

ABSTRACT
Toxin-antitoxin systems are ubiquitous in the prokaryotic world and widely distributed among chromosomes and mobile genetic elements. Several different toxin-antitoxin system types exist, but what they all have in common is that toxin activity is prevented by the cognate antitoxin. In type I toxin-antitoxin systems, toxin production is controlled by an RNA antitoxin and by structural features inherent to the toxin messenger RNA. Most type I toxins are small membrane proteins that display a variety of cellular effects. While originally discovered as modules that stabilize plasmids, chromosomal type I toxin-antitoxin systems may also stabilize prophages, or serve important functions upon certain stress conditions and contribute to population-wide survival strategies. Here, we will describe the intricate RNA-based regulation of type I toxin-antitoxin systems and discuss their potential biological functions.
KEYWORDS: toxin-antitoxin systems, RNA regulation, membrane toxins, mobile genetic elements, antibiotic persistence
INTRODUCTION
In the past few decades, there has been an explosion in the identification and characterization of toxin-antitoxin (TA) systems across bacterial species. These genetic loci encode a potential toxin that either induces growth stasis or death of the producing cell and an antitoxin that prevents this toxicity. They are categorized based on the nature of the toxin and antitoxin (protein or RNA) and the mode of antitoxin action (1, 2). While numerous TA systems have been described in much detail on the molecular level, it turned out to be more challenging to assign biological functions to these systems. The first TA systems to be discovered were located on plasmids, and it was recognized almost instantly that toxicity counteracts plasmid loss in expanding populations (2, 3). It is commonly assumed that localization of TA systems on plasmids and other mobile genetic elements (MGEs) immediately coincides with a function in MGE stabilization. But how do we explain the presence of functional TA systems in bacterial chromosomes where they occasionally occur in dozens? Unfortunately, studying biological functions of chromosomal TA systems is often complicated by experimental drawbacks, such as redundancy of systems and the absence of robust phenotypes under laboratory conditions. Despite these challenges, some functions have been assigned to these systems over the years. One of them is the defense against bacteriophages, viruses that infect bacteria, which is currently a rapidly expanding field in microbiology (4). Another function, which was also one of the drivers for the increased attention of these systems, is the formation of persister cells: a subpopulation of cells that survive an otherwise lethal dose of antibiotics (5). The production of specific toxins from select TA systems has been shown to contribute to persister cell formation (6, 7); however, there are conflicting data for other systems and the universality of these findings is unclear (8). Clearly, disclosing the secret of chromosomal TA systems is still an ongoing challenge. Within this review, we give an overview about the different TA system types, and specifically examine the type I TA families with a focus on RNA-based regulation. We discuss biological functions and present examples of how regulation of type I TA systems is tied to functionality. Finally, we give an overview about pore-forming type I toxins and their involvement in antibiotic persistence.
CATEGORIZATION OF TOXIN-ANTITOXIN SYSTEMS
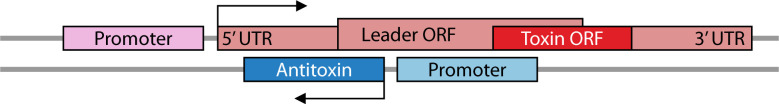
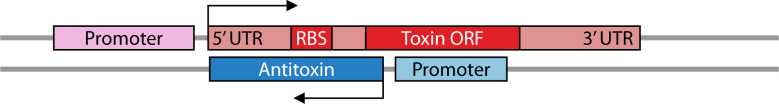
TA systems can be categorized into eight types [summarized in Fig. 1, reviewed in reference (2)]. All toxins are proteins with the exception of those from type VIII systems. The antitoxins of types I, III, and VIII are RNAs, while all other antitoxins are proteins. Type I RNA antitoxins (discussed more in depth below; Table 1) base pair to toxin mRNAs via sequence complementarity, decreasing translation of toxins that are often under 60 amino acids in length. Type III antitoxins, however, are RNAs that bind the actual toxin protein, preventing its activity. A recently described type VIII antitoxin acts in concert with a protein to prevent the activity of a toxin RNA (9). For another case, the RNA antitoxin appears not to need a protein partner to repress the toxic RNA (10).
Fig 1.

Categorizations of toxin-antitoxin systems. For types II, IV, VIII, and tripartite, dashed lines represent alternative pathways for antitoxin repression depending on the specific class examined. Please see text for details.
TABLE 1.
Different mechanisms of type I toxin repression by antitoxins along with their general genetic organization and confirmed examplesa
| Antitoxin regulation | Genetic organization | Confirmed examples | References |
|---|---|---|---|
Translational block via interaction at upstream open reading frame (ORF)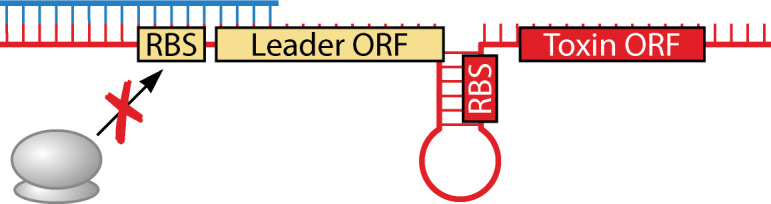
|
|
hok/sok | (11, 12) |
Translational block via direct sequestration of ribosome binding site (RBS)
|

|
fst/RNAII | (13, 14) |

|
ibs/sib | (15, 16) | |
|
aapA1/isoA1 | (17) | |
Translational block via indirect sequestration of RBS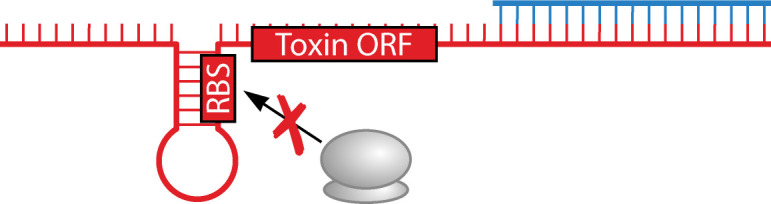
|

|
bsrG/SR4 | (18, 19) |
|
dinQ/agrB | (20, 21) | |
Translational block via sequestration of ribosome standby site (RSS) 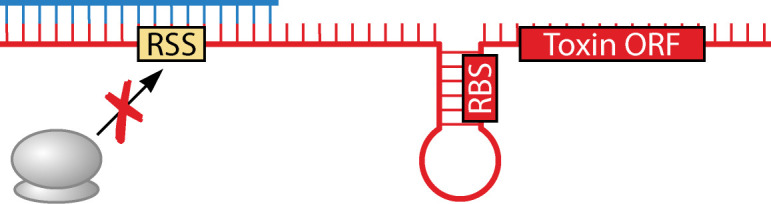
|
|
tisB/istR-1 | (22, 23) |
Translational block via mRNA degradation by RNase III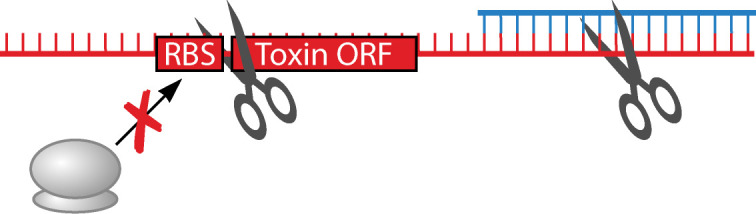
|

|
txpA/ratA | (24, 25) |
Red represents features associated with the toxin, while blue represents features associated with the antitoxin.
The antitoxins of types II, IV, V, VI, and VII, however, are all protein antitoxins, but their mode of toxin repression is variable (2). Types II, VI, and VII antitoxins physically interact with and/or act on the toxin protein. Type II systems are well-documented across plasmids and bacterial and archaeal chromosomes; interested readers can find some excellent reviews describing these gene pairs in greater depth (26, 27). Binding of type II antitoxins inhibits the biochemical activity of their toxin partner. This is in contrast to the type VI antitoxins which bind their cognate toxins, and act as proteolytic adaptors, targeting the toxin for degradation (28). Note that a separate category, type VII, has been proposed for those antitoxins that post-translationally modify their toxins through a variety of possible mechanisms (29–32). Type IV antitoxins do not bind the toxin but serve as antagonists to toxic activity by interacting with the toxin target (33–36). In contrast to those above, type V antitoxins actually target the toxin mRNA, preventing accumulation of the toxin protein; though only one example has been thoroughly described, there may be more uncharacterized (37).
Beyond classification: new spins on toxin-antitoxin biology
The above examples fall within the traditional categories; however, there are other variations that have not been officially classified (Fig. 1). For our purposes, we highlight several variants below to demonstrate the breadth of diversity that exists.
Some newly described TA systems are similar to the type II loci. For example, there are systems comprised of three protein members that are often called tripartite systems. The third component of these systems represses toxin gene transcription or stabilizes the antitoxin [for a review, see reference (1)]. Other variants include intragenic antitoxins, where the protein antitoxin is encoded within the toxin open reading frame (ORF). One example of an intragenic antitoxin is found within rpn, which encodes a recombination promoting protein within bacterial mobile elements. The full-length protein, RpnL, is toxic, but the smaller, intragenic encoded RpnS protein can bind to RpnL repressing its toxicity (38).
Recent work has identified a TA system within retrons, genetic regions that encode a reverse transcriptase and a stable ssDNA multi-copy single-stranded DNA (msDNA) that are linked to phage protection (39, 40). Retron-Sen2 of Salmonella Typhimurium encodes a toxin RcaT (39). The toxic activity can be neutralized through interactions with both the reverse transcriptase and the msDNA. However, data suggest that during phage infection, methylation of the msDNA allows for toxin escape, resulting in cell growth stasis, halting phage propagation.
TYPE I TOXIN-ANTITOXIN DISCOVERY
Plasmids
The discovery of TA systems, including type I systems (Table 1), came from studies of plasmid biology. Biologists recognized that plasmids were capable of partitioning and maintaining themselves within a population of cells, but the mechanisms were unknown. Researchers identified key genetic regions needed for maintenance of plasmid R1 of Escherichia coli by generating “mini” plasmids or transferring fragments to other plasmids and monitoring plasmid stability over time. Investigators successfully identified a region now referred to as a type I TA system (41, 42). Using a combination of elegant techniques, Gerdes and colleagues identified the hok (host killing) gene that encoded the 52 amino acid Hok protein. Production of the Hok protein resulted in “ghost” cells that lacked R1 (43, 44). They also mapped within the same region the suppressor of killing (sok) which blocked the effects of the toxin. Extensive studies followed (described below) concluding that the Sok RNA antitoxin base paired to complementary sequences within the hok mRNA, preventing its translation. Similar systems (flm and srnB) were also identified within the F plasmid and plasmid R483 (pnd) (45, 46).
Studies of pAD1 plasmid maintenance in the Gram-positive bacterium Enterococcus faecalis also revealed a type I system, fst/RNAII (47, 48). The par region of pAD1 encodes the 33 amino acid protein Fst (faecalis plasmid-stabilizing toxin; mRNA is RNAI) and the convergent antitoxin RNAII (13, 47–49).
Homologs to the above systems were subsequently found encoded within bacterial chromosomes, including numerous copies of hok/sok within E. coli chromosomes and other enteric species (7, 50, 51). Early studies also identified relF and gef that encode Hok-like proteins within the chromosomes of enteric bacteria (44, 52–54). Similarly, homologs to fst/RNAII have been identified across Gram-positive bacterial species (54–57). It should be noted though that both the expression and function of these chromosomally encoded homologs can be different than those found on plasmids.
Novel chromosomally encoded families
Unlike hok/sok and fst/RNAII, several TA systems have been identified exclusively on bacterial chromosomes (Table 1). Novel chromosomally encoded type I loci were discovered because they possessed similarities to previously described plasmid systems. They encode a small potentially toxic protein but whose translation is prevented by mRNA binding to an antitoxin RNA. For many of these gene pairs, there is little or no clear homology to plasmid sequences, so their origins are unknown. The toxicity of many of these proteins has been observed only if overproduced from a multicopy plasmid. Kawano et al. characterized the first novel system, ldr/rdl, within E. coli MG1655 which possesses four copies (A–D). Toxicity due to overproduction of the LdrD protein can be repressed by co-expression of the RdlD antitoxin (58). Soon after, the tisB/istR locus was described as encoding the LexA-repressed toxin TisB and its RNA antitoxin IstR-1 (22).
Additional type I TA families of E. coli and related enteric species were discovered due to unexpected overexpression phenotypes of novel genes, observed base pairing potential between two RNAs, identification of small RNAs encoded directly antisense to each other, and so forth. Some examples identified within E. coli include ibs/sibs, dinQ/agr, shoB/ohsC, timP/timR, and others (15, 20, 54, 59, 60). Serendipitous discovery also identified gene pairs that mimicked toxin/antitoxin systems in the Gram-positive bacterium Bacillus subtilis, with many encoded within prophage elements including txpA/ratA, bsrG/SR4, bsrE/SR5, yonT-yoyJ/SR6, and bsrH/as-bsrH (18, 24, 25, 54, 57, 61, 62). Chromosomally encoded systems have also been characterized in Helicobacter pylori, which include aapA1/isoA1 and homologs of the ibs/sibs in E. coli (17, 63).
Identification and characterization of novel type I systems
Unlike the numerous and broadly distributed type II systems, chromosomally encoded type I systems are fewer in number and not found as broadly across bacterial species. This is likely due to the challenges in identifying homologs of known type I pairs as well as identifying novel families. Many chromosomal pairs were identified serendipitously (as indicated above and below) and there have only been a few directed searches to find new families (54, 64). Several databases have been put forth as well to aid in discovery, including TASmania (65), TADB 3.0 (66), and T1TAdb (67), with T1TAdb solely focused on type I TA discovery, both novel as well as homologs to previously identified systems.
A major hindrance to discovery is the small nature of a type I toxin. As has been well-summarized for bacterial small proteins, they are difficult to predict given their lack of predictable, characterizable biochemical domains (68, 69). For other TA systems, the proteins (toxin and/or antitoxin) possess specific domains that are highly conserved and identifiable. The toxin proteins of other systems also tend to be 100 amino acids or longer, allowing for greater success using bioinformatic approaches to find homologs. Small proteins are difficult to identify via homology: small changes in amino acid sequence can result in a “loss” of a hit. This is exemplified by H. pylori which encodes Ibs-like type I toxin proteins first described in E. coli (15). The H. pylori Ibs-like proteins (aapC1/2, aapD) were not identified via a PSI-BLAST approach but researchers examining RNA-Seq results noted small proteins, similar in size and sequence to E. coli Ibs (54, 63). However, with more refined approaches, in particular RNA-Seq, RNA interaction by ligation and sequencing (RIL-Seq), and improved proteomic analyses, new systems will be discovered.
Identification of the inherent function of chromosomally encoded type I pairs is also challenging. Functional studies of type I toxin proteins are problematic due to their small size and hydrophobicity. Given the high hydrophobicity, overproduction of type I toxins results in their accumulation in the cytoplasmic membrane. This in turn makes protein isolation challenging for structural analyses as well as identifying interacting partners. Given their inherent toxicity, acquiring significant protein amounts for additional biochemical analyses is also problematic. The use of global biochemical approaches has limitations: small proteins are often missed due to size cut-offs inherent with SDS-PAGE. Genetic screens can also be biased against small RNAs and small proteins. Mainly, as they are often (i) not annotated and thus looked over, (ii) many times, deletions/disruptions of these genes lack severe phenotypes. To combat these limitations, researchers often test “known phenotypes” of well-described type I toxins as a starting point. Below, we propose that better understanding of toxin and antitoxin production at the endogenous level can provide clues for more directed studies.
REGULATION OF TYPE I TOXIN-ANTITOXINS
All type I antitoxins are RNAs that bind to their cognate toxin mRNA through complementary base pairing, repressing toxin expression. This region of complementation can vary in length and, for those examined, often occurs independently of known chaperones like Hfq (15, 18, 24, 70–72). There are significant differences though in the repressive effects of the antitoxin, as described below. Despite this, seemingly unrelated type I TA classes often exhibit several analogous regulatory mechanisms (Table 1; described below). Therefore, understanding how one class is regulated can help us understand how other classes may be as well.
