

Abstract
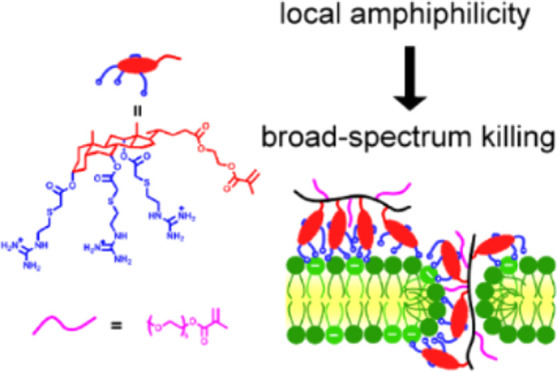
In this study, we report the design of a new guanylated, cholic-acid-based monomer (GM) to combat antimicrobial resistance. The microbial activity stems from the interfacial amphiphilicity of cholic acid, while guanidine shows a strong association with phosphate, which promotes binding to membrane phospholipids. The monomer showed strong antimicrobial activity; however, surprisingly, homopolymers synthesized by photoiniferter reversible addition–fragmentation chain-transfer (RAFT) polymerization of GM completely lost their activity likely due to the conformation of the polymer. In contrast, the design of GM copolymers with poly(ethylene glycol) methyl ether methacrylate (PEGMA) or 2-hydroxyethyl methacrylate (HEMA) allowed recovery of their antimicrobial activity. Due to the existence of cholesterol in cell membranes, hemolysis was highly dependent on the content of GM incorporated. This study highlights the unique and intriguing properties of this novel amphiphilic monomer and its polymers, providing valuable insights into the development of more potent antimicrobial materials.
Introduction
Antimicrobial resistance (AMR) caused by both Gram-positive and Gram-negative bacteria is a growing threat to public health worldwide. It is estimated that in 2019, there were 4.95 million AMR-related deaths, among which 1.27 million deaths could be directly attributed to AMR, making AMR the third leading cause of death after ischemic heart disease and stroke.1 The misuse of antibiotics both in the community and the agricultural industry accelerates the spread of AMR,2 requiring greater regulation and the development of new antibiotics. Such development has stagnated due to both scientific challenges and low economic incentives.3 Despite improvements in new antibacterial drug development over the past decade, it is still sparse compared to other diseases in fields such as oncology.4 To keep up with the development of AMR, new therapeutic strategies are urgently required.
Antimicrobial peptides (AMPs) are part of the natural defense line against pathogen inactivation and thus have drawn attention as alternatives to antibiotics due to broad bacterial susceptibility.5 AMPs are positively charged amphiphilic molecules that can selectively bind to bacteria and kill them through membrane disruption.6 Due to the high production cost of peptides by gene-modified microorganisms or solid-phase peptide synthesis, synthetic polymers similar to AMPs have been extensively explored as potential alternatives. Numerous studies have shown that the combination of hydrophobic and cationic functionalities is vital for compromising the bacterial membrane.7
Amphiphilicity can be introduced through naturally occurring amphiphilic structures. Cholic acid, a steroid found in mammals and vertebrates, has rigid hydrophobic tetracyclic rings and three hydrophilic hydroxyl groups on its concave face. This forms a facial amphiphilic structure similar to AMPs.8 Such amphiphilicity endows cholic acid derivatives with the ability to penetrate bacterial membranes, making them a useful antimicrobial material. For example, ceragenins were developed as antimicrobial materials using a cholic acid scaffold and appended amine groups.9 Polymers with cholic acid pendants have also been reported.10
Arginine-rich peptides such as TAT (HIV-1 Trans-Activator of Transcription) have been found to have membrane translocation properties.11 Compared to other cationic groups such as the primary amine in lysine or the imidazole unit in histidine, the guanidine group is thought to be the key to these translocation properties, as it can form a very stable bidentate hydrogen bond with phosphate and sulfate.12 Inspired by this, several guanidine-based polymers have been investigated for their antimicrobial activities.8,13
In this study, we designed a novel guanidine-cholic-acid-based monomer (GM) as an AMP mimic molecule. Using photoiniferter reversible addition–fragmentation chain-transfer (RAFT) polymerization, a series of homopolymers and copolymers were synthesized, and their antibacterial activity against both Gram-positive and Gram-negative bacteria was investigated. Despite the high potency of the monomer, the resultant homopolymers were found to lose their activity possibly due to steroid ring stacking, which reduced their amphiphilicity. However, polymeric antimicrobial activity was recovered through copolymerization with hydrophilic monomers such as poly(ethylene glycol) methyl ether methacrylate (PEGMA) and 2-hydroxyethyl methacrylate (HEMA). The copolymers with more hydrophilic PEGMA showed strong activity against both Gram-positive and Gram-negative bacteria, with the exception of Gram-negative bacteria rich in lipopolysaccharides (LPS). Interestingly, cell hemolysis, which reflects mammalian cell toxicity, varied with the amount of GM incorporated into the copolymers, likely due to the similarity of the cholic acid ring structure with cholesterol. This allowed us to lower the toxicity of copolymers by decreasing the amount of GM incorporated while maintaining the antimicrobial activity. This work demonstrates that tuning the selectivity of these systems for bacteria over human cells could be achieved based on the copolymer design. We believe that these systems illustrate interesting trends for optimizing antimicrobial activity and have a strong potential for further application as antimicrobial materials.
Results and Discussion
Synthesis of Boc-Protected GM Monomer
The Boc-protected guanidine, cholic acid-based monomer was synthesized via three steps (Scheme 1). First, the methacrylate group was introduced through the standard esterification of HEMA (II) and cholic acid with EDC (1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide) and DMAP (4-dimethylaminopyridine). Second, the three hydroxyl groups were esterified with bromoacetyl bromide (III) to introduce the bromide handles for further functionalization. With extensive trials, we found that the three hydroxy groups could not be fully converted in one-step even with a significant excess of bromoacetyl bromide; however, the reaction reached full conversion even for the most sterically hindered and less reactive hydroxyl groups after repeating the reaction 3 times. The ideal base in this reaction was found to be potassium carbonate (for example, triethylamine reacted with the resultant bromide rendering a quaternary amine). Third, the resulting bromides (Tri-Br-MAECA) were subsequently substituted by thiol (IV) to afford the tri-substituted Boc-protected guanidine-capped (2-methacryloyloxy) ethyl cholate (Tri-Boc-Gua-MAECA), which was compatible with RAFT polymerization. The structure of Tri-Boc-Gua-MAECA was confirmed by NMR (Figures S6–S9) and mass spectrometry (Figure S10). A primary amine counterpart, designed to act as a control, was also synthesized in a similar way, which was confirmed by NMR (Figure S11).
