

Abstract
INTRODUCTION
Plasma has been proposed as an alternative to cerebrospinal fluid (CSF) for measuring Alzheimer's disease (AD) biomarkers, but no studies have analyzed in detail which biofluid is more informative for genetics studies of AD.
METHOD
Eleven proteins associated with AD (α‐synuclein, apolipoprotein E [apoE], CLU, GFAP, GRN, NfL, NRGN, SNAP‐25, TREM2, VILIP‐1, YKL‐40) were assessed in plasma (n = 2317) and CSF (n = 3107). Both plasma and CSF genome‐wide association study (GWAS) analyses were performed for each protein, followed by functional annotation. Additional characterization for each biomarker included calculation of correlations and predictive power.
RESULTS
Eighteen plasma protein quantitative train loci (pQTLs) associated with 10 proteins and 16 CSF pQTLs associated with 9 proteins were identified. Plasma and CSF shared some genetic loci, but protein levels between tissues correlated weakly. CSF protein levels better associated with AD compared to plasma.
DISCUSSION
The present results indicate that CSF is more informative than plasma for genetic studies in AD.
Highlights
The identification of novel protein quantitative trait loci (pQTLs) in both plasma and cerebrospinal fluid (CSF).
Plasma and CSF levels of neurodegeneration‐related proteins correlated weakly.
CSF is more informative than plasma for genetic studies of Alzheimer's disease (AD).
Neurofilament light (NfL), triggering receptor expressed on myeloid cells 2 (TREM2), and chitinase‐3‐like protein 1 (YKL‐40) tend to show relatively strong inter‐tissue associations.
A novel signal in the apolipoprotein E (APOE) region was identified, which is an eQTL for APOC1.
Keywords: Alzheimer's disease, biomarkers, CSF, genomics, neurodegenerative disease, plasma, protein quantitative trait loci
1. BACKGROUND
Alzheimer's diseases (AD) is the most common cause of dementia, with an estimated 6.7 million individuals impacted in the United States alone. 1 AD is a neurodegenerative disease that has several hallmarks, including the deposition of extracellular amyloid plaques and formation of intracellular neurofibrillary tangles. 2 Although some recently approved treatments are able to slow the disease progression, no treatment exists to reverse or stop the progression of AD despite the plethora of clinical trials that have been implemented in recent history. 2 This is due, in part, to the high rate of comorbidities in patients with AD and because the disease is advanced by the time patients develop symptoms. 2 Biomarkers could be used to identify comorbidities and diagnose AD earlier to improve clinical trial outcomes. 2 , 3
Cerebrospinal fluid (CSF)–derived biomarkers can be used for evaluating several aspects of AD pathology including synaptic pathology, glial cell pathology, and comorbidities (note that some of these biomarkers are not AD specific). CSF biomarkers that are commonly used include the ratio of amyloid beta‐42 (Aß42) to amyloid beta‐40 (Aß40), phosphorylated tau (p‐tau), and total tau (t‐tau). 2 Levels of neurodegeneration can be assessed using neurofilament light (NfL), with visinin‐like protein 1 (VILIP‐1) being investigated as another such marker. 2 , 4 Other CSF biomarkers are emerging to further increase AD diagnostic accuracy. One new set of biomarkers being evaluated for potential clinical use to measure central nervous system (CNS) synaptic damage include neurogranin (NRGN) and synaptosomal‐associated protein 25 (SNAP‐25). 2 , 5 It is important to assess synaptic pathology because it occurs relatively early in the symptomatic disease process and increased levels of synaptic pathology correlate with cognitive decline. 2 Another set of biomarkers under evaluation are those that are indicators of glial cell pathology, including glial fibrillary acidic protein (GFAP), triggering receptor expressed on myeloid cells 2 (TREM2), and chitinase‐3‐like protein 1 (YKL‐40). 2 Glial cells appear to be a critical component of AD pathology, supporting the importance of such biomarkers. 2 In addition to AD biomarkers, biomarkers of other diseases are being assessed to aid in differentiating AD from other similar neurodegenerative diseases and to assess comorbidities, including α‐synuclein, progranulin (GRN), and TAR DNA‐binding protein 43 (TDP‐43). 2 , 3 , 6 CSF biomarkers are used because CSF interacts directly with the CNS extracellular space and can thus offer a representative picture of CNS pathologies. 4 However, there are limitations to using CSF as the tissue source for biomarkers; the procedure is perceived by some as invasive, which can lead to difficulty in recruitment of individuals to AD clinical trials.
One alternative biofluid being pursued for AD biomarkers is blood plasma due to its ease of access and low risk of complications. 2 In addition, advancements in assay sensitivity now enable accurate measurement of many brain‐derived proteins that are present in blood at low concentrations. 2 However, the blood–brain barrier (BBB) complicates using plasma as a potential source of AD biomarkers, since protein transfer between the brain blood supply to the peripheral circulation is heavily regulated. 7 Despite this, some plasma biomarkers such as p‐tau do have strong associations with brain pathology. 2 Thus researchers have been assessing which plasma protein levels correlate best with CSF and amyloid biomarkers to optimize which plasma proteins should be studied for clinical application for AD and other neurodegenerative diseases. 4 At the genetic level, a previous study from Bradley and coauthors 8 found genetic overlap between plasma and CSF in protein quantitative trait loci (pQTLs) of key AD genes. Research has shown, however, that plasma biomarkers tend to differentiate AD cases and controls less effectively than CSF biomarkers, with the exception of some recent highly accurate tests, which means further research is required to identify potentially clinically relevant plasma biomarkers. 3 , 9
The present study has several goals related to forwarding AD research. The main goals are to identify the genetic basis of several key AD‐related proteins and determine which tissue is more informative for genetic studies of AD. Here we will use proteomic data already generated in a large number of well‐characterized samples from the Knight Alzheimer's Disease Research Center (Knight‐ADRC) and external cohorts. By identifying pQTLs that are associated with AD, we can identify potential AD causal loci and thus potential treatment targets.
2. METHODS
2.1. Cohorts
2.1.1. Ethics statement
The ethics committee and institutional review board of Washington University School of Medicine in St. Louis approved this study. All participants provided informed consent for all data used in the present study.
2.1.2. Plasma and CSF cohorts
For the plasma analyses, 2317 European individuals were included from the Knight‐ADRC. The age range of these individuals was 27 to 104 years. The sample was 45.84% male.
For the CSF analyses, 3107 individuals of European descent were included from six different cohorts: Knight‐ADRC 10 , 11 , 12 (n = 805, 46.7% male), the Alzheimer's Disease Neuroimaging Initiative (ADNI; n = 689; 58.2% male), Dominantly‐Inherited Alzheimer Network (DIAN; n = 193; 48.4% male), Barcelona‐1 13 (n = 197; 52.3% male), Fundació Alzheimer's Center Barcelona (FACE; n = 438; 41.1% male), and Parkinson's Progression Marker Initiative (PPMI; n = 785; 55.1% male).
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using PubMed and Google Scholar. Researchers have been investigating less invasive methods than lumbar puncture for assessing neurodegenerative disease biomarkers. Plasma has been proposed as an alternative tissue for obtaining such biomarkers, but insufficient evidence exists to support its use.
Interpretation: The present research found that cerebrospinal fluid (CSF) and plasma levels of key Alzheimer's disease (AD) proteins correlate poorly despite overlap in genetic protein quantitative trait loci (pQTLs). CSF is a better source of AD biomarkers in general, although the use of plasma neurofilament light (NfL) levels as a proxy for CSF NfL levels is supported. These results will contribute to the identification of optimal AD biomarkers.
Future directions: (1) investigate the significance of the APOC1 expression in plasma apolipoprotein E (APOE) expression levels, (2) assess the biological importance of the novel pQTLs identified in the present study, and (3) extend the present analyses to other AD genes. The APOE region was identified, which is an expression quantitative trait locus (eQTL) for APOC1.
Knight‐ADRC
The predecessor of the Knight‐ADRC at Washington University began in 1979 to study cognition in participants as they age to improve understanding of the aging process, as well as diseases of aging. The Knight‐ADRC itself grew from this preceding study and was established in 1985 with a grant from the National Institute on Aging (NIA). Its ultimate goal is to foster and facilitate research on AD and related dementias (ADRDs) and to identify treatments and/or cures for AD. Eligibility criteria for the clinical cohort followed by the Knight‐ADRC include age 65 years of age or older, cognitively unimpaired or impaired at baseline, availability of a study partner, willingness to undergo longitudinal imaging and CSF studies, and no medical or psychiatric condition that precludes longitudinal participation. Participation in the study is voluntary and free for the research participant. Participants undergo annual clinical and cognitive assessments using a uniform protocol, provide blood samples at baseline and every 2 years thereafter, and have imaging studies and CSF collection at baseline and every 3 years thereafter. The Knight‐ADRC has studied 5510 unique individuals, of whom 2426 were clinically diagnosed with AD dementia and 2156 were cognitively unimpaired. Of these participants, 82.47% are European American individuals. Both CSF and plasma samples from this cohort were used for the present study. Plasma was drawn from about 3798 of the participants, and 1650 of these individuals have longitudinal plasma data available. CSF was collected by lumbar puncture from 1058 participants.
The ADNI
The ADNI is an ongoing, observational study of the various facets of AD progression in thousands of individuals aged 55 to 99 years of age with mild cognitive impairment (MCI), AD, or no dementia. The study was initiated in 2002. Data have been collected from 57 different sites in North America. There are four major versions of ADNI, and in each iteration participants have multiple types of data collected including clinical, psychometric, genetic, and imaging data. The goal of the study is to identify primary and/or secondary treatments for AD. CSF was collected from participants via lumbar puncture.
Barcelona‐1
This cohort is a collection of ≈300 individuals that were collected by the University Hospital Mutua de Terrassa in Barcelona, Spain. 14 The study has collected control individuals and individuals with memory issues ranging from MCI to AD. CSF was collected from participants using lumbar puncture. 13
The DIAN
The DIAN is a project that was initiated in Washington University in St. Louis in 2008, which studies autosomal dominant AD (ADAD), a less common type of AD. Samples were collected from many different sites across the world, including sites in the United States, Asia, Europe, Australia, and South America. Participants who were eligible for inclusion in the study were at least 18 years of age; without severe cognitive issues; able to find two partners that could offer additional information about the participant; and English speakers. Although the goal is to better understand ADAD, the project aims to develop primary and/or secondary treatments for any type of AD. Data were collected from participants in the form of interviews, memory tests, imaging, and post‐mortem brain autopsy. Both plasma and CSF samples were collected from participants, although only CSF samples were used for the present study. CSF was collected by lumbar puncture from participants.
