

Abstract
The elimination of large genomic regions has been enabled by the advent of site-specific nucleases. However, as the intended deletions get larger, the efficiency of successful engineering decreases to a point where it is not feasible to retrieve edited cells due to the rarity of on-target events. To address this issue, we developed a system called molecular alteration of chromosomes with engineered tandem elements (MACHETE). MACHETE is a CRISPR–Cas9-based system involving two stages: the initial insertion of a bicistronic positive/negative selection cassette to the locus of interest. This is followed by the introduction of single-guide RNAs flanking the knockin cassette to engineer the intended deletion, where only cells that have lost the locus survive the negative selection. In contrast to other approaches optimizing the activity of sequence-specific nucleases, MACHETE selects for the deletion event itself, thus greatly enriching for cells with the engineered alteration. The procedure routinely takes 4–6 weeks from design to selection of polyclonal populations bearing the deletion of interest. We have successfully deployed MACHETE to engineer deletions of up to 45 Mb, as well as the rapid creation of allelic series to map the relevant activities within a locus. This protocol details the design and step-by-step procedure to engineer megabase-sized deletions in cells of interest, with potential application for cancer genetics, transcriptional regulation, genome architecture and beyond.
Introduction
Genetic approaches have been fundamental to dissect the contribution of specific regions of the genome to biological phenotypes. Indeed, the development of methods to disrupt specific sequences have been instrumental to understand the role of distinct loci in development, physiology and disease. Among genetic approaches, zinc finger nucleases1, transcription activator-like effector nucleases2 and CRISPR–Cas9 (clustered regularly interspaced short palindromic repeats-directed Cas9)3 have been used for sequence-specific manipulation of the genome across multiple model organisms and cellular systems4-9. The most frequent use of nucleases has been the introduction of double-strand breaks (DSBs), which disrupt the target DNA sequence by engaging error-prone nonhomologous end joining. Among these technologies, the ease of design and implementation of CRISPR–Cas9 has enabled knockouts of individual genes to be created on a genome-wide scale across models and experimental settings7,10-12.
Despite the many advances in understanding the contribution of individual genes to cellular phenotypes, dissecting the function of larger regions of DNA has remained a challenge. This is due to the higher complexity of eliminating regions of the genome when comparing with sequence disruptions through nonhomologous end joining. Current approaches to study the role of large genomic regions rely on the simultaneous introduction of two sequence-specific DSBs, which when occur in the same DNA molecule will lead to the excision of the intervening region and eliminate the intended sequence. However, there is an inverse relation between the distance of the DSBs with the frequency of the deletion in the population of targeted cells13. These engineered events become increasingly rare when trying to model megabase-sized deletions such as the ones observed in cancer genomes. Similar limitations occur in studies aimed at dissecting the function of large DNA regions in transcriptional regulation and/or genome organization.
Development of the protocol
To circumvent the inherent inefficiency of retrieving cells with large deletions, we devised an approach termed MACHETE (molecular alteration of chromosomes with engineered tandem elements)14. The basic premise of MACHETE is that the removal of a region of interest will give cells a selective growth advantage independently of the elements within the targeted locus. To achieve this, we use CRISPR–Cas9-mediated homology-directed repair (HDR) to introduce an inducible suicide cassette into the locus of interest, which is positively selected by a linked antibiotic resistance or fluorescence gene (Fig. 1). The integration of this tandem inducible suicide and selection cassette creates a molecular handle to enrich or deplete targeted cells as needed. Upon CRISPR-mediated introduction of cassette-flanking DSBs, only cells that eliminate the locus have a route to escape negative selection, greatly enriching for cells with the deletion of interest (Fig. 1a). There are several unique features of MACHETE: first, since the cassette and locus are deleted in a sequence-specific manner via CRISPR–Cas9, our method eliminates cells containing off-target integrations of the cassette. Second, at the end of the protocol, all exogenous elements are removed from the cell, allowing for iterative engineering of deletions (Fig. 1b). Third, the high level of enrichment allows engineering of deletions of vastly different sizes across a locus, allowing refined mapping of megabase-sized regions (Fig. 1c).
Fig. 1 ∣. MACHETE allows engineering large genomic deletions.
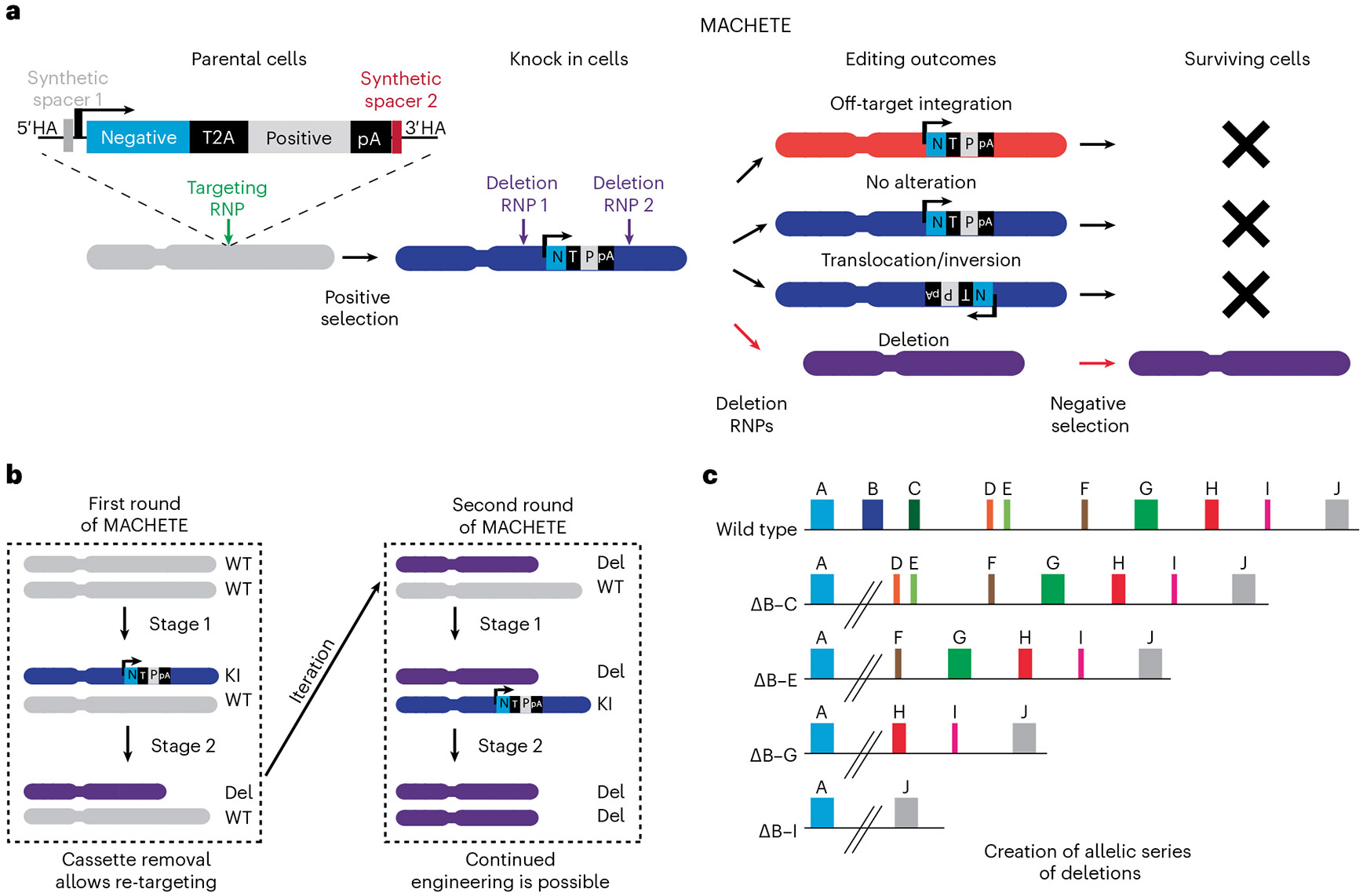
a, A schematic outline of the MACHETE protocol. A dual selection cassette is integrated via CRISPR HDR into the locus of interest. Cells with stable integration of the cassette are enriched via positive selection, ensuring the presence of the suicide cassette in the population. These KI cells are then used to engineer the intended deletions, which is followed by negative selection. Only those cells that excised the locus will survive the negative selection, greatly enriching for cells with deletions. The cassette excision occurs in a sequence-specific manner, thus eliminating cells with off-target integrations of the cassette. b, MACHETE can be iterated. Given that all exogenous elements are removed at the end of the procedure, deletions (Del) can be sequentially engineered either in the same locus or at a different one. c, MACHETE enables the creation of allelic series of deletions of a locus of interest. Using cells with a KI of the dual selection cassette in the locus of interest, MACHETE allows creating allelic series of deletions of different size (ΔB–C, loss of the region spanning genes B and C; ΔB–E, loss of the region spanning genes B and E; etc.). Image in a adapted from ref. 14, Springer Nature Ltd.
