

Abstract
Lycopodium alkaloid complanadine A, isolated by Kobayashi et al. in 2000, is a complex and unsymmetrical dimer of lycodine. Biologically, it is a novel and promising lead compound for the development of new treatment for neurodegenerative disorders and persistent pain management. Herein, we reported a concise synthesis of complanadine A using a pyrrole-to-pyridine molecular editing strategy. The use of a nucleophilic pyrrole as the precursor of the desired pyridine enabled an efficient and one-pot construction of the tetracyclic core skeleton of complanadine A and lycodine. The pyrrole group was then converted to a 3-chloropyridine via the Ciamician-Dennstedt one carbon ring expansion. A subsequent C–H arylation between the 3-chloropyridine and a pyridine N-oxide formed the unsymmetrical dimer, which was then advanced to complanadine A. Overall, from a readily available known compound, total synthesis of complanadine A was achieved in 11 steps. The pyrrole-to-pyridine molecular editing strategy enabled us to significantly enhance the overall synthetic efficiency. Additionally, as demonstrated by a Suzuki-Miyaura cross coupling, the 3-chloropyridine product from the Ciamician-Dennstedt rearrangement is amenable for further derivatization, offering an opportunity for simplified analog synthesis.
Keywords: total synthesis, molecular editing, alkaloid, complanadine, ring expansion
Graphical Abstract
Lycopodium alkaloids, due to their diverse chemical structures and remarkable biological functions, have garnered significant attention from the synthetic and medicinal communities.1 Among them, Huperzine A, discovered in 1980s, has entered human clinical trial for treating Alzheimer’s disease as a potent acetylcholinesterase inhibitor.2 Continued efforts in the isolation and study of the Lycopodium alkaloids led to the isolation of complanadine A (1, Figure 1A) by Kobayashi and coworkers.3 Complanadine A was structurally characterized as an unsymmetrical dimer of lycodine (4) via a C2–C3’ linkage. Together with complanadine A, mono-oxidized analog complanadine B (2) and partially reduced analog complanadine D (3) were discovered as well.4 In addition to these unsymmetrical lycodine dimer derivatives, lycopladines G (5) and F (6) with an amino acid derived appendages at the pyridine C3 position were also identified.5 Biologically, Kobayashi et al. discovered that complanadine A has promising activity in enhancing the mRNA expression for NGF biosynthesis in 1321N1 human astrocytoma cells and NGF production in human glial cells. Later, Siegel and coworkers, after completing their total synthesis of complanadine A,6 identified it as a selective agonist for the Mas-related G protein-coupled receptor X2 (MrgprX2). MrgprX2 is highly expressed in neurons and functions as a modulator of pain.7 These biological discoveries render complanadine A and its analogs potential lead compounds for the development of new treatment for neurological disorders and persistent pain management.
Figure 1.
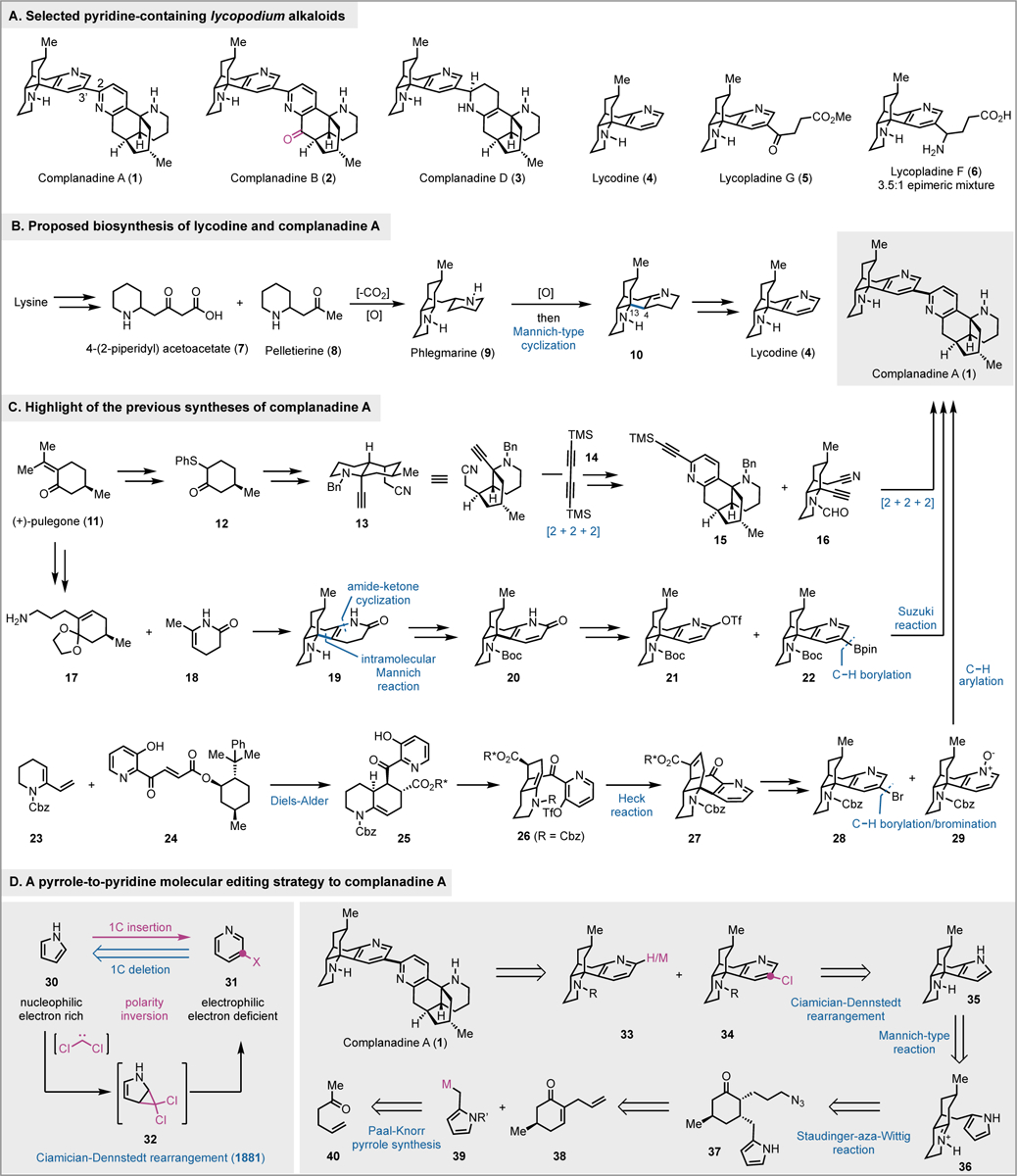
Complanadine natural products, biosynthesis, prior total syntheses, and our synthetic plan.
Biosynthetically (Figure 1B),8 lycopodium alkaloids are generated from lysine, which can be converted to 4-(2-piperidyl) acetoacetate (7) and pelletierine (8), two building blocks for the synthesis of phlegmarine (9). Phlegmarine is a key intermediate in lycopodium alkaloid biosynthesis. After a series of oxidations and an intramolecular Mannich-type cyclization, phlegmarine can be advanced to tetracyclic intermediate 10, from which lycodine and complanadines can be reached. The C2–C3’ linkage of the complanadines could be formed via intermolecular enamine-imine Mannich reaction. Since its isolation, complanadine A has become an attractive target molecule for total synthesis. Previously, three total syntheses of complanadine A have been achieved (Figure 1C). The groups of Siegel6 and Sarpong9 reported their elegant total syntheses of complanadine A at the same time in 2010. Both of their total syntheses started from the chiral pool molecule (+)-pulegone (11). Siegel and coworkers utilized two Co-mediated alkyne [2+2+2] cyclizations to build the C2–C3’ bipyridine moiety. This creative strategy enabled them to complete the total synthesis of complanadine A in 18 steps (longest linear sequence, LLS). In the Sarpong synthesis, they used a biomimetic tandem 1,4-addition/Mannich cyclization/amide-ketone condensation to unite intermediates 17 and 18 and achieve a highly efficient synthesis of tetracyclic intermediate 19, which was further advanced to 21 and 22 for a Suzuki cross coupling to complete their 15-step (LLS) total synthesis. Notably, a C–H borylation was used to prepare 22.10 Following Siegel and Sarpong’s total syntheses, in 2013, Tsukano et al. reported their total synthesis of complanadine A in 20 LLS steps.11 Their synthesis features an intermolecular Diels-Alder reaction (23+24→25) and an intramolecular Heck reaction to access tetracyclic intermediate 27, which was further advanced to 28 and 29 for a C–H arylation followed by a one-pot pyridine N-oxide reduction and deprotection to reach complanadine A.
