

Abstract
Background
Benazepril exhibits a dose‐dependent effect on biomarkers of the circulating renin‐angiotensin‐aldosterone system (RAAS) in dogs.
Hypothesis/Objectives
To characterize the dose‐exposure‐response relationship of a fixed‐dose combination product including benazepril and spironolactone (CARDALIS®) on RAAS biomarkers in dogs.
Animals
Eighteen purpose‐bred healthy beagle dogs.
Methods
Three groups of 6 dogs received different doses of CARDALIS® for 14 days following induction of RAAS activation by feeding a low‐sodium diet: (a) benazepril 0.25 mg/kg + spironolactone 2 mg/kg PO q24h (label dose); (b) benazepril 0.25 mg/kg + spironolactone 2 mg/kg PO q12h; or (c) benazepril 0.5 mg/kg + spironolactone 4 mg/kg PO q12h. Blood samples were collected at baseline and serial time intervals after CARDALIS® dosing to measure serum RAAS biomarkers and plasma concentrations of active drug metabolites. Time‐weighted averages for serum RAAS biomarkers after CARDALIS® dosing at steady state were compared between dosage groups using Wilcoxon rank‐sum testing.
Results
Compared to the label dose, the highest dose of CARDALIS® was associated with a 30% decrease in angiotensin II (P = .03), 94% increase in angiotensin 1‐7 (P = .03), 71% decrease in surrogate activity of ACE (P = .002), and 116% increase in circulating aldosterone (P = .02). CARDALIS® was well‐tolerated at all doses with no clinically relevant changes in renal values or serum electrolytes.
Conclusions and Clinical Importance
The combined CARDALIS® product leads to dose‐dependent alterations of RAAS metabolites. These results could help inform clinical trials in dogs with heart disease.
Keywords: angiotensin‐converting enzyme inhibitor, benazeprilat, canine, canrenone, mineralocorticoid receptor antagonist, TMS
Abbreviations
- AA2
marker of adrenal responsiveness to angiotensin II (ratio of aldosterone to angiotensin II)
- ACEi
angiotensin converting enzyme inhibitor
- ACE‐S
marker of angiotensin‐converting enzyme activity (ratio of angiotensin II to angiotensin I)
- ACVIM
American College of Veterinary Internal Medicine
- Ang
angiotensin
- AUC
area under the curve
- CHF
congestive heart failure
- CI
confidence interval
- LC‐MS/MS
liquid chromatography with tandem mass spectrometry
- MMVD
myxomatous mitral valve disease
- MRA
mineralocorticoid receptor antagonist
- PK
pharmacokinetic
- PRA‐S
marker of plasma renin concentration (sum of angiotensin I and angiotensin II)
- RAAS
renin‐angiotensin‐aldosterone system
- TMS
7‐alpha‐thiomethylspironolactone
- TWA
time‐weighted average
1. INTRODUCTION
Drugs that mitigate the renin‐angiotensin‐aldosterone system (RAAS), including angiotensin‐converting enzyme inhibitors (ACEi) such as benazepril and mineralocorticoid receptor antagonists (MRA) such as spironolactone, are often prescribed to dogs with cardiac disease. 1 , 2 The optimal dose of these drugs remains unknown.
Prescription of ACEi improved short‐term outcome 3 , 4 and long‐term survival compared to placebo 5 , 6 in dogs with congestive heart failure (CHF) secondary to myxomatous mitral valve disease (MMVD). Another study 7 did not demonstrate benefit of ACEi in dogs with CHF secondary to MMVD treated with concurrent pimobendan and high‐dose furosemide. In dogs with advanced preclinical MMVD (American College of Veterinary Internal Medicine [ACVIM] stage B2), one study demonstrated that prescription of ACEi improved CHF‐free survival and all‐cause mortality compared to placebo, 8 whereas other studies showed no benefit of ACEi in prolonging time to CHF when administered as monotherapy 9 or in combination with spironolactone. 10 Several recent lines of evidence suggest that the RAAS‐mitigating properties of the ACEi benazepril are dose‐dependent 11 , 12 and that higher ACEi doses might be associated with improved clinical outcomes. 13
The MRA spironolactone has survival benefit when added to furosemide and benazepril in dogs with CHF secondary to MMVD in 2 study samples with specific clinical and therapeutic characteristics, 14 , 15 but has no demonstrated benefit in delaying the onset of CHF in stage B2 MMVD. 10 Dose‐dependent differences in spironolactone's effect on RAAS biomarkers was not evident when comparing the label dose of 2 mg/kg PO q24h to 4 mg/kg PO q24h. 16
CARDALIS® is a commercially available combination drug formulated to provide a fixed dose of 0.25 mg/kg of benazepril and 2 mg/kg of spironolactone every 24 hours. The product was designed to combine benazepril and spironolactone to improve compliance and convenience of administration for dog owners. CARDALIS® is approved in both Europe and the United States for the treatment of dogs with CHF secondary to MMVD. Although the individual components of this fixed‐dose drug combination have been separately assessed for dose‐dependent effects on the RAAS fingerprint, no previous studies have investigated the dose‐dependent effects of the combination product on the circulating RAAS.
The primary objective of this study was to evaluate the dose‐response relationship of CARDALIS® on biomarkers of the classical and alternative RAAS pathways in healthy dogs with experimentally induced RAAS activation. We evaluated CARDALIS® at 3 dosages: (a) 0.25 mg/kg benazepril + 2 mg/kg spironolactone PO q24h (label dose q24h), (b) 0.25 mg/kg benazepril + 2 mg/kg spironolactone PO q12h (label dose q12h), and (c) 0.5 mg/kg benazepril + 4 mg/kg spironolactone PO q12h (double label dose q12h), with the latter dose corresponding to the dose of ACEi currently advocated by consensus guidelines. 2 We hypothesized that the effects of CARDALIS® on the RAAS fingerprint would be dose‐dependent, and that higher doses of CARDALIS® would result in superior suppression of the classical RAAS and upregulation of the alternative RAAS. Secondary objectives were to assess for adverse effects of higher CARDALIS® doses and to measure plasma levels of drug metabolites (benazeprilat, canrenone, and 7‐alpha‐thiomethylspironolactone [TMS]) to correlate drug exposure to biomarker data.
2. MATERIALS AND METHODS
2.1. Animals
The experimental procedures were approved by the Institutional Animal Care and Use Committee at Iowa State University (protocol number IACUC 23‐035). The study was performed according to the relevant guidelines and regulations, and methods are reported according to ARRIVE guidelines.
Eighteen purpose‐bred beagle dogs (9 castrated males, 9 spayed females; pair‐housed in opposite‐sex pairs), aged 17‐32 months, were used for the study. Dog pairs were randomized by body weight into 1 of 3 treatment groups (3 groups of 6 dogs each) receiving different dosages of CARDALIS® in a parallel design (see Figure 1). The dogs were part of a research colony owned by Iowa State University Laboratory Animal Resources and were housed in the Laboratory Animal Resources unit. Housing conditions were standardized with an ambient temperature of 18°C, a 12‐hour light cycle (07:00 to 19:00), and access to water ad libitum. Outside the study period, the dogs were fed a diet of Purina LabDiet Laboratory Canine Diet once daily at approximately 09:00. General health of all dogs was assessed before the study through a physical examination and routine laboratory screening (complete blood count, serum biochemical analysis).