Scheme 1. Synthetic Scheme for the Synthesis of Boc-Protected Guanidine-Functionalized Monomer through: (a) Esterification of Cholic Acid (I) with HEMA (II) to Form (2-Methacryloyloxy) Ethyl Cholate (MAECA); (b) Subsequent Esterification with Bromoacetyl Bromide (III) to Form Tri-Substituted Bromo-Ended (2-Methacryloyloxy) Ethyl Cholate (Tri-Br-MAECA); and (c) Substitution with Thiol (IV) to Yield Tri-Substituted Boc-Protected Guanidine-Capped (2-Methacryloyloxy) Ethyl Cholate (Tri-Boc-Gua-MAECA).

Polymerization and Characterization
For the polymerization of this novel guanidine-functionalized monomer, 4-cyano-4-[(dodecyl-sulfanylthiocarbonyl)-sulfanyl] pentanoic acid (CDTPA) was chosen as the RAFT agent due to its extensive use in both conventional thermoinitiated polymerization and photoiniferter RAFT polymerization, where it effectively initiates and controls the polymerization of methacrylates.14 The use of a more conventional initiator, azobis(isobutyronitrile) (AIBN), at 65 °C for 24 h resulted in limited polymerization with less than 30% conversion. This was likely due to the size of the monomer (∼1599 Da), which dramatically slowed the rate of polymerization. Fortunately, photopolymerization was found to be performed at a much faster rate. Thus, all polymerizations were conducted using a handmade light-emitting diode (LED) light source (commercial stripe light (60 LEDs/m, 12 V, 3 m total length, 14.4 W) wrapped around a plastic 15 cm diameter cylinder).15 A fan was also employed to control the ambient temperature.
To demonstrate that the photolysis of CDTPA could initiate RAFT polymerization, five separate polymerizations were conducted under the same conditions ([M] = 0.125 M, [RAFT] = 6.25 mM in dimethylformamide (DMF), room temperature, blue light, [M] refers to the monomer concentration, and [RAFT] refers to the CDTPA concentration) and terminated at different times. The results were measured by 1H NMR and are shown in Figure S42. A pseudo-first-order kinetic reaction was confirmed, indicating controlled radical concentrations throughout the polymerization. This proved that CDTPA was suitable for photoiniferter RAFT polymerization. However, the dispersity (Đ) measured via size exclusion chromatography (SEC) was found to be around 3.1 (Table S1), indicating broader molecular weight distributions suggesting the polymerization was not well controlled.
To achieve better control over the dispersity, a higher concentration of CDTPA was used. As shown in Table S1, doubling the concentration of CDTPA resulted in a decrease in dispersity from 3.12 to 1.35. This is because CDTPA served as both the photoinitiator for initiation and the chain-transfer agent (CTA) for controlling the polymerization. At lower concentrations, most of the CDTPA is converted into radicals by light, whereas at higher concentrations, most CDTPA remains undecomposed and thus functions effectively as a CTA. This phenomenon was previously observed by Bai et al.16 We further explored the ideal conditions for synthesizing polymers of different lengths. When the concentration of monomer was lowered, the rate of polymerization was reduced, resulting in a very low conversion, low dispersity, and ultimately, a shorter polymer. On the other hand, increasing the concentration of CDTPA yielded higher monomer conversion while maintaining relatively lower dispersity.
A series of Boc-protected homopolymers, PBG3, PBG6, and PBG14, were then synthesized via photoiniferter RAFT polymerization (Table 1 and Figures S12–S14). The guanidine group was recovered by the deprotection of the Boc group using trifluoroacetic acid (TFA), which was confirmed by 1H NMR (Figure S17–S19) and Fourier-transform infrared (FT-IR) spectroscopy (Figures S22 and S23). Similarly, a Boc-protected primary amine polymer (PBA9) was also synthesized for use as a control. All of the polymers with variable lengths (DP = 3, 6, and 14 for guanidine and 9 for primary amine) were characterized by SEC. The polymers had a narrow dispersity (Đ = 1.16–1.35), showing that the polymerization was well controlled (Table 1).
Table 1. Polymer Characterization.
| monomer/CDTPA ratio | Mn (SEC)a (kDa) | DP | Mn (theor.)b (kDa) | Đa | |
|---|---|---|---|---|---|
| PBG3 | 5 | 8.8 | 3 | 4.8 | 1.16 |
| PBG6 | 10 | 10.7 | 6 | 10.6 | 1.20 |
| PBG14 | 20 | 11.5 | 14 | 22.8 | 1.35 |
| PBA9 | 10 | 10.4 | 9 | 10.8 | 1.21 |
Mn and Đ were determined by SEC analysis in THF.
Mn and DP (degree of polymerization) were determined from the crude 1H NMR peak integration analysis.
Following deprotection, the homopolymers (PG3,6,14 (guanidine) and PA9 (amine)) were in their TFA salt form and had good solubility in water. However, when an attempt was made to dissolve the polymers into phosphate-buffered saline (PBS), at pH 7.4, precipitation occurred. We tested the polymers in either phosphate buffer or 150 mM NaCl solution and found that the precipitation only happened in phosphate buffer. This is probably due to the strong association between guanidine or primary amine groups in the polymers and phosphate ions in the PBS buffer solution. This showed that the cholic acid scaffold resulted in a stronger interaction between the amine groups and phosphate ions. Thus, in later work, we used Tris-buffered saline to dissolve the polymers instead of PBS.
Antimicrobial Activities of Homopolymers
The antimicrobial activities of the polymers and corresponding monomers were evaluated using the minimum inhibitory concentration (MIC) assay against clinically relevant Gram-positive Staphylococcus aureus 29213 and Gram-negative Escherichia coli 25922 (Figure 1). Both monomers with either guanidine (GM) or primary amines (AM) (Figure S20) resulted in low MICs against both S. aureus (16 μg/mL) and E. coli (32 μg/mL). There was no significant difference between the guanidine and amine functional groups used here.
Figure 1.
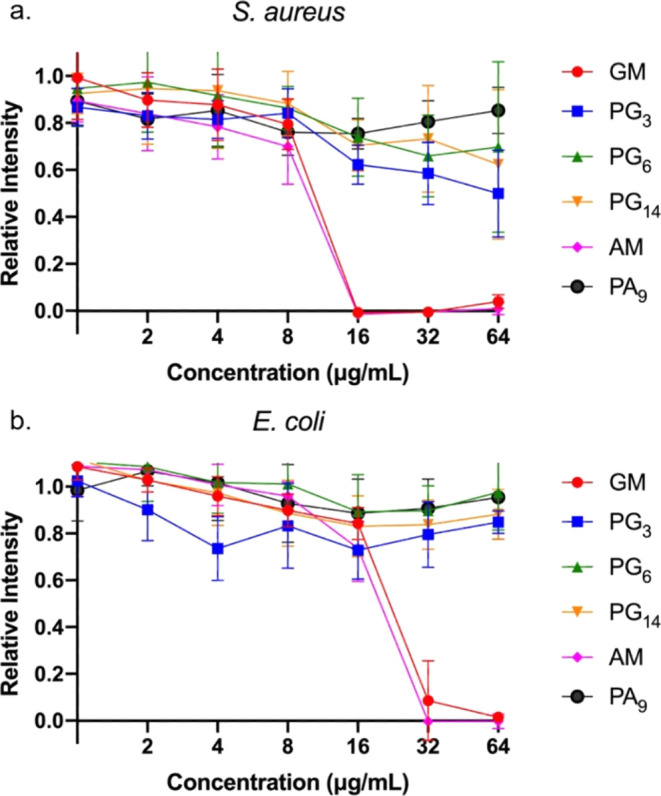
Relative bioluminescence intensity of (a) S. aureus and (b) E. coli after treatment of polymers PG3–14, PA9, and corresponding monomers GM and AM at 37 °C after 24 h.
