐seq libraries in different genetic backgrounds, we uniformly mapped insertions affecting 36 612 genes to B73v5 (Table 1; Data S1). We identified varying numbers of Mu‐tagged genes in the five different germplasms, ranging from 8396 mutated B73v5 genes in Co125 genetic background to 32 390 B73v5 genes in B73 genetic background (Figure 1a; Data S5). This bias can be partially attributed to the fact that the Mu‐seq analysis was conducted on only 576 mutagenized F2‐families in the Co125 genetic background, whereas it was performed on 4032 mutagenized F2‐families in the B73 genetic background. Of the 36 612 affected genes, 5502 (15%) were detected in BonnMu F2‐families of all 14 Mu‐seq libraries used in this study (Figure 1a). The number of overlapping genes significantly exceeded the expected count of 975 genes by chance (Table S2). This finding indicates the preference of Mu transposons for targeting specific genes across diverse inbred lines.
Figure 1.
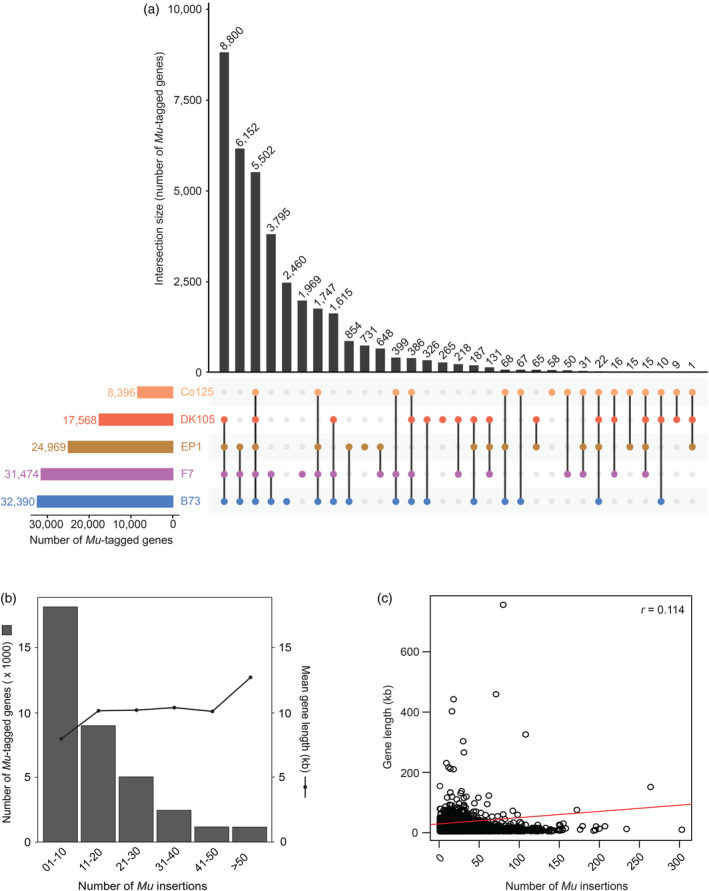
Overlap of genes affected by Mu insertions and distribution of insertions.
(a) Intersections of genes, tagged in BonnMu F2‐families of the two dent lines B73 and Co125 and three flint lines DK105, EP1, and F7, which have been mutagenized in this study. The UpSet plot displays 31 intersections. The lines connect overlapping genes among different genetic backgrounds. The total number of intersected genes is displayed above each bar.
(b) Number of tagged genes and associated mean gene length plotted against the number of Mu insertions.
(c) Distribution of the length of affected genes plotted against the number of individual Mu insertions. The calculated Pearson correlation coefficient is r = 0.114 (P < 0.001).
A higher number of 8800 overlapping genes (24%) were identified in BonnMu F2‐families of four different mutagenized germplasm: DK105, EP1, F7, and B73. A considerable proportion of Mu‐tagged genes, that is, 16 827 (46%) of 36 612 were hit in BonnMu F2‐families of Mu‐seq libraries in two or three different genetic backgrounds under analysis (Figure 1a). In summary, 85% of the Mu‐tagged genes were identified across at least two different genetic backgrounds. This result further indicated that specific genes are more prone to Mu transposon insertions and are affected consistently across multiple genetic backgrounds. Finally, the remaining 5483 genes (15%) of the affected 36 612 genes were exclusively detected in one of the mutagenized inbred lines. More precisely, the majority of 2460 genes were uniquely identified in the Mu‐seq libraries in the B73 genetic background, whereas only 58 of the tagged genes were uniquely detected in the single Mu‐seq library in Co125 genetic background (Figure 1a).
Among the 36 612 tagged genes, 4027 (11%) were identified in at least one Mu‐seq library of the mutagenized flint lines, i.e., DK105, EP1, and F7, but were not tagged in the set of affected genes identified in the dent lines B73 and Co125. Almost half of these genes, specifically 1969 of 4027 (49%), were exclusively detected in the Mu‐seq libraries in the F7 background (Figure 1a). This result can be partially explained by the fact that 2304 BonnMu F2‐families in F7 background were used for the Mu‐seq experiment, whereas only 462 and 690 BonnMu F2‐families in DK105 and EP1 backgrounds were analyzed, respectively (Table S1). Nevertheless, a considerable portion of 265 (7%) and 731 (18%) genes were uniquely tagged in BonnMu F2‐families in DK105 and EP1 backgrounds, respectively (Figure 1a). The remaining 1062 of the 4027 affected genes (26%) were overlapping in BonnMu F2‐families of two or three of the flint lines. Consequently, the genes affected by Mu insertions in flint germplasm complement the set of tagged genes identified in dent lines.
Next, we examined the distribution of all 425 924 Mu insertions within the 36 612 B73v5 genes, including insertions in promoter regions, i.e., within a 2100 bp window upstream of genes and nearby downstream regions, i.e., within a 2100 bp window downstream of genes. Among the tagged genes nearly half, i.e., 49% (17 958 of 36 612), harbored 1–10 Mu insertions, 24% (8908 of 36 612 genes) contained 11–20 insertions, and the remaining 27% (9746 of 36 612) of the genes carried at least 21 insertions (Figure 1b). Among the latter group of genes 12% (1160 of 9746) carried at least 50 insertions. One extreme example is a 5938 bp gene encoding a protein‐serine/threonine phosphatase, Zm00001eb054350, carrying 303 unique insertions. Among these insertions, 38% (115 of 303 insertions) hit the coding sequence of the gene Zm00001eb054350 (Data S1).
Subsequently, we tested whether the number of Mu insertions was positively correlated with the length of the affected genes. To this end, we calculated the mean length of the affected genes, which were grouped according to the number of insertions (Figure 1b). The five groups of genes harboring 1–50 insertions showed a comparable gene length, ranging between 7931 bp and 10 321 bp, whereas the group of genes harboring >50 insertions exhibited an increased mean length of 12 595 bp. Consequently, the computed Pearson correlation across all groups of gene sizes showed a weak positive correlation between gene size and the number of Mu insertions (r = 0.114; Figure 1c).
We obtained consistent results when only considering insertions (370 396 of 425 924, 87%) affecting genic regions, i.e., 5′ and 3′ UTRs, exons, and introns (Figure S1). These insertions covered 85% of the total affected genes (31 126 of 36 612). In this analysis, we calculated a moderate positive correlation of r = 0.147 between gene size and the number of insertion sites (Figure S1).
Moreover, we have examined 130 presumptive germinal Mu insertions in 73 genes across 125 BonnMu families in the F3‐generations. Of these, 93 insertions (72%) were confirmed by PCR/Sanger sequencing as germinal mutations. We assume that the remainder could not be confirmed either due to somatic mutations or unsuitable PCR oligonucleotide primer combinations.
The BonnMu resource provides easy‐to‐view photos of segregating F2‐families at the seedling stage accessible at the genome browser at https://jbrowse.maizegdb.org/ (Marcon et al., 2020). Among the analyzed BonnMu F2‐families various mutants were identified, such as leaf color mutants (e.g., BonnMu‐7‐C‐0336) or mutants affected in shoot development (e.g., BonnMu‐9‐G‐0034; Figure S2). The mutation rate in the 8064 F2‐families, determined by the albino and pale green leaf phenotype, was 16%. This rate aligns with previously published mutation rates (Marcon et al., 2020; Robertson, 1983), indicating a high transposon activity in the BonnMu F2‐families.
When identifying mutants from the F2‐generation of transposon‐tagged seeds of the BonnMu repository it is important to keep in mind that our resource is mainly suitable for identifying monogenic qualitative mutations (i.e., presence or absence of a trait). Such traits can be easily identified in the heterogeneous genetic of the BonnMu F2‐generation while our resource is not suitable to identify quantitative traits. For most transposon‐tagged genes in the BonnMu collection we have several independent mutant alleles, so that we can often validate observed mutant phenotypes by these alleles of the gene of interest. However, it is always advisable to backcross novel mutant phenotypes associated with specific genes for several generations to be able to study the mutant phenotype in a more homogeneous genetic background. Validation can also be done by the generation of independent CRISPR/Cas9 alleles.
Moreover, our mutant stocks still contain active Mu transposons. Hence, when we propagate our F2‐seeds to generate enough F3‐seeds for downstream experiments, we might indeed lose the insertions in our gene of interest in some instances. However, since we propagate many F2‐plants this might only affect the recovery rate of the mutants in the F3‐generation. In other words, the rate of families segregating 3:1 for wild‐type versus mutant phenotypes and also the rate of homozygous mutants (if viable) might be reduced toward homozygous wild types, but we will never lose all insertions because it is unlikely that insertions in different kernels will transpose at the same time.