FIGURE 1.
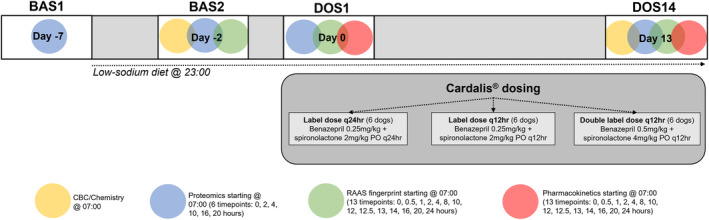
Schematic representation of experimental design and timeline of sampling. There are 4 sampling days: BAS1 (before experimental RAAS activation with low‐sodium diet), BAS2 (after 5 days of low‐sodium diet), DOS1 (after first dose of CARDALIS®), and DOS14 (after 14 days of dosing with CARDALIS®). Three groups (n = 6 dogs each) received different doses of CARDALIS®. Colored circles indicate purpose and timing of blood samples collected on each sampling day. RAAS, renin‐angiotensin‐aldosterone system.
2.2. Study design
This was a prospective study design that took place over 21 days (see Figure 1). Three groups of 6 dogs each received oral CARDALIS® at the following dosages: (a) benazepril 0.25 mg/kg + spironolactone 2 mg/kg every 24 hours (label dose q24h); (b) benazepril 0.25 mg/kg + spironolactone 2 mg/kg every 12 hours (label dose q12h); and (c) benazepril 0.5 mg/kg + spironolactone 4 mg/kg every 12 hours (double label dose q12h). These dosages were chosen to represent the label dose of CARDALIS®, the label dose given twice daily, and a dose of CARDALIS® required to reach the benazepril dosage currently recommended in consensus guidelines (0.5 mg/kg twice daily). 2
Blood sampling timepoints included 2 baseline sampling days (BAS1 [Day −7] and BAS2 [Day −2]) and 2 post‐treatment sampling days (DOS1 [Day 0] and DOS14 [Day 13]), during which blood sampling was performed over a 24‐hour period beginning at 07:00 (see Supplemental Table S1 and Figure 1). Dogs were sampled in the same order at each timepoint, and the exact sampling time was recorded.
Two days before the study period began (Day −9), the time of feeding was changed from 09:00 to 23:00. Dogs were not fed during intensive sampling to minimize the influence of feeding on pharmacodynamics of the RAAS. 17 , 18 , 19 Time of feeding was changed to 23:00 to allow sufficient time between food intake and morning dosing of CARDALIS® while limiting total time food was withheld for animal welfare reasons. At BAS1 (Day −7), blood was collected at 6 timepoints (0, 2, 4, 10, 16, and 20 hours). This blood sampling was performed for exploratory assessment of proteomic biomarkers for cardiovascular disease in dogs, which will be described in a separate report. Starting after sampling at BAS1, the dogs were fed a low‐sodium diet (Hill's Prescription h/d Heart Care; 17 mg sodium per 100 kcal, 0.08% sodium on a dry matter basis) to attain a steady activation of RAAS. 18 , 19 The dogs continued to be fed once daily at 23:00, and the h/d diet was continued throughout the remainder of the study period. The volume fed to each dog was calculated to be isocaloric with the dogs' typical diet. Any leftover food was recorded and discarded, and no other food was allowed during the study period. The dogs were not fed during 24‐hour sampling periods, but were fed at the conclusion of intensive sampling at DOS1 and DOS14 at approximately 09:00.
At BAS2 (Day −2), blood sampling included a baseline CBC and serum chemistry panel (taken at a single timepoint, 0 hour). RAAS fingerprinting was performed at 13 timepoints (0, 0.5, 1, 2, 4, 8, 12, 12.5, 13, 14, 16, 20, and 24 hours), and blood samples for proteomic analysis were also collected at the same 6 timepoints as BAS1 (0, 2, 4, 10, 16, and 20 hours).
On the first day of the dosing period (DOS1, Day 0), sampling for RAAS fingerprinting and proteomics was performed before medication dosing (0 hour, 07:00) and at the same timepoints as BAS2 over a 24‐hour period. Blood was also collected for plasma pharmacokinetics of benazepril and spironolactone at the same timepoints as for RAAS fingerprinting (see Supplemental Table S1 and Figure 1). CARDALIS® (spironolactone 25 mg and benazepril hydrochloride 2.5 mg tablets, Ceva Santé Animale) was administered orally following 0‐hour blood sampling (approximately 07:00, all dosage groups) and 12‐hour blood sampling (approximately 19:00, twice‐daily dosage groups only). The CARDALIS® dose was calculated to the nearest 1.25 mg increment. The medication was administered by study investigators with exact time of administration recorded. Kennels were examined following administration for any medications to ensure that all dogs received their medications as intended. On Days 1 through 12, CARDALIS® was administered to dogs by study investigators at 07:00 (for all dosage groups) and 19:00 (for twice‐daily dosage groups only), as described above.
On the last day of the dosing period (DOS14, Day 13), medication administration and blood sampling for RAAS, pharmacokinetics, and proteomics analyses were performed as on Day 0. Additionally, a CBC and serum chemistry panel were repeated at 0‐hour to monitor for adverse effects of CARDALIS®.
2.3. Sample collection
Venous blood samples were collected from an external jugular, lateral saphenous, or cephalic vein using a 1‐inch, 20‐ or 22‐gauge needle attached to either a 3 mL or 6 mL syringe at each timepoint. Between 1 and 4 mL of whole blood was collected at each sampling based on the specific testing required at that timepoint (see Supplemental Table S1). The most intensive sampling day was Day 13, during which a maximum of 54.5 mL was drawn from each dog over the 24‐hour period.
TABLE 1.
Summary statistics of the plasma pharmacokinetic parameters for benazeprilat, canrenone, and TMS after 14 days of treatment with CARDALIS® at 3 different dosages in 18 healthy dogs.
| Metabolite | AUC24 (ng/mL × h) | C max (ng/mL) | T max (h) | T 1/2 (h) |
|---|---|---|---|---|
| Benazeprilat | 262 (220‐474) | 43.5 (23.9‐62.1) | 2.00 (1.25‐2.00) | 3.14 (2.63‐3.65) |
| Canrenone | 1510 (842‐2060) | 87.1 (64.6‐147.0) | 2.00 (2.00‐4.00) | 9.00 (7.74‐11.50) |
| TMS | 2150 (1460‐3100) | 268 (146‐308) | 2.00 (1.00‐2.00) | 4.73 (4.36‐5.43) |
Note: Results are shown for all dosages combined, as dose‐proportionality was observed for all metabolites. AUC is calculated over 24 hours post‐dose; all other variables are calculated over 12 hours post‐dose. Data are reported as median (interquartile range).
Abbreviations: AUC24, area under the curve; C max, maximum metabolite concentration; T 1/2, elimination half‐life; T max, time of maximum metabolite concentration; TMS, 7‐alpha‐thiomethylspironolactone.
