

Summary
MYSM1 deficiency causes inherited bone marrow failure syndrome (IBMFS). We have previously identified an IBMFS patient with a homozygous pathogenic variant in MYSM1 who recovered from cytopenia due to spontaneous correction of one MYSM1 variant in the haematopoietic compartment, an event called somatic genetic rescue (SGR). The study of the genetic and biological aspects of the patient's haematopoietic/lymphopoietic system over a decade after SGR shows that one genetically corrected haematopoietic stem cell (HSC) can restore a healthy and stable haematopoietic system. This supports in vivo gene correction of HSCs as a promising treatment for IBMFS, including MYSM1 deficiency.
Keywords: bone marrow failure, haematopoiesis, MYSM1, natural gene therapy, somatic genetic rescue
INTRODUCTION
Somatic mutations naturally accumulate over time in all organs of each individual. The frequency of somatic mutation acquisition in blood cells has been estimated to be ~15–20 mutations per cell per year. 1 , 2 Although most of them are neutral, some can confer a fitness advantage that causes clonal expansion, a phenomenon known as clonal haematopoiesis of indeterminate potential (CHIP) that is associated with predisposition to haematological malignancy and increased risk of mortality. 2 On the other hand, in the context of Mendelian haematopoietic disorders, a somatic mutation may specifically mitigate the deleterious effect of the disease‐causing germline genetic defect, a phenomenon known as somatic genetic rescue (SGR). 1 While in most cases, the SGR has been shown to have partial and limited effects on the patients' haematological parameters, in rare cases it resulted in the restoration of a normal haematopoietic system and a cure of the disease, a phenomenon‐typified natural gene therapy. 1 , 2
MYSM1 functions in deubiquitinating the histone 2A ubiquitinated at position K119, an epigenetic marker for gene transcription silencing. 3 As a result, MYSM1 reverts the transcriptional repression of genes encoding ribosomal proteins and transcription factors that are essential for the maintenance of haematopoietic stem cell (HSC) quiescence and development of blood cells. 3 , 4 To our knowledge, only 12 individuals carrying biallelic pathogenic MYSM1 mutations have been reported so far, indicating the rarity of this syndrome (Table S1). We previously reported the case of a young individual carrying a homozygous c.1967A>G; p.His656Arg (H656R) MYSM1 variant associated with a reduction in the values of most blood lineages and a virtual lack of B lymphocytes. We further showed that an SGR corresponding to the genetic correction of one of the mutated alleles to wild type occurred in the patient's multipotent HSC compartment and was associated with a progressive improvement of haematopoiesis that occurred early in life. 5 Here, we report the assessment of the patient's lympho/haematopoietic features 12 years following the SGR event.
METHODS
Detailed methods are described in Supporting Information.
RESULTS AND DISCUSSION
Several tools predicted pathogenicity of the H656R MYSM1 variant (Figure S1; Table S1). Recombinant WT and H656R portion of MYSM1 containing the catalytic domain (referred to as C1‐WT and C1‐H656R, respectively) (Figure 1A) were similarly recovered from bacterial lysates (Figure 1B), indicating that the mutation does not affect protein expression/stability. However, unlike C1‐WT, C1‐H656R was devoid of in vitro deubiquitinase activity (Figure 1C), supporting the recent conclusion drawn in a mouse model that H656R MYSM1 is a loss‐of‐function mutation. 