

It has been known for over half a century that anti-nuclear antibodies (ANA) are a characteristic feature of systemic lupus erythematosus (SLE). The mechanism for this propensity towards nuclear reactivity was enigmatic until a seminal study linked dysregulated Toll-like receptor (TLR) signaling with ANA production2. Toll-like receptors are germline encoded, pattern-recognition receptors evolved to recognize conserved ligands on exogenous pathogens. However, in the context of autoimmunity, nucleic acid-containing particles derived from apoptotic cells can preferentially activate autoreactive B cells via the dual engagement of B cell receptor (BCR) and TLR signaling pathways. Of the several Myeloid differentiation primary response 88 (Myd88)-dependent TLR family members, animal studies have identified TL7 and TLR9 as the relevant receptors in lupus pathogenesis. Specifically, the single stranded RNA sensor TLR7 promotes autoantibodies against RNA and RNA-associated proteins, while autoantibodies targeting double stranded DNA (dsDNA) and chromatin require engagement of the DNA sensor TLR93.
Based on this model, one would have anticipated that deletion of TLR7 or TLR9 would each result in partial projection against autoimmunity in murine models of lupus. As predicted, genetic disruption of TLR7 function has been shown to eliminate autoimmunity in multiple independent animal models of SLE, to a similar degree as is achieved by deleting the shared adaptor molecule Myd883, 4. In contrast, TLR9 deletion exacerbates disease development despite the loss of dsDNA autoantibodies. In keeping with a dominant role for TLR7 in lupus pathogenesis, independent models of TLR7 over-expression resulted in accelerated murine SLE4.
Although these opposing pathogenic and protective roles for TLR7 and TLR9 in lupus pathogenesis have been replicated in multiple independent animal studies, whether this paradigm applies to human lupus remains uncertain. Indeed, anti-dsDNA autoantibodies, and not RNA-associated specificities, have been most closely linked to the pathogenesis of lupus nephritis, as evidenced by epidemiological links to lupus nephritis incidence and flare rate, and the observation that adoptive transfer of DNA-reactive monoclonal antibodies can promote proteinuric kidney disease in mice5, 6. While these studies suggest a direct pathogenic role for dsDNA autoantibodies, a more comprehensive assessment of immunoglobulin eluted from lupus nephritis glomeruli identified broad autoreactivity, including against several RNA-associated autoantigens7. Ultimately, despite extensive murine modeling and circumstantial evidence from human studies, it remains uncertain whether TLR7 engagement and RNA-associated autoantibodies have an actual pathogenic role in human SLE.
What did the study show?
To identify genetic variants linked with early-onset SLE, whole genome sequencing was performed on a 7-year-old child with lupus, characterized by positive ANA, autoimmune thrombocytopenia, neurologic involvement, and lupus nephritis. This revealed a de novo TLR7 p.Tyr264His (Y264H) missense mutation predicted to be damaging by bioinformatic analysis. While this mutation seemed to be a strong lupus risk candidate, this subject also carried a second heterozygous variant in RNASH2B, which is known to cause SLE when homozygous. For this reason, the authors performed a series of mechanistic studies aimed at establishing a causal role for the TLR7Y264H allele in human lupus. They first measured downstream signaling using cell lines overexpressing TLR7Y264H vs. control TLR7 alleles. These experiments demonstrated that TLR7Y264H exhibited increased NF-κB activation following guanosine or single cell RNA (ssRNA) stimulation. In keeping with enhanced signaling in vitro, computational modeling predicted increased binding of guanosine to variant TLR7. While these findings suggested that TLR7Y264H functions as a gain-of-function variant, additional animal modeling was required to confirm that this mutation is sufficient to promote lupus-like autoimmunity. For this reason, the authors used CRISPR-Cas9 gene editing to generate Tlr7Y264H knock-in mice. Notably, Tlr7Y264H mice exhibited spontaneous immune activation, including expansion of activated B and T cell populations observed in human SLE, production of class-switched autoantibodies, and the development of lupus nephritis. Importantly, intercrossing with Rnaseh2b-haploinsufficient mice only slightly increased in the lupus-associated type 1 interferon signature, without impacting other disease manifestations, suggesting that TLR7Y264H is the true causative allele.