For CBCs, 1 mL of whole blood was placed in an EDTA tube; for serum chemistry panels, 1 mL of whole blood was placed in an additive‐free tube. For RAAS fingerprinting, 1.5‐2 mL of whole blood was placed in an additive‐free tube. For pharmacokinetics (PK) of benazepril, 0.5‐1 mL of whole blood was placed in a 1 mL lithium heparin tube spiked with 11.2 uL dichlorvos in 6 mg/mL acetonitrile. For PK of spironolactone metabolites, 1 mL of whole blood was placed in a chilled lithium heparin tube with no additive and kept refrigerated during centrifugation. Samples were centrifuged at 3000 rpm for 15 minutes, and serum or plasma was transferred into cryovials that were stored at −80°C for later analysis. Blood for CBCs and serum chemistry panels was refrigerated until analysis at the Iowa State University Clinical Pathology Laboratory within 24 hours of collection. All other samples were processed immediately after sampling at each timepoint and frozen for batch analysis by the respective laboratories as described in the following sections.
2.4. RAAS fingerprint analysis
Equilibrium concentrations of AngI, AngII, Ang1‐7, Ang1‐5, and aldosterone were quantified in serum samples using liquid chromatography with tandem mass spectrometry (LC‐MS/MS) by Attoquant Diagnostic Laboratories (Vienna, Austria), using previously validated and described methods. 12 , 20 , 21 Angiotensin‐based surrogate markers for renin and angiotensin‐converting enzyme were derived from AngII and AngI levels by calculating their sum (PRA‐S) and ratio (ACE‐S), respectively. The ratio of aldosterone/Ang II (AA2) was calculated to assess AngII signaling and aldosterone response as previously described. 22
2.5. Bioanalytics of plasma metabolites
Plasma samples for benazeprilat concentration were processed and performed by the Iowa State Analytical Chemistry Laboratory using LC‐MS/MS as previously described. 12 Plasma samples for quantification of canrenone and 7‐alpha‐thiomethylspironolactone (TMS), the active metabolites of spironolactone, were analyzed by Ceva Santé Animale by LC‐MS/MS as previously described. 16 Quantification of spironolactone's active metabolites was chosen because of significantly longer half‐life compared to the parent compound, which is rapidly cleared from circulation. 23 , 24
2.6. Pharmacokinetic analysis
A preliminary PK analysis was performed using DOS14 data (presumed steady state concentrations) using a statistical moment (ie, non‐compartmental) approach using the PKNCA package from R (R version 4.3.1, R Foundation for Statistical Computing, Vienna, Austria). Standard PK parameters were generated for individual dogs for benazeprilat, canrenone and TMS concentration as follows:
Maximum plasma concentration at steady state, C max,SS
Time of maximum plasma concentration at steady state, T max,SS
Area under plasma concentration time curve for the dosing interval tau (24 hours): AUC24,SS
Plasma terminal half life, T 1/2
A linear/log trapezoidal rule was used to estimate the area under time curves. Half‐life was computed by linear regression of the logarithmic concentration vs time curve during the elimination phase. Summary statistics for the individual pharmacokinetic parameters were derived.
2.7. Statistical analysis
Statistical analyses were performed using commercial software (R version 4.3.1, R Foundation for Statistical Computing, Vienna, Austria). Linear mixed modeling was used to determine the relationship between CARDALIS® dose and exposure (AUC), with the individual dog being used as a random effect in the statistical model. Calculation of the 24‐hour time‐weighted average (TWA) for each biomarker of interest was performed by dividing the area under the effect curve (determined using the trapezoidal rule) by the observation period (24 hours). Calculation of TWA was chosen to provide an unbiased estimate of drug effects when samples are collected at irregular intervals. Unlike simple arithmetic means, TWAs account for the time between samples, ensuring that each data point contributes proportionally to the average. Time‐weighted averages were compared between dosing groups using Wilcoxon rank‐sum tests.
3. RESULTS
3.1. Pharmacokinetics of benazeprilat, canrenone, and TMS
Results of noncompartmental PK analyses performed at steady state (DOS14) are presented in Table 1. The AUC for benazeprilat exposure over the first 24 hours was significantly and positively related to dose amount (P < .001). A 1 mg increase in dose corresponded to an increase in AUC of 1.99 ng/mL × h (95% confidence interval [CI], 1.27‐2.70 in natural logarithmic scale). The AUC for canrenone exposure over the first 24 hours was significantly and positively related to dose amount (P < .001). A 1 mg increase in dose corresponded to an increase in AUC of 1.54 units (95% CI, 1.14‐1.95 in natural logarithmic scale). The AUC for TMS exposure over the first 24 hours was significantly and positively related to dose amount (P < .001). A 1 mg increase in dose corresponded to an increase in AUC of 1.50 units (95% CI, 0.80‐2.21 in natural logarithmic scale).
The time‐course of benazeprilat, canrenone, and TMS plasma concentrations for different dosage groups are displayed in Figure 2. Peak concentrations (T max) occurred approximately 2 hours after oral dosing for all metabolites. Dose‐proportionality was respected for all metabolites (P > .05 for all pairwise dose comparisons of AUC/dose).
FIGURE 2.
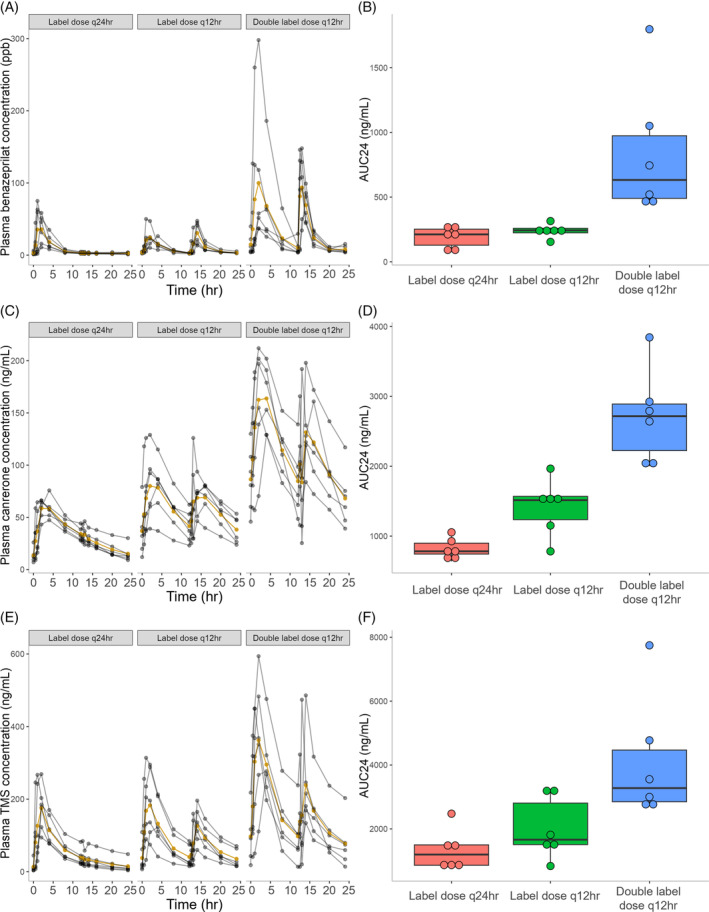
Time‐course of benazeprilat (A), canrenone (C), and 7‐alpha‐thiomethylspironolactone (TMS; E) plasma concentrations in 18 healthy dogs after the final dose of CARDALIS® administered at 3 different doses for 14 days (n = 6 dogs in each dosage group). The yellow line indicates mean plasma concentration and gray lines indicate individual dog results at each timepoint. Corresponding box‐and‐whisker plots indicate 24‐hour area under the curve of benazeprilat (B), canrenone (D), and TMS (F) concentration (ng/mL for all metabolites). The horizontal line represents median, box represents quartiles, and whiskers represent range, with outliers (>1.5 × interquartile range below Q1 or above Q3) plotted as dots. CARDALIS® dosage groups: Label dose q24h (benazepril 0.25 mg/kg + spironolactone 2 mg/kg PO q24h); label dose q12h (benazepril 0.25 mg/kg + spironolactone 2 mg/kg PO q12h); and double label dose q12h (0.5 mg/kg benazepril + 4 mg/kg spironolactone PO q12h).