Our long-term interest in neurotrophically active lycopodium alkaloids12 prompted us to embark on a total synthesis campaign of complanadine A. We communicated our concise total synthesis enabled by a novel pyrrole-to-pyridine molecular editing strategy (Figure 1D, 30→31) in 2021.13 In this Full Article, we report the details of our synthesis and highlight how late-stage molecular editing can significantly enhance the overall synthetic efficiency.14
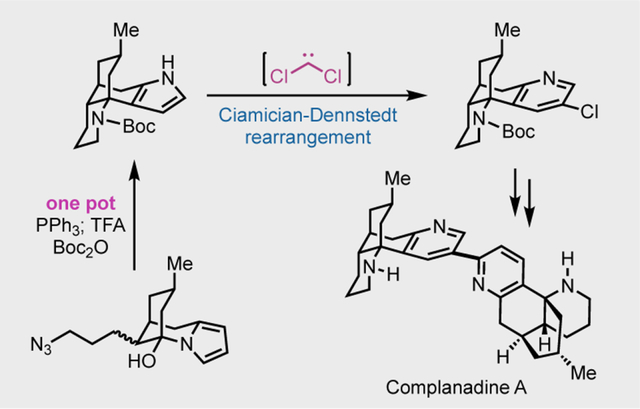
Retrosynthetically, we envisioned either a cross coupling or a C–H arylation between 33 and 34 to build the C2–C3’ heterodimeric linkage. Both 33 and 34 could be derived from the pyrrole-containing tetracyclic intermediate 35, which in forward synthesis could be transformed to 34 via the Ciamician-Dennstedt rearrangement discovered in 1881.15 The Ciamician-Dennstedt rearrangement would start with cyclopropanation of the pyrrole with an in situ generated dichlorocarbene to form a dichlorocyclopropane intermediate. Subsequent ring expansion would lead to a chloropyridine (30→32→31). The chloride could serve as a handle for the following cross coupling or C–H arylation. More importantly, the pyridine-to-pyrrole retrosynthetic analysis would allow a rapid synthesis of 35 with the desired tetracyclic core from relatively simple intermediate 37. In the forward sense, a Staudinger-aza-Wittig process would convert 37 to iminium ion 36.16 A subsequent Mannich-type cyclization with the pyrrole as the nucleophile would give 35. The use of a pyrrole at this stage is strategically important because the corresponding pyridine-containing substrate would be ineffective for the Mannich-type cyclization due to the electron deficient nature of the pyridine group. On the other hand, the pyrrole group is electron rich and highly nucleophilic and could facilitate the cyclization. The subsequent Ciamician-Dennstedt rearrangement would convert the pyrrole group to the desired 3-chloropyridine. Intermediate 37 could be potentially synthesized from 39 and readily available and known cyclohexenone 38. Given the difficulties in accessing pyrrole-derived nucleophile like 39, we designed a conjugate 1,4-addition followed by Paal-Knorr pyrrole synthesis to generate 37.
Our synthesis started from preparing known compound 38. Two approaches can be used to synthesize it in large scale. One uses cheap and abundant chiral pool molecule (R)-(+)-pulegone (11, ~1$/g), which can be converted to 38 in three steps.17 The other one uses a one-pot asymmetric organocatalytic approach to prepare 38 in one step.18 From 38, the Brown hydroboration/oxidation was used to convert its terminal olefin to a primary alcohol (41), which was subsequently protected as a benzyl ether. Notably, the Dudley’s neutral conditions worked effectively for the benzyl ether formation.19 The other acidic or basic benzyl protection protocols gave complicated results, and oxa-Michael addition was observed as one of the competing pathways. We then explored the Mukaiyama conjugated addition to enone 43 using silyl enol ether 44 derived from 40 as the nucleophile. We identified that the conjugate addition occurred smoothly with TBSOTf as the promotor.20 After a one-pot hydrolysis, product 45 was produced in 83% yield as a 1:1 mixture of diastereomers at the α position. The stereoselectivity at the β position was high, and no other stereoisomer was observed. The poor diastereoselectivity at the α position is inconsequential as both diastereomers could be converged to the same product at a later stage. Ozonolysis of the terminal olefin of 45 followed by the Paal-Knorr pyrrole synthesis gave 46,21 which unfortunately could not be isolated because the newly formed pyrrole cyclized on the ketone with its nitrogen to form hemiaminal 47 spontaneously. Since the hemiaminal formation could be reversible, we decided to move forward with 47 and believed that we should be able to release the ketone and in situ trapped it to form the iminium ion for the subsequent Mannich-type cyclization. The next was to convert the benzyl ether group to a primary azide for the Staudinger-aza-Wittig reaction. Removal of the benzyl group turned out not to be straightforward. The hydrogenolysis conditions and Lewis acid conditions we explored either gave low yield or decomposed the started material. While we were exploring the Mukaiyama conjugate addition, we noted benzyl group removal as a side pathway when TiCl4 was used.22 Thus, we decided to investigate this direction and discovered that the benzyl group could be successfully removed with excess TiCl4 at low temperature. Primary alcohol 48 was obtained as a 1.2:1 mixture of diastereomers. Mesylate formation followed by nucleophilic substitution with NaN3 gave 49 in 86% yield over two steps. Overall, this approach allowed us to prepare 49 in 8 steps from 38. While it’s scalable, this synthesis is quite lengthy and involves protection and deprotection steps. In particular, five steps were used to convert the terminal olefin to a primary azide. We wondered if we could introduce the azide at the very beginning by using the hypervalent iodine-catalyzed direct intermolecular anti-Markovnikov hydroazidation developed by Xu23 and Liu,24 which would convert 38 to 51. Meanwhile, we were aware that the introduction of the azide group at an early stage may complicate the following steps. For example, with the presence of an azide group, aza-Michael addition and/or the Schmidt-Aubé ring expansion25 could compete with the Mukaiyama conjugate addition under Lewis acidic conditions. Nevertheless, we decided to explore this approach. After further optimization of the reaction conditions developed by Xu and coworkers, we were able to realize the transformation of 38 to 51 in modest yield at gram scale with 0.5 equiv of benziodoxole 50. As we were expecting, with the primary azide group, the subsequent Mukaiyama conjugate addition turned out to be problematic. The TBSOTf conditions we established previously only delivered 52 in 26% yield with only 9% of 51 recovered. Increasing the amount of TBSOTf resulted in even lower product yield. We thus started to explore other promoters for the Mukaiyama conjugate addition and identified triflimide as an optimal one.26 The conjugate addition occurred smoothly with 30 mol% of triflimide at −78 °C. After one-pot hydrolysis with HCl, product 52 was obtained in 70% yield as a 1:1 mixture of diastereomers. Oxidative cleavage of the terminal double bond via Ozonolysis converted 52 to ketoaldehyde 53. The subsequent Paal-Knorr pyrrole synthesis was uneventful and delivered 49 in 56% yield over two steps. Overall, by introducing the azide group at an early stage, we established a 4-step synthesis of 49 from known compound 38, which is only half of the previous approach.
We then focused on building the tetracyclic core with the Staudinger-aza-Wittig-Mannich strategy. To our delight, this process was smooth and efficient. The tandem sequence started with Staudinger reduction of azide 49 with PPh3 in THF and water. Under this condition, the hemiaminal was stable and didn’t release the ketone group for the subsequent aza-Wittig reaction. Instead, the corresponding primary amine was obtained. To promote the hemiaminal hydrolysis, after concentration of the reaction mixture, trifluoroacetic acid in dichloromethane and water was added. Under the acidic conditions, iminium ion 36 was eventually formed for the next Mannich-type cyclization to provide tetracyclic product 35, which was protected as Boc carbamate 54. This one-pot procedure converted 49 to 54 in 96% yield.