4 The patient's blood cell counts that normalized a few months after SGR remained remarkably stable over an additional period of 12 years (Figure 1D). Of note for this patient who lacked B cells prior to SGR, B cell subpopulations, immunoglobulin levels, post‐vaccination serologies, post‐infectious seroconversions and Immunoglobulin heavy chain variable‐diversity‐junction repertoire were normal (Figure 1E; Figure S2). This indicates that one or a few SGR‐positive (SGR+) HSCs suffice to reconstitute and maintain a normal immuno‐haematopoietic system in a context of MYSM1 deficiency. The rapid SGR‐mediated haematopoietic reconstitution argues for a strong selective advantage of SGR+ HSCs over unmodified cells. This is in line with studies demonstrating that HSCs carrying the H656R mutation exhibit a profound intrinsic defect in haematopoiesis potential, while the bone marrow environment of Mysm1‐deficient mice does not preclude haematopoietic engraftment and development of WT HSCs. 4 , 6 The recent demonstration that MYSM1 deficiency causes proteostatic stress and ferroptosis in HSCs could explain why SGR provided such a benefit in patient' HSCs. 4 Assessment of the immune cell subsets by cytometry by time‐of‐flight (CyTOF) analysis in blood samples from the patient 12 years post‐SGR and five age‐matched healthy controls revealed overall similar profiles (Figure 2A; Figure S3). Absolute cell counts did not differ in the patient and the controls except for a slight increase in innate natural killer (NK)‐T cells (NK‐T) and mucosal‐associated invariant T cells (MAIT) populations, while the proportion of monocytes, T, B and NK cells was comparable (Figure 2B). Within the T cell population, the percentage of CD4+ T cells was significantly reduced and the CD4+/CD8+ ratio was reversed, but the absolute number of CD4+ T cells was within the normal range for age. The fractions of naive and memory T and B cells were unaffected (Figure 2C). Nevertheless, we noticed an increase in the double‐negative (DN) population (noted CD3 DN) that culminated in 21.9% of total T lymphocytes and 9.1% of total peripheral blood mononuclear cells (PBMNCs) (Figure 2B). This population consisted mainly of γδ T cells which, as the αβ T cells, exhibited a diversified T cell receptor (TCR) repertoire (Figure S2). These results indicate that, despite an increase in NK‐T and MAIT and in DN Tγδ cells, the SGR‐mediated immunohaematological reconstitution that took place more than 12 years ago gave rise to an overall normal immune system that persisted over time. Consistent with this observation, the patient had not experienced any haematological or immunological/infectious problems during these years.
FIGURE 1.
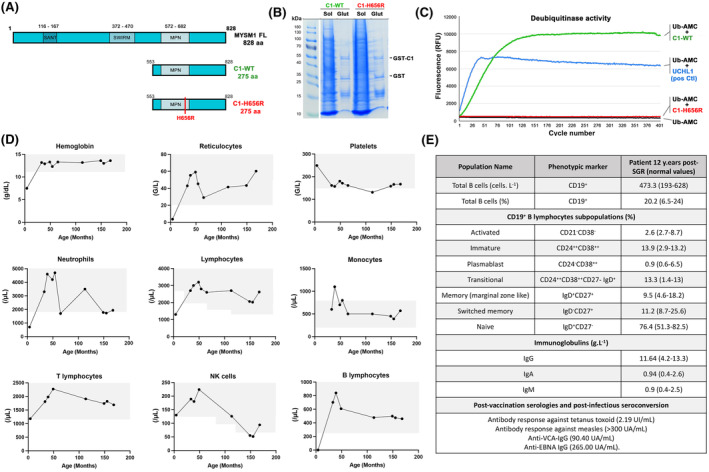
Activity of the H656R MYSM1 variant and haematological parameters in the patient over time. (A) Schematic representation of the full‐length MYSM1 protein as well as the WT and the mutated forms of the C‐terminal part of MYSM1 (noted C1‐WT and C1‐H656R, respectively) used to assess the deubiquitinase activity in vitro. (B) A 6% polyacrylamide gel stained with Coomassie blue showing fractions from a representative purification of GST‐C1‐WT and GST‐C1‐H656R used in in vitro deubiquitinase assay. Glut: Glutathione purification. (C) Fluorescence intensity measurements for C1‐WT (green line), C1‐H656R (red line), UCHL‐1 used as positive control (blue line) and Ub‐AMC alone used as background fluorescence control (black line). (D) Whole blood cell counts over time. Grey areas represent normal values obtained in age‐matched healthy subjects. (E) Immunophenotyping of the B cell compartment in the patient 12 years post‐somatic genetic rescue (SGR) and serology values post‐vaccination and post‐infection. Age‐matched normal values are indicated in brackets.
FIGURE 2.