Three dogs in the lowest dosing group had a second spike in all drug metabolites measured at the 13‐hour timepoint at DOS14, suggesting either sample mislabeling, inadvertent re‐dosing, or coprophagy. Samples from these timepoints showing unexpected PK signals were removed from further analysis of both pharmacokinetic and RAAS biomarker analysis.
3.2. RAAS biomarkers
There were no significant differences in RAAS biomarkers between dosage groups at BAS2 (Day −5) before medication administration. The 24‐hour time weighted averages (TWA) at DOS14 are shown for the different dosage groups in Figures 3 and 4. There were significant differences in 24‐hour TWA among dosage groups for all RAAS biomarkers. P‐values for all pairwise group comparisons are displayed in Figure 3. Table 2 shows percent differences in mean 24‐hour TWA for higher doses of CARDALIS® compared to the label dose.
FIGURE 3.
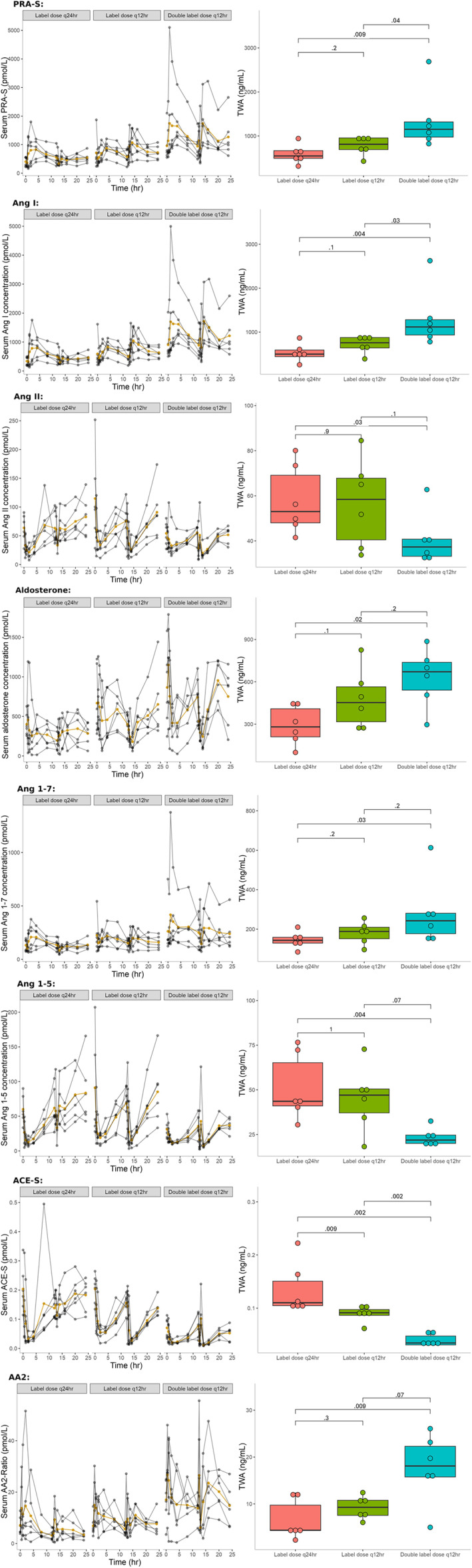
RAAS biomarker serum concentrations in 18 healthy dogs following CARDALIS® administration at 3 different doses for 14 days (n = 6 dogs in each dosage group). Spaghetti plots show 24‐hour time course of RAAS biomarkers, with yellow lines indicating mean serum biomarker concentration and gray lines indicating individual dog results. Box‐and‐whisker plots show 24‐hour time‐weighted average (TWA) for biomarker concentration. Units for time‐weighted averages of RAAS biomarkers are ng/mL; ratios are unitless. The horizontal line represents median, box represents quartiles, and whiskers represent range, with outliers plotted as dots. P‐values are shown for group comparisons of TWA. CARDALIS® dosage groups: Label dose q24h (benazepril 0.25 mg/kg + spironolactone 2 mg/kg PO q24h); label dose q12h (benazepril 0.25 mg/kg + spironolactone 2 mg/kg PO q12h); and double label dose q12h (0.5 mg/kg benazepril + 4 mg/kg spironolactone PO q12h). AA2 ratio, ratio of aldosterone to angiotensin II and measure of adrenal responsiveness to angiotensin II; ACE‐S, surrogate measure of angiotensin‐converting enzyme activity; Ang, angiotensin; PRA‐S, surrogate measure for plasma renin activity.
FIGURE 4.
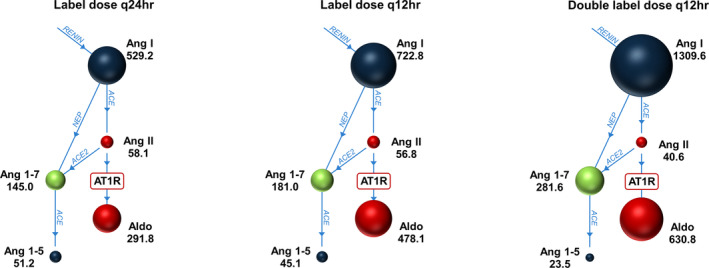
Mean values of 24‐hour time‐weighted averages of renin‐angiotensin‐aldosterone system biomarkers in 18 healthy dogs treated with 14 days of CARDALIS® at 1 of 3 doses in a parallel‐group design. Sizes of circles are proportional to media time‐weighted averages of each analyte as shown (units of time‐weighted averages are ng/mL). CARDALIS® dosage groups: Label dose q24h (benazepril 0.25 mg/kg + spironolactone 2 mg/kg PO q24h); label dose q12h (benazepril 0.25 mg/kg + spironolactone 2 mg/kg PO q12h); and double label dose q12h (0.5 mg/kg benazepril + 4 mg/kg spironolactone PO q12h). ACE, angiotensin converting enzyme; Aldo, aldosterone; Ang, angiotensin; AT1R, angiotensin type 1 receptor.
TABLE 2.