The pyrrole group served us well in constructing tetracyclic compound 54 and next needed to be converted to the chloropyridine via the Ciamician-Dennstedt rearrangement. The Ciamician-Dennstedt rearrangement was not used in total synthesis before. From the limited literature reports about the Ciamician-Dennstedt rearrangement,27 we were aware of a few potential challenges. For example, the Reimer-Tiemann formylation has been shown as a major competing pathway.28
Additionally, the dichlorocarbene tends to add on the more electron rich and more substituted double bond, but in our case, we would need the cyclopropanation to happen on the less substituted one and hoped that steric effect could help to revert the electronic effect. We first explored the use of CHCl3 and KOH to generate dichlorocarbene and were able to obtain desired product 55 in 17% yield. Indeed, the Reimer-Tiemann formylation severely competed with the Ciamician-Dennstedt rearrangement. The formylation product was obtained in 23% yield under the same conditions. After further optimization, we discovered that thermal (70 °C) release of dichlorocarbene from CCl3CO2Na performed better than the basic conditions (KOH/CHCl3). Product 55 could be obtained in 23% yield. Elevating the reaction temperature to 90 °C increased the yield to 31%. The reaction could be scaled up to 1 mmol (330 mg) of 55 with a slight drop of reaction yield (27%). While the yield needs to be further improved, the current conditions enabled us to move material forward. Additionally, the use of BnEt3N+Cl− and the power form of CCl3CO2Na were critical for the success of the ring expansion. The Boc protecting group turned out to be optimal for this process as well. Cbz-carbamate, acetamide, formamide, and methyl carbamate were less effective.
With a reliable approach to synthesize 55, we started to build the C2–C3’ bipyridine moiety. 3-Chloropyridine 55 could serve as the electrophile in the proposed cross coupling or C–H arylation. Part of the material was used to generate the corresponding nucleophile. Catalytic hydrogenolysis removed the chloride and product 56 was obtained in 83% yield. After unsuccessful attempts to functionalize the C2 position of 56 for the cross coupling approach with 55, we oxidized 56 to pyridine N-oxide 57 with mCPBA for the C–H arylation at the C2 position. Such C–H arylation was used in the Tsukano synthesis of complanadine A (Figure 1C). In their case, 3-bromopyridine 28 coupled with pyridine N-oxide 29 to build the key C2–C3’ bipyridine linkage. In our case, 3-chloropyridine 55 was expected to be less reactive in the oxidative addition step. We started with the reaction conditions used by Tsukano et al.,11 which gave 28% yield of desired product 58 together with 28% of the dechlorination byproduct 56 and 25% of recovered 55. Additionally, 68% of 57 was recycled. In order to improve the overall conversion of 55, we started to look for conditions with more electron rich ligands. We noted a protocol reported by Fagnou and coworkers,29 which was later modified by Stoltz et al. and used in their jorunnamycin synthesis.30 With the reaction conditions developed by Stoltz et al. [Pd(OAc)2, tBu2MePHBF4, Cs2CO3, and CsOPiv in toluene at 130 °C], we were delighted that C–H arylation product 58 was produced in 66% yield with a 1/3 ratio of 55/57 or 78% yield with a 1/4 ratio of 55/57. Again, the extra 57 could be recovered to avoid material loss. Subsequent reduction of the pyridine N-oxide with Pd(OH)2/C and H2 followed by removal of the Boc protecting group completed the total synthesis of complanadine A (1). Meanwhile, acid hydrolysis of the Boc protecting group of 56 gave lycodine (4)31 in excellent yield.
Additionally, since a 3-chloropyridine (cf. 55) was produced from the Ciamician-Dennstedt rearrangement of pyrrole 54, it offered an opportunity to use transition metal-catalyzed cross coupling reactions to generate synthetic analogs of complanadine A (1) for biological evaluation and comparison. For instance, Suzuki-Miyaura cross coupling between 55 and a series of N-heteroaryl boronic acid pinacol ester (ArBpin) gave a collection of simplified complanadine A analogs (59a-i, Scheme 1C) in good to excellent yield.32
Scheme 1.
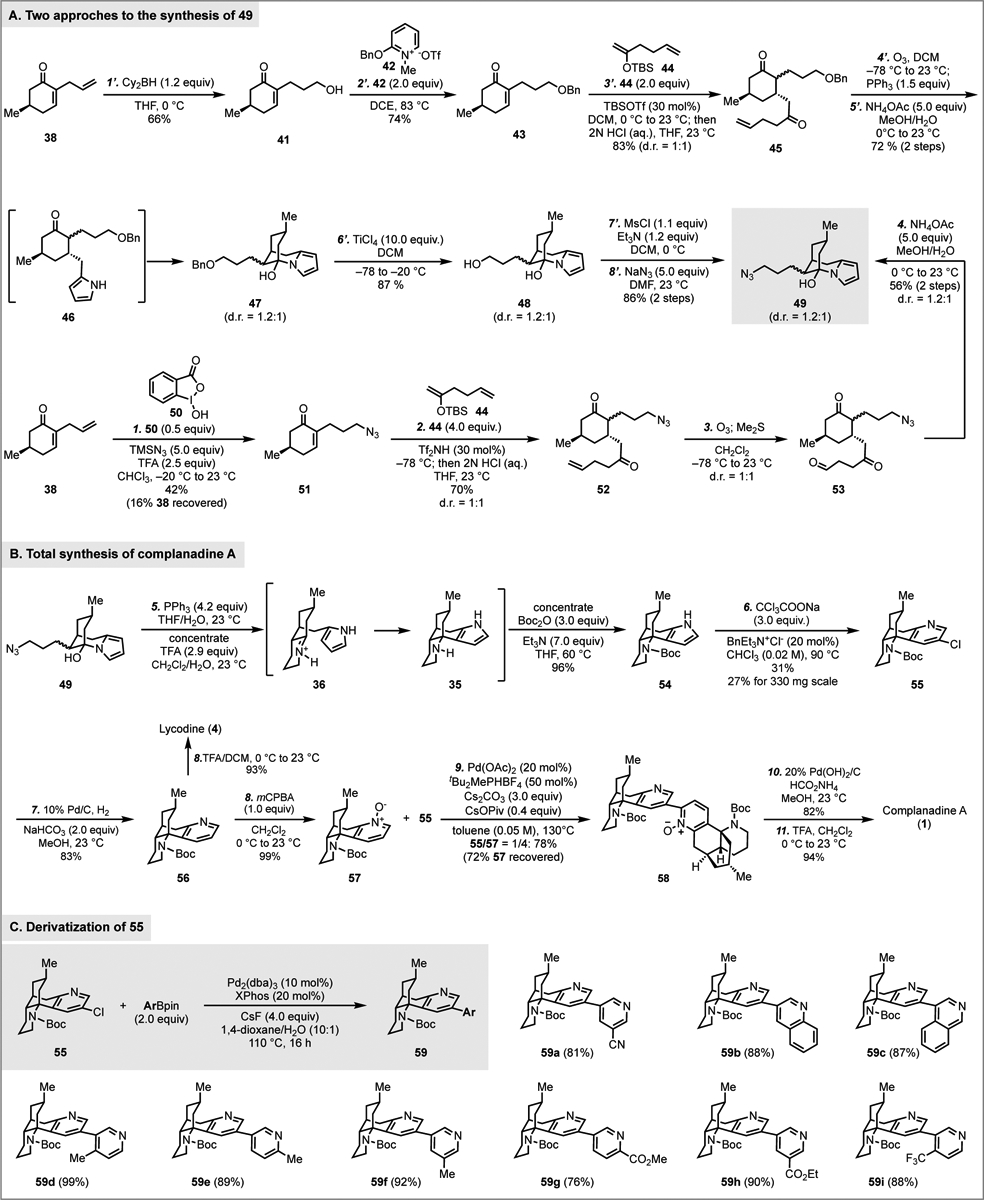
Total synthesis of complanadine A and analogs.
In summary, from readily available and known compound 38, complanadine A was synthesized in 11 steps. The concise synthesis was enabled by a pyrrole-to-pyridine molecular editing strategy. The use of a nucleophilic pyrrole as a pyridine precursor allowed a one-pot Staudinger reduction, amine-ketone condensation, and Mannich-type cyclization to rapidly construct the tetracyclic core skeleton. The pyrrole group was subsequently converted to a chloropyridine using the Ciamician-Dennstedt rearrangement. The newly introduced chloride served as a handle for the next C–H arylation to forge the C2–C3’ bipyridine linkage. It could also be functionalized by transition metal-catalyzed cross couplings such as Suzuki-Miyaura reaction to generate simplified analogs. Additionally, the iodine(III)-mediated direct intermolecular anti-Markovnikov hydroazidation significantly shortened the synthesis of key intermediate 49. This work highlights how one-carbon molecular editing can positively impact total synthesis.
