

Abstract
Current COVID-19 mRNA vaccines delivered intramuscularly (IM) induce effective systemic immunity, but with suboptimal immunity at mucosal sites, limiting their ability to impart sterilizing immunity. There is strong interest in rerouting immune responses induced in the periphery by parenteral vaccination to the portal entry site of respiratory viruses, such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), by mucosal vaccination. We previously demonstrated the combination adjuvant, NE/IVT, consisting of a nanoemulsion (NE) and an RNA-based RIG-I agonist (IVT) induces potent systemic and mucosal immune responses in protein-based SARS-CoV-2 vaccines administered intranasally (IN). Herein, we demonstrate priming IM with mRNA followed by heterologous IN boosting with NE/IVT adjuvanted recombinant antigen induces strong mucosal and systemic antibody responses and enhances antigen-specific T cell responses in mucosa-draining lymph nodes compared to IM/IM and IN/IN prime/boost regimens. While all regimens induced cross-neutralizing antibodies against divergent variants and sterilizing immunity in the lungs of challenged mice, mucosal vaccination, either as homologous prime/boost or heterologous IN boost after IM mRNA prime, was required to impart sterilizing immunity in the upper respiratory tract. Our data demonstrate the benefit of hybrid regimens whereby strong immune responses primed via IM vaccination are rerouted by IN vaccination to mucosal sites to provide optimal protection against SARS-CoV-2.
Keywords: SARS-Cov-2, prime/pull, mucosal immunization, mucosal adjuvants, intranasal vaccination, mRNA vaccines, tissue-resident memory T cells
Graphical abstract
Wong, Schotsaert, and colleagues demonstrate a rationally designed IN vaccine composed of a nanoemulsion (NE) and RIG-I agonist (IVT) adjuvant with SARS-CoV-2 subunit antigens effectively “pulls” and boosts immune responses parenterally primed by SARS-CoV-2 mRNA vaccines. This heterologous prime-pull strategy fosters enhanced local mucosal immune responses and optimally shapes protective systemic immunity.
Introduction
Multiple effective coronavirus disease 2019 (COVID-19) vaccines have been developed, which have played a pivotal role in overcoming the acute phase of the pandemic. Some of the most efficacious COVID-19 vaccines have been mRNA vaccines, which were initially administered intramuscularly (IM) as a prime/boost regimen. However, waning antibody titers and the emergence of antigenically drifted versions of the virus (i.e., Omicron variants) with mutations in the viral spike (S) protein that facilitate immune escape, have limited the duration of vaccine-induced protective immunity.1,2,3,4 Frequent booster immunizations along with introduction of updated vaccines containing mRNAs encoding for the S proteins of Omicron BA.4/5 and XBB1.5 have been deployed to address this waning immunity.5,6,7,8 These booster vaccines are also given IM and have been shown to enhance circulating B/T cell responses and improve protection from severe disease.2,9,10 However, even with these current vaccines and booster regimens, breakthrough infections and viral transmission continue to occur in fully vaccinated individuals, demonstrating that these vaccines do not confer sterilizing immunity.
For respiratory viruses such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the importance of inducing protective mucosal immune responses, including secretory antibodies and tissue-resident T cells, lies in the potential to block initial infection and viral dissemination to the lower respiratory tract (LRT). Moreover, mucosal immune responses in the respiratory tract play a critical role in preventing viral shedding and transmission and can thereby limit pandemic spread. As such, there has been marked interest in developing improved vaccines that induce robust cross-protective mucosal immunity in addition to systemic immunity. While vaccines against respiratory viruses given via the IM route poorly induce mucosal immune responses, mucosal vaccine delivery and natural infection have both been shown to induce robust mucosal immunity.11 Furthermore, hybrid immunity against SARS-CoV-2, the result of vaccine-induced immunity boosted by infection, has been suggested to provide superior protection from re-infection, even with antigenically drifted viruses.12,13 This is potentially mediated in part through re-routing and boosting vaccine-induced B and T cell responses to mucosal sites. Using this paradigm, a similar prime/pull strategy can be employed by utilizing mucosal vaccination to mimic events occurring during natural infection, to boost and re-route existing mRNA vaccine-induced immunity to the respiratory tract. While natural infection results in robust activation of local innate mucosal immune responses, achieving such responses through intranasal (IN) immunization with subunit antigens alone is challenging. However, by using rationally designed adjuvants to target immune receptor pathways activated by viral infection in the mucosa, more tailored immune responses can be induced to potentially confer similar or better outcomes as hybrid immunity.
We previously described a potent adjuvant, NE/IVT, for the mucosal delivery of vaccines.14,15,16 NE/IVT is a combination of an oil-in-water nanoemulsion (NE) and a RIG-I based agonist based on an in vitro transcribed RNA derived from Sendai virus (strain Cantell) defective interfering RNA (IVT).17 NE has established phase 1 clinical safety profiles as an IN adjuvant (NCT01354379, NCT04148118).18 The NE adjuvant induces mucosal and systemic immune responses mediated at least in part, through Toll-like receptor (TLR)2 and TLR4 activation and through NLRP3 activation via induction of immunogenic apoptosis.19,20,21 IVT is a selective RIG-I agonist and potent inducer of type I interferons (IFN-Is).17 As a combined agonist, NE/IVT can thus activate all three major innate receptor classes (TLRs, RLRs, and NLRs) necessary for induction of antiviral immune responses.14 The NE/IVT adjuvant platform has shown a good safety profile in preclinical models, is compatible with whole virus as well as recombinant protein vaccines, and induces potent systemic and mucosal immune responses when used for IN vaccination.
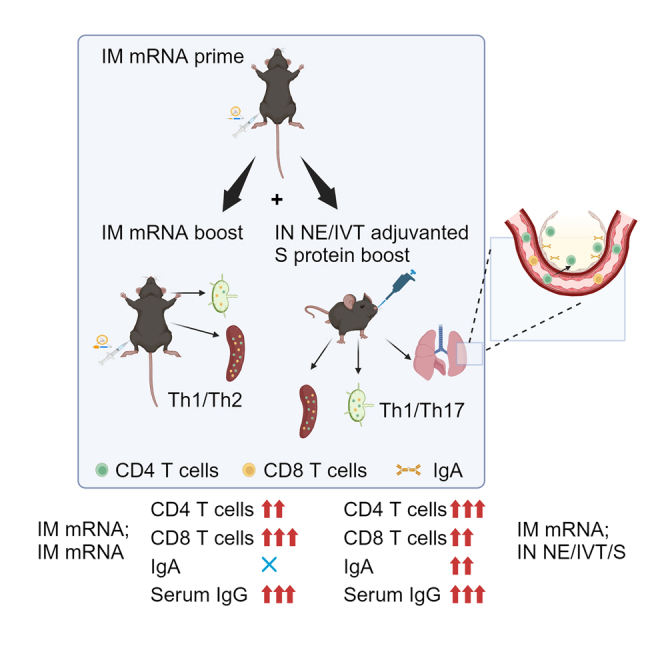
We report herein that IN recombinant SARS-CoV-2 S protein adjuvanted with NE/IVT can boost and induce (“pull”) mucosal SARS-CoV-2 spike protein-specific immune responses primed by IM mRNA vaccination with the BNT162b2 vaccine. The heterologous regimen of IM mRNA priming followed by IN NE/IVT/S boost resulted in mucosal IgA responses similar to homologous IN NE/IVT/S prime/boost vaccination, but also induced markedly enhanced T helper type 1 (Th1) polarized T cell responses in the upper respiratory tract (URT) draining lymph nodes (LNs) compared to homologous IM mRNA prime/boost and IN NE/IVT/S prime/boost. The strong mucosal antibody and T cell responses that resulted from this heterologous prime/pull vaccination strategy correlated with optimal cross-protection against various divergent variants of concern (VoCs), as reflected in protection from morbidity as well as robust viral control in both the upper and LRTs of mice challenged with divergent variants of SARS-CoV-2. In contrast, IM mRNA prime/boost could not impart sterilizing immunity in the URT. These results highlight the potential of heterologous IN boosting in harnessing the strong systemic immunity imparted by mRNA vaccines to drive potent mucosal immune responses in previously vaccinated individuals.
Results
NE/IVT-adjuvanted S protein vaccination as homologous IN prime/boost or heterologous IN boost after IM mRNA prime results in strong serum and mucosal IgG as well as mucosal IgA responses
To profile differences in immune responses induced by homologous versus heterologous prime/boost strategies, C57Bl/6 mice were given two immunizations 4 weeks apart (Figure 1A). Mice were primed IM either with BNT162b2 mRNA or PBS, or IN with wild-type (WT) S protein adjuvanted with NE (NE/S) or NE/IVT (NE/IVT/S). Two weeks after the prime (wk2), antibody responses were measured against full-length WT S protein and the receptor-binding domain (RBD), as RBD is the major target for virus-neutralizing antibodies. Priming with IM mRNA induced robust S protein-specific total IgG titers that were higher than those induced by priming with IN NE/S (by 0.5 log) or IN NE/IVT/S (by 1 log) (Figure 1B). However, RBD-specific total IgG titers were equivalent between the primed groups (Figure 1C). Mice primed with IM mRNA were then boosted either IM with mRNA, or IN with PBS, S protein alone, NE/S, or NE/IVT/S as indicated in Figure 1A. Additionally, mice primed with IN NE/S were boosted with IN NE/S, and those primed with IN NE/IVT/S were boosted with IN NE/IVT/S as homologous IN/IN prime/boost comparison groups. Finally, to examine the effect of a single IN immunization at the boost time point, mice primed with PBS were immunized with IN NE/S or NE/IVT/S. Two weeks post-boost (wk6), serum S- and RBD-specific total IgG titers increased for all groups and were highest in animals receiving IM mRNA prime/boost and for those which received IM mRNA prime followed by IN NE/S or IN NE/IVT/S boost, which resulted in equivalently high IgG titers (geometric mean titer [GMT] 2.8 × 106, 2.0 × 106, 2.8 × 106 against S protein, respectively). S-specific IgG titers for each of these groups increased by at least two logs after the boost, demonstrating the ability of IN NE/S and NE/IVT/S to boost systemic antibody responses induced by IM mRNA priming as effectively as an additional IM mRNA immunization (Figure 1D). In contrast, IM mRNA prime followed by unadjuvanted IN S protein boost only modestly increased S-specific IgG titers relative to levels induced by IM mRNA prime alone. Mice receiving two homologous IN immunizations with NE/IVT/S mounted comparable serum S-specific IgG titers as the IM mRNA prime/boost group, illustrating the ability to induce strong circulating antibody responses by IN immunization with NE/IVT/S. The inclusion of IVT with NE enhanced the magnitude of the induced S-specific IgG response compared to the singly adjuvanted NE/S group for the homologous IN prime/boost groups, confirming the synergistic activity of the combined NE/IVT adjuvant observed in our previous studies.14,15,16 All singly immunized groups (IM mRNA; IN PBS, IM PBS; IN NE/S, IM PBS; IN NE/IVT/S) induced equivalent S-specific IgG titers. While RBD-specific IgG titers were lower by approximately 1 log for all groups compared to the S-specific IgG titers, the same relative pattern between treatment groups was maintained (Figure 1E).
Figure 1.

Heterologous IM/IN prime-boost immunization induces robust S protein-specific IgG and enhances mucosal IgA production compared to homologous mRNA IM/IM prime-boost
(A) C57Bl/6 mice were given two immunizations 4-weeks apart. Mice were primed either IM with 0.4 μg of BNT162b2 mRNA or PBS, or IN with 15 μg full-length S protein with either NE or NE/IVT. Mice were then boosted IM with 0.4 μg of BNT162b2 mRNA or PBS, or IN with PBS or S protein in PBS, NE or NE/IVT as indicated. Serum antigen-specific total IgG titers against (B) WT S protein and (C) WT RBD as measured by ELISA 2 weeks after the prime immunization, and (D, E) 2 weeks after the boost immunization (wk6). (F–H) Subclass profiles for S-specific serum antibodies measured at wk6. BALF S-specific (I) IgA and (J) IgG measured at wk6. (n = 5/group; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001 by Mann-Whitney U test shown only for select groups (full statistical analysis is shown in Table S1).
IgG subclass skewing was dependent on the vaccination regimen. IM mRNA prime/boost, heterologous IM mRNA prime followed by IN NE/S or NE/IVT/S boost, as well as IN NE/IVT/S prime/boost, all resulted in high and similar antigen-specific IgG1 and IgG2b titers, with the IN NE/IVT/S prime/boost inducing slightly higher levels of IgG1 (Figures 1F and 1G). The presence of IVT in the IN NE/IVT/S prime/boost group enhanced IgG2b relative to the IN NE/S prime/boost group, consistent with the Th1 skewing properties of IVT previously observed.14,15,16 mRNA vaccination, either as homologous prime/boost or as prime followed by IN NE/S or NE/IVT/S boost was required to induce the strongest IgG2c antibody responses, inducing equally high titers (GMT 6 × 105) in these groups, which was enhanced by approximately 1 log relative to the group given NE/IVT/S prime/boost via the IN route only (Figure 1H).
