

ABSTRACT
Inspection of the genomes of bacterial pathogens indicates that their pathogenic potential relies, at least in part, on the activity of different elements that have been acquired by horizontal gene transfer from other (usually unknown) microorganisms. Similarly, in the case of resistance to antibiotics, besides mutation-driven resistance, the incorporation of novel resistance genes is a widespread evolutionary procedure for the acquisition of this phenotype. Current information in the field supports the idea that most (if not all) genes acquired by horizontal gene transfer by bacterial pathogens and contributing to their virulence potential or to antibiotic resistance originate in environmental, not human-pathogenic, microorganisms. Herein I discuss the potential functions that the genes that are dubbed virulence or antibiotic resistance genes may have in their original hosts in nonclinical, natural ecosystems. In addition, I discuss the potential bottlenecks modulating the transfer of virulence and antibiotic resistance determinants and the consequences in terms of speciation of acquiring one or another of both categories of genes. Finally, I propose that exaptation, a process by which a change of function is achieved by a change of habitat and not by changes in the element with the new functionality, is the basis of the evolution of virulence determinants and of antibiotic resistance genes.
Key words: Bacterial evolution, Horizontal gene transfer, Antibiotic resistance, Virulence, Microbiome, Bacterial pathogens, Shoort sighted evolution
INTRODUCTION
The evolution of living beings is a complex process, with a large degree of serendipity, in which the offspring displace the ancestors. Indeed, what we find in the current multicellular world, and more specifically in the animal world, are the last members of an evolutionary process; all other members in the same branch of the phylogenetic tree have disappeared. In this regard, most multicellular organisms can be considered as newcomers on Earth, which have appeared quite recently in evolutionary terms. Although there are still some progenitors that stand after the evolution of their siblings, the most common scenario for multicellular organisms is that ancestors disappear once the evolved progeny displace them (see the evolution of Homo sapiens). This type of recent evolution followed by extinction is not so frequent in the case of bacterial species, although it may have happened on some occasions (see the example of Yersinia described below). Indeed, the origin of different pathogens has been tracked to more than 100 million years ago, long before the human being (or an ancestor) was present on Earth (1). Despite this extremely long evolutionary time, which should have allowed for large diversification with the loss of ancestors, bacterial core genomes are remarkably stable. It could be expected that the allelic variants of bacterial genes should cover nearly the entire potential spectrum of synonymous mutations and even those nonsynonymous mutations without substantial associated fitness costs. However, today we can use multilocus sequence typing for distinguishing among different clones in bacterial populations, under the assumption that, at least for several of the core genome genes, fixation of mutations is not a frequent event (2). It then seems that, unless there is a major change in habitat, mutation-driven evolution is not the most important process in the speciation of bacteria in general, and in particular in the case of bacterial pathogens. A major force in such evolution, however, would be the acquisition of genetic elements (3–5), what has been dubbed evolution in quantum leaps (6). These acquired genes constitute the accessory genome of an organism and the pangenome of a given species (7).
If speciation is driven by the acquisition of novel genetic material, a full understanding of this process requires deciphering which is the origin of these elements, in particular those involved in antibiotic resistance and bacterial virulence; which are the bottlenecks involved in their transmission; and which are the consequences of the bacterial physiology of acquiring novel, adaptive traits through horizontal gene transfer (HGT).
If we take into consideration that most bacterial species evolved before the emergence of multicellularity and that the main process in their evolution is the shuffling from one bacterium to another of preexisting genes, a suitable conclusion would be that bacterial genes evolved in a unicellular world. In other words, elements that are dubbed virulence determinants or antibiotic resistance genes must have important functions besides those that they can play in human pathogens (8–12). These functions must be associated with the habitat of their original hosts, from which they have been transferred to the bacterial pathogens. Taking into consideration the ancient origin of bacterial genes, which track to long before the emergence of human beings, the functions of these determinants should have ecological value in nonclinical, environmental ecosystems where the donors of virulence determinants and antibiotic resistance elements have evolved.
DIFFERENCES IN BACTERIAL SPECIES: GENERALISTS, SPECIALISTS, AND MULTISPECIALISTS
The recent capability of exploring a large number of genomes of different isolates belonging to the same bacterial species allows determination of which parts of the genome are shared by all (or most) members of such species and which parts are specific for a subset of members of the species and thus constitute the accessory genome (13–15). In principle, this may allow us to establish different categories of microorganisms. There are species whose members present large core genomes that allow them to colonize different ecosystems. Among such ecosystems, one of them can be the human host, and because of this some opportunistic pathogens, such as Pseudomonas aeruginosa or Stenotrophomonas maltophilia, are environmental bacteria that can infect immunocompromised or debilitated patients using a set of virulence factors that are present in all members of the species (16). Indeed, it has been shown that environmental and clinical P. aeruginosa isolates are genetically and physiologically equivalent, indicating that this species does not present two phylogenetic branches, one composed of environmental microorganisms and another formed by virulent isolates (17–19). In addition, it has been shown that this microorganism can use the same virulence determinants for infecting different hosts, from plants to humans (20–22). Although some virulence determinants, such as the exotoxin ExoU, may have been acquired through HGT in P. aeruginosa (23) and HGT-acquired resistance genes are present in antibiotic-resistant isolates of this species (24, 25), most of its virulence and antibiotic resistance repertoire belongs to its core genome and is not derived from HGT-driven evolution.
Another category of pathogens is the specialists. This group comprises species in which most if not all members of the species are virulent for a few or specific hosts. Here it is worth distinguishing two groups. One is formed by pathogens, usually intracellular, presenting small genomes and for which HGT is not a major force in their evolution. Examples of this category are Mycobacterium tuberculosis and Mycobacterium leprae; these types of pathogens frequently colonize a single habitat (such as the mammal cell) and, as happens with endosymbionts (26–29), their pathway of evolution is genome reduction more than acquisition of novel traits. These microorganisms are well-adapted pathogens that have coevolved with their human hosts (30, 31). Whether or not this speciation process may end in commensalist or endosymbiotic behavior is beyond our current knowledge.
A different situation occurs with pathogens such as Yersenia pestis, which has evolved toward virulence from a nonvirulent ancestor by means of a pathway in which HGT has played a major role (32–38). Nevertheless, all members of the species contain the same HGT-acquired elements, which can be considered as belonging to the core genome of the microorganism, despite the fact that its current structure and the arrangement of genes within is the consequence of different HGT events. In addition, as in the case for the aforementioned intracellular pathogens, the evolution of this species toward infection has produced deadaptation for growing in other habitats, with the consequence that Y. pestis is a specialist, able to colonize just a small subset of different habitats, the most important ones being the different hosts involved in the transmission of and infection by this pathogen (see below). Classical professional pathogens such as Brucella melitensis and M. tuberculosis that do not present a clear habitat outside their hosts are also likely within this category.
A final category may consist of the multispecialists. This category would include species that present ecotypes each capable of colonizing a subset of habitats. One of them is Escherichia coli; this bacterial species is formed by different clonal complexes, some of them commensals (of human or animals), others virulent, and some with an environmental habitat (39–46). Each of the groups has acquired its specific properties (for instance, virulence or capability of colonizing a specific host) through the incorporation of novel traits by means of HGT. Even more, in the case of pathogenic E. coli strains, there exist different clonal complexes producing different categories of infections in different hosts, each presenting a specific repertoire of virulence determinants (47). In this case, while the species as a whole can colonize a large number of habitats, each clonal complex presents specific ecological characteristics (niche specialization), and hence the species can be considered as a multispecialist. It is worth discussing whether some of these specific clonal complexes should be considered as independent species (such as the specialist Shigella when compared with some [likely also specialist] E. coli clonal complexes) themselves or, alternatively, whether their genomic structure and ecological behavior suggests they are in the route of speciation.
Based on their ecological behavior, there are then two broad categories of bacteria: those able to colonize a small range of habitats (specialists) and those with an ample distribution among ecosystems. For the latter, two categories emerge. If all members of the species can colonize all (or most) habitats, these bacteria are generalists. However, if the species presents different ecotypes, each able to differentially colonize a small range of habitats, I propose that these bacteria should be considered as multispecialists.
SPECIATION AND SHORT-SIGHTED EVOLUTION
Evolution is frequently supposed to work as an arrow in time, where each of the selected steps is consolidated and serves for further evolution toward diversification, which leads to the emergence of novel species. This classical view of gradual evolution does not usually fit well with the evolution of bacterial pathogens (and of microorganisms in general). For this category of living beings, the most important process in speciation is HGT, a mechanism by which bacteria acquire as a whole the required traits to colonize a novel habitat (15). This evolution in quantum leaps (48) is possible only in organisms, such as bacterial pathogens, where HGT is a frequent, or at least a selectable, event. However, the fact that the development of virulence, and in several cases of antibiotic resistance, involves the acquisition of foreign DNA does not mean that mutation and intragenomic recombination are not relevant for the evolution of pathogens. As we will describe in more detail later, the acquisition by HGT of elements that allow the colonization of a novel habitat is usually followed by the selection of mutants presenting a metabolism better fitted to the characteristics of the new environment. In addition, genome reduction is expected in the case of specialized pathogens that do not require all the traits required for colonizing a large number of different niches. The speciation process in bacterial pathogens should then usually include a first HGT event that allows the pathogen to enter and colonize the new host, followed by further acquisition of novel genes and fine-tuning of the bacterial physiology by means of mutation and genome reduction (49).
It is important, however, to mention that the evolution of pathogens does not always lead to a speciation process. On several occasions, instead of an arrow, the evolution of microorganisms follows a circle in which bacteria acquire very similar mutations (or even mobile elements) when confronted with a new habitat/injury, but this adapted branch disappears when selection disappears as well (8, 50, 51). This scenario, which has been named short-sighted evolution (Fig. 1), is a frequent evolutionary pattern for generalist pathogens that can colonize different habitats as well as for pathogens confronted with strong (deadly) selective pressures such as the presence of antibiotics. They are able to evolve under the new situation, but when they return to their original habitat, they can be outcompeted by the nonevolved remaining population.