All reactions sensitive to air or moisture were conducted under argon atmosphere in dry and freshly distilled solvents under anhydrous conditions, unless otherwise noted. Anhydrous tetrahydrofuran (THF), dichloromethane (DCM), dimethylformamide (DMF) and toluene were purified by passing the pre-degassed solvents through activated alumina columns. All other solvents and reagents were used as obtained from commercial sources without further purification unless otherwise noted. Room temperature is around 23 °C. Flash column chromatography was performed using silica gel (230–400 mesh). Thin layer chromatography (TLC) was performed using glass-backed silica plates. NMR spectra were recorded on a Bruker AV-500 spectrometer at room temperature (1H at 500 MHz, and 13C at 126 MHz). Chemical shifts (δ) were given in ppm with reference to the solvent signal [1H NMR: CDCl3 (7.26), CD3OD (3.31); 13C NMR: CDCl3 (77.16), CD3OD (49.00)]. 1H NMR data were reported as follows: chemical shifts (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, m = multiplet, br = broad), coupling constant (Hz), and integration. 13C NMR data were reported in terms of chemical shift and multiplicity. High-resolution mass measurements for compound characterization were carried out using a Waters SYNAPT G2-Si system with QUANTOF analyzer or an Agilent 6550 QTOF system. IR data were recorded on a Thermo Nicolet iS50 FT-IR.
Procedures
Compounds 44, 49, 51, 52, 53, 54, 55, 56, 57, 58, 4, and 1 were reported in our previous communication.13
(R)-2-(3-hydroxypropyl)-5-methylcyclohex-2-en-1-one (41): To a stirred solution of BH3•·THF (107 mL, 1.0 M in THF, 107 mmol, 1.2 equiv) in THF (158 mL) was added dropwise cyclohexene (21.4 mL, 211.7 mmol, 2.4 equiv) under argon at 0 °C. The reaction mixture was stirred at 0 °C for 1 h. To the resulting white suspension was added a solution of enone 38 (13.28 g, 88.5 mmol, 1.0 equiv) in THF (45 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 h. Water (107 mL) and NaBO3•·4H2O (49.1 g, 318.6 mmol, 3.6 equiv) were added. The resultant mixture was allowed to stir at room temperature for 2 h. The mixture was filtered through Celite and extracted with ether (3×500 mL). The combined organic phases were washed with brine (300 mL), dried over Na2SO4, filtered and concentrated. The resulting residue was purified by flash chromatography on silica gel using hexanes/EtOAc (5:1~5:2~2:1) as eluent to give primary alcohol 41 (9.77 g, 66%) as a colorless oil.
IR (film): 3420, 2952, 2925, 2872, 1668, 1456, 1381, 1060, 929 cm−1.
1H NMR (500 MHz, CDCl3) δ 6.75 (ddd, J = 5.8, 2.5, 1.3 Hz, 1H), 3.55 (t, J = 6.1 Hz, 2H), 2.51 (ddd, J = 15.6, 3.4, 1.5 Hz, 1H), 2.42 (dt, J = 18.5, 4.9 Hz, 1H), 2.29 (t, J = 7.3 Hz, 2H), 2.24 – 2.01 (m, 4H), 1.70 – 1.60 (m, 2H), 1.06 (d, J = 6.4 Hz, 3H).
13C NMR (126 MHz, CDCl3) δ 200.7, 146.0, 139.1, 61.6, 46.6, 34.6, 32.4, 30.8, 25.3, 21.3.
HRMS (ESI): m/z Calc. for C10H17O2+ [M+H]+: 169.1223, found: 169.1225.
(R)-2-(3-(benzyloxy)propyl)-5-methylcyclohex-2-en-1-one (43): A mixture of pyridinium triflate 42 (40.6 g, 116.4 mmol, 2.0 equiv), MgO (4.66 g, 116.4 mmol, 2.0 equiv, vacuum-dried), and primary alcohol 41 (9.77 g, 58.2 mmol, 1.0 equiv) in DCE (120 mL) was heated at 83 °C under argon for 24 h. The reaction mixture was cooled to room temperature, filtered through Celite, and washed with DCM. The filtrate was concentrated, and the resulting residue was purified by flash chromatography on silica gel using hexanes/EtOAc (20:1~10:1~1:1) as eluent to give benzyl ether 43 (11.16 g, 74%) a light-yellow oil and some recovered 41 (469 mg, 5%) as a light-yellow oil.
IR (film): 3029, 2953, 2924, 2868, 1671, 1454, 1380, 1362, 1238, 1103, 1043, 1028, 904, 736, 698 cm−1.
1H NMR (500 MHz, CDCl3) δ 7.36 – 7.25 (m, 5H), 6.65 (ddd, J = 5.6, 2.7, 1.3 Hz, 1H), 4.48 (s, 2H), 3.46 (t, J = 6.4 Hz, 2H), 2.47 (ddd, J = 15.5, 3.4, 1.5 Hz, 1H), 2.41 – 2.32 (m, 1H), 2.31 – 2.23 (m, 2H), 2.21 – 2.04 (m, 2H), 2.04 – 1.96 (m, 1H), 1.76 – 1.67 (m, 2H), 1.03 (d, J = 6.4 Hz, 3H).
13C NMR (126 MHz, CDCl3) δ 199.8, 144.8, 139.0, 138.7, 128.4, 127.8, 127.6, 73.0, 69.8, 46.8, 34.5, 30.7, 28.6, 26.2, 21.3.
HRMS (ESI): m/z Calc. for C17H23O2+ [M+H]+: 259.1693, found: 259.1694.
(3S,5R)-2-(3-(benzyloxy)propyl)-5-methyl-3-(2-oxohex-5-en-1 yl)cyclohexan-1-one (45): To a solution of benzyl ether 43 (3.28 g, 12.7 mmol, 1.0 equiv) and TBS silyl enol ether 44 (2.69 g, 12.7 mmol, 1.0 equiv) in DCM (64 mL) under argon at 0 °C was added TBSOTf (290 μL, 1.27 mmol, 10 mol%). The reaction solution turned to pink and was stirred at 0 °C for 6 h. Then, additional TBS silyl enol ether 44 (1.35 g, 6.35 mmol, 0.5 equiv) in DCM (6.3 mL) was added followed by TBSOTf (290 μL, 1.27 mmol, 10 mol%). After stirring for 10 h at 0 °C, additional TBS silyl enol ether 44 (1.35 g, 6.35 mmol, 0.5 equiv) in DCM (6.3 mL) was added followed by TBSOTf (290 μL, 1.27 mmol, 10 mol%). The reaction was stirred for 9 h at 0 °C, then allowed to warm to room temperature very slowly and stirred for 22 h. The reaction was concentrated at 0 °C, then dissolved in THF (64 mL) followed by the addition of 2 M HCl aqueous solution (32 mL) at 0 °C. The reaction was allowed to warm to room temperature and stirred for 18 h. The reaction mixture was quenched with saturated NaHCO3 aqueous solution (~65 mL), extracted with EtOAc (3×150 mL), washed with brine, dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel using hexanes/EtOAc (15:1~7:1~5:1) as eluent to give 45 (3.77 g, 83%, d.r. = 1:1) as a light-yellow oil.
IR (film): 3100, 3060, 2924, 2867, 1705, 1454, 1412, 1380, 1361, 1274, 1206, 1100, 1028, 997, 912, 736, 698 cm−1.
1H NMR (500 MHz, CDCl3) δ 7.38 – 7.22 (m, 5H), 5.83 – 5.70 (m, 1H), 5.07 – 4.90 (m, 2H), 4.52 – 4.42 (m, 2H), 3.50 – 3.39 (m, 2H), 2.87 – 2.21 (m, 8H), 2.19 – 2.08 (m, 1H), 2.08 – 1.92 (m, 2H), 1.82 – 1.13 (m, 6H), 1.01 – 0.95 (m, 3H).
13C NMR (126 MHz, CDCl3) δ 214.4, 212.4, 208.9, 208.6, 138.7, 138.6, 137.02, 137.00, 128.5, 127.79, 127.75, 127.6, 115.5, 73.0, 70.3, 69.8, 55.0, 53.1, 50.4, 47.0, 46.7, 42.6, 42.5, 41.0, 38.2, 36.4, 35.4, 34.3, 30.9, 30.2, 27.9, 27.8, 27.7, 27.64, 27.57, 23.3, 22.3, 21.8.
HRMS (ESI): m/z Calc. for C23H33O3+ [M+H]+: 357.2424, found: 357.2422.
