

Summary
The role of imatinib in PDGFRA/B‐negative hypereosinophilic syndromes (HES) is controversial because of the heterogeneity of HES and the scarcity of prospective studies. We conducted a phase II clinical trial to evaluate the efficacy of imatinib in PDGFRA/B‐negative HES. Thirty‐two patients were treated with imatinib (100–400 mg daily), and the molecular basis of their response was identified using whole‐exome sequencing (WES) and whole‐transcriptome sequencing (WTS). The haematological response rate was 46.9%, with a complete haematological response (CHR) rate of 18.8%. The median time to response was 1.5 months. Among the six patients who achieved CHR, five maintained it until the 24th cycle of imatinib and one lost response after 20 months. The median progression‐free survival was 4.3 months. WES and WTS were conducted for 11 patients. The number of non‐silent mutations did not differ between responders and non‐responders. Nine differentially expressed genes, including SNORD15A, were downregulated in responders. STAT5B::RARA, PAK2::PIGX, and FIP1L1::CHIC2 fusions were identified in patients with sustained responses, and RNF130::BRAF and WNK1::KDM5A fusions were identified in non‐responders. Imatinib, along with an appropriate biomarker, could be a promising option for PDGFRA/B‐negative HES.
Keywords: hypereosinophilic syndrome, imatinib, PDGFRA/B‐negative, whole‐exome sequencing, whole‐transcriptome sequencing
INTRODUCTION
Hypereosinophilic syndromes (HES) encompass a diverse group of disorders characterized by elevated eosinophil levels and subsequent organ damage. 1 , 2 Reactive HES, caused by secondary causes, is common; after excluding this, it is important to identify primary HES with clonality. 3 With advancements in molecular diagnostic techniques, the World Health Organization and International Consensus Classification recently revised the classification systems for eosinophilic diseases. Myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusion arise from rearrangements in genes encoding tyrosine kinases such as PDGFRA, PDGFRB, and FGFR1. 4 To date, dozens of different fusion genes have been reported. 5 Identifying fusion genes is crucial in HES, as tyrosine kinase inhibitors (TKIs) are active in these cases, with imatinib proving particularly effective in patients harbouring PDGFRA or PDGFRB fusion genes. 6 , 7 , 8 , 9 In cases of idiopathic HES, where clonality and other primary causes have been excluded, corticosteroids remain the mainstay of therapy and are effective; however, resistance and side effects become problematic with long‐term use. 10 , 11 Recently, mepolizumab, a biological agent targeting interleukin‐5 (IL‐5), demonstrated efficacy in idiopathic HES. 12 , 13 , 14
Studies on the efficacy of imatinib in PDGFRA/B‐negative HES have reported response rates ranging from 9% to 60%. 15 , 16 , 17 , 18 , 19 The heterogeneous patient populations in retrospective studies may have contributed to these inconsistent results, highlighting the need for prospective studies to confirm the role of imatinib in these populations. Furthermore, patients who respond well to imatinib imply the presence of undiscovered imatinib‐sensitive genetic alterations beyond the PDGFRA/B rearrangements. Therefore, we conducted a phase II study to evaluate the efficacy and safety of imatinib in patients with PDGFRA/B‐negative HES. Additionally, we investigated the molecular characteristics of patients using whole‐exome sequencing (WES) and whole‐transcriptome sequencing (WTS).
MATERIALS AND METHODS
Study design and participants
This investigator‐initiated, open‐label, multicentre, single‐arm, prospective phase II study was conducted at three hospitals in South Korea between January 2014 and December 2020. Patients aged 20 years or older with HES were eligible if they tested negative for BCR::ABL1, FGFR1, and PDGFRB rearrangement, as well as for CHIC2 gene (located on chromosome 4q12) deletion by fluorescence in situ hybridization or RT‐PCR on bone marrow (BM). HES was defined and diagnosed based on the end‐organ manifestations that could be directly related to absolute eosinophil count (AEC) of more than 1.5 × 109/L on two or more occasions and/or tissue eosinophilia without a secondary cause. 20 All reactive causes of eosinophilia were carefully excluded prior to study enrollment. Patients who received hydroxyurea or interferon treatment within 4 weeks prior to enrollment were also excluded. Corticosteroid co‐administration was allowed if the daily dose was 15 mg/day or lower of prednisolone.
Study protocol
Imatinib mesylate was administered as a single dose, starting with 100 mg daily for the first week. Thereafter, the daily dose was increased to 400 mg at the investigator's discretion if a haematological response was not achieved. If the patient exhibited steroid dependency (requiring a concurrent prednisolone equivalent dose of >15 mg/day), they were considered non‐responsive to imatinib, and the patient was withdrawn from the study. If deemed beneficial for the patient, imatinib could be continued for a maximum of 24 cycles, with each cycle lasting 28 days. Dose modification of imatinib was performed based on the worst grade of toxicity, according to the approved protocol (Supporting Information, Methods).
Endpoints and assessment
The primary endpoint was the overall haematological response (OHR) rate. OHR was defined as the sum of the complete haematological response (CHR) and partial haematological response (PHR) rates. CHR was defined as the normalization of white blood cell counts with AEC of less than 0.7 × 109/L and the disappearance of signs of organ involvement or symptoms for ≥4 weeks. PHR was defined as a decrease in the AEC ≥50% and/or symptomatic improvement. The secondary endpoints included progression‐free survival (PFS), toxicities, and genetic characteristics related to the response. PFS was defined as the time from imatinib administration to the confirmation of no response, disease progression, or death from any cause, and the data were censored at the most recent disease assessment. Adverse events (AEs) were assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03.