Applications of the method
Genetic approaches have enabled us to identify the role of specific loci, which has been particularly focused on the biology of coding genes. MACHETE adds another tool to the genetics toolkit to facilitate understanding the biology of larger genomic regions.
The initial application of MACHETE is centered on the functional characterization of large deletions observed in cancer genomes (Fig. 2a). Given their pervasiveness and potential clinical relevance, we decided to tackle this understudied aspect of cancer genetics. Our initial focus centered on understanding the loss of chromosome 9p21.3, which is the most frequent homozygous deletion (affecting 15% of all tumors) and portends poor prognosis in the cancers that have this genetic event15. The biological consequences of 9p21.3 deletions had been ascribed to the loss of the tumor suppressor genes CDKN2A (encoding p14ARF and p16INK4A) and CDKN2B (encoding p15INK4B), whose protein products activate the RB and p53 pathways16. Thus, the classic view is that loss of 9p21.3 provides a strong proliferative advantage to cancer cells, which was validated in various cancer models. However, we and others observed that a subset of 9p21.3 deletions spanned an adjacent cluster of type I interferons (IFNs), which could account for some unexpected correlations of this deletion with alterations in tumor immunity. To dissect the role of tumor-intrinsic IFN loss, we generated the two most common 9p21.3 deletions observed in patients: a 0.4 Mb allele eliminating CDKN2A/B and a 1.3 Mb allele eliminating both CDKN2A/B and the linked cluster of 17 type I IFNs. By applying MACHETE to mouse models of pancreas cancer and melanoma, we observed that IFN-deficient cancer cells were more metastatic and resistant to immune checkpoint blockade than IFN-proficient cells. These phenotypes were dependent on intact adaptive immunity, where tumor-derived IFNs activated antigen presenting cells and CD8 T cells. To identify whether one or more tumor-derived IFNs were involved in these effects, we created an allelic series of deletions that progressively eliminated the IFN cluster. An in vivo competition assay of these alleles identified that loss of IFNε (Ifne) was a tumor-specific driver of these phenotypes in pancreas cancer14. This initial study illustrates the power of MACHETE in dissecting the complex effects that large genomic deletions can elicit on tumors (Fig. 2b).
Fig. 2 ∣. Applications of MACHETE.
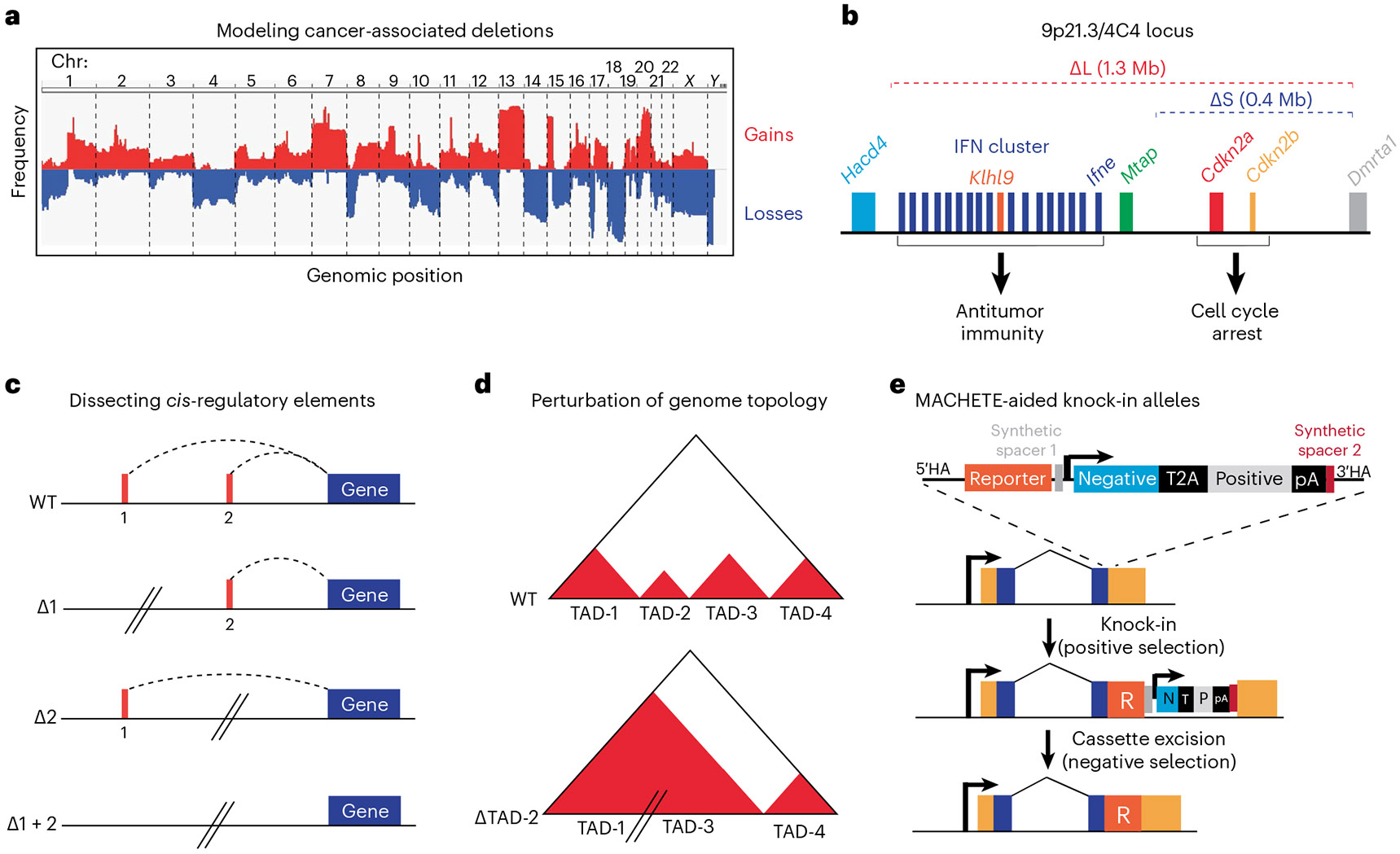
a, A schematic of copy number alteration frequency in tumors, which guides the selection of target regions to engineer in cellular models via MACHETE. b, Chromosome 9p21.3 (syntenic to mouse 4C4) coordinates cell intrinsic and extrinsic tumor suppression. MACHETE engineering of the two most common configurations of 9p21.3 loss (ΔL and ΔS) identified the contribution of tumor-derived type I IFNs in promoting immune surveillance and metastasis suppression. c, MACHETE can dissect the contribution of large cis-regulatory elements to gene regulation. d, Potential application of MACHETE to elucidate genome structure/architecture via the functional interrogation of topological associated domains (TADs). e, MACHETE-aided integration of reporters into a locus of interest. Using the strong positive/negative selection and CRISPR-mediated excision of the MACHETE cassette can aid in the integration of reporters (e.g., fluorescent proteins, recombinases, etc.) to loci that are poorly expressed.
Beyond its use in dissecting the function of deletions in cancer, we envision MACHETE will be applicable to studying the contribution of any genomic region to multiple fields (e.g., transcriptional regulation, three-dimensional genome structure, etc.). The general design principle of the approach is applicable to any cellular system where negative selection can be implemented (Fig. 2c,d).
Moreover, the MACHETE cassette can be leveraged as a tool to aid the delivery of other genetic elements into the genome. For instance, this is particularly relevant for introducing reporters into genes that are not expressed at baseline in cells of interest. The positive/negative selection then enables the excision of the MACHETE cassette to avoid the transcriptional interference of the strong exogenous promoter with the reporter-tagged endogenous gene (Fig. 2e).
