

Abstract
A library of novel π‐extended porphyrin‐hexabenzocoronene (HBC) architectures is presented. Two distinct synthetic pathways were utilized to obtain either phenyl‐ or HBC‐fused compounds. Absorption experiments reveal the species’ exciting photophysical and optoelectronic properties. Depending on the degree of π‐extension, the number of porphyrins, and their relative position, a decisive change in shape, panchromatic broadening, and red‐shifting of the absorption curves is observed. Theoretical studies give more profound insight into the molecule's electronic structures, showing vast decreases in HOMO‐LUMO energy gaps.
Keywords: π-Extension, Porphyrinoids, Fusion reaction, Scholl oxidation, Post-functionalization
The synthesis of a collection of phenyl‐ and hexabenzocoronene (HBC)‐fused porphyrin conjugates is presented. The architectures’ intriguing absorption properties were investigated, revealing the influence of the number of porphyrins, the degree of π‐extension, and their relative position in the molecule.
Introduction
The technological progress in computer sciences over the last two decades is unparalleled in its relative growth compared to any other area of today's life.[ 1 , 2 , 3 ] However, as supercomputers and comparable examples of high‐end devices become more powerful and, concomitantly, larger by the day, the components they are based on call for just the opposite. [4] Here, the problems arise due to, on the one hand, physical but also potential economic limits that will be reached in the foreseeable future. [5] Presumably, a single molecule would be the smallest imaginable building block that will still provide the structural variety required to implement specific electronic functions. Hence, the downsizing of electronic components to the molecular level is one of the biggest interdisciplinary challenges modern physics, engineering, and chemistry face. One of the ultimate goals of these collective efforts is the creation of so‐called single molecular electronics (SMEs). This means individual molecules are supposed to act as the active unit in electronic circuitry.[ 6 , 7 , 8 ] Among the most prominent molecule classes investigated in this field of research, besides oligothiophenes and carbon‐pure PAH oligomers, are porphyrins.[ 9 , 10 , 11 , 12 , 13 , 14 ] As one of nature's most famous fundamental structural motifs, the 18π‐electron macrocycle possesses intriguing optoelectronic characteristics and a high degree of modifiability regarding its shape, size, and properties.[ 15 , 16 ] Another class of compounds that gained tremendous attention in recent years due to the unique electronic perks associated with its representatives are so‐called “nanographenes”, small excerpts of the sp2‐hybridized carbon graphene lattice.[ 17 , 18 , 19 , 20 , 21 , 22 ] Many of our group's recent research interests were centered around one particular member of this group, the hexa‐peri‐hexa‐benzocoronene (HBC), whose well‐defined hexagonal‐shaped periphery can conveniently be chemically functionalized and modified in a plethora of ways.[ 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 ]
Combining both porphyrins and HBCs, in 2019, our group already gave the first insights into the geometry dependence of the electronic interaction between two porphyrins attached to a central HBC backbone. [31] Furthermore, HBC‐porphyrin geometries, including three and six porphyrins, were synthesized, and their properties were probed. [32] More recently, we discovered the possibility of applying a convenient and straightforward strategy we had previously established for tetraarylporphyrins for the π‐extension of HBC‐porphyrin conjugates.[ 33 , 34 , 35 ] Employing this method, we managed to introduce five‐membered ring motifs into the carbon scaffolds that connect the two aromatic fragments chemically and electronically while retaining the full processability of the molecules in solution (Figure 1).[ 36 , 37 , 38 , 39 , 40 , 41 , 42 ] The fused species we obtained this way did not only show exciting changes in their optoelectronic properties, including a distinctly red‐shifted absorption curve and distortion of the conjugates aromaticity, but also a significant decrease in the HOMO‐LUMO gap.[ 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 ] Especially the latter features appear to be highly relevant and beneficial for potential application in the field of SMEs.[ 8 , 52 ]
Figure 1.