Percent difference in mean 24‐hour TWA between CARDALIS® at the lowest dose (label dose q24h) compared to 2 higher doses (label dose q12h or double label dose q12h).
| Metabolite | CARDALIS® dosage comparison | |
|---|---|---|
| Label dose q12h vs label dose q24h | Double label dose q12h vs label dose q24h | |
| PRA‐S | +32.7% | +129.9% |
| AngI | +36.6% | +147.5% |
| AngII | −2.3% | −30.1% |
| Aldosterone | +63.8% | +116.2% |
| Ang1‐7 | +24.8% | +94.3% |
| Ang1‐5 | −11.8% | −54.1% |
| ACE‐S | −34.0% | −70.7% |
| AA2 | +40.6% | +169.2% |
Note: Positive percent differences indicate that higher dosages of CARDALIS® resulted in higher mean TWA values compared to the label dose q24h, while negative percent differences indicate that higher dosages of CARDALIS® resulted in lower mean TWA values compared to the label dose q24h. Percent differences that were statistically significant (P < .05) in dosage group comparisons are shown in bold. CARDALIS® dosage groups: label dose q24h (benazepril 0.25 mg/kg + spironolactone 2 mg/kg PO q24h); label dose q12h (benazepril 0.25 mg/kg + spironolactone 2 mg/kg PO q12h); and double label dose q12h (0.5 mg/kg benazepril + 4 mg/kg spironolactone PO q12h).
Abbreviations: AA2 ratio, ratio of aldosterone to angiotensin II and measure of adrenal responsiveness to angiotensin II; ACE‐S, surrogate measure of angiotensin‐converting enzyme activity; Ang, angiotensin; PRA‐S, surrogate measure for plasma renin activity.
For ACE‐S (surrogate for ACE activity), dose‐dependent differences were noted between all 3 dosage groups. Compared to dogs receiving CARDALIS® at the label dose q24h, 24‐hour TWA of ACE‐S was significantly lower in dogs from the other 2 dosage groups. For PRA‐S and AngI, 24‐hour TWA was significantly different in the group receiving double label dose q12h compared to the other 2 dosage groups. For AngII, aldosterone, Ang1‐7, and AA2, 24‐hour TWA results were significantly different only between the label dose q24h and double label dose q12h. Compared to dogs receiving the label dose q24h, dogs receiving double label dose q12h had a 30% decrease in AngII and a 94% increase in Ang1‐7 (P = .03 for both; see Figure 3 and Table 2).
A single dog in the double label dose q12h group was a visual and statistical outlier with high benazeprilat concentration in the first 12 hours of dosing (see Figure 2). This same dog also showed high outlier values of PRA‐S, AngI, and AngII (see Figure 3).
3.3. Clinicopathologic monitoring and adverse effects
No clinically relevant abnormalities were documented in complete blood count and serum chemistry during the study. Clinicopathologic values remained largely within reference ranges at BAS2 (Day −2; pre‐treatment) and DOS14 (Day 13, post‐treatment; see Table 3). Insufficient volume of blood was collected to analyze serum electrolytes for some dogs at DOS14 (see Table 3). There were no significant differences between dosage groups in percent change in clinicopathologic values pre‐ and post‐treatment (P > .05 for all group comparisons).
TABLE 3.
Selected clinicopathologic results in 18 healthy dogs before treatment (BAS2) and after 14 days of CARDALIS® administration (DOS14) at 3 different doses (n = 6 dogs in each dosage group).
| BAS2 | DOS14 | ||||||
|---|---|---|---|---|---|---|---|
| Variable | Reference range | Label dose q24h | Label dose q12h | Double label dose q12h | Label dose q24h | Label dose q12h | Double label dose q12h |
| Blood urea nitrogen | 10‐30 mg/dL | 15.0 (12.8‐16.5) | 19.0 (19‐19.8) | 17.0 (15.2‐20.0) | 9.0 (6.0‐9.8) | 11.0 (10.0‐13.0) | 10.5 (8.0‐13.8) |
| Creatinine | 0.5‐1.5 mg/dL | 0.6 (0.6‐0.6) | 0.6 (0.6‐0.7) | 0.6 (0.6‐0.7) | 0.6 (0.6‐0.6) | 0.7 (0.6‐0.7) | 0.6 (0.6‐0.8) |
| Sodium | 141‐151 mEq/L | 142.5 (142.0‐143.0) | 141.5 (141.0‐142.0) | 142.5 (141.2‐143.0) |
142.5 (142.2‐142.8) n = 2 |
140.0 (139.5‐140.5) n = 2 |
138.0 (138.0‐139.5) n = 3 |
| Chloride | 112‐121 mEq/L | 107.0 (107.0‐107.0) | 107.0 (106.2‐107.0) | 108.0 (107.2‐108.8) |
109.5 (109.2‐109.8) n = 2 |
109.0 (108.0‐110.5) n = 3 |
109.5 (108.2‐110.5) n = 4 |
| Potassium | 3.9‐5.3 mEq/L | 5.0 (4.6‐5.1) | 5.0 (4.9‐5.1) | 5.0 (4.8‐5.2) |
4.5 (4.3‐4.7) n = 2 |
4.6 (4.5‐4.7) n = 3 |
4.7 (4.6‐5.1) n = 4 |
| Total protein | 5.2‐7.1 g/dL | 6.3 (6.1‐6.5) | 6.3 (6.1‐6.4) | 6.3 (5.9‐6.3) | 5.8 (5.5‐6.0) |
5.7 (5.5‐6.0) n = 5 |
5.6 (5.5‐5.8) |
| Albumin | 2.7‐4.0 g/dL | 3.8 (3.6‐3.8) | 3.5 (3.3‐3.6) | 3.5 (3.4‐3.6) | 3.0 (2.9‐3.1) |
3.2 (2.9‐3.3) n = 5 |
2.9 (2.8‐3.0) |
| Hematocrit | 37.0‐55.0% | 55.3 (52.6‐56.5) | 53.9 (51.1‐54.6) | 52.0 (51.5‐53.2) | 52.8 (48.7‐55.5) | 50.3 (46.0‐53.1) | 49.5 (48.9‐50.0) |
Note: Data are presented as median (interquartile range). Number of dogs is indicated for variables with incomplete datasets because of insufficient volume of blood sampled. CARDALIS® dosage groups: label dose q24h (benazepril 0.25 mg/kg + spironolactone 2 mg/kg PO q24h); label dose q12h (benazepril 0.25 mg/kg + spironolactone 2 mg/kg PO q12h); and double label dose q12h (0.5 mg/kg benazepril + 4 mg/kg spironolactone PO q12h). Reference ranges are provided by the Iowa State University Clinical Pathology Laboratory.
Six dogs (2 from each dosage group) had a single incident of soft stool documented within 4 days of starting oral CARDALIS® administration. Seven dogs (5 from the label dose q24h dosage group, 2 from the label dose q12h dosage group) had an incident of vomiting documented during CARDALIS® administration (single incident in 5 dogs, 2 incidents in 2 dogs). All gastrointestinal signs were self‐limiting without treatment and all dogs consumed the entirety of the provided diet at all feedings.
4. DISCUSSION
The results of this study demonstrate a dose‐exposure‐response relationship of the commercial combination benazepril/spironolactone product (CARDALIS®) on circulating biomarkers of both the classical and alternative RAAS pathways in healthy dogs. Higher doses of CARDALIS® resulted in decreased ACE‐S (a surrogate marker of ACE activity), lower concentrations of the products of reactions catalyzed by ACE (AngII and Ang1‐5), and higher concentrations of the substrates for ACE activity (PRA‐S, AngI, Ang1‐7) when compared to lower doses. These differences were most noticeable when comparing the highest dose of CARDALIS® (double label dose q12h, which included benazepril at 0.5 mg/kg PO q12h) to the other dosage groups.