Comparison with other methods
As outlined above, the most frequent methods to engineer deletions employ sequence-specific nucleases to create two DSBs simultaneously11,13,17, where CRISPR–Cas9 is the most prominent approach18-20. Optimizations of CRISPR–Cas9 have centered on increasing the editing efficiency by either enriching for cells that incorporate the components (e.g., via linked fluorescent or antibiotic-resistance genes)21 or improving the rate at which DSBs are created (e.g., use of ribonucleoproteins, improved guide RNA scaffolds)11. However, these approaches do not circumvent the problem that most Cas9-expressing cells will not lead to the loss of the intervening region between two DSBs. To directly select for deletions, MACHETE relies on CRISPR–Cas9 to insert a selection cassette via HDR and subsequently create the intended DSBs, where negative selection robustly eliminates unedited cells.
Another approach to create deletions is the use of Cre recombinase coupled to flanking loxP sites22, and the major advantage of this strategy is the ability to create deletions in vivo. However, there are two limitations of the Cre–loxP approach: first, it requires the knock-in (KI) of two loxP sites in cis. Second, loxP sites are at fixed positions, which only allows to create a single deletion per loxP pair. Overall, despite the potential advantages of Cre–loxP or other recombinases, the complexity of the targeting procedure and relative design rigidity has precluded its widespread implementation.
Finally, several approaches have been developed to model arm- and chromosome-level aneuploidy by using synthetic telomeres or disrupting chromosome segregation23-25. These approaches are very powerful tools to study aneuploidy yet have a relatively low frequency of on-target events and are not amenable to engineer interstitial deletions.
Limitations
As MACHETE relies on CRISPR–Cas9, the limitations of this approach also apply to our method; most notably, these include challenges associated with off-target effects (OTEs) and editing efficiency.
Off-target DSBs, and the generation of unintended structural variants, are known problems associated with CRISPR–Cas9 (refs. 26,27). To mitigate this problem, for each deletion we select two pairs of single-guide (sg)RNAs with the highest efficiency and lowest predicted OTEs. Given that we target large genomic regions, there is ample room for identifying the best possible guide RNAs for any given locus of interest. The second issue, efficiency, is not directly addressed by our approach. However, since we rely on positive and negative selection for the distinct steps of MACHETE, even low-efficiency sgRNAs will be effective in our system, albeit the number of cells surviving the selection may be lower than for high-efficiency sgRNAs. Regardless, the combination of selection and the use of multiple independent guide RNAs circumvents most of the problems associated with CRISPR–Cas9.
Another limitation of MACHETE, and all other approaches to create deletions, is the limited multiplexing that can be implemented. Although negative selection enables retrieving rare cells with large deletions, in a pooled experiment smaller deletions would be the dominant outcome of the engineering, and larger deletions would be underrepresented in the initial pool. With these considerations, our experimental design of choice is to create distinct deletions independently that, if needed, can then be pooled with equal representation for downstream applications.
Depending on the application, another limitation of MACHETE is that the default outcome is heterozygous deletions. Although in the context of cancer this is not an issue since most deletions are heterozygous, this may present a problem in applications where bi-allelic targeting is needed. To address this, we have shown that MACHETE can be iterated, allowing for the generation of homozygous deletions14. However, the size of homozygous deletions will be limited by the presence of essential genes/regions within the locus.
Finally, since MACHETE relies on strong negative selection to eliminate unedited cells, currently it cannot be used for somatic in vivo engineering of deletions, which would lead to organism failure if the selection cassette is present in all tissues.
Experimental design
MACHETE is a two-stage procedure: first, an initial integration of the dual selection cassette into the locus of interest is achieved through CRISPR–Cas9 HDR and positive selection. Second, the introduction of sgRNAs is undertaken to eliminate the locus of interest followed by negative selection to enrich for cells bearing the desired deletion. Given the need for initial targeting and two rounds of selection, we recommend the application of MACHETE for deletions where the frequency of the on-target event is rare (e.g., deletions of over 1 Mb), smaller deletions where direct use of CRISPR–Cas9 has failed or when heterozygous alterations are required.
The experimental design we favor involves the generation of at least one control allele (e.g., MACHETE cassette excision and/or a focal deletion) to compare with the experimental allele. Large deletions are engineered with two distinct sets of sgRNAs. This approach ensures that all cells have gone through a similar selection process and minimizes the possibility that phenotypic differences are driven by off-target DSBs or structural variants. Of note, in our experiments, we worked with polyclonal populations due to the high efficiency of MACHETE in our systems, yet the protocol is compatible with the use of clonal populations if warranted.
In Fig. 3 we outline the experimental design and steps needed to implement MACHETE into any cellular system of interest.
Fig. 3 ∣. Design principles for the implementation of MACHETE.
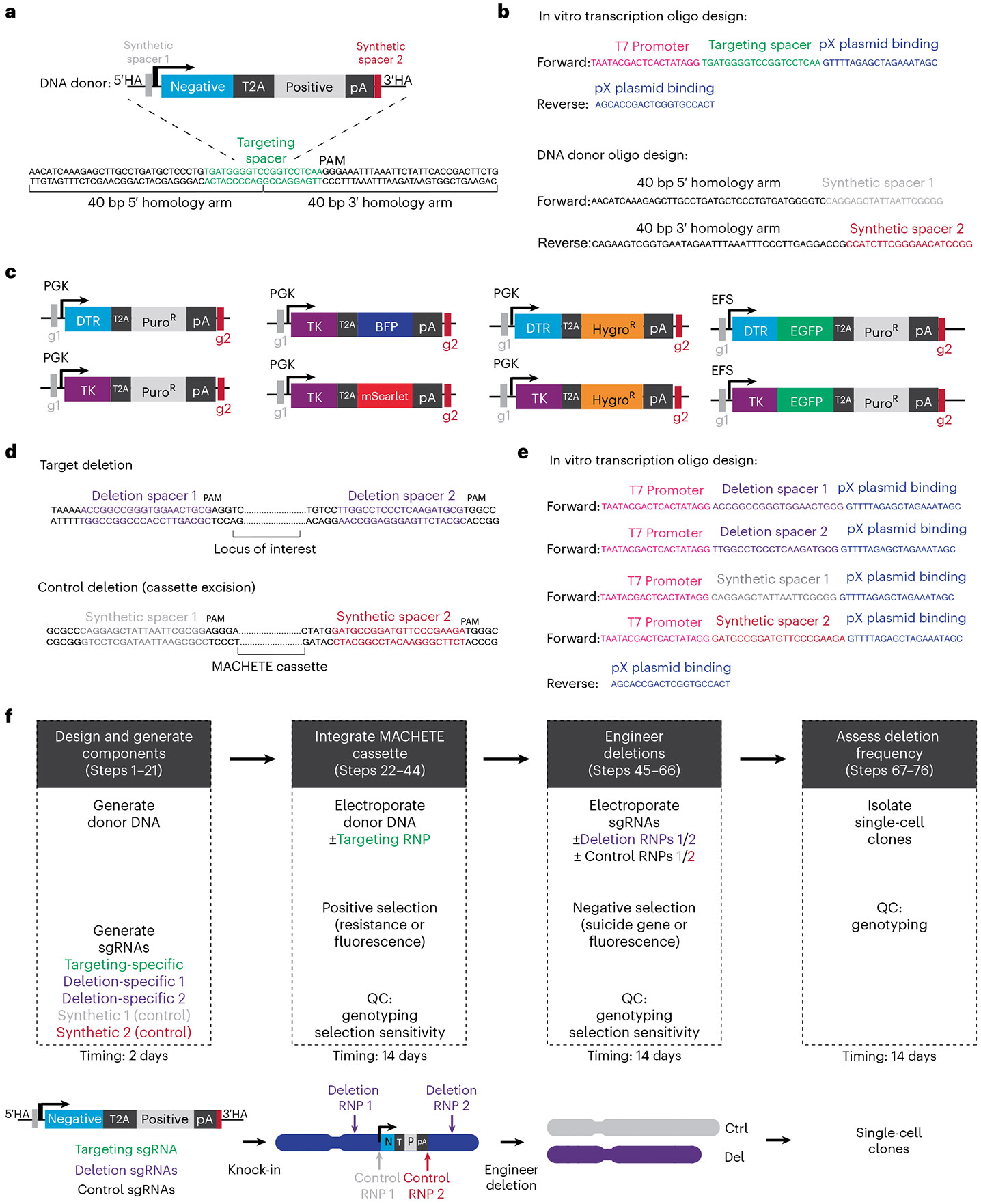
a, A schematic of a generic donor to be integrated into a region of interest. In green is highlighted the selected KI spacer sequence, and the design of the 40-bp homology arms is shown. b, The sequence of the oligonucleotides that are needed to make the targeting sgRNA and the PCR donor to the region highlighted in a. c, Available MACHETE cassettes. d, A schematic of the sequences that will define the intended deletion breakpoint (top) and the control cassette excision (bottom). In purple are highlighted the flanking spacer sequences, and in gray and dark red are highlighted the synthetic spacers to excise the MACHETE cassette. e, The sequence of the oligonucleotides that are needed to make the target and control deletion sgRNAs shown in d. f, Overview and schematic of the MACHETE protocol. QC, quality control. Image c reproduced from ref. 14, Springer Nature Ltd.
