

Abstract
Background and objectives
Statins are used for metabolic dysfunction‐associated steatotic liver disease (MASLD) (NAFLD) treatment, but their role in this context is unclear. Genetic variants of patatin‐like phospholipase domain containing 3 (PNPLA3) are associated with MASLD susceptibility and statin treatment efficacy. Access to liver biopsies before established MASLD is limited, and statins and PNPLA3 in early liver steatosis are thus difficult to study.
Methods
Liver biopsies were collected from 261 patients without known liver disease at surgery and stratified based on statin use and criteria for the metabolic syndrome (MS). Genotypes and transcript levels were measured using Illumina and Affymetrix arrays, and metabolic and lipoprotein profiles by clinical assays. Statin effects on PNPLA3, de novo lipogenesis (DNL), and lipid accumulation were further studied in vitro.
Results
The PNPLA3I148M genetic variant was associated with significantly lower hepatic levels of cholesterol synthesis‐associated transcripts. Patients with MS had significantly higher hepatic levels of MASLD and lipogenesis‐associated transcripts than non‐MS patients. Patients with MS on statin therapy had significantly higher hepatic levels of PNPLA3, acetyl‐CoA carboxylase alpha, and ATP citrate lyase, and statin use was associated with higher plasma fasting glucose, insulin, and HbA1c. Exposure of hepatocyte‐like HepG2 cells to atorvastatin promoted intracellular accumulation of triglycerides and lipogenesis‐associated transcripts. Atorvastatin‐exposure of HepG2, sterol O‐acyltransferase (SOAT) 2‐only‐HepG2, primary human hepatic stellate, and hepatic stellate cell‐like LX2 cells significantly increased levels of PNPLA3 and SREBF2‐target genes, whereas knockdown of SREBF2 attenuated this effect.
Conclusions
Collectively, these observations suggest statin‐associated regulation of PNPLA3 and DNL in liver. The potential interaction between PNPLA3 genotype and metabolic status should be considered in future studies in the context of statin therapy for MASLD.
Keywords: lipogenesis, MASLD, metabolic syndrome, NAFLD

Introduction
The metabolic dysfunction‐associated steatotic liver disease (MASLD)—formerly termed nonalcoholic fatty liver disease [1]—is the most prevalent cause of chronic liver disease worldwide, affecting an estimated one billion individuals globally [2]. Overall, 90% of patients with MASLD fulfill more than one criterion of the metabolic syndrome (MS), a cluster of clinical findings that significantly increase the risk of cardio‐metabolic diseases [3, 4].
MASLD pathophysiology typically involves excessive hepatic lipid accumulation in genetically susceptible individuals [5]. GWAS studies have uncovered strong associations between MASLD development and genetic variants of patatin‐like phospholipase domain containing 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), and glucokinase regulator (GCKR) [6]. As increased free cholesterol accumulation in the liver is a key feature of MASLD development [7, 8], statins—which inhibit cholesterol synthesis in the liver and are the first‐line therapy for dyslipidemia [9]—have been investigated in treatment of established MASLD [10, 11, 12]. The findings on statin treatment in MASLD are yet inconclusive [13]. Patients carrying the I148M mutation of PNPLA3 (PNPLA3148M)—encoding an isoleucine to methionine substitution at position 148—have a higher risk for MASLD development [14, 15] and show reduced beneficial effects on MASLD from statin therapy [16], suggesting that statin efficacy depends on genotype. PNPLA3 has lipase activity toward triglycerides in hepatocytes and retinyl esters in hepatic stellate cells (HSC) [15], and the I148M substitution confers a loss of function that promotes triglyceride accumulation in hepatocytes [15]. Although an association between PNPLA3148M and the development of MASLD has been reported [14], the links to the MS and statins remain unknown. A barrier to understanding MASLD onset and pathogenesis in the context of MS has been the limited access to liver biopsies from individuals with MS without established MASLD.
Here, we stratified patients on PNPLA3 genotype, MS, and statin use and studied markers of metabolic status in liver and plasma. The link between statin and PNPLA3 was further investigated in vitro.
Materials and methods
The study was approved by the Human Research Ethics Approval Committee of Stockholm (application numbers 2006/784‐31/1 and 2012/1633‐31/4), Sweden. Written informed consent was obtained from all participants, and the study conformed to the principles of the Declaration of Helsinki.
Patients
Liver biopsies were obtained from patients undergoing elective open‐heart surgery for ascending aortic aneurysm and/or aortic valve disease at the Karolinska University Hospital, Stockholm, Sweden, as part of the Advanced Study of Aortic Pathology (ASAP) [17, 18, 19].
Exclusion criteria
(1) Alanine aminotransferase (ALT) levels > or = 1.1 µkat/L; indicating active liver disease or hepatic dysfunction; (2) missing data on statin use or one of the five MS criteria; and (3) less than 6 h of fasting before surgery.
Liver microarray and genotyping
Gene expression was measured in liver samples using the human transcriptomic array (HTA) (Affymetrix) or the Affymetrix GeneChip Human Exon 1.0 ST array (in n = 261 and n = 144 patients for HTA and Exon 1.0 ST array, respectively) at the Karolinska Institutet Affymetrix core facility, as previously described [18]. The raw data were preprocessed using Robust Multichip Average (RMA) [20] normalization as implemented in the Affymetrix Transcriptome Analysis Console Software (for HTA) or Affymetrix Power Tools 1.10.2 package apt‐probeset‐summarize (for Exon 1.0 array). All expression measurements were log2‐transformed as part of the RMA normalization. Genotypes were determined based on the Illumina Human 610W‐Quad Bead arrays at the SNP technology platform, Uppsala University. DNA was isolated from the blood.
Plasma protein determinations
All blood samples were collected in fasting state. Blood glucose, HbA1c, and ALT levels were analyzed by certified routine assays at the Karolinska University Laboratory, Stockholm, Sweden. Insulin was measured by standard ELISA, and lipoproteins analyses were measured as described [19].
Criteria to diagnose metabolic syndrome (MS)
Waist circumference of 40 in. (101 cm) or more for men and 35 in. (90 cm) or more for women (measured across the abdomen);
Blood pressure of 130/85 mm Hg or higher;
Plasma triglyceride level above 150 mg/dL (1.7 mmol/L);
Plasma high‐density lipoprotein (HDL) level cholesterol less than 40 mg/dL (1.03 mmol/L) for men or under 50 mg/dL (1.3 mmol/L) for women;
Fasting blood glucose level above 5.6 mmol/L.
