

The title of this topic is a bit of an oxymoron—antiarrhythmic therapy is thought to be neither safe nor effective—but if you understand the medications and their mechanisms of action, you can use them both safely and effectively.
The QRS that we see on the surface electrocardiogram is the sum of the action potentials of each type of myocardial tissue (Fig. 1). A very important point is that Phase Zero (depolarization of the myocardial cell) is caused by sodium going from the outside of the cell to the inside of the cell. On the other hand, Phase Three, which is repolarization (the cell going back to the resting phase), is primarily determined by potassium going from the inside to the outside of the cell. If you block potassium from leaving the cell, you prolong repolarization and increase the action potential duration. Conversely, if you block sodium from going into the cell, you not only decrease the overall velocity of conduction, but you also cause the QRS duration to widen because you slow depolarization. If the action potential duration widens, the QT interval widens. I'd like to now focus on the 2 most common classes of antiarrhythmic medications that affect these processes.
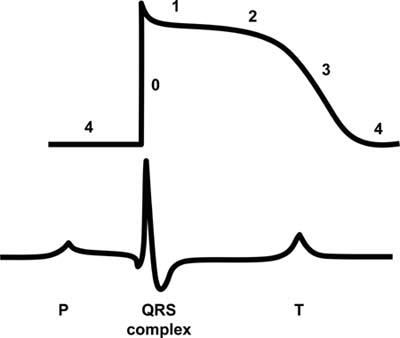
Fig. 1 Ventricular action potential
Class I: Blocking the Sodium Channel
The class I antiarrhythmics all share the ability to block the sodium channel (Fig. 2) and impair transport of the sodium ions into the cell; this has 2 major effects. First, let's look at the slowing of conduction. Slowing of conduction can be a very good thing, because if you slow conduction enough, you can terminate an arrhythmia; but you also hope that you don't cause new areas of abnormal conduction and cause pro-arrhythmic effects. So, in general, class I antiarrhythmics, which block sodium channels and slow down conduction, should be used in treating non–life-threatening arrhythmias (such as AF or SVT) in patients with structurally normal hearts, free of ischemia, infarction, valvular pathology, hypertrophy, or conduction system disease. Structural abnormalities exacerbate the risk of pro-arrhythmia. As I mentioned earlier, slowing conduction increases refractoriness and increases the likelihood of terminating a re-entrant circuit and returning to sinus rhythm.
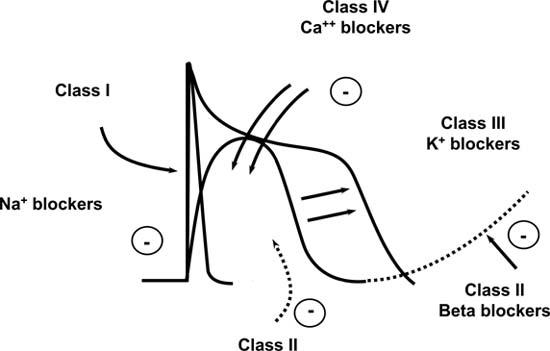
Fig. 2 Vaughan Williams classification of antiarrhythmic drugs
Another concept that I'd like to highlight is “use-dependence.” Some medications bind to their target channels more effectively when the channel is stimulated at a fast rate. In other words, these medications are more potent at faster heart rates.
Conversely, other drugs work exactly the opposite (reverse use-dependence). They bind to the channel more avidly at slower rates of activation and are more potent at slower heart rates. A drug that is use-dependent, such as lidocaine, will be very effective in terminating an arrhythmia while the patient is actually in tachycardia. Conversely, a drug that is reverse use-dependent (such as sotalol) is probably going to be very effective in preventing an arrhythmia when the heart rate is not fast, but will be very ineffective in terminating the arrhythmia once it has started.