These findings are consistent with other recent lines of evidence suggesting that the RAAS‐mitigating properties of ACEi are dose‐dependent. A retrospective study of dogs with ACVIM stage B2 and C heart disease suggested that twice daily dosing of an ACEi, compared to once daily dosing, was associated with improved survival after CHF. 13 A subsequent prospective study in healthy dogs with experimentally induced RAAS activation demonstrated a dose‐responsive effect of benazepril on biomarkers of both the classical and alternative RAAS pathways in the RAAS fingerprint, with the highest dose of benazepril studied (0.5 mg/kg) resulting in 38% lower values of AngII compared to the lowest dose studied (0.125 mg/kg). 12 A mathematical model developed from these experimental data showed that a benazepril dose of 0.5 mg/kg PO q12h was optimal in reducing the classical pathway of the RAAS (AngII) and upregulating the alternative pathway (Ang1‐7). 11 The current consensus guidelines from the ACVIM 2 recommend an ACEi dose of 0.5 mg/kg PO q12h, which aligns with the predictions of the mathematical model but is higher than the dosage used in any previous clinical trial in veterinary cardiology.
Previous clinical trials report a variable effect of ACEi in delaying the onset of CHF in dogs with asymptomatic heart disease. 8 , 9 , 10 One potential reason for these inconsistent results among studies is the wide range of ACEi doses used in clinical trials (ranging from 0.25 to 1 mg/kg per day). 3 , 4 , 5 , 6 , 8 , 9 , 10 The original label dose of benazepril in Europe (0.25 mg/kg PO q24h) was based on an early pharmacokinetic/pharmacodynamic study using ACE activity as a marker for RAAS suppression. 25 However, it is now well‐recognized that ACE activity is a poor surrogate marker to characterize the effect of RAAS‐mitigating drugs. 26 , 27 , 28 , 29 , 30 The current preferred method for investigating the effects of a drug on circulating RAAS is quantification of a RAAS fingerprint using LC‐MS/MS, 31 which allows for simultaneous evaluation of both the classical and alternative RAAS pathways. 20 , 21 The RAAS fingerprint data from the present study are aligned with the RAAS fingerprint effects of benazepril monotherapy, 12 confirming that the dose‐dependent effect of benazepril on the circulating RAAS is preserved when used in the combination product CARDALIS®.
Considering the pharmacodynamic effects of the MRA (spironolactone), the highest dose of CARDALIS® (double label dose q12h, including spironolactone at 4 mg/kg PO q12h) resulted in higher concentrations of serum aldosterone compared to the lowest dose studied (label dose q24h, including spironolactone 2 mg/kg PO q24h). This is logical given the mechanism of MRAs, which block aldosterone receptors in the distal convoluted tubule and lead to increased circulating aldosterone. These findings differ from a recent study of spironolactone monotherapy in healthy dogs, which showed no dose‐dependent differences in RAAS metabolites (including aldosterone) using spironolactone dosages of 2 mg/kg PO q24h vs 4 mg/kg PO q24h. 16 However, these results cannot be directly compared because of methodological differences between studies, including type and degree of experimental RAAS activation, dosages of spironolactone, and use of spironolactone monotherapy vs combination with ACEi.
Overall, the findings of this study suggest that the dynamics of the RAAS response to the CARDALIS® combination product are dominated by the effects of ACEi rather than MRA. For example, our results demonstrated a decrease in AngII with higher doses of CARDALIS®, which is consistent with the expected pharmacologic effect of ACEi (decreased conversion of AngI to AngII), but inconsistent with the expected pharmacologic effect of isolated MRA (displacement of aldosterone from its receptor leading to decreased plasma sodium concentration and increased renin secretion). 16 Indeed, the differences in mean ACE‐S, AngII, and Ang1‐7 between the highest and lowest doses of CARDALIS® used in this study (−71%, −30%, and +94%, respectively) are reasonably similar to differences in these same biomarkers between the highest and lowest doses of benazepril seen in our previous study (−59%, −38%, and +47%, respectively). 12 Though the specific dosages of benazepril were not equivalent between these 2 studies, the highest dose studied was 4 times higher than the lowest dose studied in both cases.
Although results of this study suggest a dose‐dependent effect of this combination product on RAAS biomarkers, this study was not designed to address the effect of CARDALIS® dosage on long‐term outcome. Ultimately, the biological effect of RAAS is not defined by the levels of biomarkers in circulation but by how these biomarkers interact with their target receptors, taking into account the specific pathophysiological conditions of the individual animal. Although our findings indicate a more beneficial RAAS peptide profile at higher doses of CARDALIS®, it is not directly implied that these variations in RAAS biomarkers translate to clinical advantages based on our data alone.
Potential adverse effects of ACEi and spironolactone include azotemia or increased serum potassium concentrations because of reduced potassium excretion. Serum biochemistry results from the present study showed no dose‐dependent or clinically relevant changes in serum electrolytes after treatment, suggesting that even the highest dosages of benazepril (0.5 mg/kg q12h) and spironolactone (4 mg/kg q12h) used in this study were well‐tolerated in these healthy dogs. This is consistent with a previous study of dogs with MMVD that showed no difference in incidence of adverse events between spironolactone 2 mg/kg q24h vs placebo when added to background therapy, which included ACEi. 32 Another study in dogs with stage B2 MMVD similarly showed no significant changes in serum sodium or potassium after a median of 15 weeks of treatment with combined ACEi (mean dose 0.54 mg/kg PO q12h) and spironolactone (mean dose 1.5 mg/kg PO q12h). 33 Several dogs in the present study from various dosage groups experienced isolated and self‐limiting gastrointestinal signs (vomiting or soft stool) during CARDALIS® administration. It is challenging to determine whether these signs were drug‐related as this study design did not include a control group, but previous studies of both ACEi and spironolactone suggest that these drugs are generally well‐tolerated with rare gastrointestinal adverse effects. 8 , 9 , 13 , 14 , 15
The results from our preliminary PK analyses reflected peak concentrations of all metabolites approximately 2 hours after dosing, with metabolite concentrations being proportional to dose administered. These findings, as well as other results of noncompartmental PK analyses, are consistent with previous PK studies of benazepril and spironolactone in dogs. 12 , 25 , 34 Three dogs in the label dose q24h group demonstrated a second increase in concentration of all 3 drug metabolites at the 13‐hour timepoint at DOS14 (after 12‐hour dosing). Potential causes for this second signal include sample mislabeling, inadvertent re‐dosing (eg, by licking another dog's saliva), or coprophagy, which was considered possible given known fecal excretion of ACEi metabolites in dogs 35 , 36 and witnessed coprophagy in this dog colony. Second‐pass metabolism was considered less likely given previous studies of benazepril PK. 25 , 37 The samples from timepoints showing unexpected PK signals were removed from further analysis of both PK and RAAS biomarker analysis.
