

Abstract
Objective
Janus kinase family members are essential for signaling by multiple cytokines, including many implicated in systemic lupus erythematosus (SLE) pathogenesis. To test whether inhibition of JAK1 signaling can be efficacious in SLE, we used a JAK1‐selective inhibitor (ABT‐317) and evaluated its ability to ameliorate disease in murine SLE.
Methods
Efficacy of ABT‐317 was evaluated using NZB/W‐F1 mice treated prophylactically and therapeutically. Primary endpoints were proteinuria, survival, and saliva production. Other endpoints included histological analysis of kidneys and salivary glands, flow cytometric analysis of splenic cell populations, and gene expression analysis by RNA sequencing in the kidneys, salivary glands, and blood. Publicly available human kidney gene transcription data were used to assess the translatability of the mouse findings.
Results
ABT‐317 was efficacious when dosed prophylactically and prevented disease for up to two months after treatment cessation. When dosed therapeutically, ABT‐317 quickly reversed severe proteinuria and restored saliva production, as well as diminished kidney and salivary gland inflammation. ABT‐317‐induced changes in glomerular morphology coincided with normalization of a human nephrotic gene signature, suggesting translatability to human lupus nephritis (LN).
Conclusion
JAK1 inhibition prevented and reversed kidney and salivary gland manifestations of murine lupus with long‐lasting effects after treatment cessation. These data, along with the presence of JAK1 and nephrotic gene signatures in human LN glomeruli, suggest that a JAK1‐selective inhibitor may be an effective therapeutic in the treatment of human SLE and LN.
INTRODUCTION
Systemic lupus erythematosus (SLE) is an autoimmune disease triggered by a loss of immunological tolerance. 1 , 2 Clinical manifestations of SLE vary considerably, likely due to differences in the operant pathogenic mechanisms. 3 Although the full scope of mechanisms that contribute to SLE pathogenesis is unknown, multiple immunological processes, or nodes, are known to contribute. Among them are (1) B cell activation to produce autoantibodies and immune complexes that can induce an inflammatory response in the tissues where they deposit, (2) effector T cell function that can induce tissue damage at sites of inflammation, and (3) the production of high levels of type I interferon (IFN‐I) by plasmacytoid dendritic cells (pDCs) that can amplify adaptive and innate immune mechanisms and can damage tissue directly. 4 The contributions of multiple nodes to the pathogenesis of SLE may contribute to the heterogeneity of disease and poor success in developing treatments. 5 , 6 To be efficacious in most patients, we hypothesize that mechanisms critical to multiple nodes must be disrupted, either with a combination of therapeutics that each target a different node or with a single therapeutic that targets multiple nodes.
JAKs are cytoplasmic tyrosine kinases that are critical for signaling through many cytokine receptors. There are four members of the JAK family: JAKs 1 through 3 and TYK2. JAK1 is of interest because it mediates signaling of multiple cytokines implicated in SLE, including interferon (IFN)‐α/β, IFNγ, interleukin (IL)‐6, IL‐2, and IL‐21. 7 , 8 The importance of JAK/STAT‐mediated signaling to SLE is further indicated by genome‐wide association studies linking TYK2, STAT1, and STAT4 to SLE. 9 , 10 , 11 Because JAK signaling impacts cytokines known to be involved with all three pathogenic SLE nodes, JAK inhibition may be more efficacious than strategies focused on only a single node.
Here, we used a JAK1 selective inhibitor, ABT‐317, 12 in a mouse model of SLE and lupus nephritis (LN) to determine if JAK inhibition could ameliorate disease. We found that ABT‐317 prevented healthy animals from developing nephritis, with a durable response out to two months after treatment cessation. Most significantly, ABT‐317 reversed severe proteinuria and decreased kidney inflammation in mice with established disease. Additionally, ABT‐317 restored saliva production and reduced lymphocytic infiltration of the salivary gland, two manifestations of Sjögren disease (SD). 13 , 14 These data, coupled with normalization of disease‐related gene signatures in both kidney and salivary glands as well as transcriptional similarities between mouse and human LN, support investigation of a JAK1‐selective inhibitor in human SLE and LN.
MATERIALS AND METHODS
Mice
Female NZB/W‐F1 mice (Jackson) at 24 through 29 weeks of age were used for all studies. Mice were housed in an association for assessment and accreditation of laboratory animal care (AAALAC) accredited specific pathogen free (SPF) facility under a 12/12‐hour light/dark cycle with food and water provided ad libitum. All studies involving animals were reviewed and approved by the AbbVie institutional animal care and use committee (IACUC).
ABT‐317
ABT‐317 is a JAK1 selective inhibitor (previously described) 12 that was synthesized at AbbVie Bioresearch Center in Worcester, Massachusetts. ABT‐317 is significantly structurally different from upadacitinib but is only slightly less potent and selective for JAK1.
Therapeutic treatment
Mice (starting at 29 weeks old; average 35 weeks old) were enrolled into treatment groups, started on treatment, and tracked for proteinuria and recovery as previously described 15 (n = 15–19). Mice exhibiting proteinuria ≥300 mg/dL before 25 weeks old were excluded from the study. Plasma was collected retro‐orbitally at time of enrollment and on day 57 for anti–double‐stranded (ds)DNA antibody measurement. ABT‐317 (60 mg/kg) was formulated and administered once daily by oral gavage. Different from the prophylactic study described in Supplementary Methods 1 in which CellCept was used as a positive control, here anti‐CD40 (201A3, 15 mg/kg) 15 was formulated in phosphate buffered saline and administered by intraperitoneal injection twice weekly as a positive control. Blood, spleens, kidneys, and salivary glands were collected from moribund animals as needed and at termination 105 days after study initiation (number of days on treatment varies due to rolling enrollment of mice). Spleens were cut and (n = 5 per group) and end pieces were used for fluorescence‐activated cell sorting (FACS) staining. Kidneys and salivary glands were processed as described previously. 15 Four experiments were conducted; data shown are from a single experiment.
Flow cytometry analysis
Staining was performed on lysed whole blood and splenocytes. Antibodies used for identifying T, B, and natural killer cell subsets can be found in Supplementary Table 1. AccuCount particles (Spherotech) were added before acquisition to obtain cell counts. Samples were acquired on a BD LSRFortessa (BD Biosciences), and data were analyzed using FlowJo software (Tree Star, Inc.).
Anti‐dsDNA antibody enzyme‐linked immunosorbent assay
Plate coating, buffers, and incubation times were as previously described. 15 Antibody titer is represented as the 50% maximum response concentration of a titration curve for each mouse.