Flecainide is the prototypical class I agent and a very potent sodium channel blocker. Blocking the sodium channel can actually cause pacing thresholds to increase. So when you have a patient who is pacemaker dependent or already has a pacemaker, and you start that patient on flecainide, be aware that the pacing threshold can increase—sometimes dramatically. As can be expected because of the underlying mechanism of action, slowing of conduction can become a substrate for malignant arrhythmias, as shown in the CAST study. You can use class I agents safely, but only in the appropriate setting, because they're very effective. The usual dosage of flecainide can start at 50 mg and go up to 150 mg b.i.d. Watch for QRS prolongation. A QRS increase of 15% to 20% indicates a pharmacologic effect and does not mean that you need to reduce the dosage, assuming that there was no underlying conduction system disease to begin with. If you have a greater than 15% to 20% increase in the QRS duration, you should reduce the dosage. Flecainide can also be used with a single oral loading dose of 300 mg (the “pill in the pocket” method) to convert atrial fibrillation. Remember that flecainide is a use-dependent drug. When you start a patient on flecainide, it is very prudent to do a treadmill stress test after the patient is fully loaded. You want to make sure that the QRS duration that increased by 15 milliseconds at rest does not end up increasing by 40 milliseconds when the patient goes out jogging, because a use-dependent drug will have more effect at faster heart rates. Flecainide is very effective in treating atrial fibrillation and atrial tachycardia. It is also very, very effective in treating triggered PVCs in a structurally normal heart. Of all the agents, it is probably one of the most potent. It carries a minimal risk of pro-arrhythmia in a structurally normal heart.
Sodium blockade may be slightly less potent with propafenone. As with flecainide, you have to watch for QRS prolongation at rest and during exercise and beware of increasing pacing thresholds. Propafenone also has a significant beta-blockade effect, and the AV nodal conduction system can be affected as well as the myocardial conduction system. So you have to be very, very careful when you use this drug on anyone who has underlying AV or ventricular conduction system disease. It is absorbed best with food. GI side effects are frequent (25%–30%), but usually transient. Some patients complain of a metallic taste on occasion when they consume dairy products. Inhibitors of the hepatic cytochrome p450 system can lead to increased levels of the drug. Because of its beta-blockade effect, propafenone can exacerbate asthma in healthy, younger patients. Both beta-blockade and the sodium channel blockade can be associated with blurred vision and paresthesias. The dosages for propafenone are 150, 225, or 300 mg b.i.d. or t.i.d. There's also a new b.i.d. sustained-release formulation.
Disopyramide is a class I agent that has the unique feature of being extremely effective for treating vagally induced atrial fibrillation, because of its anticholinergic side effects. But as can be expected, in older men it is not the ideal medication because of side effects. The incidence of proarrhythmia is about 3%, and there is an association with increased insulin secretion.
Class III: Blocking the Potassium Channel
Class III agents (Fig. 2) block the potassium channel and also prolong action potential. The action potential duration is reflected as the QT interval on the surface electrocardiogram. Therefore, class III agents have the universal effect of prolonging the QT duration and the risk of torsades.
Amiodarone is probably one of the most potent antiarrhythmic medications that we have available. In terms of its cardiac side-effect profile, it is actually a very safe, very effective ion channel blocker. The intravenous formulation is also a strong beta-blocker, and the initial effect of IV amiodarone is more prominent in terms of beta-blockade than of potassium-channel-blockade. The oral formulation also has a very long half life. Unfortunately, it also has side effects; up to 40% of patients will discontinue use within 2 years because of side effects. However, most of these side effects are dose dependent. Recently, dosages of amiodarone in common use have been decreasing, and so has the reported incidence of side effects. The most frequent side effect is GI intolerance, most often when you're loading with higher doses; as the maintenance doses are reduced, the symptoms improve. The patient needs 10 grams of oral amiodarone orally to become fully loaded. You can do this safely, if you do it gradually—for example, 400 mg per day for 3 weeks. You can do it quicker if you want to, such as 400 mg 3 times a day for 1 week. Most physicians would probably feel more comfortable monitoring patients treated with loading doses like these. Because of the homogenous blockade of potassium and other channels, the risk of torsades is relatively minor. It takes just as long for amiodarone to be removed from the system as it takes to accumulate; this becomes very important when you're trying to switch a patient from amiodarone to dofetilide.