This study had several limitations. A control group receiving a placebo was not incorporated, thus alterations in RAAS biomarkers are considered in relation to differences among dosage groups. The sample size was limited (18 dogs in total, with 6 dogs in each dosage group) and there was considerable variation in the levels of certain RAAS biomarkers across individuals, both factors that heighten the possibility of a type II error. Inter‐individual variability was evident, for example, in the unexpected second peak of drug metabolites observed at the 13‐hour mark in the label dose group, the statistical outlier with elevated benazeprilat, PRA‐S, AngI, and AngII concentrations in the double label dose q12h group, and the relatively wide range of metabolite concentrations (Table 1 and Figure 2) and RAAS‐related effects (Figure 3) across dosing groups. However, these findings align with previous literature documenting significant inter‐subject variability in the pharmacokinetics and pharmacodynamics of ACE inhibitors 11 , 12 , 18 , 38 , 39 and mineralocorticoid receptor antagonists 16 in dogs.
Because a combination drug product was studied, it was impossible to separate the individual effects of benazepril vs spironolactone on RAAS biomarkers; however, data derived from this study will be integrated with data from our research group's previous studies of benazepril and spironolactone to create and refine mathematical models accounting for individual drug signals. This study was performed in healthy dogs with experimentally induced RAAS upregulation by feeding a low‐sodium diet. Although feeding a low‐sodium diet has been shown to reliably activate RAAS in dogs, 39 results might differ in dogs with naturally occurring cardiac disease and those fed different diets. Finally, there were missing data in the post‐treatment clinicopathologic data because of insufficient blood volume. It is possible that some dogs could have experienced larger changes in electrolytes since these values were not assessed for all dogs post‐treatment.
5. CONCLUSION
The combination product CARDALIS® (benazepril/spironolactone) showed dose‐dependent effects on biomarkers of the classical and alternative RAAS in healthy dogs with experimentally induced RAAS activation. The highest dosage studied (double label dose q12h, 0.5 mg/kg benazepril + 4 mg/kg spironolactone PO q12h) resulted in the lowest concentration of AngII (suppression of the classical RAAS), the highest concentration of Ang1‐7 (upregulation of the alternative RAAS), and the highest degree of mineralocorticoid receptor blockade (indicated by the concentration of circulating aldosterone), compared to the lowest dose studied (label dose q24h, 0.25 mg/kg benazepril + 2 mg/kg spironolactone PO q24h). These data will further inform and refine mathematical models of RAAS pharmacotherapy in dogs.
CONFLICT OF INTEREST DECLARATION
Authors Emilie Guillot and Thomas Blondel are employees of Ceva Santé Animale. Authors Jessica L. Ward, Jonathan P. Mochel, and Karin Allenspach have served as consultants for Ceva Santé Animale and have received reimbursement and honoraria for consulting, expert testimony, travel, and service as key opinion leaders (KOLs). Author Oliver Domenig is an employee of Attoquant Diagnostics. Authors Elizabeth Manson and Maria Merodio have no relevant competing interests. Although Ceva Santé Animale provided funding and study drug for this research, the study logistics and sample handling were performed with randomization and by study personnel with no conflicts of interest (Elizabeth Manson and Maria Merodio).
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
IAUCUC approval was granted through Iowa State University (protocol number 23‐035).
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
Supporting information
Table S1: Schedule and timing of blood sampling during the 4 sampling days of the study period (BAS1, BAS2, DOS1, and DOS14). RAAS, renin‐angiotensin‐aldosterone system.
ACKNOWLEDGMENTS
Funding for this study was provided by Ceva Santé Animale, and this article describes the effects of a drug manufactured by Ceva Santé Animale. Open access funding provided by the Iowa State University Library.
Manson E, Ward JL, Merodio M, et al. Dose‐exposure‐response of CARDALIS® (benazepril/spironolactone) on the classical and alternative arms of the renin‐angiotensin‐aldosterone system in healthy dogs. J Vet Intern Med. 2025;39(1):e17255. doi: 10.1111/jvim.17255
REFERENCES
- 1. Franchini A, Borgarelli M, Abbott JA, et al. The longitudinal outcome of canine (K9) myxomatous mitral valve disease (LOOK‐Mitral) registry: baseline treatment characteristics. J Vet Cardiol. 2022;41:99‐120. [DOI] [PubMed] [Google Scholar]
- 2. Keene BW, Atkins CE, Bonagura JD, et al. ACVIM consensus guidelines for the diagnosis and treatment of myxomatous mitral valve disease in dogs. J Vet Intern Med. 2019;33:1127‐1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Acute and short‐term hemodynamic, echocardiographic, and clinical effects of enalapril maleate in dogs with naturally acquired heart failure: results of the Invasive Multicenter PROspective Veterinary Evaluation of Enalapril study. The IMPROVE study group. J Vet Intern Med. 1995;9:234‐242. [DOI] [PubMed] [Google Scholar]
- 4. Controlled clinical evaluation of enalapril in dogs with heart failure: results of the Cooperative Veterinary Enalapril Study Group. The COVE Study Group. J Vet Intern Med. 1995;9:243‐252. [DOI] [PubMed] [Google Scholar]
- 5. BENCH (BENazepril in Canine Heart disease) Study Group . The effect of benazepril on survival times and clinical signs of dogs with congestive heart failure: results of a multicenter, prospective, randomized, double‐blinded, placebo‐controlled, long‐term clinical trial. J Vet Cardiol. 1999;1:7‐18. [DOI] [PubMed] [Google Scholar]
- 6. Ettinger S, Benitz A, Ericsson G, et al. Effects of enalapril maleate on survival of dogs with naturally acquired heart failure. The Long‐Term Investigation of Veterinary Enalapril (LIVE) study group. J Am Vet Med Assoc. 1998;213:1573‐1577. [PubMed] [Google Scholar]
- 7. Wess G, Kresken JG, Wendt R, et al. Efficacy of adding ramipril (VAsotop) to the combination of furosemide (Lasix) and pimobendan (Vetmedin) in dogs with mitral valve degeneration: the VALVE trial. J Vet Intern Med. 2020;34:2232‐2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Atkins CE, Keene BW, Brown WA, et al. Results of the veterinary enalapril trial to prove reduction in onset of heart failure in dogs chronically treated with enalapril alone for compensated, naturally occurring mitral valve insufficiency. J Am Vet Med Assoc. 2007;231:1061‐1069. [DOI] [PubMed] [Google Scholar]
- 9. Kvart C, Häggström J, Pedersen HD, et al. Efficacy of enalapril for prevention of congestive heart failure in dogs with myxomatous valve disease and asymptomatic mitral regurgitation. J Vet Intern Med. 2002;16:80‐88. [PubMed] [Google Scholar]
- 10. Borgarelli M, Ferasin L, Lamb K, et al. DELay of Appearance of sYmptoms of canine degenerative mitral valve disease treated with spironolactone and benazepril: the DELAY study. J Vet Cardiol. 2020;27:34‐53. [DOI] [PubMed] [Google Scholar]