WES and WTS were conducted to investigate the genetic characteristics associated with the haematological response to imatinib. These tests were exclusively performed on the 11 patients who consented to the test, and blood sampling for the examination occurred prior to the initiation of imatinib. The procedures for WES and WTS are detailed in Supporting Information, Methods section.
Statistical analysis
The sample size was calculated using Simon's optimal two‐stage design. We expected a 20% OHR rate for imatinib. With a power of 80% and an alpha of 5%, 10 patients were required for the first stage. If one or more patients showed a response in the first stage, the study progressed to the second stage, which required 29 patients. Four or more responses are required for this drug to be evaluated in further studies. Considering a 10% dropout rate, 32 patients were required.
Categorical variables were compared between the two groups using Fisher's exact test or the Chi‐square test, and p‐values were two‐sided. Differences in the means or medians of the continuous variables between the two groups were assessed using the Wilcoxon rank‐sum test. Survival curves were estimated using the Kaplan–Meier method. The statistical software “R” version 4.3.1 (www.r‐project.org) was used for all statistical analyses. p‐values of <0.05 were considered statistically significant.
RESULTS
Patient and disease characteristics
A total of 32 patients were enrolled; their baseline characteristics are shown in Table 1. The median age was 50 years (range, 20–74), and 20 patients (62.5%) were men. Serum vitamin B12 and IgE levels were measured in 14 and 18 patients respectively, and were elevated in 5 and 12 patients, respectively. The median AEC and BM eosinophil % were 2.89 × 109/L (range, 1.57–13.9) and 18.4% (range, 5.2–54.9), respectively. BM findings were consistent with HES in all patients, with no evidence of mast cell infiltration, significant fibrosis, or associated haematological malignancy.
TABLE 1.
Baseline characteristics of study patients, according to haematological response.
| All patients N = 32 | Responder N = 15 | Non‐responder N = 17 | p‐Value | |
|---|---|---|---|---|
| Median age, years (range) | 50 (20–74) | 52 (20–74) | 49 (23–67) | 0.655 |
| Age >60 years, n (%) | 8 (25.0) | 4 (26.7) | 4 (23.5) | 1.000 |
| Sex, n (%) | 0.170 | |||
| Male | 20 (62.5) | 7 (46.7) | 13 (76.5) | |
| Female | 12 (37.5) | 8 (53.3) | 4 (23.5) | |
| Body mass index, median (range) | 23.7 (17.1–36.6) | 23.9 (18.9–36.6) | 23.4 (17.1–28.8) | 0.852 |
| History of eosinophilia, months, median (range) | 10.7 (2.3–183.9) | 7.8 (2.4–72.1) | 20.5 (2.3–183.9) | 0.213 |
| <6 months, n (%) | 7 (21.9) | 3 (20.0) | 4 (23.5) | 1.000 |
| 6 months to <1 year, n (%) | 11 (34.4) | 7 (46.7) | 4 (23.5) | 0.316 |
| ≥1 year, n (%) | 14 (43.8) | 5 (33.3) | 9 (52.9) | 0.448 |
| Previous treatment, n (%) | ||||
| Cyclophosphamide | 2 (6.2) | 1 (6.7) | 1 (5.9) | 1.000 |
| Hydroxyurea | 7 (21.9) | 4 (26.7) | 3 (17.6) | 0.851 |
| Corticosteroid | 31 (96.9) | 14 (93.3) | 17 (100) | 0.949 |
| Clinical manifestation, n (%) | ||||
| Heart | 2 (6.2) | 1 (6.7) | 1 (5.9) | 1.000 |
| Lung | 12 (37.5) | 5 (33.3) | 7 (41.2) | 0.927 |
| Liver | 8 (25.0) | 4 (26.7) | 4 (23.5) | 1.000 |
| Skin | 12 (37.5) | 6 (40.0) | 6 (35.3) | 1.000 |
| Lymph node enlargement | 3 (9.4) | 2 (13.3) | 1 (5.9) | 0.909 |
| Intestine | 7 (21.9) | 2 (13.3) | 5 (29.4) | 0.503 |
| Nerve | 3 (9.4) | 2 (13.3) | 1 (5.9) | 0.909 |
| Peripheral blood cell count, median (range) | ||||
| Haemoglobin, g/dL | 13.6 (10.1–16.3) | 13.0 (10.1–15.4) | 14.5 (10.6–16.3) | 0.209 |
| Platelet, ×109/L | 269 (107–493) | 234 (161–472) | 298 (107–493) | 0.051 |
| White blood cell, ×109/L | 12.0 (5.6–21.4) | 10.4 (5.6–21.4) | 12.9 (9.0–20.6) | 0.179 |
| Absolute neutrophil count | 4.56 (1.63–11.33) | 4.24 (1.63–7.35) | 4.98 (2.05–11.33) | 0.137 |
| Absolute lymphocyte count | 2.28 (0.41–4.71) | 2.00 (1.03–4.71) | 2.33 (0.41–3.92) | 0.950 |
| Absolute eosinophil count | 2.89 (1.57–13.90) | 3.00 (1.57–13.90) | 2.59 (1.60–10.76) | 1.000 |
| Conventional cytogenetics, n (%) | ||||
| Normal | 25 (78.1) | 11 (73.3) | 14 (82.4) | 0.804 |
| Y deletion | 2 (6.2) | 1 (6.7) | 1 (5.9) | |
| Not assessed | 5 (15.6) | 3 (20.0) | 2 (11.8) | |
| BM M:E ratio, median (range) | 3.6 (1.3–17.9) | 4.0 (1.9–14.0) | 3.4 (1.3–17.9) | 0.827 |
| BM blast %, median (range) | 0.6 (0–2.5) | 0.7 (0–2.0) | 0.4 (0–2.5) | 0.183 |
| BM eosinophil %, median (range) | 18.4 (5.2–54.9) | 15.6 (5.2–54.9) | 26.8 (10.3–52.7) | 0.038 |
| BM cellularity, n (%) | 0.497 | |||
| Hypercellular | 1 (3.1) | 0 (0) | 1 (5.9) | |
| Normocellular | 24 (75.0) | 13 (86.7) | 11 (64.7) | |
| Hypocellular | 4 (12.5) | 1 (6.7) | 3 (17.6) | |
| Not assessed | 3 (9.4) | 1 (6.7) | 2 (11.8) | |
Abbreviations: BM, bone marrow; HES, hypereosinophilic syndrome; M:E ratio, myeloid:erythroid ratio.