An available murine model of the human gain-of-function TLR7 allele allowed the investigators to interrogate the immune mechanisms underlying TLR7-driven lupus pathogenesis, unhindered by limited availability of human samples. These yielded several insights likely to be important for our understanding of lupus immune pathogenesis. First, using mixed bone marrow chimera models, the authors showed that B cell, and not CD4+ T cell, expression of the Tlr7Y264H allele promoted adaptive immune activation. Notably, these findings are consistent with earlier animal studies in which B cell-specific deletion of TLR7 or TLR9 was shown to fully recapitulate the phenotype of global Tlr7−/− and Tlr9−/− lupus prone mice8, emphasizing the importance of B cells in facilitating breaks in immune tolerance.
Second, the authors investigated the B cell activation pathway responsible for pathogenic plasma cell generation. It has long been appreciated that, during a humoral immune response, B cells activation can occur within germinal centers or via a parallel extra-follicular activation program. Using an elegant genetic strategy in which animals were rendered unable to form germinal centers, the authors showed that RNA-associated antibodies were still generated. Thus, in keeping with recent human immunophenotyping studies9, dysregulated TLR7 signaling appears to predominantly drive pathogenic B cell activation via an extra-follicular activation pathway.
Why is it important?
In summary, the study by Brown et al. provides an important link between the immunology insights gained from mouse models and the pathogenesis of the human SLE. Rather than being a unique feature of the mouse immune system, the identification of a rare patient with a gain-of-function TLR7 variant confirms the critical role for this receptor in human lupus pathogenesis. Moreover, this study affirmed the important role for extra-follicular B cell activation in the formation of autoantibody-producing plasma cells in SLE. Given that extra-follicular B cell activation predominantly generates short-lived plasma cells, in contrast with germinal center-derived long-lived plasma cells, this suggests that effective B cell depletion might be able to “reset” the autoimmune repertoire by eliminating the bulk of pre-formed pathogenic plasma cells. This may be particularly relevant for lupus nephritis given that B cells and T follicular helper (TFH) cells have been shown to interact in the tubulointerstitial compartment of the lupus kidney, coupled with evidence that a B cell targeted antibody exhibiting increased depletion of tissue-resident B cells successfully treated lupus nephritis.10,11 This genetic discovery also provides support for novel drugs like enpatoran, a dual TLR7/8 small molecule antagonist currently undergoing phase 2 evaluation in SLE (NCT05162586), and encourages the development of drugs like DS-7011a, a TLR7 blocking antibody now in a phase 1 study of healthy volunteers (NCT05203692). Like the remarkable success of PCSK9 inhibition for hypercholesterolemia, target identification informed by human genetics holds the promise of informing new treatments for patients with SLE, including those with lupus nephritis.
Figure 1: Identification and functional characterization of a pathogenic TLR7 mutation in early-onset SLE.