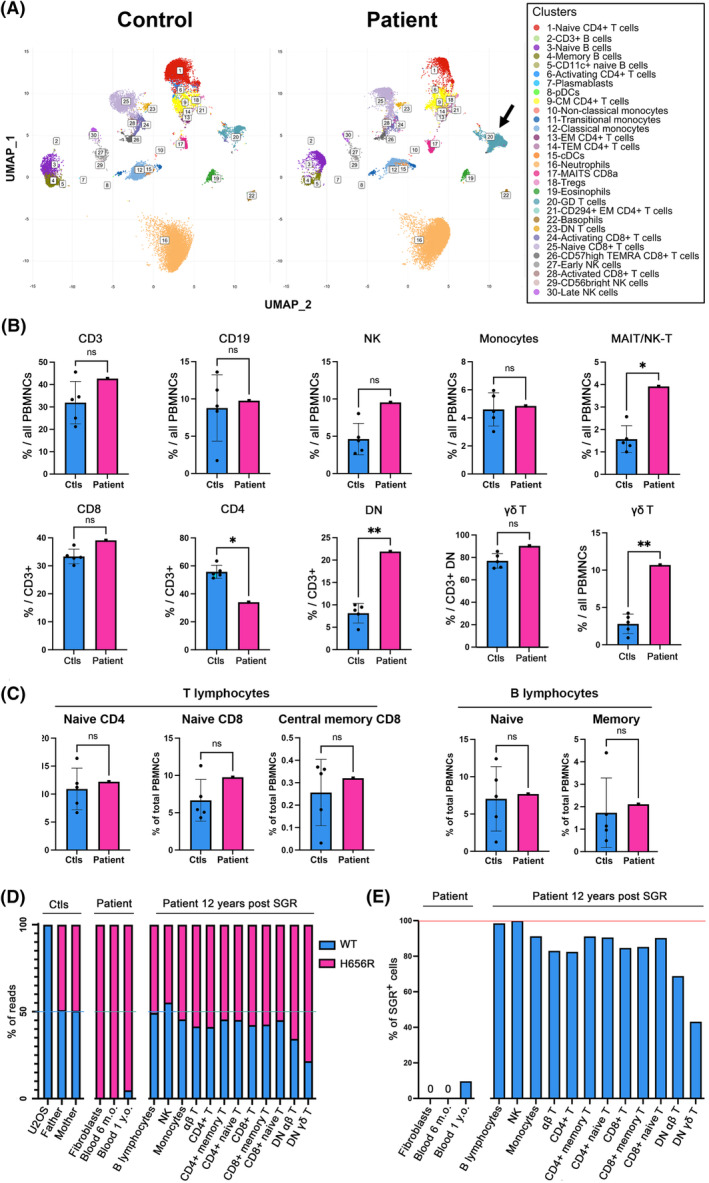
Immunological phenotype of patient's blood 12 years post‐somatic genetic rescue (SGR) and SGR detection by next‐generation sequencing (NGS). (A) Representative illustration of uniform manifold approximation and projection applied to analyse immune cell subsets from whole blood samples obtained in one age‐matched healthy control and the MYSM1‐deficient patient 12 years post‐SGR. Each cluster is visually represented by a distinct colour. The arrow highlights the T γδ cell population in patient. (B) Proportion of the immunological subsets obtained in blood from five age‐matched healthy controls and the patient 12 years post‐SGR. (C) Proportion of naive and memory B and T cell populations in the patient 12 years post‐SGR and 5 age‐matched healthy controls. (D) Graphic representation of the numbers of WT and MYSM1 c.1967A>G mutated reads obtained by NGS in the indicated cell populations. (E) Graphic representation of the estimated proportion of SGR+ cell subpopulations deduced from (D). PBMNCs, peripheral blood mononuclear cells. Statistical significance: *p < 0.05; **p < 0.01; ns, not significant.
