

ABSTRACT
Accumulating evidence suggests that phenotype switching of cancer cells is essential for therapeutic resistance. However, the immunological characteristics of drug-induced phenotype-switching melanoma cells (PSMCs) are unknown. We investigated PSMC elimination by host immunity using hyperdifferentiated melanoma model cells derived from murine B16F10 melanoma cells. Exposure of B16F10 cells to staurosporine induced a hyperdifferentiated phenotype associated with transient drug tolerance. Staurosporine-induced hyperdifferentiated B16F10 (sB16F10) cells expressed calreticulin on their surface and were phagocytosed efficiently. Furthermore, the inoculation of mice with sB16F10 cells induced immune responses against tumor-derived antigens. Despite the immunogenicity of sB16F10 cells, they activated the PD-1/PD-L1 immune checkpoint system and strongly resisted T cell-mediated tumor destruction. However, in vivo treatment with immune checkpoint inhibitors successfully eliminated the tumor. Thus, hyperdifferentiated melanoma cells have conflicting immunological properties – enhanced immunogenicity and immune evasion. Inhibiting the ability of PSMCs to evade T cell-mediated elimination might lead to complete melanoma eradication.
KEYWORDS: Anti-tumor immunity, immune checkpoint inhibitors, malignant melanoma, phenotype switching
1. Introduction
A major problem in cancer treatment is the development of resistance to therapeutic agents. Cancer chemotherapy has shifted from cytotoxic drugs to molecular-targeted drugs such as kinase inhibitors.1 However, neither of these drug types can eliminate all malignant cells and a small number of cells that survive chemotherapy can lead to future clinical relapse.2 The survival of these cancer cells is conventionally understood to result from genetic mutations in the cells.2 Recently, it has become clear that cancer cells without genetic mutations can survive in a harsh environment by phenotype switching.3 Using single-cell RNA sequencing, Rambow et al. analyzed minimal residual lesion samples derived from melanoma patients.4 They found that melanoma phenotypes could be classified into four cell states.4 This classification was further subdivided into six subtypes based on the expression of melanocyte differentiation and neural crest stem cell (NCSC) marker molecules: hyperdifferentiated, melanocytic, intermediate, starved, NCSC-like, and undifferentiated, which is now considered the standard classification.5,6 Targeted therapy induces phenotypic conversion to the highly differentiated, starved, NCSC-like, and undifferentiated types, and these phenotypes are known to be drug-resistant.5 These cells survive anticancer therapy and resume proliferation when the effects of the drugs on the microenvironment wane.6 Based on the above findings, measures to eliminate multiple phenotypes of melanoma cells are needed to develop more effective melanoma treatment protocols.
Immunotherapy, which stimulates the host immune system using immune checkpoint blocking antibodies such as anti-PD-1, has allowed significant advances in cancer treatment over the past decade.7 The anti-tumor immune response consisting of several sequential components is known as the cancer immunity cycle.8 The first step in the cancer immunity cycle is for a cancer cell to undergo immunogenic cell death (ICD), which is induced by exposure to ionizing radiation and anthracycline anticancer drugs.9 Cancer cells that undergo ICD are phagocytosed by antigen-presenting cells (APCs), a process that promotes the maturation of APCs. APCs that have phagocytosed ICD cancer cells and subsequently matured, present cancer cell-derived antigens to lymphocytes. This sequential process induces an anti-tumor lymphocyte response. In addition, it was recently reported that cancer cells that have escaped cell death from damage caused by cytotoxic agents become senescent via the irreversible arrest of cell division, and these senescent cancer cells have immunogenicity similar to that of ICD cancer cells.10 In this context, the question arises whether drug-induced phenotype-switching cells are recognized and eliminated by host immunity in the same way as senescent cells whose cellular state has been permanently altered, including the irreversible arrest of cell proliferation.
Phenotype switching has been reported in melanomas treated with BRAF/MEK kinase inhibitors.5 However, the immunological characteristics of phenotypic switching melanoma cells (PSMCs) remain incompletely characterized. No reports have examined immune responses to PSMCs using immunocompetent animal models. In this study, we generated an experimental PSMC model using staurosporine, a multi-kinase inhibitor. This model allows for the use of the traditional B16 murine melanoma cell line and several established methods to study anti-tumor immunity. We generated hyperdifferentiated melanoma model cells derived from B16F10 cells and analyzed the anti-tumor immune response against these cells to investigate their elimination by host immunity.
2. Materials and methods
2.1. Animals, plasmids, and cells
C57BL/6J mice were purchased from Charles River Laboratories Japan, Inc. (Yokohama, Japan). B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J mice (Pmel-1)11 were originally purchased from The Jackson Laboratory (Bar Harbor, ME, USA; JAX stock #005023). Eight-to-12-week-old mice were used for all experiments. Mice were housed in an appropriate animal care facility at Saitama Medical University (Saitama, Japan) and handled according to the international guidelines for experiments on animals. The Animal Care and Use Committee of Saitama Medical University (Saitama, Japan) approved the procedures for this study (approval numbers 3740 and 3742).