Toxin translational block via interaction with an upstream open reading frame
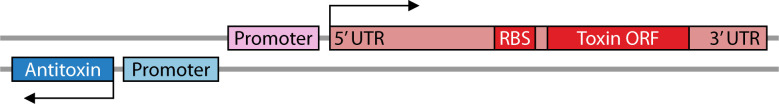
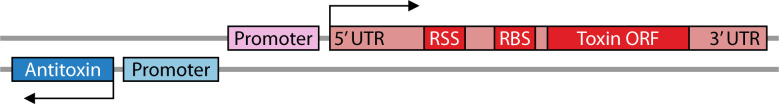
As the first type I TA described, hok/sok of the E. coli R1 plasmid has become the model for understanding type I TA regulation. Provided here is an in-depth description of hok/sok regulation such that the following classes can be described and compared in context (Table 1). The hok toxin and sok antitoxin genes are encoded in cis to each other with promoters in a face-to-face orientation (Fig. 2; Table 1). Note that the 5´ ends of the RNAs are consequently overlapping. The stable, full-length hok mRNA is translationally inactive. In order to be translated or bind Sok, the hok transcript must be first processed at its 3´ end by an unknown mechanism. This removes the fbi (fold-back-inhibition) region and leads to structural rearrangement of the hok mRNA (11, 73, 74). Sok will bind the hok mRNA at this region, leading to repression of toxin translation and degradation of toxin mRNA by RNase III (12, 75, 76). However, it is the binding of Sok to hok that inhibits translation, not the degradation by RNase III (76). Additionally, binding of Sok to hok stabilizes Sok in the absence of any known adaptor proteins (76). This RNase III independent inhibition of toxin translation is achieved through interactions with an ORF upstream of hok. Specifically, Sok binds to the ribosome binding site (RBS) of a leader peptide, mok (modulation of killing), whose translation is coupled to the translation of hok (11). This binding region is only accessible after hok mRNA processing; note that the RBS of hok is sequestered in both the full-length and processed hok transcripts. A function for Mok distinct from this regulatory interaction has not been reported. Taken together, in E. coli cells containing plasmid R1, there (i) is stable, full-length hok mRNA that cannot be translated, (ii) is free, unstable Sok, and (iii) are hok-Sok duplexes that are targeted for degradation by RNase III. However, if there is limited Sok RNA available due to plasmid loss or its degradation by RNases (12, 77), the ribosome binds the available mok RBS, allowing for Hok translation and eventual killing of plasmid-free cells via post-segregational killing (PSK) (11, 43, 44).
Fig 2.

Regulation of the hok/sok type I toxin-antitoxin system from the R1 plasmid of E. coli. Red represents features associated with the toxin, while blue represents features associated with the antitoxin. RNA structures are based on the work of reference (11). Features are not drawn to scale.
As described above, hok/sok copies exist in bacterial chromosomes, but comparatively less is known about them. Of those encoded in E. coli MG1655, four of five are believed to be inactivated via various mutations with only hokB/sokB appearing to be functional (7, 50). Some non-K12 strains of E. coli possess hokA/sokA and hokC/sokC loci (50). Like hok/sok of R1, these loci produce a stable hok mRNA that is processed at its 3´ end that maintains the conserved elements such as the fbi and an unstable Sok RNA. However, unlike hok/sok of plasmid R1, these chromosomal hok mRNAs are poorly translated and cannot confer PSK ability to plasmids (50). Native expression of these chromosomal hoks in absence of their cognate Soks was not toxic to E. coli. However, chromosomal expression of plasmid R1 hok from its native promoter was toxic to E. coli in the absence of its cognate Sok (78). These combined data suggest differences in regulation of the chromosomal copies from the plasmid homolog.
The ldr/rdl (long direct repeat/regulator detected in LDR) type I TA family of E. coli is genetically organized in a similar fashion to hok/sok. There are four copies of ldr/rdl (ldrA/rdlA through ldrD/rdlD) within the chromosome of E. coli K12 strain MG1655. Like hok, the ldrD toxin mRNA is more stable than the RdlD antitoxin RNA (58). Binding of RdlD overlaps a proposed upstream ORF coined ldrX, not ldrD (79, 80). While not confirmed, it has been hypothesized that ldrX may encode a leader peptide whose translation, like mok, is required for toxin translation (79, 80).
Toxin translational block via direct sequestration of the toxin ribosome binding site
Unlike hok/sok, the fst/RNAII type I TA system from the E. faecalis pAD1 plasmid is encoded so that they overlap at their 3´ ends (13, 47–49, 55). This organization results in a different mechanism of repression than for hok/sok (Table 1). The RNAII antitoxin binds 3´ of the fst mRNA start codon, leading to stabilization of both RNAs, unlike what is seen with the interaction of hok to Sok (81). Additional interactions occur between the RNAs at their 5´ ends, despite the genes themselves not overlapping at this region. Stable complex formation requires interactions at both the 3´ and 5´ ends of the RNAs, resulting in fst RBS sequestration by RNAII (13, 14, 82–84). Because the half-life of bound and unbound RNAII is less than bound and unbound fst, respectively, during steady state, fst and RNAII are present in approximately equimolar ratios (81). Loss of RNAII leads to toxic protein production and PSK of pAD1-free cells. For chromosomal fst/RNAII in E. faecalis, transcription of the RNAII antitoxin is reduced upon a variety of stress conditions (85). While complex formation occurs similarly to the plasmid-encoded RNAs, chromosomally encoded RNAII appears to be destabilized upon complex formation as overexpression of chromosomal fst reduces chromosomal RNAII levels (86).
Two chromosomal Fst toxin homologs, PepA1 and PepA2, have also been characterized in Staphylococcus aureus strain Newman and possess the same genetic organization of fst/RNAII (57, 87–89). The mRNA that encodes PepA1 is called sprA1, while the mRNA that encodes PepA2 is called sprA2; their respective antitoxins are SprA1AS and SprA2AS. The toxin mRNAs are constitutively expressed at low levels. However, SprA1AS is expressed in molar excess of the toxin sprA1 mRNA (35- to 90-fold excess), likely to counteract antitoxin instability (87–89). Similar to chromosomal RNAII in E. faecalis, SprA1AS and SprA2AS levels are decreased in various stress conditions while toxin mRNAs sprA1 and sprA2 levels remain relatively stable with a corresponding increase in PepA1 toxin (PepA2 was not examined) (88, 89). Interestingly, interactions between these toxin and antitoxin RNAs are only necessary at their 5´ ends for complex formation, despite possessing potential 3´ overlapping sequences (87, 89). This binding of antitoxin to toxin mRNA directly prevents ribosomal loading, thereby interfering with toxin translation.
For the type I TA family ibs/sib (induction brings stasis/short, intergenic, abundant sequences), the Sib antitoxins were originally identified within the intergenic regions of E. coli as four repeats (termed QUAD1a-d RNAs) (90) with expression of these RNAs confirmed soon after (91–94). Subsequent sequence re-evaluation identified a fifth repeat and that opposite of each QUAD sequence was encoded an 18 or 19 amino acid toxic protein referred to as ibs (A–E), and the QUADS now as sibs (Table 1) (15). Of all the copies, characterization of ibsC/sibC has been the most thorough. Base pairing between ibsC and SibC leads to repression of toxicity in a pair-specific manner (no detectable crosstalk) (15). This specificity is mediated by two variable regions, TRD1 and TRD2, that overlap the ORF and the ribosome binding site of ibsC, respectively (16). The ibsC toxin mRNA was only detectable via northern analyses upon deletion of the sibC promoter or deletion of rnc which encodes RNase III (15) (E. M. Fozo, unpublished data). This would suggest that, at least in the case of ibsC/sibC, base pairing of the antitoxin to the toxin triggers degradation of the toxin mRNA. Whether RNase III is needed to repress toxicity has not been reported. Despite difficulties in detection of ibsC mRNA in the above study, real-time quantitative PCR (qPCR) analyses indicated that the molar ratio of ibsC mRNA and SibC was equal under the conditions examined in E. coli MG1655. SibC was noted to be transcribed in excess of ibsC which may be due to decreased stability of the antitoxin to the toxin (16, 95). However, for other ibs/sib pairs, the molar ratios of the RNAs varied upon conditions, suggesting differences in possible regulation and ibs expression.
The type I TA system aapA1/isoA1 (antisense RNA-associated peptide family A/RNA inhibitor of small ORF family A) is one of six related loci in H. pylori strain 26695 identified as a potential type I TA system via analysis of the H. pylori transcriptome (63). Later confirmed to be a true type I TA system, the regulation of this locus has similarities to the other systems previously discussed (Table 1) (17). For example, the full-length transcript of aapA1 contains a sequestered RBS, rendering it translationally inactive. Specifically, the RBS of aapA1 is sequestered by its own 3´ end, which has also been suggested to occur in the aapA3 homolog (17, 96). Like hok, slow processing of the 3´ end of the full-length aapA1 is required for robust translation of toxin. However, unlike hok, processing directly enhances translation via rearrangement of the 5´ end of aapA1, resulting in an accessible RBS. This processed form can also pair to the antitoxin IsoA1, which occludes the RBS and triggers cleavage by RNase III (17, 97). This repression is robust during normal growth as the RNA levels of IsoA are in excess of 3´ processed aapA1. Additional RNases may also degrade the translationally active aapA1, though it is not known whether this processing relies on IsoA1 complex formation. Oxidative stress can reduce toxin and antitoxin promoter activity and increase levels of the 3´ processed, translationally active toxin mRNA (97). This suggests that oxidative stress may allow aapA1 escape repression by both its less stable antitoxin and its repressive 3´ end.
Toxin translational block via indirect sequestration of the toxin ribosome binding site
Like fst/RNAII, the B. subtilis bsrG/SR4 (Bacillus small RNA G /SR4) transcripts have extensive interactions between their 3´ ends, yet the repression mechanism is distinct (Table 1). Though base pairing results in RNase III degradation of bsrG, RNase III is not required for SR4 to repress bsrG toxicity (18). This is because duplex formation between the toxin and antitoxin results in structural rearrangement and sequestration of the RBS of bsrG in a stem-loop structure, preventing translation of toxin in the absence of RNase III. What is particularly noteworthy is that these RNA-RNA interactions are distal of the RBS. SR4 has been described as a dual-acting type I antitoxin as it represses toxicity both by preventing translation and by triggering degradation of the toxin mRNA (19). Regulation at the transcriptional and stability levels also play important roles. While the promoter activity of bsrG is consistent, promoter activity for SR4 can increase under select conditions. This seems to counterbalance the longer half-life of the bsrG mRNA versus the less stable SR4 (18, 98).
The dinQ/agrB (DNA-damage-inducible protein Q/arsR-gor region gene B) locus was also identified as a type I TA system in E. coli MG1655 that is sensitive to DNA damage. Transcription of toxin dinQ is repressed by LexA while the antitoxin is not, resulting in increased toxin mRNA during SOS response (Table 1) (20, 99). The full-length transcript of dinQ is not translated, but 5´ end processing results in a +44 transcript that can be translated (20, 21). This processing results in opening of the RBS that allows for active translation of dinQ (20, 21). The AgrB antitoxin binds the +44 transcript, leading to dinQ RNA structure rearrangement that sequesters the RBS. Furthermore, this binding leads to degradation of both AgrB and dinQ RNA by RNase III. Again, it is not known if RNase III is required for repression of toxicity. There is a second RNA, AgrA, that is associated with regulation of dinQ. Data support that AgrA can influence the levels of full-length dinQ mRNA, but not the +44 variant. Furthermore, deletion of the antitoxin agrB resulted in increased levels of AgrA, suggesting that these genes are interacting with each other directly or indirectly (20). This is reminiscent of how transcription of istR-2 represses transcription of antitoxin istR-1 (discussed below). However, the interactions between agrA and agrB are likely different from istR-1 and istR-2 as the agrA and agrB genes do not overlap on the chromosome.
Toxin translational block by binding to a standby ribosome binding site
The tisB/istR-1 (toxicity-induced by SOS B/inhibitor of SOS-induced toxicity by RNA) locus of E. coli is by far the best defined chromosomally encoded type I TA system (Table 1). The toxin gene tisB is part of the tisAB operon (formerly ysdAB) and is encoded divergent to the istR-1 antitoxin and another RNA, istR-2, which was originally identified via screens for small RNAs (22, 91, 94). Transcription of tisAB and istR-2 is repressed by LexA while the istR-1 antitoxin is expressed constitutively (6, 22). Though there is some basal transcription of tisAB even with LexA repression, both the 5´ untranslated region (UTR) of tisAB and the constitutive expression of IstR-1 are sufficient to repress toxin translation (22, 23). The full-length tisAB transcript is stable, but translationally inactive (23). Processing by an unknown mechanism results in cleavage of the 5´ end of the tisAB mRNA, resulting in a +42 form. This form is translatable upon binding of the ribosomal protein S1 and the 30S ribosomal subunit to a ribosome standby site (23, 100). However, IstR-1 also binds the +42 transcript, competing with S1, thereby sequestering the ribosome standby site, and triggering toxin mRNA degradation by RNase III (22, 23, 100). Despite the presence of the tisA ORF upstream of tisB and that IstR-1 binding would overlap tisA, experimental evidence suggests that tisA is not translated (22, 101). Upon DNA damage, LexA repression is relieved, resulting in enhanced tisAB transcription, increasing the levels of the toxin mRNA relative to the antitoxin RNA (6, 22). TisB is thus produced along with its subsequent effects (see below). While IstR-2 is also transcribed under these conditions, it cannot repress tisB toxicity, though it may interfere with transcription of IstR-1 (22).
Another type I TA system similar in regulation to tisB/istR-1 is zor/orz. The zor/orz class was first identified in E. coli O157:H7 EDL933 and, while prevalent in other pathogenic and commensal E. coli strains, it was not identified in lab strain MG1655. There are two zor/orz encoded in tandem in E. coli EDL933, referred to as zorO/orzO and zorP/orzP, with only a single amino acid difference between the toxin proteins. Northern analysis detected the toxin and antitoxin RNAs despite sharing a bidirectional −35 promoter element. While this arrangement likely has impacts on gene expression (see below), it has not been examined. Regulation of zorO translation appears to mirror that of tisB translation. The full-length zorO mRNA is translated poorly in vivo and in vitro (102, 103). The zorO UTR is also processed at its 5´ end (generating Δ28 zorO), which allows for robust translation of the toxin in vivo and in vitro. Like tisB, the RBS is sequestered in the processed form of zorO. A standby ribosome binding site has been proposed to exist upstream of the true RBS. The base pairing region for OrzO overlaps this putative standby site region, similar to IstR-1 binding to tisB. Following base pairing by the antitoxin, RNase III-mediated degradation of toxin mRNA occurs, though this is not required to repress zorO toxicity (102, 104).
shoB/ohsC (short hydrophobic ORF/oppression of hydrophobic ORF by sRNA), previously ryfB and ryfC, respectively, is a chromosomal-only type I TA system in E. coli (15, 59). Like tisB and dinQ, shoB is processed at its 5´ end. The full-length transcript, as assessed via reporter gene fusion, is not translated, possibly due to sequestration of the RBS in a stem structure (15, 105). Reporter gene fusions to the processed forms of shoB are translated and OhsC represses translation of these processed variants (15, 59). Thus, shoB translation appears to be regulated similarly to tisB. Even so, no work has demonstrated a standby ribosome binding site or an upstream open reading frame (similar to hok) to mediate translation of the shoB mRNA. Recent work has implicated a role for the envelope stress-sensing CpxRA two-component system in regulating shoB/ohsC at the transcriptional level. The shoB/ohsC region contains two putative CpxR binding sites (CB1 and CB2) that CpxR binds in vitro (106). However, its effects on toxin production and function have not been reported.
Degradation of the toxin-antitoxin complex via RNase III
While cleavage of toxin mRNA-antitoxin RNA complexes by RNase III has been demonstrated, it is often not required for repression of toxicity (discussed in the above sections). However, there is an exception to this rule: the txpA/ratA (toxic peptide A/RNA antitoxin A) locus identified in Bacillus subtilis. This was the first chromosomally encoded type I system described in B. subtilis. It is located within the skin prophage that is excised during late sporulation (Table 1) (24). Transcription of both the toxin txpA (formerly ygdB) and ratA antitoxin is initiated downstream of predicted σA-controlled promoters, leading to their constitutive transcription. Binding of the 3´ ends of RatA and the txpA mRNA results in cleavage of both via RNase III which is required to repress toxicity (25). Note that binding of RatA to the toxin mRNA does not result in structural rearrangement of the region upstream the start codon of txpA. In fact, the 5´ UTR of txpA is quite short compared to many other type I toxins (only 48 nt long) and it does not form secondary structures with its 3´ region. It has been suggested that the strong RBS of txpA inherently represses translation of toxin as, after ribosome recruitment, the ribosome may have difficulty escaping the RBS to initiate translation (25, 107). RNase Y, RNase J1, and PNPase are all involved in the degradation of the RatA antitoxin, with RNase Y being important for initial cleavage of RatA (25). However, this degradation of RatA via these three RNases is not impacted significantly by the presence or absence of txpA.
Unconfirmed mechanisms of toxin repression by antitoxin
The timP/timR (toxic inner membrane protein/repressor) locus was originally examined in pathogenic E. coli and Shigella dysenteriae with corresponding phenotypes attributed to the timP RNA acting as a small RNA (referred to as RyfA) (108–110). However, this gene was later confirmed to encode a type I toxin with a corresponding antitoxin (TimR) divergently encoded in Salmonella Typhimurium (60). For our purposes, timP/timR terminology will be used for Salmonella, while ryfA/ryfB terminology will be used for E. coli and Shigella. In the case of the RyfA, toxin production has not been documented (93, 94, 108, 110). In Salmonella, the timP and TimR RNAs are detected across lab conditions with TimR levels higher than timP mRNA levels (60, 111). Data support that timP RNA levels increase under specific stress conditions, but details as to how this contributes to function are not reported (111–113). Unlike for other type I TA systems, the full-length timP transcript is translated and posttranscriptional processing of the transcript has not been reported. However, despite this high potential for toxin translation, TimP is only detected from the chromosome when the antitoxin gene, timR, has been deleted (60). TimR complexes with the timP mRNA by binding the 5´ UTR of timP upstream the timP RBS. This binding does not appear to impact stability of either RNA or trigger RNase degradation as, in the absence of either gene, the half-lives of these RNAs are not significantly different (60). This suggests that repression of toxicity likely occurs through direct inhibition of translation. More work though is needed to elucidate this regulatory mechanism.