FIGURE 1.
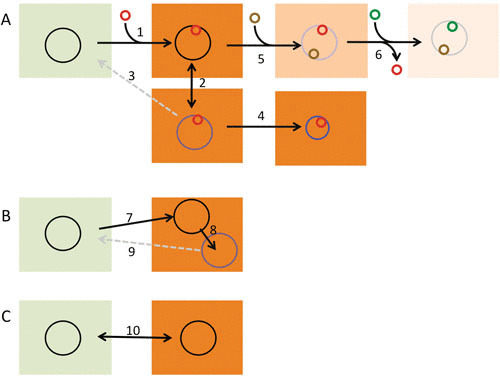
Evolutionary trajectories of bacterial pathogens. (A) The process of speciation of a pathogen (larger circles) such as Y. pestis. This process usually begins with the acquisition, by HGT, of a set of genes (red circle) that allow the shift of the pathogen’s habitat from the environment to an infected host (1). If the rate of transmission is high enough, the newborn pathogen will disseminate among different individuals (2) and evolve by different mechanisms that include mutation and eventually genome reduction (4). These evolutionary processes might cause the deadaptation of the pathogen to its original habitat, in which case the chances of the microorganism recolonizing natural ecosystems will be low (3). Once the organism is a pathogen, it can change host specificity by acquiring novel genes (5) and eventually by losing of determinants unneeded in the novel host (6). In all cases, the integration of the acquired elements into the preformed bacterial metabolic and regulatory networks will be tuned by mutation. (B) The process of short-sighted evolution of opportunistic pathogens with an environmental origin, like P. aeruginosa. These microorganisms infect patients, presenting a basal disease, using virulence determinants already encoded in their genomes (7). During chronic infection, the infective strain evolves mainly by mutation and genome rearrangements (8). However, since it only infects people with a basal disease, transmission rates are usually low, which precludes clonal expansion and further diversification. Since adaptation to the new host is of no value for colonizing the environmental habitat (9), this is a dead-end evolutionary process. (C) The evolution of pathogens such as V. cholerae that present virulence determinants with a dual role in the environment and for infections, in which case the colonization of one of these two habitats does not severely compromise the colonization of the other (10). Reproduced with permission from reference 8.
An example of this is the evolution of P. aeruginosa during long-term chronic infections in cystic fibrosis and chronic obstructive pulmonary disease patients. Different studies have shown that these patients are infected by a bacterial clone that evolves during infection and that a similar pattern of evolution is observed in most patients (52, 53). Nevertheless, although some epidemic clones have been described, particularly in countries where patients are not segregated (54–58), the primary infective strain is frequently different for each patient, and its phenotype corresponds to a nonevolved strain. This may mean that the evolved strains are outcompeted when they are released from the chronically infected host. This possibility has been explained by means of a source/sink (51) behavior of generalist pathogens. The source/sink hypothesis states that when an organism is present in a large habitat (source) and colonizes a smaller habitat (sink), the adaptation to the sink produces deadaptation to the source, in which case the deadapted (evolved) bacteria will be outcompeted when returning to the source habitat (51). This may happen in the case of cystic fibrosis or chronic obstructive pulmonary disease, in which interpatient clonal transmission is not very a frequent event in most countries. The evolved strains adapted to the infected host (sink) return to their natural ecosystem (source), where they are most likely outcompeted by the wild-type strains. However, in the case of proficient interhost transmission, the evolved strains do not return to the source and further evolution, eventually leading to speciation may happens.
Although the term “short-sighted evolution” applies mainly to mutation-driven evolution, the same situation may arise in the case of HGT. Indeed, ecologically valuable genes (such as those for antibiotic resistance) can be transferred, but the acquisition of novel genes may impose a fitness cost, a feature that has been discussed in detail for antibiotic resistance genes and much less for virulence determinants, in which case they will disappear in the absence of selection (59, 60).
It has been suggested that HGT is rare in natural ecosystems (61). However, for detecting such transfer events, enrichment and eventually fixation are needed, and for fixation, positive selection is required. It is thus possible that HGT is more common than supposed in natural ecosystems, but we do not at the moment have the tools required to quantify transient transmission events that are not fixed in the population under selection pressure.
Evolution is frequently considered as a step-way nonreturn process. However, current information supports the idea that bacteria can explore evolutionary pathways without fixing the novel acquired traits. These “futile cycles” of evolution, which have been dubbed short-sighted evolution, likely play a fundamental role in the fast adaptation of bacterial populations to strong selective pressures such as the use of antibiotics.
ORIGIN AND FUNCTIONS OF ANTIBIOTIC RESISTANCE GENES
The most commonly used definition of antibiotic resistance, and consequently of antibiotic resistance genes, comes from the clinical world. An organism is resistant if the likelihood of treatment success is low. The genes contributing to this phenotype are antibiotic resistance genes and the mutations involved resistance mutations. The finding that some genes can be transferred and confer resistance to new hosts led to the hypothesis that these genes should also play the same role in their original hosts, where they evolved before being transferred to bacterial pathogens by HGT. When looking to the origin of resistance, it has been proposed that, since antibiotic producers need to protect themselves from the action of the antimicrobials they produce, they should be the source of resistance genes acquired by bacterial pathogens (62). Nevertheless, none of the resistance genes present in antibiotic producers has been found yet in a mobile element in a bacterial pathogen. It is true that both types of microorganisms present resistance genes belonging to the same functional families, but in the few cases in which the origin of HGT-acquired resistance genes has been tracked, the original hosts harboring them were not antibiotic producers (63–65). This does not mean that antibiotic producers cannot be a source of antibiotic resistance; rather, these results expand the potential origin of a mobile resistance gene to any bacteria present on Earth (66, 67).
While the function of resistance genes in producers as antibiotic decontaminant elements seems clear (62), the situation is not the same for non-antibiotic producers. Even in the case of antibiotic-inactivating enzymes present in antibiotic producers, a double function for them has been proposed: in addition to being involved in the degradation of the produced toxic compound, they can be involved in the modification of metabolic intermediates along the biosynthetic pathway (62). Concerning nonproducers, while resistance genes can serve on occasion to circumvent the activity of antibiotic inhibitors produced by competitors (68), in several cases current information does not support such a role. This is the situation for antibiotic resistance genes present in gut commensals, since producers of the antimicrobials currently in use for clinical practice have never been found in the gut microbiota. We can cite, among several others, the multidrug resistance (MDR) efflux pump AcrAB-TolC, a major determinant of antibiotic resistance in Enterobacteriaceae, whose actual function in these microorganisms is resisting the activity of bile salts (69, 70), a set of antibacterial detergents commonly found in the gut. Another widespread family of antibiotic resistance determinants is formed by the AmpC-type β-lactamases (71). Some of them have been found in plasmids, but on several occasions ampC genes are chromosomally encoded genes that belong to the bacterial core genome. It has been described that, besides contributing to antibiotic resistance, these enzymes can be involved in the building up and the recycling of the peptidoglycan layer (72–74). The same applies for other antibiotic resistance determinants, such as some aminoglycoside-inactivating enzymes that can recognize an antibiotic substrate because of its structural similarity to a peptidoglycan intermediate, which is the actual physiological substrate of the enzyme (75).
MDR efflux pumps are the group of clinically relevant antibiotic resistance determinants (76–79) for which the functions in natural ecosystems besides resistance have been better studied. In addition to helping to resist the activity of host-produced antibacterial compounds such as bile salts or antimicrobial peptides, it has been shown that such pumps can extrude quorum-sensing signal molecules, biocides, or plant-produced compounds, which indicates that their functions go beyond resisting the action of the antimicrobials currently in use in clinical practice (80–82).
We can thus conclude that what we have dubbed antibiotic resistance determinants might have a function in their environmental original host non-related with their capability of providing resistance to industrial antibiotics. However, their original, functional substrates are similar to antibiotics and hence are capable of modifying (antibiotic-inactivating enzymes) or extruding them (efflux pumps).
As stated above, this situation expands the number of potential donors of resistance to any bacteria on Earth. Any protein able to interact with the antibiotic or with its target in such a way that the action of the antimicrobial is impaired might also be a resistance determinant, irrespectively of whether or not its original function was impeding the action of antibiotics (9, 83, 84).
ECOLOGICAL VALUE OF VIRULENCE DETERMINANTS IN NONCLINICAL ECOSYSTEMS
The understanding of the ecology and evolution toward virulence of pathogens requires determining the needs and the benefits of the adaptation to a new habitat, the infected host. As stated by Levin and Antia, “To pathogenic microparasites (viruses, bacteria, protozoa, or fungi), we and other mammals (living organisms at large) are little more than soft, thin-walled flasks of culture media” (85). In other words, the main reward of infection is gaining access to a new habitat that contains abundant and diversified nutrients (86) and where, at least in some cases (solid organ infection, prosthesis, bacteremia), the amounts of microbial competitors and of bacterial predators are low. For gaining access to the inside-host habitat, bacterial pathogens require two sets of elements. One of them is formed by those determinants required to resist the anti-infective host response. These elements include, among others, the innate immune response; the stomach’s acidic pH; the production of antimicrobial compounds such as bile salts, antimicrobial peptides, or fatty acids; the host microbiota itself, which has anticolonization properties; and, more recently, the use of antibiotics, which can be considered as an anti-infective response product of the cultural human evolution (87). In addition to interfering with the host response, bacteria must present a metabolism capable of coping with the physicochemical characteristics of the in-host habitat. To produce an infection, a bacterium requires growing at 37°C as well as at the oxygen tension and at the osmolarity of host organs. In addition, it requires making use of the nutrients present in the human body, including harboring efficient iron-uptake systems, since iron availability is scarce inside the human body (88, 89). Some of the elements may deal with both aspects required for producing a proficient infection; this is the case for bacterial proteases that are used by microbial pathogens for degrading proteins involved in defense against infection and for disrupting the extracellular matrix, which are also useful for providing nutrients such as amino acids and peptides.