B16F10 melanoma and RAW264.7 mouse macrophage cell lines obtained from the American Type Culture Collection were cultured in DMEM medium (Nacalai Tesque, Kyoto, Japan) supplemented with 10% fetal bovine serum (FBS). The DC2.4 mouse dendritic cell line, provided by Dr. Takashi Imai (National Institute of Infectious Diseases, Tokyo, Japan), was cultured in RPMI1640 medium (Nacalai Tesque) supplemented with 10% FBS. To generate B16F10 cells expressing the green fluorescent protein, ZsGreen, the lentiviral vector pLVSIN-ZsGreen (Clontech, Mountain View, CA, USA) was used. To generate B16F10 cells expressing a fluorescent ubiquitination-based cell cycle indicator (FUCCI)12 (B16F10-FUCCI), the retroviral vector pMX-Venus-hGem-IRES-mKO-hCdt (a gift from Dr. Michio Tomura, Osaka Ohtani University, Osaka, Japan) was used. pLVSIN-deltaOVA was generated by inserting deltaOVA cDNA from pcDNA3-deltaOVA (a gift from Drs. Sandra Diebold and Martin Zenke; Addgene plasmid #64595; http://n2t.net/addgene:64595.; RRID: Addgene_64595) into the EcoRI–XbaI site of the pLVSIN-CMV-Pur vector (Clontech). Lentiviral transduction was used to generate B16F10 cells expressing B16F10-ZsGreen (B16F10-ZsG) or ovalbumin (B16F10-OVA). These viral vectors were used in accordance with the manufacturer’s instructions. In addition, B16F10-OVA were cultured overnight in the presence of 100 U IFN-γ and stained with a PE-labeled monoclonal antibody that specifically reacts with the ovalbumin-derived peptide SIINFEKL bound to H-2Kb (25-D1.16, BioLegend, San Diego, CA, USA). Then, B16F10 cells expressing the H-2Kb-OVA-peptide complex on the cell surface were sorted by FACS using a FACSAria IIu (Becton Dickinson, Franklin Lakes, NJ, USA). To generate PD-L1 knockout B16F10 (B16F10-PD-L1 KO) cells, Pdcd-1L1 CRISPR/Cas9 Knockout Plasmid (Santa Cruz Biotechnology, Dallas, Tx, USA) was used.
2.2. Phenotype switching melanoma cell induction
B16F10 cells were plated in 6-well plates (2 × 105 per well) and incubated overnight (18 h). Staurosporine (100 ng/ml, Alomone labs, Jerusalem, Israel) was then added to cultures for 48 h to induce phenotype switching. An equal volume of the solvent dimethyl sulfoxide was added to the control group. After 48 hours, the medium was replaced with fresh complete medium without the reagent. Staurosporine-treated phenotype switching cells were analyzed 5–6 days after treatment.
2.3. In vitro phagocytosis assay
In vitro phagocytosis assays were performed as previously described.13 Briefly, staurosporine-treated or untreated B16F10-ZsGreen cells (B16F10-ZsG; 1 × 105/well) were cocultured with RAW264.7 or DC2.4 at a 1:1 ratio for 18 h. At the end of the incubation, the cells were harvested and stained with an Alexa 647-conjugated anti-mouse CD45 mAb (clone 30-F11, BioLegend). Phagocytosis was assessed using a flow cytometer. Colocalization of Alexa647 and ZsGreen fluorescence indicated the phagocytosis of tumor cells. Thus, the percentage of ZsGreen+ CD45+ cells among CD45+ cells was determined as the percentage of phagocytes engulfed tumors. This percentage was used as an indicator of the avidity of tumor cells to be phagocytosed.
2.4. Lymphocyte-mediated cytotoxic assay
Pmel-1 splenocytes were cultured in culture medium with interleukin (IL)-2 (175 IU/ml in KBM550 medium, KOHJIN BIO, Saitama, Japan) in the presence of H-2Db-restricted peptide hgp10025–33 (KVPRNQDWL) for 10 days and used as activated effector cells. In vitro cytotoxicity assays were performed as previously described.14
2.5. In vivo evaluation of the impact of PD-1 blockade on tumor progression
Control or staurosporine-treated hyperdifferentiated B16F10-OVA cells (1 × 105) were inoculated subcutaneously into the left flank of mice. Anti-mouse PD-1 mAb (200 μg; clone 29F.1A12; BioXCell, West Lebanon, NH, USA) was injected into the intraperitoneal cavity of mice 2, 4, 7, and 10 days following tumor inoculation, and isotype-matched mAb (rat IgG2a κ; BioXCell) was administered intraperitoneally at the same time. The tumor size (length × width × 3.14) was measured with a caliper every 2–3 days. Mice were sacrificed when the tumor area reached 470 mm2 or when ulceration occurred.
3. Results
3.1. Melanoma cells that survive treatment with low concentrations of staurosporine exhibit characteristics of hyperdifferentiated cells
The murine malignant melanoma B16 line has been widely used to analyze anti-tumor immune responses.11,15 On the basis of this, we used the B16F10 cell line as a model to evaluate the immunological aspects of surviving cells exposed to cytotoxic agents. We exposed B16F10 cells to staurosporine in graduated dilutions and assessed cytotoxicity. We found cells survived at a staurosporine concentration of 0.1 µg/ml (Figure 1a). The staurosporine sensitivity of 10 clones established from B16F10 cells was examined. The results showed no significant differences in the staurosporine sensitivity of the clones (Figure 1b). Therefore, the cell survival described above is not an expression of subclonal traits but a transient effect of the cell line responding to a low dose of staurosporine. To confirm cell survival, the fate of B16F10 cells exposed to 0.1 µg/ml staurosporine was monitored by time-lapse microscopy, which revealed the survival of very few cells (Movie S1). Phase contrast microscopy of surviving B16F10 cells 5 days after staurosporine exposure showed that the cells were enlarged and flattened, and some cells were also multinucleated (Figure 1c). These features were reminiscent of senescent cells. Furthermore, surviving cells had an apparent accumulation of senescence-associated β-galactosidase (Figure 1c).