The gene pair symE/symR (SOS-induced yjiW gene with similarity to MazE/symbiotic RNA) of E. coli MG1655, previously yjiW/ryjC, represents an unusual type I TA system, which was originally identified via two separate screens (59, 72, 99). In this case, the toxin SymE, possesses biochemical activity and its overproduction results in the cleavage of RNA. Like tisB, the symE toxin promoter contains a LexA binding site and is strongly induced by DNA damage/SOS. The antitoxin symR is transcribed under standard laboratory conditions and during SOS. SymR levels are detected in excess of symE, even after induction of SOS (72). Despite this, SymE protein is detected at later timepoints during SOS (30 min post-induction, peak at 90 min). While the exact mechanism of toxin repression by antitoxin is not known, based on their genetic organization, it is likely that the antitoxin occludes the RBS. Binding may also result in degradation of the symE mRNA, as promoter mutations of symR result in increased levels of symE (72). Further work is needed to elucidate the specific mechanism of toxin repression.
The ralR/ralA (restriction alleviation/RalR antitoxin) type I Ta system is located in the cryptic prophage rac of E. coli BW25113 (70, 114, 115). RalR toxin activity was first noted and the presence of the RNA antitoxin gene, ralA, was later confirmed (70, 116). The RNAs for both the toxin and antitoxin are detectable during exponential and stationary phase (70). RalA represses toxicity by base pairing in trans and, unlike any other type I TA system described to date, requires the RNA chaperone Hfq to repress toxicity. Hfq simultaneously allows for repression through base pairing and stabilizes the RalA RNA, protecting it from degradation. This repression of toxicity is apparently independent of RNase activity and increased RalA RNA levels do not substantially impact ralR mRNA levels (70, 117). Additional details regarding RalA repression have not yet been reported.
Questions still outstanding about regulation and potential paths forward
There are still several questions regarding the regulation of type I toxins. For example, co-precipitation of some TA transcripts with RNA chaperones Hfq and ProQ suggests that these chaperones may be important for stabilization of these RNAs (118). However, it is not known if this interaction is specific and whether this interaction is required for toxin mRNA recognition or repression. Additionally, how some type I toxin mRNAs are processed, which is needed for their translation and, in some cases, antitoxin recognition, remains unknown. Perhaps this processing is performed by multiple, redundant RNases, is the result of an unknown RNase, or is the result of self-cleavage by the toxin mRNA. For a step deemed critical for the expression of many type I toxins, it is surprising how little we know.
Historically, transcriptional control of type I toxins and their antitoxins has not been thoroughly investigated. Much of what has been reported are changes in bulk RNA levels which can be the result of changes in both promoter activity and RNA stability. The best studied to date has been for tisB/istR-1 and dinQ/agrB: the appreciation of the toxins being LexA repressed was extremely beneficial in further elucidation of potential functions, in particular for tisB.
Another understudied area is the influence of promoter organization on transcription. Unlike for the type II TA genes which are transcribed as a part of operons, type I TA genes are transcribed from their own separate promoters, thus allowing for transcriptional independence between toxin and antitoxin. However, transcription competition may exist for many type I systems given the divergent organization of their promoters. For example, face-to-face type I TA genes (Table 1) may be susceptible to RNA polymerase (RNAP) collisions and potential promoter interference as RNAP interacts with both strands of DNA during transcription: we encourage the readers to examine reviews on this topic (119–121). Regardless, the result would be a reduction of successful transcription for at least one gene. For zor/orz, the only type I TA system described to contain a bidirectional promoter with an overlapping −35 sequence, RNAP cannot bind simultaneously at both promoters. Competition for RNAP has been demonstrated for synthetic and native bacterial genes with overlapping promoters at their −10, and likely similar issues would arise with an overlapping −35 element (122, 123). Additionally, transcription of all divergent genes, whether with overlapping, face-to-face, or back-to-back promoter arrangements, is susceptible to competition through DNA torsion which leads to supercoiling [recently reviewed in reference (124)]. In the twin-supercoil domain model, RNAP during transcription creates positive supercoiling ahead and negative supercoiling behind (125). This DNA torsion can affect local structure and dynamics and can inhibit or enhance transcription of nearby genes. DNA torsion can also affect the affinity of DNA-binding proteins such as transcription factors [recently reviewed in reference (124)]. The way in which toxins and antitoxins compete for RNAP and under what conditions one “wins” over the other remain unexplored avenues of research.
The influence of heterogeneous gene expression within a population for type I TA genes is also understudied. While it has been proposed that type I toxins may only escape repression and express toxic protein in a subset of cells, little work has investigated the impact of regulation at the transcriptional or posttranscriptional level on heterogeneous expression. This is particularly difficult because expression for some genes is low, making accurate measurements at the single-cell level challenging. The development of new tools and approaches, such as the use of fluorescent reporter systems, that can do such measurements in a robust, high-throughput way is needed.
One obstacle to addressing some of these questions is our ability to detect regulatory sequences bioinformatically: by understanding when these genes are expressed, we can develop specific hypotheses regarding when they may function. Bioinformatic identification of regulatory sequences is particularly difficult for those type I TA systems in organisms where less is known about regulator sequence specificity. However, even for well-studied species such as E. coli, the success of such tools can be variable. Additionally, at the posttranscriptional level, virtual RNA folding structure tools often do not completely reflect structural changes due to things like mRNA processing that are important for regulation.
Finally, while using what is known about other type I systems to inform the targeted investigation of others can be effective, it can introduce bias in the types of regulatory mechanisms investigated. One potential way to reduce bias in these approaches is to leverage large omics-based approaches and fitness. For example, investigation of hok/sok and aapA3/isoA3 regulation using Functional AnalysiS of Toxin-Antitoxin Systems in BACteria by Deep Sequencing yielded both known and novel insights about sequence determinants relating to toxin regulation (78, 96). However, this particular approach relies on the ability of toxin genes to kill cells and, in the case of some chromosomal type I systems, it may not be feasible. Another potential way to identify known and novel regulatory sequences is to employ sequence conservation analysis across closely related strains or species, such as has been done for the type I systems tisB/istR, shoB/ohsC, and zor/orz (S. H. Shore and E. M. Fozo, unpublished data). This type of analysis had previously been performed to confirm whether known regulatory sequences were conserved but had not been utilized to investigate potential novel regulatory sequences (17). The rapidly expanding availability of full genome sequences for many species of bacteria provides us with the data necessary to explore sequence conservation in natural bacterial samples. Because this approach can be performed using publicly available data and publicly available bioinformatic tools, this method has the potential to be a quick and a freely accessible way to gain potential insights into important sequences for both regulation and function of any gene of interest, including type I TA systems.
Regardless of how we get there, there is still exciting progress to be made on the study of type I TA regulation. As the regulation of genes is often intricately tied to their function, it is important to make a concerted effort to reconsider what we know and do not know about regulation and how we best address questions for both characterized and uncharacterized type I TA classes.
CELLULAR EFFECTS AND BIOLOGICAL FUNCTIONS OF TYPE I TOXIN-ANTITOXIN SYSTEMS
Even with the detailed knowledge regarding regulatory complexity, it was common for scientists to ignore potential novel and critical biological functions for TA systems in prokaryotes. In particular, given the observed association of TA systems with plasmids and other MGEs suggests that these systems represent “selfish entities,” ensuring their own propagation by maintenance of their carriers in the course of vertical and horizontal gene transfer (HGT) (1, 3, 126). This idea is intuitive and probably applies to all MGE-associated TA systems but fails to convincingly explain the prevalence of TA systems in prokaryotic chromosomes (127–129). The fact that, in some cases, dozens of functional TA systems were retained by prokaryotic chromosomes has challenged the assumption of purely “selfish entities” and stimulated several lines of research to reveal whether they serve important functions to their hosts. In the following sections, we will discuss what is known about TA functions with a focus on type I TA systems. In contrast to type II, toxins from type I TA systems are typically small, hydrophobic proteins that target the cytoplasmic membrane and affect membrane integrity (Table 2). The concomitant cellular effects and potential biological functions will be described for some of the best-studied members.
TABLE 2.
Features of select type I toxins in Gram-positive and Gram-negative bacteria
| Toxin | Length(amino acids) | Antitoxin RNA | Localization | Cellular effects (biological function) | Species (location) | References |
|---|---|---|---|---|---|---|
| Gram-positive | ||||||
| BsrE | 30 | SR5 | Membrane | Cell lysis (prophage stabilization?) | Bacillus subtilis (P6 prophage-like element) | (61, 130, 131) |
| BsrG | 38 | SR4 | Membrane | Delocalization of cell wall systhesis machinery, membrane invaginations, cell lysis (prophage stabilization?) | Bacillus subtilis (SPβ prophage) | (19, 131, 132) |
| CD | 34–47 | RCd | Membrane | Growth inhibition (prophage stabilization) | Clostridioides difficile (phiCD630 prophage) | (133, 134) |
| Fst | 33 | RNAII | Membrane | Nucleoid condensation, cell division defects (plasmid stabilization) | Enterococcus faecalis (plasmid pAD1) | (47, 55, 135) |
| PepA1 (SprA1) | 30 | SprA1AS | Membrane | Membrane permeabilization, cytolytic activity (altruistic cell death?) | Staphylococcus aureus (pathogenicity island) | (87, 88) |
| PepA2 (SprA2) | 35 | SprA2AS | Membrane | Membrane permeabilization, cytolytic activity (altruistic cell death?) | Staphylococcus aureus (chromosome) | (89) |
| PepG1 (SprG1) | 31, 44 | SprF1 | Membrane | Membrane permeabilization, cytolytic activity (altruistic cell death?) | Staphylococcus aureus (pathogenicity island) | (136) |
| TxpA | 59 | RatA | Membrane | Cell lysis (prophage stabilization?) | Bacillus subtilis (Skin prophage) | (24, 25) |
| YonT | 58 | SR6 | Membrane | Cell lysis (prophage stabilization?) | Bacillus subtilis (SPβ prophage) | (62, 137) |
| Gram-negative | ||||||
| AapA1 | 30 | IsoA1 | Inner membrane | Morphological changes, coccoid formation | Helicobacter pylori (chromosome) | (97) |
| DinQ | 27 | AgrB | Inner membrane | Depolarization, ATP drop, nucleoid condensation (persistence?) | Escherichia coli (chromosome) | (20) |
| Hok | 52 | Sok | Inner membrane | Depolarization, cellular leakage, ghost cell formation (plasmid stabilization) | Escherichia coli (plasmid R1) | (43, 44) |
| HokB | 49 | SokB | Inner membrane | Pore formation, depolarization, ATP efflux (persistence) | Escherichia coli (chromosome) | (7, 50, 138) |
| IbsC | 18 | SibC | Inner membrane | Depolarization | Escherichia coli (chromosome) | (15) |
| LdrD | 35 | RdlD | Inner membrane | Nucleoid condensation, inhibition of translation | Escherichia coli (chromosome) | (58, 139) |
| ShoB | 26 | OhsC | Inner membrane | Depolarization | Escherichia coli (chromosome) | (15) |
| TimP | 38 | TimR | Inner membrane | Leaky membrane | Salmonella enterica (chromosome) | (60) |
| TisB | 29 | IstR-1 | Inner membrane | Pore formation, depolarization, ATP drop, protection from aminoglycosides (persistence) | Escherichia coli (chromosome) | (6, 22, 140–143) |
| ZorO | 29 | OrzO | Inner membrane | Depolarization, ATP drop, protection from aminoglycosides (persistence?) | Escherichia coli (chromosome) | (103, 104, 144) |
| RalR | 64 | RalA | Cytoplasm | DNA cleavage | Escherichia coli (cryptic prophage rac) | (70) |
| SymE | 113 | SymR | Cytoplasm | RNA cleavage, nucleoid condensation | Escherichia coli (chromosome) | (72, 145) |
Stabilization of mobile genetic elements
As already outlined further above, TA systems were originally discovered on bacterial plasmids and found to enhance plasmid propagation within expanding populations (42, 146). Due to their role in plasmid maintenance, they are often referred to as plasmid “addiction modules” (126, 147). However, TA systems have subsequently been discovered on diverse MGEs, such as prophages, superintegrons, or integrative conjugative elements (133, 148–150), which suggested a general role as MGE “maintenance modules.” Despite the diversity of TA systems found in MGEs, the mechanism by which TA systems stabilize MGEs turned out to follow a common scheme. Due to the inherent instability of the antitoxin, the antitoxin pool is quickly depleted in cells that have lost the TA carrier (i.e., the MGE), which in turn activates the stable toxin and enforces toxicity (Fig. 3A and B). The mechanism has initially been denoted as PSK (43), since some toxins have the potential to kill MGE-free cells (43, 44, 151). However, toxin-mediated growth inhibition of MGE-free cells would be sufficient to ensure that MGE-bearing cells outcompete their MGE-free siblings. Therefore, the likelihood of MGE transmission within expanding populations is enhanced by TA systems, irrespective of whether toxins kill or merely inhibit growth.
Fig 3.

Cellular effects and biological functions of type I toxin-antitoxin systems. (A) Plasmid stabilization. If the plasmid (blue) and its type I TA system is not inherited by a progeny cell, the unstable antitoxin RNA is degraded and the toxin (red) is produced. Most type I toxins are small membrane proteins that cause growth inhibition (and probably cell death). If the plasmid is retained, the antitoxin prevents toxin production and the cell is able to propagate. (B) Prophage stabilization follows the same principle as described for plasmid stabilization. The type I toxin (red) inhibits growth upon loss of the prophage (blue). (C) Morphological changes. If the type I toxin (red) is produced, it causes morphological changes, such as the transition from spiral-shaped to coccoid cells or cell division defects that are concomitant with membrane invaginations. (D) Cell death. If the type I toxin (red) is produced in sufficiently high amounts, it may induce cell lysis via membrane permeabilization. Cell death by lysis presumably represents an altruistic behavior of individual cells that benefits the remaining population. (E) Nucleoid condensation. Expression of the type I toxin (red) leads to compaction of chromosomal DNA. (F) Nucleic acid degradation. Some type I toxins (red) are cytosolic proteins that function as endonucleases and degrade either DNA or RNA.
Type I toxin-antitoxin systems and plasmid addiction
One of the first and probably best-studied examples of a plasmid-stabilizing TA system is the hok/sok system of plasmid R1 in E. coli (42). The intricate regulatory mechanisms, which have been described for hok/sok, represent a paradigm for posttranscriptional regulation in type I TA systems (see above) (80, 152). The two genes, hok and sok, are located on opposite strands within the parB region of plasmid R1 and display a fully overlapping arrangement (Table 1). The full-length hok transcript adopts a stable secondary structure with a half-life of approximately 20 min and can neither be translated nor bound by the RNA antitoxin Sok. Only the truncated hok transcript forms a translation-competent structure but is also rapidly bound by the Sok antitoxin and inactivated via RNase III processing (Fig. 2) (11, 76, 153). If E. coli cells do not inherit the R1 plasmid, the relatively unstable Sok RNA (half-life of ~30 seconds) is quickly removed from the cell, allowing the remaining pool of hok transcripts to produce Hok toxin and kill/inhibit the plasmid-free progeny cells (Fig. 3A). As a consequence, the R1 plasmid is stabilized greater than 100-fold (42). The Hok toxin is a small hydrophobic protein with a size of 52 amino acids that localizes to the inner membrane. Like other members of the Hok/Gef toxin family, it probably forms pores in the inner membrane and depletes membrane gradients, eventually causing cellular leakage and ghost cell formation (43, 44, 154) (Table 2). hok/sok systems are also present in the chromosomes of enteric bacteria, but their functions mainly remain to be elucidated (50, 51). We will, however, discuss one well-studied chromosomal member of the Hok/Gef toxin family, HokB, and its potential function in antibiotic persistence further below.
Another well-characterized example for a type I TA system with a role in plasmid addiction is fst/RNAII within the par locus of plasmid pAD1 in E. faecalis (47, 155). Fst is a membrane toxin with a size of 33 amino acids that causes cell division defects and nucleoid condensation (135) (Table 2), resulting in growth inhibition of plasmid-free cells (47) (Fig. 3A). Fst-like toxins belong to the Fst/Ldr family of type I toxins, which are widely distributed among Gram-positive bacteria (84). Given that many Fst-like toxins contribute to plasmid stabilization suggests that they are broadly utilized plasmid addiction systems (55). Interestingly, fst/RNAII systems were identified within chromosomal loci encoding proteins with a role in sugar metabolism (84). Even though an involvement of fst/RNAII systems in regulation of metabolism remains to be demonstrated, these findings likely indicate a function beyond plasmid stabilization.