Histological analysis of kidney and salivary gland sections
Methods for kidney and salivary gland processing, embedding, and sectioning were as previously described. 15 Glomerular disease and tubular dilation in the kidney and infiltrates in the salivary gland were assessed by hematoxylin and eosin (H&E). 15 Immunohistochemistry for IBA1, CD3, podoplanin, and pSTAT1/3 was performed on the Leica Bond Rx autostainer (Leica Biosystems). All reagent details can be found in Supplementary Table 2. For analysis of glomerular size, kidneys were stained for podoplanin, and the Leica Bond Polymer Refine Red Detection kit was used for detection. Images were analyzed in Visiopharm Image Analysis software, measuring podoplanin‐positive glomeruli and kidney size. pSTAT1 was stained using HIER 1 citrate buffer pH 6, Dako dual endogenous block, Dako protein block, and rabbit monoclonal anti‐pSTAT1 0.25 μg/mL; pSTAT3 was stained using HIER 2 EDTA buffer pH 9 and rabbit monoclonal anti‐pSTAT3 0.45 μg/mL. Detection and visualization were performed with goat anti‐rabbit HRP and DAB and hematoxylin counterstain, including rabbit monoclonal IgG isotype controls in each run. Slides were imaged on a brightfield whole slide scanner (P250, 3DHistotech). All histology samples were semiquantitatively scored by a pathologist according to criteria outlined in Supplementary Tables 3 through 6.
Blood, kidney, and salivary gland analysis for gene expression
Methods for processing tissues for RNA extraction and quantifying gene expression were as previously outlined. 15 All gene signatures were calculated as a single‐sample gene‐set enrichment analysis enrichment score. Type I IFN signature was defined as Ifi27, Ifi44, Ifi44l, Ifi6, and Rsad2. The 44‐gene JAK1 signature was defined using the Ingenuity Pathway Analysis database by extracting genes known to be up‐regulated by JAK1 activation and eliminating IFN‐I‐regulated genes (Supplementary Table 5). The chemokine, nephrotic, injury‐associated, and collagen gene signatures were previously defined. 15
Pilocarpine‐induced saliva collection
Saliva induction and collection was conducted as previously described 15 and performed on day 97 of the therapeutic study. Collections for proteinuric and nonproteinuric control mice were conducted 41 days before.
RESULTS
Reversal of relevant endpoints by ABT‐317
We first characterized ABT‐317 in a prophylactic dose response to assess efficacy in a spontaneous SLE model (Supplementary Methods 1 and Supplementary Figure 1). Female NZB/W‐F1 mice were treated beginning at 25 weeks of age before the onset of proteinuria. ABT‐317 was administered once daily (SID; 120 mg/kg) or twice daily (BID; 60, 30, or 10 mg/kg). CellCept was administered SID (100 mg/kg) as a separate positive control. After 16 weeks of treatment, all BID treatment regimens were converted to SID dosing, and mice followed for an additional 9 weeks (Supplementary Figure 1A), during which only mice in the 10 mg/kg cohort developed severe proteinuria (urine protein >300 mg/dL). Treatment was withdrawn at this point, whereupon we observed a dose‐dependent re‐emergence of disease. However, all mice in the 60 and 120 mg/kg dose cohorts remained nonproteinuric for as long as 9 weeks (Supplementary Figure 1A), indicating that ABT‐317 can provide long‐lasting protection from proteinuria. Survival data were reflective of proteinuria development (Supplementary Figure 1B). In addition, ABT‐317 treatment prevented the expected increase in anti‐dsDNA antibodies and reduced the number of circulating B cells by week 51 (Supplementary Figures 1C and D). B cell numbers appear to have increased somewhat during the nine weeks after the treatment phase.
Having established an efficacious dose in the prophylactic treatment study, we aimed to assess the effect of ABT‐317 on established disease. Animals were treated after the onset of severe proteinuria (two consecutive urine protein measurements of ≥300 mg/dL, 24 hours apart). The anti‐CD40 antagonist antibody 201A3 was used as a positive control. 15 Similar to anti‐CD40, ABT‐317 significantly reversed proteinuria, prolonged survival, and reduced anti‐dsDNA antibody levels compared to the vehicle at the midpoint of the study (Figure 1A–C). In addition, ABT‐317 reduced the numbers of B cells, splenic germinal center B cells, plasmablasts, and plasma cells (Figure 1D), likely explaining the decrease in anti‐dsDNA antibodies. We also observed decreases in total, activated, and central memory T cell numbers (Supplementary Figure 2), supporting the hypothesis that inhibiting JAK1 will impact multiple pathogenic nodes.
Figure 1.
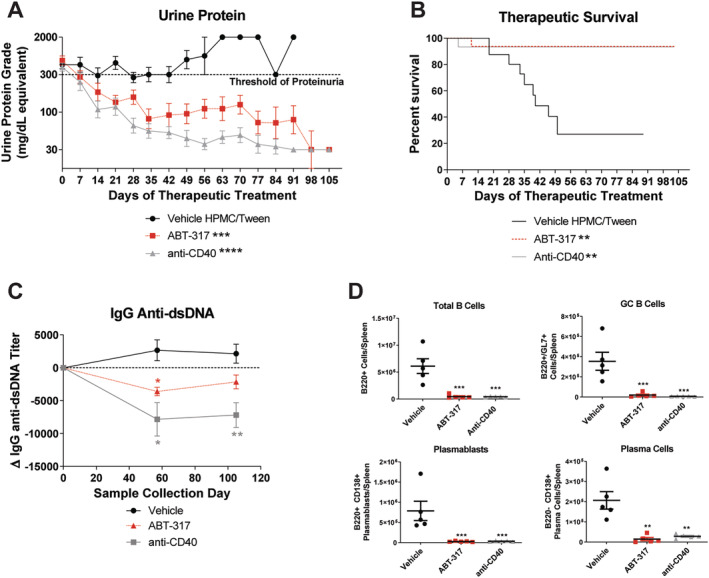
ABT‐317 reversed severe proteinuria, circulating anti‐dsDNA antibody titers, and splenic B cell subsets. Therapeutic oral treatment of female NZB/W‐F1 mice with vehicle (n = 19), 60 mg/kg SID of ABT‐317 (n = 16), or 15 mg/kg of anti‐CD40 (n = 15) began when mice became severely proteinuric. (A) The average urine protein is graphed and ABT‐317 shows reversal of proteinuria comparable to 15 mg/kg of anti‐CD40; **P < 0.01 vs vehicle ***P < 0.001; ****P < 0.0001 vs vehicle using one‐way ANOVA, Kruskall‐Wallis test with Dunn's multiple comparisons. (B) Survival percentage showing 95% survival in ABT‐317‐treated mice; **P < 0.01 vs vehicle using log‐rank Mantel‐Cox test. (C) ABT‐317 significantly reduced anti‐dsDNA antibody titers at study day 57; *P < 0.05; **P < 0.01 vs vehicle using mixed‐effects analysis with Tukey's multiple comparisons. (D) The number of B cells (B220+), splenic GC B cells (B220+/GL7+), plasmablasts (B220+/CD138+), and plasma cells (B220‐/CD138+) was determined at the end of the study by flow cytometry and significant reduction of each subset by ABT‐317 was observed; **P < 0.01; ***P < 0.001 vs vehicle using one‐way ANOVA with Dunnett's multiple comparisons. ANOVA, analysis of variance; dsDNA, double‐stranded DNA; GC, germinal center; HPMC, hydroxypropyl methylcellulose; SID, once daily.