- 11. Schneider BK, Ward J, Sotillo S, et al. Breakthrough: a first‐in‐class virtual simulator for dose optimization of ACE inhibitors in translational cardiovascular medicine. Sci Rep. 2023;13:3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sotillo S, Ward JL, Guillot E, et al. Dose–response of benazepril on biomarkers of the classical and alternative pathways of the renin–angiotensin–aldosterone system in dogs. Sci Rep. 2023;13:2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ward JL, Chou YY, Yuan L, Dorman KS, Mochel JP. Retrospective evaluation of a dose‐dependent effect of angiotensin‐converting enzyme inhibitors on long‐term outcome in dogs with cardiac disease. J Vet Intern Med. 2021;35:2102‐2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bernay F, Bland JM, Häggström J, et al. Efficacy of spironolactone on survival in dogs with naturally occurring mitral regurgitation caused by myxomatous mitral valve disease. J Vet Intern Med. 2010;24:331‐341. [DOI] [PubMed] [Google Scholar]
- 15. Coffman M, Guillot E, Blondel T, et al. Clinical efficacy of a benazepril and spironolactone combination in dogs with congestive heart failure due to myxomatous mitral valve disease: the BEnazepril Spironolactone STudy (BESST). J Vet Intern Med. 2021;35:1673‐1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Masters AK, Ward JL, Guillot E, Domenig O, Yuan L, Mochel JP. Comprehensive characterization of the effect of mineralocorticoid receptor antagonism with spironolactone on the renin‐angiotensin‐aldosterone system in healthy dogs. PLoS One. 2024;19:e0298030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mochel J, Danhof M. Chronobiology and pharmacologic modulation of the renin‐andiogensin‐aldosterone system in dogs: what have we learned? Rev Physiol Biochem Pharmacol. 2015;169:43‐69. [DOI] [PubMed] [Google Scholar]
- 18. Mochel J, Fink M, Bon C, et al. Influence of feeding schedules on the chronobiology of renin activity, urinary electrolytes and blood pressure in dogs. Chronobiol Int. 2014;31:715‐730. [DOI] [PubMed] [Google Scholar]
- 19. Mochel J, Fink M, Peyrou M, et al. Chronobiology of the renin‐angiotensin‐aldosterone system in dogs: relation to blood pressure and renal physiology. Chronobiol Int. 2013;30:1144‐1159. [DOI] [PubMed] [Google Scholar]
- 20. Domenig O, Manzel A, Grobe N, et al. Neprilysin is a mediator of alternative renin‐angiotensin‐system activation in the murine and human kidney. Sci Rep. 2016;6:33678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo Z, Poglitsch M, McWhinney BC, et al. Measurement of equilibrium angiotensin II in the diagnosis of primary aldosteronism. Clin Chem. 2020;66:483‐492. [DOI] [PubMed] [Google Scholar]
- 22. Burrello J, Buffolo F, Domenig O, et al. Renin‐angiotensin‐aldosterone system Triple‐A analysis for the screening of primary aldosteronism. Hypertens. 2020;75:163‐172. [DOI] [PubMed] [Google Scholar]
- 23. de Denus S, Leclair G, Dubé MP, et al. Spironolactone metabolite concentrations in decompensated heart failure: insights from the ATHENA‐HF trial. Eur J Heart Fail. 2020;22:1451‐1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McInnes G, Shelton J, Ramsay L, et al. Relative potency and structure activity relationships of aldosterone antagonists in healthy man: correlation with animal experience. Br J Clin Pharmacol. 1982;13:331‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. King JN, Mauron C, Kaiser G. Pharmacokinetics of the active metabolite of benazepril, benazeprilat, and inhibition of plasma angiotensin‐converting enzyme activity after single and repeated administrations to dogs. Am J Vet Res. 1995;56:1620‐1628. [PubMed] [Google Scholar]
- 26. Fyhrquist F, Saijonmaa O. Renin‐angiotensin system revisited. J Intern Med. 2008;264:224‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roig E, Perez‐Villa F, Morales M, et al. Clinical implications of increased plasma angiotensin II despite ACE inhibitor therapy in patients with congestive heart failure. Eur Heart J. 2000;21:53‐57. [DOI] [PubMed] [Google Scholar]
- 28. Ames MK, Atkins CE, Eriksson A, Hess AM. Aldosterone breakthrough in dogs with naturally occurring myxomatous mitral valve disease. J Vet Cardiol. 2017;19:218‐227. [DOI] [PubMed] [Google Scholar]
- 29. Van De Wal RMA, Plokker HWM, et al. Determinants of increased angiotensin II levels in severe chronic heart failure patients despite ACE inhibition. Int J Cardiol. 2006;106:367‐372. [DOI] [PubMed] [Google Scholar]
- 30. Geary K, Hunt M, Peach M, et al. Effects of angiotensin converting enzyme inhibition, sodium depletion, calcium, isoproterenol, and angiotensin II on renin secretion by individual renocortical cells. Endocrinology. 1992;131:1588‐1594. [DOI] [PubMed] [Google Scholar]
- 31. Ames MK, Atkins CE, Pitt B. The renin‐angiotensin‐aldosterone system and its suppression. J Vet Intern Med. 2019;33:363‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lefebvre HP, Ollivier E, Atkins CE, et al. Safety of spironolactone in dogs with chronic heart failure because of degenerative valvular disease: a population‐based, longitudinal study. J Vet Intern Med. 2013;27:1083‐1091. [DOI] [PubMed] [Google Scholar]
- 33. Thomason JD, Rockwell JE, Fallaw TK, Calvert CA. Influence of combined angiotensin‐converting enzyme inhibitors and spironolactone on serum K+, Mg 2+, and Na+ concentrations in small dogs with degenerative mitral valve disease. J Vet Cardiol. 2007;9:103‐108. [DOI] [PubMed] [Google Scholar]
- 34. Guyonnet J, Elliott J, Kaltsatos V. A preclinical pharmacokinetic and pharmacodynamic approach to determine a dose of spironolactone for treatment of congestive heart failure in dog. J Vet Pharmacol Ther. 2010;33:260‐267. [DOI] [PubMed] [Google Scholar]
- 35. Toutain PL, Lefèbvre HP. Pharmacokinetics and pharmacokinetic/pharmacodynamic relationships for angiotensin‐converting enzyme inhibitors. J Vet Pharmacol Ther. 2004;27:515‐525. [DOI] [PubMed] [Google Scholar]
- 36. Lefebvre H, Brown S, Chetboul V, King J, Pouchelon JL, Toutain P. Angiotensin‐converting enzyme inhibitors in veterinary medicine. Curr Pharm Des. 2007;13:1347‐1361. [DOI] [PubMed] [Google Scholar]
- 37. Toutain PL, Lefebvre HP, King JN. Benazeprilat disposition and effect in dogs revisited with a pharmacokinetic/pharmacodynamic modeling approach. J Pharmacol Exp Ther. 2000;292:1087‐1093. [PubMed] [Google Scholar]
- 38. Mochel JP, Teng CH, Peyrou M, Giraudel J, Danhof M, Rigel DF. Sacubitril/valsartan (LCZ696) significantly reduces aldosterone and increases cGMP circulating levels in a canine model of RAAS activation. Eur J Pharm Sci. 2019;128:103‐111. [DOI] [PubMed] [Google Scholar]
- 39. Mochel JP, Peyrou M, Fink M, et al. Capturing the dynamics of systemic renin‐angiotensin‐aldosterone system (RAAS) peptides heightens the understanding of the effect of benazepril in dogs. J Vet Pharmacol Ther. 2013;36:174‐180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Schedule and timing of blood sampling during the 4 sampling days of the study period (BAS1, BAS2, DOS1, and DOS14). RAAS, renin‐angiotensin‐aldosterone system.