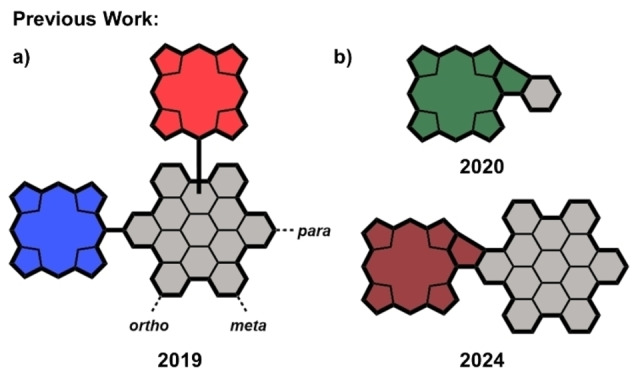
Recent advances in the field of HBC and porphyrin chemistry by the Jux group. a) Bis‐porphyrin‐HBC conjugates; [31] b) Fused porphyrins.[ 33 , 34 ]
Results and Discussion
Herein, we report a straightforward synthetic concept that further pushes the idea of π‐extension by combining porphyrins with the HBC core motif. This way, we provide insight into the effects manifold β‐meso‐fusion of porphyrins in one single molecule has on its properties. Working towards this goal, we follow two separate synthetic pathways. The first one is based on the synthesis of tailor‐made phenyl‐fused nickel porphyrin building blocks, which in a subsequent step will be introduced into the HBCs periphery via Pd‐catalyzed Suzuki cross‐coupling reactions, obtaining architectures of varying symmetry (Scheme 1). The second path involves the formation of trimesitylporphyrin fragments, which are first coupled to the HBC and subsequently further connected to its scaffold by cyclodehydrogenation under Scholl conditions.
Scheme 1.

Design of a phenyl‐fused borylated porphyrin building block 5. Reagents and conditions: a) FeCl3 (8 equiv), CH3NO2, CH2Cl2, 24 h, 0 °C→rt; b) HBpin, PdCl2(PPh3)2, NEt3, 1,2‐DCE, 24 h, 90 °C.
To this end, a collection of iodinated HBC moieties was initially synthesized following literature procedures. [31] Four distinct HBCs bearing two (6–8) and three (9) peripheral iodine atoms, respectively, were generated (Scheme 2). The phenyl‐fused porphyrin building block 5 was synthesized following a rational chain of steps to design a porphyrin with an ABAC substitution pattern. Firstly, dipyrromethane 1 was condensed with mesitaldehyde 2, yielding an trans‐A2B2 dimesitylporphyrin, which was subsequently brominated at one of the free meso‐positions and metalated with nickel. The resulting derivative was further modified by coupling it with 3,5‐di‐tert‐butyl‐phenyl boronic acid and bromination of the free remaining meso‐position. Thereafter, this final precursor 3 was subjected to oxidative Scholl conditions, leading to selective and nearly quantitative β‐meso‐fusion between the 3,5‐di‐tert‐butyl‐phenyl group and the porphyrin core. Unfortunately, in accordance with observations in the literature, the excess amount of FeCl3 used in this step leads to a partial halogen exchange reaction at the porphyrin's meso‐position, resulting in an inseparable mixture of two fused species. [35] Hence, this intermediate mixture was directly and without further separation transformed into the respective boronic ester 5 via a Miyaura‐reaction with pinacolborane in a yield of 55 % across two steps (Scheme 1). With 5 in hand, Suzuki cross‐coupling reactions were performed under conditions that have proven successful in earlier coupling experiments between porphyrins and iodinated HBCs. [31] This way, the conjugates o ‐Ph ( ortho‐Phenyl‐fused), m ‐Ph ( meta‐Phenyl‐fused), p ‐Ph ( para‐Phenyl‐fused), and tri ‐Ph ( tri ‐Phenyl‐fused) were obtained as dark green solids in good yields (Scheme 2). To the best of our knowledge, this set of molecules represents one of the first examples of successful Suzuki coupling reactions between the meso‐position of a five‐ring β‐meso‐fused porphyrin and a halogenated aromatic fragment.
Scheme 2.

Synthesis of multiple phenyl‐fused nickel‐porphyrin‐HBC conjugates via Suzuki cross‐coupling. Reagents and conditions: c) Pd(PPh3)4, Cs2CO3, toluene, DMF, 18 h, 80 °C. Ar=mesityl.