Impact of ABT‐317 on kidney inflammation and morphology
To assess the effect of ABT‐317 on established kidney inflammation, a combination of immunohistochemistry and H&E staining was used. ABT‐317‐treated mice had significantly fewer T cell (CD3+) and myeloid cell (IBA1+) infiltrates than vehicle‐treated mice at the end of the study (Figure 2A and B). They also had fewer infiltrates compared to baseline (collected at disease onset), although the difference was not significant (Figure 2B). Kidney damage, as assessed by glomerular disease and tubular dilation scores (Supplementary Table 4), 15 appeared less extensive in the ABT‐317 treated mice, though neither endpoint resulted in statistical significance versus vehicle control (Figure 2C and D). Glomerular size was assessed using podoplanin staining and quantitative image analysis. At study end, the average glomerular size in ABT‐317‐treated mice was significantly lower than in vehicle control mice (Figure 2C and E). All of these inflammatory and morphological changes were similar to those induced by anti‐CD40 treatment. Overall, ABT‐317 prevented an increase in kidney inflammation and protected mice from the accrual of further glomerular and tubular damage. Whether ABT‐317 can reverse kidney damage will require further analysis.
Figure 2.
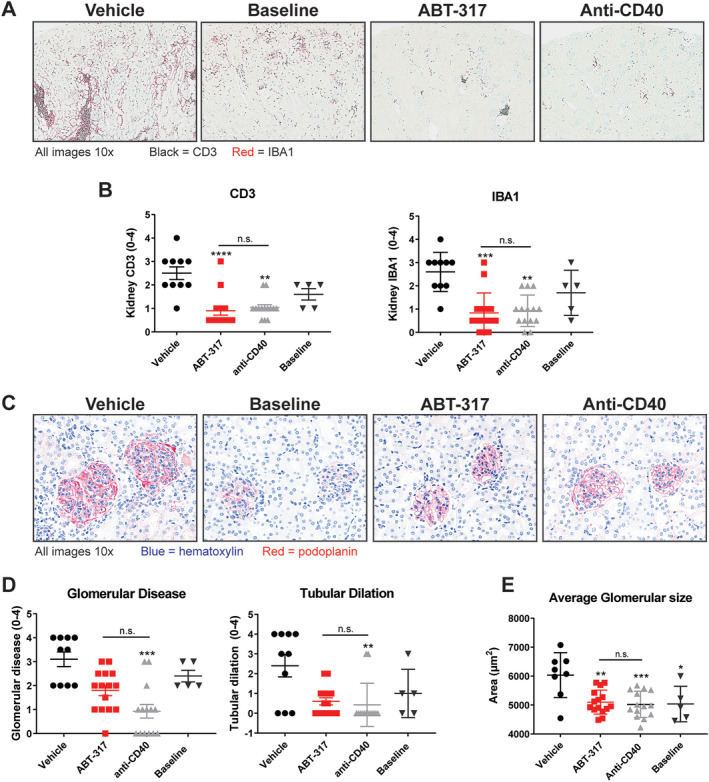
ABT‐317 reduced kidney inflammation, glomerular disease, and tubular dilation. The middle section of the kidneys was collected and fixed in formalin, followed by paraffin embedding, cutting, and staining for CD3 and IBA1. Representative baseline samples were collected at time of disease onset, whereas all other groups were collected at time of euthanasia (≥89 days on treatment). (A) Representative images of kidneys showing CD3 and IBA1 staining for T cells and myeloid cells, respectively. (B) Semiquantitative scoring of CD3 and IBA1 (n = 15) staining showed that ABT‐317 was efficacious at reducing T cells and myeloid cells, both indicators of overall inflammation. **P < 0.01; ***P < 0.001; ****P < 0.0001 vs vehicle using Kruskal‐Wallis test with Dunn's multiple comparisons. (C) Kidneys were prepared as described above but stained with H&E and podoplanin. Representative images of glomeruli and tubules show morphological changes in diseased mice. (D) Semiquantitative scoring (n = 15) revealed that, although ABT‐317 did not show significant reduction in glomerular disease and tubular dilation, results were not different from those observed with anti‐CD40, **P < 0.01; ***P < 0.001 vs vehicle using Kruskal‐Wallis test with Dunn's multiple comparisons. (E) Image analysis of podoplanin staining showed a significant decrease in average glomerular size with ABT‐317 treatment. *P < 0.05; **P < 0.01; ***P < 0.0001 vs vehicle using one‐way ANOVA with Tukey's multiple comparisons. ANOVA, analysis of variance; H&E, hematoxylin and eosin; n.s., not significant.
Normalization of disease‐relevant gene signatures
We examined the JAK1, IFN, and chemokine gene signatures, all of which are significant to SLE pathogenesis. The IFN and chemokine signature genes were defined previously. 15 The JAK1 signature consists of 44 genes assigned to the JAK1 pathway by Ingenuity Pathway Analysis and from which IFN‐regulated genes have been eliminated (Supplementary Table 7). Aside from the JAK1 signature in blood, these three signature scores were elevated in the kidneys and blood at baseline and in vehicle‐treated mice compared to prediseased mice. All signatures were normalized to predisease levels by ABT‐317 and anti‐CD40 treatment (Figure 3A and B).
Figure 3.