FACE
The Fundacion ACE (FACE) cohort is a sample that was collected by the ACE Foundation, which is a non‐profit organization located in Spain that is involved in the treatment of and research into AD. To this date FACE has collected 17,993 genetic samples from individuals with dementia, and they have more than 20,000 genetic samples in storage.
The PPMI
This PPMI cohort, funded by the Michael J. Fox Foundation for Parkinson's Research (MJFF), was created based on ongoing longitudinal observations of the progression of Parkinson's disease in ≈3000 individuals (the goal is to have a sample size of 4000 individuals). The samples have been collected from 50 different locations around the world, including cities in the United States, Europe, Israel, and Australia. The goal of this cohort is to have the largest collection of tangible data from individuals with Parkinson's disease. The study incorporates clinical, genetic, and imaging data to unveil potential improvements on modern treatments. Some of the specific areas studied include motor skills, neuropsychiatric functioning, and sleeping. Both CSF and plasma were drawn from participants in this study, along with skin and urine samples. CSF is the only tissue that was used from this cohort and was collected via standard lumbar puncture procedures. 15
2.1.3. AD genome‐wide association study (GWAS) risk summary statistics
For comparison against CSF and plasma data, the summary statistics from a previous AD study performed by Bellenguez and coauthors 16 were included for analyses. The purpose of this study was to identify and confirm loci associated with AD to better understand the physiological underpinnings of the disease. The study successfully identified 42 new loci, and confirmed 33 previous loci. This study analyzed 788,989 European individuals (111,326 individuals with AD or proxies for AD and 677,663 controls) from 11 different cohorts using fixed‐effects meta‐analysis; extensive follow‐up analyses were performed on the meta‐analysis results. Please refer to the original publication for further information. 6
For one of the present analyses, a different set of AD summary statistics from Schwartzentruber and coauthors 17 was used because Bellenguez and coauthors 16 excluded the apolipoprotein E (APOE) region from their analyses (although Bellenguez and coauthors 16 did find APOE variants that were significant at a 5 × 10−8 p‐threshold level in stage 1 of their study, before APOE was removed). Schwartzentruber and colleagues 17 used inverse‐weighted meta‐analysis of genome‐wide association study‐by‐proxy (GWASX) data, as well as genetic co‐localization analyses between AD and potential risk loci expression quantitative trait loci (eQTLs), to identify and confirm loci associated with AD; they found four novel genes and replicated 32 other loci. The study included 858 AD cases, 52,791 proxy‐AD cases, and 355,900 controls. 17
2.2. Proteins
2.2.1. Protein‐level data generation and quality control
The proteins were measured using the SOMAscan7k platform, a protein level assay that is high throughput and based on aptamers. 18 The platform can detect proteins at concentrations as low as the femtomolar level. The version of SOMAscan7k used in the present research outputs measurements for 7584 aptamers from ≈6383 unique proteins. When identifying protein concentrations, they also included 308 control proteins related to hybridization, normalization, binding specificity, and non‐human protein sequences. Protein concentrations were determined using a DNA aptamer assay that is proprietary to Somalogic. Protein concentrations were reported in relative fluorescence units. In the following normalization and quality control sections, all steps are the same for plasma and CSF unless otherwise specified.
2.2.2. Normalization of protein concentration measurements
After measuring protein concentrations, normalization of the data was performed to control for technical variation and facilitate quality control. First, hybridization control normalization was performed at the sample level. Second, the aptamers were split into three different groups based on their signal‐to‐noise ratios and processed separately for the remainder of the normalization steps. Third, the median signal normalization to calibrators was calculated, which controls for confounds for groups of wells on the same plate that have the same experimental conditions and dilution level. 18 Finally, an iterative adaptive normalization of maximum likelihood method was used to normalize the data to a reference to control for both technical and biological variance. 13
2.2.3. Quality control of protein concentration measurements
Quality control of the protein measurements was performed, using an in‐house pipeline, at both the protein aptamer level and the sample level. For protein aptamer–level quality control, aptamers were removed from the data if they met one of the following conditions: if an aptamer was found to go above the limit of detection (LOD) score threshold in >15% of the samples assessed (this quality control step was restricted to plasma), if the difference between the aptamer‐specific median scale factor and its relevant calibration scale factor was >0.5, or if the aptamer‐specific median coefficient of variation had a value >0.15. The remaining protein data were then log10 transformed and an interquartile range was calculated for these transformed values on a protein‐by‐protein basis; aptamers were removed if the aptamer‐specific median scale factor differed from the median scale factor by more than 1.5 times the interquartile range in ≥85% of samples. 13 Protein aptamers and samples with a call rate <65% were then removed from the data. Protein aptamer call rates were recalculated and protein aptamers with a call rate <85% were removed from the data. The sample call rates were then recalculated and individuals with a call rate <85% were removed.
2.3. Analyses
2.3.1. Genetic quality control
Quality control of the genetic data was performed using PLINK1.9. 19 Genetic data had already been linked to proteomics data before the proteomics in‐house quality control. 13 Imputation was performed on the genetic data using the TOPMed imputation server. 20 Before imputation, quality control was performed and variants were removed if they had: a genotyping rate <98%, minor allele frequency (MAF) ≤0.3%, and/or a Hardy‐Weinberg equilibrium (HWE) p‐value < 1 × 10−6. After imputation, further thresholds for variant inclusion were set. Variants with a HWE p‐value < 5 × 10−30 were excluded from analyses. Duplicate single‐nucleotide polymorphisms (SNPs) and duplicate samples were removed from the data. SNPs that were strand ambiguous were removed to avoid bias in the results.
Principal component (PC) analysis was performed to calculate genetic PCs, and PC1 and PC2 were plotted (Figure S1). Based on the plot, it was decided that individuals with a PC1 between −0.0025 and 0.0025, and a PC2 between −0.010 and 0.004, were European and retained for further analysis (plot visualization was used for selecting Europeans because of availability of data produced previously in the lab). An identity‐by‐descent analysis was performed to assess potential cryptic relatedness between individuals in the sample; one individual per pair with >0.2 was removed and the individual who was removed had a higher sample genotyping missingness rate within the pair.
2.3.2. Protein level GWAS
A GWAS was performed to assess plasma protein pQTLs in PLINK2, using protein aptamer levels as the dependent variables (the protein aptamers included in these analyses can be found in Table S1). The independent variables included genetic variant, age, sex, protein plate, cohort array, 10 genetic PCs, and two protein PCs. Over 10 million variants were included in the plasma and CSF analyses.
No inflation was found for any of the analyses based on QQ‐plots (Figures S2–S17). All summary statistics from this study are available on ONTIME browser. 10
2.3.3. In silico functional annotation
A scoring system was implemented to identify likely functional genes for each prioritized locus. Variants were selected for gene scoring by selecting every variant that was within 1 Mb of each trans sentinel variant that both had an r 2 ≥ 0.6 with the sentinel variant and a p‐value ≤ 5 × 10−5. Additional variants with an r 2 ≥ 0.6 with each sentinel variant were added from the Linkage Disequilibrium (LD) Proxy 1000 Genomes GRCh38 high‐coverage data. 21 Each gene at a signal was given a single point on a variant‐by‐variant basis for each time one of the following occurred: if the variant was a coding variant for that gene; if the gene was the closest gene to the variant; for each isomer of the gene in which the variant was a pQTL (from two studies that analyzed 6907 plasma and 7028 CSF proteins, respectively); and for each MetaBrain 22 or GTEx 23 CNS tissue in which that variant was an eQTL for the gene. The gene with the highest score was selected as the likely functional gene. To determine if the proposed functional genes were novel or not, we assessed both the GWAS Catalog and plasma pQTLs from the study by Ferkingstad and coauthors. 24 A gene was considered a novel pQTL if it was not associated with the respective protein in the plasma pQTL data at a p < 5 × 10−8 and if it was not mentioned as a plasma nor CNS pQTL in the GWAS Catalog.
2.3.4. Protein level correlations between tissues
Pearson correlations between CSF and plasma protein aptamer levels were calculated using the cor.test function in R to assess if there were similar levels of these aptamers between tissues. A correlation was also calculated between the plasma and CSF GWAS effect sizes for each protein aptamer separately using the cor.test function in R for the purpose of identifying protein aptamers that have similar genetic determinants between plasma and CSF. Clinically defined AD polygenic risk scores (PRSs) were correlated against the log10 protein concentrations of each protein aptamer using cor.test in R to assess protein levels that associate with increased risk of AD; the AD PRSs were calculated in PRSice‐2 25 using summary statistics from Bellenguez and coauthors. 16 Only individuals who had CSF and plasma measurements taken within 6 months of each other were included, to minimize biases due to temporal changes in measurements (n = 289). A Bonferroni correction was applied to the significance threshold for the 272 total correlations that were calculated across the entire study (p‐value threshold = 1.80 × 10−4).
2.3.5. Predicting AD status from protein levels
The program pROC was used to perform receiver‐operating characteristic (ROC) analyses to assess which proteins best predicted clinically determined AD status, and to identify which tissue showed better predictive ability of AD status. 26 For each protein aptamer in each tissue, two models were run: a covariate‐free model and a covariate‐containing model. In the covariate‐free model, an ROC analysis was run in which the outcome variable was AD status and the predictor variable was the log10 protein levels. In the covariate‐containing model, a general linear model was run in which the outcome variable was log10 protein levels and the predictor variable was the genotype of the sentinel variant for the protein; for the subsequent ROC analysis, the residuals from the general linear model were used as the predictor variable and the outcome variable was AD status.
2.3.6. Genetic co‐localization
Co‐localization analyses were performed using the coloc.abf function from the coloc package in R to identify loci that were potentially causally associated between plasma and CSF sentinel variants, as well as between plasma or CSF and clinically defined AD; the coloc.abf function assumes that there is a single causal variant. 27 For co‐localization analyses that included AD summary statistics, non‐APOE regions were assessed in the study by Bellenguez and colleagues, 16 whereas co‐localization in the APOE region was assessed using AD summary statistics from the study by Schwartzentruber and colleagues. 17 Loci were selected for colocalization analysis if a plasma top hit was within 1 Mb of a CSF top hit, or if a tissue top hit was within 1 Mb of an AD top hit. For all plasma co‐localization analyses, the variance of the protein aptamer levels was estimated by calculating the variance in protein aptamer levels of Europeans included in the analyses. We used z‐score corrected CSF data for the co‐localization analyses, so a variance of 1 was used for all CSF co‐localization analyses. A region was considered to co‐localize between two phenotypes when the posterior probability that they shared the same causal variant (PP.H4) was ≥0.8.