The benefit of our large mutant resource is that it allows screening for mutants of a large number of genes of interest in a fast and inexpensive way.
Mu insertions preferentially target the 5′ UTR of genes
Next, we investigated whether Mu transposons exhibit preferences for insertion sites within the maize genome. Maize has a complex genome comprising over 80% intergenic non‐coding sequences (Chen, Wang, et al., 2023; Haberer et al., 2020; Hufford et al., 2021). As a result, coding sequences constitute less than 20% of the genome. Specifically, the B73v5 genome can be subdivided into gene coding regions including 5′ UTRs (0.5%), exons (2%), introns (5%), 3′ UTRs (1%), and promoter regions (3.5%; Table S3) which are located upstream of the 5′ UTRs of genes. We further divided the promoter region into a core promoter (0.2%) and a proximal promoter (3.3%), referred to hereafter as promoterCore and promoterProx, respectively (Table S3). While the promoterCore, including the transcription start site, is located directly (1–100 bp) upstream of the start of the 5′ UTR, the promoterProx is 101–2100 bp upstream of the 5′ UTR. Consequently, gene‐coding regions and their associated promoter segments account for only 12% of the total maize genome (Figure 2a). The remaining 88% is composed of non‐coding sequences (Figure 2a).
Figure 2.
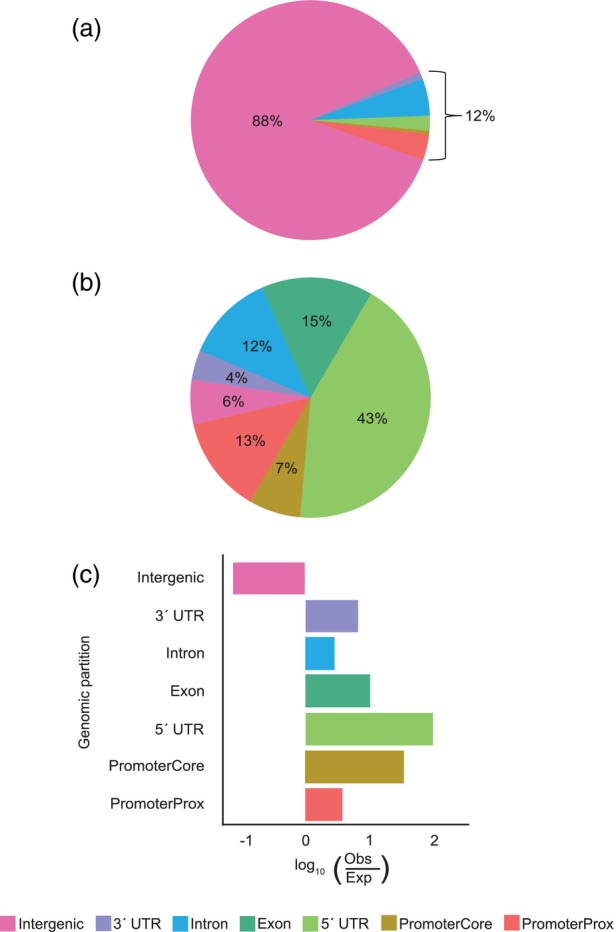
Distribution of BonnMu insertions across the maize genome.
(a) Composition of the maize genome (B73v5).
(b) Mu insertion sites across the genome.
(c) Ratio of observed and expected Mu insertions across the genome.
To explore whether Mu transposons exhibit preferences for distinct categories of the coding regions or the intergenic region of the maize genome, we investigated the set of all BonnMu insertion sites identified in the subset of seven Mu‐seq libraries consisting of 4032 BonnMu F2‐families in B73 genetic background (Data S1). Among all 774 692 somatic (not shown) and presumptive germinal insertions, a considerable proportion of 43% (331 168 insertions) affected the 5′ UTRs (Figure 2b). A comparable fraction of 15% (115 786 insertions) and 12% (95 371 insertions) tagged exons and introns of genes, while 13% (98 510 insertions) were incorporated in promoterProx sections of the maize genome. Minor fractions of insertions were identified in 3′ UTRs (4%; 27 189 insertions) and promoterCore regions (7%; 53 950 insertions). Interestingly, while 88% of the maize genome contains non‐coding regions (Figure 2a; Table S3), only 6% (52 718 insertions) of the BonnMu insertions were detected in those regions (Figure 2b). In summary, our results indicate that intergenic insertions are underrepresented, and genic regions, such as the 5′ UTRs, are frequently targeted by BonnMu insertions. To further support this finding and to account for the non‐uniformity of intergenic versus genic space, the observed and expected number of insertions per genomic partition were compared statistically using Pearson's chi‐squared tests with Yates' continuity correction. Indeed, we observed more insertions than expected for all genic and promoter partitions of the genome (Table S4; Figure 2c). For the 5′ UTRs, a number of 3824 insertions would be expected based on the genomic composition of the maize genome (Table S4). However, we discovered significantly more than expected, specifically 331 168 insertions, exceeding the anticipated count of 3824 by over 86‐fold. In contrast, we expected 684 938 BonnMu insertion sites in intergenic regions. However, only 52 718 of such insertion sites were detected (Table S4; Figure 2c), approximately 13 times fewer than expected.
BonnMu insertions correspond with gene‐dense telomeric regions
Previous studies described that Mu insertion site frequencies align with gene density (Schnable et al., 2009; Springer et al., 2018). Thus, we analyzed the distribution of BonnMu insertions in the B73 genetic background across all 10 chromosomes of maize by dividing each chromosome into 10k bins of 213 167 bp in size and counted the number of insertions per bin. At the heterochromatic centromeric regions of each chromosome, we predominantly detected bins containing less than 200 BonnMu insertions (Figure 3). In contrast, at the telomeres we frequently detected bins that carry 250–500 insertions (Figure 3), indicating that Mu elements preferentially insert into these gene‐rich regions. However, there are some exceptions, i.e., bins which are in close proximity to the telomeres of the chromosomes 5–8, but lack insertions. One extreme example is the telomeric region at the short arm of chromosome 6, illustrating a 5–6 Mb window lacking insertions (Figure 3). We observed that the absence of Mu element insertions coincides with chromosomal gaps on chromosomes 5 to 8. We investigated this using the MaizeGDB JBrowse genome browser (Woodhouse et al., 2021) and can conclude that these gaps correspond to highly heterochromatic knob regions located at the telomeric regions of these chromosomes (Ghaffari et al., 2013).
Figure 3.
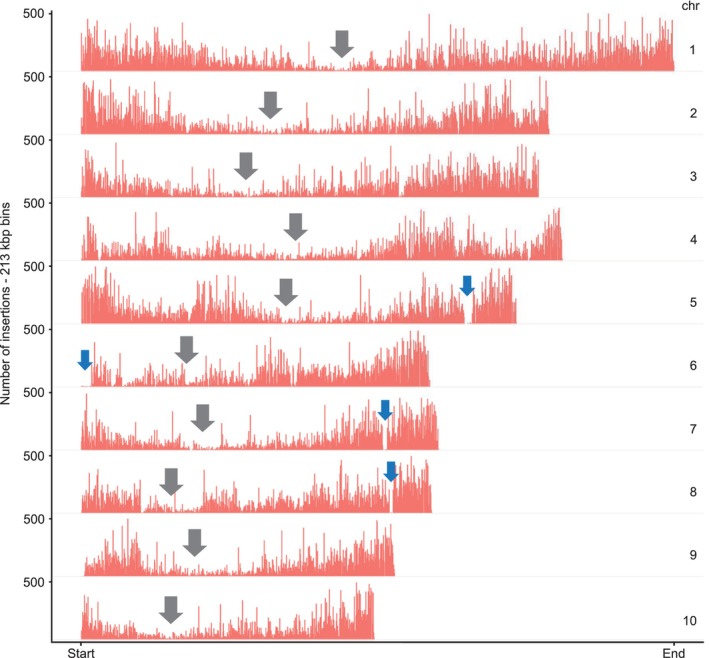
Distribution of BonnMu insertions across 10 chromosomes.
Mu insertions are predominantly found in the gene‐rich telomer regions with fewer insertions in the heterochromatic centromere regions. Gray arrows indicate the centromeric regions in each chromosome. Blue arrows indicate gaps, i.e., regions with marginal numbers of BonnMu insertions, on the arms of chromosomes 5–8. These regions correspond to highly heterochromatic knob regions (Ghaffari et al., 2013).
Spatial distribution of BonnMu insertions and chromatin accessibility across the maize genome
To gain a deeper understanding of the distribution patterns of BonnMu insertions within the genome, we investigated the relationship of the BonnMu families in B73 background to chromatin accessibility and histone modifications. To accomplish this, we utilized multiple ATAC‐seq (Assay for Transposase‐Accessible Chromatin using Sequencing) analyses, ChIP‐seq data, and published datasets related to DNA acetylation and methylation processes. ATAC‐seq is used to assess open chromatin regions on a genome‐wide scale (Buenrostro et al., 2013). In this study, we visualized the chromatin accessibility within the maize NAM (nested association mapping) population and various tissues, such as ear and leaf (Figure 4; Ricci et al., 2019). Additionally, we juxtaposed the frequency of Mu insertion sites with the genome‐wide distribution of chromatin modifications, including trimethylation of Lys‐27 of histone H3 (Makarevitch et al., 2013) and histone acetylation (Zhang et al., 2015).
Figure 4.