Next, we tested whether the immunological populations detected 12 years post‐SGR originated from cells that preexisted to the SGR (i.e. homozygous for the H656R mutation) or from a genetically corrected progenitor (i.e. carrying a WT MYSM1 allele at a heterozygous state; denoted SGR+). PCR amplification and next‐generation sequencing of the MYSM1 c.1967A>G variant in U2OS, used as WT control, recovered more than 99.99% of WT reads (Figure 2D). In the patient's father's and mother's PBMNCs, 50.8% and 50.6% of the reads were WT, respectively, while the remaining reads contained the MYSM1 variant, congruent with a heterozygous status. The MYSM1 variant was present in more than 99.98% of reads in patient's fibroblasts, confirming the homozygous status of the MYSM1 variant in this tissue (Figure 2D). Thus, the accuracy and reliability of our approach allowed us to determine the proportion of WT and mutated MYSM1 alleles in a given cell population and estimate the percentage of cells that originated from an SGR+ progenitor (Figure S4). WT reads were absent in patient's blood DNA sampled at 6 months of age while they represented 4.8% of total reads at 1 year of age, suggesting that the SGR took place in the patient's haematopoietic compartment between 6 months and 1 year of age. Patient's B and NK lymphocytes, sorted 12 years post‐SGR (Figure S5), showed almost equal proportions of WT and c.1967A>G reads, indicating that they were virtually all derived from SGR+ progenitors. A slightly lower proportion of WT reads in monocytes suggested that ~9% of them did not originate from an SGR+ precursor (Figure 2C,D). Similarly, the mutant reads were more abundant than their WT counterpart in the various T lymphocyte populations. This indicated that a substantial fraction of T lymphocytes were homozygous for the MYSM1 variant (Figure 2C,D). Remarkably, DN γδ and DN αβ T cells had lower proportion of WT reads (21.6% and 31.4%, respectively) suggesting that only ~43% of DN γδ and ~63% of DN αβ T cells were SGR+ (Figure 2C,D). These results indicate that SGR+ cells almost completely replenished the patient's immune system and persisted over time with the exception of T cells, and particularly the DN T cells, where SGR‐ cells persist (Figure S6). It is unlikely that the SGR‐ DN γδ T population represents cells that would have expanded in response to a viral infection before the SGR occurrence since the TCR gd repertoire does not highlight any clonal expansion (Figure S2). We propose that the overrepresentation of SGR‐ DN γδ T cells underlines a minor role of MYSM1 in the development and maintenance of this cell population. The SGR‐mediated haematological reconstitution in the MYSM1‐deficient patient shares some features with haematopoietic stem cell transplantation (HSCT) and autologous gene therapy. As the manipulation of HSCs and the homeostatic proliferation associated with HSCT and gene therapy can promote CHIP, 7 , 8 , 9 we wondered whether the SGR‐mediated hemato‐immunological reconstitution in the patient might have favoured CHIP. Reassuringly, genetic analysis of 65 genes associated with CHIP (Table S2) did not detect any CHIP‐associated variant nor CNV in patient's blood.
In conclusion, our study reports the full recovery of a functional haematopoietic system that persisted over a period of 12 years due to an SGR in the context of inherited bone marrow failure syndrome, a phenomenon dubbed “natural gene therapy”. It is noteworthy that since the SGR occurred in the haematopoietic compartment, the non‐haematological manifestations of the disease are not affected by the SGR. However, whether the MYSM1 SGR occurred in a unique HSC or in a hemangioblast prior to its differentiation into HSCs is unknown. It would have been interesting to analyse the phylogenetic structure of haematopoietic clones to estimate the size of the SGR+ HSC pool at the origin of haematopoietic recovery. 7 However, such an analysis requires a bone marrow sample, which is not ethically justified given the good health of the patient. Finally, as the in vivo genome editing by mRNA/lipid Cas9 strategy in HSCs has recently been shown to be efficient in mice, 10 our results predict that in vivo gene therapy of haematopoietic diseases could be efficient even with few edited cells if the correction confers a strong selective advantage, as observed in MYSM1 deficiency. Hence, our study provides evidence that MYSM1 deficiency may be an ideal condition to test the efficiency of in vivo gene therapy by mRNA/lipid transfer into the HSC compartment.
AUTHOR CONTRIBUTIONS
S.d.T. carried out the next‐generation sequencing of the SGR in the sorted cell populations. E.M. performed cell sorting. Q.R. performed CyTOF analysis. L.K. and B.F. contributed to the initial phenotypic analysis of the patient. A.M. and F.R.‐L. obtained and prepared samples for CyTOF analysis. A.E. produced the recombinant proteins and performed the in vitro deubiquitinase assay. P.H. and F.D. performed genetic analysis of CHIP. A.T. performed the T cell repertoire analysis. D.M. followed up with the patient. A.F. provided intellectual input. P.R. conceived the project and wrote the manuscript with contributions from all authors.
