

ABSTRACT
Microbiome analysis of environmental samples may represent the next frontier in environmental microbial forensics. Next-generation sequencing technologies significantly increased the available genetic data that could be used as evidentiary material. It is not clear, however, whether the microbiome can scale across institutions using forensic-based evidence due to the data resource requirements and the associated costs of maintaining these databases. A successful microbiome study is impacted by the quality of the information gathered and the steps in sample processing and data analysis. To ascertain the validity of methods and the results obtained, there needs to be a stringent procedure to validate the methods and ensure that the results are comparable and reproducible, not only within the laboratory but also between laboratories conducting similar research. Of primary importance for meaningful microbiome studies is an experimental design that leads to carefully executed, controlled, and reproducible studies. The microbiome literature contains a fair share of anecdotal descriptions of microbial community composition and “diagnostic” relative abundance of the taxa therein. These studies are now being supplemented by experimental designs that feature repeated measurements, error estimates, correlations of microbiota with covariates, and increasingly sophisticated statistical tests that enhance the robustness of data analysis and study conclusions. It is imperative to be careful, especially when carrying out attribution studies, to be fully aware of the possible biases included in a specific sample being analyzed.
INTRODUCTION
Microbiome analysis of environmental samples may represent the next frontier in environmental microbial forensics. The microbiome, defined as the sum total of all the genetic material present in a sample, contains evidence of the microbial communities in the sample at the time of collection. As such, it contains clues to past environmental events until the time the sample was collected and processed. This attribute makes the analysis of microbiomes extremely important in identifying and demonstrating the occurrence of an environmental event, be it bioterrorism or a petroleum spill.
The introduction of DNA technology in the form of fingerprinting and terminal restriction fragment analysis to microbial forensics rapidly expanded the available probative evidence that could be garnered from a contaminated site or a crime scene. Next-generation sequencing technologies significantly increased the available genetic data that could be used as evidentiary material. Next-generation sequencing of the human microbiome demonstrates that its bacterial DNA may be used to uniquely identify an individual, provide information about their life and behavioral patterns, determine the body site where a sample came from, and estimate postmortem intervals (1). Similarly, microbiome samples from the environment and/or contaminated sites can also be leveraged to address similar questions about environmental contamination events, their source, and their relative time of occurrence.
The applications of this new field in forensic science raises concerns about current methods used in sample collection, the necessary metadata associated with the sample, as well as storage, the statistical power of the sampling, and downstream sample processing. These areas of microbiome research need to be fully addressed before microbiome data can become a regularly incorporated type of evidence and possibly become a routine procedure as part of the microbial forensics toolkit.
While microbiome profiling could potentially serve as a complement to conventional environmental forensics methodology, it is not clear whether the microbiome can scale across institutions using forensic-based evidence due to the data resource requirements and the associated costs of maintaining these databases. One of the biggest challenges may be the site or subject specificity in terms of the microbiome, since data may not be easily applicable to all sites or subjects.
FROM SAMPLE ACQUISITION TO BIOINFORMATICS
A successful microbiome study is impacted by the quality of the information gathered and the steps in sample processing and data analysis. Figure 1 illustrates the salient steps in microbiome analysis.
FIGURE 1.
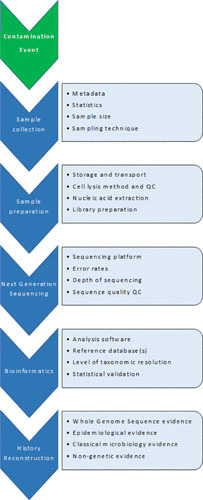
Technical factors in microbiome research that influence results and conclusions.
The rapid and exponential development of equipment, techniques, and scientific approaches to microbiome research, however, have led to a stochastic, nonstandardized approach to microbiome research. Consequently, there exists a large volume of data, gathered and interpreted in many ways, which are not readily comparable among laboratories and therefore are less useful than they otherwise could have been. In fact, some of the repositories of data may not be completely curated, raising concerns about their reliability.
To transition from basic research to environmental applications, technologies and computational methods for assessing human-, animal-, and environment-associated microbial communities must be standardized and quality controlled. This includes tools for sample collection and processing through to data generation and analysis (Fig. 1).