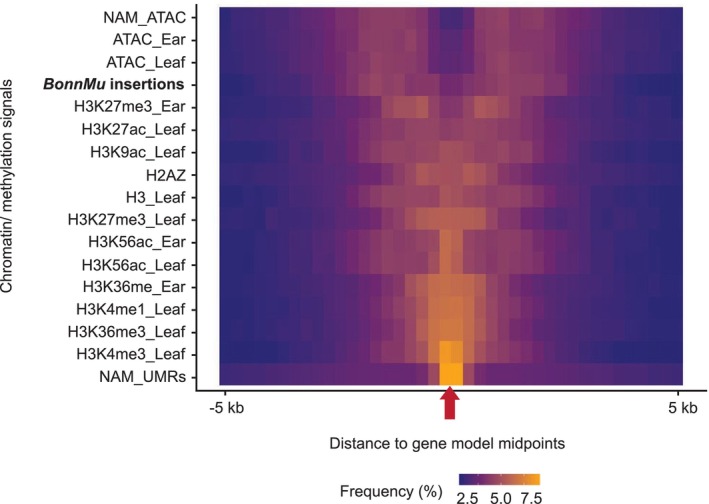
BonnMu insertions in the context of the epigenomic landscape surrounding maize genes.
Frequency distribution of BonnMu insertions, chromatin modifications, and chromatin accessibility in relation to the entire set of genes in the maize genome. The red arrow on the horizontal center of the plot indicates the midpoint of all maize gene models ignoring strand. Bins of 250 bp in size are presented on both sides of this midpoint, representing a region 5 kb upstream and 5 kb downstream of the gene midpoint. The frequency of Mu insertions, chromatin marks or accessibility signals is color‐coded (yellow = higher frequency; blue = lower frequency). NAM, nested association mapping; UMR, unmethylated regions.
In a 250 bp window around the midpoint of the maize gene models, we detected strong signals of unmethylated regions (UMRs; NAM_UMRs, Figure 4) which is in line with the high overlap of UMRs with accessible chromatin regions reported previously (Hufford et al., 2021). Similarly, distinct central enrichment at the gene model midpoints was observed for histone 3 modifications, such as trimethylations at Lys‐4 and Lys‐36 (H3K4me3_Leaf and H3K36me3_Leaf) or acetylation of Lys‐56 (H3K56ac). The frequency of these histone modifications gradually diminishes with increased distance from the gene midpoint.
A contrasting pattern was observed for accessible chromatin signals, based on ATAC‐seq datasets and BonnMu insertion features. While gradually increasing frequencies of ATAC‐seq signals and Mu insertions were identified in 250 bp windows flanking the gene midpoint, there are only a few such signals and insertions at the center of the gene. Hence, BonnMu insertions aligned well to transposase accessible chromatin signals detected in the following datasets: ATAC_Ear, ATAC_Leaf and NAM_ATAC (Figure 4). According to Figure 4, Mu transposons predominantly insert in a region flanking but not far from genes, which would include UTRs and closely adjacent regulatory sequences. This finding is in line with the preference for BonnMu insertions targeting promoterCore and 5′ UTR regions of genes (Figure 2b). Aligning partly with the pattern observed for the BonnMu insertions, H3K27me3 modifications displayed tissue‐specific differences in their distribution around gene midpoints. This can at least be partly explained by the reported observation that H3K27me3 is reported to be less coupled to chromatin accessibility than other modifications. These modifications, on average, deviate from open chromatin signals only in 15–21% of cases in a tissue‐specific manner in maize (Ricci et al., 2019).
Mapping and validation of Mu1 and Mu8 transposons
The Mu transposon system is a powerful tool for large‐scale mutagenesis in maize. Several non‐autonomous Mu species have been comprehensively characterized (Lisch, 2015). In our study, we pinpointed the potential Mu species at the 425 924 distinct presumptive germinal insertion sites, tagging 36 612 B73v5 genes (Table 1; Data S1). To identify these potential Mu species, we associated the Mu‐TIR sequence which was part of each Mu‐seq read (i.e., flanking the gene sequence of interest), to a list of known Mu species (for details see “Experimental procedures”). Due to the highly conserved nature of the TIRs in all Mu transposons and the presence of non‐specific, short TIR fragments in the Mu‐seq reads, our reliable confirmation was limited to Mu1, Mu8, or MuDR transposons. Overall, we identified 82 285 (14.9%) Mu1, 1211 (0.2%) Mu8, 80 347 (14.5%) Mu8|MuDR species. For the majority of 390 124 (70.4%) of the 425 924 insertion sites, we could not identify the respective Mu species.
For validating BonnMu insertions and corresponding Mu species, we randomly selected insertions in three distinct genes: (i) Zm00001eb052530 carrying a Mu8 insertion in the F2‐family BonnMu‐2‐A‐0982, (ii) Zm00001eb280980 and (iii) Zm00001eb256020 harboring a Mu8 and Mu1 insertion in the mutagenized F2‐families BonnMu‐7‐C‐0459 and BonnMu‐F7‐2‐F‐1001, respectively. According to Data S1 following insertion identifiers, i.e., distinct 7‐digit numbers, identifying unique insertions, were assigned to the three insertions: (i) BonnMu0031087, (ii) BonnMu0170576 and (iii) BonnMu0446992. The insertion identifier BonnMu0031087 indicates a Mu8 insertion in the single exon of the gene Zm00001eb052530 (Figure 5a), located 157 bp downstream of the A from the ATG start codon (Data S1). A PCR‐based co‐segregation analysis of 11 individual plants of the segregating F2‐family BonnMu‐2‐A‐0982, identified two plants (#1 and #11) being homozygous for the wild‐type allele. Mutants were identified as heterozygotes, so both wild‐type and mutant‐specific bands were observed (#2–#10; Figure 5b). Sanger sequencing (Sanger et al., 1977) of the Mu‐specific PCR products confirmed that the Mutator insertion in this gene was caused by a Mu8 element (Figure 5c; Data S1).
Figure 5.
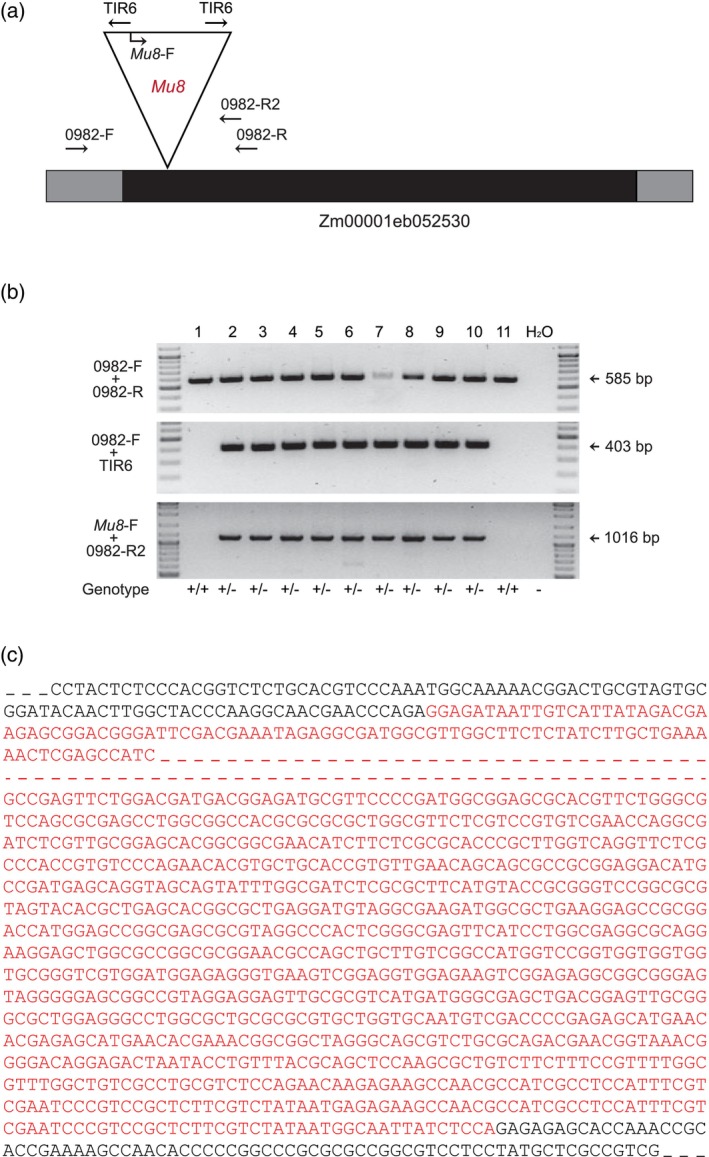
PCR‐based segregation analysis.
(a) Structure of the gene Zm00001eb052530. The single exon is illustrated as a black box and UTRs as gray boxes. The Mu8 insertion in the exon is shown as a triangle. Gene‐ and TIR‐specific primer sites are indicated as arrows.
(b) PCR segregation analysis of 11 individual plants of the segregating F2‐family BonnMu‐2‐A‐0982. Gene‐specific primers (0982‐F + 0982‐R) flanking the insertion site were combined to detect the presence of a wild‐type copy of the gene. Additionally, one gene‐specific primer along with a TIR‐specific primer (0982‐F + TIR6) were used to test for the presence of an insertion in the gene. To confirm the Mu8 insertion, a combination of a Mu8‐specific primer and a gene‐specific primer (Mu8‐F + 0982‐R2) was used. Deionized water (H2O) was used as a negative control.
(c) Confirmation of the Mu8 insertion the gene Zm00001eb052530 by Sanger sequencing. The sequence was amplified by combining two primer pairs: 0982‐F + TIR6 and Mu8‐F + 0982‐R2. The sequence represents a part of the gene with the Mu8 insertion depicted in red letters and dashed lines. The black letters represent part of the gene sequence, while dashed lines in black represent the remaining gene sequence that was not analyzed in detail.