Our second laid‐out approach aimed towards hybrid structures, including additional bonds between HBC and porphyrin. To this end, nickel porphyrin boronic ester 10 was synthesized following standard protocols (for details and references, see Scheme S6 in the Supporting Information). Subsequently, the four conjugates shown in Scheme 3 were prepared by Suzuki‐cross‐coupling reactions with the same iodinated HBC moieties 6–9 employed earlier. The following crucial cyclodehydrogenation step was supposed to be performed using standard Scholl conditions, mirroring the procedure we utilized in our earlier encounters with comparable systems. [34] The respective precursors were dissolved in CH2Cl2/CH3NO2 with iron(III)chloride added in a 60‐fold excess. Applying these conditions to the three double‐porphyrin substituted HBCs 11, 12, and 13, significant differences in reactivity were observed. For para‐derivative 12, the transformation to the double‐fused p ‐HBC ( para‐HBC‐fused) was completed within 24 h and, after a standardized work‐up, including filtration through silica, obtained as a dark purple solid in excellent yields of 94 %. Subsequent NMR measurements revealed an exclusive “cis” alignment of the newly formed bonds relative to the central HBC. For the meta‐porphyrin‐HBC 13, elongated reaction times of 72 h and purification by column chromatography to separate it from minor amounts of chlorinated side products were needed to obtain m ‐HBC ( meta‐HBC‐fused) in decent yields of 62 %. The Scholl reaction of ortho‐porphyrin‐HBC 11 proved to be even more cumbersome, with a reaction time of 148 h and a significant amount of chlorinated side products formed within this time frame. However, after column chromatography, o ‐HBC ( ortho‐HBC‐fused) could be obtained in 21 % yield (Scheme 4). For o ‐ and m ‐HBC, again, only one of the three possible regioisomers was formed, as was confirmed in NMR experiments. The observed order of reactivity can be attributed to the additional steric hindrance the compounds suffer from during the second ring‐closing reaction. The steric complications naturally rise in the order of para<meta<ortho. With this in mind, triple‐connected 14 was subjected to the same oxidative conditions, and after 72 h, triple‐fused tri ‐HBC ( tri ‐HBC‐fused) was isolated in 27 % yield by separating it from incompletely fused and chlorinated species via column chromatography (Scheme 5).
Scheme 3.

Synthesis of nickel‐porphyrin‐HBC conjugates via Suzuki cross‐coupling. Reagents and conditions: c) Pd(PPh3)4, Cs2CO3, toluene, DMF, 18 h, 80 °C. Ar=mesityl.
Scheme 4.

Reagents and conditions: FeCl3 (60 equiv), CH3NO2, CH2Cl2; b) 24 h, 0 °C→rt; c) 72 h, 0 °C→rt; d) 148 h, 0 °C→rt. Ar=mesityl.
Scheme 5.

Synthesis of triple‐fused porphyrin‐HBC tri ‐HBC. Reagents and conditions: e) FeCl3 (60 equiv), CH3NO2, CH2Cl2, 72 h, 0 °C→rt. Ar=mesityl.
Striking is the across all laid‐out cyclodehydrogenations observed regio‐selectivity. The most plausible explanation for these findings is based on reports in the literature concerning the mechanism of the reaction. In detail, Kadish, Gryko et al. performed electrochemical studies on similar β‐meso‐fused naphthyl‐porphyrins, which indicated an intramolecular biradical‐cation coupling mechanism.[ 53 , 54 ] With this in mind, the group of Osuka investigated the regioselectivity in porphyrins bearing a phenyl‐fusion motif, which is very much reminiscent of ours. They stated that considering a biradical‐cation pathway, the spin densities on the porphyrinyl‐radical cation should govern the selectivity. This was supported by results they derived from DFT calculations, which were coherent with this claim. Transferring this to our herein‐presented molecules, we have to assume the first ring fusion between porphyrin and HBC leads to a strong enough difference in spin densities in the HBC, so the second (and third) ring fusion takes place selectively. [35]
All fused conjugates were unambiguously identified by means of NMR and UV/Vis spectroscopic as well as mass spectrometric techniques. The definite regio‐geometry of the newly fused bonds in o ‐HBC, m ‐HBC, p ‐HBC, and tri ‐HBC was confirmed by NMR through analysis of correlations between chemically inequivalent carbon and hydrogen atoms.