However, none of the four homopolymers showed any activity against either bacterial strain (Figure 1). This was surprising, as it is usually observed that increasing the density of the functional groups in polymerization improves the activity due to multivalent interactions.17 Moreover, the homopolymer PG3, which only contained 3 repeating units and hence resembled an oligomer, showed no difference in activity compared to the other longer homopolymers. This is an unusual result, as one would expect such a small polymer to behave similarly to the monomer, suggesting a dramatic difference in structure between the monomer and the homopolymer.
One possible explanation for this notable loss of activity could be the loss of local amphiphilicity in the polymer. The similarity of the guanidine monomer (GM) and primary amine monomer (AM) showed that the facial amphiphilicity from cholic acid was essential to its activity. However, steroids are known to self-assemble due to their rigid tetracyclic ring system. For example, cholesterol, one of the most well-studied steroids, can be modified to endow liquid crystal properties.18 Bile acid derivates such as tripodal cholamide, on the other hand, tend to form hydrogels.19 In the case of the homopolymer, the rigid tetracyclic ring units were in proximity to each other, increasing the probability of ring stacking. Furthermore, the positively charged guanidine/amine moieties, which could prevent this ring stacking, were distanced from the cholic acid rings through their long flexible spacer. The resulting steroid ring stacking likely has a detrimental effect on the antimicrobial activity of the polymers. Similar phenomena have been observed elsewhere. For example, the Boyer group found that polymer sequence was a major contributor to antimicrobial activity.20 When the amphiphilic polymers were random, they could adopt a random coil form that was amphiphilic and membrane active (Figure S44a). However, in block polymers, micelles were formed due to self-assembly, and the separation of the cationic domain and hydrophobic domain resulted in the loss of activity (Figure S44b). Similarly, we postulate that a change of conformation whereby the tetracyclic rings stack would also form a hydrophobic core surrounded by cationic tails. The separation of charge and the hydrophobic domain may have the same impact as micelle formation, resulting in a loss of activity (Figure S44d).
Synthesis and Antimicrobial Activities of Copolymers with PEGMA and HEMA
In an attempt to revive the polymers’ activity, copolymerization with three hydrophilic monomers, PEGMAS (300 Da), PEGMAL (500 Da), and HEMA, was conducted, where S and L stand for short and long, respectively. The purpose of this copolymerization was to change the conformation of the polymer chain in solution, alleviating the potential ring stacking and thus maintaining facial amphiphilicity.
A library of copolymers with either PEGMAS or PEGMAL and HEMA were synthesized. To understand the influence of the hydrophilic comonomers, the molar ratios were tuned from 40 to 90%. The length (DP) of the polymers was around 8. All of the polymers were characterized by NMR (Figures S24–S41) and SEC (Table 2). The polymers had a higher Đ value than the homopolymers, which was possibly caused by the size differences between the two comonomers used. All of the copolymers were named using the number of repeat units as subscripts; for example, PPEGS3G5 refers to a copolymer with three 300 Da PEGMA (PEGS) units and five guanidine monomer (G) units.
Table 2. Characterization of Copolymers Synthesized with PEGMAS, HEMA, and PEGMAL.
| comonomerd ratio (%) | DP | Mn (theor.)b (kDa) | Đa | |
|---|---|---|---|---|
| PPEGS3G5c | 40 | 9 | 8.5 | 1.42 |
| PPEGS5G3 | 60 | 8 | 6.3 | 1.62 |
| PPEGS6G2 | 80 | 8 | 4.5 | 1.39 |
| PPEGS7G1 | 90 | 7 | 3.3 | 1.33 |
| PHEMA3G5 | 40 | 9 | 7.8 | 1.42 |
| PHEMA5G3 | 60 | 8 | 5.4 | 1.41 |
| PHEMA6G2 | 80 | 8 | 3.4 | 1.36 |
| PHEMA7G1 | 90 | 7 | 2.2 | 1.80 |
| PPEGL6G2 | 80 | 8 | 5.7 | 1.81 |
Đ was calculated from SEC characterization using THF as the mobile phase.
Mn and DP were calculated from the crude 1H NMR and the ratio of monomer/CDTPA.
Subscripts denote the number of repeat units.
Comonomers used other than GM, including PEGMAS, HEMA, and PEGMAL.
The antimicrobial activities of these copolymers and GM were then evaluated using their MIC against a broad spectrum of Gram-negative strains including Acinetobacter baumannii 5075, A. baumannii 5075D, A. baumannii 03–149.1, A. baumannii 03–149.2, Klebsiella pneumoniae B5055, K. pneumoniae B5055 nm, Pseudomonas aeruginosa 19147 nm, P. aeruginosa 27853, E. coli DC10B, andE. coli DH5α; and Gram-positive strains including S. aureus 29213 and Methicillin-resistantS. aureus (MRSA) 43300 (Figure 2).
Figure 2.

Heat map of MICs for GM and its copolymers showing antimicrobial activity. The bacterial strains are A. baumannii 5075, A. baumannii 5075D (colistin-resistant), A. baumannii 03–149.1 (polymyxin susceptible), A. baumannii 03–149.2 (polymyxin resistant), K. pneumoniae B5055, K. pneumoniae B5055 nm (colistin-resistant), P. aeruginosa 19147 nm (colistin-resistant), P. aeruginosa 27853, E. coli DC10B, E. coli DH5α, S. aureus 29213, and M.-ResistantS. aureus (MRSA) 43300. The MIC was determined by using a standard microbroth dilution method. In the copolymers, the monomers are depicted in different colors, GM (purple), PEGMAS (light blue), PEGMAL (dark blue), and HEMA (orange).
We first evaluated how the hydrophilic monomers affected the antimicrobial activity. In general, we observed that PEGMAS outperformed HEMA regardless of the amount incorporated. This is notable, as the content of GM in the HEMA copolymers was higher than in the PEGMAS copolymers. HEMA is not as hydrophilic as PEGMA; therefore, it forms a weaker global amphiphilic structure with more hydrophobic GM resulting in less membrane activity. PEGMAL was not as effective as PEGMAS when incorporated at 80%, which is probably due to the lower content of active GM inside the PPEGL2G6.