Similarly, we confirmed the presence of another Mu8 element in the gene Zm00001eb280980 and a Mu1 species in the gene Zm00001eb256020 by genotyping individual plants from the F2‐families BonnMu‐7‐C‐0459 and BonnMu‐F7‐2‐F‐1001, respectively (Figure S3). Subsequent confirmation involved sequencing the Mu‐specific PCR products to verify the corresponding Mu elements (data not shown).
DISCUSSION
Sequencing of 8064 Mu transposon tagged BonnMu F2‐families identified 425 924 presumptive germinal insertions, representing 36 612 (83%; Table 1) of all annotated gene models of maize. This number marks a substantial increase from the prior 57% coverage (Marcon et al., 2020), which incorporated data from the UniformMu (McCarty et al., 2013) and the ChinaMu (Liang et al., 2019) resources. As of June 30, 2023, the ChinaMu database has been updated to encompass 104 294 germinal insertions, tagging 25 948 genes, constituting a coverage of 65% (http://chinamu.jaas.ac.cn). Nearly approaching whole‐genome saturation, the BonnMu collection in maize tagged more genes than those mutagenized in rice (60%; Wang et al., 2013), but fewer than in Arabidopsis (>90% gene coverage; Alonso & Ecker, 2006). For rice and Arabidopsis different techniques were employed, such as the two‐component transposon system Ac/Ds‐based mutagenesis (van Enckevort et al., 2005), the Tos17 retrotransposon mutagenesis (Miyao et al., 2003), and the transfer‐DNA insertional mutagenesis (Alonso et al., 2003; Toki et al., 2006). To track Mu‐induced insertions in the maize genome, the BonnMu library uses the high‐throughput sequencing strategy Mu‐seq (Liu et al., 2016; McCarty et al., 2013), which has been coupled to the robust automated downstream analysis MuWU (Stöcker et al., 2022), accelerating the identification of presumptive germinal insertions.
A unique feature of the BonnMu resource is that different North American dent and European flint germplasm groups were mutagenized thereby expanding and complementing the available resource. Notably, the flint lines DK105, EP1, and F7 are adapted to the climate of central Europe (Unterseer et al., 2016), and they represent important founders for European breeding programs (Haberer et al., 2020). In this study, we demonstrated that a substantial number of 4027 genes were exclusively tagged in at least one Mu‐seq library of the mutagenized flint lines, but not in the dent lines B73 and Co125 (Figure 1a). In contrast, the mutagenized BonnMu F2‐families of the dent pool contributed 2585 Mu‐tagged genes, which remained unaffected in any of the mutagenized flint lines. It could be worth checking for genic presence‐absence variations among the genes affected by Mu insertions. Moderate genic presence‐absence variations exist among flint and dent germplasm. Some of these genes found in either the dent or the flint pool are expressed at high levels (Haberer et al., 2020), which could contribute to line‐specific adaptations to environmental impacts and hence be of interest for maize improvement and breeding. Therefore, in future studies, the effect of Mu‐tagged presence‐absence genes could be investigated to identify genotype‐specific mutations and their impact on the mutant phenotype.
It has been reported that Mu elements exhibit a pronounced preference for 5′ UTRs of genes and tend to concentrate in genomic regions with epigenetic marks of open chromatin near the transcription start site of genes (Dietrich et al., 2002; Liu et al., 2009; Marcon et al., 2020; Springer et al., 2018; Zhang et al., 2020). In support with this, we identified the vast majority of 94% of all Mu insertions in genic regions of the genome, while only 6% of the insertions targeted intergenic regions (Figure 2). Moreover, the distribution pattern of BonnMu insertions across the 10 maize chromosomes indicates that gene‐dense chromosome arms are hotspots for Mu elements, whereas the heterochromatic centromere regions harbor fewer insertions (Figure 3). This finding is consistent with previous observations that gene‐rich chromosome arms are associated with highly accumulated Mu elements, e.g., in the B73 (Schnable et al., 2009) and W22 (Springer et al., 2018) genomes.
For functional genetics experiments Mu insertions in exonic regions are typically most useful to generate strong knockdown or knockout effects (e.g., Chen, Zhao, et al., 2023; Hunter et al., 2012, 2014), because they result in frameshift mutations or premature stop codons. Mutations in the promoter region and the 5′ UTR can also be useful, because they can affect gene expression. For example, the Mu insertion alleles in multiple positions of the 5′ UTR of the gl8 gene resulted in the glossy leaf phenotype (Dietrich et al., 2002). Similarly, Mu insertions in the intron or the 3′ UTR can affect gene expression and result in a mutant phenotype. For example, several intronic Mu insertions in the knotted1 (kn1) gene lead to dominant suppressible mutations (Greene et al., 1994). Mu insertions in the 3′ UTR of a caffeoyl‐CoA O‐methyltransferase gene, a candidate for a QTL conferring pathogen resistance, unexpectedly enhanced disease resistance by increasing mRNA stability, thus elevating gene expression (Yang et al., 2017). Such findings underscore the importance of including non‐exonic insertions in the analysis of mutant phenotypes and suggest that their potential role should not be underestimated in genetic studies.
Remarkably, we pinpointed chromosomal regions, specifically on chromosomes 5–8, that exhibit minimal occurrences of BonnMu insertions. These areas could represent highly heterochromatic non‐accessible knob regions in the genome that suppress local recombination (Ghaffari et al., 2013). Knobs are multi‐megabase tandem repeat arrays, predominantly located in mid‐arm positions of chromosomes (Dawe & Hiatt, 2004). They primarily consist of two tandemly repeated DNA sequences: the 180 bp knob repeat and the 350 bp tandemly repeated element TR‐1 (Ananiev et al., 1998). However, they have not been fully sequenced or accurately represented in genome assemblies due to the challenge of assembling their long, repetitive structures. Karyotyping has identified knob loci in maize, but there is a significant discrepancy in their representation (Ghaffari et al., 2013). While the physical size of knobs extends over a million base pairs, they are represented as only a few kilobases in genome assemblies. Advances in DNA sequencing technologies have significantly improved the maize B73 genome assembly, thereby reducing the initial count of over one hundred thousand gaps to just a few thousand (Jiao et al., 2017; Schnable et al., 2009; Sun et al., 2018). In the genome of the maize inbred line Mo17, heterochromatic knob180 and TR‐1 arrays were predominantly detected in the chromosome arms of chr1L, chr4L, chr6S, chr6L, chr8L, and chr9S (Chen, Wang, et al., 2023). Similarly, the karyotype of B73 identified such tandem repeat DNA sequences, primarily on chr4L, chr5L, chr6S, chr7L, chr8L, chr9S (Ghaffari et al., 2013). These areas are roughly overlapping with the regions on chr5L, chr6S, chr7L, and chr8L in the B73 genome (Figure 3), showing a notable absence of BonnMu insertions. Since knobs represent heterochromatic repeat elements, it is likely that they are not accessible for Mu transposon integration.
As previously shown, Mu elements exhibit a preference for targeting the 5′ UTRs and transcription start sites of genes (Dietrich et al., 2002; Liang et al., 2019; Marcon et al., 2020), which corresponds to open chromatin signals (Liu et al., 2009; Springer et al., 2018). To analyze the epigenomic landscape around Mu elements, we aligned BonnMu insertions with open chromatin signals and histone modifications, obtained from published datasets (Hufford et al., 2021; Makarevitch et al., 2013; Ricci et al., 2019; Zhang et al., 2015). BonnMu insertion patterns were in line with transposase‐accessible chromatin, as demonstrated by a comparison with ATAC‐seq datasets (Ricci et al., 2019; Figure 4). Both transposase‐accessible chromatin and BonnMu insertions showed a depletion at gene model midpoints, gradually increasing in frequency in 250 bp bins away from these midpoints. The opposite frequency was found for most of the DNA methylation marks, which were predominantly found at midpoints of gene models. This finding was previously reported by a meta‐analysis of ATAC‐seq signals (Ricci et al., 2019) and by using Micrococcal Nuclease (Rodgers‐Melnick et al., 2016) and DNase‐based assays (Oka et al., 2017), respectively. Generally, the relationship between TEs and chromatin in maize has been shown to be markedly variable, with a complex interplay between DNA methylation, histone modifications, and TEs impacting gene expression in the maize genome (Noshay et al., 2019; Ricci et al., 2019; West et al., 2014; Zhao et al., 2016).
Functional genetics experiments using mutagenized maize stocks, such as BonnMu F2‐families (Marcon et al., 2020 and this study), or UniformMu stocks (McCarty et al., 2013) require the validation of Mu insertions by PCR‐based genotyping (Liu et al., 2016). This method specifically amplifies DNA located between the highly conserved TIR sequence of the Mu element and adjacent regions of the genome. The TIR6 primer, which was generated based on the TIR sequences of Mu1, Mu7, Mu3, Mu8, and their variants (Settles et al., 2004), is typically used to confirm Mu insertions (Figure 5; Figure S3). The degenerate TIR6 primer is mostly effective for PCR to amplify the different classes of autonomous and non‐autonomous Mu elements (Liu et al., 2016). Among the classes of Mu elements, Mu1, Mu8, and MuDR exhibit the highest copy numbers in the maize genome (Liu et al., 2009). Here, we identified Mu1, Mu8, and MuDR elements in 30% of the BonnMu insertions (Data S1; Figure 5; Figure S3), facilitating future genotyping of BonnMu F2‐families. For future genotyping experiments, highly specific primers with no sequence degeneration can be designed to validate the insertions of these three classes of Mu elements.