3. RESULTS
3.1. Study design
Plasma and CSF protein GWAS analyses were performed on unrelated European individuals. Eleven proteins were selected for analysis based on their known association with neurodegenerative disease: apolipoprotein E (APOE), CLU, GFAP, GRN, NfL, NRNG, α‐synuclein, SNAP‐25, TREM2, VILIP‐1, and YKL‐40. APOE, CLU, α‐synuclein, and TREM2 had multiple aptamers included from the protein measurements. Only two of the possible APOE aptamer measurements were assessed, as the other two failed the LOD filter during plasma protein quality control. The demographics of the participants can be found in Table 1.
TABLE 1.
Demographics summary by cohort.
| Tissue | Cohort | Sample Size | Mean Age (SD) | %Male | %APOE ε4+ | %AD Cases |
|---|---|---|---|---|---|---|
| Plasma | ||||||
| Knight‐ADRC | 2317 | 75.2 (10.0) | 45.8 | 39.9 | 38.1 | |
| CSF | ||||||
| ADNI | 689 | 73.7 (7.5) | 58.2 | 50.2 | 75.6 | |
| PPMI | 785 | 61.8 (9.4) | 55.1 | N/A | 0.0 | |
| FACE | 438 | 71.9 (8.3) | 41.1 | 35.6 | 54.3 | |
| DIAN | 193 | 38.6 (10.7) | 48.4 | 27.6 | 61.7 | |
| Knight‐ADRC | 805 | 71.4 (8.7) | 46.7 | 39.0 | 22.1 | |
| Barcelona‐1 | 197 | 68.8 (7.5) | 52.3 | N/A | 32.0 | |
| Total | 3107 |
Note: This table summarizes demographic information of the participants included in the plasma and CSF association analyses. The %APOE ε4+ column specifies the percentage of each cohort that included individuals with at least one APOE ε4 allele. There is some overlap between the plasma and CSF Knight‐ADRC sample. All participants were European in ancestry based on principal component analysis.
Abbreviations: AD, Alzheimer's disease; ADNI, Alzheimer's Disease Neuroimaging Initiative; ADRC, Alzheimer's Disease Research Center; CSF, cerebrospinal fluid; DIAN, Dominantly Inherited Alzheimer Network; FACE, Fundació Alzheimer's Center; PPMI, Parkinson's Progression Markers Initiative; SD, standard deviation.
The present study had several stages. First, protein aptamers were selected based on their previous association with AD and availability for analysis. Second, association analyses were run, for both plasma and CSF proteins. Third, potential functional genes were proposed for each sentinel variant (or variant in LD with the sentinel variant at r 2 ≥ 0.6) based on a score that accounted for pQTLs, eQTLs, gene proximity to each variant, and functional variant annotation. Fourth, post‐GWAS analyses were performed between plasma and CSF parameters to identify similarities between tissues, which included correlations and colocalization analyses. Fifth, both plasma and CSF data were compared against AD data to identify which tissue shared genetic loci with AD, and which tissue best predicted AD status; this included ROC analyses and co‐localization analyses. The overall study design is outlined in Figure 1A.
FIGURE 1.
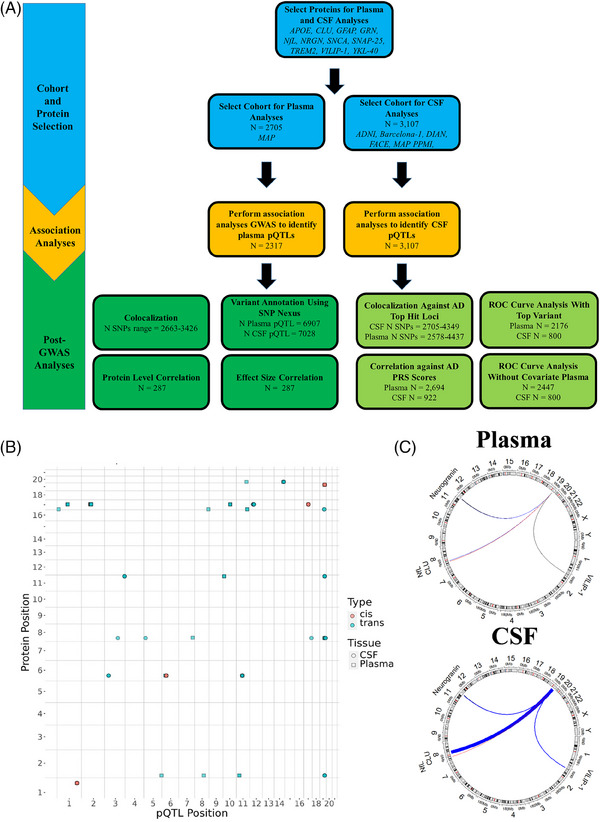
Study design flow chart and dot plot of top hits for plasma and CSF. (A) This image is a flowchart of the entire set of analyses that were performed for the present study. Proteins were selected as outcome variables based on their association with neurodegenerative disease and availability in the SOMAscan7k proteomics panel. Plasma and CSF protein measurements were used for association analyses. Several functional analyses were performed to prioritize likely functional variants and genes, compare associations between plasma and CSF (sections with dark green background in post‐GWAS analyses section), and compare associations between plasma or CSF and AD (sections with light green background in post‐GWAS analyses section). (B) This plot shows the genomic position of significant variants across all analyses with respect to the gene that codes for the respective protein aptamer. Each color indicates if a signal is cis or trans. Each shape represents the tissue in which a variant was found to be significant. (C) The Circos plots show which genes had aptamers with a top hit in the APOE region by connecting these regions with the APOE region with lines that are colored based on effect size direction (red: negative; blue: positive, black: essentially zero) and that have thickness determined by the effect size (magnitude multiplied by 10 to make the lines easier to see). The APOE region is the only unlabeled region. Protein aptamer levels were log10 transformed before analysis. All participants were European in ethnicity based on principal component analysis. AD, Alzheimer's disease; CSF, cerebrospinal fluid; GWAS, genome‐wide association study; N, sample size; pQTL: protein quantitative trait locus.
3.2. CSF and plasma pQTL findings
3.2.1. Plasma GWASs
A GWAS was performed on 2317 European individuals to determine the genetic basis of plasma protein levels for 11 proteins associated with neurodegenerative disease (Figure 1B; Table 2; Table S2; Figures S2–S17). Eighteen loci showed significant associations for 10 unique proteins (14 different aptamers; p ≤ 5 × 10−8). The only protein without any GWAS hit was α‐synuclein.
TABLE 2.
Summary of trans sentinel variants associated with plasma protein aptamer levels in Europeans.
| Plasma | CSF | AD | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Aptamer | ID | Gene Nearest Hit | Sample Size | MAF | Effect Size | p‐Value | Effect Size | p‐Value | Effect Size | p‐Value |
| CLU | X4542.24 | chr7:137280619:C:T | PTN | 2121 | 0.054 | 0.024 | 2.320 × 10−8 | −0.021 | 0.661 | 0.004 | 0.830 |
| X24941.14 | chr1:17210401:C:T | PADI1 | 1951 | 0.008 | 0.061 | 6.781 × 10−9 | 0.133 | 0.661 | −0.084 | 0.168 | |
| chr8:135875240:G:A | LINC02055 | 1987 | 0.002 | 0.120 | 2.042 × 10−9 | N/A | N/A | 0.074 | 0.535 | ||
| GFAP | X20126.19 | chr2:88954346:G:A | IGKV5‐2 | 1972 | 0.072 | 0.054 | 2.070 × 10−20 | N/A | N/A | −0.004 | 0.823 |
| GRN | X4992.49 | chr1:109275908:C:T | SORT1 | 2094 | 0.220 | −0.054 | 1.863 × 10−114 | 0.081 | 0.004 | −0.017 | 0.072 |
| chr10:71802391:T:C | CDH23 | 2094 | 0.263 | −0.020 | 1.951 × 10−16 | 0.031 | 0.224 | 0.008 | 0.402 | ||
| NFL | X10082.251 | chr19:44908684:T:C | APOE | 2116 | 0.232 | −0.138 | 6.342 × 10−259 | −0.223 | 3.548 × 10−18 | −1.202 | 1.170 × 10−881 |
| NRGN | X18303.39 | chr10:11676245:G:A | ENSG00000271046 | 2001 | 0.001 | −0.372 | 6.808 × 10−10 | 0.338 | 0.363 | 0.006 | 0.979 |
| SNAP‐25 | X13105.7 | chr14:94318281:C:T | SERPINA6 | 1905 | 0.207 | 0.036 | 4.761 × 10−22 | 0.156 | 7.413 × 10−7 | 0.011 | 0.269 |
| TREM2 | X5635.66 | chr11:60254475:G:A | MS4A4A | 2121 | 0.363 | 0.077 | 8.392 × 10−60 | 0.363 | 3.162 × 10−54 | −0.086 | 1.654 × 10−24 |
| X11851.21 | chr11:60254475:G:A | MS4A4A | 2118 | 0.363 | 0.065 | 2.242 × 10−57 | 0.366 | 1.318 × 10−57 | −0.086 | 1.654 × 10−24 | |
| X16300.4 | chr11:60254475:G:A | MS4A4A | 2111 | 0.363 | 0.074 | 2.967 × 10−37 | 0.360 | 1.549 × 10−41 | −0.086 | 1.654 × 10−24 | |
| VILIP‐1 | X13522.20 | chr5:175754615:C:A | HRH2 | 1934 | 0.004 | 0.125 | 4.207 × 10−8 | 0.118 | 0.550 | −0.122 | 0.144 |
| chr8:89002721:C:T | ENSG00000253553 | 1970 | 0.032 | 0.044 | 3.509 × 10−8 | 0.038 | 0.562 | 0.026 | 0.279 | ||
| chr11:30061301:A:G | AC124657.1 | 1970 | 0.018 | 0.061 | 8.042 × 10−9 | 0.142 | 0.170 | 0.037 | 0.299 | ||
Note: This table summarizes the variants with the lowest p‐value in each prioritized trans locus derived from the plasma GWAS. For comparison, the effect size and p‐values are shown for the same variants derived from the CSF association analyses and the Bellenguez et al., 2022 AD meta‐analysis. If missing in the Bellenguez et al., 2022 summary statistics, the Schwartzentruber et al., 2021 summary statistics were used. Sections highlighted in bold are statistically significant for CSF and/or AD at a threshold of p < 5 × 10−8. A proxy variant in LD with the top variant was used for the GRN top hit chr10:71802391:T:C (chr10:71799095:A:G; r2 = 1) AD summary statistics. Protein aptamer levels were log10 transformed before analysis. All participants were European in ethnicity based on principal component analysis. n = 2317.