Patients who fulfill the essential waist circumference criteria in addition to two or more of the other above criteria are diagnostic for MS according to the International Diabetes Federation [21, 22, 23].
Cell culture
Sterol O‐acyltransferase (SOAT) 2‐only‐HepG2 [24], HepG2, primary human HSCs (phHSCs) (LifeNet Health), and immortalized primary human hepatic stellate LX2 cells (Sigma, #SCC064) were used for in vitro experiments.
We previously generated SOAT2‐only‐HepG2 [24] because HepG2 cells, in contrast to human hepatocytes in vivo—which solely express SOAT 2—also express SOAT1. SOAT2‐only‐HepG2 cells were grown at 37°C and 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM; 1 g/L glucose and 4.5 g/L glucose, respectively) supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 µg/mL streptomycin (Thermo Scientific). We previously also developed a protocol that improves the phenotype of hepatoma cells, which become more hepatocyte‐like in terms of hepatic lipid metabolism [25]. Accordingly, SOAT2‐only‐HepG2 cells were cultured in six well‐plates as described above, but the FBS was replaced with 2% human serum for 10 days. Then, cells were incubated for 16 h in Opti‐MEM (Thermo Scientific) ± atorvastatin 5 µmol/L or dimethylsulfoxide (DMSO) used for control purpose as vehicle at 0.08% V/V. DMSO and compounds were purchased from Merck/Sigma (#2650‐100ML and #PHR1422‐1G).
HepG2 and LX2 cells were maintained in DMEM with 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37°C in air containing 5% CO2. Triglycerides were measured by triglyceride assay kit (Abcam# Ab65336) according to manufacturer instructions.
The phHSCs (n donors = 4) were thawed, seeded at the density of 1.75e4 cells per well of 12‐well plate, and maintained in DMEM supplemented with glutamax and 10% FBS according to manufacturer's instructions. phHSCs were transfected with siRNA as described below. After 48 h transfection, cells were serum‐starved for 24 h and exposed to atorvastatin or DMSO for 24 h.
RNA interference
For siRNA transfection, media was replaced with fresh DMEM + 20% FBS, 100 µL per well of 96‐well plate. siRNA/RNAiMAX (Thermo Fisher, #13778075) complexes were prepared in 100 µL of Opti‐MEM according to manufacturer's instructions using 5 pM of scrambled siRNA (Thermo Fisher, #4390843) or siRNA targeting SREBF2 (Thermo Fisher, #4390824 s27) and 0.4 µL of RNAiMAX per well. SOAT2‐only‐HepG2, HepG2, phHSC, and LX2 cells were incubated with the complexes for 24 h. Next, HepG2, phHSC, and LX2 cells were serum‐starved in DMEM for 24 h. The medium was replaced with fresh DMEM for HepG2, phHSC, and LX2 cells and for Opti‐MEM for SOAT2‐only‐HepG2 cells, and cells were exposed to atorvastatin (5 µmol/L) or DMSO for 24 h and collected for subsequent RNA or protein isolation.
RNA extraction, cDNA synthesis, and real‐time RT‐PCR
Total RNA was extracted using QIAzol (Qiagen, #79306) and transcribed into cDNA using High Capacity cDNA Reverse Transcription kit (Thermo Fisher, #4368813). mRNA levels were quantified using specific primers (Table S1), and arbitrary units were calculated by linearization of the CT values and normalized to PPIA and B2M.
Western blot
HepG2 cells were lysed in the RIPA buffer in the presence of 1× Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher, #78440) for 10 min at +4°C. Lysates were diluted in 2× Laemmli buffer, and proteins were separated using SDS–PAGE in 10% acrylamide gel. Proteins were transferred to a nitrocellulose membrane, blocked with 5% Blotting‐Grade Blocker (Bio‐Rad, #170‐6404), and stained using antibodies against SREBF2 (Cayman Chemicals, #10007663), PNPLA3 (Abcam, #ab81874), and β‐actin (Abcam, #ab8226). Donkey anti‐rabbit IgG (LI‐COR, #926‐32213) and goat anti‐mouse IgG (LI‐COR, #925‐68070) were used as secondary antibodies and signals visualized using a ChemiDoc Imager (Bio‐Rad).
Statistical analysis
Data are shown as mean ± SEM or min to max box plots with the middle line at median. Groups were compared in GraphPad Prism 9 (GraphPad Software Inc.) using an unpaired Student's t test, Mann–Whitney U test, or one‐way ANOVA followed by Fisher's least significant difference or Šídák's multiple comparisons post hoc test as indicated in the figure legends. p < 0.05 was considered significant.
Results
Patient inclusion
A total of 261 patients scheduled for aortic surgery were subjected to peroperative liver biopsy [17, 18, 19]. As MS is a key predictor of MASLD [26, 27], patients were stratified on established MS criteria [21, 22, 23]. Out of the 261 patients, 39 patients were excluded due to missing information on statin use or MS criteria, absence of sufficient fasting, or plasma ALT level above 1.1 µkat/L. Eighty‐one had ongoing statin treatment, whereas 141 did not (Fig. 1). Among statin users, 24 patients fulfilled the criteria for the MS, and 57 did not. Study subgroups and baseline characteristics are shown in Fig. 1 and Tables 1 and 2.
Fig. 1.
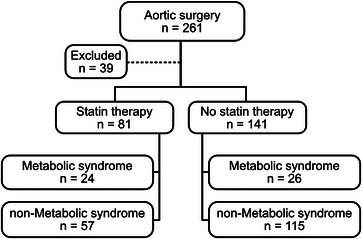
Inclusion and stratification of patients.
Table 1.
Clinical characteristics in patients without statin treatment defined as metabolic syndrome (MS) or not.
| Patients | Non‐MS (n = 115) | MS (n = 26) | p |
|---|---|---|---|
| Sex (F/M) | (26/89) | (5/21) | |
| Age | 62.4 ± 1.2 | 62.4 ± 2.4 | n.s. |
| BMI | 26 ± 0.3 | 30 ± 0.8 | <0.0001 |
| Waist circumference (cm) | 95 ± 1.0 | 107 ± 1.5 | <0.01 |
| Plasma total cholesterol (mmol/L) | 5.4 ± 0.1 | 5.5 ± 0.2 | n.s. |
| Plasma triglycerides (mmol/L) | 1.0 ± 0.05 | 1.9 ± 0.2 | <0.0001 |
| LDL cholesterol (mmol/L) | 3.5 ± 0.1 | 3.5 ± 0.2 | n.s. |
| HDL cholesterol (mmol/L) | 1.4 ± 0.04 | 1.2 ± 0.08 | <0.01 |
| Apolipoprotein B (g/L) | 1.0 ± 0.02 | 1.1 ± 0.06 | n.s. |
| Apolipoprotein A1 (g/L) | 1.6 ± 0.03 | 1.4 ± 0.06 | <0.05 |
Note: Data show mean ± SEM. Unpaired Student's t test
Abbreviations: BMI, body mass index; F, female; HDL, high density lipoprotein; LDL, low‐density lipoprotein; M, male; n.s, not significant.