The here‐discussed five‐ring β‐meso‐fusion in a porphyrin generally leads to strong alterations of the molecules’ optoelectronic and photophysical properties. Hence, the UV/Vis absorption in the obtained molecules vastly differs from that of non‐fused porphyrins. No distinct Soret‐ and Q‐band characteristics of porphyrins can be detected anymore. [31] Considering the first series of molecules o‐ /m‐/p‐/tri‐Ph, bearing the phenyl fused motif up to three times in their structure, the absorption spectra show the expected panchromatically broadened and red‐shifted absorption in the area of 400–500 nm, which is typical for this type of fusions.[ 33 , 34 , 35 ] Furthermore, the HBCs’ absorption in the lower wavelength region from 350–400 nm can be clearly identified, although they overlap with features of the fused porphyrin absorption (Figure 2).
Figure 2.

UV/Vis spectra of o ‐Ph, m ‐Ph, p ‐Ph, and tri ‐Ph (CH2Cl2).
As demonstrated in one of our group's earlier work regarding the different geometries of the double substituted HBCs, changes in the communication between porphyrins dependent on their relative orientation can be observed, resulting in slight differences between the UV curve shapes and shifts. [31] Interestingly, we find the same trend in shifts of the curve maxima as in the case for the respective series of non‐fused molecules (para>meta>ortho). For the triple‐substituted tri ‐Ph, as expected, we find a band shape that is very much identical to the one for m ‐Ph, although with increased intensities due to the additional absorption exhibited by the added porphyrin. Furthermore, with the increasing size of the molecule, a concomitant red shift of the porphyrins’ most intense absorption feature can be detected (Abs. II in Table 1). Meanwhile, the shifts of the absorptions above 500 nm stay very alike across the whole series of molecules (Abs. IV in Table 1).
Table 1.
Comparison of the four absorption wavelengths with the highest molar extinction coefficient in the UV/vis spectra of o ‐Ph, m ‐Ph, p ‐Ph, and tri ‐Ph. Highlighted are the global absorption maxima of each curve.
|
Abs. I |
Abs. II |
Abs. III |
Abs. IV |
|
|---|---|---|---|---|
|
o ‐Ph |
361 nm |
448 nm |
472 nm |
572 nm |
|
m ‐Ph |
360 nm |
452 nm |
475 nm |
572 nm |
|
p ‐Ph |
360 nm |
453 nm |
475 nm |
572 nm |
|
tri ‐Ph |
360 nm |
454 nm |
475 nm |
572 nm |
In line with our previous experiments on the β‐meso‐connection of porphyrin and HBC, an even more drastic change in absorption properties can be observed in the molecules with multiple porphyrins directly fused to the HBC. Generally, comparing this second set of molecules to the above‐discussed ones, we find a more pronounced panchromatic character and a further red‐shift of the absorption bands (Figure 3). When comparing the three geometric isomers o ‐/m‐/p‐HBC, even more substantial differences in the spectral shape can be noted. Nevertheless, p –HBC still shows the most bathochromically shifted absorption maxima at 544 nm, with a plethora of additional distinct local maxima. The total absorption range reaches up to 850 nm, well into the near‐IR area of the spectrum.
Figure 3.

UV/Vis spectra of o ‐HBC, m ‐HBC, p ‐HBC, and tri ‐HBC (CH2Cl2).
For the m ‐HBC, in comparison, the profound absorption between 500–560 nm is noticeably weaker, resulting in a global absorption maximum at 505 nm. Apart from this striking perk, the curve shape is very comparable to that of p ‐HBC. More substantial alterations can be observed in the case of o ‐HBC, where absorption in the longer wavelength region decreases even further. Spectral features in the area between 600 and 750 nm are flattened, and a new prominent contribution peaking at 484 nm emerges. However, by integrating the absorption area of 350 nm to 800 nm for all three compounds, almost identical values are obtained, meaning that the total absorption stays almost the same across the series. All in all, the spectra of the three double‐fused isomers differ more significantly from each other than is the case for the respective phenyl‐fused and non‐fused series of molecules. This indicates the strong influence that the double five‐ring formation has on the molecules, and especially on the communication between the two porphyrin moieties across the porphyrin.