(5R,7R,9S)-11-(3-(benzyloxy)propyl)-7-methyl-7,8,9,10-tetrahydro-5,9-methanopyrrolo[1,2-a]azocin-5(6H)-ol (47): A stirred solution of 45 (3.70 g, 10.4 mmol, 1.0 equiv) in DCM (90 mL) was bubbled ozone gas (30 min) at −78 °C. After the color of the mixture changed to blue, oxygen gas was bubbled to the resulting solution for 20 min, followed by addition of PPh3 (3.28 g, 12.5 mmol, 1.2 equiv) at −78 °C. The reaction was allowed to warm slowly to room temperature over 2 h and stirred at that temperature for 3 h. Additional PPh3 (0.82 g, 3.12 mmol, 0.3 equiv) was added and the reaction mixture was stirred at room temperature for 17 h. The reaction was dried with Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel using hexanes/EtOAc (5:1~5:2~2:1) as eluent to give the corresponding aldehyde as a light-yellow oil. This aldehyde was not very stable and used immediately.
To a solution of the above aldehyde (10.4 mmol, 1.0 equiv) in methanol/water (4:1, 204 mL) was added a solution of NH4OAc (4.12 g, 52.0 mmol, 5.0 equiv) in water (4 mL) under argon at 0 °C. The reaction mixture was degassed with argon for three times. The solution was stirred at 0 °C for 6 h and then at room temperature for 14 h under argon. After most methanol was removed under reduced pressure, the resulting mixture was diluted with water, extracted with DCM (3×100 mL), dried with NaSO4, filtered and concentrated. The resulting residue was purified by flash chromatography on silica gel using hexanes/EtOAc (15:1~12:1) as eluent to give pyrrole 47 (2.55 g, 72% for two steps, d.r. = 1.2:1) as a light-yellow oil.
IR (film): 3394, 3029, 2947, 2922, 2867, 1698, 1454, 1421, 1363, 1293, 1266, 1207, 1183, 1103, 1061, 1028, 963, 889, 846, 735, 698, 672 cm−1.
1H NMR (500 MHz, CDCl3) δ 7.39 – 7.22 (m, 5H), 6.95 – 6.75 (m, 1H), 6.20 – 6.10 (m, 1H), 5.83 – 5.68 (m, 1H), 4.55 – 4.42 (m, 2H), 3.59 – 3.39 (m, 2H), 3.09 – 2.94 (m, 1H), 2.85 – 2.48 (m, 1H), 2.37 – 2.28 (m, 1H), 1.96 – 1.75 (m, 3H), 1.75 – 1.20 (m, 7H), 1.15 – 0.92 (m, 1H), 0.82 – 0.76 (m, 3H).
13C NMR (126 MHz, CDCl3) δ 138.3, 138.2, 130.0, 129.7, 129.5, 128.6, 128.5, 128.0, 127.9, 127.8, 114.4, 114.2, 108.6, 108.4, 103.7, 103.5, 86.6, 85.6, 73.2, 70.4, 70.2, 49.0, 46.3, 46.1, 42.4, 42.2, 34.8, 33.0, 32.9, 30.8, 28.3, 27.8, 26.9, 26.6, 24.6, 24.2, 22.5, 21.9, 21.7.
HRMS (ESI): m/z Calc. for C22H30NO2+ [M+H]+: 340.2271, found: 340.2269.
(5R,7R,9S)-11-(3-hydroxypropyl)-7-methyl-7,8,9,10-tetrahydro-5,9-methanopyrrolo[1,2-a]azocin-5(6H)-ol (48): To a solution of pyrrole (2.11 g, 6.22 mmol, 1.0 equiv) in DCM (62 mL) at −78 °C was added TiCl4 (1.0 M in DCM, 62.2 mL, 62.2 mmol, 10.0 equiv) dropwise. The reaction mixture was allowed to slowly warm to −20 °C and stirred at that temperature for 46 h. The resulting mixture was quenched with saturated NaHCO3 aqueous solution (~300 mL) and extracted with DCM (6×60 mL). The combined organic phases were dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel using hexanes/EtOAc (4:1~2:1~2:3) as eluent to give alcohol 48 (1.35 g, 87%, d.r. = 1.2:1) as a light-yellow oil.
IR (film): 3362, 2947, 2922, 2869, 1457, 1421, 1343, 1293, 1266, 1207, 1184, 1131, 1104, 1060, 1011, 965, 895, 765, 709, 671, cm−1.
1H NMR (500 MHz, CDCl3) δ 6.92 – 6.80 (m, 1H), 6.18 – 6.10 (m, 1H), 5.80 – 5.72 (m, 1H), 3.75 – 3.51 (m, 3H), 3.10 – 2.94 (m, 1H), 2.78 – 2.49 (m, 1H), 2.39 – 2.28 (m, 1H), 2.03 – 1.17 (m, 11H), 1.16 – 0.90 (m, 1H), 0.85 – 0.74 (m, 3H).
13C NMR (126 MHz, CDCl3) δ 130.0, 129.5, 114.4, 114.3, 108.5, 108.4, 103.7, 103.5, 86.5, 85.5, 62.73, 62.69, 49.1, 46.1, 46.0, 42.4, 42.3, 34.8, 32.6, 30.8, 30.4, 26.9, 26.6, 24.6, 23.2, 22.0, 21.9, 21.7.
HRMS (ESI): m/z Calc. for C15H24NO2+ [M+H]+: 250.1802, found: 250.1802.
(5R,7R,9S)-11-(3-azidopropyl)-7-methyl-7,8,9,10-tetrahydro-5,9-methanopyrrolo[1,2-a]azocin-5(6H)-ol (49): To alcohol 48 (1.532 g, 6.15 mmol, 1.0 equiv) and Et3N (1.03 mL, 7.38 mmol, 1.2 equiv) in DCM (36 mL) at 0 °C was added methanesulfonyl chloride (520 μL, 6.77 mmol, 1.1 equiv) dropwise. The reaction mixture was stirred at the same temperature for 30 min before it was quenched with saturated NaHCO3 aqueous solution (~30 mL) and extracted with DCM (3×30 mL). The combined organic phases were dried over Na2SO4, filtered, and concentrated to give the crude mesylate which was used immediately without further purification.
To the above mesylate (6.15 mmol, 1.0 equiv) in DMF (62 mL) was added NaN3 (2.00 g, 30.75 mmol, 5.0 equiv). The reaction was stirred at room temperature for 15 h. Water (~60 mL) was added to quench the reaction and the resulting mixture was extracted with ether (3×100 mL). The combined organic phases were washed with water (3×50 mL) and brine, dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel using hexanes/EtOAc (15:1~4:1) as eluent to give 49 (1.45 g, 86% for two steps, d.r. = 1.2:1) as a light-yellow oil.
Synthesis of 3-subsituted pyridine 59a-i: To a mixture of 3-chloropyridine 55 (5.0 mg, 13.3 μmol, 1.0 equiv), the corresponding ArBpin (26.6 μmol, 2.0 equiv), Pd2(dba)3 (1.2 mg, 1.33 μmol, 10 mol%) and Xphos (1.3 mg, 2.66 μmol, 20 mol%) and CsF (8.2 mg, 53.2 μmol, 4.0 equiv) was added 1,4-dioxane (200 μL) and H2O (20 μL) under argon at room temperature. The reaction mixture was stirred at room temperature for 5 min and then heated at 110 °C for 16 h. The reaction mixture was then cooled to room temperature, quenched with brine (1.0 ml) and extracted with EtOAc (3×1 mL). The combined organic phases were dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by was purified by preparative thin layer chromatography (hexanes/acetone=4:1) to afford 59a-i.
tert-butyl (4aR,5S,10bR,12R)-9-(5-cyanopyridin-3-yl)-12-methyl-2,3,4,4a,5,6-hexahydro-1H-5,10b-propano-1,7-phenanthroline-1-carboxylate (59a, 4.8 mg, 81%, as a white foam):
IR (film): 2924.9, 1695.6, 1422.6, 1365.7, 1269.7, 1252.3, 1155.7, 975.0 cm−1.
1H NMR (400 MHz, CDCl3) δ 9.01 (d, J = 2.3 Hz, 1H), 8.89 (d, J = 2.0 Hz, 1H), 8.64 (d, J = 2.3 Hz, 1H), 8.11 (t, J = 2.1 Hz, 1H), 7.77 (d, J = 2.3 Hz, 1H), 4.19 – 4.08 (m, 1H), 3.28 (dd, J = 19.3, 7.3 Hz, 1H), 2.89 (ddd, J = 13.2, 4.0, 1.7 Hz, 1H), 2.79 (d, J = 19.3 Hz, 1H), 2.51 – 2.37 (m, 1H), 2.21 (dd, J = 7.1, 3.5 Hz, 1H), 1.96 – 1.15 (m, 18H), 0.87 (d, J = 6.4 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 159.5, 156.5, 151.33, 151.28, 145.4, 137.2, 136.7, 134.2, 132.6, 129.7, 116.4, 110.5, 80.2, 64.1, 48.3, 44.6, 43.7, 43.0, 35.0, 34.3, 28.7, 27.7, 26.7, 25.7, 22.5.