Figure 1.
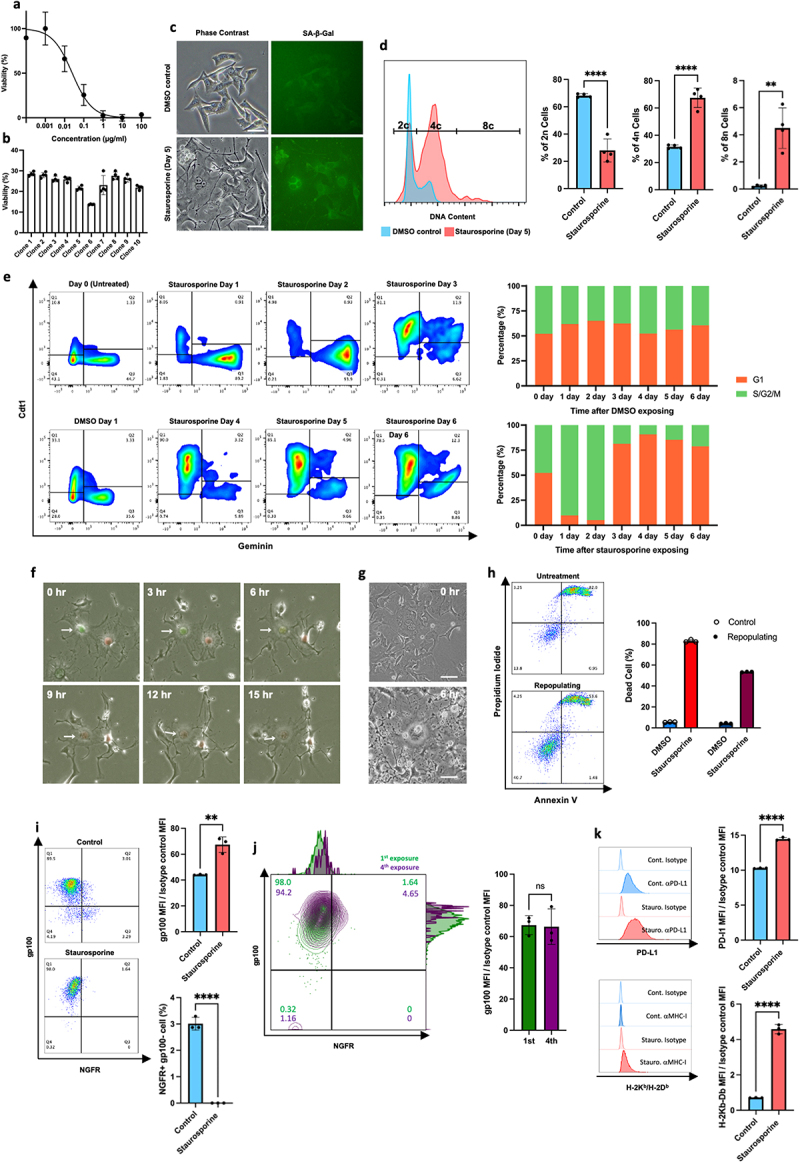
Characterization of melanoma cells surviving exposure to low dose staurosporine. (a) The percentage of viable cells 24 hours after exposure to staurosporine was measured by MTT assay. Data are expressed as the mean ± SD (n = 4). (b) The sensitivity of cell populations derived from 10 subclones isolated from parental B16F10 cells to 0.1 µg/ml staurosporine. (c) Morphological changes and senescence-associated β-galactosidase accumulation of surviving cells. Scale bars, 50 µm. (d) DNA polyploidy in surviving cells. (e) The cell cycle of surviving cells was evaluated using a FUCCI probe. Percentages indicate the fraction of G1 or S/G2/M cells for each day. (f) Representative images of mitosis slippage between days 2 and 3. Arrows indicate the timelapse images of the same cell. (g) Enlarged multinucleated cells present at the time of staurosporine re-exposure. Upper panel: before exposure. Lower panel: large multinucleated cells surviving 6 hours after exposure. (h) Recovery of staurosporine sensitivity in cells that survived staurosporine exposure and repopulated. The upper panel shows the induction of cell death by initial exposure to staurosporine and the lower panel shows the induction of cell death when repopulating cells were re-exposed to staurosporine. Right: comparison of the average percentage of cell death between initial exposure and re-exposure (n = 3). (i) The expression level of gp100 and NGFR in control and staurosporine-exposed surviving cells. Left: Representative FACS plot. Upper right: comparison of gp100 expression (n = 3). Lower right: comparison of the percentage of ngfr-positive cells (n = 3). (j) The expression level of gp100 and NGFR in the first and fourth repeated staurosporine-exposed surviving cells. Left: Representative FACS plot. Right: comparison of gp100 expression (n = 3). (g) The expression levels of PD-L1 and MHC class I in control and staurosporine-exposed surviving cells. Left: Representative FACS plot. Upper right: comparison of PD-L1 expression (n = 3). Lower right: comparison of MHC class I expression (n = 3).