Design and production of sgRNAs (Steps 1–14)
To engineer large deletions, we design guides targeting intergenic regions to minimize the potential effect on adjacent coding genes. We use the Guidescan database28,29, which has genome-wide predictions and simple format that allow the prioritization of the most specific guides with minimal OTEs. One sgRNA is used for the CRISPR–Cas9-mediated cassette KI (Fig. 3a,b), and two pairs of guide RNAs are designed to target the flanking regions (Fig. 3d,e). For MACHETE to be effective, the KI sgRNA must be located between the flanking sgRNAs, anywhere within the intervening region between the intended DSBs. We favor knocking-in the cassette closest to a specific element/gene of interest, which will enable focused mapping of that subregion and comparison with larger deletions. Given that broken-end resection of DSBs can expand in the order of several kilobases, if possible, we avoid placing the cassette too close to the intended breakpoint.
Guide RNAs are made via in vitro transcription of a PCR product bearing the T7 promoter, target spacer and tracr sequence, as previously described30. The in vitro transcribed approach reduces costs, yet chemically modified commercial guide RNAs can also be used.
Generation of the PCR donor template (Steps 15–21)
The initial step of MACHETE relies on the integration of the dual selection cassette through CRISPR–Cas9 HDR. This requires a DNA repair template around the site of the designed DSB, to which 40 bp homology arms are introduced via PCR. This PCR-based strategy precludes the need for cloning homology arms into the vector, reducing experimental time and increasing the flexibility of the approach. This size of homology arm was chosen because this length is sufficient to facilitate KIs31 and allows use of shorter oligonucelotides to reduce costs. The template for the donor PCR can be any plasmid containing positive/negative selection elements. To aid in the implementation of MACHETE, we deposited a collection of plasmids with various tandem positive and negative selection markers for use in mouse or human cells in Addgene. All plasmids have shared flanking regions, which allow the amplification of any of the cassettes of interest with the same plasmid-binding oligo sequences (Fig. 3c). These shared regions also contain distinct synthetic sgRNA binding sites absent in the mouse and human genomes, and which can be used to specifically excise the cassette. Thus, primers to amplify the donor fragment are designed as 60 mers composed of the 40 bp of homology arm and 20 bp of plasmid binding sequence. To ensure that the Cas9 ribonucleoproteins (RNPs) do not cut the donor fragment, we design primers so that the end of the 5′ homology arm is the 1–10 bp of the KI sgRNA spacer sequence; conversely, the beginning of the 3′ homology arm is the remaining 11–20 bp of the KI sgRNA spacer sequence. We optimize the amplification conditions to ensure that only the expected band is produced, which enables the donor fragment to be isolated at a high concentration via column-based PCR purification. In general, we avoid gel purification due to the reduced yield and low DNA quality associated with these approaches.
Targeting of the DNA donor into the locus of interest: electroporation, positive selection, drug sensitivity, quality control (Steps 22–45)
To introduce the cassette into the locus of interest, we use CRISPR–Cas9 RNPs and the donor DNA with 40 bp homology arms. Electroporation is used to introduce the RNP and DNA into target cells, although other delivery methods can be used. Transfected cells are left to recover for 48 h and are then selected with the antibiotic or fluorescent marker of choice. As a control, we include untransfected cells and cells receiving the donor DNA alone. After selection, surviving cells are expanded, tested for sensitivity to the negative selection and genotyped for the presence of the on-target integration of the cassette.
Generating the deletion: electroporation, negative selection, sensitivity, quality control (Steps 46–57)
To generate the deletion of interest, RNPs with two flanking guides are electroporated into cells containing the cassette. As for the initial step, if needed, the use of fluorescently-labeled Cas9 can be used to estimate delivery efficiency and isolate cells that received the RNPs. Cells are left to recover for 48 h after electroporation and are then cultured with negative selection. As controls, we use untransfected cells, or cells transfected with RNPs that specifically excise the cassette with minimal disruption of the genome (e.g., via synthetic guides in the donor DNA or guides directly flanking the cassette in the genome). We incorporated synthetic sgRNAs (absent in the mouse or human genomes) flanking the positive/negative selection cassette in all MACHETE plasmids. This allows the cassette itself to be excised without affecting the rest of the cell’s genome, which can be optionally used as a control for the procedure. These populations are compared with the cells receiving the sgRNAs used to create the deletions of interest. The cells surviving the negative selection are expanded and tested for sensitivity/resistance to the selection agents. To assess whether the deletion engineering was successful, genomic DNA is analyzed for the intended breakpoint and presence/absence of the selection cassette. Further characterization of the deletion event can be done via derivation of single-cell clones to evaluate the frequency and zygosity of the event in the population. The cells with control or experimental deletions are then used for downstream applications to assess their biological effects, where the specific phenotypic assays will be tailored to the project of interest.
Recommended controls
Quantification of background events. MACHETE enables the enrichment of deletions that can be rare within a population of cells, so it is important to ensure that the surviving cells are true deletion events and not due to incomplete selection. For this, we always include control cells that are electroporated but that do not include Cas9 RNPs for both stages of MACHETE.
Minimizing OTEs. Given the well-documented effect of CRISPR–Cas9 in creating DSBs in unintended regions, we always include two independent sets of sgRNAs to engineer every deletion of interest. This minimizes the chance that any given phenotype is driven by off target effects of specific sgRNAs. If warranted, testing for OTEs can be incorporated by assessing indels in the predicted sgRNA binding sites. However, given the ample space for sgRNA selection, all the guides we have used have no predicted off targets (with two or three mismatches). Importantly, we have not observed unintended large deletions by sparse whole-genome sequencing. In our experience the combined use of independent sgRNA pairs with the underlying features of the method ensures that OTEs are negligible.
(Optional) Cassette excision. The current design of the MACHETE plasmids includes the presence of synthetic spacer sequences flanking the selection cassette. These spacers are not present in the mouse or human genomes and enable the seamless excision of the selection cassette to obtain control cells that went through the same selection stages as the cells with larger deletions.
Materials
Biological materials
-
Cell lines: we have implemented MACHETE across a range of human and mouse cell lines (i.e., HEK293 cells, NIH3T3 cells, mouse embryonic stem cells, pancreatic ductal epithelial cells, B16F10 melanoma cells). Cell lines will be project specific and should be cultured following the manufacturer or American Type Culture Collection guidelines
▲ CAUTION The cell lines used in your research should be regularly checked to ensure that they are authentic and are not infected with mycoplasma.
Reagents
Cell culture
Cell culture media for mammalian cells used
PBS 1× (Thermo Fisher, cat. no. 14200083)
Trypsin–ethylenediaminetetraacetic acid. 0.25% (Gibco, cat. no. 25200-056)
Puromycin 10 mg/mL (Invivogen, cat. no. ant-pr-1)
Hygromycin B 50 mg/mL (Thermo Fisher, cat. no. 10687010)
-
Diphtheria toxin, unnicked, Corynebacterium diphtheriae (Sigma-Aldrich, cat. no. 322326-1MG)
▲ CAUTION Toxic when inhaled or orally. Work under a hood wearing a laboratory coat and disposable gloves.
-
Ganciclovir (Sigma-Aldrich, cat. no. 345700-50MG)
▲ CAUTION Ganciclovir is a potential cancer hazard. Work wearing a laboratory coat and disposable gloves.