Abbreviations: AD, Alzheimer's disease; CSF, cerebrospinal fluid; GFAP, glial fibrillary acidic protein; GWAS, genome‐wide association study; LD, linkage disequilibrium; MAF, minor allele frequency; NFL, neurofilament light; NRGN, neurogranin; SNAP‐25, synaptosomal‐associated protein 25; TREM2, triggering receptor expressed on myeloid cells 2; VILIP, visinin‐like protein 1..
Of the 18 loci, 13 were trans‐pQTL and 5 were cis‐pQTL associations. Cis signals were found for apoE (X2938.55: p = 4.76 × 10−19; X5312.49: p = 1.95 × 10−24), GRN (p = 3.50 × 10−9), TREM2 (p = 2.06 × 10−12) and YKL‐40 (p = 9.76 × 10−215).
For trans signals, to identify the most likely functional gene driving the association, a scoring system was implemented (see Methods; Table S3). Functional annotation in silico nominated a functional gene at the: (1) APOE locus for NfL; (2) IGKV4‐1 for GFAP, as it included a coding variant; (3) PSRC1 for GRN, as it co‐localized with eQTLs across several studies; and (4) CDH23 for GRN and (5) MS4A6A for TREM2 based on the presence of a coding variant and eQTLs. Four of the trans sentinel variants were rare (MAF <0.01): chr1:17210401:C:T and chr8:135875240:G:A associated with CLU (X24941.14), chr10:11676245:G:A associated with Ng, and chr5:175754615:C:A associated with VILIP‐1.
To determine if the signals were novel, we assessed both the GWAS Catalog and the plasma pQTLs from the Ferkingstad and coauthors 24 study. We identified one novel plasma pQTL: IGKV4‐1 for GFAP (Table S3).
3.2.2. CSF GWAS
CSF protein GWASs were performed on 3107 European samples (Figure 1B; Table 3; Figures S2–S17; Table S4). Sixteen loci showed significant association across nine unique proteins (13 aptamers; p ≤ 5 × 10−8). The two proteins without a GWAS hit were GFAP and α‐synuclein.
TABLE 3.
Summary of trans sentinel variants associated with CSF protein aptamer levels in Europeans.
| CSF | Plasma | AD | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Aptamer | ID | Gene Nearest Hit | MAF | Effect Size | p‐Value | Effect Size | p‐value | Effect Size | p‐Value |
| CLU | X24941.14 | chr19:40601522:C:T | LTBP4 | 0.007 | 1.139 | 1.072 × 10−8 | 0.022 | 0.057 | −0.073 | 0.185 |
| GRN | X4992.49 | chr12:40220632:C:T | AC079630.1, LRRK2 | 0.142 | 0.321 | 1.259 × 10−20 | N/A | N/A | −0.006 | 0.589 |
| NFL | X10082.251 | chr3:123475573:T:G | HACD2 | 0.009 | −0.477 | 3.981 × 10−8 | −0.003 | 0.878 | −0.061 | 0.117 |
| chr5:14278964:T:C | TRIO | 0.001 | −2.585 | 1.479 × 10−10 | N/A | N/A | 0.115 | 0.310 | ||
| chr17:75755876:C:T | ITGB4, GALK1 | 3.800 × 10−4 | 2.119 | 1.660 × 10−8 | N/A | N/A | −0.202 | 0.282 | ||
| chr19:44908684:T:C | APOE | 0.232 | −0.223 | 3.548 × 10−18 | −0.138 | 6.342 × 10−259 | −1.202 | 1.17 × 10−881 | ||
| NRGN | X18303.39 | chr3:190925404:G:T | OSTN | 0.370 | 0.175 | 6.310 × 10−9 | 0.002 | 0.730 | 0.009 | 0.302 |
| chr19:44888997:C:T | APOE | 0.241 | 0.265 | 5.370 × 10−16 | 0.008 | 0.147 | −0.954 | 6.64 × 10−573 | ||
| SNAP‐25 | X13105.7 | chr14:94378225:C:T | SERPINA1 | 0.249 | −0.309 | 7.244 × 10−29 | −0.031 | 9.204 × 10−21 | −0.005 | 0.618 |
| TREM2 | X5635.66 | chr3:30163366:A:G | RBMS3 | 0.058 | 0.283 | 1.230 × 10−8 | −0.026 | 0.008 | −0.007 | 0.707 |
| chr11:60177107:C:T | MS4A6A | 0.386 | 0.361 | 3.631 × 10−56 | 0.073 | 2.223 × 10−54 | −0.079 | 3.090 × 10−21 | ||
| X11851.21 | chr11:60254475:G:A | MS4A4A | 0.363 | 0.366 | 1.320 × 10−57 | 0.065 | 2.242 × 10−57 | −0.086 | 1.654 × 10−24 | |
| X16300.4 | chr11:60177107:C:T | MS4A6A | 0.386 | 0.353 | 4.467 × 10−42 | 0.068 | 6.178 × 10−32 | −0.079 | 3.090 × 10−21 | |
| VILIP‐1 | X13522.20 | chr19:44919589:G:A | APOE | 0.262 | 0.246 | 9.550 × 10−18 | −0.005 | 0.166 | 0.220 | 0.249 |
Note: This table summarizes the variants with the lowest p‐value in each prioritized trans locus derived from the CSF association analyses. For comparison, the effect size and p‐values are shown for the same variants derived from the plasma GWAS and the Bellenguez et al., 2022 AD meta‐analysis. If missing in the Bellenguez et al., 2022 summary statistics, the Schwartzentruber et al., 2021 summary statistics were used. Sections highlighted in bold are statistically significant for plasma and/or AD at a threshold of p < 5 × 10−8. A proxy variant in LD with the top variant was used for the Ng top hit chr19:44888997:C:T (chr19:44892362:A:G; r 2 = 0.750) AD summary statistics. Protein aptamer levels were log10 transformed before analysis. All participants were European in ethnicity based on principal component analysis. n = 3107.
Abbreviations: AD, Alzheimer's disease; CSF, cerebrospinal fluid; GWAS, genome‐wide association study; LD, linkage disequilibrium; MAF, minor allele frequency.
Of the 16 loci, 12 were trans‐pQTL associations and four were cis‐pQTL associations. Cis signals were found for APOE (p = 2.39 × 10−41), GRN (p = 3.16 × 10−17), TREM2 (p = 2.69 × 10−20), and YKL‐40 (p = 1.66 × 10−1452).
Of all the signals, six were rare: chr19:40601522:C:T associated with CLU (X24941.14); chr3:123475573:T:G, chr5:14278964:T:C, and chr17:75755876:C:T associated with NfL; chr6:40974457:G:A associated with TREM2 (X5635.66); and chr6:41161395:C:T associated with TREM2 (X11581.21). A Circos plot of the trans‐pQTLs for APOE can be seen in Figure 1C.
For the 12 trans signals, three were located in the APOE region, and for the remaining 9, we were able to nominate a functional gene for five of them. We nominated the following functional genes: (1) LTBP4 for CLU and (2) MS4A6A for TREM2 (all aptamers) based on the presence of a coding variant; (3) LRRK2 for GRN based on eQTL mapping; and (4) CCDC50 for NRGN, and (5) SERPINA1 for SNAP‐25 based on pQTL mapping.
Of these CSF pQTLs, four were novel and included the signals: LTBP4 with CLU; CCDC50 with NRGN; and APOE with both NRGN and VILIP‐1. Some previously identified functional genes were replicated such as the LRRK2 GRN pQTL, SERPINA1 SNAP‐25 pQTL, and MS4A6A TREM2 pQTL (Table S5).
Summary statistics for three coding variants in TREM2, in both plasma and CSF, are available in Table S6. All summary statistics from this study are available on ONTIME browser. 10
3.3. Comparison of plasma and CSF
3.3.1. Across tissue pQTL effect size correlations are weak
Correlations were calculated between the plasma and CSF pQTL effect sizes to identify genetic overlap between tissues for the same aptamer (Table S7; Figure S18; additional genetic correlation results can be found in Extended Results Section 5.1). Three different statistical significance thresholds for pQTL p‐values were used for including variants in the correlation calculations: 5 × 10−5, 5 × 10−6, and 5 × 10−8. The highest correlation coefficients were found for YKL‐40 (r = 0.89, p = 1.00 × 10−3, variants included in analysis [nVar] = 9; variant‐inclusion p‐value threshold = 5 × 10−8) and GFAP (r = 0.98, p = 0.02, nVar = 4; variant‐inclusion p‐value threshold = 5 × 10−5). No other protein showed a nominally significant correlation for effect sizes across tissues.
3.3.2. CSF and plasma protein levels are weakly correlated
Protein level correlations were calculated between CSF and plasma levels to identify proteins with similar expression levels between tissues (Figure 2;Table S8). Only two proteins showed significant correlations between plasma and CSF after Bonferroni correction for the number of tests (p = 1.80 × 10−4): YKL‐40 (r = 0.62, p = 1.71 × 10−32), and TREM2 (r ≥ 0.35, p ≤ 9.38 × 10−14).
FIGURE 2.
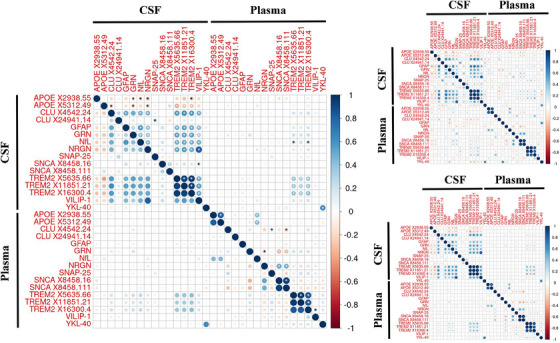
Correlation between CSF and plasma measurements of various AD‐associated protein aptamers in Europeans. These correlation plots show the correlation in protein levels between CSF and plasma for 16 different aptamers in Europeans who had both their plasma and CSF measurements taken within 6 months of each other. The left‐hand plot shows the overall correlations, whereas the right‐hand plots show correlations for AD patients (top) and controls (bottom), respectively. Larger circles indicate a stronger correlation and color indicates both the magnitude and the direction of the effect, with blue indicating a positive correlation and red indicating a negative correlation. The white asterisks indicate correlations that had p ≤ 5 × 10−8 and the black asterisks indicate correlations that has p ≤ 5 × 10−5. Protein aptamer levels were log10 transformed before analysis. All participants were European in ethnicity based on principal component analysis. AD n range = 62–71; Control n range = 201–216. AD, Alzheimer's disease; CSF, cerebrospinal fluid.