Next, we evaluated DNA polyploidy in the surviving cells. Compared with the findings in controls, there was an increase in surviving cells with duplicated DNA content, and tetraploid cells were also present (Figure 1d). The FUCCI assay12 showed that the cell cycle of surviving cells was arrested at the G2 phase in the presence of staurosporine. The removal of staurosporine led to a shift from the G2 to G1 phase, followed by a gradual increase of cells in the G2 phase (Figure 1e). During the transition from G2 arrest to G1 phase, we observed mitosis slippage – a G2 to G1 transition without cell division (Figure 1f, Movie S2). These features are similar to those of senescent cells reported by Johmura et al.16
Interestingly, surviving cells gradually resumed proliferation and appeared to have a morphology similar to that before staurosporine exposure (Movie S3). The general definition of a senescent cell is one with irreversible cell cycle arrest.17 However, because B16F10 cells that survived staurosporine exposure resumed proliferation, these cells do not fully meet the definition of senescent cells. We re-treated repopulated cells with staurosporine 15 days after the initial exposure. At this point, remaining cells that had become larger, flattened, and multinucleated following the first staurosporine exposure were mixed with cells that had repopulated with morphology similar to normal B16F10 cells (Figure 1g). The larger, multinucleated cells survived the second staurosporine exposure, but the repopulating cells were sensitive to staurosporine (Figure 1g,h). Thus, the resistant trait that survived the initial staurosporine exposure was considered transient and not a permanent trait based on a genetic mutation.
Melanoma cells have different drug sensitivities depending on their differentiation status.5,6 Therefore, we investigated the differentiation status of surviving cells based on the expression of differentiation marker molecules. Staurosporine-survivor cells were devoid of nerve growth factor receptor (NGFR), a marker molecule of dedifferentiation, and highly expressed glycoprotein 100 (gp100), a marker molecule of melanocyte differentiation (Figure 1i). This feature is consistent with hyperdifferentiated melanoma cells (HMCs) tolerant to MAP kinase inhibitors. This phenotype was also observed after four repeated exposures to staurosporine, in which cells were repopulated after exposure to staurosporine and then exposed to staurosporine again (Figure 1j).
3.2. Hyperdifferentiated melanoma cells upregulate MHC class I and PD-L1
Abnormal cells are eliminated in vivo by cytotoxic T lymphocytes (CTLs), and CTL cytotoxicity is based on the recognition of antigens presented by major histocompatibility complex (MHC) class I molecules. In contrast, immune checkpoint molecules such as PD-L1 on the surface of target cells attenuate immune cell-mediated cytotoxicity.18 To evaluate the sensitivity of surviving cells to immunological elimination, we examined the expressions of MHC class I and PD-L1 on the surfaces of staurosporine-induced hyperdifferentiated B16F10 (sB16F10) cells. Compared with the solvent control B16F10 (cB16F10) cells, sB16F10 cells showed increased MHC class I and PD-L1 expressions (Figure 1k). These results suggest that CTLs can recognize sB16F10 cells; however, PD-1–PD-L1 binding may allow sB16F10 cells to escape elimination by activated CTLs.
3.3. Efficient phagocytosis of hyperdifferentiated melanoma cells by APCs
The activation of tumor antigen-specific CTLs requires the engulfment of tumor antigens by APCs such as dendritic cells (DCs) and macrophages, and antigen presentation to T-cells by activated phagocytes.8 To investigate the possibility of HMCs as a source of tumor antigens, we examined the susceptibility of sB16F10 cells to phagocytosis.
The recognition of target cells for phagocytosis by DC and macrophages requires the exposure of “eat me” signaling molecules on the cells to be phagocytosed.19 Therefore, we examined the exposure of calreticulin, an “eat me” signaling molecule, on the cell surface of sB16F10 cells. Compared with the findings in cB16F10 cells, calreticulin exposure was significantly increased on sB16F10 cells (Figure 2a). Cocultures of RAW264.7 and DC2.4 cells showed that sB16F10 cells were phagocytosed more efficiently than control B16F10 cells (Figure 2b). Furthermore, the coculture of DC2.4 cells with sB16F10s cells led to the enhanced expressions of CD40 and CD86 on DC2.4 cells, indicative of APC activation (Figure 2c). These results suggest that sB16F10 cells can be a source of tumor antigens for the host immune system and that sB16F10-derived antigens are potentially presented to the acquired immune system.
Figure 2.