Molecular biology reagents
Locus-specific 60 mer forward oligo (5′-N40bp TGCAGGAGCTATTAATTCGC-3′), where N40bp refers to the 5′ homology arm, the remaining 20 bp bind to the 5′ sequence flanking the selection cassette in the collection of MACHETE plasmids (IDT)
Locus specific 60 mer reverse oligo: (5′-N40 GACCATCTTCGGGAACATCC-3′), where N40bp refers to the 3′ homology arm, the remaining 20 bp bind to the 3′ sequence flanking the selection cassette in the collection of MACHETE plasmids (IDT)
sgRNA specific 59 mer forward oligo: (5′-TAATACGACTCACTATAGG N20bp GTTTTAGAGCTAGAAATAGC-3′), where N20bp refers to the sgRNA/spacer sequence (IDT)
sgRNA universal reverse oligo: (5′-AGCACCGACTCGGTGCCACT-3′) (IDT)
pX330 plasmid (Addgene, plasmid no. 58778)
Plasmids containing dual selection cassettes (e.g., MACHETE plasmid collection deposited in Addgene, plasmid nos. 195282, 195283, 195284, 195285, 195286, 195288, 195289, 195290)
Q5 High-Fidelity 2× Master Mix (NEB, cat. no. M049L)
UltraPure DNAse/RNAse-Free Distilled Water (Thermo Fisher, cat. no. 10977035)
Agarose (Fisher Scientific, cat. no. BP160-500)
UltraPure DNA typing grade 50× TAE buffer (Thermo Fisher, cat. no. 24710030)
-
SYBR Safe DNA gel stain (Thermo Fisher, cat. no. S33102)
▲ CAUTION This stain is a potential cancer hazard. Work wearing a laboratory coat and disposable gloves.
QIAquick PCR purification kit (Qiagen, cat. no. 28104)
HiScribe T7 High Yield RNA synthesis kit (NEB, cat. no. E2040S)
RNA Clean & Concentrator (Zymo Research, cat. no. R1017)
TrueCut Cas9 Protein v2 (Thermo Fisher, cat. no. A36498).
(Optional) Cas9–EGFP (IDT, cat. no. 10008100) or Cy3–Cas9 (PNA Bio, cat. no. CP06)
Neon NxT Electroporation System 10 μL kit (Thermo Fisher, cat. no. N1096)
-
Crystal violet solution (Sigma-Aldrich, cat. no. V5265-500ML)
▲ CAUTION The solution contains formaldehyde and methanol, and is toxic. Work under a chemical hood while wearing a laboratory coat and disposable gloves.
GoTaq Green Master Mix (Promega, cat. no. M7122)
DNA ladder 1 kb (Thermo Fisher, cat. no. SM0311)
DNA ladder 100 bp (Thermo Fisher, cat. no. 15628019)
6× gel Orange G loading buffer (Thermo Fisher, cat. no. R0631)
DNeasy Blood and Tissue kit (Qiagen, cat. no. 69504)
-
HCl 1 M (Sigma, cat. no. 7647-01-0)
▲ CAUTION HCl is corrosive. Wear gloves and a laboratory coat when handling.
-
NaOH 1 M (Sigma, cat. no. 1310-73-2)
▲ CAUTION NaOH is corrosive. Wear gloves and a laboratory coat when handling.
Trypan blue 0.4% (Gibco, cat. no. 15250061)
Equipment
Cell culture
Cell culture incubator, 37 °C, 5% CO2
Cell culture laminar flow hood
100 cm2 tissue culture dish (Corning, cat. no. CLS430167)
6-well tissue culture plates (Corning, cat. no. CLS3506)
24-well tissue culture plates (Corning, cat. no. CLS3527)
96-well tissue culture plates (Corning, cat. no. CLS3527)
Falcon conical tubes 50 mL (Thermo Fisher, cat. no. 352070)
Falcon conical tubes 15 mL (Thermo Fisher, cat. no. 352096)
Neon Transfection System (Thermo Fisher, cat. no. NEON1SK)
TC20 automated cell counter (BioRad, cat. no. 1450102)
Cell counter slides (BioRad, cat. no. 1450003)
Benchtop Centrifuge 5810R, swinging bucket (Eppendorf)
Glass Pasteur pipettes (Thermo Fisher, cat. no. 11775098; sterilized)
Water bath, 37 °C
Molecular biology
0.2 mL PCR tube strips with caps (Axygen, cat. no. 10810721)
96-well PCR plates (BrandTech, cat. no. 10279880)
Adhesive PCR plate-sealing sheets (BrandTech, cat. no. 781390)
1.5 mL PCR tubes (Eppendorf, cat. no. 15178344)
Benchtop centrifuge 5425R, fixed rotor (Eppendorf)
T100 PCR thermal cycler (BioRad)
Agarose gel electrophoresis system (BioRad)
Chemidoc gel imaging system (BioRad)
Nanodrop 8000 spectrophotometer (Thermo Fisher)
Plate rocker (Thermo Fisher)
Reagent setup
Oligos
Resuspend oligos to 100 μM in nuclease-free water. Make a 10 μM working solution of each oligo to use in the PCR reactions. Oligos can be stored at −20 °C for over 12 months.
TAE buffer
To make 10 L of TAE 1×, dissolve 200 mL of 50× TAE in deionized water. TAE buffer is stored at room temperature (24 °C) and should be used up to 6 months after preparation.
Diphtheria toxin
Resuspend diphtheria toxin in 1 mL of PBS 1× to create a stock solution of 1 mg/mL, and store as 50 μL aliquots at −80 °C. Prepare a 50 μg/mL working solution, store at −80 °C and avoid repeated freeze–thaw cycles. Diphtheria toxin aliquots can be stored at −80 °C for at least 12 months.
Ganciclovir
Resuspend ganciclovir in distilled water and adjust solution to pH 12 with NaOH 1 M. Once ganciclovir is in solution, adjust to pH 11 with HCl 1 M to create a working solution of 10 mg/mL. Filter through 0.22 μm to sterilize the solution and make 1 mL aliquots. Store at −20 °C, and once thawed do not freeze again. Thawed solution can be kept at 4 °C for up to 4 weeks.
Equipment setup
Neon transfection system
Set the specific program for the cells of interest. Insert the pipette holder containing 3 mL of buffer E to receive the pipette with the electrode. The same holder can be used for all conditions of a given cell line, yet we swap the holder between different parental cell lines.
▲ CRITICAL If conditions are not established for the cell line of interest, follow the optimization protocol to ensure efficient delivery of components.
Procedure
sgRNA design
● TIMING 20 min per locus.
Identify the genomic coordinates of the locus of interest using a browser of choice (e.g., University of California, Santa Cruz genome browser, ENSEMBL, National Center for Biotechnology Information).
-
Use GuideScan (www.guidescan.com) to identify an sgRNA to use for the cassette KI and two pairs of flanking sgRNAs targeting the deletion breakpoints. sgRNAs should be prioritized for those with predicted lowest off-target events and highest efficiency.
▲ CRITICAL STEP Correct guide design is fundamental for the application of MACHETE.
♦ TROUBLESHOOTING
Copy the sgRNA sequence (remember to exclude the NGG PAM sequence) and insert between the T7 promoter sequence and the pX330 binding site to create a 59 mer oligo sequence. Repeat for all required sgRNAs. The primer sequence should be: TAATACGACTCACTATAGG N20bp GTTTTAGAGCTAGAAATAGC, where N20bp refers to the sgRNA spacer sequence.
Order the universal reverse primer (5′-AGCACCGACTCGGTGCCACT-3′) and the locus-specific 59 mer oligos in individual tubes at a 25 nmol scale (desalted).
sgRNA preparation
● TIMING 2 days
-
5.Setup PCR reactions to create DNA templates for the in vitro transcription. This protocol describes 30 μL per reaction, yet these can be scaled up to 100 μL per sgRNA for increased yield.
Component Volume (μL) Final concentration 2× Q5 Master Mix 15 1× 10 μM locus-specific oligo 1.5 0.5 μM 10 μM universal oligo 1.5 0.5 μM pX330 (10 ng/μL) 1.0 0.33 ng/μL Nuclease-free water 11 — -
6.Perform a PCR amplification in a thermal cycler with the following settings:
Cycle no. Denature Anneal Extend Final 1 98 °C, 2 min 2–34 98 °C, 10 s 58 °C, 10 s 72 °C, 20 s 35 72 °C, 5 min 36 16 °C, hold -
7.