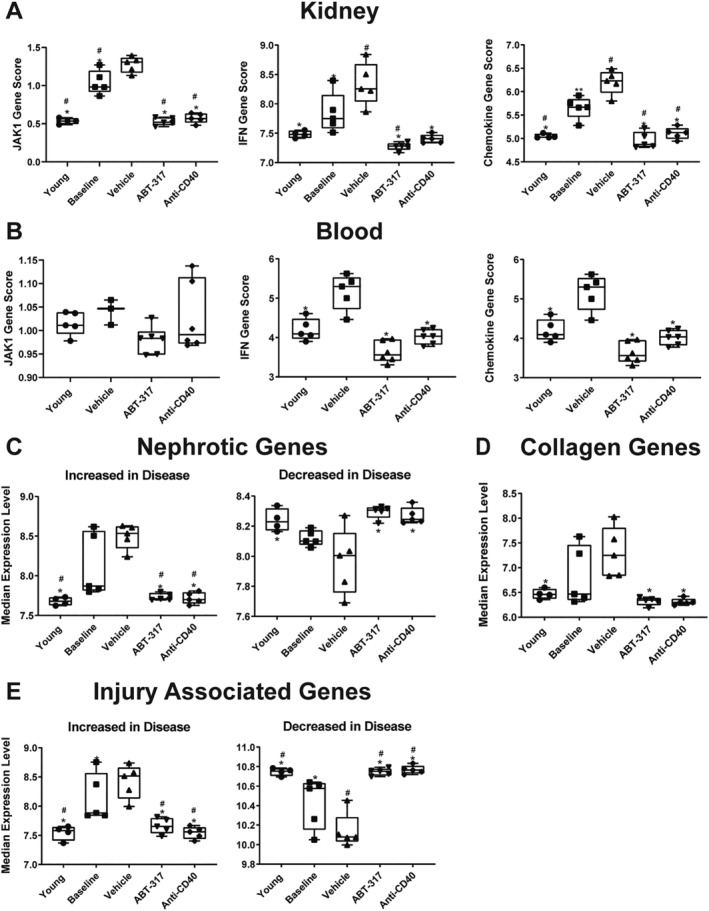
ABT‐317 normalized several SLE‐associated gene signatures in the kidney and blood. Kidney and blood were collected at the conclusion of the study and RNAseq was used to determine the JAK1 (44 genes after removal of IFN‐regulated genes), IFN (Ifi27, Ifi44, Ifi44l, and Rsad2), chemokine (31 genes up‐regulated in diseased NZB/W‐F1 kidneys [n = 15]), nephrotic (19 genes associated with nephrotic syndrome in humans), injury‐associated (13 genes encoding proteins associated with epithelial/endothelial cell injury and tubulointerstitial fibrosis), and collagen (18 genes up‐regulated in diseased NZB/W‐F1 kidneys [n = 15]) gene signatures, represented as median gene expression level. (A, B) The JAK1, IFN, and chemokine signature scores were normalized in the kidney and blood (with nonsignificant reduction of JAK1 score in blood) to scores assessed in young healthy mice after treatment with ABT‐317. (C) The nephrotic up‐ or down‐gene signature scores were normalized to scores assessed in young healthy mice with ABT‐317 treatment. (D) The collagen gene signature score was normalized to scores assessed in young healthy mice by ABT‐317 treatment. (E) The injury‐associated up‐ or down‐gene signature scores were normalized to scores assessed in young healthy mice with ABT‐317 treatment. *P < 0.05; **P < 0.01 vs vehicle; #P < 0.05 vs baseline using one‐way ANOVA with Tukey's multiple comparisons. ANOVA, analysis of variance; IFN, interferon; RNAseq, RNA sequencing; SLE, systemic lupus erythematosus.
Genes that may play a significant role in the kidney changes preceding nephritis in this model align with many of the genes implicated in human nephrotic syndrome. 16 Here, we looked at 19 genes previously defined as comprising the nephrotic gene signature 15 and found that subsets were either up‐ or down‐regulated in vehicle‐treated mice relative to young prediseased mice. ABT‐317 normalized nephrotic gene signature expression to that of young mice (Figure 3C). With one exception, these genes are not expressed by inflammatory cells, 16 arguing that the normalization of the nephrotic gene signature is not directly due to the elimination of inflammatory cells.
We also examined a group of 18 collagen genes known to be associated with fibrosis 17 and up‐regulated in kidneys of NZB/W‐F1 mice, which we used to generate a signature that was up‐regulated in vehicle‐treated mice and reduced by ABT‐317 treatment to levels observed in young mice (Figure 3D). Finally, we evaluated a group of genes expressed by glomerular or tubular cells that encode proteins associated with kidney injury (epithelial or endothelial cell injury and tubular interstitial fibrosis). 15 We used these genes to generate an injury signature score that was either elevated or decreased in vehicle‐treated mice relative to young prediseased mice and was normalized with ABT‐317 treatment to levels observed in young mice (Figure 3E). Collectively, ABT‐317 normalized multiple disease‐associated gene signatures.
Changes in disease pathway gene expression
Transcriptome‐wide profiling of the kidney and salivary gland was used to identify genes affected by ABT‐317, and differentially expressed genes were analyzed by Ingenuity Pathway Analysis 18 software to identify the specific pathways affected by disease and treatment (Supplementary Methods 2). Comparison of young prediseased and vehicle‐treated mice indicated that 80 inflammatory pathways in the kidney were up‐regulated with disease (Supplementary Figure 3A). In addition, six metabolic pathways were down‐regulated in the kidney. ABT‐317 largely normalized the expression level of all these pathways such that they resembled the levels seen in young prediseased mice (Supplementary Figure 3A), nullifying the changes in inflammatory and metabolic pathways induced by disease.
Reversal of Sjögren disease phenotype
In addition to nephritis, NZB/W‐F1 mice develop salivary gland inflammation and a decreased ability to produce saliva that coincides with the development of proteinuria and is consistent with reduced saliva production in secondary SD. 19 , 20 We reported previously that these mice produced about half as much saliva as young prediseased mice and that anti‐CD40 treatment increased saliva production from baseline. 15 H&E staining of the salivary glands and analysis by semiquantitative scoring revealed that treatment with ABT‐317 reduced overall inflammation, comparable to anti‐CD40 treatment (Figure 4A and B). Likewise, salivary gland function was restored as evidenced by a significant increase in saliva output compared to vehicle controls (Figure 4C). Much like in the kidneys, ABT‐317 treatment normalized the JAK1, IFN, and chemokine gene signatures in the salivary glands (Figure 4D). ABT‐317 also normalized the 28 inflammatory pathways identified by Ingenuity Pathway Analysis to be significantly up‐regulated in disease (Supplementary Figure 3B), therefore nullifying the changes in inflammatory pathways of salivary glands induced by disease.
Figure 4.
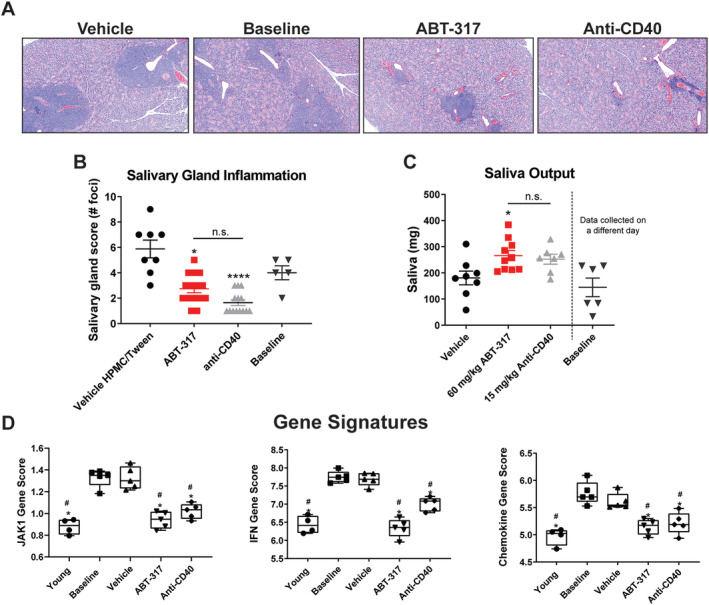
ABT‐317 reduced inflammation, restored function, and normalized SLE gene signatures in the salivary glands. Salivary glands were collected and fixed in formalin, followed by paraffin embedding, cutting, and H&E staining. Baselines were collected at time of treatment onset, whereas all other groups were collected at time of euthanasia (study day 105, ≥89 days on treatment). (A) Representative images of H&E‐stained salivary glands are shown. (B) Salivary gland images were semiquantitatively scored for inflammation (n = 15), revealing significant reduction of cellular infiltrates with ABT‐317 treatment, comparable to anti‐CD40 treatment. *P < 0.05; ****P < 0.0001 vs vehicle using Kruskal‐Wallis test with Dunn's multiple comparisons. (C) Saliva was induced by i.p. injection of pilocarpine nitrate and saliva collected using preweighed cotton swabs. The output was determined in milligrams by weighing swabs after collection. *P < 0.05 vs vehicle using one‐way ANOVA with Tukey's multiple comparisons. (D) The JAK1, IFN, and chemokine gene signature scores were normalized in the salivary gland to scores assessed in young healthy mice after treatment with ABT‐317, comparable to anti‐CD40 treatment. *P < 0.05 vs vehicle; #P < 0.05 vs baseline using one‐way ANOVA with Tukey's multiple comparisons. ANOVA, analysis of variance; H&E, hematoxylin and eosin; HPMC, hydroxypropyl methylcellulose; IFN, interferon; i.p., intraperitoneal; n.s., not significant; SLE, systemic lupus erythematosus.