Efficacy
A response evaluation was conducted for all 32 patients. Based on the best response during the study, the CHR rate was 18.8% (6/32). The PHR rate was 28.1% (9/32), resulting in an OHR rate of 46.9% (95% CI, 29.1–65.3), with the median time to response was 1.5 months (range, 0.5–4.9) (Figure 1A). Among the six patients who achieved CHR, the median time to CHR was 1.75 months (range, 1–11), with considerable variability among the patients. No differences in the baseline characteristics between responders and non‐responders were observed, except for the BM eosinophil %, which was lower in the responders (median, 15.6% vs. 26.8%, p = 0.038). Among the six patients who achieved CHR, all but one maintained CHR until the 24th cycle of imatinib; one patient lost response after 20 months of imatinib treatment. Of the five patients who sustained CHR up to the 24th cycle, three continued to maintain their CHR without additional treatment following imatinib discontinuation. Among the nine patients who achieved PHR, two lost their response at 8 and 18 months after imatinib treatment. Of the five patients who maintained PHR through the 24th cycle, three continued to sustain their response. The median PFS was 4.3 months (95% CI, 3.0–not reached), and the 6‐month PFS rate was 45.7% (95% CI, 31.2–67.1) (Figure 1B). No patient died during the study.
FIGURE 1.
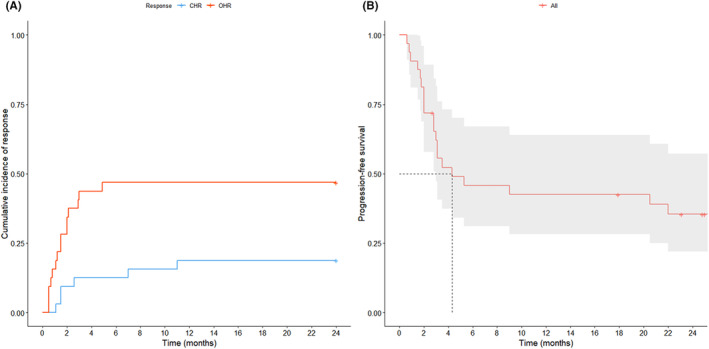
Clinical outcome of imatinib mesylate. (A) Cumulative incidence of haematological response. (B) Progression‐free survival. CHR, complete haematological response.
The dose of imatinib for responders and non‐responders was increased similarly in a stepwise manner (Figure 2; Table S1). However, the treatment duration was shorter for non‐responders, as imatinib was discontinued earlier in cases of no response. Of the 15 responders, 10 increased to the full dose (400 mg/day), and four experienced dose reduction. Twelve patients (37.5%) received concomitant corticosteroids at least once; six were non‐responders, and six were responders (including one who achieved CHR). The patient who achieved CHR initially started with prednisolone 15 mg/day, then reduced to 5 mg/day at imatinib cycle 2, and subsequently maintained at 2.5 mg/day while sustaining CHR. Ten patients (31.2%) completed the treatment for up to 24 cycles. The reasons for end‐of‐trial in the remaining 22 patients were as follows: 16 (72.7%) due to no response, 3 (13.6%) due to loss of response, 2 (9.1%) due to withdrawal of consent, and 1 (4.5%) due to an AE.
FIGURE 2.

Kinetics of the absolute eosinophil count (AEC) and dose–response relationship during 24 cycles of imatinib mesylate and oral corticosteroid (OCS) based on the response. Data points are the mean and standard deviation. (A) AEC (all patients). (B) Imatinib dose (all patients). (C) Oral prednisolone dose (12 patient who received OCS).
Toxicity
AEs occurred in 22 (68.8%) patients, with grades 1 or 2 accounting for 59.4% and grades 3 or 4 accounting for 9.4% (Table 2). The most frequently reported AEs were skin rash, myalgia or muscle cramps, and oedema, all of which were grade 1 or 2. haematological toxicity was rare, with only two cases of neutropenia: one grade 2 and one grade 3. Imatinib was abandoned in one patient because of a grade 3 complete atrioventricular block.
TABLE 2.