Use 5 μL of each PCR reaction, add 1 μL of 6× loading buffer, and run in a 2% agarose gel at 130 V for 30 min to ensure the presence of a single 110 bp band.
-
8.
Purify the remainder of the PCR product using the Qiagen PCR purification kit, following the manufacturer’s instructions. Elute in 20 μL of TE buffer 1× to increase the DNA concentration.
-
9.
Measure concentration with a Nanodrop. Store at −20 °C until needed.
■ PAUSE POINT PCR products can be stored at −20 °C for at least 12 months.
♦ TROUBLESHOOTING
-
10.Perform in vitro transcription of PCR products from Step 9 using the HiScribe T7 High Yield RNA synthesis kit, as follows (20 μL reaction per guide):
Component Volume (μL) Final concentration 10× reaction buffer 2 1× ATP 100 mM 2 10 mM CTP 100 mM 2 10 mM TTP 100 mM 2 10 mM UTP 100 mM 2 10 mM T7 RNA pol Mix 2 — DNA (500 ng) from Step 9 2 25 ng/μL Nuclease-free water 6 — -
11.
Incubate the in vitro transcription reaction at 37 °C for 4–16 h.
-
12.
Dilute 1 μL of the in vitro transcription reaction with 4 μL of nuclease-free water, add 1 μL of 6× loading buffer and run on a 2% agarose gel at 130 V for 35 min. A band of ~100 bp is expected.
♦ TROUBLESHOOTING
-
13.
Purify the transcribed sgRNAs with the Zymo RNA Clean/Concentrator kit, following the manufacturer’s instructions. Elute the sgRNAs in 20 μL of H2O to increase the concentration.
-
14.
Measure the sgRNA concentration with a Nanodrop. Aliquot and store at −80 °C. Avoid freeze–thaw cycles.
▲ CRITICAL STEP High concentration sgRNAs are needed to minimize volume and avoid disruption of the electroporation conditions.
■ PAUSE POINT sgRNAs can be stored at −80 °C for at least 12 months.
♦ TROUBLESHOOTING
HDR donor design and preparation
● TIMING 2 days
-
15.
Identify the homology arms that will be used for HDR. The approach we prefer to use is to select 30 bp upstream and 30 bp downstream of the selected sgRNA (for a total sequence of 80 bp). The 5′ homology arm would correspond to the first 40 bp and the 3′ homology arm would correspond to the second 40 bp. This design ensures the sgRNA will not cut the donor DNA, as the target site will be separated by the intervening selection cassette.
-
16.
Design and order two 60 mer oligos: one forward oligo containing the 5′ homology arm and one reverse oligo containing the 3′ homology arm. The oligos are as follows:
Forward oligo: 5′- N5′HA_40bp TGCAGGAGCTATTAATTCGC-3′, where 5′HA_40bp refers to the 40 nucleotide 5′ homology arm identified in Step 16.
Reverse oligo: 5′- N3′HA_40bp GACCATCTTCGGGAACATCC-3′, where 3′HA_40bp refers to the 40 nucleotide 3′ homology arm identified in Step 16.
-
17.Setup PCR reactions (30 μL per reaction, four reactions per locus to increase yield) to create the DNA donor for HDR:
Component Volume (μL) Final concentration 2× Q5 Master Mix 15 1× 10 μM locus-specific oligo 1.5 0.5 μM 10 μM universal oligo 1.5 0.5 μM Plasmid with positive/negative selection cassette (10 ng/μL) 1.0 0.33 ng/μL Nuclease-free water 11 — -
18.Perform a PCR amplification in a thermal cycler with the following settings:
Cycle no. Denature Anneal Extend Final 1 98 °C, 2 min 2–34 98 °C, 30 s 58 °C, 30 s 72 °C, 2 min 30 s 35 72 °C, 5 min 36 16 °C, hold -
19.
Use 5 μL of each PCR reaction, add 1 μL of 6× loading buffer and run on a 2% agarose gel at 130 V for 35 min to ensure the presence of a single 2–3 kb band (depending on the selected cassette).
♦ TROUBLESHOOTING
-
20.
Purify the remainder of the PCR product using the Qiagen PCR purification kit, following the manufacturer’s instructions. Elute in 20 μL of 1× TE buffer to increase the DNA concentration.
-
21.
Measure the DNA concentration using a Nanodrop.
▲ CRITICAL STEP The template concentration should be at least >250 ng/μL to ensure that the electroporation solution is not diluted.
■ PAUSE POINT PCR products can be stored at −20 °C for at least 12 months.
♦ TROUBLESHOOTING
MACHETE cassette KI and quality control
● TIMING 1 day for electroporation, 7–14 days for selection, 3 days for quality control
-
22.
Prepare tubes for each electroporation reaction. Mix 1 μg of recombinant Cas9 with 1 μg of sgRNA from Step 14 (KI sgRNA) per reaction and incubate at room temperature for 15 min to make the RNPs. The samples to prepare are blank (no DNA/no Cas9), Cas9 RNP alone (recombinant Cas9 + KI sgRNA from Step 14) and Cas9 RNP to be later mixed with donor DNA from Step 21 (Step 27).
-
23.
While the RNP is incubating, wash the cultured cells with PBS 1×, trypsinize and proceed to count them.
-
24.
Transfer the required number of cells (250,000 cells per condition) to a new 15 mL Falcon tube and dilute to 10 mL with PBS 1×.
-
25.
Centrifuge the cells at 300g for 3 min at 4 °C, discard the supernatant and resuspend with 10 mL of PBS 1×.
-
26.
Centrifuge the cells at 300g for 3 min at 4 °C, discard the supernatant and resuspend in Neon buffer R (10 μL of buffer/condition).
-
27.
Add the donor DNA from Step 21 to the tube with the corresponding RNP complex from Step 22.
-
28.
Add 10 μL of the cell suspension (250,000 cells) from Step 26 to each tube prepared in Steps 22 or 27: blank (from Step 22), Cas9 only (from Step 22) and RNPs + donor mix (from Step 27). Mix the suspensions by thorough pipetting.
-
29.
Add 3 mL of buffer E to the Neon electroporation plastic holder and insert it into the electroporator.
-
30.
Take 10 μL of each cell suspension with a Neon fixed-volume pipette using the electrode containing tips.
▲ CRITICAL STEP Avoid introducing bubbles into the fixed volume pipette tip, which will disrupt the electroporation.
-
31.
Insert the electrode tip into the Neon holder from Step 29 and electroporate the solution according to the cell line-specific programs.
▲ CRITICAL STEP Ensure that you have identified the correct settings to deliver DNA and RNP to your cells. If not available, perform the Neon optimization protocol to find the settings that maximize transfection while maintaining viability.
-
32.
Transfer the electroporated cells to a well in a 6-well plate and let them recover for 48 h. Repeat Steps 30–32 for each tube prepared in Step 28. (Optional) If fluorescent Cas9 (e.g., Cas9–EGFP, Cy3–Cas9) was used, check cells with a fluorescence microscope or flow cytometer after 24 h to confirm the presence of the RNPs.
-
33.
Once electroporated cells reach 60–70% confluence, wash with 1× PBS, trypsinize and passage them 1:1 into a new plate with medium supplemented with either puromycin (1 μg/mL) or hygromycin B (50 μg/mL).
▲ CRITICAL STEP Optimize the antibiotic selection conditions for each cell line. We recommend a titration curve to identify the minimal dose that completely eliminates untargeted parental cells.
-
34.
Grow the KI cells in selection media to ensure cassette activity. To keep store stocks: expand the population to three 10 cm culture plates. Once cells are 80–90% confluent, wash with 1× PBS, trypsinize and take 90% of the cell suspension to centrifuge at 300 rpm for 5 min at 4 °C. Discard the supernatant and resuspend the cell pellet of each 10 cm plate with 3 mL of cell culture medium supplemented with 10% dimethyl sulfoxide. Make 1 mL aliquots in cryotubes and freeze at −80 °C.
-
35.
Keep the remaining 10% of cells from Step 34 in culture to genotype (to ensure the on-target integration, Steps 36–41) and assess drug sensitivity (functionality of the cassette, Steps 42–45).
■ PAUSE POINT Cells can be stored at −80 °C for up to 3 months. For long-term storage, keep cells in liquid nitrogen.
♦ TROUBLESHOOTING
Genotyping
-
36.