Reduction of STAT signaling in the kidney
The normalization of the nephrotic and injury‐associated gene signatures by treatment with ABT‐317 suggests that the elevated signature scores in diseased mice is caused by cytokine signaling in glomerular and tubular cells. To identify cells actively engaged in cytokine signaling, we stained kidney sections for phosphorylated STAT1 (pSTAT1) and phosphorylated STAT3 (pSTAT3). Figure 5A shows pSTAT3 staining of inflammatory, glomerular, and tubular cells within the kidneys, suggesting that cytokine signaling is active in the kidney. pSTAT1 staining was similar (data not shown). ABT‐317 treatment significantly decreased both pSTAT levels (Figure 5B). This could be due to direct inhibition of cytokine signaling in the kidney or secondary to the reduction in T cells induced by ABT‐317 treatment. Regardless, the data are consistent with the ability of JAK1 inhibition to reverse proteinuria.
Figure 5.
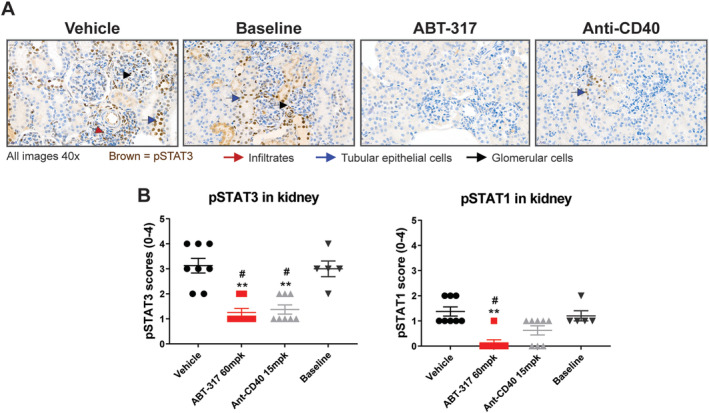
ABT‐317 reduced phospho‐STAT1 and pSTAT3 expression in the kidneys. Kidneys were collected at the conclusion of the study and stained for pSTAT1 and pSTAT3 to generate histological scores. (A) Representative images of pSTAT3 staining, showing an up‐regulation of pSTAT3 in resident glomerular and tubule epithelial cells, as well as infiltrating immune cells in baseline and vehicle‐treated mice, with reduction of PSTAT3 staining in ABT‐317 and anti‐CD40‐treated mice (B) ABT‐317 significantly reduced expression of pSTAT1 and pSTAT3 in the kidneys of NZB/W‐F1 mice. **P < 0.01 vs vehicle; #P < 0.05 vs baseline using Kruskal‐Wallis statistical test with Dunn's multiple comparisons. pSTAT1, phosphorylated STAT1; pSTAT3, phosphorylated STAT3.
Translatability of JAK1‐specific signaling in human LN
To assess the translatability of our findings to human SLE and LN, we explored whether human LN kidneys exhibited gene expression changes similar to proteinuric NZB/W‐F1 kidneys. We examined the publicly available gene expression data (GSE32591) from isolated glomerular and tubular cells from 15 healthy donors (HDs) and 32 patients with LN (Figure 6A–D). Glomerular and tubular cells were separately isolated by microdissection and subjected to transcriptomic analysis. Both cell populations from patients with LN exhibited a strong IFN signature in comparison to HDs (Figure 6A). However, in comparison to HD, LN glomeruli exhibited a significant increase in the JAK1 signature score, whereas LN tubules showed a significant decrease (Figure 6B). This suggests that either cytokine exposure or receptor expression by glomeruli and tubules is not the same.
Figure 6.
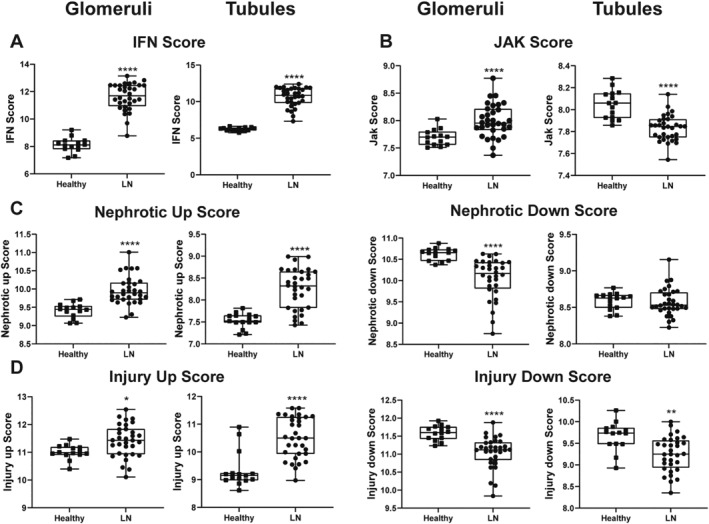
Gene signature scores in human glomeruli and tubules demonstrate model translatability. Publicly available gene expression data from isolated glomerular and tubular cells from 15 HDs and 32 patients with LN were examined for gene signatures similar to those identified in NZB/W‐F1 mice. (A) Patients with LN exhibit a significantly elevated IFN signature in glomeruli and tubules compared to HDs. (B) Patients with LN exhibit a significant increase in the JAK1 signature in glomeruli but a significant decrease in JAK1 signature in tubules compared to HDs. (C, D) Nephrotic and injury‐associated up‐ and down‐gene signatures were significantly different between LN glomeruli and tubules compared to HDs. *P < 0.05; **P < 0.01; ****P < 0.0001 vs HDs. HD, healthy donor; IFN, interferon; LN, lupus nephritis.