While the serum antigen-specific antibody levels and subclass profiles were similar between the homologous IM mRNA prime/boost, and the heterologous IM mRNA; IN NE/S and IM mRNA; IN NE/IVT/S groups, induction of mucosal IgA responses in bronchoalveolar lavage fluid (BALF) was exclusive to groups receiving IN vaccination with NE/S or NE/IVT/S, either as a boost immunization in IM mRNA primed mice, or as a homologous IN prime/boost regimen (Figure 1I). No BALF S-specific IgA was detectable in the IM mRNA prime/boost group. Moreover, no S-specific IgA was observed for mice given IM mRNA; IN S alone, highlighting the role of the NE and NE/IVT adjuvants in driving the mucosal response after IN administration. Interestingly, while a single IN NE/S or NE/IVT/S immunization induced low or no IgA, immunization with IM mRNA; IN NE/IVT/S resulted in equivalent IgA titers as homologous IN NE/IVT/S prime/boost. These results demonstrate the ability of the adjuvanted IN "pull" immunization to harness parenterally primed immune responses for driving robust mucosal immune responses. While dimeric IgA plays the predominant role in first-line defense in the mucosa of the URT, mucosal IgG also contributes to protection through transudation from the blood into the lung.22 Indeed, in contrast to the pattern observed for IgA, BALF IgG correlated with serum IgG titers, with IM mRNA prime/boost immunization inducing robust BALF antigen-specific IgG titers similar to the IM mRNA; IN NE/S and IM mRNA; IN NE/IVT/S treatment groups, as well as the IN NE/IVT/S prime/boost group (Figure 1J).
Cross-reactive serum neutralizing antibody titers reflect serum S-specific IgG binding antibody titers
We next quantified cross-reactive neutralizing antibody (nAb) titers induced in immunized C57Bl/6 mice using pseudotyped viruses carrying the S proteins of SARS-CoV-2 VoCs. In general, vaccination regimens that resulted in the highest IgG binding titers (IM mRNA prime/boost, IN NE/S and NE/IVT/S prime/boost or heterologous IM mRNA; IN NE/S or IM mRNA; IN NE/IVT/S) not only resulted in the highest nAb titers against vaccine matched ancestral virus (Figure 2A) but also against the antigenically more distant B.1.617.2 (Delta) (Figure 2B), and B.1.351 (Beta) (Figure 2C) variants. These treatment groups induced similar levels of cross-neutralizing nAbs against each variant examined, with homologous IM mRNA and homologous IN NE/IVT/S prime/boost inducing similar nAb titers as heterologous IM mRNA;IN NE/IVT/S immunization. In contrast, IM mRNA-primed mice boosted IN with unadjuvanted S alone induced the lowest nAb titers, giving similar or lower titers as the single IM mRNA immunization group against all variants tested. Neutralization potential was reduced across all vaccination groups by a similar degree (approximately 2 log) towards the more antigenically distant B.1.1.529 (Omicron BA.1) variant, maintaining the same relative pattern of nAb response magnitude observed between immunization groups as against the WT virus (Figure 2D).
Figure 2.

Heterologous IM/IN prime-boost immunization induces robust cross-neutralizing antibody responses against multiple variant viruses
Serum neutralizing antibody titers from immunized C57Bl/6 mice at week 6 after both prime/boost immunizations with the indicated adjuvant/antigen regimens were measured using pseudoviruses for the (A) WT, (B) B.1.617.2, (C) B.1.351, and (D) B.1.1.529 (BA.1) variants. (n = 5/group; ∗p < 0.05, ∗∗p < 0.01 by Mann-Whitney U test shown only for select groups (full statistical analysis is shown in Table S1).
Heterologous IM mRNA prime followed by IN NE/IVT/S boost markedly enhances Th1 and Th17 polarized antigen recall responses in spleen and cervical LNs
To evaluate T cell antigen recall responses, 2 weeks after the boost immunization, splenocytes and cervical LN (cLN) isolates were harvested from immunized mice and restimulated ex vivo with S protein (Figure 3). Heterologous boosting of IM mRNA primed mice with IN NE/S or NE/IVT/S resulted in a marked enhancement of Th1-polarized responses compared to homologous IM mRNA, IN NE/S, or IN NE/IVT/S prime/boost groups. High levels of S-specific IFN-γ were induced in splenocytes by IM mRNA vaccination boosted with either IM mRNA or IN NE/S or NE/IVT/S, with the heterologous IN-boosted groups inducing equivalent (or higher) levels of IFN-γ than the IM mRNA prime/boost group (Figure 3A). The heterologous IM mRNA; IN NE/S and IM mRNA; IN NE/IVT/S groups also demonstrated enhanced levels of IFN-γ compared to the homologous IN NE/S and IN NE/IVT/S prime/boost groups. As seen previously, inclusion of IVT in the IN NE/IVT/S prime/boost significantly enhanced the IFN-γ response relative to IN NE/S. Similar patterns were observed for antigen-specific IL-2 responses, with enhanced cytokine levels in animals primed with IM mRNA and boosted with IN NE/S or NE/IVT/S compared to those given two doses of IM mRNA, IN NE/S, or IN NE/IVT/S (Figure 3B). No significant differences were observed between vaccination groups for TNF-α in stimulated splenocytes, with all immunized groups inducing similar levels of TNF-α, with the singly immunized IN NE/S and NE/IVT/S groups exhibiting slightly reduced levels compared to their respective prime/boost groups (Figure 3C). While significant variation was observed for IP-10, IM mRNA primed animals boosted with IN NE/S or NE/IVT/S induced similar levels of IP-10 in splenocytes as the IM mRNA prime/boost group, with the exception of one mouse in each of the heterologous groups showing much higher IP-10 (5- to 10-fold relative to the IM mRNA prime/boost) (Figure S2A).
Figure 3.

Antigen recall responses assessed in splenocytes and cervical lymph node isolates from IM/IN immunized mice demonstrate enhanced Th1/Th17 profiles
Splenocytes (A–F) and cLN cellular isolates (G–L) were prepared from mice given prime/boost immunizations with the indicated adjuvant/antigen regimens two weeks after the final immunization (week 6). Cells were stimulated ex vivo with 5 μg S protein for 72 h, and levels of secreted (A, G) IFN-γ, (B, H) IL-2, (C, I) TNF-α, (D, J) IL-6, (Ε, Κ) IL-17A, and (F, L) IL-10 were measured from splenocytes and cLN respectively, by multiplex immunoassay relative to unstimulated cells. (n = 4–5/group; ∗p < 0.05, ∗∗p < 0.01 by Mann-Whitney U test shown only for select groups-(full statistical analysis is shown in Table S1).
Homologous IM mRNA administration induced significant Th2 responses in splenocytes as measured by IL-5 (Figure S2C) and IL-13 (Figure S2B) in comparison to homologous IN NE/S or IN NE/IVT/S, which did not induce detectable levels of these cytokines. Heterologous boosting of IM mRNA primed animals with IN NE/S or NE/IVT/S did not enhance these Th2 cytokines and appeared to reduce IL-5 levels compared to the groups given one or two doses of IM mRNA. While IM mRNA prime/boost induced substantial IL-6 in the spleen (greater than IN NE/S or NE/IVT/S prime/boost), priming with IM mRNA followed by boosting with IN NE/S or NE/IVT/S resulted in significantly increased IL-6 production, especially with the NE/IVT/S mucosal boost (Figure 3D).
We have established through multiple studies the robust induction of IL-17A production by IN administration of the NE adjuvant, which is further enhanced by the inclusion of IVT.14,15,16 This induced Th17 response is exclusive to the mucosal route of administration of these adjuvants. Indeed, we observed high levels of IL-17A induction in splenocytes for the IN NE/S prime/boost group, which was further enhanced approximately 2-fold in the IN NE/IVT/S prime/boost group (Figure 3E). In contrast, no IL-17A was observed for the IM mRNA prime/boost group. Interestingly, however, while the single IN immunization groups (IM PBS; IN NE/S, IM PBS; IN NE/IVT/S) induced only low levels of IL-17A (approximately 10-fold lower than the corresponding prime/boost groups), boosting IM mRNA primed animals with IN NE/S or NE/IVT/S induced high levels of IL-17A, demonstrating the ability of the IN adjuvants to boost and shape immune responses primed by the initial IM mRNA vaccination, promoting a shift in IM mRNA-primed T cell responses towards Th17. Notably, IM mRNA boosted with IN S alone did not induce significant IL-17A production. These results are significant, as Th17 responses have been shown to be a critical component of host defense at mucosal sites.23,24,25 All prime/boost immunization groups with an IM mRNA prime induced similarly high levels of IL-10 in the spleen, which was enhanced relative to the single or two dose IN NE/S or IN NE/IVT/S regimens (Figure 3F).
cLNs drain the URT and, therefore, are relevant for the assessment of local protective immunity near the portal entry sites of pulmonary pathogens and for evaluating mucosal T cell responses induced by IN immunization. IN administration of vaccines as part of a boost regimen enhanced S protein-specific cytokine responses within the cLNs, confirming effective pulling of antigen-specific immune responses to mucosal sites (Figures 3G–3L). In general, cytokine profiles in the cLN reflected the recall responses measured in spleen, but were heavily skewed by the IN vaccination regimens, resulting in greater magnification of the Th1/ Th17 polarization locally. Boosting with IN NE/IVT/S after IM mRNA prime induced markedly high antigen-specific IFN-γ levels within the cLN compared to homologous prime/boost with IM mRNA, IN NE/S, or IN NE/IVT/S (Figure 3G). Furthermore, administration of IN NE or IN NE/IVT formulations, either as a homologous prime/boost regimen or as part of a heterologous boost regimen after IM mRNA priming, also significantly enhanced antigen-specific IL-2 and IP-10 responses within the cLN compared to two doses of IM mRNA, with the highest levels observed with heterologous IM mRNA; IN NE/IVT/S vaccination (Figures 3H and S2D). Notably, IM mRNA prime/boost induced only low levels of these cytokines in the cLN. Finally, enhancement in TNF-α was also observed in the cLN for the IM mRNA; IN NE/IVT/S group as compared to the IM mRNA and IN NE/IVT/S prime/boost groups (Figure 3I). These results are of significance, as the co-production of IFN-γ, IL-2, and TNF-α has been established as a strong mediator of optimal control of viral infection and a major correlate of vaccine protection. Furthermore, these results clearly demonstrate the ability of the IN NE/IVT/S boost to drive enhanced Th1 polarized T cell responses in the local mucosal lymphoid tissue through the pulling of responses systemically primed by the IM mRNA.
Similar to the Th2 cytokine pattern in the spleen, two doses of IM mRNA induced the highest levels of IL-5 and IL-13 in the cLN, while two doses of IN NE/S or IN NE/IVT/S induced only low levels of IL-5 and no detectable IL-13 (Figures S2E and S2F). While heterologous boosting of IM mRNA primed animals with IN NE/S or NE/IVT/S resulted in higher levels of IL-5 than the homologous IN NE/S and IN NE/IVT/S prime/boost groups, the levels remained similar to or lower than those induced in the IM mRNA prime/boost group, and no significant IL-13 was detected in the heterologous prime/boost groups. These results confirm the lack of Th2 response enhancement with the adjuvanted heterologous pull immunizations. Minimal levels of S-specific IL-6 were induced in the cLNs (Figure 3J) as compared to splenocytes (Figure 3D) for all vaccination groups (two orders of magnitude lower), with the highest levels induced in animals receiving IN NE/IVT/S as part of their vaccine regimen.
The enhancement in IL-17A production seen in the splenocytes was even more pronounced in the cLN for the heterologous IM mRNA; IN NE/IVT/S group (Figure 3K). The combination of IM mRNA prime with IN NE/IVT/S boost resulted in similarly strong IL-17A responses as homologous IN NE/IVT/S prime/boost immunization. In contrast, IM mRNA prime/boost did not induce detectable IL-17A. Heterologous boost with IN NE/S was also able to enhance IL-17A in the IM mRNA primed mice; however, the inclusion of IVT in the IN boost was critical to driving maximal Th17 responses in the cLN. While IL-17A induction has been associated with immune pathology in certain contexts, it has been shown to be non-pathogenic in the context of IL-10 co-production.26,27 Indeed, IL-10 production in the cLN followed the same pattern as IL-17A, with the IN adjuvanted groups inducing the highest levels of IL-10 (Figure 3L). The lack of immune pathology was confirmed in our prior studies and the challenge studies discussed below. Overall, IM mRNA vaccination resulted in priming events that, when boosted IN with NE/IVT/S, resulted in a unique antigen-specific cytokine profile in both the spleen and in the local mucosal-draining LNs. These results demonstrate that employing an IN pull with an NE/IVT-adjuvanted vaccine after IM mRNA priming can not only drive more robust T cell responses toward SARS-CoV-2 but also tailor these responses through enhancing the Th1/ Th17 polarization.