Even when a bacterial pathogen has evolved with the human ancestors (90) or when the only modification in the pathogen is the acquisition of elements allowing a host change from another organism to humans, what can be measured is the time frame since the acquisition of virulence determinants occurred. Indeed, the evolutionary history of HGT-acquired virulence genes differs from that of ancestral (core genome) genes (4). This indicates that the primary evolution of these determinants themselves occurred before their acquisition by the pathogen through HGT. Consequently, virulence determinants may have evolved outside the infective habitat, and hence they can have (as antibiotic resistance determinants) other functions beyond conferring on bacterial pathogens the capability of infecting the human host (8).
Some of the so-called virulence factors are involved in regular processes of bacterial physiology; these include iron-uptake systems, catabolic pathways, and structures involved in cell attachment (91–94). All these traits are relevant at the point of infection, but are relevant as well in other habitats presenting similar physiological requirements; for example, iron availability is low in most ecosystems and similar nutrients as those found in the human body can be found in other ecosystems. Cell attachment is needed as well for colonizing surfaces, in particular when detaching forces are acting (as with water bodies). For instance, Vibrio cholerae requires the colonization factor GbpA for attaching to epithelial surfaces and proficiently colonizing the intestinal tract. This colonization factor allows as well the bacterial attachment to chitin-containing shells of crustaceans present in coastal waters, the natural environmental habitat of V. cholerae (95).
A different situation may occur in the case of virulence factors playing more-specific roles in bacterial/host interactions. These include, among others, specific secretion systems capable of injecting a bacterial effector or a toxin straight inside the eukaryotic cell. In this case, an effect on novel bacterial metabolic capabilities is not expected. Rather, they can be involved in general processes of interactions with multiple hosts. This is the case for P. aeruginosa, which produces a series of virulence determinants, such as siderophores, cyanide, proteases, or toxins, which are needed for infecting humans and also required for infecting plants, protozoans, worms, or insects (20–22, 96–99). If we take into consideration the evolutionary tree, it is conceivable that these virulence determinants evolved first for driving protozoan/bacterial interactions, likely before emergence of multicellularity, and have been coopted since for infecting other hosts, including humans. While prey/predator relationship might be in the basis of the evolution of these determinants, in other cases commensal interactions might play a role in their emergence. In favor of this possibility is the finding that several plant-associated bacteria present type III secretion systems, which function in mediating the plant/bacteria interactions (100). Coming back to the unicellular world, it is important to note that Legionella pneumophila, the causal agent of Legionnaires’ disease, which is able to multiply into alveolar macrophages, is also capable of replicating inside amoebas and ciliated protozoa in its natural water habitat. Further, the mechanisms used by L. pneumophila for their intracellular growth are the same in macrophages and in its unicellular environmental hosts (101). Similarly, bacterial toxins that can harm the infected human host may have evolved to play a role in mediating predator/prey interactions. This may be the situation for the Shiga toxin, which, besides being a highly relevant virulence factor, allows bacteria to evade predation by the ciliate Tetrahymena thermophila (102, 103); or the Listeria monocytogenes listeriolysin O, which allows the intracellular survival of the pathogen during infection and induces lymphocytes’ apoptosis. This major L. monocytogenes virulence determinant is needed for the survival of bacteria from predation by the bacterivorous ciliate Tetrahymena pyriformis (104).
To conclude, virulence determinants encoded in pathogenicity islands, which have been acquired by bacterial pathogens along their evolution, might be involved in intercellular interactions, including prey/predator and commensal interactions, as well as in regular aspects of bacterial metabolism in the nonclinical (environmental) ecosystems where the original hosts grow.
THE BUILDING UP OF A BACTERIAL PATHOGEN: Y. PESTIS, A RECENT CASE OF SPECIATION
As stated above, the acquisition by former nonvirulent organisms of novel traits into their genome allows for quantum leaps of evolution toward virulence (6, 48, 93). Since each pathogenic species (or clonal complex) shares the same or a very similar set of acquired virulence determinants, the evolution will be reflected in the clonal expansion of the strain that has acquired the virulence determinants (fitter in the new infective habitat than its ancestor), a situation that has been studied in the case of pathogens as Bacillus anthracis, Y. pestis, and Francisella tularensis (105).
With the description in 2011 of Yersinia pekkanenii, the genus Yersinia currently comprises 15 species (106); 3 of them, Y. pestis, Y. pseudotuberculosis, and Y. enterocolitica, are human pathogens (33, 36, 107). It has been shown that Y. pestis and Y. pseudotuberculosis diverged from Y. enterocolitica more than 40 million years ago, whereas Y. pestis is a clone that derived from Y. pseudotuberculosis less than 20,000 years ago (33, 36, 107). The successful expansion of the clone that is the origin of the Y. pestis species was caused by the incorporation into the genome of Y. pseudotuberculosis of a group of genes, the loss of others, and a final adaptation of the bacterial metabolism (Fig. 2). This novel set of determinants allowed the use by the emergent pathogen of a different transmission route through rodents and the bites of infected fleas. In addition, the evolved microorganism produced a different kind of infection than its ancestor. Together, novel transmission routes and colonization of a different habitat within the human host allowed the expansion of a clone that is at the root of the Y. pestis speciation process. Indeed, Y. pseudotuberculosis is a foodborne pathogen that produces nonfatal gastrointestinal diseases, whereas Y. pestis has been the cause of septicemic, pneumonic, and bubonic plagues, which altogether have produced around 200 million deaths, and it is transmitted through inhalation or as the consequence of the bite by an infected flea (108). This recent process of speciation has required the acquisition of some genes and the loss of others as well. Among the latter, particularly relevant is the loss of insect toxins that will kill the insect host, impeding transmission of the pathogen. A higher capability of forming biofilms inside fleas has also improved the chances for interhost transmission to humans by bites of infected insects (32). It is important to note that, in addition to improving the transmissibility of Y. pestis to humans and consequently its epidemicity, another consequence of this evolutionary process is the adaptation of Y. pestis for acting as an insects’ commensal. In addition to genome gain and loss, the evolution of Y. pestis involves the rewiring of the bacterial physiology to get a better adaptation for growing inside the host. This adaptation derives from the selection of mutants, mainly in regulatory elements (33), that are fitter for growing inside the infected host. A final step in the evolution of an organism might be the loss of those elements that are not required for colonizing the novel habitat. These processes of metabolic rewiring and genome reduction may impair the capabilities of the new virulent species for growing in the habitat of its ancestor, a situation that might further foster the speciation process.
FIGURE 2.
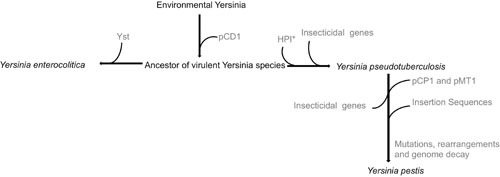
Evolution of Y. pestis. The process of Y. pestis speciation from an environmental, nonpathogenic ancestor is a good example of the evolutionary steps that are involved in the emergence of bacterial pathogens. This process began with the acquisition of the plasmid pCD1 by environmental Yersinia. This plasmid harbors genes encoding virulence determinants such as type III secretion systems and effector Yop proteins. From this ancestor of virulent Yersinia species, two branches have evolved. One diverged through the acquisition of the Yersinia stable toxin (Yst) and led to the speciation of Y. enterocolitica. This species has further evolved through acquisition and loss of genes (not shown in this figure). The other branch diverged through the acquisition of the high pathogenicity island (HPI*), which encodes an iron-uptake system and is present as well in different Enterobacteriaceae, and by the incorporation of insecticidal genes. Y. pestis is a successful clone that emerged recently from Y. pseudotuberculosis through the acquisition of the plasmids pCP1, which encodes the plasminogen activator gene, and pMT1, which allows colonization of the gut of fleas. The loss of insect toxins is an important event for the persistence of Y. pestis in its insect vectors. The acquisition of insertion sequences is the basis of the genome rearrangements and gene loss of Y. pestis. Finally, the entire process of adaptation to a new host is modulated by the mutation-driven optimization of the regulatory and metabolic networks of the pathogen. This evolutionary process is described in more detail in references 33, 37, and 105. Reproduced with permission from reference 8.
Altogether, the studies on the reconstruction of the evolution of Y. pestis support the idea that, after a first HGT step, the acquisition of other genes, together with genome reduction and selection of fitter mutants, foster the evolution of bacterial pathogens.
INSTANT EVOLUTION AND SPECIATION: TWO PATHWAYS IN THE EVOLUTION OF BACTERIAL PATHOGENS
The basic mechanisms of acquiring resistance genes and virulence factors are quite similar. It is true that virulence genes are more frequently present in large chromosomal arrangements (pathogenicity islands) and resistance genes are usually present in plasmids, but virulence plasmids and chromosomally encoded arrangements of resistance genes are also frequent, and combinations of virulence and antibiotic resistance genes in the same mobile element have been described (109–113). Despite these similarities in the genetic mechanisms of evolution, the consequences of acquiring virulence determinants or antibiotic resistance genes for the new host are completely different.
The acquisition of virulence determinants allows entry into a new habitat (the infected host). In the case that this also causes the deadaptation from the former ecosystem of the newly born pathogen, this situation is similar to geographic isolation (114, 115). This is the first step in a potential process of speciation, and hence the acquisition of virulence determinants by HGT is at the root of the speciation of bacterial pathogens (3–5).