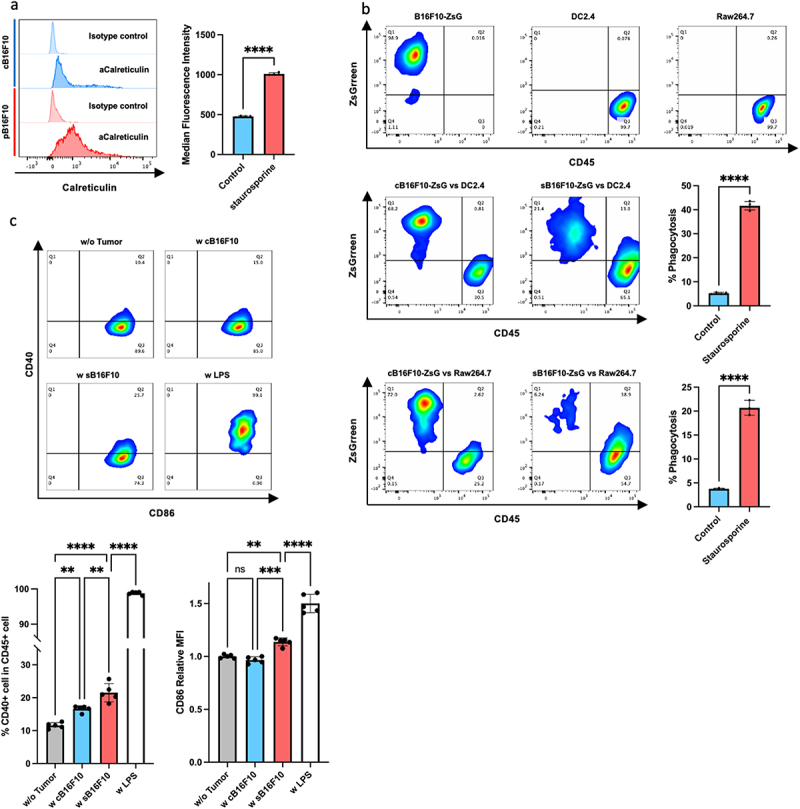
Hyperdifferentiated melanoma cells are efficiently engulfed by phagocytes and promote phagocyte activation. (a) Calreticulin exposure on the cell surface. A representative histogram (upper panel) and comparison of the mean fluorescent intensity between cB16F10 and sB16F10 cells. Data are shown as the mean ± SD in the panels (n = 4 per group). Statistical significance was determined by Student’s t-test (****p < 0.0001). (b) Flow cytometric analysis of phagocytosis by DC2.4 and RAW264.7 cells. Phagocytes were cocultured with cB16F10 or sB16F10 cells expressing ZsGreen. Representative flow cytometry plots and data quantification are shown. Data are shown as the mean ± SD in the panels (n = 4 [DC2.4] or n = 3 [RAW264.7] per group). Statistical significance was determined by Student’s t-test (****p < 0.0001). (c) Flow cytometric analysis of CD40 and CD86 (an APC activation marker) expressions by DC2.4 cocultured with cB16F10 or sB16F10 cells. Lipopolysaccharide (LPS) was used as a positive control. Representative flow cytometry plots and data quantification are shown. Data are shown in the panels as the mean ± SD (n = 3 per group). Statistical significance was determined by one-way ANOVA (**p < 0.01, ***p < 0.001, ****p < 0.0001, Dunnett’s test).
3.4. Antigens derived from hyperdifferentiated melanoma cells are recognized by the host T-cell immune system
Next, we investigated the possibility of eliciting antigen-specific T-cell responses to sB16F10-derived antigens in vivo. To analyze antigen-specific T-cell responses, we established a B16F10-OVA cell line expressing ovalbumin (OVA) as a model antigen and generated hyperdifferentiated B16F10-OVA cells by staurosporine treatment (Figure 3a). The expression of the MHC class I-OVA peptide complex on the cell surface was equivalent to that of the control and hyperdifferentiated states (Figure 3b,c). Mice were then inoculated subcutaneously with hyperdifferentiated B16F10-OVA cells or control B16F10-OVA cells. Fifteen days after inoculation, we harvested splenocytes, stimulated them with OVA antigen, and performed an IFN- ELISpot assay. Splenocytes producing IFN- in response to OVA epitope peptides were significantly increased in mice inoculated with hyperdifferentiated B16F10-OVA cells compared with the findings in mice inoculated with control B16F10-OVA cells (Figure 3d,e). These results suggest that the immunogenicity of HMCs was enhanced. Thus, HMCs might induce CTL immune responses against tumor-derived antigens.
Figure 3.