IM mRNA prime IN NE adjuvanted boost regimens enhance CD4 responses within the lung and draining LNs
The induction of high IgG2a, IgG2c, and IgA antibody titers requires efficient class switching in germinal center reactions, which require strong CD4+ T cell responses. Moreover, CD4+ T cells provide critical help to CD8+ T cell anti-viral responses, which are key mediators of protection against SARS-CoV-2 infection.28,29 Therefore, we further characterized vaccine induced CD4+ and CD8+ T cell responses in the spleen, cLNs, and lungs of immunized animals. To better observe differences between vaccine regimens, C57Bl/6 mice were given the same prime/boost regimens but at higher doses of mRNA (3 μg) and S protein (20 μg). To help distinguish circulating vs. tissue-resident T cells, mice were administered a AF488-α-CD45 antibody intravenously (i.v.) before sacrifice to label circulating cells.30 At this high mRNA dose, IM mRNA prime/boost immunization resulted in low, but significant, induction of S major histocompatibility complex (MHC) class I (VNFNFNGL) tetramer-specific CD8+ tissue-resident memory T cells (TRM) (CD69+CD103+ class I Tetramer+CD44+IV−CD3+CD8+) cells in the lungs of vaccinated mice (Figure 4A). Apart from one mouse that showed notable levels of enhancement, IM mRNA-primed animals boosted IN with NE/IVT/S did not show significant induction of tetramer+CD8+TRM’s in the lungs. In contrast, while IM mRNA prime/boost immunization did not induce S protein MHC class II-restricted (VTWFHAIHVSGTNGT) tetramer-specific CD4+ TRM’s (CD69+CD103+class II Tetramer+CD44+IV− CD3+CD4+) within the lungs, IN NE/IVT/S boosting of IM mRNA primed animals resulted in a marked increase in the percentage of antigen-specific CD4+TRMs in the lungs of vaccinated mice (Figure 4B). When examining the spleen (Figures 4C and 4D) and cervical draining LNs (Figures 4E and 4F), IM mRNA prime/boost immunization induced the highest levels of S-specific CD8+ effector memory T cells (CD69+CD62L− class I tetramer+CD44+CD3+CD8+) compared to the heterologous prime/boost groups which showed only low induction frequencies (Figures 4C and 4E). Upon examining class II Tet+CD4+ effector memory T cells (CD69+CD62L− class II tetramer+CD44+CD3+CD4+) in the spleen, we found that, while homologous IM mRNA prime/boost induced slightly higher antigen-specific CD4+ T cells, heterologous prime/boosting with IM mRNA followed by IN boost with S alone or adjuvanted with NE or NE/IVT also induced significant levels (Figure 4D). However, the impact of the adjuvanted mucosal boost was most notable in the mucosal draining LNs, in which the IM mRNA prime followed by IN boost with NE/S or NE/IVT/S markedly enhanced the induction of S-tetramer specific CD4+ T cells compared to the IM mRNA prime/boost group, as well as to the group given IM mRNA prime followed by unadjuvanted IN S boost (Figure 4F). These results demonstrate the importance of the NE and NE/IVT adjuvants in the mucosal boost vaccinations in driving local mucosal immune responses. Moreover, the enhancement in antigen-specific CD4+ T cells but not CD8+ T cells in the mucosal draining LNs with the adjuvanted mucosal boosts are consistent with the differences observed in the frequencies of induced CD8+ vs. CD4+ TRM’s within the lung for the different vaccine administration regimens.
Figure 4.

IM mRNA vaccination is important for generating robust levels of spike-specific CD8+ T cells while IN boosting enhances the frequency of spike-specific CD4+ T cells in the lung and draining LNs
Mice were given 3 μg mRNA IM, 20μg of S protein IN in NE/IVT, or PBS. Cells isolated from the lungs, spleen, and cLNs of mice given prime/boost immunizations with the indicated adjuvant/antigen regimens were incubated with MHC class I (VNFNFNGL) or MHC class II (VTWFHAIHVSGTNGT) tetramers, stained for surface markers, and analyzed by FACS. Frequencies of CD44+IV− Tet+CD69+CD103+ expressing CD8+ (A) or CD4+ (B) TRMs in the lung were quantified. (n = 3–5/group with data represented as mean ± SEM; ∗p < 0.05, ∗∗p < 0.01, by Mann-Whitney U test). Frequencies of Tet+CD44+CD69+CD62L− TEMs were similarly quantified in the spleen (C and D) and cLNs (E and F). (n = 3–5/group with data represented as mean ± SEM; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by one-way ANOVA with Tukey post hoc test shown only for select groups (full statistical analysis is shown in Table S1).
We next assessed the cytokine response of the S-specific CD4+ and CD8+ T cells generated by these vaccine regimens. In accordance with the tetramer data, IM mRNA prime/boost induced the highest frequency of IFN-γ+-expressing CD8+ T cells in the spleen upon ex vivo stimulation with S protein (Figure S5A). IN boosting of IM mRNA primed animals with IN S or NE/IVT/S also induced significant numbers of IFN-γ+ CD8+ T cells, although the frequency was slightly lower than that induced by the IM mRNA prime/boost immunization. In contrast, in the cLNs, the frequency of CD8+ T cells expressing IFN-γ+ were highest in the IM mRNA primed mice receiving heterologous IN boost with NE or NE/IVT adjuvanted antigen (Figure S5E). Cytokine expressing CD8+ T cells were low within the lungs for all vaccine regimens, even the IM mRNA prime/boost group despite the high frequency of S-tetramer specific CD8+ T cells observed for this treatment group (Figures S5I–S5K). The frequency of polyfunctional (IFN-γ+ IL-2+ TNF-α+) CD8+ T cells was also low within the spleen, cLNs, and lung (Figures S5D, S5H, and S5L). Upon analyzing CD4+ T cell cytokine responses in the spleen, we found that mice receiving IM mRNA prime/boost induced the highest frequencies of IFN-γ+, IL-2+, and TNF-α+ CD4+ T cells (Figures 5A–5C), as well as polyfunctional CD4+ T cells upon stimulation with S protein (Figure 5D). IN boost with S, NE/S, or NE/IVT/S after mRNA priming induced similar and significant frequencies of IFN-γ+, IL-2+, and TNF-α+CD4+ T cells, as well as polyfunctional CD4+ T cells, although these levels were lower than those induced by two doses of IM mRNA. Interestingly, a comparison of CD4+ IFN-γ+ responses by mean fluorescence intensity (MFI) rather than by frequency, showed equivalent MFIs for the IM mRNA prime with IN S, NE/S, or NE/IVT/S boost groups as the IM mRNA prime/boost group, demonstrating that even though the frequency of IFN-γ+ cells was slightly lower for the IM/IN groups, these cells produced high levels of IFN-γ (Figure S6). In the cLN, however, IM mRNA groups receiving IN NE/S or NE/IVT/S boost immunizations developed the strongest antigen-specific CD4+ T cell responses, with significant enhancement in the frequency of polyfunctional cells compared to groups that received a second IM mRNA dose or unadjuvanted IN S (Figures 5E–5H). While IN NE/S or NE/IVT/S in a prime/boost regimen induced IL-2- and TNF-α-expressing CD4+ T cells, IM mRNA priming was necessary to induce optimal IFN-γ expressing CD4+ T cells in the cLN, demonstrating the importance of the initial IM prime immunization in driving optimal local mucosal responses after a mucosal boost. Furthermore, while IN S boost of IM mRNA primed mice showed enhanced polyfunctional CD4+ T cell responses in the spleen comparable to the IN NE/S and NE/IVT/S boost groups, the unadjuvanted boost group showed minimal levels of polyfunctional CD4+ T cells in the cLNs, highlighting the critical role of the NE-based adjuvants in driving robust local mucosal cellular responses. Similar to the cLNs, in the lungs, groups heterologously primed with IM mRNA and then boosted IN with S, NE/S, or NE/IVT/S had similar levels of IFN-γ-expressing CD4+ T cells, which were enhanced compared to the other vaccination regimens, including IM mRNA prime/boost (Figures 5I–5L). These groups also displayed enhanced polyfunctional CD4+ T cells in the lung compared to the other vaccination regimens. Interestingly, boosting with IN S after IM mRNA priming induced higher frequencies of polyfunctional CD4+ T cells in the lung compared to boosting with IN NE/IVT/S, which was the reverse pattern observed in the cLNs, potentially suggesting differences in trafficking and kinetics at this time point between the free antigen vs. NE-formulated NE/IVT adjuvanted S protein. Finally, in accordance with the cytokine secretion data, mucosal boost of IM mRNA primed animals displayed higher frequencies of IL-17A+CD4+ T cells in the lung than the IM mRNA prime/boost group, which had no detectable response (Figure S7). However, the highest frequencies of IL-17A+CD4+ T cells were induced in the IN NE/S and IN NE/IVT/S homologous prime/boost groups. Taken together, the differences in induction of antigen-specific CD4+ vs. CD8+ responses observed in the tetramer and cytokine analyses results underscore the distinct mechanisms of the vaccination approaches, with the heterologous IM mRNA prime followed by an adjuvanted mucosal boost most effectively driving enhanced CD4+ T cell responses, especially locally within the tissue and draining LNs. These results demonstrate that this adjuvanted mucosal boost strategy represents an effective strategy to pull systemic immune responses induced by mRNA vaccination to mucosal sites.
Figure 5.

IN administration of antigen with NE adjuvants after IM mRNA priming effectively pulls antigen-specific immunity to mucosal sites
Single cell suspension were isolated from the spleen (A–D), cLNs (E–H), and lungs (I–L) of mice given prime/boost immunizations with the indicated adjuvant/antigen regimens. Mice were given 3 μg mRNA IM, and 20 μg of S protein IN in either PBS, NE, or NE/IVT. Cells were stimulated with 25 μg/mL of S protein, and antigen-specific cytokine responses were quantified in CD4+ T cells by intracellular cytokine staining and FACS analysis. (n = 4–5/group with data represented as mean ± SEM; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by one-way ANOVA with Tukey post hoc test shown only for select groups (full statistical analysis is shown in Table S1).