The UV curve of the triple‐fused species tri ‐HBC has a similar shape to that of m ‐HBC since the relative position of the individual porphyrins remains the same in both molecules. Due to the further fused molecule and the number of increased peripheral chromophores, a noticeable increase in the extinction coefficient, as well as a slight red shift of the band as a whole, is detected. The maximum absorption appears at 515 nm. Integrating the area of 350 nm to 800 nm shows an absorption increase of around 30 % compared to the double‐fused molecules.
For further characterization of the π‐extended HBC‐porphyrin molecules, absorption spectra were calculated using time‐dependent density‐functional theory (TD‐DFT) based on DFT‐optimized molecular structures (Figure 4). The observed transitions (Figure 5) are very much in line with the experimental findings and further confirm the regio‐geometry of the isomers determined by NMR. Especially for p ‐HBC, we see excellent agreement between the theoretical transitions and the experimental results for the C2v fused product, while the calculation for a fused structure with C2h symmetry shows considerably different transitions. The first optically active excitation of all four molecules consists mostly of HOMO to LUMO transitions with some mixing in of the HOMO‐1 and LUMO+1 states (see Supporting Information). The excitation energy is almost the same for the m ‐/p‐/tri‐HBC species, ranging from 1.71–1.73 eV. Interestingly, o ‐HBC has a slightly lower value of 1.62 eV, which is explained by the HOMO and LUMO levels coming closer together due to a perturbed geometry by the steric demand of the neighboring porphyrin substituents. This trend is also visible by direct inspection of the HOMO and LUMO energy levels. For m ‐/p‐/tri‐HBC, the HOMO‐LUMO gap is between 2.09–2.13 eV, while for o ‐HBC, it has a value of 1.99 eV. These findings agree well with other examples from the literature[ 17 , 21 ] and results from our earlier work on fused porphyrin‐HBC conjugates. [34]
Figure 4.

DFT‐optimized structures of a) o ‐Ph, m ‐Ph, p ‐Ph, and tri ‐Ph; b) o ‐HBC, m ‐HBC, p ‐HBC, and tri ‐HBC.
Figure 5.
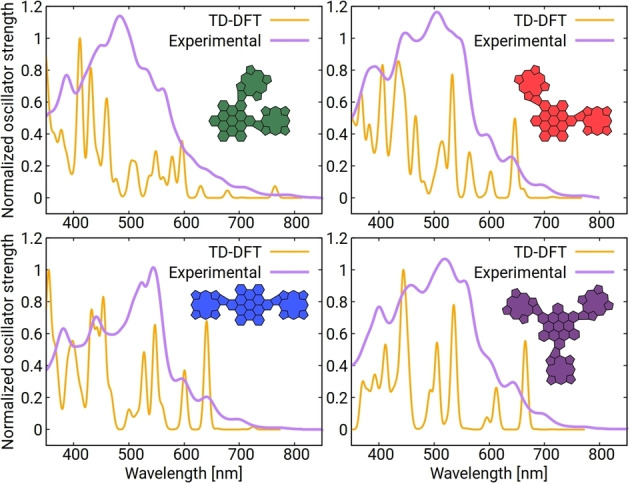
Calculated (yellow) and experimental (purple) UV/Vis absorption spectra of o ‐HBC, m ‐HBC, p ‐HBC, and tri ‐HBC (CH2Cl2). The TD‐DFT calculations were done with the B3LYP hybrid functional. Solvation effects were included by a continuum polarization model.
Continuing with the set of externally fused molecules o ‐/m‐/p‐/tri‐Ph, the energy gap is expected to increase because of a reduced π‐extension. In agreement with this expectation, the calculated HOMO‐LUMO gap energies range from 2.37–2.39 eV due to rising LUMO and declining HOMO energy levels. This trend continues when proceeding to the non‐fused reference species 11–14. Due to a considerable increase in energy of the LUMO levels they show HOMO‐LUMO gap energies of 2.79–2.84 eV. Thus, while the extension of the π‐system leads to a decrease in the energy gap when comparing the three sets of molecules, the number of substituents and their relative positions have little to no influence (Figure 4).