We then studied the ratio of incorporation. In the case of HEMA, there was a clear decrease in activity when more HEMA was incorporated. This is probably due to the amount of active GM where 94, 85, 76, and 58 wt % of the polymer were composed of GM for PHEMA3G5, PHEMA5G3, PHEMA6G2, and PHEMA7G1, respectively. In the case of PEGMAS, the strongest performance was found for PPEGS5G3 with 60% PEGMA and PPEGS6G2 with 80% PEGMA. PPEGS3G5, despite possessing more GM than PPEGS5G3, showed a slight decrease in activity, which was probably due to the inability of the reduced amount of PEGMA to prevent the ring stacking of GM. For 90% incorporation of PEGMAS, an increase in MIC was observed as GM content was reduced as more PEGMA was incorporated.
Of particular note is the fact that PEGylation recovered antimicrobial activity relative to the homopolymers; PPEGS5G3 and PPEGS6G2 outperformed the monomeric control GM in some strains tested. For example, for A. baumannii 5075D, the MIC of PPEGS5G3 was 16 μg/mL, and the MIC of PPEGS6G2 was 8 μg/mL while the MIC of GM was 32 μg/mL. For the strains where GM was not efficient, such as A. baumannii 03–149.1, A. baumannii 03–149.2, K. pneumoniae B5055, K. pneumoniae B5055 nm, and P. aeruginosa 19147 nm, our two copolymers still achieved inhibition in the range tested.
The performance of the copolymers was minimized in the case of K. pneumoniae, which has a capsule structure, an outermost layer composed of polysaccharides. In the case of A. baumannii, almost all of the copolymers and GM worked well only on A. baumannii 5075D, which is a polymyxin-dependent mutant that lacks lipopolysaccharide. Instead, phosphatidylglycerol (PG) is abundant on its outer membrane.21 Considering the fact that guanidine in GM binds strongly with phosphate ions in PBS, it is likely that the mechanism of action for both GM and the copolymers involved binding to phospholipids instead of polysaccharides. This is possibly why copolymers also work well against E. coli and S. aureus, likely through targeting PG, phosphatidylethanolamine (PE) and cardiolipins (CL) on the bacterial membrane.
Overall, our results demonstrate that the copolymerization recovered the activity of GM. Using a more hydrophilic comonomer such as PEGMA showed higher microbial activity due to stronger amphiphilicity of the whole polymer. The microbial activity was affected by the incorporation of PEGMA in two key ways; (1) above 40% PEGMA was needed to prevent steroid stacking, and (2) less PEGMA resulted in higher concentration of GM and higher activity.
Hemolysis Assay
For potential therapeutic applications, it is important to understand whether the copolymers have any detrimental effects on human cells. Therefore, hemolysis assay was conducted to evaluate the toxicity of copolymers (Figure 3). HC50, defined as the polymer concentration needed to cause 50% lysis of red blood cells, was used to indicate the toxicity of the copolymers. Both copolymers with PEGMAS or HEMA showed a dependence of hemolysis on the amount of GM incorporated where increasing incorporation of PEGMAS or HEMA resulted in decreasing toxicity. The decrease in toxicity was significant for PEGMAS, compared to the MIC, as the HC50 increased from 4 to 512 μg/mL with the increasing amount of PEGMAs. We hypothesize that GM was capable of inserting into the cell membrane better due to both association of phospholipid with guanidine and steroid stacking with cholesterol; moreover, with additional hydrophilic components, the copolymers could affect the amphiphilicity of the whole membrane structure to a greater extent. When the different hydrophilic comonomers were compared, more hydrophilic PEGMAS resulted in more hemolysis than HEMA. HEMA was found to be less membrane active in our case for both bacterial and mammalian cell membranes.
Figure 3.
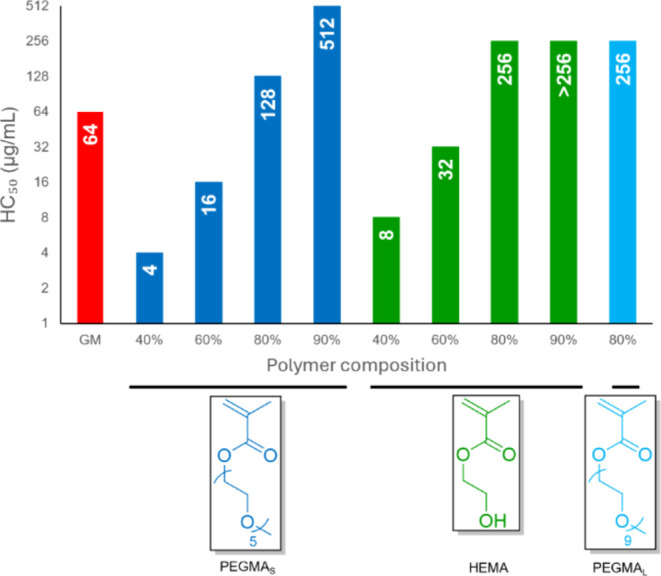
Hemolytic (HC50) activities for GM and its copolymers with PEGMAS, HEMA, and PEGMAL. HC50 is the lowest polymer concentration required to cause 50% lysis of red blood cells.
Tuning the amount of PEGMA incorporated into GM copolymers reduced the hemolysis while preserving antimicrobial activity, and thus, good selectivity (defined as HC50/MIC) was achieved. This is different from traditional cationic polymers where selectivity comes from electrostatic interactions to negatively charged bacterial membranes.22 For GM copolymers, due to the interaction of phospholipid with guanidine, the association with both bacterial and cell membranes is expected to be similar except in a bacterial membrane with abundant LPS (Figure 4a). In all ratios tested here, GM copolymers performed similarly against bacterial membranes after association (Figure 4b); however, lower GM content resulted in a significant reduction in hemolysis (Figure 4c). The best copolymer in our screen was PPEGS6G2, which had a selectivity of 16 against A. baumannii 5075D.
Figure 4.

Illustration of the interaction between copolymers of GM with different membranes. (a) LPS (lipopolysaccharide) rich Gram-negative bacterial membrane; (b) LPS sufficient bacterial membrane; and (c) mammalian cell membrane where only high GM content caused hemolysis.
Conclusions
Herein, we report an extraordinary copolymer system for the development of antimicrobial materials. The guanylated cholic-acid-based monomer showed good antimicrobial activity. However, its homopolymer lost antimicrobial activity possibly due to an unfavored conformational structure from tetracyclic ring stacking. By copolymerization with a hydrophilic monomer such as PEGMA, antimicrobial activity was recovered. The copolymers also exhibited a stronger dependence on hemolysis and showed that good selectivity could be achieved by tuning the amount of GM incorporated. This work showed that both cationic and hydrophobic components significantly contribute to their activity. These novel copolymers are promising antimicrobial agents for the treatment of both Gram-positive and Gram-negative bacteria, including antibiotic-resistant strains. This work provides an interesting platform for researchers in the field to design better antimicrobial materials in the future.