“Metadata” is a general term used to encompass all descriptors that qualify the individual site from which the environmental sample is obtained. To date, little work has been done on consensus definition of minimal sets of metadata, and international studies increase the complexity of the task due to differences in, e.g., regulatory requirements in different countries.
Sampling may perhaps be the most sensitive part of an environmental microbial forensics study. Without a representative sample, it would be very difficult to obtain reliable information; this becomes key whenever the study is one needed for legal attribution or for public health protection purposes. As mentioned previously, there are some standard protocols for obtaining representative samples, which in many cases may not be possible. This would be the case when trying to obtain an air or flowing water sample after a discrete event of, e.g., contamination.
A DNA extraction procedure is a key determinant in the success and quality of the microbiome study. This is often the most vulnerable step in assessing taxonomic composition and relative abundance in samples. Three elements play key roles in the ultimate success and reliability of the method: (i) microbial lysis, (ii) removal of contaminants, and (iii) the method of DNA recovery.
Bacterial lysis. Complex microbial communities are composed of diverse microorganisms that can differ dramatically in their resistance to lysis. Failure or inadequate DNA extraction at this first step of the procedure will lead to erroneous and/or inaccurate interpretations of the data. Microorganisms that are most resistant to lysis, such as Gram-positive cocci and methanogenic archaea, require harsher physical and chemical treatments than do Gram-negative bacteria. Conversely, harsher treatments may end up excessively denaturing the genomes of Gram-negative bacteria such as those in the Bacteroidetes group.
Contaminant removal. Environmental matrices such as fecal or soil samples often contain aromatic constituents, such as fecal sterols and humic/fulvic acids, that may coextract with DNA molecules. The latter will often inhibit enzymes and require high dilutions of extracts or further processing to allow for PCR amplification, which is required for library preparation. Higher dilutions of sample DNA might lead to reducing the concentration of DNA from certain poorly represented taxa, well below the detection limit of the method, and thus obfuscate the results.
DNA recovery mode. The classical mode of DNA recovery upon cell lysis involves alcohol precipitation (e.g., phenol-alcohol). Numerous commercially available kits have been designed and optimized for the extraction of nucleic acids from one or more sample types. Many involve mild yet thorough cell lysis conditions that involve both enzymatic (e.g., lysozyme) and mechanical disruption.
In addition to these technical factors, sample- and microbiome-specific factors further confound putative results, including taxonomic distribution and relative abundance. Such factors include:
Bacterial cell wall composition
Genome size and supercoiling
rRNA operon copy number
G+C composition
Other important factors include:
Choice of the variable region to be targeted for PCR in library preparation
Next-generation sequencing protocol
Analysis software used
QUALITY CONTROL OF METAGENOMIC ANALYSIS
To ascertain the validity of methods and the results obtained, there needs to be a stringent procedure to validate the methods and ensure that the results are comparable and reproducible, not only within the laboratory but also between laboratories conducting similar research. For this to become a reality, stringent quality control procedures need to be implemented. Quality control standards that can be used throughout the process must be incorporated in the various protocols to validate one or more of the steps in the microbiome project.
Next-generation sequencing applications are becoming increasingly popular in microbial forensics, to support microbial strain identification and for microbiome studies aimed at identifying unique environments based on microbial community composition. Currently, these observations are mainly based on taxonomic profiles as determined by comparisons of the relative abundance of operational taxonomic units from one microbiome to another. These are largely based on sequence information derived from small-subunit rRNA genes. Studies of the microbial community composition of such environments, also known as microbiomes, analyze and compare microbiomes based on the relative abundance of operational taxonomic units resulting from sequence analysis of metagenomic DNA extracted from such environments. Unfortunately, the peer-reviewed literature is replete with contradictory results on microbiome composition of the same or similar samples (2). Many of these observed contradictions may be a consequence of different methodologies for sample processing (3) or the result of sampling site and subject uniqueness and specificity. In addition, such inconsistencies may be the result of various technical factors, including sample collection and storage, specific microbiome composition of the sample, methodology of DNA extraction, library preparation, DNA sequencing platform, and bioinformatics pipeline.
All researchers know that it is critically important to start out with a proper experimental design if meaningful data are to be obtained in any experiment, but it becomes especially important for microbiome studies because of the tremendous amount of data obtained. Proper experimental design will lead to carefully executed and controlled experiments leading to reproducible results (4). As previously mentioned, there are many examples in the literature of microbial community descriptions coupled with “diagnostic” relative abundances of taxa. Studies are now being designed explicitly to take into consideration replicate analyses, error estimates, correlations with covariates, and sophisticated statistical analyses are used; this approach is leading to data and conclusions that are more reliable and comparable to other studies (5).