In our previous work on porphyrin‐HBC conjugates, we showed that the fusion of five‐membered rings at the periphery of the porphyrin introduces a strong biradicaloid character in the porphyrin core, which has a strong impact on the excited state lifetimes. [34] Therefore, we also investigated the degree of biradicaloid character for all conjugates of this study by calculating the stability of the broken symmetry singlet state (see Supporting Information). [34] We find a significant biradicaloid character in all porphyrin cores after the fusion of the five‐membered rings. However, further analysis shows that the biradicaloid character is almost independent of the relative position of the porphyrins, the position of the fused ring (at the outer periphery of the porphyrin or between porphyrin and HBC), and the number of porphyrins in the conjugate.
Conclusions
In summary, we demonstrated the synthesis of two series of novel HBC‐based compounds containing multiple β‐meso‐fusing motifs within two or more porphyrins. The laid‐out synthesis strategies follow a modular approach and produce extensively π‐extended molecules with drastically altered photophysical and optoelectronic properties compared to the respective non‐fused equivalents. A well‐defined phenyl‐fused borylated precursor was prepared to obtain four hybrids of different geometry and substitution patterns. For these conjugates involving a connection between the porphyrin and a meso‐attached 3,5‐di‐tert‐butyl‐phenyl group, we observe interesting parallels to earlier investigated porphyrin‐HBC conjugates, [31] albeit with a substantial distortion of the porphyrin's π‐system. In the case of direct five‐ring fusion between HBC and porphyrin moieties, steric hindrance reduces the rate of the second and third ring‐closure reactions. Nevertheless, four conjugates with up to three additional C−C bonds were obtained. Photophysical analysis of those molecules implies that the π‐extension greatly affects the communication between the porphyrins. DFT calculations were performed, revealing the geometries and electronic properties of all fused architectures. They show the expected decrease in the energy gaps with increasing degree of fusion and π‐system size, as well as some dependence on the geometry. Furthermore, calculated absorption spectra using TD‐DFT showed great overlap with the experimentally obtained data for the HBC‐fused conjugates. Altogether, these fundamental findings could be highly interesting for designing single molecular components in the areas of near‐IR dyes and field‐effect transistors. Deeper investigations on the biradical character and photophysical properties of the conjugates are currently ongoing in our laboratories.
Experimental Section
Details of synthesis and characterization, optical measurements, computation, and instrumentation are provided in the Supporting Information.
Conflict of Interests
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgments
Funded by the Deutsche Forschungsgemeinschaft (DFG) through SFB 953 (project number 182849149) and GRK 2423 (project number 377472739). C. O. and C. L. R. thank the Graduate School Molecular Science (GSMS) for financial support. Computational resources were provided by the Erlangen National High Performance Computing Center NHR@FAU. Open Access funding enabled and organized by Projekt DEAL.
Oleszak C., Ritterhoff C. L., Martin M. M., Meyer B., Jux N., Chem. Eur. J. 2024, 30, e202403250. 10.1002/chem.202403250
Contributor Information
Bernd Meyer, Email: bernd.meyer@fau.de.