Design genotyping oligos for each KI locus. Two PCRs are designed: an internal PCR for the cassette of interest, and a site-specific PCR by combining a genomic oligo upstream of the 5′ homology arm and an internal oligo binding to the MACHETE cassette. In general, we design PCR products between 300 and 1,000 bp.
-
37.
Collect 500,000 KI and wild-type (WT) cells and centrifuge them at 300g for 5 min at 4 °C. Extract genomic DNA from the pellet of parental and KI cells by using the Qiagen DNEasy Blood and Tissue kit, following the manufacturer’s instructions.
-
38.
Measure the genomic DNA concentration using a Nanodrop spectrophotometer.
-
39.Setup the PCR reaction using GreenTaq GO (20 μL/reaction) as follows. Include negative controls (e.g., blank, unrelated KI and/or unrelated cassette):
Component Volume (μL) Final concentration 2× Green Taq Go Mix 10 1× 10 μM locus- (on-target) or cassette-specific (internal) oligo 1.0 0.5 μM 10 μM cassette-specific oligo 1.0 0.5 μM Genomic DNA (100 ng/μL) 1.0 5 ng/μL Nuclease-free water 7.0 — -
40.Perform a PCR amplification in a thermal cycler with the following settings:
Cycle no. Denature Anneal Extend Final 1 98 °C, 2 min 2–34 98 °C, 30 s 58 °C, 30 s 72 °C, 30 s 35 72 °C, 5 min 36 16 °C, hold -
41.
Run samples on a 2% agarose gel at 130 V for 35 min, and assess the presence of on-target and internal PCR products in the KI cells.
♦ TROUBLESHOOTING
KI cell drug sensitivity
-
42.
Seed 100,000 parental or KI cells in a 6-well plate. Leave the cells untreated, or add positive selection (puromycin 1 μg/mL or hygromycin B 50 μg/mL) or negative selection (diphtheria toxin 50 ng/mL or ganciclovir 10 μg/mL). Culture cells for 48–72 h. Alternatively, in the case of nonadherent cultures, measure the relative cell survival by counting and using viability dyes (e.g., trypan blue): take a 10 μL aliquot of the cell suspension, add 10 μL of trypan blue and mix by pipetting. Transfer 10 μL of this mix into a cell counter slide and use automated counter to evaluate the fraction of living cells.
-
43.
Remove the medium and stain cells with crystal violet solution for 20 min on a plate shaker.
-
44.
Remove the crystal violet solution, and wash the plate by immersing in a container with running water ten times.
-
45.
Leave plates to dry and image in a ChemiDoc. (Optional) Plates can be imaged in a color scanner.
♦ TROUBLESHOOTING
Deletion engineering and quality control
● TIMING 1 day for electroporation, 7–14 days for selection, 14 days for quality control
-
46.
Prepare tubes for each electroporation reaction. Mix 2 μg of recombinant Cas9 with 1 μg of 5′ sgRNA and 1 μg of 3′ sgRNA (flanking sgRNAs prepared in Step 14) per reaction. Incubate this complex at room temperature for 15 min to make the RNPs. Prepare the following samples: negative controls (e.g., blank, Cas9 alone), Cas9 RNPs to engineer intended deletions and Cas9 RNPs to excise the cassette.
-
47.
Follow Steps 23–32 for the electroporation of the tubes prepared in Step 46.
-
48.
Trypsinize and passage the electroporated cells once they reach 60–70% confluence and plate into cell culture medium supplemented with negative selection: diphtheria toxin for diphtheria toxin receptor (DTR) constructs (50 ng/mL) or ganciclovir for thymidine kinase (TK) constructs (10 μg/mL).
-
49.
Expand the surviving cells under constant selection and freeze down several vials as in Step 34. Keep a fraction of cells in culture to test by genotyping (to ensure the presence of the breakpoints and loss of cassette) and drug sensitivity (functionality of the cassette).
■ PAUSE POINT Cells can be stored at −80 °C for up to 3 months. For long-term storage, keep cells in liquid nitrogen.
♦ TROUBLESHOOTING
Genotyping
-
50.
Genotype cells (from Step 49) as described in Steps 36–41.
Deleted cell drug sensitivity
-
51.
Test the sensitivity of cells (from Step 49) to selection agents as described in Steps 42–45.
Assessment of deletion frequency by single-cell clone genotyping
● TIMING 10–14 days
-
52.
Wash with 1× PBS, trypsinize and centrifuge cells with the engineered deletions. Count cells and transfer 100–200 cells to a 15 mL conical tube with 10 mL of medium. Plate 100 μL per well in each well of a 96-well plate.
-
53.
24 h later, identify wells with individual cells under the miscroscope and allow for clonal expansion. Change media every 72 h.
▲ CRITICAL STEP Using the microscope, ensure that single cells are observed in the wells that will be analyzed.
-
54.
Wash cells with PBS 1×, trypsinize and plate 10% of the cells in a well of a 24-well plate and expand in culture in the absence of any selection in standard media. Once cells reach 70–80% confluence, proceed to freeze down vials as described in Step 34.
-
55.
Transfer the remaining 90% of the cells of each clone (from Step 54) to a new Eppendorf tube with 500 μL of PBS 1× and centrifuge at 300g at 4 °C for 5 min.
-
56.
Extract genomic DNA of the pellet from Step 55 with the DNEasy Tissue and Blood collection kit, following the manufacturer’s instructions. Genotype cells following Steps 36–41.
-
57.
(Optional) If single-cell clones are needed for downstream functional assays, identify wells with the expected deletions from Step 55, further expand and freeze down a stock of cells as described in Step 34.
♦ TROUBLESHOOTING
-
58.
Use either polyclonal (from Step 49) or single-cell clones (from Step 54) with the intended deletions for phenotypic characterization of cells. The specific downstream applications to assess the phenotypes of the deletions should be tailored to the project of interest and can include molecular, cellular or in vivo assays.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1 ∣.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 2 | No guide with high efficiency or low off-target scores | Selected region contains repetitive elements | Use RepeatMasker to avoid repetitive elements. Change coordinates (1 kb upstream or downstream) |
| 9 | Low DNA concentration | Not enough template or inefficient PCR | Repeat PCR with more plasmid template or pool multiple reactions |
| 12 | Smear or no band in gel | sgRNA degradation | Repeat reaction and ensure nuclease-free conditions |
| 14 | Low sgRNA concentration | Poor in vitro transcription | Repeat reaction with more DNA template or pool multiple reactions |
| 19 | No amplification of donor DNA | Not sufficient template | Repeat reaction with more template |
| 21 | Low concentration of donor DNA | Low yield | Repeat by setting up multiple reactions per donor and pool for PCR purification |
| 35 | No cells survive positive selection | Low HDR efficiency | Repeat electroporation and scale up number of targeted cells. Alternatively, extend homology arms to 160 bp using an ultramer oligo for PCR |
| 41 | Undetectable on-target cassette integration | Genotyping PCR is not optimal (more common) or cassette is present only in off-target locus | Redesign genotyping primers. Alternatively, change KI locus |
| 45 | KI cells are resistant to negative selection | Presence of untargeted cells due to incomplete positive selection | Increase concentration of positive selection. If needed, single-cell clones can be derived |
| 49 | No differential survival between negative control and targeted cells treated with negative selection | (i) Inefficient delivery of sgRNAs. (ii) sgRNAs do not efficiently create the intended DSBs |
(i) Repeat with increased concentration of sgRNAs. (ii) Design new sgRNAs targeting the intended locus |
| 57 | Low frequency of cells with deletion | Negative selection not robust | Repeat deletion electroporation followed by more stringent negative selection. The cell number can be scaled accordingly to increase yield |
Timing
Steps 1–4, Guide selection and design: 20 min per locus
Steps 5–14, Preparation of sgRNAs: 2 days
Steps 15–21, HDR donor design and preparation: 24 h
Steps 22–45, MACHETE cassette KI and quality control: 10–14 days
Steps 46–51, Deletion engineering and quality control: 10–14 days
Steps 52–57, Assessment of deletion frequency by single-cell clone genotyping: 10–14 days
Anticipated results
The PCR product containing the T7 promoter, spacer sequence and tracr sequence is expected to be 110 bp (Fig. 4b). The concentration of purified DNA for an effective transcription should range between 60 and 80 ng/μL. The in vitro-transcribed sgRNA runs around the 100 bp marker, although in the nondenaturing agarose gel, this may be seen as a doublet for some sequences. The in vitro transcription reaction yields ~100 μg of sgRNA, which is used at 1 μg/μL to reduce the volume in the electroporation reaction.