The situation of antibiotic resistance is sharply different. From a human standpoint, resistance to antibiotics is a relevant process in the case of bacterial pathogens that have already gained access to the habitat that constitutes the human body. Resistance is needed for evasion of a deadly selective force, the presence of the antibiotic, and constitutes an instant evolutionary process (bacteria are resistant or they will die). Nevertheless, the reward is not the access to a new habitat, but just the possibility to keep multiplying in the old one, now polluted with antibiotics (87).
Although the acquisition of resistance can alter the bacterial metabolism and compensatory mutations can be selected after a first step in the evolution toward resistance (116–118), it is unsuitable that this situation should follow with a speciation process, as happens in the case of the acquisition of virulence determinants.
EXAPTATION AS A GENERAL PROCESS IN THE EVOLUTION OF BACTERIAL PATHOGENS
A common way of thinking is that evolution is a gradual process that operates through the sequential selection of minor genetic variants, each one rendering a fitter phenotype than the ancestor. Once the evolved variant is fixed, a novel genetic event occurs and the sequential selection of these improved variants is the basis of evolution. This gradual process, which explains well the selection of simple structures and traits, is more difficult to understand in the case of more complex systems that require being fully formed to be selected. One example of this is the feathers of birds. A bird can fly if it presents this complex structure. However, what are the selective forces behind the selection of intermediates between a regular hair and a feather if these evolutionary intermediates cannot be used for flying? For solving the problem of direct selection of complex systems, Gould introduced the concept of exaptation (119, 120). In the case of feathers, it is possible that their ancestors might have been gradually selected because of an improved capability (as compared with hair) for maintaining bird temperature homeostasis. This provides a potential selective force for this kind of structure. Only when the structure is complex enough will the bird will be able to fly, which constitutes an emerging property of feathers for which they were not previously selected during their evolution. The process of the acquisition of a new function is hence not driven by the selection of novel improved variants in a preexistent habitat but rather by a change of habitat where the novel selected properties can be used for a new function.
As we have seen before, antibiotic resistance genes and virulence determinants have evolved in their original hosts to play roles that are not necessarily resisting the presence of antibiotics or infecting a human host. Nevertheless, when confronted with the presence of antibiotics or when bacteria grow inside the new host, resistance and virulence are their more relevant functions. This situation fits well with the aforementioned type of adaptation in which evolution toward a new functionality happens as the consequence of a change of habitat more than because of the selection of a fitter variant.
While virulence determinants might have very similar roles in the natural environment and in the infected host, the situation is not the same in the case of antibiotic resistance genes. Indeed, a siderophore (88, 89) would produce the same effect for its bacterial host (getting iron) and a toxin may serve to kill a predator, which can be a unicellular protozoan or a macrophage, in which case the differences are more in the name of this element (virulence determinants) than in its actual function (getting nutrients and avoiding predators’ activity in a given habitat). Nevertheless, in the case of antibiotic resistance genes, when they are transferred to a new host they are decoupled from the metabolic and regulatory networks of the host where they evolved. Further, the habitat colonized by the novel host does not necessarily contain the signals and cues for which the presence of these elements was needed. As an example, it has been shown that different P. aeruginosa efflux pumps can extrude quorum-sensing signals and their metabolic intermediates, suggesting that they can modulate the quorum-sensing response in this bacterial species (121–124). If these efflux pumps are acquired through HGT by a new host that does not produce the same quorum-sensing signals, they cannot play the role for which they were selected in nature. Under these circumstances of decontextualization, the unique function that HGT-acquired antibiotic resistance determinants may play is the one for which they have been selected after their transfer to a human pathogen: resistance to antimicrobials currently used for treating infectious diseases (Fig. 3).
FIGURE 3.
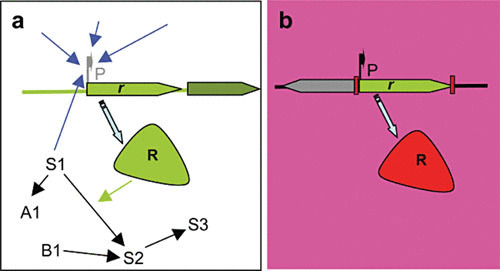
Exaptation and gene decontextualization in the evolution of antibiotic resistance. Antibiotic resistance genes (r) have evolved for millions of years located in the chromosomes of their original hosts (a). During this evolution, the expression of these determinants (R) from their promoters (P) has been finely tuned to respond to several signals that might include the response to environmental and metabolic changes (blue arrows). Besides, the determinants encoded by these genes are integrated in physiological networks, where they can play a role as metabolic enzymes. S1 to S3 represent metabolites of the same pathway, and A1 and B1 metabolites of other interconnected pathways. When these genes are integrated in gene capture (for instance, an integron) and transfer units (for instance, a plasmid), they can be transferred to a new host and submitted to strong antibiotic selective pressure (b), and they can be constitutively expressed from a strong promoter (P) present in the capture unit and therefore lack the regulatory and physiological network encountered in the original host (gene decontextualization). Under these circumstances, the only function these determinants can play is antibiotic resistance, in such a way that this functional shift is not the consequence of adaptive changes in the determinants but rather of changes in their environment (exaptation). Reproduced with permission from reference 127.
The number of genes on Earth capable of conferring resistance to antibiotics is several orders of magnitude above those acquired by human pathogens. The introduction of antibiotics for therapeutic purposes has thus produced an enrichment of a specific subset of genes whose unique function is conferring resistance to antibiotics in an exaptation process that is a product of cultural human evolution (125–127).
BOTTLENECKS IN THE ACQUISITION OF GENETIC ELEMENTS BY BACTERIAL PATHOGENS FROM ENVIRONMENTAL MICROORGANISMS
The increasing use of functional and sequence-based genomic and metagenomic approaches has demonstrated that genes capable of conferring resistance to antibiotics in a heterologous host are present in any ecosystem and in any bacteria. Although not studied in detail, a similar situation might also occur in the case of virulence determinants, which might eventually be present in nearly all metagenomes. In addition, similar virulence determinants are present in humans, animals, plants, and even protozoan pathogens. For the sake of simplicity and since information about virulence determinants in metagenomes has not been reviewed in depth, in this section I will mainly discuss the bottlenecks modulating the transfer of antibiotic resistance genes, although the conclusions are likely similar in the case of virulence determinants. In this context, it is worth mentioning that the variability of HGT-acquired antibiotic resistance genes is much lower than can be found in currently available metagenomes, which implies the existence of strong bottlenecks modulating the acquisition by bacterial pathogens of genetic elements from environmental microorganisms (11, 128).
The first one is ecological connectivity; the chances for the transfer of genetic material increase if the donor and the recipient bacteria are in the vicinity and belong to the same gene-exchange community. It is true that a chain of transfer events may allow the acquisition of an antibiotic resistance determinant by a bacterial pathogen even if the original host for this element is ecologically disconnected from this pathogen. Nevertheless, the success of this chain of transfer events would only be possible if selection operates along all steps. Because of this, although the first step in the acquisition of resistance determinants likely occurs in nonclinical, natural environments, where environmental and pathogenic microorganisms can encounter one another, the spread of these determinants occurs in places with high antibiotic loads such as hospitals and possibly other reservoirs like wastewater treatment plants, farms, and fisheries, in which selective pressure by antibiotics and other compounds, such as heavy metals or biocides, which are capable of select antibiotic resistance can be high on occasion (129–131).
The second bottleneck is the founder effect. Once a given bacterium has acquired a resistance gene, antibiotic selective pressure does not operate; bacteria are already resistant. Consequently, there is not any reward for incorporating a new gene conferring resistance to the antibiotic. If clonal expansion and HGT spread of the resistance determinant occur before such a second resistance gene is incorporated into the pathogens’ population, the first one will spread among the population and the acquisition of novel resistance genes with the same antibiotic profile will be precluded. There are some situations in which the founder effect is a partial bottleneck (132). For instance, different elements conferring resistance to the same antibiotic can be selected at different geographic locations or in different clones if this happens before the global spread of a single gene. Also, if the gene produces fitness costs (see below), extinction and reentrance episodes may occur, as well as the displacement of one resistance gene by a new, fitter one.
In the case of the acquisition of virulence determinants, the founder effect has a more drastic effect, since, as discussed above, the incorporation of traits that allow gaining access to the host may be the first step in a speciation process triggered through the clonal expansion of the clone that has acquired the novel traits. It is still possible that another clone from the same species incorporates a different subset of virulence determinants, but in this case the new evolved clone will be considered as belonging to a different clonal complex, as happens for intraintestinal and extraintestinal E. coli clones (133), which are not fully ecoequivalent. In the long term, this process can end in the speciation of the clones that have acquired different repertoires of virulence determinants.
The last bottleneck consists of fitness costs associated with the acquisition of novel genetic material (134–138). It has been suggested that these fitness costs should be the consequence of metabolic and energetic burden associated with the replication, transcription, and translation of the novel genes incorporated by the pathogen. If that is the case, the differences in fitness after incorporating one or another set of new genes will depend on the size of such genes as well as on the levels of their transcription and translation. Nevertheless, although some common trends can be found, different studies have shown that fitness costs associated with acquisition of antibiotic resistance and of foreign DNA depend on the type of gene, the environment, and even the microorganisms incorporating such genes (139–142). In addition, bacteria present mechanisms such as compensatory mutations or metabolic rewiring that ameliorate the costs of the acquisition of resistance (116, 118, 143–145). In this context, the fixation into the different members of the bacterial species of a new genetic element requires that it does not produce an unaffordable fitness cost or that fitness costs can be compensated (146). In the case of acquisition of antibiotic resistance, spread and fixation of the resistant strain requires low fitness costs in all habitats where these microorganisms can replicate, including natural, nonclinical ecosystems, where antibiotic selective pressure is low (147). However, in the case of virulence determinants, their incorporation may allow an increased fitness for colonizing the infected host at the cost of reducing the fitness for growing in their original environmental habitat. These differential fitness costs would be the basis of the speciation toward virulence of the new pathogen, which will be outcompeted by its ancestor in its original habitat while not having a competitor in the new one (infected host).