Amiodarone is an iodine-rich benzofuran derivative with a molecular structure somewhat similar to that of thyroid hormone. Almost 40% of the molecular weight of amiodarone is iodine. Hence, chronic amiodarone therapy is associated with a tremendous iodine load. Iodine is not only necessary for thyroid hormone synthesis, it also influences intrathyroidal processes. It can also penetrate other tissues and be associated with toxicities of multiple organ systems. Side effects include hyper- and hypothyroidism, congestive and inflammatory hepatopathy, and corneal deposits (benign and seen in nearly all patients). A very rare complication is idiosyncratic optic neuritis; irreversible and unpredictable, it can occur after the first dose. One in about 5,000 patients will get this. There are 2 forms of pulmonary toxicity. There's an inflammatory acute idiosyncratic condition that can occur after 2 to 3 weeks, manifesting as cough, dyspnea, and fevers; respiratory decompensation can occur. The CT scan shows specific changes, and there are indications of an acute inflammatory process. This acute response can be irreversible, somewhat like the optic neuritis. There is also a more chronic pulmonary fibrosis. This is dose dependent and occurs over longer periods of time when patients have been on amiodarone for years. Bronchial alveolar lavage can show the characteristic amiodarone-containing phagocytes. The CT scan really shows interstitial changes, and the PFTs show restrictive physiology, which is not reversible.
Skin deposits can occur and are usually benign but not usually reversible. Photosensitivity is independent of the discoloration and can also occur in these patients. Peripheral neuropathy and tremors have been noted, usually at doses of 400 mg or above. With the recent use of 200 mg a day, these toxicities have declined. Screening tests with PFTs, TSH, LFTs, chest x-ray, and an annual eye exam are standard. We usually do all studies other than the PFTs and the eye exam every 3 months.
Sotalol is less potent than amiodarone as a potassium channel blocker, but more potent as a beta-blocker. Because of its significant reverse use-dependence, it is ineffective in terminating AF but extremely effective in preventing AF. The strongest effects can occur at slower heart rates; so when you put patients on sotalol, you make sure that you get an ECG, and measure the QT interval when they're in sinus rhythm, not while they are still in atrial fibrillation. Sotalol is also very effective in VT. We have been starting at a lower dose of 80 mg b.i.d., because some patients have an idiosyncratic hyperresponsiveness to sotalol with regard to QT prolongation. Increases in QT interval are noted even with lower doses, and this usually happens after the first couple of doses. We start at 80 b.i.d. for the first day, and then work our way up to the final dosage. Dose adjustments are necessary in renal but not in hepatic dysfunction. Sotalol is not absorbed well with dairy products, and it must be taken on an empty stomach for maximal absorption.
Dofetilide is a pure potassium channel blocker and extremely effective. It is safe (relatively speaking) to use in patients with structural heart disease. The risk of torsades is about 4%; inpatient oral loading is required and you need an electrocardiogram 2 to 3 hours after each dose. There are minimal extracardiac effects, and, in this sense, dofetilide provides almost a mirror image of what you see with amiodarone. It needs to be adjusted in the presence of renal dysfunction, and there are a number of medications with which it can't be used. Amiodarone needs to be discontinued 2 months before starting dofetilide. Verapamil is contraindicated. In general, we use dofetilide only in the highly compliant and motivated patient. With dofetilide, if you miss one dose, you cannot double up on the next. It's now recommended that patients who miss 2 doses be required to come back into the hospital and be reloaded. It is also reverse use-dependent, so you need to be alert for problems in patients with slower heart rates.
Ibutilide blocks potassium channels and activates a separate background sodium channel—entirely different from the sodium channel that I was talking about earlier. It thus prolongs the QT interval. It is an extremely effective agent and probably the most effective pharmacologic means of converting atrial fibrillation and flutter. We don't use it for permanent atrial fibrillation. The incidence of torsades is 4%. Some advocate the prophylactic use of magnesium with ibutilide. Before beginning ibutilide, it is at least a good idea to check magnesium levels, in addition to all the other electrolytes. Patients need to be on a cardiac monitor for 4 hours after the administration of ibutilide. It is given as 1 mg over 10 minutes, although in the EP lab, we may give it over 5 minutes. If you see no effect, you can repeat it once, 30 minutes later.
Footnotes
Presented at the Texas Heart Institute's Sixth Symposium on Cardiac Arrhythmias: New Pharmacologic and Interventional Strategies; held at the Houstonian Hotel, Club & Spa; 19 February 2005; Houston. E-mail: mehdirazavi@mailsnare.net