HRMS (ESI): m/z Calc. for C27H33N4O2+ [M+H]+: 445.2598, found: 445.2603
tert-butyl (4aR,5S,10bR,12R)-12-methyl-9-(quinolin-3-yl)-2,3,4,4a,5,6-hexahydro-1H-5,10b-propano-1,7-phenanthroline-1-carboxylate (59b, 5.5 mg, 88%, as a colorless oil):
IR (film): 2924.0, 1697.0, 1452.6, 1364.9, 1270.0, 1155.6, 980.9, 752.4 cm−1.
1H NMR (400 MHz, CDCl3) δ 9.15 (d, J = 2.3 Hz, 1H), 8.78 (d, J = 2.3 Hz, 1H), 8.30 (d, J = 2.2 Hz, 1H), 8.15 (d, J = 8.5 Hz, 1H), 7.92 – 7.85 (m, 2H), 7.75 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.60 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 4.21 – 4.11 (m, 1H), 3.29 (dd, J = 19.1, 7.3 Hz, 1H), 2.97 – 2.88 (m, 1H), 2.79 (d, J = 19.1 Hz, 1H), 2.50 (ddd, J = 13.6, 12.2, 3.0 Hz, 1H), 2.21 (dd, J = 7.1, 3.5 Hz, 1H), 1.97 – 1.21 (m, 18H), 0.88 (d, J = 6.2 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 158.0, 156.6, 149.4, 147.7, 145.9, 136.2, 133.4, 132.6, 132.0, 130.8, 129.9, 129.5, 128.1, 128.0, 127.4, 80.1, 64.2, 48.5, 44.5, 43.8, 43.1, 35.0, 34.4, 28.7, 27.8, 26.8, 25.8, 22.6.
HRMS (ESI): m/z Calc. for C30H36N3O2+ [M+H]+: 470.2802, found: 470.2808.
tert-butyl (4aR,5S,10bR,12R)-9-(isoquinolin-4-yl)-12-methyl-2,3,4,4a,5,6-hexahydro-1H-5,10b-propano-1,7-phenanthroline-1-carboxylate (59c, 5.4 mg, 87%, as a white foam):
IR (film): 2924.4, 1697.6, 1455.9, 1390.6, 1365.3, 1269.2, 1252.5, 1155.0, 754.4 cm−1.
1H NMR (400 MHz, CDCl3) δ 9.29 (s, 1H), 8.59 (d, J = 2.2 Hz, 1H), 8.49 (s, 1H), 8.07 (d, J = 7.8 Hz, 1H), 7.89 (d, J = 8.5 Hz, 1H), 7.79 – 7.70 (m, 2H), 7.67 (ddd, J = 8.0, 6.8, 1.2 Hz, 1H), 4.15 (ddd, J = 12.5, 5.0, 2.7 Hz, 1H), 3.33 (dd, J = 19.1, 7.3 Hz, 1H), 2.94 – 2.79 (m, 2H), 2.53 (ddd, J = 13.6, 11.7, 3.3 Hz, 1H), 2.22 (dd, J = 6.6, 3.2 Hz, 1H), 1.97 – 1.85 (m, 2H), 1.84 – 1.76 (m, 1H), 1.72 – 1.19 (m, 15H), 0.91 (d, J = 6.0 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 157.7, 156.3, 152.8, 147.8, 143.1, 135.9, 135.8, 134.1, 131.23, 131.17, 129.8, 128.6, 128.4, 127.6, 124.2, 80.0, 64.2, 48.5, 44.4, 43.8, 43.1, 34.9, 34.4, 28.6, 27.8, 26.8, 25.8, 22.6.
HRMS (ESI): m/z Calc. for C30H36N3O2+ [M+H]+: 470.2802, found: 470.2809.
tert-butyl (4aR,5S,10bR,12R)-12-methyl-9-(4-methylpyridin-3-yl)-2,3,4,4a,5,6-hexahydro-1H-5,10b-propano-1,7-phenanthroline-1-carboxylate (59d, 5.7 mg, 99%, as a colorless oil):
IR (film): 2924.0, 1697.9, 1455.4, 1365.3, 1269.0, 1155.2, 966.4 cm−1.
1H NMR (400 MHz, CDCl3) δ 8.48 (d, J = 5.0 Hz, 1H), 8.45 (s, 1H), 8.40 (d, J = 2.2 Hz, 1H), 7.55 (d, J = 2.2 Hz, 1H), 7.22 (d, J = 5.0 Hz, 1H), 4.18 – 4.07 (m, 1H), 3.27 (dd, J = 19.0, 7.4 Hz, 1H), 2.87 – 2.72 (m, 2H), 2.45 (ddd, J = 13.6, 12.0, 3.0 Hz, 1H), 2.31 (s, 3H), 2.19 (dd, J = 7.1, 3.5 Hz, 1H), 2.06 – 1.22 (m, 18H), 0.87 (d, J = 6.3 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 157.5, 156.4, 150.0, 149.0, 147.4, 144.9, 135.4, 134.8, 134.3, 131.8, 125.6, 80.0, 64.1, 48.5, 44.4, 43.8, 43.1, 35.0, 34.4, 28.7, 27.8, 26.8, 25.8, 22.6, 20.1.
HRMS (ESI): m/z Calc. for C27H36N3O2+ [M+H]+: 434.2802, found: 434.2804.
tert-butyl (4aR,5S,10bR,12R)-12-methyl-9-(6-methylpyridin-3-yl)-2,3,4,4a,5,6-hexahydro-1H-5,10b-propano-1,7-phenanthroline-1-carboxylate (59e, 5.1 mg, 89%, as a white foam):
IR (film): 2924.0, 1697.8, 1676.4, 1453.4, 1365.0, 1268.8, 1251.7, 1155.7, 966.3 cm−1.
1H NMR (400 MHz, CDCl3) δ 8.70 (d, J = 2.1 Hz, 1H), 8.61 (d, J = 2.3 Hz, 1H), 7.75 (dd, J = 8.0, 2.4 Hz, 1H), 7.71 (d, J = 2.2 Hz, 1H), 7.24 (d, J = 8.1 Hz, 1H), 4.18 – 4.08 (m, 1H), 3.25 (dd, J = 19.0, 7.4 Hz, 1H), 2.85 (ddd, J = 13.1, 3.9, 1.7 Hz, 1H), 2.75 (d, J = 19.0 Hz, 1H), 2.61 (s, 3H), 2.45 (ddd, J = 13.7, 12.3, 2.9 Hz, 1H), 2.18 (dd, J = 7.1, 3.4 Hz, 1H), 2.03 – 1.19 (m, 18H), 0.86 (d, J = 6.3 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 158.1, 157.7, 156.5, 147.4, 145.7, 136.0, 134.7, 132.1, 132.0, 130.8, 123.5, 80.0, 64.2, 48.5, 44.4, 43.8, 43.1, 35.0, 34.4, 28.7, 27.8, 26.7, 25.8, 24.3, 22.6.
HRMS (ESI): m/z Calc. for C27H36N3O2+ [M+H]+: 434.2802, found: 434.2806.
tert-butyl (4aR,5S,10bR,12R)-12-methyl-9-(5-methylpyridin-3-yl)-2,3,4,4a,5,6-hexahydro-1H-5,10b-propano-1,7-phenanthroline-1-carboxylate (59f, 5.3 mg, 92%, as a colorless oil):
IR (film): 2923.8, 1697.5, 1455.3, 1365.1, 1269.4, 1252.5, 1155.0, 973.8 cm−1.
1H NMR (400 MHz, CDCl3) δ 8.63 (d, J = 2.3 Hz, 2H), 8.46 (s, 1H), 7.74 (d, J = 2.3 Hz, 1H), 7.65 (s, 1H), 4.13 (dq, J = 14.3, 2.9 Hz, 1H), 3.26 (dd, J = 19.0, 7.4 Hz, 1H), 2.89 (ddd, J = 13.1, 3.9, 1.7 Hz, 1H), 2.75 (d, J = 19.0 Hz, 1H), 2.45 (ddd, J = 13.9, 12.3, 3.0 Hz, 1H), 2.40 (s, 3H), 2.18 (dd, J = 7.2, 3.6 Hz, 1H), 1.93 – 1.20 (s, 18H), 0.86 (d, J = 6.3 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 157.9, 156.6, 149.7, 145.7, 145.3, 136.0, 134.9, 133.5, 133.2, 132.4, 131.9, 80.0, 64.2, 48.4, 44.4, 43.8, 43.1, 35.0, 34.4, 28.7, 27.8, 26.7, 25.8, 22.5, 18.6.