The issue of intra- and inter-laboratory reproducibility has always been a problem in science, and these problems are even more prevalent when it comes to sampling and sequencing data, however, this has not been systematically explored. Erroneous conclusions could be the result of these biases. Currently The Microbiome Quality Control (MBQC) project is trying to identify possible sources of variation in microbiome studies. If this can be addressed immediately, it will be possible to rapidly come up with a proper design and use of different positive and negative control strategies (6, 7).
As more research laboratories become involved in microbiome studies, the need for proper optimization as well as for standardizing sample collection and processing methods has become obvious. The latter are important to be sure of the robustness of the results (8, 9).This problem might be solved by the use of spike-in reference standards that would enable the detection of possible biases on the data output. It should be clear that any correlations, or differences detected as a result of biased data analyses will have a direct effect on the conclusions drawn on the environment and the microbiome of interest (10). Potential biases could be introduced at various stages of microbiome research including sample preparation, DNA extraction, PCR amplification and sequencing and/or data analysis. The use of internal standards including possibly mock microbial communities would most likely allow for normalization of the resulting data. The routine use of these standards would have the added benefit of obtaining quantitative data from what is now, at best a semi-quantitative, if not qualitative approach.
MICROBIAL METABOLOMICS
“Metabolomics” is the characterization of metabolites generated by one or more organisms in a given physiological and environmental context using a variety of methods such as mass spectroscopy, nuclear magnetic resonance, or other analytical methods (11). Microbial metabolomics studies the complete set of metabolites within one or more microorganisms in the environmental context in which they reside. In contrast to metagenomics, metabolomics gives an indication of the biological and metabolic processes that living organisms were carrying out at the time of sampling because it measures actual metabolites rather than DNA, which could have been present as a result of metabolic processes by microorganisms present prior to the sampling event.
Because the microbial community composition of a given environmental sample is a reflection of the environmental conditions in which the sample was found, metabolomics can provide first-hand information on the substrates that are currently being metabolized by the microbial community. This way, the metabolomics profiles of pristine and contaminated sites can be used to assesses whether or not a given environmental sample was contaminated and the contaminants responsible for such shifts in profiles.
FINAL COMMENTS
This is an exciting time for environmental microbial forensics; the price of the technology necessary for thorough analyses is becoming accessible to most laboratories, and thus the amount of data available is increasing exponentially. However, it is imperative to be careful, especially when carrying out attribution studies, if the analyst is not fully aware of the possible biases included in a specific sample being analyzed or relies on a biased database. Contamination during sampling or during analysis can be relatively easily controlled; however, the use of controls (such as seed-in standards), as previously mentioned, is key to drawing correct conclusions from the available data. The uniqueness of environmental sites and samples will play a role in the possible biases, and the analyst has to be aware of this. The use of commercially available kits is key to the standardization of, e.g., DNA extraction. However, the analyst also has to be aware of possible sample and site uniqueness that may present problems with, e.g., inhibitors. Whenever dealing with a precious sample, such as an ancient artifact, the analyst has to take into consideration many variables that will affect what is and what is not detected.