Nephrotic and injury‐associated gene expression was also different between HDs and patients with LN. Like the mouse kidney analysis, 15 the nephrotic genes from Park et al 16 that differed in expression between LN and HDs were used to determine a gene signature score. The identified signature contained 12 genes that overlapped with mouse (Supplementary Figure 4). As shown in Figure 6C, the nephrotic up‐gene signature was significantly different between LN and HD glomeruli and tubules, whereas the nephrotic down‐gene signature was significantly different only in the glomeruli. The injury‐associated up‐ and down‐gene signatures were significantly different between LN and HDs in both cell types (Figure 6D). Thus, the gene signatures associated with proteinuria that were normalized by ABT‐317 treatment in mice were altered in the glomeruli and tubules of human LN. Given the central role of the glomerulus in controlling urine protein levels, the strong JAK1 signature and altered expression of nephrotic genes suggests that a JAK1 inhibitor could be efficacious in reversing proteinuria in human LN.
DISCUSSION
JAK inhibitors have demonstrated clinical efficacy in a wide array of immunological diseases, including rheumatoid and psoriatic arthritis, atopic dermatitis, and ulcerative colitis. 21 , 22 Because the JAK–STAT pathway is crucial for signaling by a multitude of cytokines implicated in SLE pathogenesis, targeting JAK could be an effective treatment for SLE. Preclinical studies in murine SLE with tofacitinib and baricitinib have shown that JAK inhibition ameliorates disease in these animals. 23 , 24 , 25 Tofacitinib is considered a pan‐JAK inhibitor (with preference for JAK3 over 1 and 2) and baricitinib is a JAK1/2 inhibitor. 21 Here, we have described the ability of ABT‐317, a selective JAK1 inhibitor, to both prevent and reverse proteinuria, and to reduce salivary gland inflammation in the NZB/W‐F1 mouse model of SLE.
Prophylactic treatment with ABT‐317 resulted in sustained protection against proteinuria (Supplementary Figure 1A). Higher doses of ABT‐317 demonstrated a greater suppressive effect and offered long‐term protection against proteinuria following treatment cessation that was profoundly different from CellCept, a commonly used therapeutic in LN. 26 , 27 , 28 We speculate that ABT‐317 prevents and possibly reverses the activation of a disease critical mechanism(s) that results in autoreactive B cell activation and that time is required following treatment cessation to reset this mechanism. In agreement with this, all doses of ABT‐317 reduced the number of circulating B cells and prevented the increase in anti‐dsDNA antibodies (Supplementary Figure 2C and D). This is also consistent with the efficacy of prophylactic treatment with anti‐CD20 and the subsequent five‐month delay in disease development after B cell depletion, 29 as well as with the drug‐free remission induced in patients with SLE depleted of B cells by CD19 CAR T cell treatments that lasts long after B cell reconstitution. 30
Although prophylactic efficacy with ABT‐317 demonstrates proof of concept for using a JAK1 inhibitor in SLE, the ability to reverse established disease is a crucial characteristic for a potential SLE treatment. Few therapeutics have demonstrated reversal of severe proteinuria in mice. We previously reported therapeutic efficacy using the anti‐CD40 antagonist antibody, 201A3. 15 Other efficacious molecules tested in similar therapeutic models include an anti‐CD40L Fab′ fragment (CDP7657) 31 and the topoisomerase I inhibitor irinotecan. 32 , 33 Here, we have demonstrated the ability of ABT‐317 to reverse proteinuria comparably to anti‐CD40 (Figure 1A), with a subsequent survival rate of approximately 95% (Figure 1B). Treatment‐induced reduction of splenic B and T cell populations (Figure 1D and Supplementary Figure 2) likely contributed to the overall reduction of pathogenic B and T cell–driven inflammation, resulting in restoration of kidney function and validating the efficacy of JAK1 inhibition in reversing established murine disease.
JAK1 inhibition should affect signaling by multiple cytokines involved in SLE, 7 , 8 and we hypothesize that ABT‐317's efficacy in this model is driven by its combined impact on the three pathogenic nodes described above. For example, inhibition of IL‐6 and IL‐21 signaling would affect the B cell node by impacting plasma cell differentiation, consistent with the observed reduction of anti‐dsDNA titers and splenic germinal center B cells, plasmablasts, and plasma cells versus vehicle‐treated mice (Figures 1C and D). Although circulating IFN‐I levels are not elevated in NZB/W‐F1 mice, ABT‐317 likely still affects the IFN‐I node based on the observed elevated IFN‐I gene signature in blood, kidney, and salivary gland that was normalized by therapeutic ABT‐317 treatment (Figures 3A and B, Figure 4D). Finally, the T cell node could be affected by inhibition of cytokines such as IL‐2, IL‐6, IL‐7, and IL‐21, accounting in part for the observed reduction of total splenic, activated, and central memory T cells in therapeutically treated mice (Supplementary Figure 2). IL‐2 is required for regulatory T cell (Treg) differentiation but the effect of ABT‐317 on Treg cells is unclear. Because ABT‐317 reversed disease, any loss of Treg function may have been offset by the profound anti‐inflammatory effect. Thus, ABT‐317's efficacy is likely due to the combined inhibition of multiple cytokines affecting multiple pathogenic mechanisms.
ABT‐317's effect on the kidney is significant. Histological analysis revealed significantly fewer inflammatory cells in the kidneys of ABT‐317 treated mice compared to diseased animals (Figure 2A and B). Consequently, glomerular size was reduced (Figure 2C and E) with an overall lesser extent of glomerular disease and tubular dilation (Figure 2D). The positive effects on kidney inflammation and morphology could be traced to ABT‐317's efficacy at a gene expression level. The JAK1, IFN, and chemokine gene signatures were normalized by ABT‐317 to reflect those of young, healthy mice in both the kidneys and blood (Figure 3A and B), and RNA sequencing analysis of kidneys indicates that the active inflammatory pathways at baseline are silenced by ABT‐317 treatment (Supplementary Fig. 3A). It is possible that inflammatory cells are migrating out of the tissues, as suggested by the reduction of the chemokine signature (Figure 3A and B), or that apoptosis is occurring because the IL‐2 family cytokines provide T cell survival signals and are JAK1‐dependent. 34 , 35
Although the elimination of inflammatory cells from the kidney may be necessary for proteinuria reversal, we previously proposed that it is not sufficient because prednisolone eliminated inflammatory cells but did not reverse proteinuria. 15 We think that restoring normal expression of genes linked to human nephrotic syndrome is likely critical to the reversal of severe proteinuria in NZB/W‐F1 mice. Most of these genes are expressed by podocytes, which are cells crucial to the integrity of the glomerular filtration barrier. Restoring the glomerular filtration barrier and glomerular morphology may rely on normalizing expression of these genes. 15 Treatment with a JAK1 inhibitor had the same effect as anti‐CD40 on glomerular morphology (Figure 2C–E) and nephrotic gene expression (Figure 3C) suggesting that they affect the same pathogenic pathway. Our findings suggest that the ability of JAK1 inhibition to reverse proteinuria and restore glomerular function is dependent on restoring function of the kidney parenchymal cells.