HRMS (ESI): m/z Calc. for C27H36N3O2+ [M+H]+: 434.2802, found: 434.2808.
tert-butyl (4aR,5S,10bR,12R)-9-(6-(methoxycarbonyl)pyridin-3-yl)-12-methyl-2,3,4,4a,5,6-hexahydro-1H-5,10b-propano-1,7-phenanthroline-1-carboxylate (59g, 4.8 mg, 76%, as a white foam):
IR (film): 2924.6, 1723.9, 1697.5, 1454.4, 1437.2, 1364.8, 1312.3, 1277.8, 1238.0, 1155.6, 966.6 cm−1.
1H NMR (400 MHz, CDCl3) δ 8.94 (dd, J = 2.3, 0.8 Hz, 1H), 8.67 (d, J = 2.3 Hz, 1H), 8.23 (dd, J = 8.2, 0.8 Hz, 1H), 8.00 (dd, J = 8.1, 2.3 Hz, 1H), 7.78 (d, J = 2.3 Hz, 1H), 4.19 – 4.10 (m, 1H), 4.04 (s, 3H), 3.27 (dd, J = 19.2, 7.4 Hz, 1H), 2.88 (ddd, J = 13.2, 3.9, 1.7 Hz, 1H), 2.77 (d, J = 19.2 Hz, 1H), 2.50 – 2.39 (m, 1H), 2.19 (dd, J = 6.8, 3.1 Hz, 1H), 1.95 – 1.16 (m, 18H), 0.86 (d, J = 6.4 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 165.6, 159.1, 156.5, 148.1, 147.1, 145.9, 137.0, 136.4, 135.2, 132.6, 130.8, 125.5, 80.12, 64.1, 53.2, 48.4, 44.4, 43.8, 43.0, 35.1, 34.4, 28.7, 27.8, 26.7, 25.7, 22.5.
HRMS (ESI): m/z Calc. for C28H36N3O4+ [M+H]+: 478.2700, found: 478.2706.
tert-butyl (4aR,5S,10bR,12R)-9-(5-(ethoxycarbonyl)pyridin-3-yl)-12-methyl-2,3,4,4a,5,6-hexahydro-1H-5,10b-propano-1,7-phenanthroline-1-carboxylate (59h, 5.9 mg, 90%, as a colorless oil):
IR (film): 2924.8, 1726.1, 1697.9, 1427.7, 1365.9, 1311.9, 1298.7, 1281.7, 1264.3, 1155.6, 1115.6, 973.4, 767.6 cm−1.
1H NMR (400 MHz, CDCl3) δ 9.22 (d, J = 2.0 Hz, 1H), 8.96 (d, J = 2.3 Hz, 1H), 8.67 (d, J = 2.3 Hz, 1H), 8.45 (t, J = 2.2 Hz, 1H), 7.78 (d, J = 2.3 Hz, 1H), 4.45 (q, J = 7.1 Hz, 2H), 4.19 – 4.10 (m, 1H), 3.27 (dd, J = 19.1, 7.3 Hz, 1H), 2.88 (ddd, J = 13.2, 3.9, 1.7 Hz, 1H), 2.77 (d, J = 19.1 Hz, 1H), 2.45 (ddd, J = 13.7, 12.2, 3.0 Hz, 1H), 2.19 (dd, J = 7.0, 3.4 Hz, 1H), 1.94 – 1.49 (m, 15H), 1.43 (t, J = 7.1 Hz, 3H), 1.39 – 1.20 (m, 3H), 0.87 (d, J = 6.3 Hz, 3H).
13C NMR (101 MHz, CDCl3) δ 165.2, 158.6, 156.5, 151.6, 150.1, 145.8, 136.3, 135.1, 133.6, 132.5, 130.9, 126.6, 80.1, 64.1, 61.8, 48.4, 44.5, 43.8, 43.1, 35.0, 34.4, 28.7, 27.8, 26.7, 25.8, 22.5, 14.4.
HRMS (ESI): m/z Calc. for C29H38N3O4+ [M+H]+: 492.2857, found: 492.2866.
tert-butyl (4aR,5S,10bR,12R)-12-methyl-9-(4-(trifluoromethyl)pyridin-3-yl)-2,3,4,4a,5,6-hexahydro-1H-5,10b-propano-1,7-phenanthroline-1-carboxylate (59i, 5.7 mg, 88%, as a colorless oil):
IR (film): 2925.6, 1698.2, 1455.9, 1407.4, 1365.9, 1318.3, 1269.9, 1250.7, 1181.1, 1156.8, 1140.7, 1066.8, 966.6, 659.6 cm−1.
1H NMR (600 MHz, CDCl3) δ 8.82 (d, J = 5.1 Hz, 1H), 8.70 (s, 1H), 8.39 (d, J = 2.3 Hz, 1H), 7.64 (d, J = 5.1 Hz, 1H), 7.62 (s, 1H), 4.12 (ddd, J = 14.5, 5.2, 2.8 Hz, 1H), 3.29 (dd, J = 19.0, 7.4 Hz, 1H), 2.84 – 2.73 (m, 2H), 2.42 (ddd, J = 13.8, 12.4, 2.6 Hz, 1H), 2.19 (dd, J = 7.0, 3.5 Hz, 1H), 1.92 – 1.82 (m, 2H), 1.80 – 1.49 (m, 4H), 1.44 (s, 9H), 1.40 – 1.19 (m, 3H), 0.87 (d, J = 6.4 Hz, 3H).
13C NMR (151 MHz, CDCl3) δ 158.7, 156.2, 152.6, 150.2, 146.9, 136.4 (q, J = 31.8 Hz), 135.5, 135.1, 132.2, 130.0, 122.9 (q, J = 275.1 Hz), 119.9 (q, J = 5.0 Hz), 79.9, 64.0, 48.4, 44.3, 43.8, 43.1, 35.1, 34.4, 28.6, 27.9, 26.8, 25.8, 22.6.
19F NMR (565 MHz, CDCl3) δ −59.0.
HRMS (ESI): m/z Calc. for C27H33F3N3O2+ [M+H]+: 488.2519, found: 488.2527.
Supplementary Material
Acknowledgment
We thank Prof. Corinna Schindler and Prof. Mark Levin for editing the Special Issue “Advances in Skeletal Editing and Rearrangement Reactions”.
Funding Information
This work was supported by NIH GM128570.
Biographies

Brandon S. Martin received a B.S. in chemistry from Eastern Illinois University in 2014. Subsequently, he began graduate study at Purdue University under the supervision of Professor Mingji Dai and received his Ph.D. in 2021. He is currently a scientist in medicinal chemistry at Ferring Pharmaceuticals in San Diego, California.

Donghui Ma received his B.S. degree from Northwest A&F University in 2009. He then pursued his graduate study under the supervision of Prof. Xuegong She at Lanzhou University. After earning his Ph.D. degree in 2015, he conducted postdoctoral research in Georgia State University, University at Albany - State University of New York, The University of Texas at San Antonio, and Purdue University. He is currently a postdoctoral fellow in Professor Mingji Dai’s group at Emory University focusing on the synthesis of biologically active natural products.

Takeru Saito received his B.S. degree from Purdue University in chemical engineering in 2020. During his time at Purdue University, he worked as an undergraduate student researcher in the lab of Prof. Mingji Dai. He is currently a graduate student at University of Massachusetts Boston, working in the lab of Professor Wei Zhang. He is also a graduate student co-op at GSK, working with their Encoded Library Technologies group in Cambridge, Massachusetts.

Katelyn S. Gallagher started her undergraduate study in 2018 at Purdue University majoring biochemistry. In the summer of 2019, she started research in Professor Mingji Dai’s Group, working on natural product total synthesis. She graduated from Purdue with a Bachelor of Science in Chemistry in May of 2022. In the fall of 2022, she embarked her graduate study in the Reisman Group at the California Institute of Technology continuing research in total synthesis and synthetic methodologies.