FUNDING INFORMATION
This work has been supported by institutional grants from Institut national de la santé et de la recherche médicale (INSERM); Ligue Nationale contre le Cancer (Equipe Labellisée La Ligue ‘Ligue 2023’ to P.R.), Institut national du Cancer (INCa), the Agence National de la Recherche (grant nos. ANR‐18‐CE17‐0001 ‘Action’; and ANR‐22‐CE15‐0047‐02 ‘BREAK‐ITP’), the Fondation pour la recherche Médicale (FRM: EQU202103012670) and state funding from the Agence Nationale de la Recherche under ‘Investissements d'avenir’ programme (Institut Hospitalo‐Universitaire ANR‐10‐IAHU‐01, Recherche Hospitalo‐Universitaire, grant no. ANR‐18‐RHUS‐0010). This study contributes to the IdEx Université de Paris ANR‐18‐IDEX‐0001PR. Q.R. received an Institut Imagine MD‐PhD fellowship and a Société Nationale Française de Médecine Interne (SNFMI) fellowship. P.R. is a scientist from the Centre National de la Recherche Scientifique (CNRS).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
The study and protocols comply with the 1975 Declaration of Helsinki, as well as with the local legislation and ethical guidelines from the Comité de Protection des Personnes de l'ile de France and the French advisory committee on data processing in medical research. The INSERM Institutional Review Board also approved this study.
PATIENT CONSENT STATEMENT
Informed and written consent were obtained from healthy donors and the patient.
PERMISSION TO REPRODUCE MATERIAL FROM OTHER SOURCES
Not applicable.
CLINICAL TRIAL REGISTRATION (INCLUDING TRIAL NUMBER)
Not applicable.
Supporting information
Data S1.
ACKNOWLEDGEMENTS
The authors thank the patient and his family for their contribution to this study. We thank the staff from the bioinformatics and genomics core facility of the Imagine Institute for technical assistance as well as the CyPS platform from La Pitié‐Salpêtrière hospital and Sorbonne University.
de Tocqueville S, Martin E, Riller Q, Kermasson L, France B, Magérus A, et al. Long‐term assessment of haematological recovery following somatic genetic rescue in a MYSM1‐deficient patient: Implications for in vivo gene therapy. Br J Haematol. 2024;205(6):2349–2354. 10.1111/bjh.19744
Emmanuel Martin and Quentin Riller equal participation.
DATA AVAILABILITY STATEMENT
All data are available upon request to Patrick Revy.
REFERENCES
- 1. Revy P, Kannengiesser C, Fischer A. Somatic genetic rescue in Mendelian haematopoietic diseases. Nat Rev Genet. 2019;20:582–598. [DOI] [PubMed] [Google Scholar]
- 2. Mitchell E, Spencer Chapman M, Williams N, Dawson KJ, Mende N, Calderbank EF, et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature. 2022;606:343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fiore A, Liang Y, Lin YH, Tung J, Wang H, Langlais D, et al. Deubiquitinase MYSM1 in the hematopoietic system and beyond: a current review. Int J Mol Sci. 2020;21:3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao J, Jia Y, Mahmut D, Deik AA, Jeanfavre S, Clish CB, et al. Human hematopoietic stem cell vulnerability to ferroptosis. Cell. 2023;186:732–747. e716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Le Guen T, Touzot F, Andre‐Schmutz I, Lagresle‐Peyrou C, France B, Kermasson L, et al. An in vivo genetic reversion highlights the crucial role of Myb‐like, SWIRM, and MPN domains 1 (MYSM1) in human hematopoiesis and lymphocyte differentiation. J Allergy Clin Immunol. 2015;136:1619–1626. [DOI] [PubMed] [Google Scholar]
- 6. Petrov JC, Nijnik A. MYSM1 expression in the bone marrow niche is not essential for hematopoietic maintenance. Exp Hematol. 2017;47:76–82. [DOI] [PubMed] [Google Scholar]
- 7. Campbell P, Chapman MS, Wilk M, Boettcher S, Mitchell E, Dawson K, et al. Clonal dynamics after allogeneic haematopoietic cell transplantation using genome‐wide somatic mutations. Durham, NC: Research Square; 2023. Accessed April 28, 2023. https://www.researchsquare.com/article/rs‐2868644/v1 [Google Scholar]
- 8. Fabre MA, de Almeida JG, Fiorillo E, Mitchell E, Damaskou A, Rak J, et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature. 2022;606:335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. White SL, Lee TD, Toy T, Carroll JE, Polsky L, Campo Fernandez B, et al. Evaluation of clonal hematopoiesis in pediatric ADA‐SCID gene therapy participants. Blood Adv. 2022;6:5732–5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Breda L, Papp TE, Triebwasser MP, Yadegari A, Fedorky MT, Tanaka N, et al. In vivo hematopoietic stem cell modification by mRNA delivery. Science. 2023;381:436–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
All data are available upon request to Patrick Revy.