The observation that TA systems cause plasmid addiction immediately suggests their “selfishness,” as it has already been suggested for restriction-modification systems (156). The bacteria become addicted to the presence of the antitoxin, even though the plasmid that provides the antitoxin represents a cellular burden. Assuming, however, that the plasmid provides important functions, maintenance of vertical plasmid transmission via TA systems is a clear advantage for the host (157). Furthermore, TA systems may also avoid the loss of beneficial plasmids when the host is invaded by a competitor plasmid via HGT. If the competitor plasmid belongs to the same incompatibility group but lacks the TA system, it is eliminated from the population via PSK of progeny cells (158). But how can bacteria eliminate a disadvantageous plasmid once they get addicted to it? One possibility is the acquisition of a chromosomal TA system that shares sufficient homology with the plasmid-based system. In such a scenario, the chromosomal antitoxin neutralizes the plasmid-derived toxin in plasmid-free progeny cells and terminates plasmid addiction. This phenomenon has been denoted anti-addiction and suggests that TA systems are involved in genome-plasmid conflicts (126, 157). It may also explain why free-living bacteria, which are regularly invaded by foreign DNA, have accumulated many chromosomal TA systems (128). The outstanding question is of course whether the “domesticated” chromosomal systems serve other important functions to their hosts.
Association of type I toxin-antitoxin systems with prophages
In lysogenic bacteria, the genomes of bacteriophages are often integrated into the chromosome where they are preserved as prophages and transmitted vertically to daughter cells (Fig. 3B). Alternatively, prophages may also exist as plasmids that are transmitted by both vertical and horizontal gene transfer. In the case of the P1 phage of E. coli, it was observed that the plasmid prophage is not easily eliminated from the bacterial population. The so-called “curing” of prophages is a rare event (loss rate of ~10−5 per cell per generation), and the underlying element for prophage stabilization turned out to be a type II TA system, denoted phd/doc (150). In prophage-free progeny cells, the antitoxin Phd will be slowly depleted by the ClpXP protease, avoiding further cell divisions due to liberation of the Doc toxin (159).
Detailed knowledge on type I TA systems with a role in prophage stabilization is just slowly accumulating. In the Gram-positive bacterium Clostridioides difficile, several type I TA systems have been identified within the phiCD630 prophage regions and denoted CD/RCd systems (133, 134) (Table 2). The membrane-targeting CD toxins stabilize the phiCD630 prophages due to growth inhibition of prophage-free cells (133) (Fig. 3B). Likewise, in B. subtilis, several type I TA systems are present within prophage elements (Table 2). The corresponding membrane toxins, such as BsrE, BsrG, TxpA, and YonT, potentially cause cell lysis (24, 61, 62, 132), and it was proposed that they maintain prophages via killing of prophage-free cells (137). It can, therefore, be expected that prophage stabilization by type I TA systems is a common mechanism that contributes to the intricate relationship between lysogenic bacteria and their phages.
Type I toxins that cause morphological changes and cell lysis
Pathogenic bacteria have developed manifold strategies to adapt to the specific site of infection within their hosts. Type I TA systems may help to counteract certain stresses or support survival of the bacterial population during infection. For example, H. pylori colonizes the human stomach and resides in the gastric mucosa, where it is challenged by reactive oxygen species (ROS) produced by the host (160). It was shown that H. pylori depletes the RNA antitoxin IsoA1 in response to oxidative stress, which increases the likelihood of AapA1 synthesis. AapA1 is a membrane toxin with a size of 30 amino acids (Table 2) that supports the morphological transformation from spiral-shaped to coccoid cells (97) (Fig. 3C). Experimental evidence supports the view that coccoid cells are viable but “dormant” forms of H. pylori. In such a dormant state, which is reminiscent of a persister state (see further below), H. pylori might withstand stress and even escape the immune response of the host (161). Hence, AapA1 is part of the H. pylori survival strategy within the stomach and potentially contributes to persistent infections.
The plasmid-stabilizing toxin Fst from E. faecalis was demonstrated to cause morphological changes, which were associated with cell division defects, such as missing cell wall bands, aberrant division furrows, and invaginations of cell filaments (162) (Fig. 3C). In the case of Fst, and in contrast to other membrane toxins, severe membrane permeabilization was not observed. It seems likely that the predominant role of Fst is disturbance of cell division (Table 2), which in turn inhibits plasmid-free cells and contributes to plasmid stabilization in clonal populations (47, 135, 162), as described above.
Further interesting observations concerning morphological changes stem from the bsrG/SR4 type I TA system located in the SPβ prophage region of B. subtilis (Table 2). The BsrG membrane toxin (38 amino acids) was shown to cause delocalization of the cell wall synthesis machinery, which was accompanied by membrane invaginations (132) (Fig. 3C). The morphological changes were followed by cell lysis in a process that depended on the autolysins LytC and LytD, and the bacterial cytoskeletal protein MreB (132). Hypothetically, BsrG-mediated cell lysis provides an efficient means to stabilize the SPβ prophage. Whether other membrane toxins from B. subtilis prophage regions (i.e., BsrE, TxpA, YonT; Table 2) cause morphological changes that precede cell lysis remains to be demonstrated. It is tempting to speculate that only those cells that produce sufficiently high toxin amounts are subject to cell lysis, while other cells remain within a state of growth inhibition that is probably reversible and marked by harmless morphological changes and cell division defects.
Cell lysis may represent a strategy that benefits pathogenic bacteria during infection. In S. aureus strain Newman, the type I TA systems sprA1/sprA1AS and sprG1/sprF1 are located within pathogenicity islands, whereas the sprA2/sprA2AS locus is in the core genome (87–89, 136). The corresponding toxins PepA1 (SprA1), PepA2 (SprA2), and PepG1 (SprG1) were associated with cytolytic effects (Table 2), which presumably support virulence and the spread of infection. Depletion of the antitoxin RNA de-represses the toxin mRNA and enables production of toxin, which has the potential to cause cell death of the producing S. aureus cells via membrane permeabilization (Fig. 3D) but may also have hemolytic activity and antimicrobial activity against competing bacteria (87–89, 136). In the case of sprA1/sprA1AS, the antitoxin RNA SprA1AS is depleted upon acidic pH or oxidative stress, conditions that are predominant in phagolysosomes of host immune cells. The authors speculated that PepA1 production is triggered in phagolysosomes, leading to lysis of most PepA1-producing cells, release of PepA1 toxins, and damage of the host membrane. Hence, the PepA1-induced cell death of S. aureus represents an “altruistic behavior” that benefits the surviving cells by promoting their escape from immune cells and spreading into the host (88). In the case of sprA2/sprA2AS, the antitoxin SprA2AS is depleted in response to osmotic stress and starvation. As speculated for PepA1, PepA2-induced cell death is presumably altruistic and increases the success of the population by promoting cytotoxic effects against host cells (89). The SprG1 mRNA encodes two peptides with lengths of 31 and 44 amino acids, designated PepG1 toxins (Table 2). Both toxins trigger S. aureus cell death by lysis (Fig. 3D) and are secreted (136). While the longer toxin is more active against host cells, the shorter toxin is more active against competing bacteria. As for PepA1 and PepA2, cell death and release of PepG1 toxins may represent an advantageous strategy during infections, as toxin-producing cells sacrifice themselves for the benefit of the remaining population.
Type I toxins that affect nucleic acids
A recurrent observation upon expression of toxin genes from type I TA systems concerns compaction of the nucleoid, also referred to as nucleoid condensation (Fig. 3E). Nucleoid condensation was observed, among others, for Fst, LdrD, and DinQ (20, 58, 135) (Table 2). It is so far unknown which mechanism is responsible for nucleoid condensation in response to these membrane toxins, but it appears likely that condensation is a secondary effect. The manifestation of nucleoid condensation probably depends on an unknown toxin target (58), is a downstream effect of inhibition of protein biosynthesis (139), or involves a component of the cytoskeleton (135). Whether nucleoid condensation itself contributes to toxin-mediated growth inhibition or affects DNA repair process, as speculated for DinQ (20), remains to be demonstrated in the future.
The type I toxins discussed so far are targeted toward the cytoplasmic membrane and represent small, hydrophobic proteins with a size below 60 amino acids (Table 2). There are, however, exceptions to this rule. The RalR toxin from the ralR/ralA TA system, which is located in the cryptic prophage rac of E. coli, has a size of 64 amino acids and remains in the cytoplasm. It acts as an endonuclease that cleaves both methylated and unmethylated DNA (70) (Fig. 3F). It was observed that expression of ralR inhibits growth and provides protection against fosfomycin, an antibiotic that inhibits peptidoglycan biosynthesis (70). Whether there is a direct link between DNA cleavage and protection against an inhibitor of cell wall biosynthesis remains unknown.
Another type I toxin with a cytoplasmic localization is SymE from the symE/symR system in E. coli (Table 2). The symE gene is induced in response to DNA damage (SOS response) (99). The SymE toxin has a size of 113 amino acids and was originally identified as an endoribonuclease that cleaves and recycles damaged RNAs (72) (Fig. 3F). However, global analyses challenged the endoribonucleolytic activity of SymE and rather suggested that SymE is a DNA-binding toxin with the potential to cause nucleoid condensation (145). In contrast to the membrane toxins described further above, nucleoid condensation might be a direct function of SymE due to its DNA-binding properties. Whether these findings exclude an endoribonucleolytic activity or whether SymE is a toxin with dual functions remains to be elucidated.
TYPE I TOXIN-ANTITOXIN SYSTEMS AND THEIR CONTRIBUTION TO ANTIBIOTIC PERSISTENCE
Persistence is characterized by a fraction of cells that display a transient state of antibiotic tolerance. The persister state itself is marked by reduced activity of major cellular processes and halted cell growth. We will introduce antibiotic persistence and shortly summarize what is known about the factors that potentially induce the persister state. Finally, we will discuss the involvement of TA systems with a focus on pore-forming type I toxins.
How bacteria counteract antibiotics: resistance versus persistence
Microorganisms produce an unprecedented diversity of secondary metabolites, many of which provide ecologically important activities. A prominent example are antimicrobial compounds (e.g., antibiotics) that are used as weapons in the ongoing warfare between microorganisms in their natural habitats (163). Due to the pervasive threat posed by antibiotics, bacteria have developed manifold strategies to counteract the action of antibiotics, resulting in full or partial resilience of populations.
Most notorious for antibiotic resilience is the occurrence of resistance, which is the genetically acquired capability to not only survive but also thrive in the presence of antibiotics. Bacteria may develop a chromosomal mutation that leads to a modified structure of the antibiotic target molecule, thereby making the antibiotic ineffective. Alternatively, bacteria may acquire genes that provide resistance by enzymatic inactivation or exclusion/export of the antibiotic. Since resistance genes are eventually transferred to bacterial pathogens by HGT, they represent a major challenge to our healthcare system (164). Resistant strains usually have three features: (i) the minimum inhibitory concentration of the respective antibiotic is increased in comparison to a susceptible strain, (ii) the resistant phenotype is inherited by progeny cells, and (iii) the whole population shows the resistant phenotype [on a note, transient resistance in a subpopulation of cells, denoted heteroresistance, is occasionally observed but not further discussed here (165)]. As a consequence, the entire population of cells of a resistant strain continues growing in the presence of the antibiotic, even at concentrations that effectively kill susceptible strains (Fig. 4A).
Fig 4.

Persistence caused by type I toxin-antitoxin systems. (A) Illustration of antibiotic killing kinetics (left) and corresponding bacterial populations (right). Susceptible cells are rapidly killed (gray), while resistant cells continue growing (blue) in the presence of drugs (e.g., bactericidal antibiotics). If persister cells are present, a biphasic killing curve emerges due to long-term antibiotic tolerance of the persister subpopulation (red). (B) Induction of persistence by type I toxins. An active cell has a polarized membrane (as indicated by protons at the outside of its cytoplasmic membrane). The proton gradient is used by ATPases to produce ATP (top middle). Active cells are susceptible and rapidly killed when exposed to drugs (top left). Some type I toxins are pore-forming membrane toxins (red) with the potential to promote a drug-tolerant persister state. Type I toxins may impede drug uptake due to depolarization of the cytoplasmic membrane (upper right). In addition, depolarization inhibits ATP production and leads to ATP depletion (bottom right). Alternatively, type I toxins form pores that are capable of promoting ATP efflux (bottom left). Decreasing ATP levels cause cellular inactivity, which prevents killing by drugs.
In 1944, Joseph Bigger observed that penicillin-treated Staphylococcus cultures retained a small fraction of surviving cells even after several days of treatment. He called these survivors “persister cells” and concluded that they are in a non-growing state and thus tolerate antibiotics for a long period of time (166, 167). A hallmark of persistence is the biphasic killing curve that occurs upon antibiotic treatment due to the heterogeneous nature of the populations. The susceptible subpopulation (usually the majority) is rapidly killed by the antibiotic, while the persister subpopulation (usually a minor fraction) tolerates even high doses of the antibiotic for an extended period and is therefore killed at a much slower rate (Fig. 4A). Persister cells strongly contribute to long-term survival of bacterial populations, as they withstand antibiotics for several days and probably even longer. This clearly sets them apart from short-term tolerant cells, which tolerate antibiotics only for several hours (168, 169). Importantly, recultivation of the surviving persister fraction gives rise to a heterogeneous population with the same features as the original population (168, 170). Hence, the persister state is of transient nature, suggesting that changes in gene expression determine the phenotypic alterations (171).
Even though persistence is a phenomenon that is restricted to a small subpopulation of cells, it potentially has big consequences in the context of bacterial infections. Due to their recalcitrance toward antibiotics, the emergence of persister cells is associated with treatment failure of cystic fibrosis patients suffering from Pseudomonas aeruginosa infections (172), or patients infected by Mycobacterium tuberculosis and Candida albicans (173, 174). The failure to efficiently eradicate persister cells by antibiotics leads to recurrent infections and causes major problems in clinical settings (175). Furthermore, repeated antibiotic therapy selects for pathogenic strains with “high persistence” (Hip) phenotypes (i.e., increased persister frequencies) (172–174), which eventually serves as melting pot for the development of resistance (176–178). Bacterial biofilms represent another complicating issue because biofilm environments not only support persister formation but also physically protect persister cells from the immune system and antibiotics, resulting in tremendous treatment failure (179).
Persistence has been known for decades and has been intensively studied for the past 20 years. However, there is still no unifying model that describes the generation or physiological state of persister cells. The central question still is how a persister state is established. Many factors have been suggested to play a determining role in the persister formation process, including the stationary-phase response and its regulators RpoS and the alarmone (p)ppGpp (180–187), oxidative stress (188–191), low membrane potential and adenosine triphosphate (ATP) depletion (192–195), inhibition of core processes via nutrient limitation (196), bacteriostatic agents (197), and “errors and glitches” (also known as “persistence as stuff happens”) (198). What these factors have in common is their association with growth inhibition. Since many toxins from TA systems inhibit growth, they have been suggested early on as bona fide persistence factors. However, this view has been challenged during the last years and is still subject to an ongoing scientific debate.
The controversial dogma of toxin-induced persistence
After the initial discovery of persister cells in 1944 by Bigger (166, 167), it was almost 40 years later until a molecular factor was suggested that had the potential to induce the persister state. Moyed and Bertrand (199) applied chemical mutagenesis to isolate E. coli mutants with a Hip phenotype. Specifically, the hipA7 mutant showed higher persister frequencies when treated with diverse cell wall-inhibiting antibiotics (199). Subsequent work showed that HipA was the toxin moiety of the type II TA system hipAB, and that the hipA7 allele increased persister frequencies by 10- to 10,000-fold, even when different classes of antibiotics were applied (200–203). Since toxins were already known to impair essential cellular processes, it was tempting to generalize the idea that chromosomal TA systems were involved in establishment of an antibiotic-tolerant state. And indeed, subsequent observations supported the “toxin-induced-persistence” model. First, it was shown that several TA systems were upregulated in isolated persister fractions of E. coli and M. tuberculosis (204–206). Furthermore, some toxin deletion strains had lower persister frequencies under specific experimental conditions (6, 140, 181, 207, 208), and plasmid-borne overexpression of almost all tested toxins led to a sharp increase in persister frequencies, regardless of which antibiotic was applied (6, 202, 208, 209).
Even though the “toxin-induced-persistence” model is intriguing, it was frequently challenged because different laboratories produced conflicting results concerning phenotypes and activation of toxins (8, 210, 211). As it stands right now, the community seems divided, with some scientists doubting the involvement of toxins in the persister formation process. Clearly, TA systems are dispensable for persistence to occur (196, 212, 213), but this does not refute them as contributing factors.
Pore-forming type I toxins affect antibiotic persistence via ATP depletion
In bacteria with respiratory activity, generation of ATP is mainly catalyzed by ATP synthase. This multi-subunit protein complex is fueled by the proton motive force (PMF), an electrochemical gradient across the inner membrane (Fig. 4B). Since ATP is the most important energy source for a plethora of cellular processes, PMF dissipation and ATP depletion have been associated with growth inhibition and establishment of a persister state (6, 140, 141, 192, 193). Some toxins from type I TA systems have the potential to directly compromise the PMF and thereby interfere with ATP production (Fig. 4B). Potentially, these type I toxins form oligomeric structures that resemble narrow pores (or channels), which are compatible with passage of ions across the inner membrane. Since ions follow their gradients, the PMF is easily discharged by such ion-selective pores (141, 214, 215). Indeed, when potential sensitive probes were applied, a reduction in the membrane potential, which is a good indicator for a compromised PMF, was observed for several type I toxins, including TisB, HokB, DinQ, ZorO, ShoB, and IbsC in E. coli (7, 15, 20, 103, 140, 141, 216). Reduction in the membrane potential was usually paralleled by a drop in intracellular ATP levels (103, 141, 142). Interestingly, it was suggested that mature HokB pores have an increased diameter, causing direct leakage of ATP to the exterior (138) (Fig. 4B). Whether this is a common mechanism needs to be clarified, but as it stands right now, most type I toxins only indirectly deplete ATP via formation of narrow pores that dissipate the PMF. Regardless of how ATP is depleted (directly or indirectly), the resulting energy deprivation is expected to slow down major cellular processes, protect antibiotic targets, and induce a persister state (5, 217) (Fig. 4B). An involvement in persister formation was hence suggested for some type I toxins, such as HokB and TisB (6, 7, 140).