It is worth mentioning that crossing of mutant alleles into multiple genetic inbred backgrounds can aid in detecting genetic variation in the mutant phenotype expression between the different inbred lines. Moreover, as demonstrated by comparisons of the genome sequences of the flint inbred lines DK105, EP1, and F7 used in this study and the dent inbred line B73, genetic presence/absence variations strongly support flint and dent as distinctive germplasm (Haberer et al., 2020). Hence, while hundreds of genes are present only in flint or dent inbred lines there are also many genes unique to individual flint or dent inbred lines. Hence, the BonnMu resource covering multiple maize genotypes could also be used to study inbred line‐specific mutations that are not present in other inbred lines.
In summary, the BonnMu resource has undergone significant expansion, providing a comprehensive assortment of Mu‐seq libraries that encompass diverse genetic backgrounds. The genetic diversity represented by the different genetic backgrounds in BonnMu enables the identification of genotype‐specific mutations in the future. The ability to tag and identify almost every gene in the maize genome is particularly noteworthy, as it enhances the resource's utility for researchers conducting functional genomic studies. Details on how to order the BonnMu F2‐families are summarized on our website at: https://www.bonnmu.uni‐bonn.de.
EXPERIMENTAL PROCEDURES
Plant material
Mutagenized BonnMu F2‐families were generated in field nurseries at the University of Bonn (Germany), Chile, Hawaii, and Mexico in the years 2014–2022 as previously described (Marcon et al., 2020). Briefly, we obtained the F1‐population by crossing a Mu‐active stock (i.e., Mu 4 per se; provided by Patrick Schnable, Iowa State University, USA) into five distinct inbred lines: B73, Co125, DK105, EP1, and F7. After obtaining the F1‐population, the F2‐population, segregating for recessive mutations, was generated by self‐pollinating all plants of the F1‐generation. The BonnMu F2‐families comprise a genetic background of 50% inbred line (e.g., B73) and 50% a Mu‐active stock. The Mu‐active stock is a hybrid derived from crossing various Mu stocks, referred to as Mu 4 per se (Robertson, 1983). For example, Mu 2 per se represents the F1‐generation resulting from the cross‐pollination of two distinct Mu stocks, and crossing two Mu 2 per se stocks produces the Mu 4 per se generation (Robertson, 1983). The genetic background of the Mu 2 per se stocks is unknown.
Construction of Mu‐seq libraries
BonnMu libraries were constructed using the Mu‐seq method (Liu et al., 2016; Marcon et al., 2020; McCarty et al., 2013). Briefly, we pooled a total of 6912 BonnMu families in 12 Mu‐seq libraries in the genetic backgrounds of B73, Co125, DK105, EP1, and F7 (Table S1). A 2‐dimensional 24 × 24 grid design was utilized to pool 576 BonnMu F2‐families per library (Marcon et al., 2020). For each Mu‐seq library construction, we germinated eight seeds per family using a paper roll system (Hetz et al., 1996). We incubated the seedlings in a climate chamber with a photoperiod of 16 h (28°C, 2700 lux) and a dark period of 8 h (21°C) at 70% humidity. At 10–12 days after germination, the leaf samples were harvested and pooled based on the 24 × 24 grid design. The samples were taken from at least three seedlings of each F2‐family. To ensure the presence of at least one mutant allele per Mu‐tagged gene within the 3–8 germinated plants per F2‐family, the probability of 99% was calculated using dbinom() and dhyper() functions in R (Table S5; Liu et al., 2016; Marcon et al., 2020; McCarty et al., 2013; R Core Team, 2021). For the precise identification of presumptive heritable insertions at the intersections of rows and columns in the grid, leaf samples from independent somatic cell lineages, i.e., alternate leaves of each seedling per family, were sampled in one distinct row and one distinct column pool. By using this method, somatic insertions appeared in most instances only in a single axis of the grid and were subsequently excluded from downstream analyses. The harvested samples were frozen in liquid nitrogen and kept at −80°C before use. For each library, the frozen leaf samples were ground manually using pre‐cooled mortars and pestles.
After isolation of genomic DNA from each pool according to Nalini et al. (2003), the genomic DNA was randomly sheared using a Bioruptor® Pico sonication device (Diagenode, Liège, Belgium) at 2 sec‐on/2 sec‐off setting for 2–4 cycles to obtain the fragment sizes of about 1 kb. The size of fragmented genomic DNA was analyzed by agarose gel electrophoresis after sonication. The randomly sheared genomic DNA fragments had single‐stranded overhangs, which were attentively filled in using an enzyme mix (Quick Blunting™ Kit; Thermo Fisher, Schwerte, North Rhine‐Westphalia, Germany). This process generated blunt ends, facilitating the subsequent ligation of a double‐stranded universal (U) adapter.
The Mu‐flanking amplicons were subsequently enriched through a ligation‐mediated PCR (PCR‐I), using a Mu‐TIR‐specific primer and a specific primer for the ligated U adapter. Then, the fragments were incorporated with a part of an Illumina sequencing adapter and a TIR sequence in the PCR‐II. To minimize the number of very short Mu‐flanking fragments, PCR‐II products were purified using a CleanNGS magnetic bead‐based clean‐up system (CleanNA, Krefeld, North Rhine‐Westphalia, Germany). The final PCR‐III integrates the remaining sequencing adapters and 6 bp barcodes which enabled multiplexing of the 48 pools. Quality and quantity of each Mu‐seq library were assessed by using a Bioanalyzer with a DNA 7500 chip (Agilent Technologies, Waldbronn, Baden‐Württemberg, Germany) to obtain the required concentrations for sequencing. The multiplexed Mu‐seq libraries were subjected to paired‐end sequencing with a read length of 150 base pairs (bp) using the HiSeq X Ten sequencing system. The raw sequencing data were stored at the Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) under BioProject accession number PRJNA914277.
MuWU: Identification of Mu insertion sites in B73, Co125, DK105, EP1, and F7
Mu‐Seq reads were processed using an automated processing pipeline, referred to as Mu‐Seq Workflow Utility (MuWU; Stöcker et al., 2022). Briefly, Mu insertion sites were detected based on the characteristic 9 bp target site duplications at the insertion flanking regions of Mu transposons. Combined with the grid design, this allowed the differentiation between presumptive germinal and somatic insertion events. After mapping all Mu‐seq reads to the B73v5 reference genome (Zm‐B73‐REFERENCE‐NAM‐5.0; Zm00001eb.1; Gage et al., 2020), insertions were associated with specific genomic loci. In detail, we considered Mu insertion sites in 5′ and 3′ untranslated regions (UTRs) of genes, exons and introns, the ≤2100 bp upstream promoter regions and ≤2100 bp downstream regions of genes (Data S1). With the release of MuWU v1.5, we added the capability to determine the specific classes of Mu1, Mu8, and MuDR elements for a detected insertion. Details of the implementation and required data are outlined in the software's GitHub repository (https://github.com/groupschoof/MuWU). Finally, we generated an output table of presumptive germinal insertion events, that included additional information on each event such as genomic location, associated information based on genome annotations, and the most likely class of Mu element (Data S1).
Furthermore, we investigated the presumptive germinal insertion sites by aligning Mu‐seq reads from the 3456 sequenced BonnMu F2‐families in the genetic backgrounds of the flint cultivars DK105, EP1, and F7 to their corresponding genomes: DK105 (Zm00016a.1; https://download.maizegdb.org/Zm‐DK105‐REFERENCE‐TUM‐1.0/), EP1 (Zm00010a.1; https://download.maizegdb.org/Zm‐EP1‐REFERENCE‐TUM‐1.0) and F7 (Zm00011a.1; https://download.maizegdb.org/Zm‐F7‐REFERENCE‐TUM‐1.0; Haberer et al., 2020). The respective output tables list presumptive germinal Mu insertions and affected genes (DK105: Data S2; EP1: Data S3; F7: Data S4).
Downstream analysis of BonnMu insertion sites
To investigate the presence of Mu insertions in different libraries/genotypes, a presence/absence intersection matrix based on GeneIDs was created. An Upset plot was generated using the UpSetR package (Conway et al., 2017) in R, providing a visual representation of the intersections among the 14 Mu‐seq libraries and five genotypes. Correlations between the number of insertions and the length of the affected genes were calculated based on Pearson correlation coefficient (r) in R v4.3.1 (R Core Team, 2021). To further visualize the distribution of Mu insertions across various genomic partitions, i.e., exons, introns, UTRs, and promoter regions, we analyzed the seven Mu‐seq libraries in B73 background. To determine if the Mu insertion sites align with gene density, the distribution of BonnMu insertions in B73 background was aligned with the genes in each chromosome using the MaizeGDB JBrowse genome browser (Woodhouse et al., 2021). In addition, we employed the published ATAC‐seq (Ricci et al., 2019), ChIP‐seq (Makarevitch et al., 2013; Zhang et al., 2015), NAM‐ATAC and NAM‐UMRs datasets (Hufford et al., 2021) to investigate chromatin accessibility and histone modifications in relation to BonnMu insertions. We used the GenomicDistributions R package (Kupkova et al., 2022) to analyze distributions and create visualizations.