In addition, correlations were calculated stratified by AD status and trends similar to the overall correlations were observed. CSF versus plasma YKL‐40 correlations were consistent across tissues between cases and controls (controls: r = 0.64, p = 5.81 × 10−26; cases: r = 0.58, p = 7.25 × 10−8). However, TREM2 showed protein correlations across tissues in controls (r ≥ 0.35, p ≤ 1.38 × 10−7) but not cases (r ≥ 0.21, p ≤ 0.08; Table S8). In addition, the across‐tissue NRGN protein correlation was six times larger in AD patients (r = 0.37, p = 1.00 × 10−3) than in controls (r = 0.06, p = 0.40; Fisher's z = 2.34, p = 0.02). NfL also showed a higher correlation in AD patients, (r = 0.27, p = 0.03) than in controls (r = 0.07, p = 0.28; Fisher's z = 1.37, p = 0.17), although the difference was not significant.
3.3.3. CSF and plasma pQTLs are shared except in the APOE region
Colocalization analysis was performed to assess overlap in signals between plasma and CSF protein levels (Table 4). The loci for GRN, NfL, SNAP‐25, TREM2, and YKL‐40 in CSF and plasma co‐localized (PP.H4 range = 0.87–1.00); for TREM2, the exception is that TREM2 (X16300.4) failed to co‐localize with the other two TREM2 aptamers on chromosome 6.
TABLE 4.
Co‐localization analysis between CSF and plasma aptamers in Europeans.
| Plasma Gene | Plasma Aptamer | Plasma Hit | CSF Gene | CSF Aptamer | CSF Hit | Number of Variants | PP.H4 |
|---|---|---|---|---|---|---|---|
| APOE | X2938.55 | chr19:44913034:C:T | APOE | X2938.55 | chr19:44908684:T:C | 3112 | 0.031 |
| APOE | X5312.49 | chr19:44908684:T:C | 3112 | 0.018 | |||
| NfL | X10082.251 | chr19:44908684:T:C | 3112 | 0.018 | |||
| NRGN | X18303.39 | chr19:44888997:C:T | 3071 | 0.006 | |||
| VILIP‐1 | X13522.20 | chr19:44919589:G:A | 3090 | 0.004 | |||
| X5312.49 | chr19:44908822:C:T | APOE | X2938.55 | chr19:44908684:T:C | 3128 | 0.008 | |
| APOE | X5312.49 | chr19:44908684:T:C | 3128 | 0.000 | |||
| NfL | X10082.251 | chr19:44908684:T:C | 3128 | 0.000 | |||
| NRGN | X18303.39 | chr19:44888997:C:T | 3087 | 0.000 | |||
| VILIP‐1 | X13522.20 | chr19:44919589:G:A | 3076 | 0.000 | |||
| GRN | X4992.49 | chr17:44352876:C:T | GRN | X4992.49 | chr17:44352876:C:T | 2192 | 1.000 |
| NFL | X10082.251 | chr19:44908684:T:C | APOE | X2938.55 | chr19:44908684:T:C | 3129 | 1.000 |
| APOE | X5312.49 | chr19:44908684:T:C | 3129 | 1.000 | |||
| NFL | X10082.251 | chr19:44908684:T:C | 3129 | 1.000 | |||
| NRGN | X18303.39 | chr19:44888997:C:T | 3088 | 0.997 | |||
| VILIP‐1 | X13522.20 | chr19:44919589:G:A | 3074 | 0.997 | |||
| SNAP‐25 | X13105.7 | chr14:94318281:C:T | SNAP‐25 | X13105.7 | chr14:94378225:C:T | 3727 | 0.994 |
| TREM2 | X5635.66 | chr11:60254475:G:A | TREM2 | X11851.21 | chr11:60177107:C:T | 2663 | 0.999 |
| TREM2 | X16300.4 | chr11:60177107:C:T | 2663 | 0.984 | |||
| TREM2 | X5635.66 | chr11:60177107:C:T | 2663 | 0.874 | |||
| X11851.21 | chr11:60254475:G:A | TREM2 | X11851.21 | chr11:60177107:C:T | 2663 | 0.999 | |
| TREM2 | X16300.4 | chr11:60177107:C:T | 2663 | 0.984 | |||
| TREM2 | X5635.66 | chr11:60177107:C:T | 2663 | 0.874 | |||
| X16300.4 | chr6:41161469:C:T | TREM2 | X11851.21 | chr6:41161395:C:T | 3426 | 0.037 | |
| TREM2 | X16300.4 | chr6:41161469:C:T | 3426 | 1.000 | |||
| TREM2 | X5635.66 | chr6:40974457:G:A | 2842 | 0.484 | |||
| chr11:60254475:G:A | TREM2 | X11851.21 | chr11:60177107:C:T | 2663 | 0.999 | ||
| TREM2 | X16300.4 | chr11:60177107:C:T | 2663 | 0.984 | |||
| TREM2 | X5635.66 | chr11:60177107:C:T | 2663 | 0.874 | |||
| YKL‐40 | X11104.13 | chr1:203183049:C:T | YKL‐40 | X11104.13 | chr1:203183673:T:C | 2766 | 0.996 |
Note: This table shows the posterior probabilities that a causal variant was shared between plasma and CSF in loci that were prioritized in both plasma and CSF. The PP.H4 column contains the prior probability that two phenotypes share a causal gene hit based on a coloc.abf analysis. Co‐localization analysis was performed between a plasma and CSF sentinel variant if they were within 1 Mb of each other. Values highlighted with bold are statistically significant at a PP.H4 ≥ 0.8. The AD top association data were derived from Bellenguez et al., 2022. All participants were European based on principal component analysis. CSF n = 3107; Plasma n = 2317.
Abbreviations: AD, Alzheimer's disease; CSF, cerebrospinal fluid; NFL, neurofilament light; NRGN, neurogranin; PP.H4, posterior probability of sharing causative variant; SNAP‐25, synaptosomal‐associated protein 25; TREM2, triggering receptor expressed on myeloid cells 2; YKL‐40, chitinase‐3‐like protein 1; VILIP, visinin‐like protein 1.
Both plasma APOE cis‐pQTLs identified in this study failed to demonstrate colocalization with all APOE region CSF pQTLs identified in this study (PP.H4 ≤ 3.1%; includes pQTLs for APOE, NfL, NRGN, VILIP‐1). These two plasma APOE pQTL sentinel variants also had low LD with all of the APOE region CSF pQTL sentinel variants (r 2 range = 0.01–0.12). However, the plasma NfL APOE region pQTL co‐localized with all APOE region CSF pQTLs (PP.H4 range = 0.99–1.00).
3.4. Plasma and CSF biomarkers as quantitative traits for genetics studies for AD
3.4.1. AD top hits co‐localize with CSF sentinel variants more frequently than plasma sentinel variants
Co‐localization was assessed between clinical AD risk loci and both plasma and CSF pQTLs for the non‐APOE regions using the study by Bellenguez and coauthors, 16 as the APOE region data were not available in this data set (Table 5; Table S9; Extended Results Section 5.2). AD sentinel variants co localized with both plasma and CSF for the following loci: the GRN AD hit associated with a GRN pQTL and the MS4A4A and TREM2 AD hits associated with TREM2 pQTLs.
TABLE 5.
Co‐localization analysis between CSF or plasma and AD in Europeans.
| Plasma | CSF | ||||||
|---|---|---|---|---|---|---|---|
| Gene | Aptamer | Variant ID | Gene Nearest Hit | Number of Variants | PP.H4 | Number of Variants | PP.H4 |
| APOE | X2938.55 | chr19:44913034:C:T | APOE | 3030 | 0.017 | 2905 | 1.000 |
| chr19:44908684:T:C | APOE | 3023 | 0.017 | 2897 | 1.000 | ||
| X5312.49 | chr19:44908822:C:T | APOE | 3024 | 6.960 × 10−16 | 2898 | 1.000 | |
| chr19:44908684:T:C | APOE | 3023 | 6.960 × 10−16 | 2897 | 1.000 | ||
| CLU | X24941.14 | chr19:40601522:C:T | LTBP4 | 3446 | 0.010 | 3487 | 0.007 |
| GRN | X4992.49 | chr1:109275908:C:T | CELSR2 | 3105 | 0.027 | 3093 | 0.018 |
| X4992.49 | chr17:44352876:C:T | GRN | 2705 | 1.000 | 2578 | 1.000 | |
| NFL | X10082.251 | chr5:14278964:T:C | TRIO | 3633 | 0.033 | 3746 | 0.082 |
| chr19:44908684:T:C | APOE | 3023 | 1.000 | 2897 | 1.000 | ||
| NRGN | X18303.39 | chr10:11676245:G:A | ENSG00000271046 | 3988 | 0.029 | 4269 | 0.014 |
| chr19:44888997:C:T | APOE | 3048 | 0.044 | 2945 | 1.000 | ||
| TREM2 | X5635.66 | chr6:40974457:G:A | UNC5CL | 4349 | 0.999 | 4437 | 0.848 |
| chr11:60177107:C:T | MS4A6A | 3425 | 0.920 | 3404 | 1.000 | ||
| chr11:60254475:G:A | MS4A4A | 3424 | 0.920 | 3328 | 1.000 | ||
| X11851.21 | chr6:41161395:C:T | TREM2 | 4157 | 0.065 | 4185 | 0.555 | |
| chr11:60177107:C:T | MS4A6A | 3425 | 0.999 | 3404 | 1.000 | ||
| chr11:60254475:G:A | MS4A4A | 3424 | 0.999 | 3328 | 1.000 | ||
| X16300.4 | chr6:41161469:C:T | TREM2 | 4157 | 0.223 | 4187 | 0.539 | |
| chr11:60177107:C:T | MS4A6A | 3428 | 0.994 | 3404 | 1.000 | ||
| chr11:60254475:G:A | MS4A4A | 3427 | 0.990 | 3328 | 1.000 | ||
| VILIP‐1 | X13522.20 | chr19:44919589:G:A | APOE | 3017 | 0.054 | 2899 | 1.000 |
Note: This table shows the posterior probabilities that a causal variant was shared between plasma or CSF and AD. PP.H4 represents the probability that two phenotypes share a causal gene hit based on a coloc.abf analysis. Co‐localization analysis was performed between a plasma, or a CSF, and an AD sentinel variant if they were within 1 Mb of each other. Values highlighted with bold are statistically significant at a PP.H4 ≥ 0.8 The AD top association data were derived from Bellenguez et al., 2022. All participants were European based on principal component analysis. CSF n = 3107; Plasma n = 2317.