Norbert Jux, Email: norbert.jux@fau.de.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Grumbling E., Horowitz M., Quantum Computing, National Academies Press, Washington, D. C. 2019. [Google Scholar]
- 2. Gyongyosi L., Imre S., Comput. Sci. Rev. 2019, 31, 51. [Google Scholar]
- 3. Shalf J., Philos. Transact. A, Math. Phys. Eng. Sci. 2020, 378, 20190061. [DOI] [PubMed] [Google Scholar]
- 4. Razavi B., Fundamentals of Microelectronics. with Robotics and Bioengineering applications, Wiley, Hoboken: 2021. [Google Scholar]
- 5. Schaller R. R., IEEE Spectrum 1997, 34, 52. [Google Scholar]
- 6. Aviram A., Ratner M. A., Chem. Phys. Lett. 1974, 29, 277. [Google Scholar]
- 7. Choi H., Mody C. C., Soc. Stud. Sci. 2009, 39, 11. [Google Scholar]
- 8. Su T. A., Neupane M., Steigerwald M. L., Venkataraman L., Nuckolls C., Nat. Rev. Mater. 2016, 1, 16002. [Google Scholar]
- 9. Leary E., Limburg B., Alanazy A., Sangtarash S., Grace I., Swada K., Esdaile L. J., Noori M., González M. T., Rubio-Bollinger G. et al., J. Am. Chem. Soc. 2018, 140, 12877. [DOI] [PubMed] [Google Scholar]
- 10. Limburg B., Thomas J. O., Holloway G., Sadeghi H., Sangtarash S., Hou I. C.-Y., Cremers J., Narita A., Müllen K., Lambert C. J. et al., Adv. Funct. Mater. 2018, 28, 1803629. [Google Scholar]
- 11. Mol J. A., Lau C. S., Lewis W. J. M., Sadeghi H., Roche C., Cnossen A., Warner J. H., Lambert C. J., Anderson H. L., Briggs G. A. D., Nanoscale 2015, 7, 13181. [DOI] [PubMed] [Google Scholar]
- 12. Schosser W. M., Hsu C., Zwick P., Beltako K., Dulić D., Mayor M., van der Zant H. S. J., Pauly F., Nanoscale 2022, 14, 984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zwick P., Dulić D., van der Zant H. S. J., Mayor M., Nanoscale 2021, 13, 15500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shi Y., Zhang F., Linhardt R. J., Dyes Pigm. 2021, 188, 109136. [Google Scholar]
- 15. Yang J., Yoon M.-C., Yoo H., Kim P., Kim D., Chem. Soc. Rev. 2012, 41, 4808. [DOI] [PubMed] [Google Scholar]
- 16.K. M. Kadish, K. M. Smith, R. Guilard, Handbook of Porphyrin Science, World Scientific Publishing Company, Singapore, 2010.
- 17. Borissov A., Maurya Y. K., Moshniaha L., Wong W.-S., Żyła-Karwowska M., Stępień M., Chem. Rev. 2022, 122, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gu Y., Qiu Z., Müllen K., J. Am. Chem. Soc. 2022, 144, 11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mishra S., Yao X., Chen Q., Eimre K., Gröning O., Ortiz R., Di Giovannantonio M., Sancho-García J. C., Fernández-Rossier J., Pignedoli C. A. et al., Nat. Chem. 2021, 13, 581. [DOI] [PubMed] [Google Scholar]
- 20. Pozo I., Guitián E., Pérez D., Peña D., Acc. Chem. Res. 2019, 52, 2472. [DOI] [PubMed] [Google Scholar]
- 21. Stępień M., Gońka E., Żyła M., Sprutta N., Chem. Rev. 2017, 117, 3479. [DOI] [PubMed] [Google Scholar]
- 22. Wang X.-Y., Yao X., Narita A., Müllen K., Acc. Chem. Res. 2019, 52, 2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuznetsov A. E., Pure Appl. Chem. 2022, 94, 747. [Google Scholar]
- 24. Kuznetsov A. E., Comput. Theor. Chem. 2020, 1188, 112973. [Google Scholar]
- 25. Englert J. M., Malig J., Zamolo V. A., Hirsch A., Jux N., Chem. Commun. 2013, 49, 4827. [DOI] [PubMed] [Google Scholar]
- 26. Lungerich D., Hitzenberger J. F., Marcia M., Hampel F., Drewello T., Jux N., Angew. Chem. Int. Ed. Engl. 2014, 53, 12231. [DOI] [PubMed] [Google Scholar]
- 27. Lungerich D., Reger D., Hölzel H., Riedel R., Martin M. M. J. C., Hampel F., Jux N., Angew. Chem. Int. Ed. Engl. 2016, 55, 5602. [DOI] [PubMed] [Google Scholar]