Because secondary SD is a common comorbidity for patients with SLE 36 , 37 and the NZB/W‐F1 model is validated for studying both SLE and SD, 15 , 19 , 20 we investigated salivary gland manifestations in addition to SLE endpoints. Although many of the preclinical studies for SD have been conducted in mice or modalities other than the NZB/W‐F1 model, 38 , 39 , 40 , 41 , 42 data have been generated to suggest that STAT3 signaling may be an important mechanism in disease. 41 , 42 For example, metformin indirectly inhibits STAT3 signaling through AMP‐activated protein kinase activation 18 , 41 , 43 and demonstrated reduction of salivary gland infiltrates and significant improvement in salivary output in NOD/ShiLtJ mice. 41 Here, we have demonstrated that therapeutic administration of ABT‐317 significantly reduced salivary gland infiltrates and restored saliva output in NZB/W‐F1 mice (Figure 4A–C) while also normalizing the salivary gland JAK1, IFN, and chemokine gene signatures (Figure 4D) and silencing active inflammatory pathways (Supplementary Figure 3B). Although we did not stain the salivary glands for pSTAT3, we can infer that a similar reduction as seen in the kidneys (Figure 5A and B) occurred in the salivary glands, resulting in efficacy. Collectively, these data represent a novel finding with a JAK1 inhibitor that may be translatable to the clinic.
Baricitinib showed promising results in a phase II trial in SLE, with significant improvement in disease activity over placebo, as measured by Systemic Lupus Erythematosus Disease Activity Index 2000 and Systemic Lupus Erythematosus Responder Index 4, driven by the number of tender and swollen joints and rash. 44 Unfortunately, baricitinib failed to meet its primary endpoints in a phase III SLE trial, though any positive effect of baricitinib in this trial may have been masked by a high placebo rate or inability to dose to full JAK occupancy. 45 , 46 Our analysis of gene expression in microdissected human glomeruli and tubules (Figure 6) supports the use of a JAK1 inhibitor in LN. Although the IFN‐I signature is present in both the glomeruli and tubules, consistent with systemic exposure, the JAK1 gene signature is evident only in the glomeruli. In addition, human LN glomeruli exhibit an altered nephrotic gene signature and thus, as with NZB/W‐F1 mice, we suggest that JAK1 inhibition in human LN will block JAK1‐mediated cytokine signaling and normalize the nephrotic gene signature leading to the reversal of proteinuria.
In summary, we have shown efficacy with a JAK1 selective inhibitor in the NZB/W‐F1 murine model of SLE. ABT‐317 both prevented the development of proteinuria and reversed established severe proteinuria. Moreover, ABT‐317 restored salivary gland function by reducing cellular infiltrates, representing a novel finding with a JAK1 inhibitor that may be translatable to the clinic. Finally, we observed similarities between mice and humans on disease‐ and pathway‐relevant gene signatures, suggesting the potential for translatability to the clinic. Indeed, upadacitinib, a JAK1‐selective inhibitor met its phase II primary endpoint and is currently in a phase III trial. Altogether, a JAK1‐selective inhibitor fulfills our criteria for a successful therapeutic in SLE and may be efficacious in the treatment of established human disease.
AUTHOR CONTRIBUTIONS
All authors contributed to at least one of the following manuscript preparation roles: conceptualization AND/OR methodology, software, investigation, formal analysis, data curation, visualization, and validation AND drafting or reviewing/editing the final draft. As corresponding author, Ms. Twomey confirms that all authors have provided the final approval of the version to be published, and takes responsibility for the affirmations regarding article submission (eg, not under consideration by another journal), the integrity of the data presented, and the statements regarding compliance with institutional review board/Helsinki Declaration requirements.
ROLE OF THE STUDY SPONSOR
AbbVie participated in the interpretation of data, review, and approval of the publication. Publication of this article was contingent upon approval by AbbVie, Inc.
Supporting information
Disclosure form:
Appendix S1. Supporting Information.
Supported by AbbVie, Inc.
Additional supplementary information cited in this article can be found online in the Supporting Information section (https://acrjournals.onlinelibrary.wiley.com/doi/10.1002/acr2.11745).
Author disclosures are available at https://onlinelibrary.wiley.com/doi/10.1002/acr2.11745.
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1. Yaniv G, Twig G, Shor DB, et al. A volcanic explosion of autoantibodies in systemic lupus erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmun Rev 2015;14:75–79. [DOI] [PubMed] [Google Scholar]
- 2. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997;40:1725. [DOI] [PubMed] [Google Scholar]
- 3. Ceccarelli F, Perricone C, Borgiani P, et al. Genetic factors in systemic lupus erythematosus: contribution to disease phenotype. J Immunol Res 2015;2015:745647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mohan C, Putterman C. Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nat Rev Nephrol 2015;11:329–341. [DOI] [PubMed] [Google Scholar]
- 5. Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 2003;197:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Han GM, Chen SL, Shen N, et al. Analysis of gene expression profiles in human systemic lupus erythematosus using oligonucleotide microarray. Genes Immun 2003;4:177–186. [DOI] [PubMed] [Google Scholar]
- 7. Kawasaki M, Fujishiro M, Yamaguchi A, et al. Possible role of the JAK/STAT pathways in the regulation of T cell‐interferon related genes in systemic lupus erythematosus. Lupus 2011;20:1231–1239. [DOI] [PubMed] [Google Scholar]
- 8. Ohl K, Tenbrock K. Inflammatory cytokines in systemic lupus erythematosus. J Biomed Biotechnol 2011;2011:432595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bolin K, Sandling JK, Zickert A, et al. Association of STAT4 polymorphism with severe renal insufficiency in lupus nephritis. PLoS One 2013;8:e84450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen L, Morris DL, Vyse TJ. Genetic advances in systemic lupus erythematosus: an update. Curr Opin Rheumatol 2017;29:423–433. [DOI] [PubMed] [Google Scholar]
- 11. Zheng J, Yin J, Huang R, et al. Meta‐analysis reveals an association of STAT4 polymorphisms with systemic autoimmune disorders and anti‐dsDNA antibody. Hum Immunol 2013;74:986–992. [DOI] [PubMed] [Google Scholar]
- 12. Ge T, Jhala G, Fynch S, et al. The JAK1 Selective inhibitor ABT 317 blocks signaling through interferon‐γ and common γ chain cytokine receptors to reverse autoimmune diabetes in NOD mice. Front Immunol 2020;11:588543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Patel R, Shahane A. The epidemiology of Sjögren's syndrome. Clin Epidemiol 2014;6:247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alani H, Henty JR, Thompson NL, et al. Systematic review and meta‐analysis of the epidemiology of polyautoimmunity in Sjögren's syndrome (secondary Sjögren's syndrome) focusing on autoimmune rheumatic diseases. Scand J Rheumatol 2018;47:141–154. [DOI] [PubMed] [Google Scholar]