Abbreviations: AD, Alzheimer's disease; CSF, cerebrospinal fluid; NFL, neurofilament light; NRGN, neurogranin; PP.H4, posterior probability of sharing a causative variant; TREM2, triggering receptor expressed on myeloid cells 2; VILIP, visinin‐like protein 1.
Although AD top hits co‐localized with GRN sentinel variants in the GRN region on chromosome 17 in both tissues, the GRN trans‐pQTL in the chr1 PSRC1 region failed to co‐localize with AD hits in both plasma (PP.H4 = 0.03) and CSF (PP.H4 = 0.02). However, this chr1 pQTL is close to the SORT1 gene that was prioritized in the study by Bellenguez et al. 16
Co‐localization analysis was also performed between AD and protein level sentinel variants in the APOE locus using the data set from Schwartzentruber and coauthors 17 (Table 5; Table 9). AD top hits co‐localized with all proteins with a CSF pQTL on chromosome 19: APOE, NfL, NRGN, and VILIP‐1. In plasma, only the NfL sentinel variant co‐localized with an AD top hit. Thus in the APOE region, CSF and AD top hits co‐localized more frequently than plasma and AD top hits.
3.4.2. CSF protein levels predict AD status more accurately than plasma protein levels
ROC analyses were performed to identify the predictive value of plasma and CSF protein levels on clinical AD status (Tables S10, S11; Figure 3). The p‐value threshold for significance was set at p ≤ 0.003 based on a Bonferroni correction. CSF protein levels were significantly better predictors of AD status than plasma protein levels for GFAP, GRN, NfL, NRGN, TREM2 (X16300.4), and VILIP‐1, whereas plasma protein levels of none of the assessed proteins were significantly better predictors of AD status compared to CSF protein levels (Tables S10, S11; Figure 3; Figure S19). These results were replicated when performing the ROC analyses on only individuals who had both CSF and plasma measurements within a 6 month period (Figure S19). The largest differences in area under the curve (AUC) between plasma and CSF were found for NfL (ΔAUC in plasma compared to CSF = −0.15, p = 2.33 × 10−5), VILIP‐1 (ΔAUC = −0.13, p = 2.09 × 10−8), NRGN (ΔAUC = −0.11, p = 2.91 × 10−12), and GRN (ΔAUC = −0.07, p = 3.00 × 10−3). NfL protein levels were the most accurate predictor of AD status in both plasma (AUC = 0.60) and CSF (AUC = 0.76). TREM2 levels showed similar AD status predictive accuracy between plasma and CSF for two of TREM2 aptamers, whereas TREM2 (X16300.4) showed a significantly higher predictive ability in CSF (CSF AUC = 0.59) than plasma (plasma AUC = 0.50; p = 0.001).
FIGURE 3.
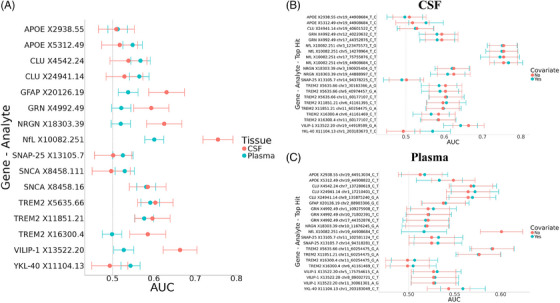
Predictive ability of protein aptamer levels on AD status in CSF and plasma in Europeans. (A) This dot plot shows the predictive ability of protein aptamer levels on AD status in plasma and CSF without covariates included. (B) This dot plot shows the predictive ability of protein aptamer levels on AD status in CSF with a top hit included as a covariate. The top hits were included by regressing protein aptamer levels on top hit genotype and then running an ROC analysis with AD status as the predictor and the residuals of the protein aptamer‐top hit model as the predictor. (C) This dot plots shows the predictive ability of protein aptamer levels on AD status in plasma with a top hit included as a covariate. For Plots B and C, the no‐covariate model is plotted for comparison and color indicates the presence or lack of a top hit covariate. Error bars represent 95% confidence intervals of the AUC. Protein aptamer levels were log10 transformed before analysis. All participants were European in ethnicity based on principal component analysis. CSF n = 800; Plasma n = 2176. AD, Alzheimer's disease; AUC, area under the curve; CSF, cerebrospinal fluid; ROC, receiver‐operating characteristic.
To assess if adding genetics to the prediction models improved the predictive power of these proteins, a separate set of ROC analyses was performed that controlled for the top hit genotype in each locus (Table S11). Only one of the ROC analyses resulted in a significant difference in predictive ability between the covariate‐free and top hit‐containing models; when controlling for the sentinel variant chr19:44908684:T:C, plasma NfL showed a decrease in AD status predictive ability (∆AUC = −0.08, p = 9.68 × 10−7). The CSF YKL‐40 protein level predictive abilities showed a similar change in AUC when controlling for chr1:203183673:T:C (∆AUC = 0.08, p = 0.02).
3.4.3. AD PRS correlates weakly with both plasma and CSF protein levels
Correlations for both plasma and CSF protein levels with six clinical AD PRS scores (with or without APOE included and using three different variant‐inclusions thresholds) were performed to determine if the overall AD genetic architecture correlates with protein levels (Table S12; Figure S20). Most of the correlations failed to reach significance. Plasma NfL showed correlation with an AD PRS only when correlated against the PRS that included APOE (5 × 10−5 threshold: r = −0.33, p = 1.10 × 10−65; 5 × 10−8 threshold: r = −0.38, p = 5.55 × 10−91).
TREM2 protein levels correlated with several of the PRSs across both tissues (Table S12; Figure S20; plasma: r ≤ −0.08, p ≤ 3.38 × 10−5; CSF: r ≤ −0.14, p ≤ 3.73 × 10−5). CSF levels of TREM2 protein aptamers tended to correlate with the PRS about three times as strongly as the plasma levels of these aptamers across statistical significance‐inclusion thresholds for the APOE‐including PRS. However, correlations were relatively similar with the non‐APOE PRS between TREM2 plasma and CSF protein aptamers. As a note, TREM2 (X11851.21) showed a four times stronger correlation with the 5 × 10−8 non‐APOE PRS in plasma (r = −0.12, p = 4.45 × 10−9) compared to CSF (r = −0.03, p = 0.41)
4. DISCUSSION
The goal of the present study was to (1) identify potential functional genes that act as pQTLs for AD‐relevant proteins in two key diagnostic tissues; (2) compare protein levels between CSF and plasma, and (3) determine what tissue protein levels represent better endophenotypes for genetic studies.
Previous studies found overlapping and tissue‐specific pQTLs between plasma and CSF, 10 , 28 which was also found in this study. pQTLs were found for most of the assessed proteins in both plasma and CSF, with a total of 18 loci in plasma and 16 in CSF. Six of the loci were shared across tissues for the same protein (two trans: MS4A4A with TREM2 and APOE with NfL; four cis: GRN, SNAP‐25, TREM2, YKL‐40) indicating that shared genetic regulators across multiple tissues, 8 and suggesting some genetic overlap across tissues, although that overlap seems to be driven by either pleiotropic regions (APOE) or cis‐signals. These data also suggest that some plasma‐derived biomarkers of AD may be appropriate for genetic studies of AD.
A notable finding is with respect to the two independent signals in plasma for apoE protein levels in the APOE locus (chr19:44913034:C:T, p = 4.77 × 10−19; chr19:44908822:C:T, p = 1.95 × 10−24; r 2 = 0.04). One of these variants (chr19:44913034:C:T) has weak LD with both APOE ε2 (r 2 = 0.04) and APOE4 ε4 (r 2 = 0.12), and does not co‐localize with the either of these alleles, suggesting that this is an independent signal. The finding is further notable because this variant is also associated with AD risk. 29 Functional in silico analyses indicated that the variant is an eQTL for APOC1, which is 3′’ from APOE, in the brain cortex; thus, we propose that APOC1 is the functional gene for this plasma APOE signal. A recent study demonstrated that the APOE locus was also associated with sTREM2 levels. 30 Wang and colleagues 31 performed QTL mapping and functional analyses, and for sTREM2 they reported that NECTIN2, not APOE or APOC1, was driving the association with sTREM2. The variant identified in the present study (chr19:44913034:C:T) is not in LD with the variant they found to be associated with sTREM2 (rs11666329; r 2 = 2.00 × 10−3). This indicates that the signal we have identified is yet another independent signal in the APOE region that is associated with AD‐related phenotypes. Previous studies also indicate that other genes in this region, beyond APOE, such as NECTIN2 30 or APOC1, 32 may be implicated in AD. The mechanism by which APOC1 affects APOE levels could be through its inhibitory interactions in plasma with APOE, and other apolipoproteins, 33 such that decreased function could lead to excessive APOE activity that leads to excessive deposition of lipids in cells. Taken together, these results emphasize that it is possible to have independent cis‐signals in the same locus across tissues, pointing to tissue‐specific regulatory mechanisms.
One novel plasma pQTL and four novel CSF pQTLs were identified. The functional genes nominated for some of these pQTLs are known to be part of biological and/or pathological pathways. With respect to the novel plasma pQTL, the IGKV4‐1 gene on chromosome 2, proposed as the functional gene for the GFAP pQTL, is implicated in the immune system and is associated with the complement cascade and B‐cell receptor signaling. 34 The genes that are most co‐expressed with IGKV4‐1 are associated with the microglia pathogen phagocytosis pathway (WP3937), and tend to be microglia enriched, 34 suggesting that that IGKV4‐1 is associated with both AD and GFAP expression through immune activation.
The proposed functional gene for the novel CSF NRGN pQTL, CCDC50, has been found to be associated with AD, indicating a potential biological pathway linking CCDC50 neurogranin (NRGN), and AD. 35 LTBP4, the proposed functional gene for the CLU pQTL locus on chromosome 19 (over 4 Mb from APOE), has been found to be associated with brain volume 35 and Duchenne muscular dystrophy. 36 Thus we have identified pQTLs that have biological and functional relevance to neurodegenerative disease that should be pursued further.