Table 2.
Clinical characteristics in patients who fulfilled the criteria for the metabolic syndrome (ms), stratified on statin use.
| Patients | No statin (n = 26) | Statin (n = 24) | p |
|---|---|---|---|
| Sex (F/M) | (5/19) | (6/18) | |
| Age | 62.3 ± 2.4 | 67.7 ± 2.1 | n.s. |
| BMI | 30.0 ± 0.8 | 31.8 ± 0.8 | n.s. |
| Waist circumference (cm) | 107 ± 1.5 | 114 ± 4.2 | n.s. |
| Plasma total cholesterol, mmol/L | 5.5 ± 0.2 | 4.7 ± 0.1 | <0.01 |
| Plasma triglycerides, mmol/L | 1.9 ± 0.2 | 1.9 ± 0.2 | n.s. |
| LDL cholesterol, mmol/L | 3.5 ± 0.2 | 2.6 ± 0.1 | <0.001 |
| HDL cholesterol, mmol/L | 1.2 ± 0.08 | 1.1 ± 0.09 | n.s. |
| Apolipoprotein B, g/L | 1.1 ± 0.06 | 1.0 ± 0.03 | <0.05 |
| Apolipoprotein A1, g/L | 1.4 ± 0.06 | 1.4 ± 0.05 | n.s. |
Note: Data show mean ± SEM. Unpaired Student's t test.
Abbreviations: BMI, body mass index; F, female; HDL, high density lipoprotein; LDL, low‐density lipoprotein; M, male; n.s, not significant.
The PNPLA3I148M genetic variant was associated with higher hepatic levels of MASLD‐associated and lower hepatic levels of cholesterol‐synthesis‐associated transcripts
The genetic variant PNPLA3 I148M is associated with MASLD development and severity [15]. We grouped patients based on PNPLA3 genotype (i.e., homozygote PNPLA3 148II, heterozygote PNPLA3 148M/I, and homozygote PNPLA3 148MM) and investigated hepatic transcript levels of the MASLD‐associated PNPLA3, TM6SF2, GCKR, and membrane bound O‐acyltransferase domain containing 7 (MBOAT7) using Illumina and Affymetrix arrays. We observed no significant difference in PNPLA3 levels between genotype groups (Fig. 2a). Levels of TM6SF2, GCKR, and MBOAT7 were significantly higher in the PNPLA3148MM compared with the PNPLA3148II group (Fig. 2b). Interestingly, in patients without statin therapy, the PNPLA3148MM group had significantly lower transcript levels of cholesterol synthesis genes 3‐hydroxy‐3‐methylglutaryl‐CoA synthase (HMGCS) and 3‐hydroxy‐3‐methylglutaryl‐CoA reductase (HMGCR) compared to the PNPLA3148II group (Fig. 2c). No significant difference was observed in SREBF2 between groups (Fig. 2d). Because there was an association between PNPLA3 genetic variants and levels of key cholesterol synthesis genes, and previous studies linked PNPLA3 genetic variants with the efficiency of statin therapy in MASLD [16], we next investigated whether statin treatment was associated with differences in PNPLA3 levels.
Fig. 2.
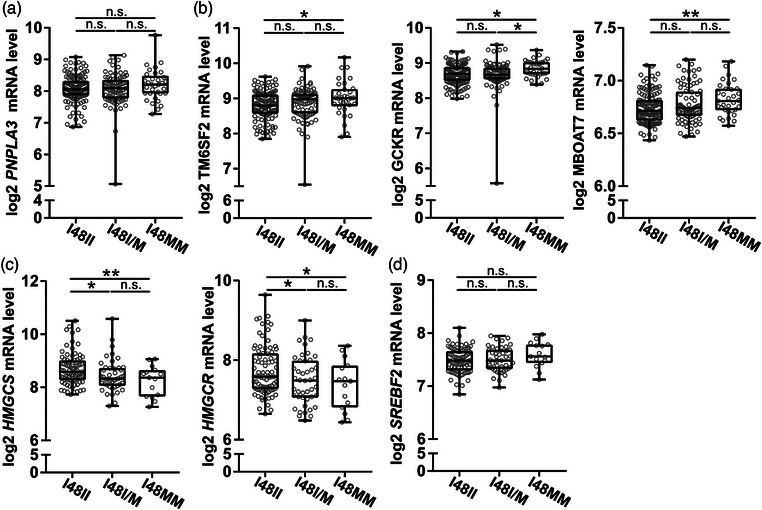
PNPLA3I148M was associated with altered hepatic levels of metabolic dysfunction–associated steatotic liver disease (MASLD)‐associated and cholesterol synthesis–associated transcripts. Liver biopsy homogenates from individuals without known liver disease were analyzed using human transcriptome array and grouped by genotype for the PNPLA3I148M. (a and b) Transcript levels of MASLD associated genes patatin‐like phospholipase domain containing 3(PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), glucokinase regulator (GCKR), and membrane bound O‐acyltransferase domain containing 7 (MBOAT7) (c) Transcript levels of cholesterol synthesis associated genes, in patients without statin therapy, 3‐hydroxy‐3‐methylglutaryl‐CoA synthase (HMGCS), 3‐hydroxy‐3‐methylglutaryl‐CoA reductase (HMGCR) and (d) SREBF2. 148II—homozygote for the enzymatically active allele, 148I/M—heterozygote, and 148MM—homozygote for the enzymatically inactive alleles. Data are expressed as min to max box plots with middle line at median, each dot represents one patient. One‐way ANOVA, Fisher's least significant difference (LSD) test. n.s., not significant. *p < 0.05 and **p < 0.01 (n = 15–83/group).