REFERENCES
- 1.Metcalf JL, Xu ZZ, Bouslimani A, Dorrestein P, Carter DO, Knight R. 2017. Microbiome tools for forensic science. Trends Biotechnol 35:814–823. [PubMed] [DOI] [PubMed] [Google Scholar]
- 2.Stämmler F, Gläsner J, Hiergeist A, Holler E, Weber D, Oefner PJ, Gessner A, Spang R. 2016. Adjusting microbiome profiles for differences in microbial load by spike-in bacteria. Microbiome 4:28. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorzelak MA, Gill SK, Tasnim N, Ahmadi-Vand Z, Jay M, Gibson DL. 2015. Methods for improving human gut microbiome data by reducing variability through sample processing and storage of stool. PLoS One 10:e0134802. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, Caporaso JG, Knight R, Ley RE. 2014. Conducting a microbiome study. Cell 158:250–262. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.La Rosa PS, Brooks JP, Deych E, Boone EL, Edwards DJ, Wang Q, Sodergren E, Weinstock G, Shannon WD. 2012. Hypothesis testing and power calculations for taxonomic-based human microbiome data. PLoS One 7:e52078. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sinha R, Abnet CC, White O, Knight R, Huttenhower C. 2015. The microbiome quality control project: baseline study design and future directions. Genome Biol 16:276. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hiergeist A, Reischl U, Gessner A, Priority Program 1656 Intestinal Microbiota Consortium/Quality Assessment Participants. 2016. Multicenter quality assessment of 16S ribosomal DNA-sequencing for microbiome analyses reveals high inter-center variability. Int J Med Microbiol 306:334–342. [PubMed] [DOI] [PubMed] [Google Scholar]
- 8.Kim D, Hofstaedter CE, Zhao C, Mattei L, Tanes C, Clarke E, Lauder A, Sherrill-Mix S, Chehoud C, Kelsen J, Conrad M, Collman RG, Baldassano R, Bushman FD, Bittinger K. 2017. Optimizing methods and dodging pitfalls in microbiome research. Microbiome 5:52. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Endrullat C, Glökler J, Franke P, Frohme M. 2016. Standardization and quality management in next-generation sequencing. Appl Transl Genomics 10:2–9. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tourlousse DM, Yoshiike S, Ohashi A, Matsukura S, Noda N, Sekiguchi Y. 2017. Synthetic spike-in standards for high-throughput 16S rRNA gene amplicon sequencing. Nucleic Acids Res 45:e23–e23. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castillo-Peinado LS, Luque de Castro MD. 2016. Present and foreseeable future of metabolomics in forensic analysis. Anal Chim Acta 925:1–15. [PubMed] [DOI] [PubMed] [Google Scholar]
- 12.Alvarez AJ, Khanna M, Toranzos GA, Stotzky G. 1998. Amplification of DNA bound on clay minerals. Mol Ecol 7:775–778. [Google Scholar]
- 13.Alvarez AJ, Yumet GM, Santiago CL, Toranzos GA. 1996. Stability of manipulated plasmid DNA in aquatic environments. Environ Toxicol Water Qual 11:129–135. [Google Scholar]
- 14.Bohmann K, Evans A, Gilbert MT, Carvalho GR, Creer S, Knapp M, Yu DW, de Bruyn M. 2014. Environmental DNA for wildlife biology and biodiversity monitoring. Trends Ecol Evol 29:358–367. (Erratum, 10.1016/j.tree.2014.05.012.) [DOI] [PubMed] [Google Scholar]
- 15.Budowle B, Murch R, Chakraborty R. 2005. Microbial forensics: the next forensic challenge. Int J Legal Med 119:317–330. [PubMed] [DOI] [PubMed] [Google Scholar]
- 16.Budowle B. 2003. Defining a new forensic discipline: microbial forensics. Profiles DNA 6:7–10. [Google Scholar]
- 17.Cano RJ, Rivera-Perez J, Toranzos GA, Santiago-Rodriguez TM, Narganes-Storde YM, Chanlatte-Baik L, García-Roldán E, Bunkley-Williams L, Massey SE. 2014. Paleomicrobiology: revealing fecal microbiomes of ancient indigenous cultures. PLoS One 9:e106833. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toranzos GA, Santiago-Rodriguez TM, Cano RJ, Fornaciari G. 2017. Proper authentication of ancient DNA is essential, yes; but so are undogmatic approaches. FEMS Microbiol Ecol 93:fix043. [PubMed] [DOI] [PubMed] [Google Scholar]
- 19.Patrício AR, Herbst LH, Duarte A, Vélez-Zuazo X, Santos Loureiro N, Pereira N, Tavares L, Toranzos GA. 2012. Global phylogeography and evolution of chelonid fibropapilloma-associated herpesvirus 1. J Gen Virol 93:1035–1045. [PubMed] [DOI] [PubMed] [Google Scholar]
- 20.Piñar G, Piombino-Mascali D, Maixner F, Zink A, Sterflinger K. 2013. Microbial survey of the mummies from the Capuchin Catacombs of Palermo, Italy: biodeterioration risk and contamination of the indoor air. FEMS Microbiol Ecol 86:341–356. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.von Wintzingerode F, Göbel UB, Stackebrandt E. 1997. Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev 21:213–229. [PubMed] [DOI] [PubMed] [Google Scholar]