The number of genes capable of conferring antibiotic resistance present in available metagenomes is much larger than those currently acquired by human pathogens (83, 84, 129, 148, 149). This finding supports the premise that bottlenecks, such as the above-discussed ecological connectivity, founder effect, and fitness costs, modulate the acquisition, fixation, and spread of HGT-acquired genes in bacterial populations.
ACKNOWLEDGMENTS
My laboratory is supported by grants from the Spanish Ministry of Economy and Competitiveness (BIO2014-54507-R and JPI Water StARE JPIW2013-089-C02-01) and from the Instituto de Salud Carlos III (Spanish Network for Research on Infectious Diseases) (RD16/0016/0011).
REFERENCES
- 1.Ochman H, Groisman EA. 1994. The origin and evolution of species differences in Escherichia coli and Salmonella typhimurium. EXS 69:479–493. 10.1007/978-3-0348-7527-1_27. [DOI] [PubMed] [Google Scholar]
- 2.Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 95:3140–3145. 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Achtman M, Wagner M. 2008. Microbial diversity and the genetic nature of microbial species. Nat Rev Microbiol 6:431–440. 10.1038/nrmicro1872. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Ochman H, Lawrence JG, Groisman EA. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304. 10.1038/35012500. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Wiedenbeck J, Cohan FM. 2011. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol Rev 35:957–976. 10.1111/j.1574-6976.2011.00292.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 6.Groisman EA, Ochman H. 1996. Pathogenicity islands: bacterial evolution in quantum leaps. Cell 87:791–794. 10.1016/S0092-8674(00)81985-6. [PubMed] [DOI] [PubMed] [Google Scholar]
- 7.Rouli L, Merhej V, Fournier PE, Raoult D. 2015. The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect 7:72–85. 10.1016/j.nmni.2015.06.005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martínez JL. 2013. Bacterial pathogens: from natural ecosystems to human hosts. Environ Microbiol 15:325–333. 10.1111/j.1462-2920.2012.02837.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 9.Martínez JL. 2008. Antibiotics and antibiotic resistance genes in natural environments. Science 321:365–367. 10.1126/science.1159483. [PubMed] [DOI] [PubMed] [Google Scholar]
- 10.Martínez JL, Baquero F, Andersson DI. 2007. Predicting antibiotic resistance. Nat Rev Microbiol 5:958–965. 10.1038/nrmicro1796. [PubMed] [DOI] [PubMed] [Google Scholar]
- 11.Martinez JL, Fajardo A, Garmendia L, Hernandez A, Linares JF, Martínez-Solano L, Sánchez MB. 2009. A global view of antibiotic resistance. FEMS Microbiol Rev 33:44–65. 10.1111/j.1574-6976.2008.00142.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 12.Fajardo A, Linares JF, Martínez JL. 2009. Towards an ecological approach to antibiotics and antibiotic resistance genes. Clin Microbiol Infect 15(Suppl 1):14–16. 10.1111/j.1469-0691.2008.02688.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 13.Lukjancenko O, Wassenaar TM, Ussery DW. 2010. Comparison of 61 sequenced Escherichia coli genomes. Microb Ecol 60:708–720. 10.1007/s00248-010-9717-3. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levin BR, Bergstrom CT. 2000. Bacteria are different: observations, interpretations, speculations, and opinions about the mechanisms of adaptive evolution in prokaryotes. Proc Natl Acad Sci U S A 97:6981–6985. 10.1073/pnas.97.13.6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hacker J, Kaper JB. 2000. Pathogenicity islands and the evolution of microbes. Annu Rev Microbiol 54:641–679. 10.1146/annurev.micro.54.1.641. [PubMed] [DOI] [PubMed] [Google Scholar]
- 16.Berg G, Martinez JL. 2015. Friends or foes: can we make a distinction between beneficial and harmful strains of the Stenotrophomonas maltophilia complex? Front Microbiol 6:241. 10.3389/fmicb.2015.00241. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alonso A, Rojo F, Martínez JL. 1999. Environmental and clinical isolates of Pseudomonas aeruginosa show pathogenic and biodegradative properties irrespective of their origin. Environ Microbiol 1:421–430. 10.1046/j.1462-2920.1999.00052.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 18.Wiehlmann L, Wagner G, Cramer N, Siebert B, Gudowius P, Morales G, Köhler T, van Delden C, Weinel C, Slickers P, Tümmler B. 2007. Population structure of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 104:8101–8106. 10.1073/pnas.0609213104. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morales G, Wiehlmann L, Gudowius P, van Delden C, Tümmler B, Martínez JL, Rojo F. 2004. Structure of Pseudomonas aeruginosa populations analyzed by single nucleotide polymorphism and pulsed-field gel electrophoresis genotyping. J Bacteriol 186:4228–4237. 10.1128/JB.186.13.4228-4237.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahme LG, Ausubel FM, Cao H, Drenkard E, Goumnerov BC, Lau GW, Mahajan-Miklos S, Plotnikova J, Tan MW, Tsongalis J, Walendziewicz CL, Tompkins RG. 2000. Plants and animals share functionally common bacterial virulence factors. Proc Natl Acad Sci U S A 97:8815–8821. 10.1073/pnas.97.16.8815. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahajan-Miklos S, Rahme LG, Ausubel FM. 2000. Elucidating the molecular mechanisms of bacterial virulence using non-mammalian hosts. Mol Microbiol 37:981–988. 10.1046/j.1365-2958.2000.02056.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 22.Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899–1902. 10.1126/science.7604262. [PubMed] [DOI] [PubMed] [Google Scholar]
- 23.Wolfgang MC, Kulasekara BR, Liang X, Boyd D, Wu K, Yang Q, Miyada CG, Lory S. 2003. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 100:8484–8489. 10.1073/pnas.0832438100. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Libisch B, Gacs M, Csiszár K, Muzslay M, Rókusz L, Füzi M. 2004. Isolation of an integron-borne blaVIM-4 type metallo-β-lactamase gene from a carbapenem-resistant Pseudomonas aeruginosa clinical isolate in Hungary. Antimicrob Agents Chemother 48:3576–3578. 10.1128/AAC.48.9.3576-3578.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee K, Lim JB, Yum JH, Yong D, Chong Y, Kim JM, Livermore DM. 2002. blaVIM-2 cassette-containing novel integrons in metallo-β-lactamase-producing Pseudomonas aeruginosa and Pseudomonas putida isolates disseminated in a Korean hospital. Antimicrob Agents Chemother 46:1053–1058. 10.1128/AAC.46.4.1053-1058.2002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yizhak K, Tuller T, Papp B, Ruppin E. 2011. Metabolic modeling of endosymbiont genome reduction on a temporal scale. Mol Syst Biol 7:479. 10.1038/msb.2011.11. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pérez-Brocal V, Gil R, Ramos S, Lamelas A, Postigo M, Michelena JM, Silva FJ, Moya A, Latorre A. 2006. A small microbial genome: the end of a long symbiotic relationship? Science 314:312–313. 10.1126/science.1130441. [PubMed] [DOI] [PubMed] [Google Scholar]
- 28.Gil R, Latorre A, Moya A. 2004. Bacterial endosymbionts of insects: insights from comparative genomics. Environ Microbiol 6:1109–1122. 10.1111/j.1462-2920.2004.00691.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 29.Tamas I, Klasson L, Canbäck B, Näslund AK, Eriksson AS, Wernegreen JJ, Sandström JP, Moran NA, Andersson SG. 2002. 50 million years of genomic stasis in endosymbiotic bacteria. Science 296:2376–2379. 10.1126/science.1071278. [PubMed] [DOI] [PubMed] [Google Scholar]
- 30.Brites D, Gagneux S. 2015. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol Rev 264:6–24. 10.1111/imr.12264. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gagneux S. 2012. Host-pathogen coevolution in human tuberculosis. Philos Trans R Soc Lond B Biol Sci 367:850–859. 10.1098/rstb.2011.0316. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chouikha I, Hinnebusch BJ. 2012. Yersinia—flea interactions and the evolution of the arthropod-borne transmission route of plague. Curr Opin Microbiol 15:239–246. 10.1016/j.mib.2012.02.003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou D, Yang R. 2009. Molecular Darwinian evolution of virulence in Yersinia pestis. Infect Immun 77:2242–2250. 10.1128/IAI.01477-08. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lesic B, Carniel E. 2005. Horizontal transfer of the high-pathogenicity island of Yersinia pseudotuberculosis. J Bacteriol 187:3352–3358. 10.1128/JB.187.10.3352-3358.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou D, Han Y, Song Y, Tong Z, Wang J, Guo Z, Pei D, Pang X, Zhai J, Li M, Cui B, Qi Z, Jin L, Dai R, Du Z, Bao J, Zhang X, Yu J, Wang J, Huang P, Yang R. 2004. DNA microarray analysis of genome dynamics in Yersinia pestis: insights into bacterial genome microevolution and niche adaptation. J Bacteriol 186:5138–5146. 10.1128/JB.186.15.5138-5146.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Achtman M, Morelli G, Zhu P, Wirth T, Diehl I, Kusecek B, Vogler AJ, Wagner DM, Allender CJ, Easterday WR, Chenal-Francisque V, Worsham P, Thomson NR, Parkhill J, Lindler LE, Carniel E, Keim P. 2004. Microevolution and history of the plague bacillus, Yersinia pestis. Proc Natl Acad Sci U S A 101:17837–17842. 10.1073/pnas.0408026101. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wren BW. 2003. The yersiniae—a model genus to study the rapid evolution of bacterial pathogens. Nat Rev Microbiol 1:55–64. 10.1038/nrmicro730. [PubMed] [DOI] [PubMed] [Google Scholar]