Summary of adverse event.
| Adverse events | Grade 1–2 | Grade 3–4 | Total |
|---|---|---|---|
| Haematological toxicity, n (%) | |||
| Neutropenia | 0 | 2 (6.2) | 2 (6.2) |
| Non‐haematological toxicity, n (%) | |||
| Oedema | 7 (21.9) | 0 | 7 (21.9) |
| Skin rash | 7 (21.9) | 0 | 7 (21.9) |
| Myalgia/muscle cramps | 7 (21.9) | 0 | 7 (21.9) |
| Abdominal pain | 6 (18.8) | 0 | 6 (18.8) |
| Nausea/vomiting | 4 (12.5) | 0 | 4 (12.5) |
| Fatigue | 4 (12.5) | 0 | 4 (12.5) |
| Infections | 3 (9.4) | 0 | 3 (9.4) |
| Headache | 2 (6.2) | 0 | 2 (6.2) |
| Alopecia | 2 (6.2) | 0 | 2 (6.2) |
| Anxiety | 2 (6.2) | 0 | 2 (6.2) |
| Diarrhoea/constipation | 1 (3.1) | 0 | 1 (3.1) |
| Chest pain | 1 (3.1) | 0 | 1 (3.1) |
| Pleural effusion | 0 | 1 (3.1) | 1 (3.1) |
| Peripheral neuropathy | 1 (3.1) | 0 | 1 (3.1) |
| Blurred vision | 1 (3.1) | 0 | 1 (3.1) |
| Complete atrioventricular block | 0 | 1 (3.1) | 1 (3.1) |
WES and WTS analysis
WES and WTS were performed on 11 patients (five responders and six non‐responders). WES datasets were analysed to explore the mutational profiles of the responders and non‐responders. The median coverage of depth was a median of 159.9X (137.6–239.7X) for HES and 148.1X (107.2–175.6X) for matched normal genomes (Table S2). A total of 121 somatic mutations (113 single‐nucleotide variants and eight indels) were identified in the HES genome (Table S3). There were no significant differences in the number of non‐silent mutations between responders (median of eight mutations, range 3–11) and non‐responders (median of six mutations, range 3–10) (Figure 3A). Mutations in PCDHB13 and SIGLEC16 were recurrently detected in non‐responders (two of six, 33%), whereas mutations in DSPP and HRNR were recurrently detected in responders (two of five, 40%).
FIGURE 3.

Genetic characteristics related to the response. (A) Whole‐exome sequencing analysis between responders and non‐responders. (B) Heatmap of differentially expressed genes between responders and non‐responders.
Transcriptome analysis was conducted using 11 WTS datasets. The heatmap of the differentially expressed genes (DEGs) showed a discrepancy bewteen the responders and non‐responders (Figure 3B). A total of 12 genes was deregulated in the responder group compared with the non‐responder group (adjusted p‐value <0.05, log2 fold change >1); three genes were relatively upregulated, and nine genes were relatively downregulated in the responders (Table 3). DEGs upregulated in responders were enriched for the hallmark heme metabolism‐related pathway in GESA (adjusted p‐value <0.05, normalized enrichment score >1.3). In addition, we identified seven fusion transcripts reported in the fusion database, 21 but none of the PDGFRA fusion transcripts were observed. STAT5B::RARA fusion was identified in one patient with PHR, and PAK2::PIGX and FIP1L1::CHIC2 fusions were identified in one patient with CHR. Both patients maintained their responses throughout the study. RNF130::BRAF and WNK1::KDM5A fusion were identified in the non‐responder group.
TABLE 3.
Results of RNA sequencing analysis.
| Dysregulated genes in the responders as compared with the non‐responders | |||
|---|---|---|---|
| Gene | Log2 fold change | p‐Value | FDR |
| Upregulated (N = 3) | |||
| ABC7‐43041300I9.1 | 22.384 | <0.001 | <0.001 |
| RP11‐734I18.1 | 4.924 | <0.001 | 0.008 |
| RFPL4AL1 | 6.400 | <0.001 | 0.020 |
| Downregulated (N = 9) | |||
| RP11‐34P13.15 | −25.496 | <0.001 | <0.001 |
| SNORD15A | −23.347 | <0.001 | <0.001 |
| AC132186.1 | −21.718 | <0.001 | <0.001 |
| PMS2P2 | −1.697 | <0.001 | 0.006 |
| HOXA2 | −2.033 | <0.001 | 0.007 |
| RNF17 | −6.538 | <0.001 | 0.013 |
| GTF2IRD2P1 | −4.600 | <0.001 | 0.013 |
| SNORA53 | −5.915 | <0.001 | 0.020 |
| DKK3 | −3.165 | <0.001 | 0.028 |
| List of fusion transcripts identified by whole‐transcriptome sequencing | ||||||||
|---|---|---|---|---|---|---|---|---|
| Gene1 | Gene2 | chr1 | Breakpoint1 | str1 | chr2 | Breakpoint2 | str2 | Fusion type |
| Responder | ||||||||
| RARA | STAT5B | 17 | 38 474 700 | + | 17 | 40 369 482 | − | UTR/CDS (truncated) |
| PIGX | PAK2 | 3 | 196 731 093 | + | 3 | 196 782 627 | + | Intra‐chromosomal |
| CHIC2 | FIP1L1 | 4 | 54 930 349 | − | 4 | 54 292 039 | + | In‐frame |
| Non‐responder | ||||||||
| PIGX | PAK2 | 3 | 196 731 093 | + | 3 | 196 782 627 | + | Intra‐chromosomal |
| WNK1 | KDM5A | 12 | 863 490 | + | 12 | 495 140 | − | In‐frame |
| BPTF | PITPNC1 | 17 | 67 875 019 | + | 17 | 67 578 185 | + | Intra‐chromosomal |
| RNF130 | BRAF | 5 | 179 440 061 | − | 7 | 140 508 795 | − | In‐frame |
Abbreviations: CDS, coding sequence; chr, chromosome; FDR, false discovery rate; str, strand; UTR, untranslated region.