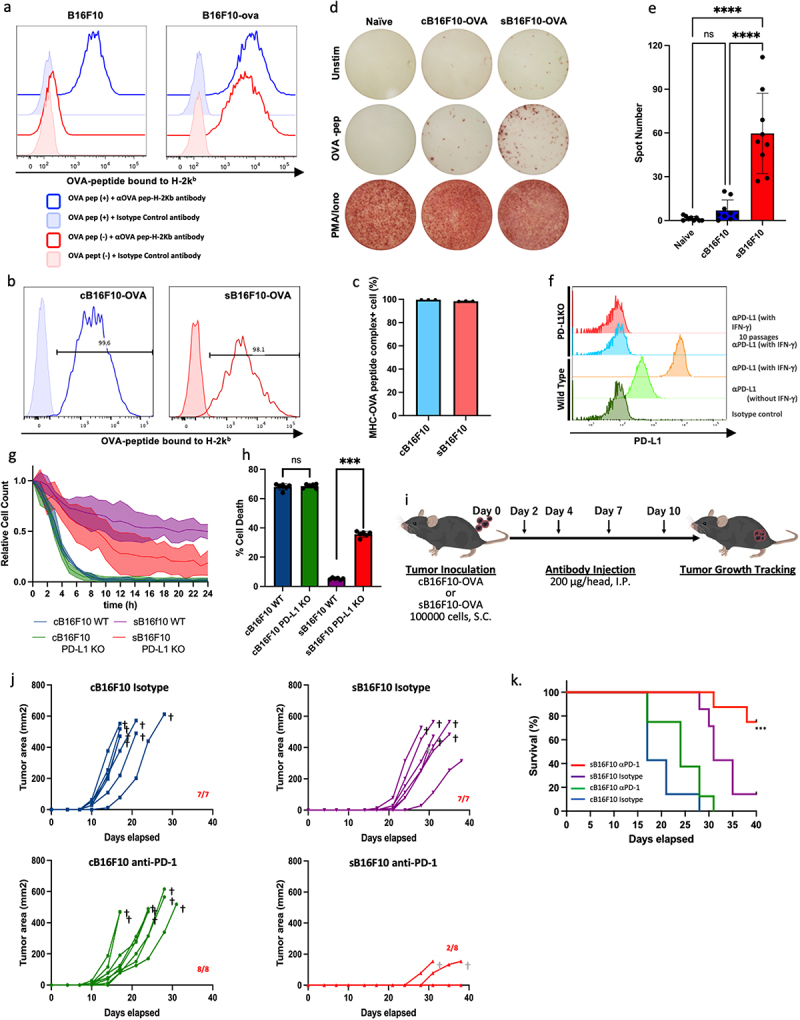
Hyperdifferentiated melanoma cells elicit antitumor immune responses. (a) Establishment of B16F10-OVA. Histograms show the cell surface expression of ova-derived epitope peptide-mhc complexes. (b) and (c) comparison of the expressions of cell surface ova-peptide-mhc complexes in control and staurosporine-exposed surviving B16F10-OVA cells. (d) and (e) an ELISpot assay was used to evaluate tumor-derived antigen-specific immune responses. Representative ELISpot images (d) and data quantification (e) are shown. Data are shown as the mean ± SD in the right panels (n = 9 per group). Statistical significance was determined by one-way ANOVA (****p < 0.0001, Dunnett’s test). (f) Establishment of a PD-L1 knockout B16F10 cell line. Histograms show the expression of PD-L1 on the cell surface of B16F10 cells 48 hours after culture in the presence of 100 U ifn-γ. (g) Measurement of the immunological elimination of cB16F10 or sB16F10 cells by CTLs derived from pmel-1 mice over time. The cell counts are shown relative to the initial time of measurement. (h) Altered susceptibility of sB16F10 cells to T cell mediated cytotoxicity in the presence or absence of PD-L1. Data are shown as the mean ± SD. Statistical significance was determined by Student’s t-test (***p < 0.001). (i) C57BL/6J mice were subcutaneously inoculated with cB16F10-ova or pB16F10-ova cells (1 × 105 cells/head) followed by the intraperitoneal administration of anti-PD-1 mAbs or isotype mAb as a control. The effect of immune checkpoint inhibition on the tumor growth (j) and survival time (k) of tumor-bearing mice. In (k), the data were analyzed by log-rank test (***p < 0.001). The images in (c) are from TogoTV (https://togotv.Dbcls.jp © 2016 DBCLS TogoTV, CC-BY-4.0 https://creativecommons.Org/licenses/by/4.0/deed.Ja).
3.5. Hyperdifferentiated melanoma cells escape CTL-mediated cytokilling via the PD-1/PD-L1 signaling pathway
We examined whether tumor antigen-specific activated CTLs eliminated HMCs. First, gp100-specific CTLs derived from Pmel-1 mice were cocultured with sB16F10 or cB16F10 cells, and the cytokilling of CTLs was monitored by live cell imaging. gp100-specific CTLs killed most tumor cells when targeting cB16F10 cells (Figure 3g and Movie S4). When sB16F10 cells were used as the target cell, the CTLs did kill the target cells (Figure 3g and Movie S5). To evaluate the influence of PD-L1 on the evasion of sB16F10 killing by CTLs, we performed the same experiments as above using PD-L1 knockout (KO) B16F10 cells (Figure 3f) as target cells. The results showed that PD-L1-KO sB16F10 cells were eliminated by Pmel-1 CTL (Figure 3g and Movie S6). The CTL sensitivity of PD-L1-KO sB16F10 cells was significantly higher than that of wild-type sB16F10 cells, even under conventional CTL assay conditions, in which effector cells and target cells were cocultured for 4 hours (Figure 3h). These results suggest that the PD-1/PD-L1 signaling pathway has a critical role in the immune escape of HMCs.
3.6. Immune checkpoint blockade inhibits tumor formation caused by hyperdifferentiated melanoma cells
We investigated the differences in tumor growth between control and hyperdifferentiated melanomas in vivo using a mouse transplantation tumor model. We also examined tumor growth inhibition using an anti-PD-1 antibody, which functions as an immune checkpoint inhibitor (Figure 3i). When sB16F10 cells or cB16F10 cells were implanted subcutaneously into mice inoculated with an isotype control antibody, palpable tumors developed in sB16F10-transplanted mice compared with the findings in cB16F10-transplanted mice (Figure 3j). However, the time from tumor onset to death was 9.1 ± 2.85 days in cB16F10-transplanted mice compared with 10.7 ± 2.75 days in sB16F10-transplanted mice, which did not reach statistical significance. This suggests that the delay in tumorigenesis of sB16F10 cells represents the time it takes for growth-arrested HMCs to resume proliferation in vivo. Finally, when using hyperdifferentiated and control B16F10 cells, tumor development was observed in all mice transplanted with tumor cells.