Statin treatment was associated with higher levels of hepatic PNPLA3 and DNL‐associated transcripts in MS patients
Patients were stratified on the presence of MS and statin use. Transcript levels of PNPLA3 and TM6SF2 were significantly higher in MS patients compared with the non‐MS group, but there was no significant difference in GCKR and MBOAT7 (Fig. 3a,b). Transcript levels of key enzymes in hepatic de novo lipogenesis (DNL)—including ATP citrate lyase (ACLY), acetyl‐CoA carboxylase alpha (ACACA), and stearoyl‐CoA desaturase (SCD)—were also higher in patients with MS compared with the non‐MS group (Fig. 3c). No significant difference was observed in SREBF1c levels between groups (Fig. 3d). Thus, in patients without statin therapy, levels of DNL‐ and MASLD‐associated hepatic transcripts were higher in MS compared to non‐MS patients.
Fig. 3.
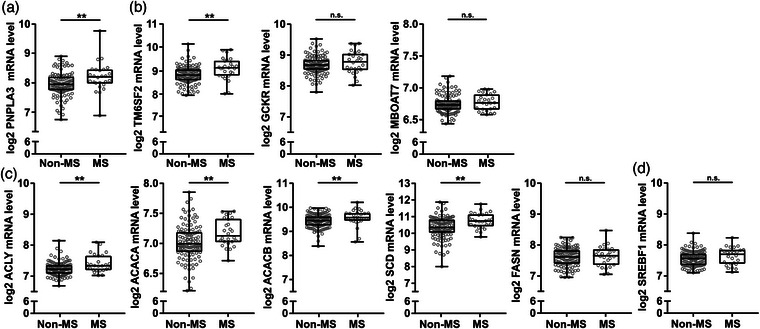
Hepatic levels of metabolic dysfunction–associated steatotic liver disease (MASLD)‐ and de novo lipogenesis (DNL)‐associated transcripts were higher in human metabolic syndrome patients. Liver biopsy homogenates from individuals without known liver disease and without statin therapy were analyzed using human transcriptome array and grouped by meeting criteria for the metabolic syndrome (MS) or not (non‐MS). (a and b) Transcript levels of MASLD associated genes patatin‐like phospholipase domain containing 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), glucokinase regulator (GCKR), and membrane bound O‐acyltransferase domain containing 7 (MBOAT7). (c) Transcript levels of key enzymes involved in DNL ATP citrate lyase (ACLY), acetyl‐CoA carboxylase alpha (ACACA), ACACB, stearoyl‐CoA desaturase (SCD), and FASN. (d) Transcript levels of SREBF1 transcriptional regulator of DNL associated genes. Data are expressed as min to max box plots with middle line at median, each dot represents one patient. Mann Whitney U test. n.s., not significant. *p < 0.05, **p < 0.01, and ***p < 0.001 (n = 26–115/group).
Levels of PNPLA3 were significantly higher in the statin‐treated group (Fig. 4a). In contrast, transcript levels of TM6SF2, GCKR, and MBOAT7 were not significantly different between groups (Fig. 4b). Congruent with ongoing statin treatment [28, 29], patients in the “statin treatment group” showed significantly higher transcript levels of SREBF2 and the SREBF2‐regulated genes HMGCS, HMGCR, and LDL receptor (LDLR) in liver biopsies as compared with patients without statin therapy (Fig. 4c).
Fig. 4.
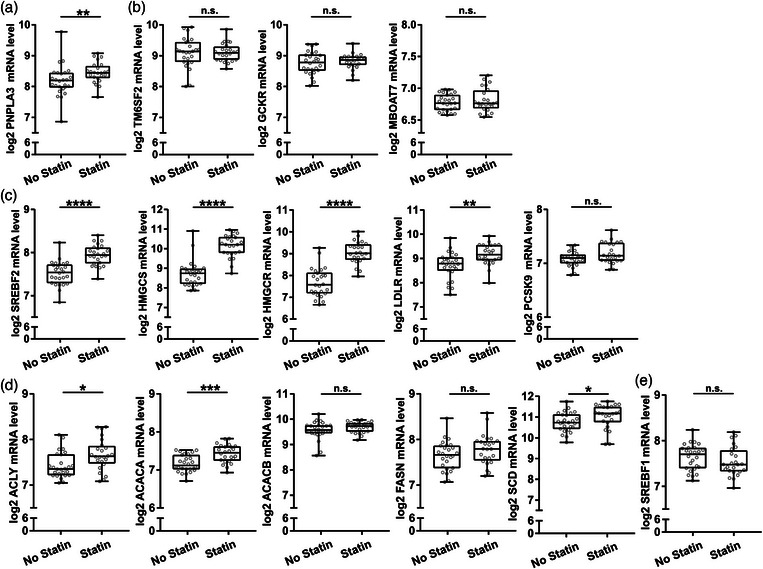
Hepatic expression of metabolic dysfunction‐associated steatotic liver disease (MASLD)‐ and de novo lipogenesis (DNL)‐associated genes in patients with metabolic syndrome was elevated in the statin‐treated group. Liver biopsy homogenates from individuals without known liver disease were analyzed using human transcriptome array. (a and b) Transcript levels of MASLD associated genes patatin‐like phospholipase domain containing 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), glucokinase regulator (GCKR), and membrane bound O‐acyltransferase domain containing 7 (MBOAT7). (c) Transcript levels of key regulators of hepatic cholesterol metabolism (SREBF2, 3‐hydroxy‐3‐methylglutaryl‐CoA synthase (HMGCS1), 3‐hydroxy‐3‐methylglutaryl‐coa reductase (HMGCR), low‐density lipoprotein receptor (LDLR), and PCSK9) in metabolic syndrome (MS) patients with or without statin treatment. (d and e) Transcript levels of transcriptionally regulated key enzymes involved in DNL ATP citrate lyase (ACLY), acetyl‐CoA carboxylase alpha (ACACA), ACACB, stearoyl‐CoA desaturase (SCD), FASN, and SREBF1. Data are expressed as min to max box plots with the middle line at the median, each dot representing one patient. Mann Whitney U test. n.s., not significant. *p < 0.05, **p < 0.01, ****p < 0.0001 (n = 24–26/group).
Plasma levels of LDL‐cholesterol and apolipoprotein B were lower in the statin‐treated group (Table 2), further supporting ongoing statin therapy. Patients with MS with ongoing statin treatment also showed significantly higher hepatic transcript levels of the key DNL enzymes—that is, ACLY, ACACA, and SCD—as compared with MS patients without statin treatment (Fig. 4d). No significant difference was observed in SREBF1c levels between groups (Fig. 4d).