- 38.Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A 96:14043–14048. 10.1073/pnas.96.24.14043. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le Gall T, Clermont O, Gouriou S, Picard B, Nassif X, Denamur E, Tenaillon O. 2007. Extraintestinal virulence is a coincidental by-product of commensalism in B2 phylogenetic group Escherichia coli strains. Mol Biol Evol 24:2373–2384. 10.1093/molbev/msm172. [PubMed] [DOI] [PubMed] [Google Scholar]
- 40.Smati M, Clermont O, Bleibtreu A, Fourreau F, David A, Daubié AS, Hignard C, Loison O, Picard B, Denamur E. 2015. Quantitative analysis of commensal Escherichia coli populations reveals host-specific enterotypes at the intra-species level. MicrobiologyOpen 4:604–615. 10.1002/mbo3.266. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Lin K. 2012. A phylogenomic analysis of Escherichia coli/Shigella group: implications of genomic features associated with pathogenicity and ecological adaptation. BMC Evol Biol 12:174. 10.1186/1471-2148-12-174. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alteri CJ, Mobley HL. 2012. Escherichia coli physiology and metabolism dictates adaptation to diverse host microenvironments. Curr Opin Microbiol 15:3–9. 10.1016/j.mib.2011.12.004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carlos C, Pires MM, Stoppe NC, Hachich EM, Sato MI, Gomes TA, Amaral LA, Ottoboni LM. 2010. Escherichia coli phylogenetic group determination and its application in the identification of the major animal source of fecal contamination. BMC Microbiol 10:161. 10.1186/1471-2180-10-161. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo C, Walk ST, Gordon DM, Feldgarden M, Tiedje JM, Konstantinidis KT. 2011. Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc Natl Acad Sci U S A 108:7200–7205. 10.1073/pnas.1015622108. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tenaillon O, Skurnik D, Picard B, Denamur E. 2010. The population genetics of commensal Escherichia coli. Nat Rev Microbiol 8:207–217. 10.1038/nrmicro2298. [PubMed] [DOI] [PubMed] [Google Scholar]
- 46.Milkman R. 1997. Recombination and population structure in Escherichia coli. Genetics 146:745–750. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H, Achtman M. 2006. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol 60:1136–1151. 10.1111/j.1365-2958.2006.05172.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heesemann J. 2004. Darwin’s principle of divergence revisited: small steps and quantum leaps set the path of microbial evolution. Int J Med Microbiol 294:65–66. 10.1016/j.ijmm.2004.06.012. [PubMed] [DOI] [PubMed] [Google Scholar]
- 49.Ghosh AR. 2013. Appraisal of microbial evolution to commensalism and pathogenicity in humans. Clin Med Insights Gastroenterol 6:1–12. 10.4137/CGast.S11858. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levin BR, Bull JJ. 1994. Short-sighted evolution and the virulence of pathogenic microorganisms. Trends Microbiol 2:76–81. 10.1016/0966-842X(94)90538-X. [PubMed] [DOI] [PubMed] [Google Scholar]
- 51.Sokurenko EV, Gomulkiewicz R, Dykhuizen DE. 2006. Source-sink dynamics of virulence evolution. Nat Rev Microbiol 4:548–555. 10.1038/nrmicro1446. [PubMed] [DOI] [PubMed] [Google Scholar]
- 52.Martínez-Solano L, Macia MD, Fajardo A, Oliver A, Martinez JL. 2008. Chronic Pseudomonas aeruginosa infection in chronic obstructive pulmonary disease. Clin Infect Dis 47:1526–1533. 10.1086/593186. [PubMed] [DOI] [PubMed] [Google Scholar]
- 53.Oliver A, Cantón R, Campo P, Baquero F, Blázquez J. 2000. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288:1251–1253. 10.1126/science.288.5469.1251. [PubMed] [DOI] [PubMed] [Google Scholar]
- 54.van Mansfeld R, de Vrankrijker A, Brimicombe R, Heijerman H, Teding van Berkhout F, Spitoni C, Grave S, van der Ent C, Wolfs T, Willems R, Bonten M. 2016. The effect of strict segregation on Pseudomonas aeruginosa in cystic fibrosis patients. PLoS One 11:e0157189. 10.1371/journal.pone.0157189. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wiehlmann L, Cramer N, Tümmler B. 2015. Habitat-associated skew of clone abundance in the Pseudomonas aeruginosa population. Environ Microbiol Rep 7:955–960. 10.1111/1758-2229.12340. [PubMed] [DOI] [PubMed] [Google Scholar]
- 56.Oliver A, Mulet X, López-Causapé C, Juan C. 2015. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist Updat 21–22:41–59. 10.1016/j.drup.2015.08.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 57.de Vrankrijker AM, Brimicombe RW, Wolfs TF, Heijerman HG, van Mansfeld R, van Berkhout FT, Willems RJ, Bonten MJ, van der Ent CK. 2011. Clinical impact of a highly prevalent Pseudomonas aeruginosa clone in Dutch cystic fibrosis patients. Clin Microbiol Infect 17:382–385. 10.1111/j.1469-0691.2010.03295.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 58.van Mansfeld R, Willems R, Brimicombe R, Heijerman H, van Berkhout FT, Wolfs T, van der Ent C, Bonten M. 2009. Pseudomonas aeruginosa genotype prevalence in Dutch cystic fibrosis patients and age dependency of colonization by various P. aeruginosa sequence types. J Clin Microbiol 47:4096–4101. 10.1128/JCM.01462-09. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.San Millan A, Toll-Riera M, Qi Q, MacLean RC. 2015. Interactions between horizontally acquired genes create a fitness cost in Pseudomonas aeruginosa. Nat Commun 6:6845. 10.1038/ncomms7845. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andersson DI, Hughes D. 2010. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol 8:260–271. 10.1038/nrmicro2319. [PubMed] [DOI] [PubMed] [Google Scholar]
- 61.Forsberg KJ, Patel S, Gibson MK, Lauber CL, Knight R, Fierer N, Dantas G. 2014. Bacterial phylogeny structures soil resistomes across habitats. Nature 509:612–616. 10.1038/nature13377. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benveniste R, Davies J. 1973. Aminoglycoside antibiotic-inactivating enzymes in actinomycetes similar to those present in clinical isolates of antibiotic-resistant bacteria. Proc Natl Acad Sci U S A 70:2276–2280. 10.1073/pnas.70.8.2276. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Poirel L, Rodriguez-Martinez JM, Mammeri H, Liard A, Nordmann P. 2005. Origin of plasmid-mediated quinolone resistance determinant QnrA. Antimicrob Agents Chemother 49:3523–3525. 10.1128/AAC.49.8.3523-3525.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Humeniuk C, Arlet G, Gautier V, Grimont P, Labia R, Philippon A. 2002. β-Lactamases of Kluyvera ascorbata, probable progenitors of some plasmid-encoded CTX-M types. Antimicrob Agents Chemother 46:3045–3049. 10.1128/AAC.46.9.3045-3049.2002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoon EJ, Goussard S, Touchon M, Krizova L, Cerqueira G, Murphy C, Lambert T, Grillot-Courvalin C, Nemec A, Courvalin P. 2014. Origin in Acinetobacter guillouiae and dissemination of the aminoglycoside-modifying enzyme Aph(3′)-VI. mBio 5:e01972-e14. 10.1128/mBio.01972-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wright GD. 2010. The antibiotic resistome. Expert Opin Drug Discov 5:779–788. 10.1517/17460441.2010.497535. [PubMed] [DOI] [PubMed] [Google Scholar]
- 67.D’Costa VM, McGrann KM, Hughes DW, Wright GD. 2006. Sampling the antibiotic resistome. Science 311:374–377. 10.1126/science.1120800. [PubMed] [DOI] [PubMed] [Google Scholar]
- 68.Laskaris P, Tolba S, Calvo-Bado L, Wellington EM. 2010. Coevolution of antibiotic production and counter-resistance in soil bacteria. Environ Microbiol 12:783–796. 10.1111/j.1462-2920.2009.02125.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 69.Thanassi DG, Cheng LW, Nikaido H. 1997. Active efflux of bile salts by Escherichia coli. J Bacteriol 179:2512–2518. 10.1128/jb.179.8.2512-2518.1997. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. 1995. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol 16:45–55. 10.1111/j.1365-2958.1995.tb02390.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 71.Jacoby GA. 2009. AmpC β-lactamases. Clin Microbiol Rev 22:161–182. 10.1128/CMR.00036-08. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morosini MI, Ayala JA, Baquero F, Martínez JL, Blázquez J. 2000. Biological cost of AmpC production for Salmonella enterica serotype Typhimurium. Antimicrob Agents Chemother 44:3137–3143. 10.1128/AAC.44.11.3137-3143.2000. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wiedemann B, Pfeifle D, Wiegand I, Janas E. 1998. β-Lactamase induction and cell wall recycling in gram-negative bacteria. Drug Resist Updat 1:223–226. 10.1016/S1368-7646(98)80002-2. [DOI] [PubMed] [Google Scholar]
- 74.Henderson TA, Young KD, Denome SA, Elf PK. 1997. AmpC and AmpH, proteins related to the class C β-lactamases, bind penicillin and contribute to the normal morphology of Escherichia coli. J Bacteriol 179:6112–6121. 10.1128/jb.179.19.6112-6121.1997. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Macinga DR, Rather PN. 1999. The chromosomal 2′-N-acetyltransferase of Providencia stuartii: physiological functions and genetic regulation. Front Biosci 4:D132–D140. 10.2741/Macinga. [PubMed] [DOI] [PubMed] [Google Scholar]