SOS-dependent persister formation: the TisB paradigm
While overexpression of toxins usually has strong effects, deletion of toxin genes often needs specific experimental conditions to produce phenotypes. Thus, knowing the inducing condition for a toxin is beneficial to study its phenotypic consequences under physiological conditions. In E. coli MG1655, the toxin gene tisB is under LexA control and strongly induced under DNA damage/SOS conditions (6, 22, 218). When tisB was deleted, application of the DNA-damaging fluoroquinolone antibiotic ciprofloxacin led to a ~ 10-fold reduction in persister frequencies (6, 140). Interestingly, under these experimental conditions, a TisB-dependent reduction in the membrane potential was observed only in a fraction of cells, indicating heterogeneous tisB expression levels among the population (140). Consistent with the posttranscriptional tisB regulation described further above, chromosomal deletion of the antitoxin RNA (ΔistR-1) and shortening of the tisB 5´ UTR (Δ1–41) caused strong TisB synthesis upon ciprofloxacin treatment (219). As a result, the fraction of depolarized cells was increased, ATP levels were reduced, translation was inhibited, and persister frequencies increased up to 100-fold (140, 219). These data suggest that TisB potentially affects persistence through pore formation (141, 214, 215), dissipation of the PMF, and ATP depletion (Fig. 4B). In addition, the sophisticated posttranscriptional regulation of tisB (and other type I toxins) sets tight thresholds and restricts toxin production to specific stress conditions (140, 220).
PMF dissipation and ATP depletion are not the only observable consequences of TisB production. Overexpression of TisB (and other type I toxins) led to the production of ROS, which was also confirmed in a wild-type background after treatment with fluoroquinolone antibiotics (216, 221). Interestingly, ROS production itself was associated with persister formation (188–191). Recent observations also suggest that TisB is the major molecular determinant of protein aggregation and cytoplasmic condensation in response to fluoroquinolone antibiotics (216, 222). We conclude that disruption of membrane functioning by TisB likely causes primary (PMF dissipation) and secondary effects (ATP depletion, ROS production, protein aggregation, cytoplasmic condensation), which all have the potential to induce a persister state. However, in wild-type cultures, TisB-dependent effects are often only apparent after extended fluoroquinolone treatments, indicating that TisB is only slowly produced over time (140, 216, 221). These observations stimulate the view that the primary function of TisB is not necessarily induction but rather stabilization of a persister state. Potentially, TisB production and its concomitant effects (primary and secondary) determine dormancy depth and avoid premature awakening of persister cells (221, 223).
A new turn in type I toxin functionality: reduced uptake of toxic compounds
Cellular inactivity through ATP depletion is probably an indirect and rather delayed consequence of PMF dissipation by pore-forming type I toxins. A more direct and immediate consequence is the interference with membrane transport and drug uptake (Fig. 4B), which was recently suggested for TisB and other membrane toxins. TisB was shown to impair membrane transport and to prevent uptake of select toxic compounds, such as the aminoglycoside antibiotic gentamicin (143). Similarly, for the type I toxin ZorO of E. coli O157:H7 strain EDL933, overexpression of the zorO gene led to a shortened lag phase in sublethal levels of the aminoglycoside antibiotic kanamycin. However, a point mutation that rendered the translated ZorO protein non-toxic also conferred the same phenotype, suggesting that the mechanism of ZorO toxicity was separate from the mechanism of shortened lag (103, 144). Another study applied a library of randomized DNA sequences, which led to the identification of a synthetic, hydrophobic peptide, Arp1, that reduced the membrane potential and increased the resistance to aminoglycosides (224). Accordingly, type I toxins may affect antibiotic resilience by both supporting dormancy and preventing drug uptake via reduction of membrane potential. Hypothetically, these two functions are not strictly tethered and depend on the given toxin features and/or expression levels.
Open questions about type I toxin functions
It is usually assumed that type I toxins act by themselves via pore formation or other oligomeric structures. However, it is a so far unexplored possibility if type I toxins have interaction partners and modulate the function of membrane complexes, as observed for other small proteins (225). In this regard, it also remains unknown whether the charged amino acids, which are often essential for type I toxin functionality (103, 221, 222), solely contribute to oligomerization (214, 215) or are involved in the interaction with protein partners.
Another intriguing question concerns RNA editing of toxin mRNAs. In E. coli, it was observed that adenosine (A) to inosine (I) editing of the hokB mRNA recoded a tyrosine into a cysteine codon, which enhanced HokB toxicity (226). The editing rate increased with high cell densities, implying that a change in toxicity is potentially important under growth-limiting conditions. Whether similar mechanisms apply to other toxin mRNAs needs to be elucidated. The observation that synthetic, toxic peptides can be selected from randomized DNA sequences (224) suggests that type I toxins can be used as blueprints for the development of antimicrobial agents (227).
Many type I toxin functions have been deduced from strains with artificial expression constructs that might be prone to generating abnormal effects. It is a foremost task to identify the specific factors for toxin activation, which may help to define the physiologically relevant conditions for detection of authentic phenotypes (6, 181). However, even with the knowledge about the physiologically relevant condition, different laboratories sometimes obtain conflicting results when using slightly different treatment schemes (6, 140, 210, 216, 222). Revealing the biological reason for these inconsistencies under seemingly concordant conditions might contribute to our understanding of toxin-related phenotypes. Regarding type I TA systems and persistence, the idea that type I toxins determine dormancy depth and avoid premature awakening of persister cells (223) is intriguing and needs further attention. Finally, most type I TA systems have been studied in pure cultures, often using strains that are devoid of their phages. Studying type I TA systems in the context of their natural competitors might shed new light on these systems and reveal further relevant biological functions.
ACKNOWLEDGMENTS
We thank Patrick Lane of ScEYEnce Studios for enhancement of figures.
F.H.L. and B.A.B. would like to acknowledge support from the German Research Council (DFG) in the framework of the SPP 2002 (BE 5210/3-2). S.F.H.S. and E.M.F. would like to acknowledge support from the NIH via R15GM106291 and the UTK Office of Research, Innovation and Economic Development.
Contributor Information
Elizabeth M. Fozo, Email: efozo@utk.edu.
Bork A. Berghoff, Email: Bork.A.Berghoff@mikro.bio.uni-giessen.de.
Gregory J. Phillips, University of Georgia, Athens, Georgia, USA
REFERENCES
- 1. Harms A, Brodersen DE, Mitarai N, Gerdes K. 2018. Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol Cell 70:768–784. doi: 10.1016/j.molcel.2018.01.003 [DOI] [PubMed] [Google Scholar]
- 2. Jurėnas D, Fraikin N, Goormaghtigh F, Van Melderen L. 2022. Biology and evolution of bacterial toxin-antitoxin systems. Nat Rev Microbiol 20:335–350. doi: 10.1038/s41579-021-00661-1 [DOI] [PubMed] [Google Scholar]
- 3. Hayes F. 2003. Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science 301:1496–1499. doi: 10.1126/science.1088157 [DOI] [PubMed] [Google Scholar]
- 4. Leroux M, Laub MT. 2022. Toxin-antitoxin systems as phage defense elements. Annu Rev Microbiol 76:21–43. doi: 10.1146/annurev-micro-020722-013730 [DOI] [PubMed] [Google Scholar]
- 5. Lewis K. 2010. Persister cells. Annu Rev Microbiol 64:357–372. doi: 10.1146/annurev.micro.112408.134306 [DOI] [PubMed] [Google Scholar]
- 6. Dörr T, Vulić M, Lewis K. 2010. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol 8:e1000317. doi: 10.1371/journal.pbio.1000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Verstraeten N, Knapen WJ, Kint CI, Liebens V, Van den Bergh B, Dewachter L, Michiels JE, Fu Q, David CC, Fierro AC, Marchal K, Beirlant J, Versées W, Hofkens J, Jansen M, Fauvart M, Michiels J. 2015. Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol Cell 59:9–21. doi: 10.1016/j.molcel.2015.05.011 [DOI] [PubMed] [Google Scholar]
- 8. Van Melderen L, Wood TK. 2017. Commentary: what is the link between stringent response, endoribonuclease encoding type II toxin-antitoxin systems and persistence? Front Microbiol 8:191. doi: 10.3389/fmicb.2017.00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li M, Gong L, Cheng F, Yu H, Zhao D, Wang R, Wang T, Zhang S, Zhou J, Shmakov SA, Koonin EV, Xiang H. 2021. Toxin-antitoxin RNA pairs safeguard CRISPR-Cas systems. Science 372:eabe5601. doi: 10.1126/science.abe5601 [DOI] [PubMed] [Google Scholar]
- 10. Choi JS, Kim W, Suk S, Park H, Bak G, Yoon J, Lee Y. 2018. The small RNA, SdsR, acts as a novel type of toxin in Escherichia coli. RNA Biol 15:1319–1335. doi: 10.1080/15476286.2018.1532252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Franch T, Gultyaev AP, Gerdes K. 1997. Programmed cell death by hok/sok of plasmid R1: processing at the hok mRNA 3’-end triggers structural rearrangements that allow translation and antisense RNA binding. J Mol Biol 273:38–51. doi: 10.1006/jmbi.1997.1294 [DOI] [PubMed] [Google Scholar]
- 12. Gerdes K, Thisted T, Martinussen J. 1990. Mechanism of post-segregational killing by the hok/sok system of plasmid R1: sok antisense RNA regulates formation of a hok mRNA species correlated with killing of plasmid-free cells. Mol Microbiol 4:1807–1818. doi: 10.1111/j.1365-2958.1990.tb02029.x [DOI] [PubMed] [Google Scholar]
- 13. Greenfield TJ, Ehli E, Kirshenmann T, Franch T, Gerdes K, Weaver KE. 2000. The antisense RNA of the par locus of pAD1 regulates the expression of a 33-amino-acid toxic peptide by an unusual mechanism. Mol Microbiol 37:652–660. doi: 10.1046/j.1365-2958.2000.02035.x [DOI] [PubMed] [Google Scholar]
- 14. Shokeen S, Patel S, Greenfield TJ, Brinkman C, Weaver KE. 2008. Translational regulation by an intramolecular stem-loop is required for intermolecular RNA regulation of the par addiction module. J Bacteriol 190:6076–6083. doi: 10.1128/JB.00660-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fozo EM, Kawano M, Fontaine F, Kaya Y, Mendieta KS, Jones KL, Ocampo A, Rudd KE, Storz G. 2008. Repression of small toxic protein synthesis by the Sib and OhsC small RNAs. Mol Microbiol 70:1076–1093. doi: 10.1111/j.1365-2958.2008.06394.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Han K, Kim KS, Bak G, Park H, Lee Y. 2010. Recognition and discrimination of target mRNAs by Sib RNAs, a cis-encoded sRNA family. Nucleic Acids Res 38:5851–5866. doi: 10.1093/nar/gkq292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arnion H, Korkut DN, Masachis Gelo S, Chabas S, Reignier J, Iost I, Darfeuille F. 2017. Mechanistic insights into type I toxin antitoxin systems in Helicobacter pylori: the importance of mRNA folding in controlling toxin expression. Nucleic Acids Res 45:4782–4795. doi: 10.1093/nar/gkw1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jahn N, Preis H, Wiedemann C, Brantl S. 2012. BsrG/SR4 from Bacillus subtilis--the first temperature-dependent type I toxin-antitoxin system. Mol Microbiol 83:579–598. doi: 10.1111/j.1365-2958.2011.07952.x [DOI] [PubMed] [Google Scholar]
- 19. Jahn N, Brantl S. 2013. One antitoxin-two functions: SR4 controls toxin mRNA decay and translation. Nucleic Acids Res 41:9870–9880. doi: 10.1093/nar/gkt735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weel-Sneve R, Kristiansen KI, Odsbu I, Dalhus B, Booth J, Rognes T, Skarstad K, Bjørås M. 2013. Single transmembrane peptide DinQ modulates membrane-dependent activities. PLoS Genet 9:e1003260. doi: 10.1371/journal.pgen.1003260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kristiansen KI, Weel-Sneve R, Booth JA, Bjørås M. 2016. Mutually exclusive RNA secondary structures regulate translation initiation of DinQ in Escherichia coli. RNA 22:1739–1749. doi: 10.1261/rna.058461.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vogel J, Argaman L, Wagner EGH, Altuvia S. 2004. The small RNA IstR inhibits synthesis of an SOS-induced toxic peptide. Curr Biol 14:2271–2276. doi: 10.1016/j.cub.2004.12.003 [DOI] [PubMed] [Google Scholar]
- 23. Darfeuille F, Unoson C, Vogel J, Wagner EGH. 2007. An antisense RNA inhibits translation by competing with standby ribosomes. Mol Cell 26:381–392. doi: 10.1016/j.molcel.2007.04.003 [DOI] [PubMed] [Google Scholar]
- 24. Silvaggi JM, Perkins JB, Losick R. 2005. Small untranslated RNA antitoxin in Bacillus subtilis. J Bacteriol 187:6641–6650. doi: 10.1128/JB.187.19.6641-6650.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Durand S, Gilet L, Condon C. 2012. The essential function of B. subtilis RNase III is to silence foreign toxin genes. PLoS Genet 8:e1003181. doi: 10.1371/journal.pgen.1003181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jurėnas D, Van Melderen L. 2020. The variety in the common theme of translation inhibition by type II toxin-antitoxin systems. Front Genet 11:262. doi: 10.3389/fgene.2020.00262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fraikin N, Goormaghtigh F, Melderen L. 2020. Type II toxin-antitoxin systems: evolution and revolutions. J Bacteriol 202:e00763. doi: 10.1128/JB.00763-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aakre CD, Phung TN, Huang D, Laub MT. 2013. A bacterial toxin inhibits DNA replication elongation through a direct interaction with the β sliding clamp. Mol Cell 52:617–628. doi: 10.1016/j.molcel.2013.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang X, Yao J, Sun YC, Wood TK. 2021. Type VII toxin/antitoxin classification system for antitoxins that enzymatically neutralize toxins. Trends Microbiol 29:388–393. doi: 10.1016/j.tim.2020.12.001 [DOI] [PubMed] [Google Scholar]
- 30. Yao J, Zhen X, Tang K, Liu T, Xu X, Chen Z, Guo Y, Liu X, Wood TK, Ouyang S, Wang X. 2020. Novel polyadenylylation-dependent neutralization mechanism of the HEPN/MNT toxin/antitoxin system. Nucleic Acids Res 48:11054–11067. doi: 10.1093/nar/gkaa855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marimon O, Teixeira JMC, Cordeiro TN, Soo VWC, Wood TL, Mayzel M, Amata I, García J, Morera A, Gay M, Vilaseca M, Orekhov VY, Wood TK, Pons M. 2016. An oxygen-sensitive toxin–antitoxin system. Nat Commun 7:13634. doi: 10.1038/ncomms13634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu X, Gao X, Zhu K, Yin H, Mao X, Wojdyla JA, Qin B, Huang H, Wang M, Sun Y-C, Cui S. 2020. Characterization of a toxin-antitoxin system in Mycobacterium tuberculosis suggests neutralization by phosphorylation as the antitoxicity mechanism. Commun Biol 3:216. doi: 10.1038/s42003-020-0941-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brown JM, Shaw KJ. 2003. A novel family of Escherichia coli toxin-antitoxin gene pairs. J Bacteriol 185:6600–6608. doi: 10.1128/JB.185.22.6600-6608.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tan Q, Awano N, Inouye M. 2011. YeeV is an Escherichia coli toxin that inhibits cell division by targeting the cytoskeleton proteins, FtsZ and MreB. Mol Microbiol 79:109–118. doi: 10.1111/j.1365-2958.2010.07433.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heller DM, Tavag M, Hochschild A. 2017. CbtA toxin of Escherichia coli inhibits cell division and cell elongation via direct and independent interactions with FtsZ and MreB. PLoS Genet 13:e1007007. doi: 10.1371/journal.pgen.1007007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Masuda H, Tan Q, Awano N, Wu KP, Inouye M. 2012. YeeU enhances the bundling of cytoskeletal polymers of MreB and FtsZ, antagonizing the CbtA (YeeV) toxicity in Escherichia coli. Mol Microbiol 84:979–989. doi: 10.1111/j.1365-2958.2012.08068.x [DOI] [PubMed] [Google Scholar]