Patients with ongoing statin treatment who did not fulfill MS criteria also showed significantly higher hepatic transcript levels of PNPLA3 (Fig. S1a) and the key DNL enzymes as compared with patients without statin treatment who did not fulfill MS criteria (Fig. S1c). No significant difference was observed in SREBF1c levels between the groups (Fig. S1d). As expected, patients on statins showed significantly increased transcript levels of SREBF2 and the SREBF2‐regulated genes in liver biopsies (Fig. S1e).
In vitro, hepatocyte‐like HepG2 cells showed significant induction of ACLY, ACACA, and SCD and a significant reduction of SREBF1c following atorvastatin exposure (Fig. S2a,b). No significant difference was observed in ACACB and FASN levels following atorvastatin exposure (Fig. S2a). In‐line with this, we observed higher levels of triglycerides in HepG2 cells following atorvastatin exposure, supporting a physiological effect of the statin‐induced increase of DNL transcripts (Fig. S2c).
We were unable to measure liver fat accumulation as an indication of DNL in our clinical cohort. Instead, we proceeded to compare key metabolic indicators between statin‐treated and non‐statin‐treated MS and non‐MS patients. Patients who fulfilled MS criteria and were on statin therapy showed significantly higher fasting glucose, insulin, and HbA1c levels as compared with MS patients without statin therapy (Fig. 5a). When patients with diabetes were excluded from the analysis, levels of fasting glucose and insulin were still significantly higher in statin‐treated patients compared with patients without statin treatment who fulfilled MS criteria (Fig. 5b). However, in patients who did not fulfill the criteria for MS, there was no significant difference in levels of fasting glucose, insulin, or HbA1c between statin‐treatment groups (Fig. 5a). Together, the findings suggest that statin therapy may promote hepatic DNL and aggravated hyperglycemic features in MS patients in this cohort.
Fig. 5.
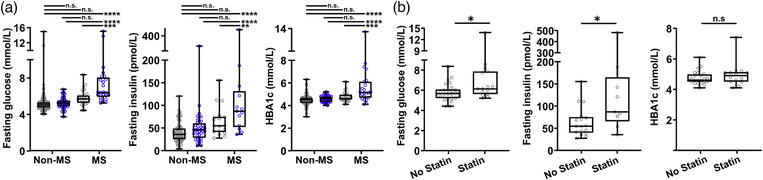
Statin treatment associated with hyperglycemic signs in patients with metabolic syndrome. (a) Plasma levels of fasting blood glucose (n = 57–115 and 24–26/treatment group, insulin (n = 39–70 and 13–17/treatment group) and HbA1c (n = 51–95 and 22–24/treatment group) in metabolic and non‐metabolic syndrome patients treated with (blue dots) or without (gray dots) statins. Data are expressed as min to max box plots, and each dot represents one patient. One‐way ANOVA with Šídák's multiple comparisons test. (b) Plasma levels of fasting blood glucose (n = 15–26/treatment group), insulin (n = 9–17/treatment group) and HbA1c (n = 14–24/treatment group) in metabolic syndrome (MS) patients, after exclusion of individuals diagnosed with diabetes, treated with or without statins. Data are expressed as min to max box plots, and each dot represents one patient. Student's t test. n.s., not significant. *p < 0.05.
Statin regulation of PNPLA3 in liver cells required SREBF2
To investigate the mechanism of statin regulation of PNPLA3, we turned to cell culture and exposed four different cell models relevant in the context of liver pathophysiology—hepatocellular carcinoma (HepG2) cells, SOAT2‐only‐HepG2 cells, immortalized phHSC line (LX2) cells, and phHSC—to atorvastatin in vitro. In HepG2, LX2, and phHSC cells, atorvastatin significantly increased levels of transcripts involved in hepatic cholesterol metabolism—including SREBF2, HMGCR, PCSK9, and LDLR (Figs. 6, 7, and Fig. S3a). Consistently, both HMGCR and PCSK9 were upregulated in SOAT2‐only‐HepG2 cells (Fig. 8a). In‐line with the observations in human liver biopsies (Fig. 4a, Fig. S1a), PNPLA3 levels were significantly higher in all four cell models exposed to atorvastatin in vitro as compared with vehicle (Figs. 6, 7, 8 and Fig. S3b).
Fig. 6.
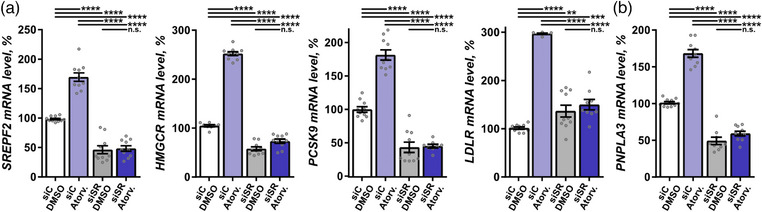
Statin regulation of patatin‐like phospholipase domain containing 3 (PNPLA3) in liver cells required SREBF2. (a and b) HepG2 was exposed to atorvastatin (Atrov.) or vehicle dimethylsulfoxide (DMSO), with and without SREBF2 knockdown, and the mRNA levels of SREBF2, 3‐hydroxy‐3‐methylglutaryl‐CoA reductase (HMGCR), PCSK9, low‐density lipoprotein receptor (LDLR), and PNPLA3 were quantified using qPCR. Data are expressed as mean ± SEM. One‐way ANOVA, Fisher's least significant difference (LSD) post hoc test. n.s., not significant; siC, scrambled siRNA; siSR, SREBF2 siRNA. **p < 0.01, ***p < 0.001, and ****p < 0.0001 (n = 10).
Fig. 7.
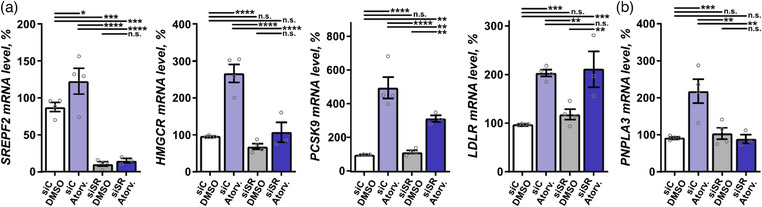
Statin regulation of patatin‐like phospholipase domain containing 3 (PNPLA3) in primary human HSC required SREBF2. (a and b) Primary human hepatic stellate cells (HSC) were exposed to atorvastatin (Atrov.) or vehicle dimethylsulfoxide (DMSO), with and without SREBF2 knockdown, and the mRNA levels of SREBF2, 3‐hydroxy‐3‐methylglutaryl‐CoA reductase (HMGCR), PCSK9, low‐density lipoprotein receptor (LDLR), and PNPLA3 were quantified using qPCR. Data are expressed as mean ± SEM. One‐way ANOVA, Fisher's least significant difference (LSD) post hoc test. n.s., not significant; siC, scrambled siRNA; siSR, SREBF2 siRNA. **p < 0.01, ***p < 0.001, and ****p < 0.0001 (n = 3–4).