Mingji Dai received his B.S. degree from Peking University in 2002. After two years’ research with Professors Zhen Yang and Jiahua Chen in the same university, he ventured to New York City in 2004 and pursued graduate study under the guidance of Professor Samuel J. Danishefsky at Columbia University. After earning his Ph.D. degree in 2009, he took a postdoctoral position in the laboratory of Professor Stuart L. Schreiber at Harvard University and the Broad Institute. In August 2012, he began his independent career as an assistant professor in the Chemistry Department and Center for Cancer Research of Purdue University. He was promoted to associate professor with tenure in 2018 and full professor in 2020. In August 2022, he moved to Emory University and is currently the Asa Griggs Candler Professor of Chemistry at Emory University. His lab focuses on developing new strategies and methodologies for the synthesis of complex natural products and other medicinally important molecules.
Footnotes
Supporting Information
The NMR spectra of new compounds are provided in the Supporting Information.
Conflict of Interest
The authors declare no conflict of interest.
References
- (1).Ma X; Gang DR Nat. Prod. Rep 2004, 21, 752. [DOI] [PubMed] [Google Scholar]
- (2).(a) Zhang H-Y Acta Pharmacol. Sin 2012, 33, 1170–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Herzon SB; Tun MKM J. Exp. Pharmacol 2012, 4, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kobayashi J; Hirasawa Y; Yoshida N; Morita H Tetrahedron Lett 2000, 41, 9069. [Google Scholar]
- (4).(a) Morita H; Ishiuchi K; Haganuma A; Hoshino T; Obara Y; Nakahata N; Kobayashi J Tetrahedron 2005, 61, 1955. [Google Scholar]; (b) Ishiuchi K; Kubota T; Ishiyama H; Hayashi S; Shibata T; Mori K; Obara Y; Nakahata N; Kobayashi J Bioorg. Med. Chem 2011, 19, 749. [DOI] [PubMed] [Google Scholar]; (c) Ishiuchi K; Kubota T; Mikami Y; Obara Y; Nakahata N; Kobayashi J Bioorg. Med. Chem 2007, 15, 413. [DOI] [PubMed] [Google Scholar]
- (5).(a) Ishiuchi K; Kubota T; Hayashi S; Shibata T; Kobayashi J Tetrahedron Lett 2009, 50, 4221. [Google Scholar]; (b) Yeap JS-Y; Lim K-H; Yong K-T; Lim S-H; Kam T-S; Low Y-Y J. Nat. Prod 2019, 82, 324. [DOI] [PubMed] [Google Scholar]; (c) Haley HMS; Payer SE; Papidocha SM; Clemens S; Nyenhuis J; Sarpong R J. Am. Chem. Soc 2021, 143, 4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Yuan CX; Chang CT; Axelrod A; Siegel D J. Am. Chem. Soc 2010, 132, 5924. [DOI] [PubMed] [Google Scholar]; (b) Yuan CX; Chang CT; Siegel D J. Org. Chem 2013, 78, 5647. [DOI] [PubMed] [Google Scholar]
- (7).Johnson T; Siegel D Bioorg. Med. Chem. Lett 2014, 24, 3512. [DOI] [PubMed] [Google Scholar]
- (8).Wang J; Zhang Z-K; Jiang F-F; Qi B-W; Ding N; Hnin SYY; Liu X; Li J; Wang X-H, Tu P-F; Abe I; Morita H; Shi S-P Org. Lett 2020, 22, 8725. [DOI] [PubMed] [Google Scholar]
- (9).(a) Fischer DF; Sarpong R J. Am. Chem. Soc 2010, 132, 5926. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Newton JN; Fischer DF; Sarpong R Angew. Chem. Int. Ed 2013, 52, 1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Ishiyama T; Takagi J; Ishida K; Miyaura N; Anastasi NR; Hartwig JF J. Am. Chem. Soc 2002, 124, 390. [DOI] [PubMed] [Google Scholar]; (b) Cho J-Y; Tse MK; Holmes D; Maleczka RE Jr.; Smith MR Science 2002, 295, 305. [DOI] [PubMed] [Google Scholar]
- (11).(a) Zhao L; Tsukano C; Kwon E; Takemoto Y; Hirama M Angew. Chem. Int. Ed 2013, 52, 1722. [DOI] [PubMed] [Google Scholar]
- (12).Yang Y; Haskins CW; Zhang W; Low PL; Dai M Angew. Chem. Int. Ed 2014, 53, 3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ma D; Martin BS; Gallagher KS; Saito T; Dai M J. Am. Chem. Soc 2021, 143, 16383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jurczyk J; Woo J; Kim SF; Dherange BD; Sarpong R; Levin MD Nat. Syn 2022, 1, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ciamician GL; Dennstedt M Ber. Dtsch. Chem. Ges 1881, 14, 1153. [Google Scholar]
- (16).(a) Yang Y; Bai Y; Sun S; Dai M Org. Lett 2014, 16, 6216. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nakahara K; Hirano K; Maehata R; Kita Y; Fujioka H Org. Lett 2011, 13, 2015. [DOI] [PubMed] [Google Scholar]; (c) Chen J; Forsyth CJ Org. Lett 2003, 5, 1281. [DOI] [PubMed] [Google Scholar]
- (17).Meng L J. Org. Chem 2016, 81, 7784. [DOI] [PubMed] [Google Scholar]
- (18).Linghu X; Kennedy-Smith JJ; Toste FD Angew. Chem. Int. Ed 2007, 46, 7671. [DOI] [PubMed] [Google Scholar]
- (19).(a) Poon KWC; House SE; Dudley GB Synlett 2005, 3142. [Google Scholar]; (b) Poon KWC; Dudley GB J. Org. Chem 2006, 71, 3923. [DOI] [PubMed] [Google Scholar]
- (20).Del Bel M; Rovira A; Guerrero CA J. Am. Chem. Soc 2013, 135, 12188. [DOI] [PubMed] [Google Scholar]
- (21).Thangaraj M; Gaykar RN; Roy T; Biju AT J. Org. Chem 2017, 82, 4470. [DOI] [PubMed] [Google Scholar]
- (22).Sudhakar G; Kadam VD; Bayya S; Pranitha G; Jagadeesh B Org. Lett 2011, 13, 5452. [DOI] [PubMed] [Google Scholar]
- (23).Li H; Shen S-J; Zhu C-L; Xu H J. Am. Chem. Soc 2019, 141, 9415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li X; Chen P; Liu G Sci. China Chem 2019, 62, 1537. [Google Scholar]
- (25).Aube J; Milligan GL J. Am. Chem. Soc 1991, 113, 8965. [Google Scholar]
- (26).Zhao W; Sun J Chem. Rev 2018, 118, 10439. [DOI] [PubMed] [Google Scholar]
- (27).(a) Dhanak D; Reese C J. Chem. Soc., Perkin Trans 1 1987, 2829. [Google Scholar]; (b) Raheem IT; Thiara PS Jacobsen EN Org. Lett 2008, 10, 1577. [DOI] [PubMed] [Google Scholar]; (c) Dherange BD; Kelly PQ; Liles JP; Sigman MS; Levin MD J. Am. Chem. Soc 2021, 143, 11337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wynberg H Chem. Rev 1960, 60, 169. [Google Scholar]
- (29).Campeau L-C; Schipper DJ; Fagnou K J. Am. Chem. Soc 2008,130, 3266. [DOI] [PubMed] [Google Scholar]
- (30).Welin ER; Ngamnithiporn A; Klatte M; Lapointe G; Pototschnig GM; McDermott MSJ; Conklin D; Gilmore CD; Tadross PM; Haley CK; Negoro K; Glibstrup E; Grünanger CU; Allan KM; Virgil SC; Slamon DK; Stoltz BM Science 2019, 363, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).(a) Heathcock CH; Kleinman EF; Binkley ES J. Am. Chem. Soc 1982, 104, 1054. [Google Scholar]; (b) Zhao L; Tsukano C; Kown E; Shirakawa H; Kaneko S; Takemoto Y; Hiram M Chem. Eur. J 2017, 23, 802. [DOI] [PubMed] [Google Scholar]; (c) Azuma M; Yoshikawa T; Kogure N; Kitajima M; Takayama H J. Am. Chem. Soc 2014, 136, 11618. [DOI] [PubMed] [Google Scholar]
- (32).Hu Y; Shi H; Zhou M; Ren Q; Zhu W; Zhang W; Zhang Z; Zhou C; Liu Y; Ding X; Shen HC; Yan SF; Dey F; Wu W; Zhai G; Zhou Z; Xu Z; Ji Y; Lv H; Jiang T; Wang W; Xu Y; Vercruysse M; Yao X; Mao Y; Yu X; Bradley K; Tan X J. Med. Chem 2020, 63, 9623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.