- 37. Wang X, Lord DM, Cheng HY, Osbourne DO, Hong SH, Sanchez-Torres V, Quiroga C, Zheng K, Herrmann T, Peti W, Benedik MJ, Page R, Wood TK. 2012. A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat Chem Biol 8:855–861. doi: 10.1038/nchembio.1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhong A, Jiang X, Hickman AB, Klier K, Teodoro GIC, Dyda F, Laub MT, Storz G. 2023. Toxic antiphage defense proteins inhibited by Intragenic antitoxin proteins. Proc Natl Acad Sci U S A 120:e2307382120. doi: 10.1073/pnas.2307382120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bobonis J, Mitosch K, Mateus A, Karcher N, Kritikos G, Selkrig J, Zietek M, Monzon V, Pfalz B, Garcia-Santamarina S, Galardini M, Sueki A, Kobayashi C, Stein F, Bateman A, Zeller G, Savitski MM, Elfenbein JR, Andrews-Polymenis HL, Typas A. 2022. Bacterial retrons encode phage-defending tripartite toxin-antitoxin systems. Nature 609:144–150. doi: 10.1038/s41586-022-05091-4 [DOI] [PubMed] [Google Scholar]
- 40. Millman A, Bernheim A, Stokar-Avihail A, Fedorenko T, Voichek M, Leavitt A, Oppenheimer-Shaanan Y, Sorek R. 2020. Bacterial retrons function in anti-phage defense. Cell 183:1551–1561. doi: 10.1016/j.cell.2020.09.065 [DOI] [PubMed] [Google Scholar]
- 41. Nordström K, Molin S, Aagaard-Hansen H. 1980. Partitioning of plasmid R1 in Escherichia coli. I. Kinetics of loss of plasmid derivatives deleted of the par region. Plasmid 4:215–227. doi: 10.1016/0147-619x(80)90011-6 [DOI] [PubMed] [Google Scholar]
- 42. Gerdes K, Larsen JEL, Molin S. 1985. Stable inheritance of Plasmid R1 requires two different Loci. J Bacteriol 161:292–298. doi: 10.1128/jb.161.1.292-298.1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gerdes K, Rasmussen PB, Molin S. 1986. Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc Natl Acad Sci U S A 83:3116–3120. doi: 10.1073/pnas.83.10.3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gerdes K, Bech FW, Jørgensen ST, Løbner-Olesen A, Rasmussen PB, Atlung T, Boe L, Karlstrom O, Molin S, von Meyenburg K. 1986. Mechanism of postsegregational killing by the hok gene product of the parB system of plasmid R1 and its homology with the relF gene product of the E. coli relB operon. EMBO J 5:2023–2029. doi: 10.1002/j.1460-2075.1986.tb04459.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Loh SM, Cram DS, Skurray RA. 1988. Nucleotide sequence and transcriptional analysis of a third function (Flm) involved in F-plasmid maintenance. Gene 66:259–268. doi: 10.1016/0378-1119(88)90362-9 [DOI] [PubMed] [Google Scholar]
- 46. Nielsen AK, Thorsted P, Thisted T, Wagner EGH, Gerdes K. 1991. The rifampicin-inducible genes srnB from F and pnd from R483 are regulated by antisense RNAs and mediate plasmid maintenance by killing of plasmid-free segregants. Mol Microbiol 5:1961–1973. doi: 10.1111/j.1365-2958.1991.tb00818.x [DOI] [PubMed] [Google Scholar]
- 47. Weaver KE, Tritle DJ. 1994. Identification and characterization of an Enterococcus faecalis plasmid pAD1-encoded stability determinant which produces two small RNA molecules necessary for its function. Plasmid 32:168–181. doi: 10.1006/plas.1994.1053 [DOI] [PubMed] [Google Scholar]
- 48. Weaver KE, Jensen KD, Colwell A, Sriram SI. 1996. Functional analysis of the Enterococcus faecalis plasmid pAD1-encoded stability determinant par. Mol Microbiol 20:53–63. doi: 10.1111/j.1365-2958.1996.tb02488.x [DOI] [PubMed] [Google Scholar]
- 49. Weaver KE, Clewell DB, An F. 1993. Identification, characterization, and nucleotide sequence of a region of Enterococcus faecalis pheromone-responsive plasmid pAD1 capable of autonomous replication. J Bacteriol 175:1900–1909. doi: 10.1128/jb.175.7.1900-1909.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pedersen K, Gerdes K. 1999. Multiple hok genes on the chromosome of Escherichia coli. Mol Microbiol 32:1090–1102. doi: 10.1046/j.1365-2958.1999.01431.x [DOI] [PubMed] [Google Scholar]
- 51. Faridani OR, Nikravesh A, Pandey DP, Gerdes K, Good L. 2006. Competitive inhibition of natural antisense Sok-RNA interactions activates Hok-mediated cell killing in Escherichia coli. Nucleic Acids Res 34:5915–5922. doi: 10.1093/nar/gkl750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bech FW, Jørgensen ST, Diderichsen B, Karlström OH. 1985. Sequence of the relB transcription unit from Escherichia coli and identification of the relB gene. EMBO J 4:1059–1066. doi: 10.1002/j.1460-2075.1985.tb03739.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Poulsen LK, Larsen NW, Molin S, Andersson P. 1989. A family of genes encoding a cell-killing function may be conserved in all gram-negative bacteria. Mol Microbiol 3:1463–1472. doi: 10.1111/j.1365-2958.1989.tb00131.x [DOI] [PubMed] [Google Scholar]
- 54. Fozo EM, Makarova KS, Shabalina SA, Yutin N, Koonin EV, Storz G. 2010. Abundance of type I toxin-antitoxin systems in bacteria: searches for new candidates and discovery of novel families. Nucleic Acids Res 38:3743–3759. doi: 10.1093/nar/gkq054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Weaver KE, Reddy SG, Brinkman CL, Patel S, Bayles KW, Endres JL. 2009. Identification and characterization of a family of toxin-antitoxin systems related to the Enterococcus faecalis plasmid pAD1 par addiction module. Microbiology (Reading) 155:2930–2940. doi: 10.1099/mic.0.030932-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koyanagi S, Lévesque CM. 2013. Characterization of a Streptococcus mutans intergenic region containing a small toxic peptide and its cis-encoded antisense small RNA antitoxin. PLoS One 8:e54291. doi: 10.1371/journal.pone.0054291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wen J, Fozo EM. 2014. sRNA antitoxins: more than one way to repress a toxin. Toxins (Basel) 6:2310–2335. doi: 10.3390/toxins6082310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kawano M, Oshima T, Kasai H, Mori H. 2002. Molecular characterization of long direct repeat (LDR) sequences expressing a stable mRNA encoding for a 35-amino-acid cell-killing peptide and a cis-encoded small antisense RNA in Escherichia coli. Mol Microbiol 45:333–349. doi: 10.1046/j.1365-2958.2002.03042.x [DOI] [PubMed] [Google Scholar]
- 59. Kawano M, Reynolds AA, Miranda-Rios J, Storz G. 2005. Detection of 5’- and 3’-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli. Nucleic Acids Res 33:1040–1050. doi: 10.1093/nar/gki256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Andresen L, Martínez-Burgo Y, Nilsson Zangelin J, Rizvanovic A, Holmqvist E. 2020. The small toxic Salmonella protein TimP targets the cytoplasmic membrane and is repressed by the small RNA TimR. mBio 11:1–16. doi: 10.1128/mBio.01659-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Müller P, Jahn N, Ring C, Maiwald C, Neubert R, Meißner C, Brantl S. 2016. A multistress responsive type I toxin-antitoxin system: BsrE/SR5 from the B. subtilis chromosome. RNA Biol 13:511–523. doi: 10.1080/15476286.2016.1156288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Reif C, Löser C, Brantl S. 2018. Bacillus subtilis type I antitoxin SR6 promotes degradation of toxin yonT mRNA and is required to prevent toxic yoyJ overexpression. Toxins (Basel) 10:74. doi: 10.3390/toxins10020074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermüller J, Reinhardt R, Stadler PF, Vogel J. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464:250–255. doi: 10.1038/nature08756 [DOI] [PubMed] [Google Scholar]
- 64. Coray DS, Wheeler NE, Heinemann JA, Gardner PP. 2017. Why so narrow: distribution of anti-sense regulated, type I toxin-antitoxin systems compared with type II and type III systems. RNA Biol 14:275–280. doi: 10.1080/15476286.2016.1272747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Akarsu H, Bordes P, Mansour M, Bigot DJ, Genevaux P, Falquet L. 2019. TASmania: a bacterial toxin-antitoxin systems database. PLoS Comput Biol 15:e1006946. doi: 10.1371/journal.pcbi.1006946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Guan J, Chen Y, Goh YX, Wang M, Tai C, Deng Z, Song J, Ou HY. 2024. TADB 3.0: an updated database of bacterial toxin-antitoxin loci and associated mobile genetic elements. Nucleic Acids Res 52:D784–D790. doi: 10.1093/nar/gkad962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tourasse NJ, Darfeuille F. 2021. T1Tadb: the database of type I toxin-antitoxin systems. RNA 27:1471–1481. doi: 10.1261/rna.078802.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hobbs EC, Fontaine F, Yin X, Storz G. 2011. An expanding universe of small proteins. Curr Opin Microbiol 14:167–173. doi: 10.1016/j.mib.2011.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fuchs S, Engelmann S. 2023. Small proteins in bacteria - big challenges in prediction and identification. Proteomics 23:e2200421. doi: 10.1002/pmic.202200421 [DOI] [PubMed] [Google Scholar]
- 70. Guo Y, Quiroga C, Chen Q, McAnulty MJ, Benedik MJ, Wood TK, Wang X. 2014. RalR (a DNase) and RalA (a small RNA) form a type I toxin-antitoxin system in Escherichia coli. Nucleic Acids Res 42:6448–6462. doi: 10.1093/nar/gku279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136:615–628. doi: 10.1016/j.cell.2009.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kawano M, Aravind L, Storz G. 2007. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol Microbiol 64:738–754. doi: 10.1111/j.1365-2958.2007.05688.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Franch T, Gerdes K. 1996. Programmed cell death in bacteria: translational repression by mRNA end-pairing. Mol Microbiol 21:1049–1060. doi: 10.1046/j.1365-2958.1996.771431.x [DOI] [PubMed] [Google Scholar]
- 74. Møller-Jensen J, Franch T, Gerdes K. 2001. Temporal translational control by a metastable RNA structure. J Biol Chem 276:35707–35713. doi: 10.1074/jbc.M105347200 [DOI] [PubMed] [Google Scholar]
- 75. Thisted T, Gerdes K. 1992. Mechanism of post-segregational killing by the hok/sok system of plasmid R1: Sok antisense RNA regulates hok gene expression indirectly through the overlapping mok gene. J Mol Biol 223:41–54. doi: 10.1016/0022-2836(92)90714-u [DOI] [PubMed] [Google Scholar]
- 76. Gerdes K, Nielsen A, Thorsted P, Wagner EG. 1992. Mechanism of killer gene activation. Antisense RNA-dependent RNase III cleavage ensures rapid turn-over of the stable hok, srnB and pndA effector messenger RNAs. J Mol Biol 226:637–649. doi: 10.1016/0022-2836(92)90621-p [DOI] [PubMed] [Google Scholar]
- 77. Dam Mikkelsen N, Gerdes K. 1997. Sok antisense RNA from plasmid R1 is functionally Inactivated by RNase E and polyadenylated by poly(A) polymerase I. Mol Microbiol 26:311–320. doi: 10.1046/j.1365-2958.1997.5751936.x [DOI] [PubMed] [Google Scholar]
- 78. Le Rhun A, Tourasse NJ, Bonabal S, Iost I, Boissier F, Darfeuille F. 2023. Profiling the intragenic toxicity determinants of toxin-antitoxin systems: revisiting hok/Sok regulation. Nucleic Acids Res 51:E4–E4. doi: 10.1093/nar/gkac940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kawano M. 2012. Divergently overlapping cis-encoded antisense RNA regulating toxin-antitoxin systems from E. coli: hok/sok, ldr/rdl, symE/symR. RNA Biol 9:1520–1527. doi: 10.4161/rna.22757 [DOI] [PubMed] [Google Scholar]
- 80. Gerdes K, Wagner EGH. 2007. RNA antitoxins. Curr Opin Microbiol 10:117–124. doi: 10.1016/j.mib.2007.03.003 [DOI] [PubMed] [Google Scholar]
- 81. Weaver KE, Ehli EA, Nelson JS, Patel S. 2004. Antisense RNA regulation by stable complex formation in the Enterococcus faecalis plasmid pAD1 par addiction system. J Bacteriol 186:6400–6408. doi: 10.1128/JB.186.19.6400-6408.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Greenfield TJ, Weaver KE. 2000. Antisense RNA regulation of the pAD1 par post-segregational killing system requires interaction at the 5’ and 3’ ends of the RNAs. Mol Microbiol 37:661–670. doi: 10.1046/j.1365-2958.2000.02034.x [DOI] [PubMed] [Google Scholar]
- 83. Greenfield TJ, Franch T, Gerdes K, Weaver KE. 2001. Antisense RNA regulation of the par post-segregational killing system: structural analysis and mechanism of binding of the antisense RNA, RNAII and its target, RNAI. Mol Microbiol 42:527–537. doi: 10.1046/j.1365-2958.2001.02663.x [DOI] [PubMed] [Google Scholar]
- 84. Weaver K. 2020. The Fst/Ldr family of type I TA system toxins: potential roles in stress response, metabolism and pathogenesis. Toxins (Basel) 12:474. doi: 10.3390/toxins12080474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Michaux C, Hartke A, Martini C, Reiss S, Albrecht D, Budin-Verneuil A, Sanguinetti M, Engelmann S, Hain T, Verneuil N, Giard JC. 2014. Involvement of Enterococcus faecalis small RNAs in stress response and virulence. Infect Immun 82:3599–3611. doi: 10.1128/IAI.01900-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Weaver KE, Chen Y, Miiller EM, Johnson JN, Dangler AA, Manias DA, Clem AM, Schjodt DJ, Dunny GM. 2017. Examination of Enterococcus faecalis toxin-antitoxin system toxin Fst function utilizing a pheromone-inducible expression vector with tight repression and broad dynamic range. J Bacteriol 199:e00065-17. doi: 10.1128/JB.00065-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sayed N, Jousselin A, Felden B. 2011. A cis-antisense RNA acts in trans in Staphylococcus aureus to control translation of a human cytolytic peptide. Nat Struct Mol Biol 19:105–112. doi: 10.1038/nsmb.2193 [DOI] [PubMed] [Google Scholar]
- 88. Sayed N, Nonin-Lecomte S, Réty S, Felden B. 2012. Functional and structural insights of a Staphylococcus aureus apoptotic-like membrane peptide from a toxin-antitoxin module. J Biol Chem 287:43454–43463. doi: 10.1074/jbc.M112.402693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Germain-Amiot N, Augagneur Y, Camberlein E, Nicolas I, Lecureur V, Rouillon A, Felden B. 2019. A novel Staphylococcus aureus cis-trans type I toxin-antitoxin module with dual effects on bacteria and host cells. Nucleic Acids Res 47:1759–1773. doi: 10.1093/nar/gky1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rudd KE. 1999. Novel Intergenic repeats of Escherichia coli K-12. Res Microbiol 150:653–664. doi: 10.1016/s0923-2508(99)00126-6 [DOI] [PubMed] [Google Scholar]
- 91. Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EGH, Margalit H, Altuvia S. 2001. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol 11:941–950. doi: 10.1016/s0960-9822(01)00270-6 [DOI] [PubMed] [Google Scholar]
- 92. Hershberg R, Altuvia S, Margalit H. 2003. A survey of small RNA-encoding genes in Escherichia coli. Nucleic Acids Res 31:1813–1820. doi: 10.1093/nar/gkg297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rivas E, Klein RJ, Jones TA, Eddy SR. 2001. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr Biol 11:1369–1373. doi: 10.1016/s0960-9822(01)00401-8 [DOI] [PubMed] [Google Scholar]
- 94. Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S. 2001. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev 15:1637–1651. doi: 10.1101/gad.901001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jahanshahi S, Li Y. 2020. An effective method for quantifying RNA expression of IbsC-SibC, a type I toxin-antitoxin system in Escherichia coli. Chembiochem 21:3120–3130. doi: 10.1002/cbic.202000280 [DOI] [PubMed] [Google Scholar]
- 96. Masachis S, Tourasse NJ, Lays C, Faucher M, Chabas S, Iost I, Darfeuille F. 2019. A genetic selection reveals functional metastable structures embedded in a toxin-encoding mRNA. Elife 8:e47549. doi: 10.7554/eLife.47549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. El Mortaji L, Tejada-Arranz A, Rifflet A, Boneca IG, Pehau-Arnaudet G, Radicella JP, Marsin S, De Reuse H. 2020. A peptide of a type I toxin-antitoxin system induces Helicobacter pylori morphological transformation from spiral shape to coccoids. Proc Natl Acad Sci U S A 117:31398–31409. doi: 10.1073/pnas.2016195117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Jahn N, Brantl S. 2016. Heat-shock-induced refolding entails rapid degradation of bsrG toxin mRNA by RNases Y and J1. Microbiology (Reading) 162:590–599. doi: 10.1099/mic.0.000247 [DOI] [PubMed] [Google Scholar]