- 15. Perper SJ, Westmoreland SV, Karman J, et al. Treatment with a CD40 antagonist antibody reverses severe proteinuria and loss of saliva production and restores glomerular morphology in murine systemic lupus erythematosus. J Immunol 2019;203:58–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park J, Shrestha R, Qiu C, et al. Single‐cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 2018;360:758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tveita AA, Ninomiya Y, Sado Y, et al. Development of lupus nephritis is associated with qualitative changes in the glomerular collagen IV matrix composition. Lupus 2009;18:355–360. [DOI] [PubMed] [Google Scholar]
- 18. Vasamsetti SB, Karnewar S, Kanugula AK, et al. Metformin inhibits monocyte‐to‐macrophage differentiation via AMPK‐mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes 2014;64:2028–2041. [DOI] [PubMed] [Google Scholar]
- 19. Kessler HS. A laboratory model for Sjögren's syndrome. Am J Pathol 1968;52:671–685. [PMC free article] [PubMed] [Google Scholar]
- 20. Bagavant H, Michrowska A, Deshmukh US. The NZB/WF1 mouse model for Sjögren's syndrome: a historical perspective and lessons learned. Autoimmun Rev 2020;19:102686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu C, Kieltyka J, Fleischmann R, et al. A decade of JAK inhibitors: what have we learned and what may be the future? Arthritis Rheumatol 2021;73:2166–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shawky AM, Almalki FA, Abdalla AN, et al. A comprehensive overview of globally approved JAK inhibitors. Pharmaceutics 2022;14:1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ripoll E, de Ramon L, Draibe Bordignon J, et al. JAK3‐STAT pathway blocking benefits in experimental lupus nephritis. Arthritis Res Ther 2016;18:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou M, Guo C, Li X, et al. JAK/STAT signaling controls the fate of CD8+CD103+ tissue‐resident memory T cell in lupus nephritis. J Autoimmun 2020;109:102424. [DOI] [PubMed] [Google Scholar]
- 25. Lee J, Park Y, Jang SG, et al. Baricitinib attenuates autoimmune phenotype and podocyte injury in a murine model of systemic lupus erythematosus. Front Immunol 2021;12:704526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Avasare R, Drexler Y, Caster DJ, et al. Management of lupus nephritis: new treatments and updated guidelines. Kidney360 2023;4:1503–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chan T‐M, Tse K‐C, Tang CS‐O, et al. Long‐Term study of mycophenolate mofetil as continuous induction and maintenance treatment for diffuse proliferative lupus nephritis. J Am Soc Nephrol 2005;16:1076–1084. [DOI] [PubMed] [Google Scholar]
- 28. Chan TM, Li FK, Tang CS, et al; Hong Kong‐Guangzhou Nephrology Study Group . Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. N Engl J Med 2000;343:1156–1162. [DOI] [PubMed] [Google Scholar]
- 29. Bekar KW, Owen T, Dunn R, et al. Prolonged effects of short‐term anti‐CD20 B cell depletion therapy in murine systemic lupus erythematosus. Arthritis Rheum 2010;62:2443–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mackensen A, Müller F, Mougiakakos D, et al. Anti‐CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med 2022;28:2124–2132. [DOI] [PubMed] [Google Scholar]
- 31. Shock A, Burkly L, Wakefield I, et al. CDP7657, an anti‐CD40L antibody lacking an Fc domain, inhibits CD40L‐dependent immune responses without thrombotic complications: an in vivo study. Arthritis Res Ther 2015;17:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frese‐Schaper M, Keil A, Steiner SK, et al. Low‐dose irinotecan improves advanced lupus nephritis in mice potentially by changing DNA relaxation and anti‐double‐stranded DNA binding. Arthritis Rheumatol 2014;66:2259–2269. [DOI] [PubMed] [Google Scholar]
- 33. Frese‐Schaper M, Zbaeren J, Gugger M, et al. Reversal of established lupus nephritis and prolonged survival of New Zealand black x New Zealand white mice treated with the topoisomerase I inhibitor irinotecan. J Immunol 2010;184:2175–2182. [DOI] [PubMed] [Google Scholar]
- 34. Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci 2018;27:1984–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev 2009;228:273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Theander E, Jacobsson LTH. Relationship of Sjögren's syndrome to other connective tissue and autoimmune disorders. Rheum Dis Clin North Am 2008;34:935–947. [DOI] [PubMed] [Google Scholar]
- 37. Szanto A, Szodoray P, Kiss E, et al. Clinical, serologic, and genetic profiles of patients with associated Sjögren's syndrome and systemic lupus erythematosus. Hum Immunol 2006;67:924–930. [DOI] [PubMed] [Google Scholar]
- 38. Lavoie TN, Lee BH, Nguyen CQ. Current concepts: mouse models of Sjögren's syndrome. J Biomed Biotechnol 2011;2011:549107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu Y, Li C, Wang S, et al. Human umbilical cord mesenchymal stem cells confer potent immunosuppressive effects in Sjögren's syndrome by inducing regulatory T cells. Mod Rheumatol 2021;31:186–196. [DOI] [PubMed] [Google Scholar]
- 40. Gong B, Zheng L, Huang W, et al. Murine embryonic mesenchymal stem cells attenuated xerostomia in Sjögren‐like mice via improving salivary gland epithelial cell structure and secretory function. Int J Clin Exp Pathol 2020;13:954–963. [PMC free article] [PubMed] [Google Scholar]
- 41. Kim J‐W, Kim S‐M, Park J‐S, et al. Metformin improves salivary gland inflammation and hypofunction in murine Sjögren's syndrome. Arthritis Res Ther 2019;21:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Charras A, Arvaniti P, Le Dantec C, et al. JAK inhibitors and oxidative stress control. Front Immunol 2019;10:2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nerstedt A, Johansson A, Andersson CX, et al. AMP‐activated protein kinase inhibits IL‐6‐stimulated inflammatory response in human liver cells by suppressing phosphorylation of signal transducer and activator of transcription 3 (STAT3). Diabetologia 2010;53:2406–2416. [DOI] [PubMed] [Google Scholar]
- 44. Wallace DJ, Furie RA, Tanaka Y, et al. Baricitinib for systemic lupus erythematosus: a double‐blind, randomised, placebo‐controlled, phase 2 trial. Lancet 2018;392:222–231. [DOI] [PubMed] [Google Scholar]
- 45. Morand EF, Vital EM, Petri M, et al. Baricitinib for systemic lupus erythematosus: a double‐blind, randomised, placebo‐controlled, phase 3 trial (SLE‐BRAVE‐I). Lancet 2023;401:1001–1010. [DOI] [PubMed] [Google Scholar]
- 46. Petri M, Bruce IN, Dörner T, et al. Baricitinib for systemic lupus erythematosus: a double‐blind, randomised, placebo‐controlled, phase 3 trial (SLE‐BRAVE‐II). Lancet 2023;401:1011–1019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure form:
Appendix S1. Supporting Information.
Data Availability Statement
Research data are not shared.