Identification of Mu species per insertion
With the release of MuWU v1.5 (https://github.com/tgstoecker/MuWU) we have incorporated a new feature facilitating the identification of sub‐types or specific elements within the detected Mu insertions. This works by supplying a set of sequences which are specific to a particular subtype/ element of the particular transposon in question (Liu et al., 2009). For this feature the raw input reads have to contain this sequence. However, it has to be considered that such sequences (in our usage of MuWU) are cut/trimmed during an analysis run. Specifically, for the BonnMu libraries we use a 12‐fold degenerate TIR primer which is trimmed away as a prerequisite before the alignment step. Therefore, based on the subtype/ element sequence association, all matching raw reads are sorted into files for the specific subtype/ element as a process uncoupled from the normal MuWU workflow. Once the insertions are identified, we associate them via their corresponding reads with all respective subtypes/elements. Since the TIR sequence of any particular Mu element can vary between the left and right end of the transposon, both the “_L” (left side) and the “_R” (right side) sequence has to be considered. For each insertion we looked at the TIR section of all aligned reads and counted the exact matches to each subtype sequence that has been supplied to the MuWU software. In many cases this resulted unclear and contradictory subtyping since usage of the 12‐fold degenerate primer is prone to errors. For our insertion tables, we therefore added a quality filter that only categorizes an insertion if the majority of reads align with a specific Mu subtype. We further only included information of subtyping on Mu1, Mu8, and MuDR as we tested these and were able to confirm subtyping with PCR‐based co‐segregation analysis.
While a priori‐guided determination of known Mu elements in our analysis is valuable to further understand the landscape of Mu insertions, we also support investigation of putative novel elements. Insertions which cannot be associated with a supplied species type (no match to supplied TIR sequences) are additionally investigated. Their reads are extracted and clustered and their redundancy is removed. This, in theory, allows for the detection of putative novel types or elements that were not considered by the user/ sequencing steps and can be further investigated.
Confirmation of Mu insertions by PCR
We performed PCR‐based confirmation of Mu insertions using the following BonnMu F2‐families: BonnMu‐2‐A‐0982, BonnMu‐7‐C‐0458, and BonnMu‐F7‐2‐F‐1001. To this end, 12–30 seeds per segregating BonnMu F2‐family were germinated using the paper roll system (Hetz et al., 1996). Leaf samples were harvested 10 days after germination and genomic DNA was isolated according to Nalini et al. (2003). Gene‐specific primers flanking the Mu insertion sites were designed using the Primer‐BLAST online tool (https://www.ncbi.nlm.nih.gov/tools/primer‐blast/). To genotype the plants, three different combinations of primers were used in separate reactions: (i) gene‐specific forward and reverse primer to detect the presence of the gene copy, (ii) gene‐specific forward and TIR6 primer, and (iii) gene‐specific reverse and TIR6 primer, both to detect the presence of Mu insertions. Primer sequences are provided in Table S6. The PCR was performed using Phusion™ High‐Fidelity DNA Polymerase (Thermo Fisher). The PCR products from the individual plants that showed the presence of Mu insertions were subjected to Sanger sequencing (Sanger et al., 1977). The resulting sequences were then analyzed using BioEdit software (Hall, 1999) to confirm the presence and specific locations of the Mu insertions.
ACCESSION NUMBERS
Raw sequencing data of Mu‐seq libraries are stored at the Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra, https://www.ncbi.nlm.nih.gov/sra/PRJNA608624) with the accession number PRJNA608624 and PRJNA914277.
AUTHOR CONTRIBUTIONS
CM and FH established the research project. CM, YNW and AB produced the Mu‐seq libraries. CM, YNW and XD phenotyped the BonnMu F2‐families and generated seedling photos for public access via MaizeGDB.org. TS, MP, AK, H‐PP and HS conducted the bioinformatics analyses. YNW and TS interpreted the data and drafted the article. CM and FH contributed in data interpretation and assisted to draft the article. All authors endorsed the final draft of the article.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
OPEN RESEARCH BADGES
This article has earned Open Data and Open Materials badges. Data and materials are available at http://www.ncbi.nlm.nih.gov/sra/PRJNA914277, https://jbrowse.maizegdb.org/?loc=chr2%3A1..243675191&tracks=gene_models_official%2CBonnMu2024&highlight= and https://github.com/groupschoof/MuWU.
Supporting information
Data S1. Number of Mu insertions and affected genes among all BonnMu F2‐families in B73, Co125, DK105, EP1, and F7 genetic background. During the MuWU analysis Mu‐seq reads were mapped to the genome of B73v5.
Data S2. Number of Mu insertions and affected genes among BonnMu F2‐families in DK105 genetic background. During the MuWU analysis Mu‐seq reads were mapped to the genome of DK105.
Data S3. Number of Mu insertions and affected genes among BonnMu F2‐families in EP1 genetic background. During the MuWU analysis Mu‐seq reads were mapped to the genome of EP1.
Data S4. Number of Mu insertions and affected genes among BonnMu F2‐families in F7 genetic background. During the MuWU analysis Mu‐seq reads were mapped to the genome of F7.
Data S5. Presence (1)/ absence (0) matrix to generate the Upset plot shown in Figure 1(a).
Figure S1. Number of genes affected by Mu insertions and distribution of insertions. (a) Number of tagged genes and associated mean gene length plotted against the number of Mu insertions. Only insertions in 5′ and 3′ UTRs, exons, and introns of genes were considered. (b) Distribution of affected gene lengths plotted against the number of individual Mu insertions. The calculated Pearson correlation coefficient is r = 0.136 (P < 0.001). The number of insertions >50 ranges from 51 to 255 Mu insertions per gene.
Figure S2. Exemplary seedling mutants segregating from two BonnMu F2‐families. (a) Pale green leaf mutants from BonnMu‐7‐C‐0336 and (b) mutants affected in shoot development from BonnMu‐9‐G‐0034.
Figure S3. Confirmation of Mu species by PCR. (a) Simplified gene model of Zm00001eb280980 carrying a Mu8 element in its 5′ UTR and PCR segregation analysis of 13 segregating plants of the BonnMu‐7‐C‐0459 family (lower picture). Genotyping of 13 individual plants using gene‐specific primers 0459‐F + 0459‐R and Mu‐specific primers 0459‐F + TIR6 and 0459‐R + TIR6 identified 10 plants as heterozygotes (−/+: #1–3; #5–6; #8–12) and three plants as homozygous wild types (+/+: #4; #7; #13). (b) Simplified gene model of Zm00001eb256020 tagged by a Mu1 element in an intron. PCR segregation analysis of BonnMu‐F7‐2‐F‐1001 family (lower panel) identified seven of the segregating plants as heterozygotes (+/−: #1–3; #7–8; #10–11) and five plants as homozygous wild types (+/+: #4–6; #9; #12). Exons in the upper panels of (a) and (b) are illustrated as black boxes and UTRs as gray boxes. The Mu insertions are shown as triangles. Gene‐ and MuTIR‐specific primer sites are indicated as arrows (F/R = gene‐specific forward and reverse primers).
Table S1. BonnMu F2‐families used for Mu‐seq library construction. Per library, 576 mutagenized F2‐families were pooled.
Table S2. Number of observed and expected Mu tagged genes among the Mu insertional libraries in various inbred lines (related to Figure 1).
Table S3. Partition of the B73v5 genome (related to Figure 2a).
Table S4. Number of observed and expected Mu insertions across the B73v5 genome (related to Figure 2c).
Table S5. Calculated probability of obtaining at least one mutant allele per tagged gene among 3–8 germinated plants per BonnMu F2‐family. The left panel, highlighted in gray, shows probabilities for scenarios with eight germinated seedlings per F2‐family. The right panel represents probabilities when only three out of the eight seedlings per F2‐family germinated and were subsequently harvested. WT: wild type, mut.: mutant.
Table S6. List of oligonucleotide primers used for PCR‐based genotyping of segregating plants of three BonnMu F2‐families.
ACKNOWLEDGEMENTS
We thank Christa Schulz and Helmut Rehkopf (University of Bonn, Germany) for technical assistance. We thank Patrick Schnable (Iowa State University, USA) for providing the Mu‐active stock. This work was funded by the Deutsche Forschungsgemeinschaft (DFG) grant MA8427/1‐1 to CM.
Contributor Information
Frank Hochholdinger, Email: frank.hochholdinger@uni-bonn.de.
Caroline Marcon, Email: marcon@uni-bonn.de.
DATA AVAILABILITY STATEMENT
Raw sequencing data of Mu‐seq libraries are stored at the Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra, https://www.ncbi.nlm.nih.gov/sra/PRJNA608624) with the accession number PRJNA608624 and PRJNA914277.