- 99. Fernández De Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, Ohmori H, Woodgate R. 2000. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol Microbiol 35:1560–1572. doi: 10.1046/j.1365-2958.2000.01826.x [DOI] [PubMed] [Google Scholar]
- 100. Romilly C, Deindl S, Wagner EGH. 2019. The ribosomal protein S1-dependent standby site in tisB mRNA consists of a single-stranded region and a 5′ structure element. Proc Natl Acad Sci U S A 116:15901–15906. doi: 10.1073/pnas.1904309116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Weel-Sneve R, Bjørås M, Kristiansen KI. 2008. Overexpression of the LexA-regulated tisAB RNA in E. coli inhibits SOS functions; implications for regulation of the SOS response. Nucleic Acids Res 36:6249–6259. doi: 10.1093/nar/gkn633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wen Jia, Harp JR, Fozo EM. 2017. The 5΄ UTR of the type I toxin ZorO can both inhibit and enhance translation. Nucleic Acids Res 45:4006–4020. doi: 10.1093/nar/gkw1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Bogati B, Shore SFH, Nipper TD, Stoiculescu O, Fozo EM. 2022. Charged amino acids contribute to ZorO toxicity. Toxins (Basel) 15:32. doi: 10.3390/toxins15010032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wen J, Won D, Fozo EM. 2014. The ZorO-Orzo type I toxin-antitoxin locus: repression by the OrzO antitoxin. Nucleic Acids Res 42:1930–1946. doi: 10.1093/nar/gkt1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fozo EM. 2012. New type I toxin-antitoxin families from “wild” and laboratory strains of E. coli: Ibs-Sib, ShoB-OhsC and Zor-Orz. RNA Biol 9:1504–1512. doi: 10.4161/rna.22568 [DOI] [PubMed] [Google Scholar]
- 106. Zhao Z, Xu Y, Jiang B, Qi Q, Tang Y-J, Xian M, Wang J, Zhao G. 2022. Systematic identification of CpxRA-regulated genes and their roles in Escherichia coli stress response. mSystems 7:e0041922. doi: 10.1128/msystems.00419-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Daou-Chabo R, Mathy N, Bénard L, Condon C. 2009. Ribosomes initiating translation of the Hbs mRNA protect it from 5’-To-3’ exoribonucleolytic degradation by RNase J1. Mol Microbiol 71:1538–1550. doi: 10.1111/j.1365-2958.2009.06620.x [DOI] [PubMed] [Google Scholar]
- 108. Fris ME, Broach WH, Klim SE, Coschigano PW, Carroll RK, Caswell CC, Murphy ER. 2017. Sibling sRNA Ryfa1 influences Shigella dysenteriae pathogenesis. Genes (Basel) 8:50. doi: 10.3390/genes8020050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ranjith K, Ramchiary J, Prakash JSS, Arunasri K, Sharma S, Shivaji S. 2019. Gene targets in ocular pathogenic Escherichia coli for mitigation of biofilm formation to overcome antibiotic resistance. Front Microbiol 10:1308. doi: 10.3389/fmicb.2019.01308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Bessaiah H, Pokharel P, Loucif H, Kulbay M, Sasseville C, Habouria H, Houle S, Bernier J, Massé É, Van Grevenynghe J, Dozois CM. 2021. The RyfA small RNA regulates oxidative and osmotic stress responses and virulence in uropathogenic Escherichia coli. PLoS Pathog 17:e1009617. doi: 10.1371/journal.ppat.1009617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kröger C, Colgan A, Srikumar S, Händler K, Sivasankaran SK, Hammarlöf DL, Canals R, Grissom JE, Conway T, Hokamp K, Hinton JCD. 2013. An infection-relevant transcriptomic compendium for Salmonella enterica serovar Typhimurium. Cell Host Microbe 14:683–695. doi: 10.1016/j.chom.2013.11.010 [DOI] [PubMed] [Google Scholar]
- 112. Ramachandran VK, Shearer N, Thompson A. 2014. The primary transcriptome of Salmonella enterica serovar Typhimurium and its dependence on ppGpp during late stationary phase. PLoS One 9:e92690. doi: 10.1371/journal.pone.0092690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Deng X, Li Z, Zhang W. 2012. Transcriptome sequencing of Salmonella enterica serovar Enteritidis under desiccation and starvation stress in peanut oil. Food Microbiol 30:311–315. doi: 10.1016/j.fm.2011.11.001 [DOI] [PubMed] [Google Scholar]
- 114. Zabeau M, Friedman S, Van Montagu M, Schell J. 1980. The ral gene of phage lambda. I. Identification of a non-essential gene that modulates restriction and modification in E. coli. Mol Gen Genet 179:63–73. doi: 10.1007/BF00268447 [DOI] [PubMed] [Google Scholar]
- 115. Liu X, Li Y, Guo Y, Zeng Z, Li B, Wood TK, Cai X, Wang X. 2015. Physiological function of rac prophage during biofilm formation and regulation of rac excision in Escherichia coli K-12. Sci Rep 5:16074. doi: 10.1038/srep16074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sevin EW, Barloy-Hubler F. 2007. RASTA-bacteria: a web-based tool for identifying toxin-antitoxin loci in prokaryotes. Genome Biol 8:R155. doi: 10.1186/gb-2007-8-8-r155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Altuvia Y, Bar A, Reiss N, Karavani E, Argaman L, Margalit H. 2018. In vivo cleavage rules and target repertoire of RNase III in Escherichia coli. Nucleic Acids Res 46:10380–10394. doi: 10.1093/nar/gky684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Melamed S, Adams PP, Zhang A, Zhang H, Storz G. 2020. RNA-RNA interactomes of ProQ and Hfq reveal overlapping and competing roles. Mol Cell 77:411–425. doi: 10.1016/j.molcel.2019.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sneppen K, Dodd IB, Shearwin KE, Palmer AC, Schubert RA, Callen BP, Egan JB. 2005. A mathematical model for transcriptional interference by RNA polymerase traffic in Escherichia coli. J Mol Biol 346:399–409. doi: 10.1016/j.jmb.2004.11.075 [DOI] [PubMed] [Google Scholar]
- 120. Courtney CM, Chatterjee A. 2014. Cis-antisense RNA and transcriptional interference: coupled layers of gene regulation. J Gene Ther 2:9. doi: 10.13188/2381-3326.1000004 [DOI] [Google Scholar]
- 121. Wang L. 2024. RNA polymerase collisions and their role in transcription. Transcription 15:1–10. doi: 10.1080/21541264.2024.2316972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Warman EA, Forrest D, Guest T, Haycocks J, Wade JT, Grainger DC. 2021. Widespread divergent transcription from bacterial and archaeal promoters is a consequence of DNA-sequence symmetry. Nat Microbiol 6:746–756. doi: 10.1038/s41564-021-00898-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Christoffersen CA, Brickman TJ, Hook-Barnard I, McIntosh MA. 2001. Regulatory architecture of the iron-regulated fepD-ybdA bidirectional promoter region in Escherichia coli. J Bacteriol 183:2059–2070. doi: 10.1128/JB.183.6.2059-2070.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Teves SS, Henikoff S. 2014. DNA torsion as a feedback mediator of transcription and Chromatin dynamics. Nucleus 5:211–218. doi: 10.4161/nucl.29086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Liu LF, Wang JC. 1987. Supercoiling of the DNA template during transcription. Proc Natl Acad Sci U S A 84:7024–7027. doi: 10.1073/pnas.84.20.7024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Ramisetty BCM, Santhosh RS. 2016. Horizontal gene transfer of chromosomal type II toxin–antitoxin systems of Escherichia coli. FEMS Microbiol Lett 363:238. doi: 10.1093/femsle/fnv238 [DOI] [PubMed] [Google Scholar]
- 127. Grønlund H, Gerdes K. 1999. Toxin-antitoxin systems homologous with relBE of Escherichia coli plasmid P307 are ubiquitous in prokaryotes. J Mol Biol 285:1401–1415. doi: 10.1006/jmbi.1998.2416 [DOI] [PubMed] [Google Scholar]
- 128. Pandey DP, Gerdes K. 2005. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res 33:966–976. doi: 10.1093/nar/gki201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Leplae R, Geeraerts D, Hallez R, Guglielmini J, Drèze P, Van Melderen L. 2011. Diversity of bacterial type II toxin–antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res 39:5513–5525. doi: 10.1093/nar/gkr131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Meißner C, Jahn N, Brantl S. 2016. In vitro characterization of the type I toxin-antitoxin system bsrE/SR5 from Bacillus subtilis. J Biol Chem 291:560–571. doi: 10.1074/jbc.M115.697524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Saito S, Kakeshita H, Nakamura K. 2009. Novel small RNA-encoding genes in the intergenic regions of Bacillus subtilis. Gene 428:2–8. doi: 10.1016/j.gene.2008.09.024 [DOI] [PubMed] [Google Scholar]
- 132. Jahn N, Brantl S, Strahl H. 2015. Against the mainstream: Tt membrane-associated type I toxin BsrG from Bacillus subtilis interferes with cell envelope biosynthesis without increasing membrane permeability. Mol Microbiol 98:651–666. doi: 10.1111/mmi.13146 [DOI] [PubMed] [Google Scholar]
- 133. Peltier J, Hamiot A, Garneau JR, Boudry P, Maikova A, Hajnsdorf E, Fortier L-C, Dupuy B, Soutourina O. 2020. Type I toxin-antitoxin systems contribute to the maintenance of mobile genetic elements in Clostridioides difficile. Commun Biol 3:718. doi: 10.1038/s42003-020-01448-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Maikova A, Peltier J, Boudry P, Hajnsdorf E, Kint N, Monot M, Poquet I, Martin-Verstraete I, Dupuy B, Soutourina O. 2018. Discovery of new type I toxin-antitoxin systems adjacent to CRISPR arrays in Clostridium difficile. Nucleic Acids Res 46:4733–4751. doi: 10.1093/nar/gky124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Patel S, Weaver KE. 2006. Addiction toxin Fst has unique effects on chromosome segregation and cell division in Enterococcus faecalis and Bacillus subtilis. J Bacteriol 188:5374–5384. doi: 10.1128/JB.00513-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Pinel-Marie ML, Brielle R, Felden B. 2014. Dual toxic-peptide-coding Staphylococcus aureus RNA under antisense regulation targets host cells and bacterial rivals unequally. Cell Rep 7:424–435. doi: 10.1016/j.celrep.2014.03.012 [DOI] [PubMed] [Google Scholar]
- 137. Durand S, Jahn N, Condon C, Brantl S. 2012. Type I toxin-antitoxin systems in Bacillus subtilis. RNA Biol 9:1491–1497. doi: 10.4161/rna.22358 [DOI] [PubMed] [Google Scholar]
- 138. Wilmaerts D, Bayoumi M, Dewachter L, Knapen W, Mika JT, Hofkens J, Dedecker P, Maglia G, Verstraeten N, Michiels J. 2018. The persistence-inducing toxin HokB forms dynamic pores that cause ATP leakage. mBio 9:e00744-18. doi: 10.1128/mBio.00744-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Pulido S, Rückert H, Falsone SF, Göbl C, Meyer NH, Zangger K. 2023. The membrane-binding bacterial toxin long direct repeat D inhibits protein translation. Biophys Chem 298:107040. doi: 10.1016/j.bpc.2023.107040 [DOI] [PubMed] [Google Scholar]
- 140. Berghoff BA, Hoekzema M, Aulbach L, Wagner EGH. 2017. Two regulatory RNA elements affect TisB-dependent depolarization and persister formation. Mol Microbiol 103:1020–1033. doi: 10.1111/mmi.13607 [DOI] [PubMed] [Google Scholar]
- 141. Gurnev PA, Ortenberg R, Dörr T, Lewis K, Bezrukov SM. 2012. Persister-promoting bacterial toxin TisB produces anion-selective pores in planar lipid bilayers. FEBS Lett 586:2529–2534. doi: 10.1016/j.febslet.2012.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Unoson C, Wagner EGH. 2008. A small SOS-induced toxin is targeted against the inner membrane in Escherichia coli. Mol Microbiol 70:258–270. doi: 10.1111/j.1365-2958.2008.06416.x [DOI] [PubMed] [Google Scholar]
- 143. Su W-L, Bredèche M-F, Dion S, Dauverd J, Condamine B, Gutierrez A, Denamur E, Matic I. 2022. TisB protein protects Escherichia coli cells suffering massive DNA damage from environmental toxic compounds. mBio 13:e0038522. doi: 10.1128/mbio.00385-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Bogati B, Wadsworth N, Barrera F, Fozo EM. 2022. Improved growth of Escherichia coli in aminoglycoside antibiotics by the zor-orz toxin-antitoxin system . J Bacteriol 204:JB0040721. doi: 10.1128/JB.00407-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Thompson MK, Nocedal I, Culviner PH, Zhang T, Gozzi KR, Laub MT. 2022. Escherichia coli SymE is a DNA-binding protein that can condense the nucleoid. Mol Microbiol 117:851–870. doi: 10.1111/mmi.14877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ogura T, Hiraga S. 1983. Mini-F Plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci U S A 80:4784–4788. doi: 10.1073/pnas.80.15.4784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Engelberg-Kulka H, Glaser G. 1999. Addiction modules and programmed cell death and antideath in bacterial cultures. Annu Rev Microbiol 53:43–70. doi: 10.1146/annurev.micro.53.1.43 [DOI] [PubMed] [Google Scholar]
- 148. Iqbal N, Guérout AM, Krin E, Le Roux F, Mazel D. 2015. Comprehensive functional analysis of the 18 Vibrio cholerae N16961 toxin-antitoxin systems substantiates their role in stabilizing the superintegron. J Bacteriol 197:2150–2159. doi: 10.1128/JB.00108-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Wozniak RAF, Waldor MK. 2009. A toxin–antitoxin system promotes the maintenance of an integrative conjugative element. PLoS Genet 5:e1000439. doi: 10.1371/journal.pgen.1000439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Lehnherr H, Maguin E, Jafri S, Yarmolinsky MB. 1993. Plasmid addiction genes of bacteriophage P1: doc, which causes cell death on curing of prophage, and phd, which prevents host death when prophage is retained. J Mol Biol 233:414–428. doi: 10.1006/jmbi.1993.1521 [DOI] [PubMed] [Google Scholar]
- 151. Critchlow SE, O’Dea MH, Howells AJ, Couturier M, Gellert M, Maxwell A. 1997. The interaction of the F Plasmid killer protein, Ccdb, with DNA gyrase: induction of DNA cleavage and blocking of transcription. J Mol Biol 273:826–839. doi: 10.1006/jmbi.1997.1357 [DOI] [PubMed] [Google Scholar]
- 152. Berghoff BA, Wagner EGH. 2017. RNA-based regulation in type I toxin–antitoxin systems and its implication for bacterial persistence. Curr Genet 63:1011–1016. doi: 10.1007/s00294-017-0710-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Thisted T, Sørensen NS, Gerdes K. 1995. Mechanism of post-segregational killing: secondary structure analysis of the entire Hok mRNA from plasmid R1 suggests a fold-back structure that prevents translation and antisense RNA binding. J Mol Biol 247:859–873. doi: 10.1006/jmbi.1995.0186 [DOI] [PubMed] [Google Scholar]
- 154. Wilmaerts D, De Loose P-J, Vercauteren S, De Smedt S, Verstraeten N, Michiels J. 2021. Functional analysis of cysteine residues of the Hok/Gef type I toxins in Escherichia coli. FEMS Microbiol Lett 368:69. doi: 10.1093/femsle/fnab069 [DOI] [PubMed] [Google Scholar]
- 155. Weaver KE, Clewell DB. 1989. Construction of Enterococcus faecalis pAD1 miniplasmids: identification of a minimal pheromone response regulatory region and evaluation of a novel pheromone-dependent growth inhibition. Plasmid 22:106–119. doi: 10.1016/0147-619x(89)90020-6 [DOI] [PubMed] [Google Scholar]
- 156. Naito T, Kusano K, Kobayashi I. 1995. Selfish behavior of restriction-modification systems. Science 267:897–899. doi: 10.1126/science.7846533 [DOI] [PubMed] [Google Scholar]
- 157. Van Melderen L, Saavedra De Bast M. 2009. Bacterial toxin–antitoxin systems: more than selfish entities? PLoS Genet 5:e1000437. doi: 10.1371/journal.pgen.1000437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Cooper TF, Heinemann JA. 2000. Postsegregational killing does not increase plasmid stability but acts to mediate the exclusion of competing plasmids. Proc Natl Acad Sci U S A 97:12643–12648. doi: 10.1073/pnas.220077897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Lehnherr H, Yarmolinsky MB. 1995. Addiction protein Phd of plasmid prophage P1 is a substrate of the ClpXP serine protease of Escherichia coli. Proc Natl Acad Sci U S A 92:3274–3277. doi: 10.1073/pnas.92.8.3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Butcher LD, den Hartog G, Ernst PB, Crowe SE. 2017. Oxidative stress resulting from Helicobacter pylori infection contributes to gastric carcinogenesis. Cell Mol Gastroenterol Hepatol 3:316–322. doi: 10.1016/j.jcmgh.2017.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Chaput C, Ecobichon C, Cayet N, Girardin SE, Werts C, Guadagnini S, Prévost MC, Mengin-Lecreulx D, Labigne A, Boneca IG. 2006. Role of AmiA in the morphological transition of Helicobacter pylori and in immune escape. PLoS Pathog 2:e97. doi: 10.1371/journal.ppat.0020097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Weaver KE, Weaver DM, Wells CL, Waters CM, Gardner ME, Ehli EA. 2003. Enterococcus faecalis plasmid pAD1-encoded Fst toxin affects membrane permeability and alters cellular responses to lantibiotics. J Bacteriol 185:2169–2177. doi: 10.1128/JB.185.7.2169-2177.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Granato ET, Meiller-Legrand TA, Foster KR. 2019. The evolution and ecology of bacterial warfare. Curr Biol 29:R521–R537. doi: 10.1016/j.cub.2019.04.024 [DOI] [PubMed] [Google Scholar]