- 76.Piddock LJ. 2006. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin Microbiol Rev 19:382–402. 10.1128/CMR.19.2.382-402.2006. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vila J, Martínez JL. 2008. Clinical impact of the over-expression of efflux pump in nonfermentative Gram-negative bacilli, development of efflux pump inhibitors. Curr Drug Targets 9:797–807. 10.2174/138945008785747806. [PubMed] [DOI] [PubMed] [Google Scholar]
- 78.Li XZ, Plésiat P, Nikaido H. 2015. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin Microbiol Rev 28:337–418. 10.1128/CMR.00117-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hernando-Amado S, Blanco P, Alcalde-Rico M, Corona F, Reales-Calderón JA, Sánchez MB, Martínez JL. 2016. Multidrug efflux pumps as main players in intrinsic and acquired resistance to antimicrobials. Drug Resist Updat 28:13–27. 10.1016/j.drup.2016.06.007. [PubMed] [DOI] [PubMed] [Google Scholar]
- 80.Piddock LJ. 2006. Multidrug-resistance efflux pumps—not just for resistance. Nat Rev Microbiol 4:629–636. 10.1038/nrmicro1464. [PubMed] [DOI] [PubMed] [Google Scholar]
- 81.Alvarez-Ortega C, Olivares J, Martínez JL. 2013. RND multidrug efflux pumps: what are they good for? Front Microbiol 4:7. 10.3389/fmicb.2013.00007. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martinez JL, Sánchez MB, Martínez-Solano L, Hernandez A, Garmendia L, Fajardo A, Alvarez-Ortega C. 2009. Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol Rev 33:430–449. 10.1111/j.1574-6976.2008.00157.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 83.Martínez JL, Coque TM, Baquero F. 2015. Prioritizing risks of antibiotic resistance genes in all metagenomes. Nat Rev Microbiol 13:396. 10.1038/nrmicro3399-c2. [PubMed] [DOI] [PubMed] [Google Scholar]
- 84.Martínez JL, Coque TM, Baquero F. 2015. What is a resistance gene? Ranking risk in resistomes. Nat Rev Microbiol 13:116–123. 10.1038/nrmicro3399. [PubMed] [DOI] [PubMed] [Google Scholar]
- 85.Levin BR, Antia R. 2001. Why we don’t get sick: the within-host population dynamics of bacterial infections. Science 292:1112–1115. 10.1126/science.1058879. [DOI] [PubMed] [Google Scholar]
- 86.Eisenreich W, Dandekar T, Heesemann J, Goebel W. 2010. Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat Rev Microbiol 8:401–412. 10.1038/nrmicro2351. [PubMed] [DOI] [PubMed] [Google Scholar]
- 87.Martínez JL, Baquero F. 2002. Interactions among strategies associated with bacterial infection: pathogenicity, epidemicity, and antibiotic resistance. Clin Microbiol Rev 15:647–679. 10.1128/CMR.15.4.647-679.2002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martínez JL, Delgado-Iribarren A, Baquero F. 1990. Mechanisms of iron acquisition and bacterial virulence. FEMS Microbiol Rev 6:45–56. 10.1016/0378-1097(90)90522-R. [PubMed] [DOI] [PubMed] [Google Scholar]
- 89.de Lorenzo V, Martinez JL. 1988. Aerobactin production as a virulence factor: a reevaluation. Eur J Clin Microbiol Infect Dis 7:621–629. 10.1007/BF01964239. [PubMed] [DOI] [PubMed] [Google Scholar]
- 90.Trueba G, Dunthorn M. 2012. Many neglected tropical diseases may have originated in the Paleolithic or before: new insights from genetics. PLoS Negl Trop Dis 6:e1393. 10.1371/journal.pntd.0001393. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chouikha I, Germon P, Brée A, Gilot P, Moulin-Schouleur M, Schouler C. 2006. A selC-associated genomic island of the extraintestinal avian pathogenic Escherichia coli strain BEN2908 is involved in carbohydrate uptake and virulence. J Bacteriol 188:977–987. 10.1128/JB.188.3.977-987.2006. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luck SN, Turner SA, Rajakumar K, Sakellaris H, Adler B. 2001. Ferric dicitrate transport system (Fec) of Shigella flexneri 2a YSH6000 is encoded on a novel pathogenicity island carrying multiple antibiotic resistance genes. Infect Immun 69:6012–6021. 10.1128/IAI.69.10.6012-6021.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hacker J, Carniel E. 2001. Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep 2:376–381. 10.1093/embo-reports/kve097. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schubert S, Rakin A, Karch H, Carniel E, Heesemann J. 1998. Prevalence of the “high-pathogenicity island” of Yersinia species among Escherichia coli strains that are pathogenic to humans. Infect Immun 66:480–485. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kirn TJ, Jude BA, Taylor RK. 2005. A colonization factor links Vibrio cholerae environmental survival and human infection. Nature 438:863–866. 10.1038/nature04249. [PubMed] [DOI] [PubMed] [Google Scholar]
- 96.Miyata S, Casey M, Frank DW, Ausubel FM, Drenkard E. 2003. Use of the Galleria mellonella caterpillar as a model host to study the role of the type III secretion system in Pseudomonas aeruginosa pathogenesis. Infect Immun 71:2404–2413. 10.1128/IAI.71.5.2404-2413.2003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mahajan-Miklos S, Tan MW, Rahme LG, Ausubel FM. 1999. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 96:47–56. 10.1016/S0092-8674(00)80958-7. [PubMed] [DOI] [PubMed] [Google Scholar]
- 98.Carilla-Latorre S, Calvo-Garrido J, Bloomfield G, Skelton J, Kay RR, Ivens A, Martinez JL, Escalante R. 2008. Dictyostelium transcriptional responses to Pseudomonas aeruginosa: common and specific effects from PAO1 and PA14 strains. BMC Microbiol 8:109. 10.1186/1471-2180-8-109. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cosson P, Zulianello L, Join-Lambert O, Faurisson F, Gebbie L, Benghezal M, Van Delden C, Curty LK, Köhler T. 2002. Pseudomonas aeruginosa virulence analyzed in a Dictyostelium discoideum host system. J Bacteriol 184:3027–3033. 10.1128/JB.184.11.3027-3033.2002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hueck CJ. 1998. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev 62:379–433. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gao LY, Harb OS, Abu Kwaik Y. 1997. Utilization of similar mechanisms by Legionella pneumophila to parasitize two evolutionarily distant host cells, mammalian macrophages and protozoa. Infect Immun 65:4738–4746. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lainhart W, Stolfa G, Koudelka GB. 2009. Shiga toxin as a bacterial defense against a eukaryotic predator, Tetrahymena thermophila. J Bacteriol 191:5116–5122. 10.1128/JB.00508-09. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Steinberg KM, Levin BR. 2007. Grazing protozoa and the evolution of the Escherichia coli O157:H7 Shiga toxin-encoding prophage. Proc Biol Sci 274:1921–1929. 10.1098/rspb.2007.0245. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pushkareva VI, Ermolaeva SA. 2010. Listeria monocytogenes virulence factor Listeriolysin O favors bacterial growth in co-culture with the ciliate Tetrahymena pyriformis, causes protozoan encystment and promotes bacterial survival inside cysts. BMC Microbiol 10:26. 10.1186/1471-2180-10-26. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Keim PS, Wagner DM. 2009. Humans and evolutionary and ecological forces shaped the phylogeography of recently emerged diseases. Nat Rev Microbiol 7:813–821. 10.1038/nrmicro2219. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Murros-Kontiainen A, Johansson P, Niskanen T, Fredriksson-Ahomaa M, Korkeala H, Björkroth J. 2011. Yersinia pekkanenii sp. nov. Int J Syst Evol Microbiol 61:2363–2367. 10.1099/ijs.0.019984-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 107.Morelli G, Song Y, Mazzoni CJ, Eppinger M, Roumagnac P, Wagner DM, Feldkamp M, Kusecek B, Vogler AJ, Li Y, Cui Y, Thomson NR, Jombart T, Leblois R, Lichtner P, Rahalison L, Petersen JM, Balloux F, Keim P, Wirth T, Ravel J, Yang R, Carniel E, Achtman M. 2010. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat Genet 42:1140–1143. 10.1038/ng.705. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Perry RD, Fetherston JD. 1997. Yersinia pestis—etiologic agent of plague. Clin Microbiol Rev 10:35–66. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ramirez MS, Traglia GM, Lin DL, Tran T, Tolmasky ME. 2014. Plasmid-mediated antibiotic resistance and virulence in Gram-negatives: the Klebsiella pneumoniae paradigm. Microbiol Spectr 2:PLAS-0016-2013. 10.1128/microbiolspec.PLAS-0016-2013. [DOI] [PubMed] [Google Scholar]
- 110.Colonna B, Ranucci L, Fradiani PA, Casalino M, Calconi A, Nicoletti M. 1992. Organization of aerobactin, hemolysin, and antibacterial resistance genes in lactose-negative Escherichia coli strains of serotype O4 isolated from children with diarrhea. Infect Immun 60:5224–5231. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Darfeuille-Michaud A, Jallat C, Aubel D, Sirot D, Rich C, Sirot J, Joly B. 1992. R-plasmid-encoded adhesive factor in Klebsiella pneumoniae strains responsible for human nosocomial infections. Infect Immun 60:44–55. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Delgado-Iribarren A, Martinez-Suarez J, Baquero F, Perez-Diaz JC, Martinez JL. 1987. Aerobactin-producing multi-resistance plasmids. J Antimicrob Chemother 19:552–553. 10.1093/jac/19.4.552. [PubMed] [DOI] [PubMed] [Google Scholar]