Plasma and CSF shared pQTLs in several loci, as reported previously. 10 , 28 Co‐localization analyses indicated that the same variant was driving the signal in both plasma and CSF for GRN, NfL, SNAP‐25, TREM2, and YKL‐40 (Table 4). There was a lack of co‐localization between plasma and CSF pQTLs in the APOE locus as discussed above. CLU, another key AD protein, and NRGN also had trans pQTLs on different chromosomes between tissues. These data indicate that there are likely differing tissue‐specific regulatory mechanisms that regulate protein levels.
Previous studies have evaluated protein correlations between plasma and CSF biomarkers of AD. 37 , 38 , 39 , 40 , 41 , 42 , 43 Some of these studies indicate a poor correlation between plasma and CSF biomarkers, 39 and some indicate a strong correlation, 41 but these studies only looked at one or two biomarkers. In our study, only two of the assessed proteins (TREM2 r ≥ 0.349; YLK‐40 r = 0.62) showed significant correlations in protein levels between plasma and CSF. This differs from previous studies, including some that found plasma NfL correlated relatively strongly with CSF NfL levels (r = 0.64‐0.86; sample size 78‐188), 38 , 40 , 43 and another study that found a poor correlation between sTREM2 levels between plasma and CSF (r = 0.04; n = 180) 44 ; however, these other studies were done in smaller sample sizes. Furthermore, proteins within the same tissue tended to correlate stronger than the same protein across tissues (see Extended Results Section 5.3).
In addition, we found that in some cases the protein correlation across tissue depend on disease status. NRGN levels showed a six times greater correlation across tissues in patients with AD than in controls. A higher correlation in cases indicates that expression levels are becoming increasingly similar between tissues as AD progresses, which is important because CSF NRGN has been linked to AD progression. 45 On the other hand, the TREM2 across‐tissue correlation was found in controls but not cases, and the across‐tissue correlation for YKL‐40 was not affected by disease status. 4 These changes in the across‐tissue protein correlations might reflect changes in the biology of the disease or even inform us about what disease stage those biomarkers may be tagging. Based on this, we would hypothesize that sTREM2 is tagging early stages (correlation only on controls) and NRGN is tagging later stages or tagging progression (correlation only in cases). However, additional studies and comparison are needed to fully determine what specific phenotype and biological process these biomarkers are capturing.
It is important to compare plasma and CSF proteins as biomarkers of AD. Previous studies perform such analyses but many of these studies look only at Aß, tau, and/or NfL, only in one tissue, and very few have compared across tissues. The present results indicate that, for the assessed proteins, CSF biomarkers would be more effective in determining AD status. However, it is important to note that this study did not include p‐tau217, which has been shown to be a very good plasma biomarker. 46 In our analyses, the CSF protein levels would predict AD status more accurately than the plasma‐derived version. Despite this, NfL showcased its effectiveness as an AD biomarker, as it was the most accurate predictor of AD status across all assessed proteins in both CSF (AUC = 0.76; max non‐NfL AUC = 0.66 [VILIP‐1]) and plasma (AUC = 0.60; max non‐NfL AUC = 0.59 [TREM2]). Combining NfL with other biomarkers, such as p‐tau181 or 217, to predict AD status may have higher predictive power than each marker alone.
There are some limitations to consider in our study. The present study analyzed several proteins associated with AD, but did not include Aß (Aß40, 42, 38) or tau proteoforms (p‐tau181, 217, 231), which are the most reliable fluid biomarkers for AD in both CSF and plasma. 47 However, these proteoforms are unavailable in the SOMAscan7K platform that was used for analysis. Another limitation of our study is that participants were considered to have AD based on clinical status and not biomarker evaluation. Biomarker status was available for the CSF samples and has been used before in previous studies stratifying by biomarker. 10 , 11 , 13 , 48 However, this was not the case for plasma, as the proteins necessary to perform the amyloid/tau/neurodegeneration (ATN) classification were not available. Note that we also did not assess the possibility of confounding from other neurodegenerative diseases. Another limitation is related to the lack of correlation between plasma and CSF GFAP levels of GFAP was identified, which is different from a previous study that found a significant correlation in levels (r = 0.40). 43 This inconsistency with previous studies might be due to known protein measurement issues with the SOMAscan7K assay when compared to immunoassays. 13
The results of the present study are compelling as they demonstrate that, despite substantial overlap in genetic pQTLs, CSF and plasma protein levels do not correlate significantly. In addition, novel pQTLs were identified in both plasma and CSF that should be further investigated to identify potential biological pathways involved in AD, and to potentially identify AD treatment targets. CSF protein levels are better predictors of AD status across many of the assessed proteins, indicating that CSF is the preferred tissue to draw AD biomarkers.
CONFLICT OF INTEREST STATEMENT
Randall Bateman: R.B. receives grants from many different institutions including the National Institute on Aging (NIA), the Alzheimer's Association (AA), and Biogen. R.B. has equity interest in, and receives income from, C2N Diagnostics. R.B. receives drugs and services from Eisai, Janssen, and Hoffman La Roche. Carlos Cruchaga: C.C. has received research support from GLAXOSMITHKLINE (GSK) and EISAI. The funders of the study had no role in the collection, analysis, or interpretation of data; in the writing of the report; or in the decision to submit the paper for publication. C.C. is a member of the advisory board for Circular Genomics and owns stocks. DIAN: DIAN is funded by National Institutes of Health (NIH) Grant #5U19AG032438 and the AA grant – SG‐20‐690363. Priyanka Gorijala: P.G. has no conflicts to declare. Menghan Liu: M.L. has no conflicts to declare. Thomas W. Marsh: T.W.M. has no conflicts to declare. John C. Morris: J.C.M is funded by NIH grants # P30 AG066444; P01AG003991; P01AG026276; neither Dr. Morris nor his family owns stock or has equity interest (outside of mutual funds or other externally directed accounts) in any pharmaceutical or biotechnology company. Pau Pastor: P.T. has no conflicts to declare. Susan E. Schindler: S.E.S. has no conflicts to declare. Yun Ju Sung: has no conflicts to declare. Jigyasha Timsina: J.T. has no conflicts to declare. Daniel Western: D.W. has no conflicts to declare. Chengran Yang: C.Y. has no conflicts to declare. Author disclosures are available in the supporting information.
CONSENT STATEMENT
The ethics committee and institutional review board of Washington University School of Medicine in St. Louis approved this study. All participants provided informed consent for all data used in the present study.
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
We thank all the participants and their families, as such research would not be possible without their contributions. We would also like to thank all the researchers and staff who collected and prepared the data analyzed in the present study. This work was supported by grants from the National Institutes of Health (NIH): R01AG044546 (C.C.), P30AG066444 (J.C.M.), P01AG003991 (J.C.M.), P01AG026276 (J.C.M.), RF1AG053303 (C.C.), RF1AG058501 (C.C.), U01AG058922 (C.C.), RF1AG074007 (Y.J.S.), R00AG062723 (Laura Ibañez [L.I.]), P30 AG066515 (T.W.C., M.D.G.); the Chan Zuckerberg Initiative (CZI); the Michael J. Fox Foundation (L.I.; C.C.), the Department of Defense (L.I.‐ W81XWH2010849 A); the Alzheimer's Association Zenith Fellows Award (ZEN‐22‐848604, awarded to C.C.); and the Bright Focus Foundation (A2021033S, LI). GlaxoSmithKline (GSK) provided funding to support the analyses performed in this study. The recruitment and clinical characterization of research participants at Washington University were supported by NIH P30AG066444 (J.C.M.), P01AG03991 (J.C.M.), and P01AG026276 (J.C.M.). This work was supported by access to equipment made possible by the Hope Center for Neurological Disorders, the NeuroGenomics and Informatics Center (NGI: https://neurogenomics.wustl.edu/), and the Departments of Neurology and Psychiatry at Washington University School of Medicine. DIAN: Data collection and sharing for this project was supported by The Dominantly Inherited Alzheimer Network (DIAN, U19AG032438) funded by the National Institute on Aging (NIA), the Alzheimer's Association (SG‐20‐690363‐DIAN), the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Nseurological Research (FLENI), partial support by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), Spanish Institute of Health Carlos III (ISCIII), Canadian Institutes of Health Research (CIHR), Canadian Consortium of Neurodegeneration and Aging, Brain Canada Foundation, and Fonds de Recherche du Québec Santé. This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. We acknowledge the altruism of the participants and their families and contributions of the DIAN research and support staff at each of the participating sites for their contributions to this study. ADNI: Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) NIH Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the NIA, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. ADC Olink proteomic data is part of the neurodegeneration research program of Amsterdam Neuroscience and was supported by: Alzheimer Nederland (WE.03‐2018‐05, MC and CT) and Selfridges Group Foundation (NR170065, MC and CT). M.C. is supported by the attraction talent fellowship of Comunidad de Madrid (2018‐T2/BMD‐11885) and “PROYECTOS I+D+I – 2020”‐ Retos de investigación from the Ministerio Español de Ciencia e innovación (PID2020‐115613RA‐I00). Alzheimer Center Amsterdam is supported by Stichting Alzheimer Nederland and Stichting VUmc fonds. Research of CET is supported by the European Commission (Marie Curie International Training Network, grant agreement No 860197 (MIRIADE), Innovative Medicines Initiatives 3TR (Horizon 2020, grant no 831434) EPND (IMI 2 Joint Undertaking (JU) under grant agreement No. 101034344grant no) and JPND (bPRIDE), National MS Society (Progressive MS alliance) and Health Holland, the Dutch Research Council (ZonMW), Alzheimer Drug Discovery Foundation, The Selfridges Group Foundation, Alzheimer Netherlands, Alzheimer Association. CT is recipient of ABOARD, which is a public‐private partnership receiving funding from ZonMW (#73305095007) and Health∼Holland, Topsector Life Sciences & Health (PPP‐allowance; #LSHM20106). ABOARD also receives funding from Edwin Bouw Fonds and Gieskes‐Strijbisfonds. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Marsh TW, Western D, Timsina J, et al. A genetic and proteomic comparison of key AD biomarkers across tissues. Alzheimer's Dement. 2024;20:6423–6440. 10.1002/alz.14139