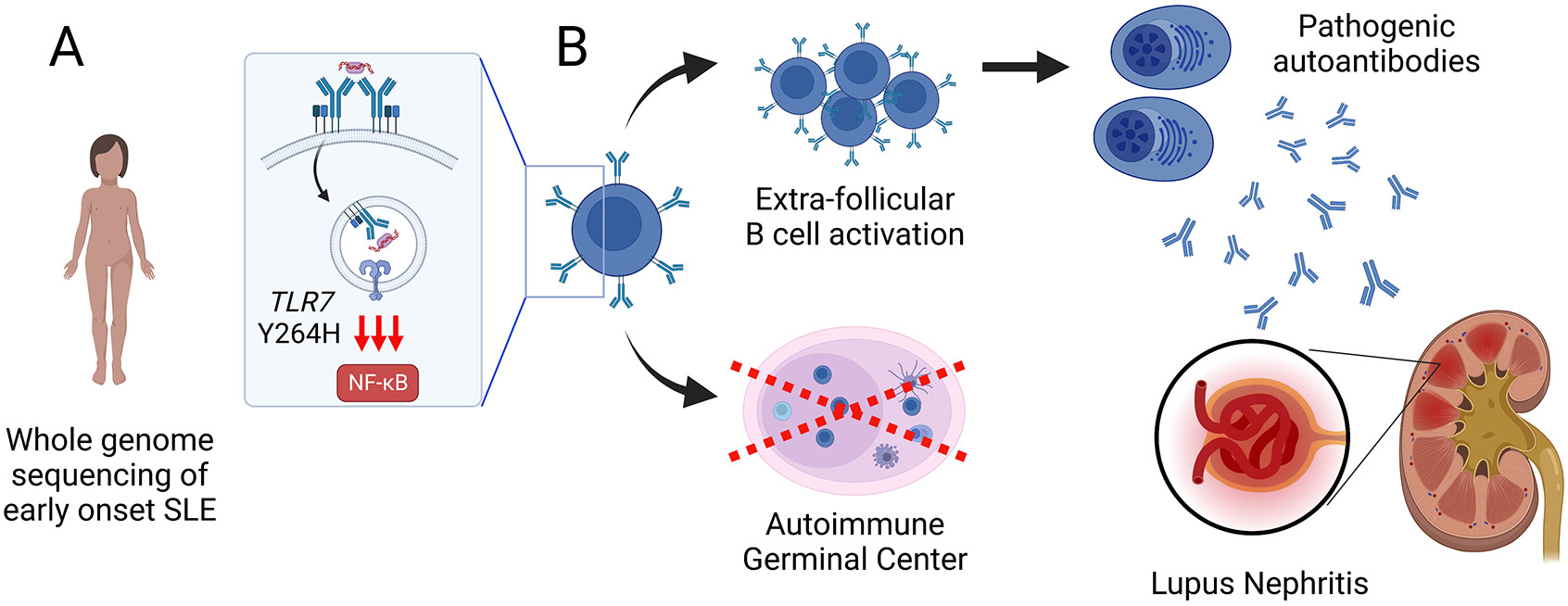
(A) Using whole genome sequencing, Brown et al. identified a de novo TLR7Y264H missense mutation in a pediatric patient with early onset lupus nephritis. After binding self-antigens derived from apoptotic cells, autoreactive B cells traffic nucleic acid-containing autoantigens to the endosomal receptors TLR7 and TLR9, resulting in dual BCR/TLR-mediated B cell activation. By increasing activation by guanosine-containing self-ligands, TLR7Y264H promotes pathogenic B cell activation and the development of SLE. (B) Parallel extra-follicular and germinal center-dependent B cell activation pathways contribute to the production of autoantibody-producing plasma cells during humoral autoimmunity. Surprisingly, autoantibodies persisted in Tlr7Y264H mice genetically incapable of forming germinal centers, indicating that extra-follicular B cell activation is the predominant pathway leading to autoantibody formation and the development of immune complex-mediated lupus nephritis. Created with BioRender.com.
Acknowledgments:
This work was support by National Institutes of Health grants: R01AR073938 and R01AR075813 (to S.W.J) and R01AR071947 (to B.H.R). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Disclosures
The authors declare that they have no competing interests.
References
- 1.Brown GJ, Canete PF, Wang H, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature 2022; 605: 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leadbetter EA, Rifkin IR, Hohlbaum AM, et al. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 2002; 416: 603–607. [DOI] [PubMed] [Google Scholar]
- 3.Christensen SR, Shupe J, Nickerson K, et al. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006; 25: 417–428. [DOI] [PubMed] [Google Scholar]
- 4.Rawlings DJ, Schwartz MA, Jackson SW, et al. Integration of B cell responses through Toll-like receptors and antigen receptors. Nature reviews Immunology 2012; 12: 282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isenberg DA, Manson JJ, Ehrenstein MR, et al. Fifty years of anti-ds DNA antibodies: are we approaching journey's end? Rheumatology (Oxford) 2007; 46: 1052–1056. [DOI] [PubMed] [Google Scholar]
- 6.Ehrenstein MR, Katz DR, Griffiths MH, et al. Human IgG anti-DNA antibodies deposit in kidneys and induce proteinuria in SCID mice. Kidney Int 1995; 48: 705–711. [DOI] [PubMed] [Google Scholar]
- 7.Mannik M, Merrill CE, Stamps LD, et al. Multiple autoantibodies form the glomerular immune deposits in patients with systemic lupus erythematosus. J Rheumatol 2003; 30: 1495–1504. [PubMed] [Google Scholar]
- 8.Jackson SW, Scharping NE, Kolhatkar NS, et al. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol 2014; 192: 4525–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenks SA, Cashman KS, Zumaquero E, et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 2020; 52: 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang A, Clark MR, Ko K. Cellular aspects of the pathogenesis of lupus nephritis. Curr Opin Rheumatol 2021; 33:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furie RA, Aroca G, Cascino MD, et al. B-cell depletion with obinutuzumab for the treatment of proliferative lupus nephritis: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis 2022; 81:100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]