REFERENCES
- Alonso, J.M. & Ecker, J.R. (2006) Moving forward in reverse: genetic technologies to enable genome‐wide phenomic screens in Arabidopsis . Nature Reviews Genetics, 7, 524–536. Available from: 10.1038/nrg1893 [DOI] [PubMed] [Google Scholar]
- Alonso, J.M. , Stepanova, A.N. , Leisse, T.J. , Kim, C.J. , Chen, H. , Shinn, P. et al. (2003) Genome‐wide insertional mutagenesis of Arabidopsis thaliana . Science, 301, 653–657. Available from: 10.1126/science.1086391 [DOI] [PubMed] [Google Scholar]
- Ananiev, E.V. , Phillips, R.L. & Rines, H.W. (1998) A knob‐associated tandem repeat in maize capable of forming fold‐back DNA segments: are chromosome knobs megatransposons? Proceedings of the National Academy of Sciences of the United States of America, 95, 10785–10790. Available from: 10.1073/pnas.95.18.10785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro, J.D. , Giresi, P.G. , Zaba, L.C. , Chang, H.Y. & Greenleaf, W.J. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nature Methods, 10, 1213–1218. Available from: 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candela, H. & Hake, S. (2008) The art and design of genetic screens: maize. Nature Reviews Genetics, 9, 192–203. Available from: 10.1038/nrg2291 [DOI] [PubMed] [Google Scholar]
- Chen, C. , Zhao, Y. , Tabor, G. , Nian, H. , Phillips, J. , Wolters, P. et al. (2023) A leucine‐rich repeat receptor kinase gene confers quantitative susceptibility to maize southern leaf blight. New Phytologist, 238, 1182–1197. Available from: 10.1111/nph.18781 [DOI] [PubMed] [Google Scholar]
- Chen, J. , Wang, Z. , Tan, K. , Huang, W. , Shi, J. , Li, T. et al. (2023) A complete telomere‐to‐telomere assembly of the maize genome. Nature Genetics, 55, 1221–1231. Available from: 10.1038/s41588-023-01419-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway, J.R. , Lex, A. & Gehlenborg, N. (2017) UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics, 33, 2938–2940. Available from: 10.1093/bioinformatics/btx364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, D. , Jin, L. , Huo, Z. , Yan, S. , Ma, Z. , Qi, W. et al. (2021) Maize pentatricopeptide repeat protein DEK53 is required for mitochondrial RNA editing at multiple sites and seed development. Journal of Experimental Botany, 71, 6246–6261. Available from: 10.1093/JXB/ERAA348 [DOI] [PubMed] [Google Scholar]
- Dawe, R.K. & Hiatt, E.N. (2004) Plant neocentromeres: fast, focused, and driven. Chromosome Research, 12, 655–669. Available from: 10.1023/B:CHRO.0000036607.74671.db [DOI] [PubMed] [Google Scholar]
- Dietrich, C.R. , Cui, F. , Packila, M.L. , Li, J. , Ashlock, D.A. , Nikolau, B.J. et al. (2002) Maize Mu transposons are targeted to the 5′ untranslated region of the gl8 gene and sequences flanking Mu target‐site duplications exhibit nonrandom nucleotide composition throughout the genome. Genetics, 160, 697–716. Available from: 10.1093/genetics/160.2.697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage, J.L. , Monier, B. , Giri, A. & Buckler, E.S. (2020) Ten years of the maize nested association mapping population: impact, limitations, and future directions. The Plant Cell, 32, 2083–2093. Available from: 10.1105/tpc.19.00951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaffari, R. , Cannon, E.K.S. , Kanizay, L.B. , Lawrence, C.J. & Dawe, R.K. (2013) Maize chromosomal knobs are located in gene‐dense areas and suppress local recombination. Chromosoma, 122, 67–75. Available from: 10.1007/s00412-012-0391-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene, B. , Walko, R. & Hake, S. (1994) Mutator insertions in an intron of the maize knotted1 gene result in dominant suppressible mutations. Genetics, 138, 1275–1285. Available from: 10.1093/genetics/138.4.1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberer, G. , Kamal, N. , Bauer, E. , Gundlach, H. , Fischer, I. , Seidel, M.A. et al. (2020) European maize genomes highlight intraspecies variation in repeat and gene content. Nature Genetics, 52, 950–957. Available from: 10.1038/s41588-020-0671-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, T.A. (1999) BioEdit: a user‐friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98. [Google Scholar]
- Hetz, W. , Hochholdinger, F. , Schwall, M. & Feix, G. (1996) Isolation and characterization of rtcs, a maize mutant deficient in the formation of nodal roots. The Plant Journal, 10, 845–857. Available from: 10.1046/j.1365-313X.1996.10050845.x [DOI] [Google Scholar]
- Hufford, M.B. , Seetharam, A.S. , Woodhouse, M.R. , Chougule, K.M. , Ou, S. , Liu, J. et al. (2021) De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science, 373, 655–662. Available from: 10.1126/science.abg5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter, C.T. , Hill Kirienko, D. , Sylvester, A.W. , Peter, G.F. , McCarty, D.R. & Koch, K.E. (2012) Cellulose synthase‐like D1 is integral to normal cell division, expansion, and leaf development in maize. Plant Physiology, 158, 708–724. Available from: 10.1104/pp.111.188466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter, C.T. , Suzuki, M. , Saunders, J. , Wu, S. , Tasi, A. , McCarty, D.R. et al. (2014) Phenotype to genotype using forward‐genetic Mu‐seq for identification and functional classification of maize mutants. Frontiers in Plant Science, 4, 545. Available from: 10.3389/fpls.2013.00545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, Y. , Peluso, P. , Shi, J. , Liang, T. , Stitzer, M.C. , Wang, B. et al. (2017) Improved maize reference genome with single‐molecule technologies. Nature, 546, 524–527. Available from: 10.1038/nature22971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupkova, K. , Mosquera, J.V. , Smith, J.P. , Stolarczyk, M. , Danehy, T.L. , Lawson, J.T. et al. (2022) GenomicDistributions: fast analysis of genomic intervals with Bioconductor. BMC Genomics, 23, 299. Available from: 10.1186/s12864-022-08467-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Hey, S. , Liu, S. , Liu, Q. , McNinch, C. , Hu, H.C. et al. (2016) Characterization of maize roothairless6 which encodes a D‐type cellulose synthase and controls the switch from bulge formation to tip growth. Scientific Reports, 6, 34395. Available from: 10.1038/srep34395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, L. , Zhou, L. , Tang, Y. , Li, N. , Song, T. , Shao, W. et al. (2019) A sequence‐indexed Mutator insertional library for maize functional genomics study. Plant Physiology, 181(4), 1404–1414. Available from: 10.1104/pp.19.00894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch, D. (2002) Mutator transposons. Trends in Plant Science, 7, 498–504. Available from: 10.1016/S1360-1385(02)02347-6 [DOI] [PubMed] [Google Scholar]
- Lisch, D. (2015) Mutator and MULE transposons. Microbiology Spectrum, 3, MDNA3‐0034‐2014. Available from: 10.1128/microbiolspec.MDNA3-0032-2014 [DOI] [PubMed] [Google Scholar]
- Liu, P. , McCarty, D.R. & Koch, K.E. (2016) Transposon mutagenesis and analysis of mutants in UniformMu maize (Zea mays). Current Protocols in Plant Biology, 1, 451–465. Available from: 10.1002/cppb.20029 [DOI] [PubMed] [Google Scholar]
- Liu, S. , Yeh, C.T. , Ji, T. , Ying, K. , Wu, H. , Tang, H.M. et al. (2009) Mu transposon insertion sites and meiotic recombination events co‐localize with epigenetic marks for open chromatin across the maize genome. PLoS Genetics, 5, e1000733. Available from: 10.1371/journal.pgen.1000733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarevitch, I. , Eichten, S.R. , Briskine, R. , Waters, A.J. , Danilevskaya, O.N. , Meeley, R.B. et al. (2013) Genomic distribution of maize facultative heterochromatin marked by trimethylation of H3K27. The Plant Cell, 25, 780–793. Available from: 10.1105/tpc.112.106427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcon, C. , Altrogge, L. , Win, Y.N. , Stöcker, T. , Gardiner, J.M. , Portwood, J.L., II et al. (2020) BonnMu: a sequence‐indexed resource of transposon‐induced maize mutations for functional genomics studies. Plant Physiology, 184, 620–631. Available from: 10.1104/pp.20.00478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty, D.R. , Latshaw, S. , Wu, S. , Suzuki, M. , Hunter, C.T. , Avigne, W.T. et al. (2013) Mu‐seq: sequence‐based mapping and identification of transposon induced mutations. PLoS One, 8, e77172. Available from: 10.1371/journal.pone.0077172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock, B. (1951) Mutable loci in maize. Carnegie Institution of Washington Yearbook, 50, 174–181. [Google Scholar]
- Miyao, A. , Tanaka, K. , Murata, K. , Sawaki, H. , Takeda, S. , Abe, K. et al. (2003) Target site specificity of the Tos17 retrotransposon shows a preference for insertion within genes and against insertion in retrotransposon‐rich regions of the genome. The Plant Cell, 15, 1771–1780. Available from: 10.1105/tpc.012559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalini, E. , Bhagwat, S. & Jawali, N. (2003) A simple method for isolation of DNA from plants suitable for long‐term storage and DNA marker analysis. BARC Newsletter, 249, 208–214. [Google Scholar]
- Nestler, J. , Liu, S. , Wen, T.J. , Paschold, A. , Marcon, C. , Tang, H.M. et al. (2014) Roothairless5, which functions in maize (Zea mays L.) root hair initiation and elongation encodes a monocot‐specific NADPH oxidase. The Plant Journal, 79, 729–740. Available from: 10.1111/tpj.12578 [DOI] [PubMed] [Google Scholar]
- Noshay, J.M. , Anderson, S.N. , Zhou, P. , Ji, L. , Ricci, W. , Lu, Z. et al. (2019) Monitoring the interplay between transposable element families and DNA methylation in maize. PLoS Genetics, 15, e1008291. Available from: 10.1371/journal.pgen.1008291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka, R. , Zicola, J. , Weber, B. , Anderson, S.N. , Hodgman, C. , Gent, J.I. et al. (2017) Genome‐wide mapping of transcriptional enhancer candidates using DNA and chromatin features in maize. Genome Biology, 18, 137. Available from: 10.1186/s13059-017-1273-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2021) R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Ricci, W.A. , Lu, Z. , Ji, L. , Marand, A.P. , Ethridge, C.L. , Murphy, N.G. et al. (2019) Widespread long‐range cis‐regulatory elements in the maize genome. Nature Plants, 5, 1237–1249. Available from: 10.1038/s41477-019-0547-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, D.S. (1978) Characterization of a mutator system in maize. Mutation Research, Fundamental and Molecular Mechanisms of Mutagenesis, 51, 21–28. Available from: 10.1016/0027-5107(78)90004-0 [DOI] [Google Scholar]