- 113.Martínez-Suárez JV, Martínez JL, López de Goicoechea MJ, Pérez-Díaz JC, Baquero F, Meseguer M, Liñares J. 1987. Acquisition of antibiotic resistance plasmids in vivo by extraintestinal Salmonella spp. J Antimicrob Chemother 20:452–453. 10.1093/jac/20.3.452. [PubMed] [DOI] [PubMed] [Google Scholar]
- 114.Bentley SD, Parkhill J. 2015. Genomic perspectives on the evolution and spread of bacterial pathogens. Proc Biol Sci 282:20150488. 10.1098/rspb.2015.0488. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.de la Cruz F, Davies J. 2000. Horizontal gene transfer and the origin of species: lessons from bacteria. Trends Microbiol 8:128–133. 10.1016/S0966-842X(00)01703-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 116.Olivares J, Álvarez-Ortega C, Martínez JL. 2014. Metabolic compensation of fitness costs associated with overexpression of the multidrug efflux pump MexEF-OprN in Pseudomonas aeruginosa. Antimicrob Agents Chemother 58:3904–3913. 10.1128/AAC.00121-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schulz zur Wiesch P, Engelstädter J, Bonhoeffer S. 2010. Compensation of fitness costs and reversibility of antibiotic resistance mutations. Antimicrob Agents Chemother 54:2085–2095. 10.1128/AAC.01460-09. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Andersson DI. 2006. The biological cost of mutational antibiotic resistance: any practical conclusions? Curr Opin Microbiol 9:461–465. 10.1016/j.mib.2006.07.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 119.Gould SJ, Lloyd EA. 1999. Individuality and adaptation across levels of selection: how shall we name and generalize the unit of Darwinism? Proc Natl Acad Sci U S A 96:11904–11909. 10.1073/pnas.96.21.11904. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gould SJ, Vrba S. 1982. Exaptation: a missing term in the science of form. Paleobiology 8:4–15. 10.1017/S0094837300004310. [DOI] [Google Scholar]
- 121.Olivares J, Alvarez-Ortega C, Linares JF, Rojo F, Köhler T, Martínez JL. 2012. Overproduction of the multidrug efflux pump MexEF-OprN does not impair Pseudomonas aeruginosa fitness in competition tests, but produces specific changes in bacterial regulatory networks. Environ Microbiol 14:1968–1981. 10.1111/j.1462-2920.2012.02727.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 122.Lamarche MG, Déziel E. 2011. MexEF-OprN efflux pump exports the Pseudomonas quinolone signal (PQS) precursor HHQ (4-hydroxy-2-heptylquinoline). PLoS One 6:e24310. 10.1371/journal.pone.0024310. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Köhler T, van Delden C, Curty LK, Hamzehpour MM, Pechere JC. 2001. Overexpression of the MexEF-OprN multidrug efflux system affects cell-to-cell signaling in Pseudomonas aeruginosa. J Bacteriol 183:5213–5222. 10.1128/JB.183.18.5213-5222.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Evans K, Passador L, Srikumar R, Tsang E, Nezezon J, Poole K. 1998. Influence of the MexAB-OprM multidrug efflux system on quorum sensing in Pseudomonas aeruginosa. J Bacteriol 180:5443–5447. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Martínez JL. 2012. Natural antibiotic resistance and contamination by antibiotic resistance determinants: the two ages in the evolution of resistance to antimicrobials. Front Microbiol 3:1. 10.3389/fmicb.2012.00001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Martinez JL. 2009. The role of natural environments in the evolution of resistance traits in pathogenic bacteria. Proc Biol Sci 276:2521–2530. 10.1098/rspb.2009.0320. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Baquero F, Alvarez-Ortega C, Martinez JL. 2009. Ecology and evolution of antibiotic resistance. Environ Microbiol Rep 1:469–476. 10.1111/j.1758-2229.2009.00053.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 128.Martínez JL. 2012. Bottlenecks in the transferability of antibiotic resistance from natural ecosystems to human bacterial pathogens. Front Microbiol 2:265. 10.3389/fmicb.2011.00265. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Berendonk TU, Manaia CM, Merlin C, Fatta-Kassinos D, Cytryn E, Walsh F, Bürgmann H, Sørum H, Norström M, Pons MN, Kreuzinger N, Huovinen P, Stefani S, Schwartz T, Kisand V, Baquero F, Martinez JL. 2015. Tackling antibiotic resistance: the environmental framework. Nat Rev Microbiol 13:310–317. 10.1038/nrmicro3439. [PubMed] [DOI] [PubMed] [Google Scholar]
- 130.Baquero F, Martínez JL, Cantón R. 2008. Antibiotics and antibiotic resistance in water environments. Curr Opin Biotechnol 19:260–265. 10.1016/j.copbio.2008.05.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 131.Cabello FC, Godfrey HP, Tomova A, Ivanova L, Dölz H, Millanao A, Buschmann AH. 2013. Antimicrobial use in aquaculture re-examined: its relevance to antimicrobial resistance and to animal and human health. Environ Microbiol 15:1917–1942. 10.1111/1462-2920.12134. [PubMed] [DOI] [PubMed] [Google Scholar]
- 132.Chen MY, Lira F, Liang HQ, Wu RT, Duan JH, Liao XP, Martínez JL, Liu YH, Sun J. 2016. Multilevel selection of bcrABDR-mediated bacitracin resistance in Enterococcus faecalis from chicken farms. Sci Rep 6:34895. 10.1038/srep34895. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Köhler CD, Dobrindt U. 2011. What defines extraintestinal pathogenic Escherichia coli? Int J Med Microbiol 301:642–647. 10.1016/j.ijmm.2011.09.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 134.San Millan A, Toll-Riera M, Qi Q, MacLean RC. 2015. Interactions between horizontally acquired genes create a fitness cost in Pseudomonas aeruginosa. Nat Commun 6:6845. 10.1038/ncomms7845. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Baltrus DA. 2013. Exploring the costs of horizontal gene transfer. Trends Ecol Evol 28:489–495. 10.1016/j.tree.2013.04.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 136.Starikova I, Harms K, Haugen P, Lunde TT, Primicerio R, Samuelsen Ø, Nielsen KM, Johnsen PJ. 2012. A trade-off between the fitness cost of functional integrases and long-term stability of integrons. PLoS Pathog 8:e1003043. 10.1371/journal.ppat.1003043. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Park C, Zhang J. 2012. High expression hampers horizontal gene transfer. Genome Biol Evol 4:523–532. 10.1093/gbe/evs030. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Johnsen PJ, Levin BR. 2010. Adjusting to alien genes. Mol Microbiol 75:1061–1063. 10.1111/j.1365-2958.2010.07075.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 139.Knöppel A, Lind PA, Lustig U, Näsvall J, Andersson DI. 2014. Minor fitness costs in an experimental model of horizontal gene transfer in bacteria. Mol Biol Evol 31:1220–1227. 10.1093/molbev/msu076. [PubMed] [DOI] [PubMed] [Google Scholar]
- 140.Schaufler K, Semmler T, Pickard DJ, de Toro M, de la Cruz F, Wieler LH, Ewers C, Guenther S. 2016. Carriage of extended-spectrum beta-lactamase-plasmids does not reduce fitness but enhances virulence in some strains of pandemic E. coli lineages. Front Microbiol 7:336. 10.3389/fmicb.2016.00336. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sánchez MB, Martínez JL. 2012. Differential epigenetic compatibility of qnr antibiotic resistance determinants with the chromosome of Escherichia coli. PLoS One 7:e35149. 10.1371/journal.pone.0035149. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Björkman J, Nagaev I, Berg OG, Hughes D, Andersson DI. 2000. Effects of environment on compensatory mutations to ameliorate costs of antibiotic resistance. Science 287:1479–1482. 10.1126/science.287.5457.1479. [PubMed] [DOI] [PubMed] [Google Scholar]
- 143.Handel A, Regoes RR, Antia R. 2006. The role of compensatory mutations in the emergence of drug resistance. PLoS Comput Biol 2:e137. 10.1371/journal.pcbi.0020137. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Maisnier-Patin S, Berg OG, Liljas L, Andersson DI. 2002. Compensatory adaptation to the deleterious effect of antibiotic resistance in Salmonella typhimurium. Mol Microbiol 46:355–366. 10.1046/j.1365-2958.2002.03173.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 145.Böttger EC, Springer B, Pletschette M, Sander P. 1998. Fitness of antibiotic-resistant microorganisms and compensatory mutations. Nat Med 4:1343–1344. 10.1038/3906. [PubMed] [DOI] [PubMed] [Google Scholar]
- 146.Hernando-Amado S, Sanz-García F, Blanco P, Martínez JL. 2017. Fitness costs associated with the acquisition of antibiotic resistance. Essays Biochem 61:37–48. 10.1042/EBC20160057. [PubMed] [DOI] [PubMed] [Google Scholar]
- 147.Martínez JL, Baquero F. 2014. Emergence and spread of antibiotic resistance: setting a parameter space. Ups J Med Sci 119:68–77. 10.3109/03009734.2014.901444. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fitzpatrick D, Walsh F. 2016. Antibiotic resistance genes across a wide variety of metagenomes. FEMS Microbiol Ecol 92:92. 10.1093/femsec/fiv168. [DOI] [PubMed] [Google Scholar]
- 149.Hu Y, Yang X, Qin J, Lu N, Cheng G, Wu N, Pan Y, Li J, Zhu L, Wang X, Meng Z, Zhao F, Liu D, Ma J, Qin N, Xiang C, Xiao Y, Li L, Yang H, Wang J, Yang R, Gao GF, Wang J, Zhu B. 2013. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun 4:2151. 10.1038/ncomms3151. [PubMed] [DOI] [PubMed] [Google Scholar]