Heterologous IM/IN prime-boost immunization induces robust virus-neutralizing antibody titers in 129S1 and K18-hACE2 mice and results in more optimal protection in the upper respiratory tract
To examine the effects of genetic background on induced immune responses, we next repeated vaccination with the heterologous prime/boost regimens in 129S1 and K18-hACE2 mice. The 129S1 strain was selected for vaccination/challenge studies as they are WT mice that have shown to be more susceptible to morbidity after experimental infection with SARS-CoV-2 viruses with the N501Y mutation, such as B.1.351.31 In contrast, SARS-CoV-2 Omicron lineages do not efficiently infect WT mice but can replicate in the respiratory tract of transgenic K18-hACE2 mice, which overexpress human ACE2 receptor in the epithelia. Thus, we used this transgenic model for protective efficacy studies with Omicron BA.5 challenge. Prime immunization with IM mRNA, IN NE/S, IN NE/IVT/S induced equivalent S-specific serum IgG in 129S1 mice, giving similar titers as primed C57Bl/6 and K18-hACE2 mice (Figures 1B and S1). Boosting of IM mRNA primed animals with IM mRNA, IN NE/S, or IN NE/IVT/S resulted in high serum total binding IgG against WT S protein in both genetic backgrounds (Figures 6A and 6F). In both 129S1 and K18-hACE2 mice, comparable induction of high S-specific IgG titers with the IM mRNA prime/boost was observed as for the heterologous IM mRNA prime with IN NE/S or NE/IVT/S boost, with these titers being enhanced compared to the IN NE/S and IN NE/IVT/S prime/boost, and IM mRNA; IN S groups. Overall, an identical pattern for S-specific IgG titers between immunization groups was observed for the 129S1, K18-hACE2, and C57Bl/6 mice. Furthermore, for comparison we assessed whether prime/boost immunization with S protein adjuvanted with the MF59-equivalent IM adjuvant, Addavax (Advx), would induce similar immune responses. IM Advx/S prime/boost resulted in similar S-specific IgG as the IM mRNA prime/boost group in both 129S1 and K18-hACE2 strains (Figures 6A and 6F). Interestingly, while the IM mRNA; IN NE/S and IM mRNA; IN NE/IVT/S groups induced similarly robust neutralization titers as the IM mRNA prime/boost groups in 129S1, K18-hACE2, and C57Bl/6 mice against ancestral virus, a more distinct enhancement in the breadth of viral neutralization with these heterologous IM/IN groups was observed in the 129S1 mice compared to the other two strains when measured against B.1.351, BA.1, and especially against BA.4/5 (Figures 6B–6E and 6G–6J). For example in the 129S1 mice, IM mRNA; IN NE/IVT/S treatment resulted in a 1 log enhancement in B.1.351 and BA.1 neutralization compared to the IM mRNA prime/boost group. In 129S1 mice, all vaccine groups that included a prime/boost had detectable BA.4/5-specific nAbs with the exception of the IM mRNA prime/boost group in which half of the mice failed to show detectable neutralization (Figure 6E). In K18-hACE2 mice, the same vaccination regimens resulted in less efficient induction of BA.4/5-specific nAb titers, with only the group that was primed with IM mRNA followed by IN NE/IVT/S boost inducing significant nAb titers in all of the animals within the immunization group (Figure 6J). Thus, while the general trends are the same among the three mouse strains, there are nuanced differences that highlight the importance of comparing responses in the context of different genetic backgrounds. Finally, while IM Advx/S prime/boost induced similar total binding S-specific IgG titers as IM mRNA prime/boost, IN NE/IVT prime/boost, and IM mRNA prime with IN NE/S or NE/IVT/S boost, the IM Advx/S group displayed lower viral neutralization titers overall compared to these treatment groups. For example, in K18-hACE2 mice, the majority of IM Advx/S prime/boost mice showed no detectable neutralization of BA.4/5, pointing to the reduced breath of the antibody response (Figure 6J). To evaluate protection against cross-variant viral challenge, we then infected 129S1 mice with 104 plaque-forming units (PFU) of B.1.351 (Figures 7A and 7B) and K18-hACE2 mice with 104 PFU of BA.5 (Figures 7C and 7D) SARS-CoV-2. Lungs as well as nasal turbinates (NTs) were harvested at 4 days post-infection (d.p.i.) for viral load determination by plaque assay. B.1.351 contains the N501Y mutation, which allows it to replicate in WT 129S1 mice, causing up to 10% body weight loss.31 In contrast, while SARS-CoV-2 Omicron variants, including BA.5, can replicate in the respiratory tract of transgenic K18-hACE2 mice, infection is mainly characterized by the absence of overt morbidity.32 Nonetheless, it provides a useful model for evaluating the breadth of immune protection with virus titer reduction as a surrogate of protection. In B.1.351 challenged 129S1 mice, IM mRNA prime/boost resulted in complete protection in the lungs (LRT) with the absence of replicating virus (Figure 7A). However, the IM mRNA prime/boost vaccination failed to prevent viral replication in the nasal turbinates (URT) (Figure 7B). In contrast, IM mRNA prime with IN adjuvanted S booster vaccination (NE/S, NE/IVT/S) demonstrated effective viral control in both the LRT and URT, with undetectable replicating virus in both the lungs and NTs of challenged mice. The IN NE/IVT/S prime/boost regimen also conferred complete protection in both the LRT and URT, highlighting the critical role of mucosal immunization in promoting URT protection. IN NE/S prime/boost immunization also blocked viral replication in the NTs and conferred significant protection in the lungs, with most mice showing no detectable viral replication in the lungs. However, two out of five mice in this treatment group exhibited modest breakthrough viral replication just above the limit of detection, supporting the advantage of the NE/IVT combined adjuvant. Similar to other studies studying the prime/pull approach with IM mRNA prime followed by unadjuvanted IN S protein boost, we also observed significant protection with this vaccine regimen; however, one of the five mice in this group displayed detectable viral titers in both the lungs and NTs.33 While IM Advx/S prime/boost offered a significant degree of protection as compared to the unvaccinated PBS control group, the Advx group showed the highest viral load in the lungs compared to all the other groups receiving two immunizations. Moreover, all mice in the Advx group demonstrated high viral titers in the NTs that were not significantly different from the unvaccinated control. These results further support the role of IN vaccination in promoting sterilizing immune responses in the URT. Notably, a single immunization with IN NE/S or NE/IVT/S did not confer significant protection in the lungs, yielding similar viral titers as the unvaccinated PBS group. However, a few animals in these groups did show a lack of viral replication in the NTs, while the others had similar titers as the unvaccinated control. Thus, the more complete protective effects observed in the URT for the heterologous immunization groups is attributable to the combined effects of both the IM mRNA prime and NE-based IN pull components.
Figure 6.

Heterologous IM/IN prime-boost immunization induces robust virus-neutralizing antibody titers in 129S1 and K18-hACE mice
129S1 and K18-hACE2 mice were vaccinated twice 4 weeks apart. Mice were primed either IM with 0.4 μg of BNT162b2 mRNA or PBS, or IN with 15 μg full-length S protein with either NE or NE/IVT. Mice were then boosted 4 weeks later IM with 0.4 μg of BNT162b2 mRNA, or IN with 15 μg S with PBS, NE or NE/IVT as indicated. Groups receiving two immunizations with IM Advx/S or PBS were included for comparison. Serum (A) IgG titers against WT S-protein, and nAb titers against (B) WT, (C) B.1.351, (D) B.1.1.529 (BA.1), and (E) BA.4/5 variant PSVs were measured 2 weeks after the boost immunization (wk6). K18-hACE2 transgenic mice were similarly primed either IM with 0.4 μg of BNT162b2 mRNA, or PBS, or IN with 15 μg S with either NE or NE/IVT. Mice were boosted 4 weeks later IM with 0.4 μg of BNT162b2 mRNA or PBS, or IN with 15 μg S with PBS, NE or NE/IVT as indicated. Groups receiving two immunizations with IM Advx/S or IN PBS were included for comparison. Serum (F) IgG titers against WT S-protein, and nAb titers against (G) WT, (H) B.1.351, (I) B.1.1.529 (BA.1), and (J) BA.4/5 variant PSVs were measured 2 weeks after the boost immunization (wk6). (n = 4–5/group; ∗p < 0.05, ∗∗p < 0.01 by Mann-Whitney U test shown only for select groups (full statistical analysis is shown in Table S1).
Figure 7.

Heterologous IM/IN prime/pull and IN/IN immunization strategies provide sterilizing immunity upon heterologous challenge in both the upper and lower respiratory tracts in contrast to IM/IM immunization with BNT162b2 mRNA or Addavax/S
129S1 mice were primed either IM with 0.4 μg of mRNA or PBS, or IN with 15 μg S with either NE or NE/IVT. Mice were boosted 4 weeks later IM with 0.4 μg mRNA, or IN with 15 μg S with PBS, NE or NE/IVT as indicated. Groups receiving two immunizations with IM Advx/S or IN PBS were included for comparison. 3 weeks post-boost, mice were challenged IN with 104 PFU B.1.351, and viral titers were measured at 4 d.p.i. in the (A) lungs and (B) nasal turbinates. K18-hACE2 mice were similarly vaccinated and then challenged IN with 104 pfu BA.5. Viral titers were measured at 4 d.p.i. in the (C) lungs and (D) nasal turbinates. Each data point represents the average of replicate counts for each animal (n = 4–5/group; ∗p < 0.05, ∗∗p < 0.01 by Mann-Whitney U test). Cytokine and chemokine levels in lung homogenate were also measured at 4 d.p.i. by multiplex immunoassay from the immunized (E) 129S1 mice challenged with B.1.351, and the (F) K18-hACE2 mice challenged with BA.5. Individual cytokine/chemokine levels were normalized to the cytokine/chemokine range and then normalized based on multiplication with log2 fold changes to normalize expression changes. Heatmap shows expression changes for the mean of each group. Individual cytokine levels and statistical analyses are provided in Figures S7 and S8.
Similar effective viral control was observed for the IM mRNA prime, IN NE/S or IN NE/IVT/S boosted K18-hACE2 mice challenged with BA.5, with no viral replication detected in the lungs or NTs (Figures 7C and 7D). Consistent with the results from the B.1.351 challenge in 129S1 mice, IM mRNA prime/boost also showed strong protection in the lungs after BA.5 challenge in vaccinated K18-hACE2 mice, as only one of the five mice had detectable viral titers just above the detection limit. These mice also displayed higher levels of viral replication in the NTs compared to the IM mRNA prime, IN-adjuvanted S boosted groups showing low, but detectable viral titers in two of the five mice. IM Advx/S prime/boost also provided some degree of protection in the lungs of BA.5 challenged K18-hACE2 mice but had two of the five mice with viral titers that were close those of unvaccinated controls. Moreover, no protection against BA.5 challenge in the NTs was observed in this group compared to unvaccinated controls. Notably, overall viral titers were lower for the BA.5 challenged K18-hACE2 mice even for the unimmunized control group as compared to B.1.351-challenged 129S1 mice, reflecting the poor infectivity of the Omicron variants in mouse models as we and others have previously observed.34,35 As with the B.1.351 challenge model, IM mRNA prime, IN S only boost also provided effective protection, with no virus detectable in either the LRT or the URT for the BA.5 challenge model, which has reduced replication efficiency in mice. Thus, while the IM mRNA, IN-adjuvanted S groups showed the most complete cross-variant sterilizing immune responses throughout the respiratory tract, such hybrid immunization approaches show a benefit in promoting protective mucosal immune responses, even with unadjuvanted antigen alone delivered IN. Host immune responses upon infection reflect disease course and pathogenesis, or lack thereof, due to vaccine-mediated protection. Thus, we measured cytokine levels in lung homogenates at 4 d.p.i. for the vaccinated and challenged 129S1 and K18-hACE2 mice (Figures 7E and 7F; individual cytokine data are provided in Figures S8 and S9). In 129S1 mice, pro-inflammatory innate cytokines/chemokines MIP-1α, MIP-1β, IP-10, MIP-2α, MCP-1, MCP-3, RANTES, GRO-α, IL-6, and TNFα were elevated in mice that showed breakthrough infection after receiving single vaccinations with either IN NE/S or IN NE/IVT/S, as well as with IM prime/boost with Advx/S or in the negative control group that received IN PBS twice (Figure 7E). The T cell cytokines IFN-γ, IL-18, IL-22, and IL-10 were also increased in these groups, suggesting strong immune activation to control viral infection. Similarly, IM mRNA-primed mice boosted IN with S alone or with NE/S had elevated innate cytokine/chemokine responses, although with reduced adaptive immune cytokines. In contrast, 129S1 mice given homologous IM mRNA or IN NE/IVT/S prime/boost immunizations, as well as mice given heterologous IM mRNA prime followed by IN NE/IVT/S boost had very low levels of both innate and adaptive chemokines/cytokines in the lungs after challenge, consistent with the effective viral control observed in these groups. Furthermore, 129S1 mice that received IM Advx/S showed elevated Th2 cytokines IL-4, IL-5, IL-13, and eotaxin, which seemed to be higher than the unvaccinated group, reflecting the Th2 bias of the adjuvant. Unlike B.1.351 infection in 129S1 mice, which resulted in strong host immune responses, BA.5 infection resulted in overall lower immune responses in K18-hACE2 mice, reflecting the better replication efficiency and pathogenesis of B.1.351 compared to BA.5 (Figure 7F). In K18-hACE2 mice, breakthrough infection in mice singly vaccinated with IM mRNA resulted in the strongest induction of the innate pro-inflammatory cytokines/chemokines, MIP-1α, MIP-1β, IP-10, MCP-1, MCP-3, and IL-6, along with the cytokines TNF-α, IFN-γ, and IL-18. In contrast, similar to the 129S1 mice, groups primed with IM mRNA, boosted either IM with mRNA or IN with NE/S or NE/IVT/S, showed minimal induction of these inflammatory chemokines and cytokines, reflecting the effective viral control in these groups. Interestingly, IM mRNA prime followed by IN S alone showed a similar pro-inflammatory chemokine/cytokine profile as the single IM mRNA immunization group, albeit slightly reduced, highlighting the importance of the IN NE and NE/IVT adjuvants in mediating optimal protection. Overall, cytokine/chemokine profiles are elevated in groups for which replicating virus could be detected in lungs or NTs at 4 d.p.i., and chemokine/cytokine responses are skewed both by adjuvant type and vaccination routes.