Fig. 8.
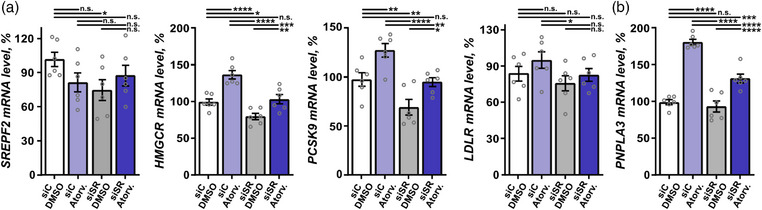
Statin regulation of patatin‐like phospholipase domain containing 3 (PNPLA3) in sterol O‐acyltransferase (SOAT) 2‐only‐HepG2 cells involved SREBF2. (a and b) SOAT2‐only‐HepG2 cells were exposed to atorvastatin (Atrov.) or vehicle (DMSO), with and without SREBF2 knockdown, and the mRNA levels of SREBF2, 3‐hydroxy‐3‐methylglutaryl‐CoA reductase (HMGCR), PCSK9, low‐density lipoprotein receptor (LDLR), and PNPLA3 were quantified using qPCR. Data are expressed as mean ± SEM. One‐way ANOVA, Fisher's least significant difference (LSD) post hoc test. n.s., not significant; siC, scrambled siRNA; siSR, SREBF2 siRNA. **p < 0.01, ***p < 0.001, and ****p < 0.0001 (n = 3–4).
As SREBF2 is a key transcription factor in the cholesterol synthesis pathway targeted by statins [28, 29], we investigated the effect of SREBF2 in the context of statin‐regulated PNPLA3 transcription. SREBF2 was knocked down using siRNA (Fig. S4) in the four cell models: HepG2, SOAT2‐only‐HepG2, LX2 cells, and phHSC. SREBF2 knockdown reduced basal expression of PNPLA3 by ∼50% in HepG2 cells and by ∼20% in LX2 cells (Fig. 6b, Fig. S3b). Cultures were subsequently exposed to atorvastatin or vehicle. Knockdown of SREBF2 attenuated the atorvastatin‐dependent upregulation of PNPLA3 along with SREBF2, HMGCR, and PCSK9 in HepG2, LX2, and phHSC cells; LDLR in HepG2 and LX2 cells (Figs. 6, 7, Figs. S3a,b and S4); and HMGCR and PCSK9 in SOAT2‐only‐HepG2 cells (Fig. 8a). Taken together, atorvastatin‐mediated regulation of PNPLA3 required SREBF2.
Discussion
Here, we observed that patients without known liver disease who fulfill the criteria for MS showed higher hepatic levels of MASLD‐ and DNL‐associated transcripts. Statin treatment was associated with higher levels of hepatic PNPLA3 and lipogenesis‐associated transcripts in patients without known liver disease, regardless of whether they fulfilled the criteria for MS. Interestingly, statin regulation of PNPLA3 required SREBF2 in human hepatocyte‐like cells and primary HSCs in vitro.
An important function of the liver is to synthesize fatty acids from non‐lipid precursors such as glucose, so‐called DNL. Patients with MS frequently show increased liver fat accumulation [30], which can progress into MASLD [26]. Hepatic DNL‐associated genes are elevated in MASLD patients [31], and enhanced DNL significantly contributes to liver fat accumulation in MASLD [32]. Although established MASLD has been extensively studied, the molecular mechanisms that underlie the early hepatic fat accumulation in patients with MS are not well understood.
The MS‐associated elevation of hepatic DNL‐associated transcripts observed here suggests that DNL is linked to MASLD development in MS. Of note, patients in this cohort with MS treated with statins showed higher glucose, insulin, and HBA1c levels compared to MS non‐statin users. These observations were supported by statin‐induced elevation of DNL‐associated transcripts and corresponding significant accumulation of triglycerides in cell cultures in this study. Collectively, these findings suggest an additional potential mechanism for the proposed possibility of a diabetogenic effect of statins [1, 32, 33, 34].
Our finding in our cohort that statin‐treated patients had higher levels of hepatic PNPLA3 is interesting in the context of previous genetic studies of MASLD—in particular, that carriers of PNPLA3148M do not benefit from statin therapy [16]: Previous studies showed that hepatic overexpression of human PNPLA3148M, but not PNPLA3148I, promoted MASLD in mice [35]. Congruently, silencing PNPLA3148M after its initial overexpression ablated fatty liver disease [36]. Together, the findings suggest that statin‐mediated upregulation of hepatic PNPLA3148M might contribute to MASLD progression.
There was increased hepatic expression of MASLD‐associated genes in homozygous PNPLA3148MM individuals, potentially resulting from the metabolic consequences of loss of PNPLA3 hydrolase activity. PNPLA3148MM individuals also had lower levels of key cholesterol synthesis‐associated transcripts, hinting at higher intracellular basal cholesterol levels, and suggesting an interaction between PNPLA3148MM and the intracellular cholesterol homeostasis. As statin therapy is known to inhibit intracellular cholesterol synthesis, it is plausible that there is an interaction between statin therapy and PNPLA3148MM. Accordingly, it will be important to pursue further functional studies on the effect of the PNPLA3 genotype on hepatic cholesterol hemostasis in the context of statin therapy.
PNPLA3 is known to be regulated by SREBF1 [37]. The overexpression of both Srebf1 and Srebf2 in livers of transgenic mice promotes Pnpla3, and Pnpla3 upregulation was reported to be 25‐fold stronger following Srebf1 overexpression [38]. Statins promote SREBF2 nuclear translocation and DNA binding, but statins are not known to directly regulate PNPLA3. The findings here that statin treatment was associated with an elevation of PNPLA3 in the liver and that exposures of hepatocyte‐like SOAT2‐only‐HepG2, HepG2, phHSC, and HSC‐like LX2 cells to atorvastatin in vitro upregulated PNPLA3 in an SREBF2‐dependent manner, imply that both SREBF1 and SREBF2 can regulate PNPLA3 in human liver cells.