- Robertson, D.S. (1983) A possible dose‐dependent inactivation of mutator (mu) in maize. Molecular and General Genetics, 191, 86–90. Available from: 10.1007/BF00330894 [DOI] [Google Scholar]
- Rodgers‐Melnick, E. , Vera, D.L. , Bass, H.W. & Buckler, E.S. (2016) Open chromatin reveals the functional maize genome. Proceedings of the National Academy of Sciences of the United States of America, 113, E3177–E3184. Available from: 10.1073/pnas.1525244113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger, F. , Nicklen, S. & Coulson, A.R. (1977) DNA sequencing with chain‐terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America, 74, 5463–5467. Available from: 10.1073/pnas.74.12.5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable, J.C. & Freeling, M. (2011) Genes identified by visible mutant phenotypes show increased bias toward one of two subgenomes of maize. PLoS One, 6, e17855. Available from: 10.1371/journal.pone.0017855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable, P.S. , Ware, D. , Fulton, R.S. , Stein, J.C. , Wei, F. , Pasternak, S. et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science, 326, 1112–1115. Available from: 10.1126/science.1178534 [DOI] [PubMed] [Google Scholar]
- Settles, A.M. , Latshaw, S. & McCarty, D.R. (2004) Molecular analysis of high‐copy insertion sites in maize. Nucleic Acids Research, 32, e54. Available from: 10.1093/nar/gnh052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer, N.M. , Anderson, S.N. , Andorf, C.M. , Ahern, K.R. , Bai, F. , Barad, O. et al. (2018) The maize W22 genome provides a foundation for functional genomics and transposon biology. Nature Genetics, 50, 1282–1288. Available from: 10.1038/s41588-018-0158-0 [DOI] [PubMed] [Google Scholar]
- Stöcker, T. , Altrogge, L. , Marcon, C. , Win, Y.N. , Hochholdinger, F. & Schoof, H. (2022) MuWU: mutant‐seq library analysis and annotation. Bioinformatics, 38, 837–838. Available from: 10.1093/bioinformatics/btab679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, S. , Zhou, Y. , Chen, J. , Shi, J. , Zhao, H. , Song, W. et al. (2018) Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nature Genetics, 50, 1289–1295. Available from: 10.1038/s41588-018-0182-0 [DOI] [PubMed] [Google Scholar]
- Tan, B.C. , Chen, Z. , Shen, Y. , Zhang, Y. , Lai, J. & Sun, S.S.M. (2011) Identification of an active new mutator transposable element in maize. G3: Genes, Genomes, Genetics, 1, 293–302. Available from: 10.1534/g3.111.000398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toki, S. , Hara, N. , Ono, K. , Onodera, H. , Tagiri, A. , Oka, S. et al. (2006) Early infection of scutellum tissue with agrobacterium allows high‐speed transformation of rice. The Plant Journal, 47, 969–976. Available from: 10.1111/j.1365-313X.2006.02836.x [DOI] [PubMed] [Google Scholar]
- Unterseer, S. , Pophaly, S.D. , Peis, R. , Westermeier, P. , Mayer, M. , Seidel, M.A. et al. (2016) A comprehensive study of the genomic differentiation between temperate dent and flint maize. Genome Biology, 17, 137. Available from: 10.1186/s13059-016-1009-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Enckevort, L.J. , Droc, G. , Piffanelli, P. , Greco, R. , Gagneur, C. , Weber, C. et al. (2005) EU‐OSTID: a collection of transposon insertional mutants for functional genomics in rice. Plant Molecular Biology, 59, 99–110. Available from: 10.1007/s11103-005-8532-6 [DOI] [PubMed] [Google Scholar]
- Wang, N. , Long, T. , Yao, W. , Xiong, L. , Zhang, Q. & Wu, C. (2013) Mutant resources for the functional analysis of the rice genome. Molecular Plant, 6, 596–604. Available from: 10.1093/mp/sss142 [DOI] [PubMed] [Google Scholar]
- West, P.T. , Li, Q. , Ji, L. , Eichten, S.R. , Song, J. , Vaughn, M.W. et al. (2014) Genomic distribution of H3K9me2 and DNA methylation in a maize genome. PLoS One, 9, e105267. Available from: 10.1371/journal.pone.0105267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse, M.R. , Cannon, E.K. , Portwood, J.L., II , Harper, L.C. , Gardiner, J.M. , Schaeffer, M.L. et al. (2021) A pan‐genomic approach to genome databases using maize as a model system. BMC Plant Biology, 21, 385. Available from: 10.1186/s12870-021-03173-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C. , Tai, H. , Saleem, M. , Ludwig, Y. , Majer, C. , Berendzen, K.W. et al. (2015) Cooperative action of the paralogous maize lateral organ boundaries (LOB) domain proteins RTCS and RTCL in shoot‐borne root formation. New Phytologist, 207, 1123–1133. Available from: 10.1111/nph.13420 [DOI] [PubMed] [Google Scholar]
- Yang, Q. , He, Y. , Kabahuma, M. , Chaya, T. , Kelly, A. , Borrego, E. et al. (2017) A gene encoding maize caffeoyl‐CoA O‐methyltransferase confers quantitative resistance to multiple pathogens. Nature Genetics, 49, 1364–1372. Available from: 10.1038/ng.3919 [DOI] [PubMed] [Google Scholar]
- Zhang, W. , Garcia, N. , Feng, Y. , Zhao, H. & Messing, J. (2015) Genome‐wide histone acetylation correlates with active transcription in maize. Genomics, 106, 214–220. Available from: 10.1016/j.ygeno.2015.05.005 [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Zhao, M. , McCarty, D.R. & Lisch, D. (2020) Transposable elements employ distinct integration strategies with respect to transcriptional landscapes in eukaryotic genomes. Nucleic Acids Research, 48, 6685–6698. Available from: 10.1093/nar/gkaa370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, H. , Zhu, X. , Wang, K. , Gent, J.I. , Zhang, W. , Dawe, K.R. et al. (2016) Gene expression and chromatin modifications associated with maize centromeres. G3: Genes, Genomes, Genetics, 6, 183–192. Available from: 10.1534/g3.115.022764 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Number of Mu insertions and affected genes among all BonnMu F2‐families in B73, Co125, DK105, EP1, and F7 genetic background. During the MuWU analysis Mu‐seq reads were mapped to the genome of B73v5.
Data S2. Number of Mu insertions and affected genes among BonnMu F2‐families in DK105 genetic background. During the MuWU analysis Mu‐seq reads were mapped to the genome of DK105.
Data S3. Number of Mu insertions and affected genes among BonnMu F2‐families in EP1 genetic background. During the MuWU analysis Mu‐seq reads were mapped to the genome of EP1.
Data S4. Number of Mu insertions and affected genes among BonnMu F2‐families in F7 genetic background. During the MuWU analysis Mu‐seq reads were mapped to the genome of F7.
Data S5. Presence (1)/ absence (0) matrix to generate the Upset plot shown in Figure 1(a).
Figure S1. Number of genes affected by Mu insertions and distribution of insertions. (a) Number of tagged genes and associated mean gene length plotted against the number of Mu insertions. Only insertions in 5′ and 3′ UTRs, exons, and introns of genes were considered. (b) Distribution of affected gene lengths plotted against the number of individual Mu insertions. The calculated Pearson correlation coefficient is r = 0.136 (P < 0.001). The number of insertions >50 ranges from 51 to 255 Mu insertions per gene.
Figure S2. Exemplary seedling mutants segregating from two BonnMu F2‐families. (a) Pale green leaf mutants from BonnMu‐7‐C‐0336 and (b) mutants affected in shoot development from BonnMu‐9‐G‐0034.
Figure S3. Confirmation of Mu species by PCR. (a) Simplified gene model of Zm00001eb280980 carrying a Mu8 element in its 5′ UTR and PCR segregation analysis of 13 segregating plants of the BonnMu‐7‐C‐0459 family (lower picture). Genotyping of 13 individual plants using gene‐specific primers 0459‐F + 0459‐R and Mu‐specific primers 0459‐F + TIR6 and 0459‐R + TIR6 identified 10 plants as heterozygotes (−/+: #1–3; #5–6; #8–12) and three plants as homozygous wild types (+/+: #4; #7; #13). (b) Simplified gene model of Zm00001eb256020 tagged by a Mu1 element in an intron. PCR segregation analysis of BonnMu‐F7‐2‐F‐1001 family (lower panel) identified seven of the segregating plants as heterozygotes (+/−: #1–3; #7–8; #10–11) and five plants as homozygous wild types (+/+: #4–6; #9; #12). Exons in the upper panels of (a) and (b) are illustrated as black boxes and UTRs as gray boxes. The Mu insertions are shown as triangles. Gene‐ and MuTIR‐specific primer sites are indicated as arrows (F/R = gene‐specific forward and reverse primers).
Table S1. BonnMu F2‐families used for Mu‐seq library construction. Per library, 576 mutagenized F2‐families were pooled.
Table S2. Number of observed and expected Mu tagged genes among the Mu insertional libraries in various inbred lines (related to Figure 1).
Table S3. Partition of the B73v5 genome (related to Figure 2a).
Table S4. Number of observed and expected Mu insertions across the B73v5 genome (related to Figure 2c).
Table S5. Calculated probability of obtaining at least one mutant allele per tagged gene among 3–8 germinated plants per BonnMu F2‐family. The left panel, highlighted in gray, shows probabilities for scenarios with eight germinated seedlings per F2‐family. The right panel represents probabilities when only three out of the eight seedlings per F2‐family germinated and were subsequently harvested. WT: wild type, mut.: mutant.
Table S6. List of oligonucleotide primers used for PCR‐based genotyping of segregating plants of three BonnMu F2‐families.
Data Availability Statement
Raw sequencing data of Mu‐seq libraries are stored at the Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra, https://www.ncbi.nlm.nih.gov/sra/PRJNA608624) with the accession number PRJNA608624 and PRJNA914277.