DISCUSSION
In this prospective phase II trial, we present data on the efficacy of imatinib mesylate in 32 patients with PDGFRA/B‐negative HES, along with genetic information obtained through WES and WTS. The haematological response rate to imatinib was 46.9%, with a CHR rate of 18.8%, and the response was stable. Acceptable and manageable toxicity profiles were observed.
In PDGFRA/B‐positive HES, the CHR to imatinib is well‐established at 99%, demonstrating its effectiveness. 19 , 22 , 23 Following the report by Cools et al. that imatinib may also be effective in patients with PDGFRA/B‐negative HES, 24 subsequent studies have evaluated the efficacy of imatinib in this population; however, the response rates varied. Two previous prospective phase II trials of imatinib included 15 and 8 patients with PDGFRA/B‐negative HES, with haematological response rates of 40% and 50%, respectively. 17 , 18 In contrast, in a prospective study involving 18 patients with PDGFRA‐negative HES, 16 and in another prospective study involving 36 patients with PDGFRA/B‐negative HES, 19 the haematological response rates were 16% and 25%, respectively. This discrepancy among studies is likely attributable to the relatively small sample size and heterogeneous patient selection. Therefore, this study aimed to evaluate the efficacy of imatinib in a sufficient number of patients. The median time to haematological response was 1.5 months, indicating a prompt response, which is in line with previous studies. 17 , 18 , 19 Furthermore, 12 of the 15 responders maintained their response, indicating that sustained response, in addition to rapid response, can be expected in a significant proportion of responders.
The rapid and durable response observed in some patients with PDGFRA/B‐negative HES suggests the presence of unknown imatinib‐sensitive genetic alterations. Although we excluded CHIC2 deletion and PDGFRB rearrangements, the possibility of other fusion partners with PDGFRA remains, as several have been reported. Curtis et al. also reported the discovery of two novel fusion genes (STRN::PDGFRA fusion for t(2;4) and ETV6::PDGFRA for t(4;12)). 25 Our data suggest several fusion genes that might be associated with the response to imatinib. The STAT5B::RARA fusion gene, identified in one of our imatinib responders, has been previously reported in patients with acute promyelocytic leukaemia. 26 It has been reported that the STAT5B::RARA fusion can lead to the activation of the STAT5B transcription factor. 27 Imatinib has been reported to have an inhibitory effect on the phosphorylation of STAT5, suggesting that the STAT5 pathway might be a potential target for imatinib. 28 The RNF130::BRAF fusion induces constitutive activation of the kinase domain of BRAF. 29 As a result, the downstream MAPK signalling pathway is aberrantly activated, potentially diminishing the efficacy of imatinib. 30 , 31 , 32 In addition, several studies have reported that when WNK1 is fused with other genes, it increases the expression of the fused gene. 33 , 34 KDM5A is associated with the resistance to TKIs. 35 , 36 Therefore, the WNK1::KDM5A fusion observed in a non‐responder in our study might have contributed to the non‐responsiveness to imatinib. In our study, the downregulation of nine genes, including SNORD15A, and the upregulation of three genes were associated with a favourable response to imatinib. Fujita et al. reported the potential involvement of small nucleolar RNAs, including SNORD17 and SNORD15A, in telomere biology. 37 Considering the dose‐dependent inhibition of telomerase activity by imatinib, our study aligns with previous reports, suggesting that the inhibition of cell proliferation by suppressing telomerase activity, not mediated by tyrosine kinases, could be another mode of action of imatinib. 38 , 39 Further research is required regarding the perspective of fusion analysis through RNA sequencing.
Recently, given the pivotal role of IL‐5 in eosinophil activation, biological agents targeting IL‐5 or its receptor, such as mepolizumab and benralizumab, have become the focus of increased investigation. 40 , 41 Although the most appropriate dose and predictive biomarkers for response have not yet been clearly identified, mepolizumab has significantly reduced the onset and risk of flares in PDGFRA‐negative HES while maintaining manageable AEs. 12 , 42 , 43 Benralizumab has also demonstrated efficacy in reducing AEC in PDGFRA‐negative HES, and a phase III trial is currently underway. 44 Therefore, in PDGFRA/B‐negative HES, imatinib should be considered with appropriate patient selection. Unlike monoclonal antibodies such as mepolizumab, which target cell surface antigens, imatinib, as a TKI, targets the underlying disease mechanism. Thus, imatinib can be effective when disease progression is associated with the pathways it modulates. However, there is a scarcity of research on genetic alterations associated with imatinib response in PDGFRA/B‐negative HES, and detailed genomic analyses beyond PDGFR fusion are indeed limited in clinical settings. Therefore, predicting the imatinib response based on clinical characteristics can be beneficial. In our study, no differences in baseline characteristics, including AEC, were observed between responders and non‐responders, which is consistent with the findings of previous studies. 18 , 19 However, responders had a significantly lower % of BM eosinophils. Whether the difference in BM eosinophil levels is a result of genetic alterations related to the imatinib response remains unclear. Khoury et al. reported that features of primary myeloid HES might serve as surrogate markers for imatinib response in PDGFRA‐negative HES. 45 Further research on predictive biomarkers is warranted. On the other hand, considering that most responders achieved rapid response, a short imatinib trial may easily identify patients who would benefit from imatinib. Intermesoli et al. also reported that a short trial of imatinib (100 mg/day for 2 weeks) can rapidly confirm imatinib‐sensitive HES. 15
This study has several limitations. First, the sample sizes used in WES and WTS were small. This could have resulted in low statistical power. Furthermore, due to limited sample availability, independent assays for the identified fusion genes could not be conducted; therefore, information regarding their functional relevance is missing. Second, pathological confirmation through tissue biopsy was not performed in the assessment of haematological response.