- 164. Alekshun MN, Levy SB. 2007. Molecular mechanisms of antibacterial multidrug resistance. Cell 128:1037–1050. doi: 10.1016/j.cell.2007.03.004 [DOI] [PubMed] [Google Scholar]
- 165. Andersson DI, Nicoloff H, Hjort K. 2019. Mechanisms and clinical relevance of bacterial heteroresistance. Nat Rev Microbiol 17:479–496. doi: 10.1038/s41579-019-0218-1 [DOI] [PubMed] [Google Scholar]
- 166. Bigger JW. 1944. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. The Lancet 244:497–500. doi: 10.1016/S0140-6736(00)74210-3 [DOI] [Google Scholar]
- 167. Bigger JW. 1944. The Bactericidal action of penicillin on Staphylococcus pyogenes. Ir J Med Sci 19:553–568. doi: 10.1007/BF02948386 [DOI] [Google Scholar]
- 168. Hossain T, Singh A, Butzin NC. 2023. Escherichia coli cells are primed for survival before lethal antibiotic stress. Microbiol Spectr 11:e0121923. doi: 10.1128/spectrum.01219-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Svenningsen MS, Svenningsen SL, Sørensen MA, Mitarai N. 2022. Existence of log-phase Escherichia coli persisters and lasting memory of a starvation pulse. Life Sci Alliance 5:e202101076. doi: 10.26508/lsa.202101076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Balaban Nathalie Q, Helaine S, Lewis K, Ackermann M, Aldridge B, Andersson DI, Brynildsen MP, Bumann D, Camilli A, Collins JJ, Dehio C, Fortune S, Ghigo J-M, Hardt W-D, Harms A, Heinemann M, Hung DT, Jenal U, Levin BR, Michiels J, Storz G, Tan M-W, Tenson T, Van Melderen L, Zinkernagel A. 2019. Definitions and guidelines for research on antibiotic persistence. Nat Rev Microbiol 17:441–448. doi: 10.1038/s41579-019-0196-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Balaban N.Q, Merrin J, Chait R, Kowalik L, Leibler S. 2004. Bacterial persistence as a phenotypic switch. Science 305:1622–1625. doi: 10.1126/science.1099390 [DOI] [PubMed] [Google Scholar]
- 172. Mulcahy LR, Burns JL, Lory S, Lewis K. 2010. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J Bacteriol 192:6191–6199. doi: 10.1128/JB.01651-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Torrey HL, Keren I, Via LE, Lee JS, Lewis K. 2016. High persister mutants in Mycobacterium tuberculosis. PLoS One 11:e0155127. doi: 10.1371/journal.pone.0155127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Lafleur MD, Qi Q, Lewis K. 2010. Patients with long-term oral carriage harbor high-persister mutants of Candida albicans. Antimicrob Agents Chemother 54:39–44. doi: 10.1128/AAC.00860-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Fauvart M, De Groote VN, Michiels J. 2011. Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J Med Microbiol 60:699–709. doi: 10.1099/jmm.0.030932-0 [DOI] [PubMed] [Google Scholar]
- 176. Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ. 2017. Antibiotic tolerance facilitates the evolution of resistance. Science 355:826–830. doi: 10.1126/science.aaj2191 [DOI] [PubMed] [Google Scholar]
- 177. Liu J, Gefen O, Ronin I, Bar-Meir M, Balaban NQ. 2020. Effect of tolerance on the evolution of antibiotic resistance under drug combinations. Science 367:200–204. doi: 10.1126/science.aay3041 [DOI] [PubMed] [Google Scholar]
- 178. Windels EM, Michiels JE, Fauvart M, Wenseleers T, Van den Bergh B, Michiels J. 2019. Bacterial persistence promotes the evolution of antibiotic resistance by increasing survival and mutation rates. ISME J 13:1239–1251. doi: 10.1038/s41396-019-0344-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Lewis K. 2008. Multidrug tolerance of biofilms and persister cells. Curr Top Microbiol Immunol 322:107–131. doi: 10.1007/978-3-540-75418-3_6 [DOI] [PubMed] [Google Scholar]
- 180. Korch SB, Henderson TA, Hill TM. 2003. Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol Microbiol 50:1199–1213. doi: 10.1046/j.1365-2958.2003.03779.x [DOI] [PubMed] [Google Scholar]
- 181. Helaine S, Cheverton AM, Watson KG, Faure LM, Matthews SA, Holden DW. 2014. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 343:204–208. doi: 10.1126/science.1244705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Svenningsen MS, Veress A, Harms A, Mitarai N, Semsey S. 2019. Birth and resuscitation of (p)ppGpp induced antibiotic tolerant persister cells. Sci Rep 9:6056. doi: 10.1038/s41598-019-42403-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Liu X, Wang P, Shi Y, Cui Y, Li S, Wu Dong G, Li J, Hao M, Zhai Y, Zhou D, Liu W, Wang A, Jin Y. 2023. (P)ppGpp synthetase Rsh participates in rifampicin tolerance of persister cells in Brucella abortus in vitro. Microb Pathog 183:106310. doi: 10.1016/j.micpath.2023.106310 [DOI] [PubMed] [Google Scholar]
- 184. Khakimova M, Ahlgren HG, Harrison JJ, English AM, Nguyen D. 2013. The stringent response controls catalases in Pseudomonas aeruginosa and is required for hydrogen peroxide and antibiotic tolerance. J Bacteriol 195:2011–2020. doi: 10.1128/JB.02061-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Nguyen D, Joshi-Datar A, Lepine F, Bauerle E, Olakanmi O, Beer K, McKay G, Siehnel R, Schafhauser J, Wang Y, Britigan BE, Singh PK. 2011. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334:982–986. doi: 10.1126/science.1211037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Murakami K, Ono T, Viducic D, Kayama S, Mori M, Hirota K, Nemoto K, Miyake Y. 2005. Role for rpoS gene of Pseudomonas aeruginosa in antibiotic tolerance. FEMS Microbiol Lett 242:161–167. doi: 10.1016/j.femsle.2004.11.005 [DOI] [PubMed] [Google Scholar]
- 187. Stewart PS, Franklin MJ, Williamson KS, Folsom JP, Boegli L, James GA. 2015. Contribution of stress responses to antibiotic tolerance in Pseudomonas aeruginosa biofilms. Antimicrob Agents Chemother 59:3838–3847. doi: 10.1128/AAC.00433-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Wu Y, Vulić M, Keren I, Lewis K. 2012. Role of oxidative stress in persister tolerance. Antimicrob Agents Chemother 56:4922–4926. doi: 10.1128/AAC.00921-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Hernandez-Morfa M, Reinoso-Vizcaíno NM, Olivero NB, Zappia VE, Cortes PR, Jaime A, Echenique J. 2022. Host cell oxidative stress promotes intracellular fluoroquinolone persisters of Streptococcus pneumoniae. Microbiol Spectr 10:e0436422. doi: 10.1128/spectrum.04364-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Wang T, El Meouche I, Dunlop MJ. 2017. Bacterial persistence induced by salicylate via reactive oxygen species. Sci Rep 7:43839. doi: 10.1038/srep43839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Peyrusson F, Nguyen TK, Najdovski T, Van Bambeke F. 2022. Host cell oxidative stress induces dormant Staphylococcus aureus persisters. Microbiol Spectr 10:e0231321. doi: 10.1128/spectrum.02313-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Shan Y, Brown Gandt A, Rowe SE, Deisinger JP, Conlon BP, Lewis K. 2017. ATP-dependent persister formation in Escherichia coli. mBio 8:e02267-16. doi: 10.1128/mBio.02267-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Conlon BP, Rowe SE, Gandt AB, Nuxoll AS, Donegan NP, Zalis EA, Clair G, Adkins JN, Cheung AL, Lewis K. 2016. Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat Microbiol 1:16051. doi: 10.1038/nmicrobiol.2016.51 [DOI] [PubMed] [Google Scholar]
- 194. Manuse S, Shan Y, Canas-Duarte SJ, Bakshi S, Sun W-S, Mori H, Paulsson J, Lewis K. 2021. Bacterial persisters are a stochastically formed subpopulation of low-energy cells. PLoS Biol 19:e3001194. doi: 10.1371/journal.pbio.3001194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Zalis EA, Nuxoll AS, Manuse S, Clair G, Radlinski LC, Conlon BP, Adkins J, Lewis K. 2019. Stochastic variation in expression of the tricarboxylic acid cycle produces persister cells. mBio 10:e01930-19. doi: 10.1128/mBio.01930-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Pontes MH, Groisman EA. 2019. Slow growth determines nonheritable antibiotic resistance in Salmonella enterica. Sci Signal 12:eaax3938. doi: 10.1126/scisignal.aax3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Kwan BW, Valenta JA, Benedik MJ, Wood TK. 2013. Arrested protein synthesis increases persister-like cell formation. Antimicrob Agents Chemother 57:1468–1473. doi: 10.1128/AAC.02135-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Johnson PJT, Levin BR. 2013. Pharmacodynamics, population dynamics, and the evolution of persistence in Staphylococcus aureus. PLoS Genet 9:e1003123. doi: 10.1371/journal.pgen.1003123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Moyed HS, Bertrand KP. 1983. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J Bacteriol 155:768–775. doi: 10.1128/jb.155.2.768-775.1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Black DS, Irwin B, Moyed HS. 1994. Autoregulation of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J Bacteriol 176:4081–4091. doi: 10.1128/jb.176.13.4081-4091.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Black DS, Kelly AJ, Mardis MJ, Moyed HS. 1991. Structure and organization of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J Bacteriol 173:5732–5739. doi: 10.1128/jb.173.18.5732-5739.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Korch SB, Hill TM. 2006. Ectopic overexpression of wild-type and mutant hipA genes in Escherichia coli: effects on macromolecular synthesis and persister formation. J Bacteriol 188:3826–3836. doi: 10.1128/JB.01740-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K. 2004. Persister cells and tolerance to antimicrobials. FEMS Microbiol Lett 230:13–18. doi: 10.1016/S0378-1097(03)00856-5 [DOI] [PubMed] [Google Scholar]
- 204. Keren I, Shah D, Spoering A, Kaldalu N, Lewis K. 2004. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli . J Bacteriol 186:8172–8180. doi: 10.1128/JB.186.24.8172-8180.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K. 2006. Persisters: a distinct physiological state of E. coli. BMC Microbiol 6:53. doi: 10.1186/1471-2180-6-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Keren I, Minami S, Rubin E, Lewis K. 2011. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio 2:e00100-11. doi: 10.1128/mBio.00100-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Harrison JJ, Wade WD, Akierman S, Vacchi-Suzzi C, Stremick CA, Turner RJ, Ceri H. 2009. The chromosomal toxin gene yafQ is a determinant of multidrug tolerance for Escherichia coli growing in a biofilm. Antimicrob Agents Chemother 53:2253–2258. doi: 10.1128/AAC.00043-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Singh R, Barry CE, Boshoff HIM. 2010. The three RelE homologs of Mycobacterium tuberculosis have individual, drug-specific effects on bacterial antibiotic tolerance. J Bacteriol 192:1279–1291. doi: 10.1128/JB.01285-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Chowdhury N, Kwan BW, Wood TK. 2016. Persistence increases in the absence of the alarmone guanosine tetraphosphate by reducing cell growth. Sci Rep 6:20519. doi: 10.1038/srep20519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Goormaghtigh F, Van Melderen L. 2019. Single-cell imaging and characterization of Escherichia coli persister cells to ofloxacin in exponential cultures. Sci Adv 5:eaav9462. doi: 10.1126/sciadv.aav9462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. LeRoux M, Culviner PH, Liu YJ, Littlehale ML, Laub MT. 2020. Stress can induce transcription of toxin-antitoxin systems without activating toxin. Mol Cell 79:280–292. doi: 10.1016/j.molcel.2020.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Goormaghtigh F, Fraikin N, Putrinš M, Hallaert T, Hauryliuk V, Garcia-Pino A, Sjödin A, Kasvandik S, Udekwu K, Tenson T, Kaldalu N, Van Melderen L. 2018. Reassessing the role of type II toxin-antitoxin systems in formation of Escherichia coli type II persister cells. mBio 9:e00640-18. doi: 10.1128/mBio.00640-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Hossain T, Deter HS, Peters EJ, Butzin NC. 2021. Antibiotic tolerance, persistence, and resistance of the evolved minimal cell, Mycoplasma mycoides JCVI-Syn3B. iScience 24:102391. doi: 10.1016/j.isci.2021.102391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Steinbrecher T, Prock S, Reichert J, Wadhwani P, Zimpfer B, Bürck J, Berditsch M, Elstner M, Ulrich AS. 2012. Peptide-lipid interactions of the stress-response peptide TisB that induces bacterial persistence. Biophys J 103:1460–1469. doi: 10.1016/j.bpj.2012.07.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Schneider V, Wadhwani P, Reichert J, Bürck J, Elstner M, Ulrich AS, Kubař T. 2019. Tetrameric charge-zipper assembly of the TisB peptide in membranes—computer simulation and experiment. J Phys Chem B 123:1770–1779. doi: 10.1021/acs.jpcb.8b12087 [DOI] [PubMed] [Google Scholar]
- 216. Cayron J, Oms T, Schlechtweg T, Zedek S, Van Melderen L. 2024. TisB protein is the single molecular determinant underlying multiple downstream effects of ofloxacin in Escherichia coli. Sci Adv 10:eadk1577. doi: 10.1126/sciadv.adk1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Wagner EGH, Unoson C. 2012. The toxin-antitoxin system tisB-istR1: expression, regulation, and biological role in persister phenotypes. RNA Biol 9:1513–1519. doi: 10.4161/rna.22578 [DOI] [PubMed] [Google Scholar]
- 218. Berghoff BA, Karlsson T, Källman T, Wagner EGH, Grabherr MG. 2017. RNA-sequence data normalization through in silico prediction of reference genes: the bacterial response to DNA damage as case study. BioData Min 10:30. doi: 10.1186/s13040-017-0150-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Edelmann D, Leinberger FH, Schmid NE, Oberpaul M, Schäberle TF, Berghoff BA. 2021. Elevated expression of toxin TisB protects persister cells against ciprofloxacin but enhances susceptibility to mitomycin C. Microorganisms 9:943. doi: 10.3390/microorganisms9050943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Edelmann D, Oberpaul M, Schäberle TF, Berghoff BA. 2021. Post‐transcriptional deregulation of the tisB/istR‐1 toxin–antitoxin system promotes SOS ‐independent persister formation in Escherichia coli. Environ Microbiol Rep 13:159–168. doi: 10.1111/1758-2229.12919 [DOI] [PubMed] [Google Scholar]
- 221. Edelmann D, Berghoff BA. 2019. Type I toxin-dependent generation of superoxide affects the persister life cycle of Escherichia coli. Sci Rep 9:14256. doi: 10.1038/s41598-019-50668-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Leinberger FH, Cassidy L, Edelmann D, Schmid NE, Blumenkamp P, Schmidt S, Natriashvili A, Ulbrich MH, Tholey A, Koch H-G, Berghoff BA. 2024. Protein aggregation is a consequence of the dormancy-inducing membrane toxin TisB in Escherichia Coli. bioRxiv. doi: 10.1101/2024.02.22.581605 [DOI] [PMC free article] [PubMed]
- 223. Edelmann D, Berghoff BA. 2022. A shift in perspective: a role for the type I toxin TisB as persistence-stabilizing factor. Front Microbiol 13:871699. doi: 10.3389/fmicb.2022.871699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Knopp M, Gudmundsdottir JS, Nilsson T, König F, Warsi O, Rajer F, Ädelroth P, Andersson DI. 2019. De novo emergence of peptides that confer antibiotic resistance. mBio 10:e00837-19. doi: 10.1128/mBio.00837-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Hemm MR, Weaver J, Storz G. 2020. Escherichia coli small proteome. EcoSal Plus 9. doi: 10.1128/ecosalplus.esp-0031-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Bar-Yaacov D, Mordret E, Towers R, Biniashvili T, Soyris C, Schwartz S, Dahan O, Pilpel Y. 2017. RNA editing in bacteria recodes multiple proteins and regulates an evolutionarily conserved toxin-antitoxin system. Genome Res 27:1696–1703. doi: 10.1101/gr.222760.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Booth JA, Suganthan R, Gaustad P, Bjørås M. 2015. Development of DinQ from Escherichia coli as an anti-cell-envelope antibiotic. Int J Antimicrob Agents 45:196–197. doi: 10.1016/j.ijantimicag.2014.10.005 [DOI] [PubMed] [Google Scholar]