Virus-neutralizing serum antibodies induced through heterologous vaccination provides partial protection both in the lower and upper respiratory tracts, but is insufficient for complete viral control
To obtain additional mechanistic insights into the key components contributing to the protective efficacy of the different vaccine regimens, we examined the role of serum antibodies in mediating viral control for select treatment groups. The 129S1 donor mice were immunized with the same vaccine dosages and intervals as in Figure 7, with IM mRNA prime/boost or IN NE/IVT/S prime/boost, and compared to groups immunized with an IM mRNA prime followed by boost with IN S alone or adjuvanted with NE/IVT (Figures 8 and S10). Two weeks post boost, 150 μL serum pooled according to vaccination group was passively transferred IP to naive donor mice. 24 h after the transfer, mice were challenged IN with 104 B.1.351, and viral loads in the lungs and NTs were quantified at 4 d.p.i. Compared to mice given serum from unvaccinated PBS controls, mice receiving serum from all vaccination groups tested showed reductions in viral titers in the lungs. Groups receiving serum from the IM mRNA prime/boost and IM mRNA; IN NE/IVT/S groups showed the largest and equivalent reductions in viral load (approximately 1 log reduction) (Figure 8A). Groups receiving serum from the IM mRNA; IN S and IN NE/IVT/S prime/boost groups also showed some reductions in viral titers in the lung compared to the PBS control, however, these differences were not statistically significant. Upon examining viral titers in the NTs, interestingly, mice receiving serum from the heterologous IM mRNA prime with IN S or IN NE/IVT/S boost groups showed the lowest viral titers (approximately 1 log reduction), and therefore allowed control of virus replication, while serum from the IM mRNA prime/boost group did not appear to provide significant reductions compared to the PBS control (Figure 8B). The IN NE/IVT/S prime/boost group also demonstrated notable decreases in viral load in the NTs of some receiver mice; however, these changes were not statistically significant from the PBS control. These results suggest that the protection conferred through the heterologous IM mRNA; IN NE/IVT/S vaccination strategy is not mediated through serum antibody responses alone, and requires the additional presence of other cellular factors, including T cells and other immune effector cells activated by these primed T cell responses, as well as local mucosal immune responses to provide the robust viral control observed in both the upper and lower respiratory tracts, particularly against divergent variants capable of evading antibody neutralization.
Figure 8.

Passive serum transfer provides some but not complete protection against cross-variant challenge
129S1 mice were immunized with IM mRNA prime/boost, IN NE/IVT/S prime/boost, or with IM mRNA prime followed by IN boost with S alone or adjuvanted with NE/IVT at a 4-week interval. Vaccine doses consisted of 0.4μg of mRNA and 15 μg S protein. At 2 weeks post-boost, 150 μL of sera pooled from each treatment group was passively transferred to naive mice, which were subsequently IN challenged with 104 pfu of B.1.351. Viral titers in the (A) lungs and (B) nasal turbinates were measured at 4 d.p.i. by plaque assay. (n = 4–5/group; ∗p < 0.05, ∗∗p < 0.01 by Mann-Whitney U test.
Discussion
Currently licensed IM-administered mRNA vaccines are efficient inducers of systemic nAbs and circulating T cell responses, but are poor inducers of protective mucosal immune responses. However, evidence has suggested that “hybrid immunity” acquired through IM vaccination followed by natural respiratory infection provides more complete protection both systemically and at mucosal sites.34,35,36 In an effort to emulate this hybrid immunity, we assessed whether boosting IM mRNA-primed animals via the IN route with NE/IVT-adjuvanted S protein could confer robust immunity through driving more optimal humoral and cellular immune responses, in both the periphery and at mucosal sites.
Humoral responses in serum were compared between various heterologous IM/IN and homologous IM/IM and IN/IN immunization regimens. In C57Bl/6 mice, priming with IM mRNA induced higher antigen-specific IgG than the IN NE or NE/IVT/S. However, after boost vaccination IM mRNA prime/boost, IN NE/IVT/S prime/boost and IM mRNA prime followed by heterologous boost with IN NE/S or NE/IVT/S resulted in equally robust S- and RBD-specific IgG titers. In contrast, minimal increase in S-specific IgG was observed in IM mRNA primed animals boosted IN with unadjuvanted S alone, demonstrating the role of the NE adjuvants in driving an optimal boost effect for IN immunization. Similar effects were observed in mice of different genetic backgrounds. Furthermore, heterologous boosting with IN NE/S or NE/IVT/S induced robust cross-nAb responses across WT, B.1.617.2, B.1.351, and B.1.1.529 variants, which were enhanced compared to those induced by IM mRNA; IN S alone in IM mRNA-primed C57Bl/6 and K18-hACE2 mice, confirming the effectiveness of the NE adjuvants in IN booster vaccines. The same relative patterns for nAbs were maintained in 129S1 mice; however, the IM mRNA; IN NE/IVT/S immunization showed more distinct enhancement in viral cross-neutralizing antibodies compared to IM mRNA prime/boost. Such nuanced differences in the 129S1 mice may reflect the comparatively lower levels of IFN-α production observed by others in C57Bl/6 and K18-hACE2 mice.37,38 Taken together, these results may support the ability of the IN adjuvants to improve the breadth of the humoral immune response compared to homologous mRNA prime/boost regimens.
We previously showed that the combination of NE and IVT results in a more Th1-polarized host immune response compared to the balanced Th1/Th2 response induced with NE alone, which is reflected in the antigen-specific IgG2b/IgG1 and IgG2c/IgG1 ratios in serum.14 Induction of IgG2c is significant due to its role in Fc-mediated effector functions, including antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement activation, which have shown to be key contributors to SARS-CoV-2 immunity.39 IN NE/IVT/S prime/boost induced higher IgG2b and IgG2c titers compared to IN NE/S prime/boost, confirming the Th1 skewing properties of the IVT. However, IM mRNA prime followed by IM mRNA boost or IN boost with NE/S or NE/IVT/S resulted in similar IgG2b and significantly greater IgG2c levels relative to IN NE/IVT/S prime/boost immunization, suggesting that priming with IM mRNA can enhance the Th1 polarization induced by IN NE/IVT. Indeed, T cell antigen-recall assessment in splenocytes and cLN isolates from vaccinated mice revealed dramatic enhancement of Th1 cytokine production by the heterologous IM mRNA; IN NE/IVT/S vaccination regimen compared to IM mRNA prime/boost, with high levels of secreted antigen-specific IFN-γ, IL-2, TNF-α, and IP-10 observed in response to S protein stimulation. Levels of these cytokines were especially enhanced in the local mucosal draining LNs. For example, IM mRNA; IN NE/IVT/S resulted in a nearly 20-fold enhancement in antigen-specific IFN-γ production in the cLN compared to IM mRNA prime/boost, demonstrating the crosstalk between immune responses primed in the periphery by the mRNA and those triggered within the mucosa by the NE/IVT-adjuvanted boost. Notably, IN boosting of IM mRNA-primed T cell responses with NE/S or NE/IVT/S induced significantly higher percentages of S-specific polyfunctional IFN-γ+IL-2+TNF-α+CD4+ T cells in the cLN compared to homologous IM mRNA, IN NE/S, or IN NE/IVT/S prime/boost regimens. This is significant, as polyfunctional IFN-γ+IL-2+TNF-α+T cells have been shown to be a strong predictive indicator of potent antiviral T cell responses. In contrast with the Th1 cytokines, heterologous boosting of IM mRNA-primed animals with IN NE/S or NE/IVT/S did not enhance Th2 cytokine responses relative to the IM mRNA prime/boost group. CD4+ T cells in secondary lymphoid tissues are required for class switching to result in the production of IgG and IgA. Therefore, the optimal induction of CD4+ T cell responses in cLNs is expected to promote class switching events in mucosal associated lymphoid tissues like the cLNs, which correlates with the higher levels of IgA observed in heterologously boosted groups. Interestingly, while all three IM mRNA prime, IN boost groups (S alone, NE/S, and NE/IVT/S) induced higher frequencies of polyfunctional CD4+ T cells in the lung compared to IM mRNA prime/boost, the IN S alone boost did not significantly enhance these cells within the cLNs, which required the NE or NE/IVT adjuvants for optimal enhancement. These results highlight the enhanced mucosal cellular responses induced by this mucosal booster strategy and underscore the role of the NE adjuvants in potentiating these responses. Interestingly, IN boosting of IM mRNA-primed T cell responses with NE/IVT/S induced significantly higher percentages of S-specific CD4+TRMs within the lung compared to IM mRNA prime/boost, as well as CD4+TEMs in the cLNs, but induced lower CD8+TRMs in the lung and lower CD8+TEMs in the cLNs, pointing to potential key mechanistic differences between the protection conferred through heterologous IM/IN vs. homologous IM/IM (or IN/IN) vaccination strategies.
Th17 cells in the context of IL-10 have been shown to be critical in promoting high and sustained levels of IgA production at mucosal sites, particularly the lung, and in the establishment of resident memory T cells.25 Induction of IL-17A is exclusive to the IN route of NE or NE/IVT administration and is a key component of NE/IVT-mediated immunity. Interestingly, boosting IM mRNA primed mice with IN NE/IVT/S resulted in equivalent levels of IL-17A (and IL-10) production in the cLN as two immunizations with IN NE/IVT/S, which was higher than that of mice given only one IN NE/IVT/S immunization. Accordingly, we found that the IN NE/S or NE/IVT/S boost of IM mRNA primed mice resulted in increased frequencies of IL-17A-secreting CD4T cells in the lung. These findings highlight the ability to use parenteral prime immunizations to set the stage for induction of more robust local mucosal responses upon mucosal pull immunization.
Consistent with IL-17A production, when induction of mucosal S-specific IgA in BALF was considered, we found that IN immunization, either as a homologous prime/boost or as part of a heterologous IM prime/IN boost regimen, was required to obtain detectable S-specific IgA. IM mRNA prime/boost did not induce detectable S-specific IgA, while IN NE/S and IN NE/IVT/S prime/boost resulted in robust IgA responses. Interestingly, while single IN NE/S or IN NE/IVT/S immunizations induced only low levels of mucosal IgA, equivalently high antigen-specific IgA was induced upon boosting IM mRNA-primed animals with IN NE/IVT/S to similar levels as those induced by two immunizations with IN NE/IVT/S. These results further show the potential of the NE/IVT adjuvanted pull approach to drive improved mucosal immune responses initially primed by IM mRNA vaccination. It is well described that IgG can exudate from the blood into the lung and indeed, groups with highest serum S-binding titers also had the highest S-specific binding IgG titers in BALF. This suggests that deep lung antibody-mediated protection during infection may also be provided by IgG and not only sIgA, although aside from their shared ability to mediate viral neutralization, they can provide protection via different mechanisms. IgA-mediated protection can involve removal of immune complexes through immune exclusion, as well as triggering FcαR1-mediated immune mechanisms like respiratory burst from neutrophils.40 IgG-mediated protection can also occur through activation of ADCCs and ADCPs through Fcγ receptor-mediated mechanisms.41 Thus, protection in the respiratory tract provided by IgA and IgG likely occurs through a combination of mechanisms, including direct virus neutralization, opsonization, and the activation of Fc receptor-dependent mechanisms.