Despite these limitations, our study is the first prospective trial of imatinib in patients with PDGFRA/B‐negative HES. Our study addresses the long‐standing problem that retrospective studies with a small number of patients may not adequately evaluate the efficacy of imatinib. Furthermore, we identified several DEGs and fusion transcripts that may be associated with the response to imatinib. Given the rarity of genomic analyses related to the imatinib response in PDGFRA/B‐negative HES, the present study holds value as it suggests further research on potential genomic biomarkers.
In conclusion, imatinib demonstrated favourable outcomes in some patients with PDGFRA/B‐negative HES. The responses were rapid and sustained among the responders, with good compliance. Several DEGs and fusion transcripts have been suggested to be potentially associated with the imatinib response. Further comprehensive investigations with larger sample sizes are required to validate the predictive genetic biomarkers for imatinib in PDGFRA/B‐negative HES.
AUTHOR CONTRIBUTIONS
Dong Hyun Kim: Data curation, formal analysis, investigation, writing—original draft, writing—review and editing. Seokhyeon Kim: Formal analysis, investigation, writing—original draft. Seonyang Park, Ja Min Byun, Junshik Hong, Dong‐Yeop Shin, Inho Kim, Soo Mee Bang, Jeong‐Ok Lee, Ji Yun Lee, Sang‐A Kim, Ki Hwan Kim: Investigation, writing—review and editing. Yeun‐Jun Chung: Methodology, writing—review and editing. Seung‐Hyun Jung: Methodology, supervision, writing—original draft. Youngil Koh, Sung‐Soo Yoon: Conceptualization, investigation, supervision, writing—review and editing. All the authors checked and approved the final version of the manuscript.
FUNDING INFORMATION
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (Ministry of Science, ICT, and Future Planning) (grant number NRF‐2021R1A4A2001553). Imatinib mesylate was provided free of charge by Boryung Pharma (Korea). This study is an investigator‐sponsored trial, and the study was conducted independently of the funder. The funder had no role in study design, data collection, data analysis, or data interpretation.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
ETHICS STATEMENT
This trial was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice Guidelines, and National Policies on Bioethics and Human Biological Specimens. The study protocol was approved by the Institutional Review Board of each participating institution.
PATIENT CONSENT STATEMENT
Written informed consent was obtained from all patients before their participation.
CLINICAL TRIAL REGISTRATION (INCLUDING TRIAL NUMBER)
This trial was approved and registered with the Korean Ministry of Food and Drug Safety (No. 20130196318).
Supporting information
Data S1.
ACKNOWLEDGEMENTS
We thank the participating patients and their families, all study coinvestigators, and research coordinators. We also appreciate the support of the Basic Medical Science Facilitation Program through the Catholic Medical Center of the Catholic University of Korea, funded by the Catholic Education Foundation.
Kim DH, Kim S, Park S, Byun JM, Hong J, Shin D‐Y, et al. Phase II trial of imatinib mesylate in patients with PDGFRA/B‐negative hypereosinophilic syndrome. Br J Haematol. 2024;205(6):2305–2314. 10.1111/bjh.19828
Dong Hyun Kim and Seokhyeon Kim contribute equally.
Contributor Information
Seung‐Hyun Jung, Email: hyun@catholic.ac.kr.
Youngil Koh, Email: go01@snu.ac.kr.
Sung‐Soo Yoon, Email: ssysmc@snu.ac.kr.