REFERENCES
- 1. 2023 Alzheimer's disease facts and figures. Alzheimers Dement. 2023;19(4):1598‐1695. doi: 10.1002/alz.13016 [DOI] [PubMed] [Google Scholar]
- 2. Simrén J, Elmgren A, Blennow K, Zetterberg H. Chapter six—fluid biomarkers in Alzheimer's disease. Adv Clin Chem. 2023:249‐281. [DOI] [PubMed] [Google Scholar]
- 3. Knopman DS, Amieva H, Petersen RC, et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7(1):33. doi: 10.1038/s41572-021-00269-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Molinuevo JL, Ayton S, Batrla R, et al. Current state of Alzheimer's fluid biomarkers. Acta Neuropathol. 2018;136(6):821‐853. doi: 10.1007/s00401-018-1932-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schindler SE, Li Y, Todd KW, et al. Emerging cerebrospinal fluid biomarkers in autosomal dominant Alzheimer's disease. Alzheimers Dement. 2019;15(5):655‐665. doi: 10.1016/j.jalz.2018.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bellenguez C, Kucukali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54(4):412‐436. doi: 10.1038/s41588-022-01024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood‐brain barrier: from physiology to disease and back. Physiol Rev. 2019;99(1):21‐78. doi: 10.1152/physrev.00050.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bradley J, Gorijala P, Schindler SE, et al. Genetic architecture of plasma Alzheimer disease biomarkers. Hum Mol Genet. 2023;32(15):2532‐2543. doi: 10.1093/hmg/ddad087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barthélemy NR, Salvadó G, Schindler S, et al. Highly accurate blood test for Alzheimer's disease comparable or superior to clinical CSF tests. Nat Med. 2024:30:1085‐1095. doi: 10.1038/s41591-024-02869-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cruchaga C, Western D, Timsina J, et al. Proteogenomic analysis of human cerebrospinal fluid identifies neurologically relevant regulation and informs causal proteins for Alzheimer's disease. Res Sq. 2023. doi: 10.21203/rs.3.rs-2814616/v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ali M, Timsina J, Wang L, et al. Multi‐cohort cerebrospinal fluid proteomics identifies robust molecular signatures for asymptomatic and symptomatic Alzheimer's disease. Alzheimers Dementia. 2023;19(S15):e077212. doi: 10.1002/alz.077212 [DOI] [Google Scholar]
- 12. Yang CGP, Timsina J, Wang L, et al. European and African stratified plasma protein‐QTL and metabolite‐QTL analyses identify ancestry‐specific T2D effector proteins and metabolites. AJHG. 2023. [Google Scholar]
- 13. Timsina J, Gomez‐Fonseca D, Wang L, et al. Comparative analysis of Alzheimer's disease cerebrospinal fluid biomarkers measurement by multiplex SOMAscan platform and immunoassay‐based approach. J Alzheimers Dis. 2022;89(1):193‐207. doi: 10.3233/JAD-220399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Álvarez I, Diez‐Fairen M, Aguilar M, et al. Added value of cerebrospinal fluid multimarker analysis in diagnosis and progression of dementia. Eur J Neurol. 2021;28(4):1142‐1152. doi: 10.1111/ene.14658 [DOI] [PubMed] [Google Scholar]
- 15. Initiative PPM. The Parkinson Progression Marker Initiative (PPMI). Prog Neurobiol. 2011;95(4):629‐635. doi: 10.1016/j.pneurobio.2011.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54(4):412‐436. doi: 10.1038/s41588-022-01024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schwartzentruber J, Cooper S, Liu JZ, et al. Genome‐wide meta‐analysis, fine‐mapping and integrative prioritization implicate new Alzheimer's disease risk genes. Nat Genet. 2021;53(3):392‐402. doi: 10.1038/s41588-020-00776-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Candia J, Daya GN, Tanaka T, Ferrucci L, Walker KA. Assessment of variability in the plasma 7k SomaScan proteomics assay. Sci Rep. 2022;12(1):17147. doi: 10.1038/s41598-022-22116-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Purcell S, Neale B, Todd‐Brown K, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81(3):559‐575. doi: 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590(7845):290‐299. doi: 10.1038/s41586-021-03205-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Machiela MJ, Chanock SJ. LDlink: a web‐based application for exploring population‐specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31(21):3555‐3557. doi: 10.1093/bioinformatics/btv402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Klein N, Tsai EA, Vochteloo M, et al. Brain expression quantitative trait locus and network analyses reveal downstream effects and putative drivers for brain‐related diseases. Nat Genet. 2023;55(3):377‐388. doi: 10.1038/s41588-023-01300-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318‐1330. doi: 10.1126/science.aaz1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ferkingstad E, Sulem P, Atlason BA, et al. Large‐scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53(12):1712‐1721. doi: 10.1038/s41588-021-00978-w [DOI] [PubMed] [Google Scholar]
- 25. Choi SW, O'Reilly PF. PRSice‐2: polygenic risk score software for biobank‐scale data. GigaScience. 2019;8(7):giz082. doi: 10.1093/gigascience/giz082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robin X, Turck N, Hainard A, et al. pROC: an open‐source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011;12:77. doi: 10.1186/1471-2105-12-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wallace C. A more accurate method for colocalisation analysis allowing for multiple causal variants. PLoS Genet. 2021;17(9):e1009440. doi: 10.1371/journal.pgen.1009440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang C, Farias FHG, Ibanez L, et al. Genomic atlas of the proteome from brain, CSF and plasma prioritizes proteins implicated in neurological disorders. Nat Neurosci. 2021;24(9):1302‐1312. doi: 10.1038/s41593-021-00886-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marioni RE, Harris SE, Zhang Q, et al. GWAS on family history of Alzheimer's disease. Transl Psychiatry. 2018;8(1):99. doi: 10.1038/s41398-018-0150-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lihua W, Niko‐Petteri N, Daniel W, et al. Proteo‐genomics of soluble TREM2 in cerebrospinal fluid provides novel insights and identifies novel modulators for Alzheimer's disease. medRxiv. 2023:2023061423291409. doi: 10.1101/2023.06.14.23291409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang L, Nykänen N‐P, Western D, et al. Proteo‐genomics of soluble TREM2 in cerebrospinal fluid provides novel insights and identifies novel modulators for Alzheimer's disease. Mol Neurodegen. 2024;19(1):1. doi: 10.1186/s13024-023-00687-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou Q, Peng D, Yuan X, et al. APOE and APOC1 gene polymorphisms are associated with cognitive impairment progression in Chinese patients with late‐onset Alzheimer's disease. Neural Regen Res. 2014;9(6):653‐660. doi: 10.4103/1673-5374.130117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fuior EV, Gafencu AV. Apolipoprotein C1: its pleiotropic effects in lipid metabolism and beyond. Int J Mol Sci. 2019;20(23). doi: 10.3390/ijms20235939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie Z, Bailey A, Kuleshov MV, et al. Gene set knowledge discovery with enrichr. Curr Protoc. 2021;1(3):e90. doi: 10.1002/cpz1.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sollis E, Mosaku A, Abid A, et al. The NHGRI‐EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 2023;51(D1):D977‐d985. doi: 10.1093/nar/gkac1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stelzer G, Rosen N, Plaschkes I, et al. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics. 2016;54:1.30.1‐1.30.33. doi: 10.1002/cpbi.5 [DOI] [PubMed] [Google Scholar]
- 37. Bakker L, Köhler S, Eussen SJPM, et al. Correlations between kynurenines in plasma and CSF, and their relation to markers of Alzheimer's disease pathology. Brain Behav Immun. 2023;111:312‐319. doi: 10.1016/j.bbi.2023.04.015 [DOI] [PubMed] [Google Scholar]
- 38. Álvarez‐Sánchez L, Peña‐Bautista C, Ferré‐González L, et al. Assessment of plasma and cerebrospinal fluid biomarkers in different stages of Alzheimer's disease and frontotemporal dementia. Int J Mol Sci. 2023;24(2):1226. doi: 10.3390/ijms24021226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Le Bastard N, Aerts L, Leurs J, Blomme W, De Deyn PP, Engelborghs S. No correlation between time‐linked plasma and CSF Aβ levels. Neurochem Int. 2009;55(8):820‐825. doi: 10.1016/j.neuint.2009.08.006 [DOI] [PubMed] [Google Scholar]
- 40. Vrillon A, Ashton NJ, Karikari TK, et al. Comparison of CSF and plasma NfL and pNfH for Alzheimer's disease diagnosis: a memory clinic study. J Neurol. 2023;271(3):1297‐1310. doi: 10.1007/s00415-023-12066-6 [DOI] [PubMed] [Google Scholar]
- 41. Martínez‐Dubarbie F, Guerra‐Ruiz A, López‐García S, et al. Accuracy of plasma Aβ40, Aβ42, and p‐tau181 to detect CSF Alzheimer's pathological changes in cognitively unimpaired subjects using the Lumipulse automated platform. Alzheimers Res Ther. 2023;15(1):163. doi: 10.1186/s13195-023-01319-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Minaya MA, Mahali S, Iyer AK, et al. Conserved gene signatures shared among MAPT mutations reveal defects in calcium signaling. Front Mol Biosci. 2023;10:1051494. doi: 10.3389/fmolb.2023.1051494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Parvizi T, König T, Wurm R, et al. Real‐world applicability of glial fibrillary acidic protein and neurofilament light chain in Alzheimer's disease. Front Aging Neurosci. 2022;14:887498. doi: 10.3389/fnagi.2022.887498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Piccio L, Deming Y, Del‐Águila JL, et al. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016;131(6):925‐933. doi: 10.1007/s00401-016-1533-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Portelius E, Zetterberg H, Skillbäck T, et al. Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer's disease. Brain. 2015;138(11):3373‐3385. doi: 10.1093/brain/awv267. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rehman H, Ang TFA, Tao Q, et al. Comparison of commonly measured plasma and cerebrospinal fluid proteins and their significance for the characterization of cognitive impairment status. J Alzheimers Disease. 2024;97:621‐633. doi: 10.3233/JAD-230837 [DOI] [PubMed] [Google Scholar]
- 47. Kang JH, Korecka M, Lee EB, et al. Alzheimer disease biomarkers: moving from CSF to plasma for reliable detection of amyloid and tau pathology. Clin Chem. 2023;69(11):1247‐1259. doi: 10.1093/clinchem/hvad139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang C, Western D, Yang C, et al. Unique genetic architecture of CSF and brain metabolites pinpoints the novel targets for the traits of human wellness. Res Sq. 2023. doi: 10.21203/rs.3.rs-2923409/v1 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information