As the N501Y mutation of B.1.351 allows it to infect WT mice, we used this variant to challenge 129S1 mice for evaluation of the impact of different vaccination strategies on viral replication and virus-host responses. Complete viral control in the lungs was only observed for IM mRNA prime/boost mice and IM mRNA primed mice receiving a boost immunization through the IN route. These results demonstrate that induction of potent antibody responses through IM, IN or a combination of both can effectively prevent viral replication in the lungs. Prime/boost with IM Advx/S did not impart sterilizing immunity in the lung, despite having similar nAb titers against the B.1.351 variant as the IM mRNA; IN S group and similar serum S-specific serum IgG titers against the ancestral S protein as groups, which did demonstrate full viral control. Thus, additional components besides serum nAbs are key contributors to sterilizing immunity in the LRT. When viral replication in the URT (nasal turbinates) was examined, we found that a mucosal boost vaccination was required to prevent viral replication. Despite full control of viral replication in the LRT for the IM mRNA prime/boost group, viral titers were still detectable in the URT. Moreover, replicating virus was detected in the URT of all mice vaccinated with IM Advx/S prime/boost. Full control of viral replication in the URT thus correlated with induction of mucosal IgA and local cellular responses by the mucosal booster strategies. Indeed, passive serum transfer from IM mRNA; IN NE/IVT/S-vaccinated mice into naive receiver mice provided only a modest degree of protection from heterologous viral challenge both in the LRT and URT. Thus, complete viral control mediated by the IM/IN vaccination regimen requires the additional presence other immune effector cells and likely mucosal antibody responses, especially for protection against a divergent variant. As real-world studies have demonstrated, robust circulating and tissue-resident cellular immune effectors become increasingly important as viral evolution continues to facilitate escape from antibody neutralization.42
We observed that host immune responses to infection are influenced by the type of adjuvanted vaccination, even if complete viral control was observed. Heterologous boosting of IM mRNA primed mice IN with NE/S or NE/IVT/S in 129S1 mice markedly suppressed induction of major inflammatory markers associated with severe disease upon challenge with B.1.351, particularly in the IN NE/IVT/S-boosted group. These findings confirm the protection afforded by these vaccination strategies. Similarly, homologous IM mRNA and IN NE/IVT/S prime/boost groups showed a similar cytokine/chemokine profile after infection in the lung, with effective prevention of virally induced inflammatory responses typically associated with more severe disease.37 Boosting IM mRNA-primed mice with IN/S alone or homologous IN prime/boosting with NE/S also reduced inflammatory responses to infection compared to singly vaccinated animals and PBS control mice; however, in accordance with the incomplete protection observed in these groups as assessed by viral titers, this suppression was only partial. Increases in cytokines associated with type II polarization (IL-4, IL-5, and IL-13) in lungs after infection were seen in mice that received IM Advx-adjuvanted vaccine. Promotion of Th2-driven vaccine responses thus seem to translate to type II host immune responses in the lungs of infected mice. This is an observation that we also have reported recently for mouse experiments performed with Advx-adjuvanted licensed influenza vaccines and influenza virus challenge.43
There is currently no optimal animal model for studying infection with Omicron SARS-CoV-2 variants, as they give attenuated pathology even in transgenic K18-hACE2 mice.32 Nevertheless, the K18-hACE2 infection model provides a good representation of mild Omicron SARS-CoV-2 infection for testing vaccine effectiveness when virus titers are considered in the URT and LRT. Similar to observations with B.1.351 infection in 129S1 mice, a mucosal boost vaccination of IM mRNA-primed animals was required to obtain sterilizing immunity in both the URT and LRT in BA.5-challenged K18-hACE2 mice. In contrast, IM mRNA given once or as a prime/boost as well as IM Advx/S prime/boost were insufficient. In the K18-hACE2 model, mice that received only a single IM mRNA immunization showed a typical macrophage inflammatory profile upon BA.5 infection, whereas groups receiving heterologous IM mRNA prime, IN NE/S, or NE/IVT/S boost effectively prevented induction of this inflammatory profile, consistent with the complete viral control offered by these groups. If vaccination can prevent replication of virus in the NTs, this is seen as a first step in efficient interference with virus transmission. Taken together, these results strongly demonstrate that heterologous IN boost vaccination is able to induce optimal mucosal immunity that together with systemic immunity effectively controls viral replication in the URT. Our ongoing experiments in hamster transmission models will be important for validating this prime/pull vaccination strategy also blocks viral transmission. While it is difficult to make direct comparisons with other studies that have shown the potential of mucosal boosting with spike protein alone in the context of SARS-CoV-2 mRNA vaccine priming, our results show significant enhancement in humoral and cellular immune correlates with the addition of our NE/IVT adjuvant system compared to unadjuvanted S protein alone in IN boost immunizations.33 The NE/IVT adjuvant was necessary for driving robust local responses in the mucosal lymphoid tissue and the lung and was essential for optimally polarizing these local cellular immune responses. Furthermore, while these studies demonstrated reductions in viral titer in the lungs and NTs with the unadjuvanted mucosal boost against the vaccine-matched challenge variant, vaccination was still infection permissive, consistent with what we observed for the unadjuvanted IN boost. In contrast, the NE/IVT adjuvanted IN boost imparted complete viral control in both the LRT and URT against significantly divergent challenge variants. Additionally, other mucosal immunization strategies with adenovirus (Ad)-based vaccines have also shown promising efficacy in mouse models and non-human primates.44,45,46 However, a recent clinical study examining the ChAdOx-1 vector in an IN prime/boost vaccine or as an IN boost for IM mRNA-immunized subjects showed discouraging responses systemically and mucosally, likely due to poor infectivity of the ChAdOx1 vector in the human respiratory epithelium.47 While other Ad-based vectors are being explored as IN boosts, vector-based immunity may present an additional hurdle for additional IN boost immunizations.
The ability to induce cross-variant sterilizing immunity within the respiratory tract is critical both for limiting viral transmission and disease progression, particularly as new SARS-CoV-2 variants continue to emerge. We herein demonstrated that an IM mRNA-based prime followed by a mucosal pull with a rationally designed adjuvant and recombinant protein is an effective strategy for inducing robust, tailored, and cross-sterilizing antibody and T cell responses both systemically and locally within the mucosa of the upper and lower respiratory tracts. Given the relatively rapid waning of immunity now widely observed with the IM mRNA vaccines, and the durable immune responses induced by the IN NE/IVT adjuvant, heterologous boosting with IN NE/IVT/S may also improve the immune response longevity of current vaccines.16 Finally, we expect that the mucosal responses observed herein will be further enhanced and strengthened with additional mucosal booster vaccinations, given that at least two mucosal immunizations are required for optimal immune responses with NE/IVT as a homologous vaccine regimen and that prolonged antigen exposure in the mucosa has been shown to drive optimal resident-memory T cell responses. This, as well as the effects of prior infection on the prime/pull approach, will be important questions to explore in future studies, especially given that the majority of the population has now acquired a degree of immunity through a combination of IM vaccinations and respiratory infections with COVID-19.12 In conclusion, as annual booster vaccinations continue to be required for COVID-19, our work underscores the value of a mucosal boost vaccination and highlights the promising potential of the IN NE/IVT adjuvant for inducing more complete hybrid immune responses in previously vaccinated subjects.
Materials and methods
Adjuvants and antigen
NE was produced by emulsifying cetylpyridinium chloride and Tween 80 at a 1:6 (w/w) ratio, with ethanol (200 proof), super refined soybean oil (Croda) and water using a high-speed homogenizer as previously described.48,49 The sequence and synthesis of IVT RNA has previously been described in detail.17 Briefly, IVT was in vitro transcribed using a HiScribe T7 High Yield RNA synthesis kit (New England Biolabs) followed by DNAse I clean-up with a TURBO DNA-free kit (Thermo Fisher Scientific), and purification with an RNeasy purification kit (Qiagen). Recombinant WT SARS-CoV-2 full-length S protein and RBD (aa319-545) (derived from Wuhan-Hu-1) with C-terminal His tags were produced in Expi293F or ExpiCHO cells, respectively, and purified by the University of Michigan Center for Structural Biology as previously described.50 Addavax (MF59 similar) was obtained from Invivogen, and the BNT162b2 mRNA vaccine (Pfizer) was obtained through the National Institutes of Health (NIH) SAVE program.
Cell lines
Vero E6 cells (ATCC) were maintained in DMEM supplemented with 10% heat inactivated fetal bovine serum (HI FBS) and 1× non-essential amino acids (NEAAs). HEK293T cells expressing hACE2 (293T-hACE2) were obtained from BEI resources and maintained in HEK293T medium (DMEM with 4 mM L-glutamine, 4,500 mg/L L-glucose, 1 mM sodium pyruvate, and 1,500 mg/L sodium bicarbonate, 10% HI FBS and 100 IU penicillin, and 100 μg/mL streptomycin).
Viruses
SARS-CoV-2 clinical isolate USA-WA1/2020 (BEI resources; NR-52281), and B.1.351 and BA.5 variant viruses were propagated in Vero E6 cells or Vero-TMPRSS2. All viral stocks were verified by deep sequencing. All work with authentic SARS-CoV-2 viruses were performed in certified BSL3 or ABSL3 facilities in accordance with institutional safety and biosecurity procedures.
Lentivirus pseudotyped virus
Generation of pseudotyped lentiviruses (PSVs) expressing the SARS-CoV-2 S proteins from WT, B.1.351, B.1.617.2, and B.1.1.529 (BA.1), and BA.4/5 variants harboring GFP and luciferase reporter genes was performed as previously described for the WT PSV.51 Plasmids carrying the full-length SARS-CoV-2 S protein from each variant containing a C-terminal 19 amino acid deletion to remove the ER retention signal were used for pseudotyping (Invivogen). Viral titers (TU/mL) across variants were determined by measuring PSV transduction of GFP in 293T-hACE2 cells.
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committees at the University of Michigan and Icahn School of Medicine at Mount Sinai and were carried out in accordance with these guidelines. The 6- to 8-week-old female C57Bl/6, 129S1 (Jackson Laboratory), or K18-hACE2 mice (bred in-house) were housed in specific pathogen-free conditions. Mice were acclimated for 2 weeks before initiation of each study. For challenge studies, mice were transferred to ABSL3 facilities 1 week before viral challenge.
Immunization
Mice were anesthetized using isoflurane in a IMPAC6 precision vaporizer. For IN immunization, mice were given 12 μL (6 μL/nare) of each vaccine formulation (which ensures retention within the nasal cavity), and for IM immunization, the vaccine was delivered in a 50-μL volume into the thigh muscle of the hind limbs. Each group received prime and boost immunizations at a 4-week interval. For C57Bl/6 mice, mice were primed IM with 0.4 μg BNT162b2 mRNA (Pfizer/BioNTech). Mice were boosted either through the IM route with the same dose of mRNA, or through the IN route with PBS or 15 μg of WT S protein in PBS, 20% NE (w/v) (NE/S), or 20% NE with 0.5 μg IVT (NE/IVT/S). Immune responses were compared to mice given homologous prime/boost immunizations with IN NE/S, IN NE/IVT/S, or IM 50% Advx/S with the same amount of S protein. Comparison groups primed with IM PBS and boosted with IN NE/S or NE/IVT/S were also included. Select immunization regimens using the same adjuvant/antigen doses were chosen for evaluation in 129S1 and K18-hACE2 mice.
Serum was obtained by saphenous vein bleeding 2 and 4 weeks after the prime, and by cardiac puncture at the end of the experiment at week 8. BALF was obtained by lung lavage with 0.8 mL PBS containing protease inhibitors at week 8.
ELISA
Immunograde 96-well ELISA plates (Midsci) were coated with 100 ng S protein or RBD in 50 μL PBS/well overnight at 4°C, and then blocked in 5% non-fat dry milk/PBS for 1 h at 37°C. Sera (or BAL) from immunized mice were serially diluted in PBSB (PBS/0.1% BSA). Blocking buffer was removed, and serum dilutions were added and incubated for 2 h at 37°C followed by overnight incubation at 4°C. Plates were washed with PBST (0.05% Tween 20), and alkaline phosphatase conjugated secondary antibodies diluted in PBSB were added (goat-anti-mouse IgG, IgG1, IgG2b, and IgG2c from Jackson Immuno Research Laboratories; goat-anti-mouse IgA [Catalog: 62–6722] from Thermo Fisher Scientific), and incubated 1h at 37°C. Plates were washed with PBST, and developed by incubation with p-nitrophenyl phosphate substrate in diethanolamine (Thermo Fisher Scientific) at room temperature (RT). Absorbance was measured at 405 nm, and titers were determined using a cutoff defined by the sum of the average absorbance at the lowest dilution of naive serum and two times the standard deviation.
Pseudovirus microneutralization assays
Pseudovirus (PSV) microneutralization assays were performed as previously described.16 Briefly, 1.25 × 104 HEK293T-hACE2 cells/well were seeded overnight on 96-well tissue culture plates. Sera from immunized mice were serially diluted by a factor of three, starting at a dilution of 1:30 in HEK293T medium. 50 μL of the diluted sera was then added to 50 μL of PSVs (40,000 TU/mL) and incubated for 1 h at 37°C. The PSV titer used across variant PSVs was selected based on the titer of WT PSV giving >100,000 RLUs above background. The virus/serum mixture was then added to the cells and incubated for 3 d at 37°C. Infection media was removed, and luminescence was measured by addition of 25 μL BrightGlo in 25 μL PBS. Neutralization titers were determined as the dilution at which the luminescence remained below the luminescence of the (virus only control-uninfected control)/2. Samples with undetectable neutralization were designated as having a titer of 10°. Neutralization assays with these PSVs have been demonstrated by us and numerous groups to be representative of authentic virus neutralization assays.15,52
Tissue isolation and single cell suspension preparation
Two weeks post-boost (week 8), mice were i.v. injected through the tail vein with 3 μg of α-CD45-AF488 antibody (Biolegend) 3 months before sacrifice to discriminate resident from circulating T cells.30 The left lung lobe was harvested, and single-cell suspensions were prepared by mincing with surgical scissors, followed by incubation in 3 mL of digestion media (RPMI, 10% HI FBS, 2 mM L-glutamine, 1% NEAA, 1 mM sodium pyruvate, 50 μM 2-mercaptoethanol, 100 IU penicillin, 100 μg/mL streptomycin with 1 mg/mL collagenase A [Roche], 20 U/mL of DNAse-I [Roche]) followed by incubation at 37°C for 1 h with shaking. Tissue dissociation was continued by passages through an 18G needle and filtering through a cell strainer. Cells were incubated in ACK lysis buffer for 5 minutes at RT and washed with PBS. Methods for splenocyte and cLN lymphocyte preparation have previously been described.16 All cells were resuspended in T cell media (DMEM, 5% HI FBS, 2 mM L-glutamine, 1% NEAA, 1 mM sodium pyruvate, 10 mM MOPS, 50 μM 2-mercaptoethanol, 100 IU penicillin, 100 μg/mL streptomycin) for further downstream analysis.