DATA AVAILABILITY STATEMENT
The datasets generated during the current study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Valent P, Klion AD, Roufosse F, Simon D, Metzgeroth G, Leiferman KM, et al. Proposed refined diagnostic criteria and classification of eosinophil disorders and related syndromes. Allergy. 2023;78(1):47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang SA, Orazi A, Gotlib J, Reiter A, Tzankov A, Hasserjian RP, et al. The international consensus classification of eosinophilic disorders and systemic mastocytosis. Am J Hematol. 2023;98(8):1286–1306. [DOI] [PubMed] [Google Scholar]
- 3. Caminati M, Brussino L, Carlucci M, Carlucci P, Carpagnano LF, Caruso C, et al. Managing patients with hypereosinophilic syndrome: a statement from the Italian Society of Allergy, Asthma, and Clinical Immunology (SIAAIC). Cells. 2024;13(14):1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shomali W, Gotlib J. World Health Organization and international consensus classification of eosinophilic disorders: 2024 update on diagnosis, risk stratification, and management. Am J Hematol. 2024;99(5):946–968. [DOI] [PubMed] [Google Scholar]
- 5. Metzgeroth G, Steiner L, Naumann N, Lübke J, Kreil S, Fabarius A, et al. Myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions: reevaluation of the defining characteristics in a registry‐based cohort. Leukemia. 2023;37(9):1860–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gleich GJ, Leiferman KM, Pardanani A, Tefferi A, Butterfield JH. Treatment of hypereosinophilic syndrome with imatinib mesilate. Lancet. 2002;359(9317):1577–1578. [DOI] [PubMed] [Google Scholar]
- 7. Metzgeroth G, Schwaab J, Naumann N, Jawhar M, Haferlach T, Fabarius A, et al. Treatment‐free remission in FIP1L1‐PDGFRA–positive myeloid/lymphoid neoplasms with eosinophilia after imatinib discontinuation. Blood Adv. 2020;4(3):440–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jawhar M, Naumann N, Schwaab J, Baurmann H, Casper J, Dang TA, et al. Imatinib in myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRB in chronic or blast phase. Ann Hematol. 2017;96:1463–1470. [DOI] [PubMed] [Google Scholar]
- 9. Cheah CY, Burbury K, Apperley JF, Huguet F, Pitini V, Gardembas M, et al. Patients with myeloid malignancies bearing PDGFRB fusion genes achieve durable long‐term remissions with imatinib. Blood. 2014;123(23):3574–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cogan E, Roufosse F. Clinical management of the hypereosinophilic syndromes. Expert Rev Hematol. 2012;5(3):275–290. [DOI] [PubMed] [Google Scholar]
- 11. Klion AD. How I treat hypereosinophilic syndromes. Blood. 2015;126(9):1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roufosse F, Kahn J‐E, Rothenberg ME, Wardlaw AJ, Klion AD, Kirby SY, et al. Efficacy and safety of mepolizumab in hypereosinophilic syndrome: a phase III, randomized, placebo‐controlled trial. J Allergy Clin Immunol. 2020;146(6):1397–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuang FL, Fay MP, Ware J, Wetzler L, Holland‐Thomas N, Brown T, et al. Long‐term clinical outcomes of high‐dose mepolizumab treatment for hypereosinophilic syndrome. J Allergy Clin Immunol Pract. 2018;6(5):1518–1527. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gleich GJ, Roufosse F, Chupp G, Faguer S, Walz B, Reiter A, et al. Safety and efficacy of mepolizumab in hypereosinophilic syndrome: an open‐label extension study. J Allergy Clin Immunol Pract. 2021;9(12):4431–4440. e1. [DOI] [PubMed] [Google Scholar]
- 15. Intermesoli T, Delaini F, Acerboni S, Salmoiraghi S, Spinelli O, Guerini V, et al. A short low‐dose imatinib trial allows rapid identification of responsive patients in hypereosinophilic syndromes. Br J Haematol. 2009;147(5):681–685. [DOI] [PubMed] [Google Scholar]
- 16. Jain N, Cortes J, Quintás‐Cardama A, Manshouri T, Luthra R, Garcia‐Manero G, et al. Imatinib has limited therapeutic activity for hypereosinophilic syndrome patients with unknown or negative PDGFRα mutation status. Leuk Res. 2009;33(6):837–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Helbig G, Hus M, Hałasz M, Dudziński M, Więcławek A, Stachowicz M, et al. Imatinib mesylate may induce long‐term clinical response in FIP1L1‐PDGFRα‐negative hypereosinophilic syndrome. Med Oncol. 2012;29:1073–1076. [DOI] [PubMed] [Google Scholar]
- 18. Metzgeroth G, Walz C, Erben P, Popp H, Schmitt‐Graeff A, Haferlach C, et al. Safety and efficacy of imatinib in chronic eosinophilic leukaemia and hypereosinophilic syndrome–a phase‐II study. Br J Haematol. 2008;143(5):707–715. [DOI] [PubMed] [Google Scholar]
- 19. Baccarani M, Cilloni D, Rondoni M, Ottaviani E, Messa F, Merante S, et al. The efficacy of imatinib mesylate in patients with FIP1L1‐PDGFRα‐positive hypereosinophilic syndrome. Results of a multicenter prospective study. Haematologica. 2007;92(9):1173–1179. [DOI] [PubMed] [Google Scholar]
- 20. Valent P, Klion AD, Horny H‐P, Roufosse F, Gotlib J, Weller PF, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130(3):607–612. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jang YE, Jang I, Kim S, Cho S, Kim D, Kim K, et al. ChimerDB 4.0: an updated and expanded database of fusion genes. Nucleic Acids Res. 2020;48(D1):D817–D824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gotlib J. Tyrosine kinase inhibitors in the treatment of eosinophilic neoplasms and systemic mastocytosis. Hematol/Oncol Clin. 2017;31(4):643–661. [DOI] [PubMed] [Google Scholar]