In cB16F10-transplanted mice, we observed a trend toward slightly slower tumor progression in response to anti-PD-1 antibody treatment compared with that in response to isotype control antibody administration (Figure 3k). However, all mice were euthanized because of tumor progression. In contrast, we observed tumor development in only two of eight mice transplanted with sB16F10 cells and treated with anti-PD-1 antibody. Tumor progression was slower in the anti-PD-1-treated sB16F10-transplanted mice than in the other groups (Figure 3j,k). However, the tumor-bearing anti-PD-1-treated sB16F10-transplanted mice all died before the tumor size reached the endpoint for determining euthanasia (Figure 3j,k).
4. Discussion
The present study provides information about the immunological characteristics of certain types of HMCs that remain in the cancer cell population after targeted therapy, leading to cancer recurrence. Similar to our observations, the upregulation of MHC class I and PD-L1 have been reported in human melanoma cells following their hyperdifferentiation upon exposure to BRAF inhibitors.20 An immunocompetent mouse model with implanted tumors is beneficial when investigating the impact of such hyperdifferentiated melanoma cells on the host immune system. However, experimental studies that mimic human melanoma hyperdifferentiated cells are not easy because murine immunogenic melanoma cell lines that respond to clinically-used targeted drugs such as BRAFi/MEKi are not a standard type of cell line. Our results suggest how to overcome the above challenges. Using mice inoculated with HMCs induced by staurosporine, we observed the activation of HMC-derived antigen-specific T cells. These results suggest that the immunogenicity of HMCs was improved compared with those before drug exposure. However, HMCs also had an enhanced ability to evade elimination by T cells. This suggested they might escape elimination by host immunity and remain in vivo. We found that inhibiting the ability of HMCs to evade elimination by T cells through immune checkpoint inhibitory therapy using anti-PD-1 antibodies, led to the elimination of HMCs. This elimination of HMCs by immune checkpoint inhibition is reminiscent of the prolonged treatment-free survival often observed in patients treated with immune checkpoint inhibitors.21
The cascade of effects of HMC elimination by immune checkpoint inhibition begins with the priming of CD8+ T cells by APCs. The exposure of calreticulin, an “eat me” signal, on the surface of cells undergoing apoptosis induced by cytotoxic drugs is a well-known feature of immunogenic cancer cell death.22 In the present study, exposure to staurosporine enhanced calreticulin exposure on the surface of live HMCs. This increased calreticulin exposure by living cells was also reported for cancer cells undergoing cellular senescence.10 The cells we observed were considered to be in a different cellular state than senescent cells because their growth arrest was not irreversible. Nonetheless, the increased calreticulin exposure was similar to the findings of Marin et al. regarding senescent cells.10 This calreticulin exposure induced by cytotoxic stress can render HMCs more susceptible to phagocytosis by APCs, triggering the priming of CD8+ T cells to HMC-derived antigens.
In the history of tumor immunology, numerous studies have described the importance of immunogenic cell death as an initiator of the anti-tumor immune response. However, Sriram et al. demonstrated that damaged surviving cells were more immunogenic by comparing the immunogenicity of dead cells with that of damaged surviving cells.23,24 Given the findings of Sriram et al. as well as the activation of anti-tumor immunity by senescent cells reported by Marin et al. as well as our observations in this study, immunogenic cell death as well as damaged surviving cells may have an essential role in triggering immune responses against tumor cell-derived antigens.
As a result of phagocytosis by APCs, a variety of HMC-derived antigens are incorporated into the host immune system, leading to the priming of an acquired immune response with a broad immune repertoire. The unleashing of this multivalent immune response by immune checkpoint inhibitors may have led to the inhibitory tumor growth effect in vivo, as shown in Figure 3j. The actual impact of the presence or absence of HMCs on the abundance of the immune repertoire is an exciting subject for future studies.
A more detailed immunological characterization of PSMCs will provide the basis for a strategy to immunologically eliminate PSMCs, which is the key to therapeutic resistance. Targeting melanoma phenotypic plasticity is an attractive strategy that will ultimately reduce the emergence of therapeutic resistance.
Supplementary Material
Acknowledgments
We would like to acknowledge the excellent service provided by the Saitama Medical University Biomedical Research Center. We thank J. Ludovic Croxford, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript. Finally, we thank Ms. Kanae Hino and all the members of the Department of Microbiology for their continuous support and helpful discussions.
Funding Statement
This work was supported by JSPS KAKENHI Grant Numbers [21K08333], [22K08437] and [24K11482].
Disclosure statement
YA, SH, MM, AN, and YH have no financial interests. TM has financial interests in Denka Co., Ltd.