Antigen recall response
T cell antigen recall response was assessed by cytokine release in cell isolates from the spleen and cLN of immunized mice 2 weeks post-boost (week 8). For antigen recall, isolated cells were plated at 8 × 105 cells/well and stimulated with 5 μg (25 μg/mL) S protein (WT) in T cell media for 72 h at 37°C. Secreted cytokines (IFN-γ, IL-2, IP-10, IL-5, IL-6, IL-13, IL-10, IL-17A, and TNF-α) were measured relative to unstimulated cells in supernatants using a Milliplex MAP Magnetic Mouse Cytokine/Chemokine multiplex immunoassay (EMD Millipore).
Flow cytometry
For analysis of S protein-specific CD8+ T cells, 2 × 106 cells isolated from the lungs or 1 × 106 cells from the spleen or cLNs were stained with LIVE/DEAD Fixable Yellow (Thermo Fisher Scientific) in PBS/2mM EDTA for 20 m at 4°C, washed, and then incubated with Fc block (Biolegend) in FACS buffer (PBS, 1% FBS, 2mM EDTA) for 30 m at 4°C. Cells were stained with the H2kb-VNFNFNGL-PE tetramer (obtained from the NIH Tetramer Core Facility) and surface marker antibodies for 1 h at 4°C, and then washed and fixed in IC fixation buffer (Thermo Fisher Scientific). For analysis of S-specific CD4+ T cells, 1 × 106 cells isolated from the lung, spleen, or cLNs were pre-incubated with Fc block (Biolegend) in T cell media for 30 m at 37°C and then incubated with the I-Ab -VTWFHAIHVSGTNGT-PE tetramer (obtained from the NIH Tetramer Core Facility) for 1 h at 37°C. Cells were then washed and stained for surface markers in FACS buffer for 30 m at 4°C, followed by staining with LIVE/DEAD Fixable Aqua for 20 m at 4°C before fixation. For T cell intracellular cytokine analysis, 1 × 106 cells isolated from the lungs, spleen, or cLNs were stimulated with 25 μg/mL S protein (WT) in T cell media for 24 h at 37°C, with brefeldin A (Biolegend) added during the last 6 h. Cells were stained with LIVE/DEAD Fixable Yellow or Aqua in PBS/2 mM EDTA for 20 m at 4°C, washed, and then incubated with Fc block for 10 m at 4°C before staining for surface T cell makers (CD3, CD4, and CD8) for 30 minutes at 4°C. Cells were washed, fixed in IC fixation buffer 30 m at RT and permeabilized 30 m at 4°C before staining with antibodies for intracellular cytokines in permeabilization buffer for 30 minutes at 4°C. Cells were washed with permeabilization buffer and resuspended in PBS/2 mM EDTA. Antibody details and specific staining conditions are provided in Table S2. FACS was performed on a Novocyte3000 flow cytometer and analyzed using FlowJo software.
Viral challenge
Immunized mice were challenged 3 weeks post-boost (week 7). We challenged 129S1 mice IN with 104 PFU B.1.351, and K18-hACE2 mice were challenged IN with 104 PFU BA.5 delivered in 30 μL PBS. Mice were sacrificed 4 d.p.i.. Lungs were harvested in 500 μL of PBS, and NTs were collected. Homogenates were prepared for virus titration by plaque assay as previously described, and cytokine profiling post-challenge was performed on homogenate using a Milliplex MAP Magnetic Mouse Cytokine/Chemokine multiplex immunoassay (EMD Millipore).53
Passive transfer
Serum was collected from vaccinated mice 2 weeks post-boost and pooled for each group. Naive 129S1 receiver mice were IP injected with 150 μL serum. Efficient serum transfer was confirmed 24 h later by submandibular bleeding and measuring S-specific IgG in the serum. Mice were then challenged IN with 104 PFU of B.1.351 as described above. Lung and NT viral titers were evaluated at 4 d.p.i.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 9 (GraphPad Software). Comparisons were performed by Mann-Whitney U test or one-way ANOVA with Tukey post hoc test as indicated.
Data and code availability
All data are provided in the main text, figures, or the supplemental information, and raw data files are available upon request. Requests for reagents or cell lines used in this study can directed to the corresponding authors.
Acknowledgments
SARS-CoV-2 work in the laboratory of M.S. is supported by NIH/NIAID R01AI160706 and NIH/NIDDK R01DK130425. S.C.P has received support from the Korea Health Industry Development Institute (KHIDI) for research under the Biomedical Global Talent Nurturing Program (HI22C2101). Work in the laboratory of P.T.W. is supported by NIH/NIAID R01AI160706 and NIH/NIAID R21AI176069. This study was also partly funded by CRIPT (Center for Research on Influenza Pathogenesis and Transmission), a NIH NIAID-funded Center of Excellence for Influenza Research and Response (CEIRR, contract number 75N93021C00014), and by NIH/NIAID contract 75N93019C00046 and U19AI135972 to AG-S. We thank Richard Cadagan for technical support. The enhanced Emerging Pathogens Facility at ISMMS is a NIHBSL3/BSL3/ABSL3 facility that is part of the BSL-3 Biocontainment CoRE supported by subsidies from the Dean’s Office and by NIH NIAID G20AI174733 (to R.A.A.). We thank Dr. Wendy Fonseca Aguilar for scientific input, Julianne d’Acunzo for experimental assistance, the U of M vector core for producing the lentivirus pseudovirus and for providing technical input on assays, Joel Whitfield and the Rogel Cancer Center Immunology Core for multiplex assay support, as well as the U of M Center for Structural Biology for producing recombinant proteins. The CSB acknowledges support from the U-M Biosciences Initiative and the U-M Rogel Cancer Center.
Author contributions
P.T.W., M.S., G.L., and M.J.W. conceived of the studies and performed experiments, data analysis, and interpretation. D.K., L.A.C., J.J.O., S.P., V.Y., P.W., M.F., J.J.L., and K.W.J. performed experiments and data analysis. P.T.W., M.S., G.L., and M.J.W. contributed to the writing of the manuscript, and J.J.O., A.G.S., and J.R.B. provided key scientific input.
Declaration of interests
P.T.W., M.S., J.R.B., and A.G.-S. are co-inventors on a patent for NE/IVT. The A.G.-S laboratory has received research support from GSK, Pfizer, Senhwa Biosciences, Kenall Manufacturing, Blade Therapeutics, Avimex, Johnson & Johnson, Dynavax, 7Hills Pharma, Pharmamar, ImmunityBio, Accurius, Nanocomposix, Hexamer, N-fold LLC, Model Medicines, Atea Pharma, Applied Biological Laboratories and Merck, outside of the reported work. A.G.-S. has consulting agreements with the following companies involving cash and/or stock: Castlevax, Amovir, Vivaldi Biosciences, Contrafect, 7Hills Pharma, Avimex, Pagoda, Accurius, Esperovax, Farmak, Applied Biological Laboratories, Pharmamar, CureLab Oncology, CureLab Veterinary, Synairgen, Paratus, Pfizer and Prosetta, outside of the reported work. A.G.-S. has been an invited speaker in meeting events organized by Seqirus, Janssen, Abbott, and Astrazeneca. A.G.-S. is inventor on patents and patent applications on the use of antivirals and vaccines for the treatment and prevention of virus infections and cancer, owned by the Icahn School of Medicine at Mount Sinai, New York. The M.S. laboratory received unrelated research support as sponsored research agreements from ArgenX BV, Phio Pharmaceuticals, 7Hills Pharma LLC and Moderna.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2024.10.016.
Contributor Information
Pamela T. Wong, Email: ptw@umich.edu.
Michael Schotsaert, Email: michael.schotsaert@mssm.edu.
Supplemental information
References
- 1.Cele S., Jackson L., Khoury D.S., Khan K., Moyo-Gwete T., Tegally H., San J.E., Cromer D., Scheepers C., Amoako D.G., et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature. 2022;602:654–656. doi: 10.1038/s41586-021-04387-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tseng H.F., Ackerson B.K., Sy L.S., Tubert J.E., Luo Y., Qiu S., Lee G.S., Bruxvoort K.J., Ku J.H., Florea A., et al. mRNA-1273 bivalent (original and Omicron) COVID-19 vaccine effectiveness against COVID-19 outcomes in the United States. Nat. Commun. 2023;14:5851. doi: 10.1038/s41467-023-41537-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arunachalam P.S., Lai L., Samaha H., Feng Y., Hu M., Hui H.S.-Y., Wali B., Ellis M., Davis-Gardner M.E., Huerta C., et al. Durability of immune responses to mRNA booster vaccination against COVID-19. J. Clin. Invest. 2023;133 doi: 10.1172/JCI167955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goel R.R., Painter M.M., Apostolidis S.A., Mathew D., Meng W., Rosenfeld A.M., Lundgreen K.A., Reynaldi A., Khoury D.S., Pattekar A., et al. mRNA vaccines induce durable immune memory to SARS-CoV-2 and variants of concern. Science. 2021;374 doi: 10.1126/science.abm0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Florea A., Sy L.S., Qian L., Ackerson B.K., Luo Y., Tubert J.E., Lee G.S., Ku J.H., Bruxvoort K.J., Talarico C.A., et al. Effectiveness of Messenger RNA-1273 Vaccine Booster Against Coronavirus Disease 2019 in Immunocompetent Adults. Clin. Infect. Dis. 2023;76:252–262. doi: 10.1093/cid/ciac785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrews N., Stowe J., Kirsebom F., Toffa S., Sachdeva R., Gower C., Ramsay M., Lopez Bernal J. Effectiveness of COVID-19 booster vaccines against COVID-19-related symptoms, hospitalization and death in England. Nat. Med. 2022;28:831–837. doi: 10.1038/s41591-022-01699-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis-Gardner M.E., Lai L., Wali B., Samaha H., Solis D., Lee M., Porter-Morrison A., Hentenaar I.T., Yamamoto F., Godbole S., et al. Neutralization against BA.2.75.2, BQ.1.1, and XBB from mRNA Bivalent Booster. N. Engl. J. Med. 2023;388:183–185. doi: 10.1056/NEJMc2214293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marking U., Bladh O., Aguilera K., Yang Y., Greilert Norin N., Blom K., Hober S., Klingström J., Havervall S., Åberg M., et al. Humoral immune responses to the monovalent XBB.1.5-adapted BNT162b2 mRNA booster in Sweden. Lancet Infect. Dis. 2024;24:e80–e81. doi: 10.1016/S1473-3099(23)00779-X. [DOI] [PubMed] [Google Scholar]
- 9.Traut C.C., Blankson J.N. Bivalent mRNA vaccine-elicited SARS-CoV-2 specific T cells recognise the omicron XBB sublineage. Lancet Microbe. 2023;4 doi: 10.1016/S2666-5247(23)00105-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tartof S.Y., Slezak J.M., Frankland T.B., Puzniak L., Hong V., Ackerson B.K., Stern J.A., Simmons S., Jodar L., McLaughlin J.M. BNT162b2 XBB1.5-adapted Vaccine and COVID-19 Hospital Admissions and Ambulatory Visits in US Adults. medRxiv. 2023 doi: 10.1101/2023.12.24.23300512. Preprint at. [DOI] [Google Scholar]
- 11.Lavelle E.C., Ward R.W. Mucosal vaccines — fortifying the frontiers. Nat. Rev. Immunol. 2022;22:236–250. doi: 10.1038/s41577-021-00583-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lasrado N., Barouch D.H. SARS-CoV-2 Hybrid Immunity: The Best of Both Worlds. J. Infect. Dis. 2023;228:1311–1313. doi: 10.1093/infdis/jiad353. [DOI] [PubMed] [Google Scholar]
- 13.Bates T.A., Leier H.C., McBride S.K., Schoen D., Lyski Z.L., Lee D.X., Messer W.B., Curlin M.E., Tafesse F.G. An extended interval between vaccination and infection enhances hybrid immunity against SARS-CoV-2 variants. JCI Insight. 2023;8 doi: 10.1172/jci.insight.165265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong P.T., Goff P.H., Sun R.J., Ruge M.J., Ermler M.E., Sebring A., O’Konek J.J., Landers J.J., Janczak K.W., Sun W., Baker J.R., Jr. Combined Intranasal Nanoemulsion and RIG-I Activating RNA Adjuvants Enhance Mucosal, Humoral, and Cellular Immunity to Influenza Virus. Mol. Pharm. 2021;18:679–698. doi: 10.1021/acs.molpharmaceut.0c00315. [DOI] [PubMed] [Google Scholar]