- 23. Helbig G, Stella‐Holowiecka B, Grosicki S, Bober G, Krawczyk M, Wojnar J, et al. The results of imatinib therapy for patients with primary eosinophilic disorders. Eur J Haematol. 2006;76(6):535–536. [DOI] [PubMed] [Google Scholar]
- 24. Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348(13):1201–1214. [DOI] [PubMed] [Google Scholar]
- 25. Curtis CE, Grand FH, Musto P, Clark A, Murphy J, Perla G, et al. Two novel imatinib‐responsive PDGFRA fusion genes in chronic eosinophilic leukaemia. Br J Haematol. 2007;138(1):77–81. [DOI] [PubMed] [Google Scholar]
- 26. Peterson JF, He RR, Nayer H, Cuevo RS, Smadbeck JB, Vasmatzis G, et al. Characterization of a rarely reported STAT5B/RARA gene fusion in a young adult with newly diagnosed acute promyelocytic leukemia with resistance to ATRA therapy. Cancer Genet. 2019;237:51–54. [DOI] [PubMed] [Google Scholar]
- 27. Arnould C, Philippe C, Bourdon V, Grégoire MJ, Berger R, Jonveaux P. The signal transducer and activator of transcription STAT5b gene is a new partner of retinoic acid receptor α in acute promyelocytic‐like leukaemia. Hum Mol Genet. 1999;8(9):1741–1749. [DOI] [PubMed] [Google Scholar]
- 28. Thiant S, Moutuou M, Laflamme P, Sidi Boumedine R, Leboeuf DM, Busque L, et al. Imatinib mesylate inhibits STAT5 phosphorylation in response to IL‐7 and promotes T cell lymphopenia in chronic myelogenous leukemia patients. Blood Cancer J. 2017;7(4):e551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jones DT, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz HJ, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45(8):927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jones DT, Gronych J, Lichter P, Witt O, Pfister SM. MAPK pathway activation in pilocytic astrocytoma. Cell Mol Life Sci. 2012;69:1799–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li F, Huynh H, Li X, Ruddy DA, Wang Y, Ong R, et al. FGFR‐mediated reactivation of MAPK signaling attenuates antitumor effects of imatinib in gastrointestinal stromal tumors. Cancer Discov. 2015;5(4):438–451. [DOI] [PubMed] [Google Scholar]
- 32. Suzuki M, Abe A, Imagama S, Nomura Y, Tanizaki R, Minami Y, et al. BCR‐ABL‐independent and RAS/MAPK pathway‐dependent form of imatinib resistance in Ph‐positive acute lymphoblastic leukemia cell line with activation of EphB4. Eur J Haematol. 2010;84(3):229–238. [DOI] [PubMed] [Google Scholar]
- 33. Costa V, Esposito R, Ziviello C, Sepe R, Bim LV, Cacciola NA, et al. New somatic mutations and WNK1‐B4GALNT3 gene fusion in papillary thyroid carcinoma. Oncotarget. 2015;6(13):11242–11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Y, Liu T, Li N, Wang T, Pu Y, Lin R. Identification of a novel WNK1–ROS1 fusion in a lung adenocarcinoma sensitive to crizotinib. Lung Cancer. 2019;129:92–94. [DOI] [PubMed] [Google Scholar]
- 35. Yang G‐J, Zhu M‐H, Lu X‐J, Liu YJ, Lu JF, Leung CH, et al. The emerging role of KDM5A in human cancer. J Hematol Oncol. 2021;14(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. White JC, Pucci P, Crea F. The role of histone lysine demethylases in cancer cells' resistance to tyrosine kinase inhibitors. Cancer Drug Resist. 2019;2(2):326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fujita T, Yuno M, Okuzaki D, Ohki R, Fujii H. Identification of non‐coding RNAs associated with telomeres using a combination of enChIP and RNA sequencing. PLoS One. 2015;10(4):e0123387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tauchi T, Nakajima A, Sashida G, Shimamoto T, Ohyashiki JH, Abe K, et al. Inhibition of human telomerase enhances the effect of the tyrosine kinase inhibitor, imatinib, in BCR‐ABL‐positive leukemia cells. Clin Cancer Res. 2002;8(11):3341–3347. [PubMed] [Google Scholar]
- 39. Uziel O, Fenig E, Nordenberg J, Beery E, Reshef H, Sandbank J, et al. Imatinib mesylate (Gleevec) downregulates telomerase activity and inhibits proliferation in telomerase‐expressing cell lines. Br J Cancer. 2005;92(10):1881–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iurlo A, Cattaneo D. Biologic therapies for hypereosinophilic disorders: from tyrosine kinase inhibitors to monoclonal antibodies. Towards an increasingly customized management? Blood Rev. 2023;58:101014. [DOI] [PubMed] [Google Scholar]
- 41. Harish A, Schwartz SA. Targeted anti‐IL‐5 therapies and future therapeutics for hypereosinophilic syndrome and rare eosinophilic conditions. Clin Rev Allergy Immunol. 2020;59(2):231–247. [DOI] [PubMed] [Google Scholar]
- 42. Rothenberg ME, Klion AD, Roufosse FE, Kahn JE, Weller PF, Simon HU, et al. Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N Engl J Med. 2008;358(12):1215–1228. [DOI] [PubMed] [Google Scholar]
- 43. Roufosse FE, Kahn J‐E, Gleich GJ, Schwartz LB, Singh AD, Rosenwasser LJ, et al. Long‐term safety of mepolizumab for the treatment of hypereosinophilic syndromes. J Allergy Clin Immunol. 2013;131(2):461–467. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kuang FL, Legrand F, Makiya M, Ware JA, Wetzler L, Brown T, et al. Benralizumab for PDGFRA‐negative hypereosinophilic syndrome. N Engl J Med. 2019;380(14):1336–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Khoury P, Desmond R, Pabon A, Holland‐Thomas N, Ware JM, Arthur DC, et al. Clinical features predict responsiveness to imatinib in platelet‐derived growth factor receptor‐alpha‐negative hypereosinophilic syndrome. Allergy. 2016;71(6):803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
The datasets generated during the current study are available from the corresponding author upon reasonable request.