As both hepatic cholesterol and triglyceride levels contribute to MASLD progression, hepatic cholesterol‐lowering alone likely does not fully explain all the previously reported positive changes of statin therapy in MASLD [10, 11, 12]. Statins have been shown to improve MASLD in experimental models and patients [12, 39, 40] but were not helpful in patients carrying PNPLA3148M [16]. The association between statins and PNPLA3 found here suggests a possible mechanism for the observed differential effects of statins on MASLD, that is, through upregulating PNPLA3 expression. The effects of the PNPLA3 upregulation are variant‐dependent according to experimental studies [35, 36]. Accordingly, we speculate that PNPLA3 and DNL upregulation with statins may have differential outcomes dependent on genotype and metabolic status. Based on what we know today, one possibility is that the effect of statins in hepatocytes and HSC will depend on patient genotype and metabolic status, ultimately shaping MASLD progression.
More studies are thus needed to investigate the interaction between statins and the PNPLA3 148M polymorphism, and whether alternative lipid‐lowering drugs such as PCSK9 inhibitors should be considered for treating dyslipidemia in this group of patients.
In conclusion, the findings here indicate that the interaction between the PNPLA3 genotype and metabolic status should be considered in the context of statin therapy for MASLD.
Conflict of interest statement
OA, MP, and PP are cofounders and co‐owners of Lipoprotein Research Stockholm AB. PSO is a shareholder of Emune AB.
Supporting information
Supplementary Table 1. Primers.
Figure S1. Hepatic levels of PNPLA3 and DNL associated transcripts in non‐metabolic syndrome patients were higher in the statin‐treated group.
Figure S2. Hepatic DNL‐associated transcript levels following HepG2 exposure of atorvastatin.
Figure S3. Statin regulation of PNPLA3 required SREBF2.
Figure S4: Statin regulation of PNPLA3 required SREBF2.
Acknowledgments
OA was supported by Novo Nordisk fellowship and Stiftelsen Professor Nanna Svartz fond. GDN is supported by Progetti di Rilevante Interesse Nazionale [PRIN 2017 K55HLC], Ricerca Finalizzata, Ministry of Health [RF‐2019‐12370896], PNRR Missione 4 [Progetto CN3‐National Center for Gene Therapy and Drugs based on RNA Technology], PNRR Missione 4 [Progetto MUSA‐Multilayered Urban Sustainability Action], PNRR Missione 6 [PNRR‐MAD‐2022‐12375913], and EUROPEAID [173691/DD/ACT/XK Nanokos]. PE and HB are supported by the Swedish Research Council [2020‐01442]; the Swedish Heart‐Lung Foundation [20180451]; and a donation by Mr. Fredrik Lundberg. PSO was supported by the Swedish Research Council (2021‐00500, 2020‐02583), the Heart‐Lung Foundation (20230006, 20210524, 20200827), MedTechLabs, and NovoNordisk.
Ahmed O, Shavva VS, Tarnawski L, et al. Statin‐associated regulation of hepatic PNPLA3 in patients without known liver disease. J Intern Med. 2025;297:47–59.
References
- 1. Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multi‐society Delphi consensus statement on new fatty liver disease nomenclature. Ann Hepatol. 2023;29:101133. 10.1016/j.aohep.2023.101133 [DOI] [PubMed] [Google Scholar]
- 2. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184:2537–2564. [DOI] [PubMed] [Google Scholar]
- 3. Samson SL, Garber AJ. Metabolic syndrome. Endocrinol Metab Clin North Am. 2014;43:1–23. [DOI] [PubMed] [Google Scholar]
- 4. Almeda‐Valdes P, Aguilar‐Olivos N, Uribe M, Mendez‐Sanchez N. Common features of the metabolic syndrome and nonalcoholic fatty liver disease. Rev Recent Clin Trials. 2014;9:148–158. [DOI] [PubMed] [Google Scholar]
- 5. Lindén D, Romeo S. Therapeutic opportunities for the treatment of NASH with genetically validated targets. J Hepatol. 2023;. 79:1056–1064. 10.1016/j.jhep.2023.05.007 [DOI] [PubMed] [Google Scholar]
- 6. Sookoian S, Pirola CJ. Genetic predisposition in nonalcoholic fatty liver disease. Clin Mol Hepatol. 2017;23:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Min H‐Ki, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Rooyen DM, Larter CZ, Haigh WG, Yeh MM, Ioannou G, Kuver R, et al. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology. 2011;141:1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111–188. [DOI] [PubMed] [Google Scholar]
- 10. Athyros VG, Tziomalos K, Gossios TD, Griva T, Anagnostis P, Kargiotis K, et al. Safety and efficacy of long‐term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) study: a post‐hoc analysis. Lancet (London, England). 2010;376:1916–1922. [DOI] [PubMed] [Google Scholar]
- 11. Sfikas G, Psallas M, Koumaras C, Imprialos K, Perdikakis E, Doumas M, et al. Prevalence, diagnosis, and treatment with 3 different statins of non‐alcoholic fatty liver disease/non‐alcoholic steatohepatitis in military personnel. Do genetics play a role? Curr Vasc Pharmacol. 2021;19:572–581. [DOI] [PubMed] [Google Scholar]
- 12. Fatima K, Moeed A, Waqar E, Atif AR, Kamran A, Rizvi H, et al. Efficacy of statins in treatment and development of non‐alcoholic fatty liver disease and steatohepatitis: a systematic review and meta‐analysis. Clin Res Hepatol Gastroenterol. 2022;46:101816. [DOI] [PubMed] [Google Scholar]
- 13. Ayada I, Van Kleef LA, Zhang H, Liu K, Li P, Abozaid YJ, et al. Dissecting the multifaceted impact of statin use on fatty liver disease: a multidimensional study. EBioMedicine. 2023;87:104392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trépo E, Romeo S, Zucman‐Rossi J, Nahon P. PNPLA3 gene in liver diseases. J Hepatol. 2016;65:399–412. [DOI] [PubMed] [Google Scholar]
- 16. Dongiovanni P, Petta S, Mannisto V, Mancina RM, Pipitone R, Karja V, et al. Statin use and non‐alcoholic steatohepatitis in at risk individuals. J Hepatol. 2015;63:705–712. [DOI] [PubMed] [Google Scholar]