Materials and Methods
Materials
4-cyano-4-[(dodecyl-sulfanylthiocarbonyl)-sulfanyl] pentanoic acid (CDTPA), 4-(dimethylamino) pyridine (DMAP), triethylamine (TEA), cholic acid, 2-hydroxyethyl methacrylate (HEMA), bromoacetyl bromide, potassium carbonate, cysteamine hydrochloride, S-methylisothiourea hemisulfate, calcium chloride, trifluoroacetic acid (TFA), hydrochloride acid (37%), sodium chloride (NaCl), potassium chloride, potassium phosphate monobasic, and sodium phosphate dibasic were purchased from Sigma-Aldrich and used without further purification. N-ethyl-N′-(3-(dimethylamino)propyl) carbodiimide hydrochloride (EDC-HCl) was purchased from Oakwood Chemical, and di-tert-butyl dicarbonate was purchased from Alfa Aesar. Tris(hydroxymethyl) aminomethane (Tris) and all of the solvents used were purchased from Chem Supply. S-methylisothiourea hydrochloride was prepared from hemisulfate salt by the addition of calcium chloride and removal of the precipitate. Tris-buffered saline (10 mM pH 7.4) was prepared using Tris (10 mM 1.211 g) and NaCl (150 mM 8.766 g) dissolved into approximately 1 L of DI water, and pH was adjusted using 1 M HCl solution to pH 7.4. Cation-adjusted Mueller–Hinton broth (MHB) powder was purchased from Thermo Fisher and used to prepare the microbial broths according to the manufacturer’s instructions. S. aureus 29213, M.-ResistantS. aureus (MRSA) 43300, E. coli 25922, E. coli DH5α, E. coli DC10B, P. aeruginosa 27853, P. aeruginosa 19147 nm (colistin-resistant), A. baumannii 5075, A. baumannii 5075D (colistin-resistant), A. baumannii 03–149.1 (polymyxin susceptible), A. baumannii 03–149.2 (polymyxin resistant), and K. pneumoniae B5055 and K. pneumoniae B5055 nm (colistin-resistant) were used for the antimicrobial study.
Characterization
1H NMR spectroscopy data was collected on a Bruker 400 MHz NMR spectrometer using deuterated chloroform (CDCl3) or deuterium oxide (D2O) as the solvent. Chemical shifts were referenced to residual protons in the deuterated NMR solvent. NMR data analysis was performed on Topspin software. Size exclusion chromatography (SEC) was performed on a Waters Alliance system equipped with an Alliance 2695 Separations Module (integrated quaternary solvent delivery, solvent degasser, and autosampler system), a Waters column heater module, a Waters 2414 RDI refractive index detector, a Waters PDA 2996 photodiode array detector (210 to 400 nm at 1.2 nm), and 4× Agilent PL-Gel columns (3× PL-Gel Mixed C (5 μm) and 1× PL-Gel Mixed E (3 μm) columns), each 300 × 7.8 mm2, providing an effective molar mass range of 200–2 × 106 Da. Tetrahydrofuran (THF) high purity solvent (HPLC grade) was prefiltered through aluminum oxide (90 active neutral, 70–230 mesh) with 0.45 μm filter, and 0.1 g/L 2,6-ditert-butyl-4-methylphenol (BHT) was added as an inhibitor. The filtered THF-containing BHT was purged slowly with nitrogen gas and used as an eluent with a flow rate of 1 mL/min at 30 °C. Number (Mn) and weight-average (Mw) molar masses were evaluated using Waters Empower-3 software. The SEC columns were calibrated with low dispersity polystyrene (PSt) standards (Polymer Laboratories) ranging from 580 to 7,500,000 g mol–1, and molar masses are reported as PSt equivalents. A third-order polynomial was used to fit the log Mp vs time calibration curve, which was near linear across the molar mass ranges. Fourier-transform infrared (FT-IR) spectra were collected on a Bruker Tensor 27 FT-IR spectrometer. The samples were prepared by the KBr disc technique, and the scan range was 4000–400 cm–1. Mass spectrometric analysis was performed on a Thermo Scientific Q Exactive mass spectrometer fitted with a HESI-II ion source. Positive and negative ion electrospray mass spectra were recorded in an appropriate mass range set for 140,000 mass resolution. The probe was used with a 0.3 mL/min flow of methanol. The nitrogen nebulizing/desolvation gas used for vaporization was heated to 350 °C in these experiments. The sheath gas flow rate was set to 25 °C and the auxiliary gas flow rate to 10 (both arbitrary units). The spray voltage was 3.50 kV, and the capillary temperature was 300 °C.
Synthesis of (2-methacryloyloxy) Ethyl Cholate (MAECA)
To a solution of cholic acid (8.16 g, 20 mmol), EDC-HCl (7.63 g, 40 mmol), and DMAP (0.48 g, 4 mmol) in anhydrous THF (100 mL), HEMA (4.88 mL, 40 mmol) was added under a nitrogen atmosphere. After addition, the reaction mixture was connected to dried air to prevent polymerization. The reaction mixture was allowed to react for 3 days. The reaction mixture was evaporated and dissolved in dichloromethane (DCM), washed with water (3×) and brine (1×), and then the organic phase was dried over anhydrous MgSO4. After removing the solvent under vacuum, the reaction mixture was dissolved in ethanol and purified by precipitation in water 3 times to afford the product as a white solid (9.3 g, 89% yield). 1H NMR (400 MHz, CDCl3): δ 6.13 (s, 1H), 5.60 (s, 1H), 4.33 (br, 4H), 3.97 (s, 1H), 3.85 (s, 1H), 3.46 (br, 1H), 1.95 (s, 3H), 0.99 (d, 3H), 0.89 (s, 3H), 0.68 (s, 3H). The fully assigned 1H NMR spectra are shown in Figure S1.
Synthesis of Tri-Br-MAECA
To a solution of MAECA (7.2 g, 13.8 mmol) and K2CO3 (23 g, 166 mmol) in anhydrous DCM (100 mL) at 0 °C, bromoacetyl bromide (7.2 mL, 83 mmol) was added dropwise under a nitrogen atmosphere. The reaction mixture was warmed up to room temperature and allowed to react overnight. The reaction mixture was sequentially washed with water (1×), saturated NaHCO3 (2×) and brine (1×), and then the organic phase was dried over anhydrous MgSO4. After removal of the solvent under vacuum, the product was afforded without further purification as a brown liquid (10 g, 82% yield). 1H NMR (400 MHz, CDCl3): δ 6.12 (s, 1H), 5.60 (s, 1H), 5.17 (s, 1H), 5.01 (s, 1H), 4.64 (br, 1H), 4.33 (br, 4H), 3.78 (br, 6H), 1.99 (s, 3H), 0.94 (s, 3H), 0.85 (d, 3H), 0.75 (s, 3H). The fully assigned 1H NMR spectra are shown in Figure S2.