Author contributions
YA and YH conceived the study, designed and performed the experiments, analyzed the data, and produced the figures; YA, YH, and TM wrote the manuscript; SH and MM performed image acquisition; AN and TM helped with experimental design; and TM provided modified cell resources. All authors read and approved the final version of the manuscript.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. This study did not generate/analyze datasets.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2024.2437211
References
- 1.Bhullar KS, Lagarón NO, McGowan EM, Parmar I, Jha A, Hubbard BP, Rupasinghe HPV.. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer. 2018;17(1):48. doi: 10.1186/s12943-018-0804-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen P, Cross D, Jänne PA.. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov. 2021;20(7):551–9. doi: 10.1038/s41573-021-00195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boumahdi S, de Sauvage FJ. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov. 2020;19(1):39–56. doi: 10.1038/s41573-019-0044-1. [DOI] [PubMed] [Google Scholar]
- 4.Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaele M, Karras P, Brown D, Chang YH, Debiec-Rychter M, et al. Toward minimal residual disease-directed therapy in melanoma. Cell. 2018;174(4):843–855.e19. doi: 10.1016/j.cell.2018.06.025. [DOI] [PubMed] [Google Scholar]
- 5.Rambow F, Marine J-C, Goding CR. Melanoma plasticity and phenotypic diversity: therapeutic barriers and opportunities. Genes Dev. 2019;33(19–20):1295–1318. doi: 10.1101/gad.329771.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arozarena I, Wellbrock C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat Rev Cancer. 2019;19(7):377–391. doi: 10.1038/s41568-019-0154-4. [DOI] [PubMed] [Google Scholar]
- 7.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 9.Kroemer G, Galassi C, Zitvogel L, Galluzzi L. Immunogenic cell stress and death. Nat Immunol. 2022;23(4):487–500. doi: 10.1038/s41590-022-01132-2. [DOI] [PubMed] [Google Scholar]
- 10.Marin I, Boix O, Garcia-Garijo A, Sirois I, Caballe A, Zarzuela E, Ruano I, Attolini CSO, Prats N, López-Domínguez JA, et al. Cellular senescence is immunogenic and promotes antitumor immunity. Cancer Discov. 2023;13(2):410–431. doi: 10.1158/2159-8290.CD-22-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198(4):569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132(3):487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- 13.Inoue S, Horiuchi Y, Setoyama Y, Takeuchi Y, Beck Y, Murakami T, Odaka A. Immune checkpoint inhibition followed by tumor infiltration of dendritic cells in murine neuro-2a neuroblastoma. J Surg Res. 2020;253:201–213. doi: 10.1016/j.jss.2020.03.059. [DOI] [PubMed] [Google Scholar]
- 14.Horiuchi Y, Nakamura A, Imai T, Murakami T, Hiller K. Infection of tumor cells with Salmonella typhimurium mimics immunogenic cell death and elicits tumor-specific immune responses. PNAS Nexus. 2024;3(1):gad484. doi: 10.1093/pnasnexus/pgad484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji Y, Abrams N, Zhu W, Salinas E, Yu Z, Palmer DC, Jailwala P, Franco Z, Roychoudhuri R, Stahlberg E, et al. Identification of the genomic insertion site of Pmel-1 TCR α and β transgenes by next-generation sequencing. PLOS ONE. 2014;9(5):e96650. doi: 10.1371/journal.pone.0096650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johmura Y, Shimada M, Misaki T, Naiki-Ito A, Miyoshi H, Motoyama N, Ohtani N, Hara E, Nakamura M, Morita A, et al. Necessary and sufficient role for a mitosis skip in senescence induction. Mol Cell. 2014;55(1):73–84. doi: 10.1016/j.molcel.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Chaib S, Tchkonia T, Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med. 2022;28(8):1556–1568. doi: 10.1038/s41591-022-01923-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault M-C, Trevino TN, Azimi CS, Scheer AK, Randolph HE, Thompson TW, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest. 2018;128(10):4654–4668. doi: 10.1172/JCI99317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khalaji A. Don’t eat me/eat me signals as a novel strategy in cancer immunotherapy. Heliyon. 2023;9:e20507. doi: 10.1016/j.heliyon.2023.e20507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harbers FN, Thier B, Stupia S, Zhu S, Schwamborn M, Peller V, Chauvistré H, Crivello P, Fleischhauer K, Roesch A, et al. Melanoma differentiation trajectories determine sensitivity toward pre-existing CD8+ tumor-infiltrating lymphocytes. J Invest Dermatol. 2021;141(10):2480–2489. doi: 10.1016/j.jid.2021.03.013. [DOI] [PubMed] [Google Scholar]
- 21.Regan MM, Mantia CM, Werner L, Tarhini AA, Larkin J, Stephen Hodi F, Wolchok J, Postow MA, Stwalley B, Moshyk A, et al. Treatment-free survival over extended follow-up of patients with advanced melanoma treated with immune checkpoint inhibitors in CheckMate 067. J Immunother Cancer. 2021;9(11):e003743. doi: 10.1136/jitc-2021-003743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petroni G, Buqué A, Zitvogel L, Kroemer G, Galluzzi L. Immunomodulation by targeted anticancer agents. Cancer Cell. 2021;39(3):310–345. doi: 10.1016/j.ccell.2020.11.009. [DOI] [PubMed] [Google Scholar]
- 23.Sriram G, Milling LE, Chen J-K, Kong YW, Joughin BA, Abraham W, Swartwout S, Handly ED, Irvine DJ, Yaffe MB, et al. The injury response to DNA damage in live tumor cells promotes antitumor immunity. Sci Signal. 2021;14(705):eabc4764. doi: 10.1126/scisignal.abc4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sriram G, Emmons TR, Milling LE, Irvine DJ, Yaffe MB. Immunogenic cell stress and injury versus immunogenic cell death: implications for improving cancer treatment with immune checkpoint blockade. Mol Cell Oncol. 2022;9(1):2039038. doi: 10.1080/23723556.2022.2039038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials. This study did not generate/analyze datasets.