- 17. Jackson V, Petrini J, Caidahl K, Eriksson MJ, Liska J, Eriksson P, et al. Bicuspid aortic valve leaflet morphology in relation to aortic root morphology: a study of 300 patients undergoing open‐heart surgery. Eur J Cardio‐Thoracic Surg. 2011;40:e118–e124. [DOI] [PubMed] [Google Scholar]
- 18. Folkersen L, van't Hooft F, Chernogubova E, Agardh HE, Hansson GK, Hedin U, et al. Association of genetic risk variants with expression of proximal genes identifies novel susceptibility genes for cardiovascular disease. Circ Cardiovasc Genet. 2010;3:365–373. [DOI] [PubMed] [Google Scholar]
- 19. Folkersen L, Wågsäter D, Paloschi V, Jackson V, Petrini J, Kurtovic S, et al. Unraveling divergent gene expression profiles in bicuspid and tricuspid aortic valve patients with thoracic aortic dilatation: the ASAP study. Mol Med. 2011;17:1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bolstad BM, Irizarry RA, Åstrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. [DOI] [PubMed] [Google Scholar]
- 21. Alberti K, Zimmet P, Shaw J. The metabolic syndrome—a new worldwide definition. Lancet (London, England). 2005;366:1059–1062. [DOI] [PubMed] [Google Scholar]
- 22. Zimmet P, Alberti KG, Kaufman F, Tajima N, Silink M, Arslanian S, et al. The metabolic syndrome in children and adolescents—an IDF consensus report. Pediatr Diabetes. 2007;8:299–306. [DOI] [PubMed] [Google Scholar]
- 23. Weihe P, Weihrauch‐Blüher S Metabolic syndrome in children and adolescents: diagnostic criteria, therapeutic options and perspectives. Curr Obes Rep. 2019;8:472–479. [DOI] [PubMed] [Google Scholar]
- 24. Pramfalk C, Jakobsson T, Verzijl CRC, Minniti ME, Obensa C, Ripamonti F, et al. Generation of new hepatocyte‐like in vitro models better resembling human lipid metabolism. Biochim Biochim Biophys Acta Mol Cell Biol Lipids. 2020; 1865: 158659. 10.1016/j.bbalip.2020.158659 [DOI] [PubMed] [Google Scholar]
- 25. Pramfalk C, Larsson L, Härdfeldt J, Eriksson M, Parini P Culturing of HepG2 cells with human serum improve their functionality and suitability in studies of lipid metabolism. Biochim Biophys Acta Mol Cell Biol Lipids. 2016;1861: 51–59. 10.1016/j.bbalip.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 26. Al Rifai M, Silverman MG, Nasir K, Budoff MJ, Blankstein R, Szklo M, et al. The association of nonalcoholic fatty liver disease, obesity, and metabolic syndrome, with systemic inflammation and subclinical atherosclerosis: the multi‐ethnic study of atherosclerosis (MESA). Atherosclerosis. 2015;239:629–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–923. [DOI] [PubMed] [Google Scholar]
- 28. Ahmed O, Littmann K, Gustafsson U, Pramfalk C, Öörni K, Larsson L, et al. Ezetimibe in combination with simvastatin reduces remnant cholesterol without affecting biliary lipid concentrations in gallstone patients. J Am Heart Assoc. 2018;7:e009876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parini P, Gustafsson U, Davis MA, Larsson L, Einarsson C, Wilson M, et al. Cholesterol synthesis inhibition elicits an integrated molecular response in human livers including decreased ACAT2. Arterioscler Thromb Vasc Biol. 2008;28:1200–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kotronen A, Westerbacka J, Bergholm R, PietiläInen KH, Yki‐JäRvinen H. Liver fat in the metabolic syndrome. J Clin Endocrinol Metab. 2007;92:3490–3497. [DOI] [PubMed] [Google Scholar]
- 31. Kohjima M, Enjoji M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, et al. Re‐evaluation of fatty acid metabolism‐related gene expression in nonalcoholic fatty liver disease. Int J Mol Med. 2007;20:351–358. [PubMed] [Google Scholar]
- 32. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Casula M, Mozzanica F, Scotti L, Tragni E, Pirillo A, Corrao G, et al. Statin use and risk of new‐onset diabetes: a meta‐analysis of observational studies. Nutr Metab Cardiovasc Dis. 2017;27:396–406. [DOI] [PubMed] [Google Scholar]
- 34. Galicia‐Garcia U, Jebari S, Larrea‐Sebal A, Uribe KB, Siddiqi H, Ostolaza H, et al. Statin treatment‐induced development of type 2 diabetes: from clinical evidence to mechanistic insights. Int J Mol Sci. 2020;21:4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122:4130–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Banini BA, Kumar DP, Cazanave S, Seneshaw M, Mirshahi F, Santhekadur PK, et al. Identification of a metabolic, transcriptomic, and molecular signature Patatin‐like phospholipase domain containing 3‐mediated acceleration of steatohepatitis. Hepatology. 2021;73:1290–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liang H, Xu J, Xu F, Liu H, Yuan D, Yuan S, et al. The SRE motif in the human PNPLA3 promoter (‐97 to ‐88 bp) mediates transactivational effects of SREBP‐1c. J Cell Physiol. 2015;230:2224–2232. [DOI] [PubMed] [Google Scholar]
- 38. Huang Y, He S, Li JZ, Seo Y‐K, Osborne TF, Cohen JC, et al. A feed‐forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci USA. 2010;107:7892–7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Rooyen DM, Gan LT, Yeh MM, Haigh WG, Larter CZ, Ioannou G, et al. Pharmacological cholesterol lowering reverses fibrotic NASH in obese, diabetic mice with metabolic syndrome. J Hepatol. 2013;59:144–152. [DOI] [PubMed] [Google Scholar]
- 40. Hassan R, Tammam SN, Safy SEl, Abdel‐Halim M, Asimakopoulou A, Weiskirchen R, et al. Prevention of hepatic stellate cell activation using JQ1‐ and atorvastatin‐loaded chitosan nanoparticles as a promising approach in therapy of liver fibrosis. Eur J Pharm Biopharm. 2019;134:96–106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Primers.
Figure S1. Hepatic levels of PNPLA3 and DNL associated transcripts in non‐metabolic syndrome patients were higher in the statin‐treated group.
Figure S2. Hepatic DNL‐associated transcript levels following HepG2 exposure of atorvastatin.
Figure S3. Statin regulation of PNPLA3 required SREBF2.
Figure S4: Statin regulation of PNPLA3 required SREBF2.