- 15.Jangra S., Landers J.J., Rathnasinghe R., O’Konek J.J., Janczak K.W., Cascalho M., Kennedy A.A., Tai A.W., Baker J.R., Schotsaert M., Wong P.T. A Combination Adjuvant for the Induction of Potent Antiviral Immune Responses for a Recombinant SARS-CoV-2 Protein Vaccine. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.729189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jangra S., Landers J.J., Laghlali G., Rathnasinghe R., Warang P., Park S.-C., O’Konek J.J., Singh G., Janczak K.W., García-Sastre A., et al. Multicomponent intranasal adjuvant for mucosal and durable systemic SARS-CoV-2 immunity in young and aged mice. NPJ Vaccin. 2023;8:96. doi: 10.1038/s41541-023-00691-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel J.R., Jain A., Chou Y.y., Baum A., Ha T., García-Sastre A. ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type-I interferon. EMBO Rep. 2013;14:780–787. doi: 10.1038/embor.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stanberry L.R., Simon J.K., Johnson C., Robinson P.L., Morry J., Flack M.R., Gracon S., Myc A., Hamouda T., Baker J.R. Safety and immunogenicity of a novel nanoemulsion mucosal adjuvant W805EC combined with approved seasonal influenza antigens. Vaccine. 2012;30:307–316. doi: 10.1016/j.vaccine.2011.10.094. [DOI] [PubMed] [Google Scholar]
- 19.Bielinska A.U., Makidon P.E., Janczak K.W., Blanco L.P., Swanson B., Smith D.M., Pham T., Szabo Z., Kukowska-Latallo J.F., Baker J.R. Distinct pathways of humoral and cellular immunity induced with the mucosal administration of a nanoemulsion adjuvant. J. Immunol. 2014;192:2722–2733. doi: 10.4049/jimmunol.1301424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myc A., Kukowska-Latallo J.F., Smith D.M., Passmore C., Pham T., Wong P., Bielinska A.U., Baker J.R. Nanoemulsion nasal adjuvant W₈₀5EC induces dendritic cell engulfment of antigen-primed epithelial cells. Vaccine. 2013;31:1072–1079. doi: 10.1016/j.vaccine.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orzechowska B.U., Kukowska-Latallo J.F., Coulter A.D., Szabo Z., Gamian A., Myc A. Nanoemulsion-based mucosal adjuvant induces apoptosis in human epithelial cells. Vaccine. 2015;33:2289–2296. doi: 10.1016/j.vaccine.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renegar K.B., Small P.A., Jr., Boykins L.G., Wright P.F. Role of IgA versus IgG in the Control of Influenza Viral Infection in the Murine Respiratory Tract1. J. Immunol. 2004;173:1978–1986. doi: 10.4049/jimmunol.173.3.1978. [DOI] [PubMed] [Google Scholar]
- 23.Guglani L., Khader S.A. Th17 cytokines in mucosal immunity and inflammation. Curr. Opin. HIV AIDS. 2010;5:120–127. doi: 10.1097/COH.0b013e328335c2f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills K.H.G. IL-17 and IL-17-producing cells in protection versus pathology. Nat. Rev. Immunol. 2023;23:38–54. doi: 10.1038/s41577-022-00746-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christensen D., Mortensen R., Rosenkrands I., Dietrich J., Andersen P. Vaccine-induced Th17 cells are established as resident memory cells in the lung and promote local IgA responses. Mucosal Immunol. 2017;10:260–270. doi: 10.1038/mi.2016.28. [DOI] [PubMed] [Google Scholar]
- 26.Maroof A., Yorgensen Y.M., Li Y., Evans J.T. Intranasal vaccination promotes detrimental Th17-mediated immunity against influenza infection. Plos Pathog. 2014;10 doi: 10.1371/journal.ppat.1003875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paiva I.A., Badolato-Corrêa J., Familiar-Macedo D., de-Oliveira-Pinto L.M. Th17 Cells in Viral Infections-Friend or Foe? Cells. 2021;10:1159. doi: 10.3390/cells10051159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kingstad-Bakke B., Lee W., Chandrasekar S.S., Gasper D.J., Salas-Quinchucua C., Cleven T., Sullivan J.A., Talaat A., Osorio J.E., Suresh M. Vaccine-induced systemic and mucosal T cell immunity to SARS-CoV-2 viral variants. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2118312119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J., Yu J., McMahan K., Jacob-Dolan C., He X., Giffin V., Wu C., Sciacca M., Powers O., Nampanya F., et al. CD8 T cells contribute to vaccine protection against SARS-CoV-2 in macaques. Sci. Immunol. 2022;7 doi: 10.1126/sciimmunol.abq7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson K.G., Mayer-Barber K., Sung H., Beura L., James B.R., Taylor J.J., Qunaj L., Griffith T.S., Vezys V., Barber D.L., Masopust D. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 2014;9:209–222. doi: 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rathnasinghe R., Jangra S., Ye C., Cupic A., Singh G., Martínez-Romero C., Mulder L.C.F., Kehrer T., Yildiz S., Choi A., et al. Characterization of SARS-CoV-2 Spike mutations important for infection of mice and escape from human immune sera. Nat. Commun. 2022;13:3921. doi: 10.1038/s41467-022-30763-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halfmann P.J., Iida S., Iwatsuki-Horimoto K., Maemura T., Kiso M., Scheaffer S.M., Darling T.L., Joshi A., Loeber S., Singh G., et al. SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature. 2022;603:687–692. doi: 10.1038/s41586-022-04441-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mao T., Israelow B., Peña-Hernández M.A., Suberi A., Zhou L., Luyten S., Reschke M., Dong H., Homer R.J., Saltzman W.M., Iwasaki A. Unadjuvanted intranasal spike vaccine elicits protective mucosal immunity against sarbecoviruses. Science. 2022;378 doi: 10.1126/science.abo2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hornsby H., Nicols A.R., Longet S., Liu C., Tomic A., Angyal A., Kronsteiner B., Tyerman J.K., Tipton T., Zhang P., et al. Omicron infection following vaccination enhances a broad spectrum of immune responses dependent on infection history. Nat. Commun. 2023;14:5065. doi: 10.1038/s41467-023-40592-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puhach O., Bellon M., Adea K., Bekliz M., Hosszu-Fellous K., Sattonnet P., Hulo N., Kaiser L., Eckerle I., Meyer B. SARS-CoV-2 convalescence and hybrid immunity elicits mucosal immune responses. EBioMedicine. 2023;98 doi: 10.1016/j.ebiom.2023.104893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bobrovitz N., Ware H., Ma X., Li Z., Hosseini R., Cao C., Selemon A., Whelan M., Premji Z., Issa H., et al. Protective effectiveness of previous SARS-CoV-2 infection and hybrid immunity against the omicron variant and severe disease: a systematic review and meta-regression. Lancet Infect. Dis. 2023;23:556–567. doi: 10.1016/S1473-3099(22)00801-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robertson S.J., Bedard O., McNally K.L., Shaia C., Clancy C.S., Lewis M., Broeckel R.M., Chiramel A.I., Shannon J.G., Sturdevant G.L., et al. Genetically diverse mouse models of SARS-CoV-2 infection reproduce clinical variation in type I interferon and cytokine responses in COVID-19. Nat. Commun. 2023;14:4481. doi: 10.1038/s41467-023-40076-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clough B., Finethy R., Khan R.T., Fisch D., Jordan S., Patel H., Coers J., Frickel E.-M. C57BL/6 and 129 inbred mouse strains differ in Gbp2 and Gbp2b expression in response to inflammatory stimuli in vivo. Wellcome Open Res. 2019;4:124. doi: 10.12688/wellcomeopenres.15329.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang A., Stacey H.D., D’Agostino M.R., Tugg Y., Marzok A., Miller M.S. Beyond neutralization: Fc-dependent antibody effector functions in SARS-CoV-2 infection. Nat. Rev. Immunol. 2023;23:381–396. doi: 10.1038/s41577-022-00813-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y., Jin L., Chen T. The Effects of Secretory IgA in the Mucosal Immune System. Biomed. Res. Int. 2020;2020 doi: 10.1155/2020/2032057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gunn B.M., Bai S. Building a better antibody through the Fc: advances and challenges in harnessing antibody Fc effector functions for antiviral protection. Hum. Vaccin. Immunother. 2021;17:4328–4344. doi: 10.1080/21645515.2021.1976580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riou C., Keeton R., Moyo-Gwete T., Hermanus T., Kgagudi P., Baguma R., Valley-Omar Z., Smith M., Tegally H., Doolabh D., et al. Escape from recognition of SARS-CoV-2 variant spike epitopes but overall preservation of T cell immunity. Sci. Transl. Med. 2022;14:eabj6824. doi: 10.1126/scitranslmed.abj6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jangra S., Lamoot A., Singh G., Laghlali G., Chen Y., Yz T., García-Sastre A., De Geest B.G., Schotsaert M. Lipid nanoparticle composition for adjuvant formulation modulates disease after influenza virus infection in QIV vaccinated mice. bioRxiv. 2024 doi: 10.1101/2024.01.14.575599. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang J., Zeng C., Cox T.M., Li C., Son Y.M., Cheon I.S., Wu Y., Behl S., Taylor J.J., Chakaraborty R., et al. Respiratory mucosal immunity against SARS-CoV-2 after mRNA vaccination. Sci. Immunol. 2022;7 doi: 10.1126/sciimmunol.add4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lapuente D., Fuchs J., Willar J., Vieira Antão A., Eberlein V., Uhlig N., Issmail L., Schmidt A., Oltmanns F., Peter A.S., et al. Protective mucosal immunity against SARS-CoV-2 after heterologous systemic prime-mucosal boost immunization. Nat. Commun. 2021;12:6871. doi: 10.1038/s41467-021-27063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gagne M., Flynn B.J., Andrew S.F., Marquez J., Flebbe D.R., Mychalowych A., Lamb E., Davis-Gardner M.E., Burnett M.R., Serebryannyy L.A., et al. Mucosal adenovirus vaccine boosting elicits IgA and durably prevents XBB.1.16 infection in nonhuman primates. Nat. Immunol. 2024;25:1913–1927. doi: 10.1038/s41590-024-01951-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Madhavan M., Ritchie A.J., Aboagye J., Jenkin D., Provstgaad-Morys S., Tarbet I., Woods D., Davies S., Baker M., Platt A., et al. Tolerability and immunogenicity of an intranasally-administered adenovirus-vectored COVID-19 vaccine: An open-label partially-randomised ascending dose phase I trial. EBioMedicine. 2022;85 doi: 10.1016/j.ebiom.2022.104298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong P.T., Wang S.H., Ciotti S., Makidon P.E., Smith D.M., Fan Y., Schuler C.F., Baker J.R. Formulation and characterization of nanoemulsion intranasal adjuvants: effects of surfactant composition on mucoadhesion and immunogenicity. Mol. Pharm. 2014;11:531–544. doi: 10.1021/mp4005029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong P.T., Leroueil P.R., Smith D.M., Ciotti S., Bielinska A.U., Janczak K.W., Mullen C.H., Groom J.V., Taylor E.M., Passmore C., et al. Formulation, high throughput in vitro screening and in vivo functional characterization of nanoemulsion-based intranasal vaccine adjuvants. PLoS One. 2015;10 doi: 10.1371/journal.pone.0126120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herrera N.G., Morano N.C., Celikgil A., Georgiev G.I., Malonis R.J., Lee J.H., Tong K., Vergnolle O., Massimi A.B., Yen L.Y., et al. Characterization of the SARS-CoV-2 S Protein: Biophysical, Biochemical, Structural, and Antigenic Analysis. ACS Omega. 2021;6:85–102. doi: 10.1021/acsomega.0c03512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J., et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020;11:1620. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang Y., Borisov O., Kee J.J., Carpp L.N., Wrin T., Cai S., Sarzotti-Kelsoe M., McDanal C., Eaton A., Pajon R., et al. Calibration of two validated SARS-CoV-2 pseudovirus neutralization assays for COVID-19 vaccine evaluation. Sci. Rep. 2021;11 doi: 10.1038/s41598-021-03154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rathnasinghe R., Strohmeier S., Amanat F., Gillespie V.L., Krammer F., García-Sastre A., Coughlan L., Schotsaert M., Uccellini M.B. Comparison of transgenic and adenovirus hACE2 mouse models for SARS-CoV-2 infection. Emerg. Microbes Infect. 2020;9:2433–2445. doi: 10.1080/22221751.2020.1838955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are provided in the main text, figures, or the supplemental information, and raw data files are available upon request. Requests for reagents or cell lines used in this study can directed to the corresponding authors.