Synthesis of N,N′-di-Boc-S-Methylisothiourea (Boc-Isothiourea)
To a solution of S-methylisothiourea hydrochloride (7.25 g, 57.5 mmol) in methanol (100 mL), di-tert-butyl dicarbonate (27.3 g, 125 mmol) was added in one portion, and then TEA (8.37 mL, 60 mmol) dissolved in methanol (10 mL) was added dropwise. The reaction mixture was allowed to react for 2.5 h. The reaction mixture was evaporated and dissolved in hexane, washed with water (2×) and brine (1×), and then the organic phase was dried over anhydrous MgSO4. After removal of the solvent under vacuum, the product was afforded without further purification as a white solid (12.54 g, 75% yield). 1H NMR (400 MHz, CDCl3): δ 2.40 (s, 3H), 1.52 (s, 18H). The fully assigned 1H NMR spectra are shown in Figure S3.
Synthesis of Tert-Butyl-(Tert-Butoxycarbonylamino) (2-Thioethylamino) Methylenecarbamate (Boc-Gua-Thiol)
To a solution of cysteamine hydrochloride (2.34 g, 20 mmol) and Boc-isothiourea (3 g, 10 mmol) in methanol (40 mL) was added TEA (2.88 mL, 20 mmol) under a nitrogen atmosphere. The reaction mixture was allowed to react for 3 h. DCM was added to the reaction mixture which was sequentially washed with water (2×), and brine (1×), and then the organic phase was dried over anhydrous MgSO4. After the solvent was removed by blowing nitrogen, the reaction mixture was purified by silica gel column chromatography (2% diethyl ether/DCM) to afford the product as a white solid (2.1 g, 64% yield). 1H NMR (400 MHz, CDCl3): δ 11.49 (s, 1H), 8.66 (br, 1H), 3.64 (dd, 2H), 2.73 (dd, 2H) 1.50 (s, 18H), 1.42 (t, 1H). The fully assigned 1H NMR spectra are shown in Figure S4.
Synthesis of 2-(Boc-Smino) Ethanethiol
To a solution of cysteamine hydrochloride (3.4 g, 30 mmol) in methanol (30 mL), di-tert-butyl dicarbonate (6.5 g, 30 mmol) was added in one portion, and then TEA (4.2 mL, 30 mmol) was added under nitrogen atmosphere. The reaction mixture was allowed to react for 3 h. DCM was added to the reaction mixture, which was sequentially washed with water (2×), and brine (1×), and then, the organic phase was dried over anhydrous MgSO4. After removing the solvent by blowing nitrogen, the intermediate (containing corresponding disulfide) was afforded without further purification as a colorless liquid (4.3 g, 81% yield). 1H NMR (400 MHz, CDCl3): δ 4.94 (br, 1H), 3.30 (dd, 2H), 2.64 (dd, 2H), 1.43 (s, 9H), 1.34 (s, 1H). The fully assigned 1H NMR spectra are shown in Figure S5.
Synthesis of Tri-Boc-Gua-MAECA
Tri-Br-MAECA (3.72 g, 4.2 mmol), Boc-Gua-thiol (5.4 g, 16.9 mmol), and K2CO3 (1.16 g, 8.4 mmol) were dissolved in DMF (80 mL) under nitrogen atmosphere. The reaction mixture was allowed to react overnight. DCM was added to the reaction mixture, which was sequentially washed with water (2×) and brine (1×), and then, the organic phase was dried over anhydrous MgSO4. After removal of the solvent under vacuum, the reaction mixture was purified by silica gel column chromatography (30% ethyl acetate/hexane) to afford the product as a colorless solid (5.2 g, 77% yield). 1H NMR (400 MHz, CDCl3): δ 11.47 (s, 3H), 8.61 (br, 3H), 6.12 (s, 1H), 5.59 (t, 1H) 5.13 (s, 1H), 4.96 (s, 1H), 4.60 (br, 1H), 4.33 (br, 4H), 3.70 (br, 6H), 3.29 (br, 4H), 3.23 (s, 2H), 2.92 (t, 2H), 2.85 (br, 4H), 1.94 (s, 3H), 1.49 (s, 54H), 0.92 (s, 3H), 0.84 (d, 3H), 0.73 (s, 3H). The fully assigned 1H NMR spectra and mass spectra are shown in Figure S6–10.
Synthesis of Tri-Boc-Ami-MAECA
Br-MAECA (3.08 g, 3.5 mmol), 2-(Boc-amino) ethanethiol (2.48 g, 14.0 mmol), and K2CO3 (0.96 g, 7.0 mmol) were dissolved in DMF (15 mL) under nitrogen atmosphere. The reaction mixture was allowed to react overnight. DCM was added to the reaction mixture, which was sequentially washed with water (2×) and brine (1×), and then, the organic phase was dried over anhydrous MgSO4. After removing the solvent under vacuum, the reaction mixture was purified by silica gel column chromatography (40% ethyl acetate/hexane) to afford the product as a colorless solid (2.5 g, 61% yield). 1H NMR (400 MHz, CDCl3): δ 6.12 (s, 1H), 5.59 (t, 1H) 5.13 (br, 4H), 4.97 (s, 1H), 4.60 (br, 1H), 4.33 (br, 4H), 3.36 (br, 6H), 3.29 (br, 6H), 2.82 (t, 2H), 2.76 (br, 4H), 1.94 (s, 3H), 1.44 (s, 27H), 0.93 (s, 3H), 0.84 (d, 3H), 0.74 (s, 3H). The fully assigned 1H NMR spectra are shown in Figure S11.
Polymerization of Tri-Boc-Gua-MAECA and Tri-Boc-Ami-MAECA to Produce Polymers (PBG3–14 and PBA9)
The synthesis of PBG3 is described below, and all other polymers were synthesized similarly. Tri-Boc-Gua-MAECA (300 mg, 0.186 mmol) and 4-cyano-4-(phenyl carbonothioylthio) pentanoate (15.1 mg, 0.037 mmol) were dissolved in DMF (0.75 mL) and transferred into a Schlenk flask with a magnetic stirrer bar. The flask was degassed by three freeze–pump–thaw cycles (5, 10, and 15 min, respectively). The reaction mixture was stirred at room temperature for 16 h under blue light irradiation. The reaction was terminated by exposing the reaction mixture to air. The mixture was purified by repeated dissolution–precipitation process using DCM/methanol 3 times to give a colorless solid (49 mg, 15% yield). The fully assigned 1H NMR spectra and SEC data are shown in Figure S12–S15.
Deprotection of Boc Group of Monomers or Polymers to Produce Corresponding Guanidium or Amine
Deprotection of Tri-Boc-Gua-MAECA is described below, and all other Boc deprotections were carried out similarly. Tri-Boc-Gua-MAECA (100 mg, 0.0625 mmol) was dissolved in DCM (5.2 mL), TFA (0.575 mL, 7.51 mmol) was added, and the reaction mixture was allowed to react at room temperature overnight. After the removal of solvent in vacuo, a white waxy solid was obtained as the deprotected monomer. The monomer was subsequently dissolved in water and lyophilized to yield the product (88.26 mg, 84% yield). The fully assigned 1H NMR spectra are shown in Figures S16–S21 and S33–S41.