- 28. Martin M. M., Dusold C., Hirsch A., Jux N., J. Porphyrins Phthalocyanines 2020, 24, 268. [Google Scholar]
- 29. Martin M. M., Hampel F., Jux N., Chem. Eur. J. 2020, 26, 10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martin M. M., Jux N., J. Porphyrins Phthalocyanines 2018, 22, 454. [Google Scholar]
- 31. Martin M. M., Lungerich D., Haines P., Hampel F., Jux N., Angew. Chem. Int. Ed. Engl. 2019, 58, 8932. [DOI] [PubMed] [Google Scholar]
- 32. Martin M. M., Lungerich D., Hampel F., Langer J., Ronson T. K., Jux N., Chem. Eur. J. 2019, 25, 15083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martin M. M., Oleszak C., Hampel F., Jux N., Eur. J. Org. Chem. 2020, 2020, 6758. [Google Scholar]
- 34.C. Oleszak, P. R. Schol, C. L. Ritterhoff, M. Krug, M. M. Martin, Y. Bo, B. Meyer, T. Clark, D. M. Guldi, N. Jux, Angew. Chem., Int. Ed. Engl. 2024, e202409363. [DOI] [PubMed]
- 35. Fukui N., Lee S.-K., Kato K., Shimizu D., Tanaka T., Lee S., Yorimitsu H., Kim D., Osuka A., Chem. Sci. 2016, 7, 4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fukui N., Cha W., Shimizu D., Oh J., Furukawa K., Yorimitsu H., Kim D., Osuka A., Chem. Sci. 2017, 8, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao H., Wang Y., Chen Q., Liu Y., Gao Y., Müllen K., Li S., Narita A., Adv. Sci. 2024, 11, e2309131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang P., Yu C., Yin Y., Droste J., Klabunde S., Hansen M. R., Mai Y., Chem. Eur. J. 2020, 26, 16497. [DOI] [PubMed] [Google Scholar]
- 39. Lewtak J. P., Koszarna B., Charyton M. K., Gryko D. T., J. Porphyrins Phthalocyanines 2020, 24, 448. [Google Scholar]
- 40. Davis N. K. S., Thompson A. L., Anderson H. L., J. Am. Chem. Soc. 2011, 133, 30. [DOI] [PubMed] [Google Scholar]
- 41. Chen Q., Lodi A., Zhang H., Gee A., Wang H. I., Kong F., Clarke M., Edmondson M., Hart J., O'Shea J. N. et al., Nat. Chem. 2024, 16, 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Q., Brambilla L., Daukiya L., Mali K. S., de Feyter S., Tommasini M., Müllen K., Narita A., Angew. Chem. Int. Ed. Engl. 2018, 57, 11233. [DOI] [PubMed] [Google Scholar]
- 43. Ota K., Tanaka T., Osuka A., Org. Lett. 2014, 16, 2974. [DOI] [PubMed] [Google Scholar]
- 44. Wiengarten A., Lloyd J. A., Seufert K., Reichert J., Auwärter W., Han R., Duncan D. A., Allegretti F., Fischer S., Oh S. C. et al., Chem. Eur. J. 2015, 21, 12285. [DOI] [PubMed] [Google Scholar]
- 45. Shen D.-M., Liu C., Chen Q.-Y., J. Org. Chem. 2006, 71, 6508. [DOI] [PubMed] [Google Scholar]
- 46.D.-M. Shen, C. Liu, Q.-Y. Chen, Chem. Commun. 2005, 4982. [DOI] [PubMed]
- 47. Mitsushige Y., Yamaguchi S., Lee B. S., Sung Y. M., Kuhri S., Schierl C. A., Guldi D. M., Kim D., Matsuo Y., J. Am. Chem. Soc. 2012, 134, 16540. [DOI] [PubMed] [Google Scholar]
- 48. Lash T. D., Smith B. E., Melquist M. J., Godfrey B. A., J. Org. Chem. 2011, 76, 5335. [DOI] [PubMed] [Google Scholar]
- 49. Jiao C., Huang K.-W., Chi C., Wu J., J. Org. Chem. 2011, 76, 661. [DOI] [PubMed] [Google Scholar]
- 50. Fukui N., Cha W.-Y., Lee S., Tokuji S., Kim D., Yorimitsu H., Osuka A., Angew. Chem. Int. Ed. Engl. 2013, 52, 9728. [DOI] [PubMed] [Google Scholar]
- 51.S. Fox, R. W. Boyle, Chem. Commun. 2004, 1322.
- 52. Breslow R., F. W. Foss Jr. , J. Phys. Condens. Matter 2008, 20, 374104. [Google Scholar]
- 53. Yamane O., Sugiura K., Miyasaka H., Nakamura K., Fujimoto T., Nakamura K., Kaneda T., Sakata Y., Yamashita M., Chem. Lett. 2004, 33, 40. [Google Scholar]
- 54. Fang Y., Koszelewski D., Kadish K. M., Gryko D. T., Chem. Commun. 2014, 50, 8864. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.