Copolymerization of Tri-Boc-Gua-MAECA with Other Monomers to Produce Polymers (PPEGSBG, PHEMABG, and PPEGLBG)
Synthesis of PPEGS3BG5 is described below, and all other polymers were synthesized similarly. Tri-Boc-Gua-MAECA (200 mg, 0.119 mmol), CDTPA (8.4 mg, 0.021 mmol), and PEGMAS (25 mg, 0.083 mmol) were dissolved in DMF (0.84 mL) and transferred into a Schlenk flask with a magnetic stirrer bar. The flask was degassed by three freeze–pump–thaw cycles (5, 10, and 15 min, respectively). The reaction mixture was stirred at room temperature for 16 h under blue light irradiation. The reaction was terminated by exposing the reaction mixture to air. The mixture was purified by repeated dissolution–precipitation process using DCM/ether 3 times to give a colorless solid (187 mg, 80% yield). The fully assigned 1H NMR spectra and SEC data are shown in Figures S24–S32.
Antimicrobial Testing
The minimum inhibitory concentration (MIC), defined as the lowest polymer concentration tested to completely prevent visible bacterial growth, and minimum bactericidal concentration (MBC), defined as the lowest concentration to achieve bactericidal killing (99.9% reduction in the initial inoculum), were determined using a standard microbroth dilution method according to Clinical and Laboratory Standards Institute (CLSI) guidelines.
Initial MIC Assay of S. aureus 29213 and E. coli 25922
To prepare the inoculums, a colony of either bacterium was directly transferred from a nutrient agar plate into 10 mL of Mueller–Hinton broth (MHB – cation adjusted) bacterial growth media and incubated at 37 °C overnight. The concentration of the bacteria was measured by the optical density at 600 nm (OD600) and adjusted to approximately 1 × 108 cfu/mL (colony-forming units per mL). Culture suspension was further diluted by 100-fold to 1 × 106 cfu/mL in MHB media. Serial 2-fold dilutions of the nanoparticles in Tris-buffered saline (pH 7.4) were prepared to obtain the desired concentration range. An aliquot of 100 μL of the nanoparticle solution was added to each well of a 96-well flat-bottom plate (Corning, Sigma-Aldrich) followed by the addition of 100 μL of bacterial suspension (1 × 106 cfu/mL). The solution only containing bacterial suspension and Tris-saline buffer (pH 7.4) served as positive controls, and solution only containing MHB media and the nanoparticles served as a negative control. The plates were incubated for 24 h at 37 °C under constant shaking at 75 rpm. The growth was measured by OD600 using a 96-well plate reader (Byonoy absorbance 96, Byonoy). Triplicates were carried out for each condition.
MIC Assay
The strains were inoculated and prepared according to the same procedure described above. 100 μL of MHB solution containing the polymer or monomer in serial 2-fold dilutions was placed into each well of a 96-well microplate followed by the addition of an equal volume of microbial suspension. The plates were incubated for 16–20 h at 37 °C. The MIC was taken as the concentration of the sample at which no visible microbial growth was observed with unaided eyes. The experiments were repeated twice.
MBC Assay
After the MIC assay, an aliquot of 10 μL of bacterial suspension from each clear well of the MIC study was plated onto LB agar plates. After incubation at 37 °C for 24 h, MBC values were determined by the lowest concentration of tested reagents that resulted in no growth of bacteria on the agar plates.
Hemolysis Assay
Heparinized blood from adult Sprague–Dawley rats (Biospecimen service, Monash Animal Research Platform) was collected 3 h before the assay. One milliliter of blood was diluted in 49 mL of prechilled phosphate buffer saline (PBS, 10 mM, pH 7.4) and washed several times by centrifugation until the supernatant was clear (10 min at 3000 rpm at 4 °C). After the supernatant was carefully removed, red blood cells were diluted in Tris-buffered saline (10 mM, pH 7.4) to obtain a blood suspension of 4% (v/v) final concentration. The polymers were dissolved in Tris-buffered saline at specific concentrations ranging from 2.0 to 1024 μg/mL, and 100 μL of polymer solutions at different concentrations were added to the diluted blood suspension (4% v/v, 100 μL) in a V-shaped clear-bottom 96-well plate (in triplicates for each concentration). Blood suspension was also mixed with Tris-buffered saline and 0.2% Triton X-100 (in triplicate) to obtain negative and positive controls. The plate was incubated at 37 °C for 1 h. The plate was then spun at 3000 rpm at room temperature, and 100 μL of the supernatant was carefully transferred to a flat, clear-bottom 96-well plate. The ultraviolet (UV) absorbances were measured at 540 nm using a plate reader (Clariostar, BMG), and hemolysis percentage was calculated from the following equation. The experiment was repeated 3 times independently.
Acknowledgments
We would like to acknowledge funding through the ARC Research Council Discovery Program (DP240102642) and the National Health and Medical Research Council (NHMRC) (ideas grant APP 2029218).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c08266.
Details of 1H NMR; SEC; FT-IR; mass spectrum; and additional bacterial and hemolysis testing are provided (DOCX)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Murray C. J. L.; Ikuta K. S.; Sharara F.; Swetschinski L.; Aguilar G. R.; Gray A.; Han C.; Bisignano C.; Rao P.; Wool E.; et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022, 399 (10325), 629–655. 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Prestinaci F.; Pezzotti P.; Pantosti A. Antimicrobial resistance: a global multifaceted phenomenon. Pathog. Global Health 2015, 109 (7), 309–318. 10.1179/2047773215Y.0000000030. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dadgostar P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. 10.2147/IDR.S234610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca I.; Akova M.; Baquero F.; Carlet J.; Cavaleri M.; Coenen S.; Cohen J.; Findlay D.; Gyssens I.; Heure O. E.; et al. The global threat of antimicrobial resistance: science for intervention. New Microbes New Infect. 2015, 6, 22–29. 10.1016/j.nmni.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler M. S.; Henderson I. R.; Capon R. J.; Blaskovich M. A. T. Antibiotics in the clinical pipeline as of December 2022. J. Antibiot. 2023, 76 (8), 431–473. 10.1038/s41429-023-00629-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magana M.; Pushpanathan M.; Santos A. L.; Leanse L.; Fernandez M.; Ioannidis A.; Giulianotti M. A.; Apidianakis Y.; Bradfute S.; Ferguson A. L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20 (9), e216–e230. 10.1016/S1473-3099(20)30327-3. [DOI] [PubMed] [Google Scholar]
- Lei J.; Sun L.; Huang S.; Zhu C.; Li P.; He J.; Mackey V.; Coy D. H.; He Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11 (7), 3919–3931. [PMC free article] [PubMed] [Google Scholar]
- a Salas-Ambrosio P.; Tronnet A.; Verhaeghe P.; Bonduelle C. Synthetic Polypeptide Polymers as Simplified Analogues of Antimicrobial Peptides. Biomacromolecules 2021, 22 (1), 57–75. 10.1021/acs.biomac.0c00797. [DOI] [PubMed] [Google Scholar]; b Mowery B. P.; Lee S. E.; Kissounko D. A.; Epand R. F.; Epand R. M.; Weisblum B.; Stahl S. S.; Gellman S. H. Mimicry of Antimicrobial Host-Defense Peptides by Random Copolymers. J. Am. Chem. Soc. 2007, 129 (50), 15474–15476. 10.1021/ja077288d. [DOI] [PubMed] [Google Scholar]; c Kuroda K.; DeGrado W. F. Amphiphilic Polymethacrylate Derivatives as Antimicrobial Agents. J. Am. Chem. Soc. 2005, 127 (12), 4128–4129. 10.1021/ja044205+. [DOI] [PubMed] [Google Scholar]; d Ilker M. F.; Nüsslein K.; Tew G. N.; Coughlin E. B. Tuning the Hemolytic and Antibacterial Activities of Amphiphilic Polynorbornene Derivatives. J. Am. Chem. Soc. 2004, 126 (48), 15870–15875. 10.1021/ja045664d. [DOI] [PubMed] [Google Scholar]
- Chin W.; Zhong G.; Pu Q.; Yang C.; Lou W.; De Sessions P. F.; Periaswamy B.; Lee A.; Liang Z. C.; Ding X.; et al. A macromolecular approach to eradicate multidrug resistant bacterial infections while mitigating drug resistance onset. Nat. Commun. 2018, 9 (1), 917 10.1038/s41467-018-03325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.; Wang Y.; Le M.; Chen K.-F.; Jia Y.-G. Recent Progress in Bile Acid-Based Antimicrobials. Bioconjugate Chem. 2021, 32 (3), 395–410. 10.1021/acs.bioconjchem.0c00642. [DOI] [PubMed] [Google Scholar]
- Lai X.-Z.; Feng Y.; Pollard J.; Chin J. N.; Rybak M. J.; Bucki R.; Epand R. F.; Epand R. M.; Savage P. B. Ceragenins: Cholic Acid-Based Mimics of Antimicrobial Peptides. Acc. Chem. Res. 2008, 41 (10), 1233–1240. 10.1021/ar700270t. [DOI] [PubMed] [Google Scholar]
- Rahman M. A.; Bam M.; Luat E.; Jui M. S.; Ganewatta M. S.; Shokfai T.; Nagarkatti M.; Decho A. W.; Tang C. Macromolecular-clustered facial amphiphilic antimicrobials. Nat. Commun. 2018, 9 (1), 5231 10.1038/s41467-018-07651-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt N.; Mishra A.; Lai G. H.; Wong G. C. L. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010, 584 (9), 1806–1813. 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- a Mitchell D. J.; Steinman L.; Kim D. T.; Fathman C. G.; Rothbard J. B. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56 (5), 318–325. 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]; b Locock K. E. S.; Michl T. D.; Valentin J. D. P.; Vasilev K.; Hayball J. D.; Qu Y.; Traven A.; Griesser H. J.; Meagher L.; Haeussler M. Guanylated Polymethacrylates: A Class of Potent Antimicrobial Polymers with Low Hemolytic Activity. Biomacromolecules 2013, 14 (11), 4021–4031. 10.1021/bm401128r. [DOI] [PubMed] [Google Scholar]; c Gabriel G. J.; Madkour A. E.; Dabkowski J. M.; Nelson C. F.; Nusslein K.; Tew G. N. Synthetic Mimic of Antimicrobial Peptide with Nonmembrane-Disrupting Antibacterial Properties. Biomacromolecules 2008, 9 (11), 2980–2983. 10.1021/bm800855t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Shanmugam S.; Corrigan N. A.; Boyer C.. Catalyst-Free Visible Light-Induced RAFT Photopolymerization. In Controlled Radical Polymerization: Mechanisms, ACS Symposium Series; American Chemical Society, 2015; Vol. 1187, pp 247–267. [Google Scholar]
- a McKenzie T. G.; Fu Q.; Wong E. H. H.; Dunstan D. E.; Qiao G. G. Visible Light Mediated Controlled Radical Polymerization in the Absence of Exogenous Radical Sources or Catalysts. Macromolecules 2015, 48 (12), 3864–3872. 10.1021/acs.macromol.5b00965. [DOI] [Google Scholar]; b Allison-Logan S.; Karimi F.; Sun Y.; McKenzie T. G.; Nothling M. D.; Bryant G.; Qiao G. G. Highly Living Stars via Core-First Photo-RAFT Polymerization: Exploitation for Ultra-High Molecular Weight Star Synthesis. Acs Macro Lett. 2019, 8 (10), 1291–1295. 10.1021/acsmacrolett.9b00643. [DOI] [PubMed] [Google Scholar]
- Wang H.; Li Q.; Dai J.; Du F.; Zheng H.; Bai R. Real-Time and in Situ Investigation of “Living”/Controlled Photopolymerization in the Presence of a Trithiocarbonate. Macromolecules 2013, 46 (7), 2576–2582. 10.1021/ma400208j. [DOI] [Google Scholar]
- Liu S. P.; Zhou L.; Lakshminarayanan R.; Beuerman R. W. Multivalent Antimicrobial Peptides as Therapeutics: Design Principles and Structural Diversities. Int. J. Pept. Res. Ther. 2010, 16 (3), 199–213. 10.1007/s10989-010-9230-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercole F.; Whittaker M. R.; Quinn J. F.; Davis T. P. Cholesterol Modified Self-Assemblies and Their Application to Nanomedicine. Biomacromolecules 2015, 16 (7), 1886–1914. 10.1021/acs.biomac.5b00550. [DOI] [PubMed] [Google Scholar]
- Svobodová H.; Noponen V.; Kolehmainen E.; Sievänen E. Recent advances in steroidal supramolecular gels. Rsc Adv. 2012, 2 (12), 4985–5007. 10.1039/c2ra01343f. [DOI] [Google Scholar]
- Judzewitsch P. R.; Nguyen T.-K.; Shanmugam S.; Wong E. H. H.; Boyer C. Towards Sequence-Controlled Antimicrobial Polymers: Effect of Polymer Block Order on Antimicrobial Activity. Angew. Chem., Int. Ed. 2018, 57 (17), 4559–4564. 10.1002/anie.201713036. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Lu J.; Han M.-L.; Jiang X.; Azad M. A. K.; Patil N. A.; Lin Y.-W.; Zhao J.; Hu Y.; Yu H. H.; et al. Polymyxins Bind to the Cell Surface of Unculturable Acinetobacter baumannii and Cause Unique Dependent Resistance. Adv. Sci. 2020, 7 (15), 2000704 10.1002/advs.202000704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham P.; Oliver S.; Boyer C. Design of Antimicrobial Polymers. Macromol. Chem. Phys. 2023, 224 (3), 2200226 10.1002/macp.202200226. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.