Fig. 4 ∣. Expected results.
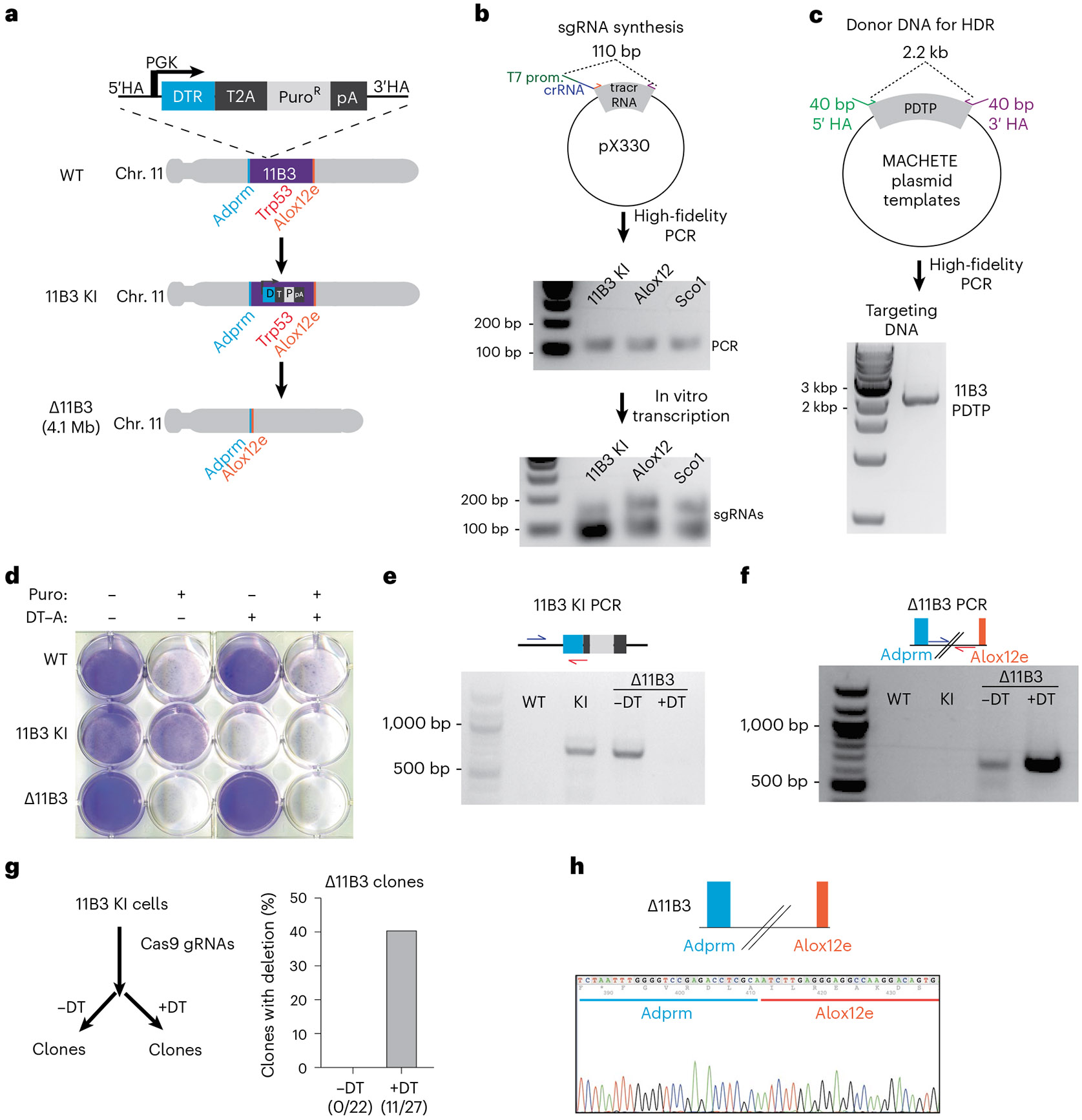
a, A schematic representation of the design to create a 4 Mb deletion of the mouse chromosome (Chr.) 11B3 locus containing the tumor suppressor Trp53 gene. b, A schematic of the procedure to synthesize KI and deletion sgRNAs for the murine 11B3 locus. Representative agarose gel of the PCR products used for in vitro transcription (top). Representative agarose gel of the transcribed sgRNAs, which can appear as doublets in these nondenaturing agarose gels (bottom). c, Schematic and representative images of the procedure to create the DNA donor for CRISPR-mediated HDR of a PGK DTR T2A Puro cassette into the 11B3 locus. A representative agarose gel of the DNA donor containing the MACHETE cassette and flanking homology arms used for HDR. d, Crystal violet staining of parental (WT), KI and deleted (Δ11B3) cells in the presence or absence of positive (puromycin, 1 μg/mL) and negative (diphtheria toxin, 50 ng/mL) selection. e, A PCR of the integration of the MACHETE cassette in the 11B3 locus. This was tested in the following cells: parental (WT), KI, unselected deleted (Δ11B3 − DT–A), and selected deleted (Δ11B3 + DT–A). f, A PCR of the engineered breakpoint of the 4.1 Mb Δ11B3 deletion. This was tested in the following cells: parental (WT), KI, unselected deleted (Δ11B3 − DT–A), and selected deleted (Δ11B3 + DT–A). g, Frequency of the intended Δ11B3 deletion in the population of unselected (− DT-A) and negatively selected (+ DT–A) cells. h, Representative Sanger sequencing of the Δ11B3 breakpoint PCR confirming the intended junction in genomic DNA. Images b–h reproduced from ref. 14, Springer Nature Ltd.
The PCR-based donor DNA will give a 2–3 kb band depending on the cassette of choice (Fig. 4c). Gel purification should be avoided given the low yield of extraction. Electroporation conditions should be optimized for each cell line, although there are available conditions for most of the commonly used cell types. The first electroporation is done with 250,000 cells per condition, receiving 1 μg of Cas9, 1 μg of sgRNA and 0.5–1.0 μg of donor DNA. We have observed little to no toxicity of the electroporation per se, and cells recover in less than 24 h. For the positive selection step, negative controls (blank or no sgRNAs) will be eliminated, whereas as conditions receiving RNP + donor will generate cells bearing the selection marker. The speed at which selection occurs depends on the sensitivity of untargeted cells to the antibiotic and the doubling time of the resistant cells under selection. On average, it takes ~7 days to be able to expand sufficient cells for downstream targeting and quality control (via genotyping and drug sensitivity assays).
Having confirmed the presence of the on-target integration and sensitivity to negative selection (Fig. 4d), these stable KI cells can be stored long term and used for any deletions involving the targeted locus. Importantly, we keep cells under constant selection to ensure that the cassette is retained. In our experience, the rate-limiting step of the MACHETE protocol is the integration of the cassette given the low efficiency of HDR, yet the use of strong selection enables even rare events to be highly enriched.
With the established KI lines, the procedure of deletion engineering is straightforward: electroporation of Cas9 RNPs with flanking guides, followed by negative selection. Cell populations are then characterized by genotyping the presence/absence of the KI cassette and appearance of the intended breakpoints (Fig. 4e,f). We have observed that the size of the deletion is inversely correlated with the number of cells surviving the negative selection. Our data show that for deletions of more than 4 Mb, the use of MACHETE becomes enabling, where negative selection is required to detect the breakpoint event in clones (Fig. 4g). Importantly, we have not detected off-target integrations in cells with deletions (tested via genotyping using internal cassette primers in cells that survived negative selection). As for positive selection, the speed of recovery of the cells will be related to the doubling time of the surviving population.
Our studies have relied on polyclonal populations, yet depending on the downstream application users may prefer to rely on single-cell clones, whose characterization and expansion we have incorporated into the assessment of deletion frequency (Fig. 4g,h).
Key points.
MACHETE is a clustered regularly interspaced short palindromic repeats directed Cas9-based, two-stage system for creating megabase-sized genomic deletions: first, a bicistronic positive/negative selection cassette is inserted into the locus of interest, then single-guide RNAs are added to engineer the intended deletion. Only cells that have lost the locus survive the negative selection, thus greatly enriching for cells with deletions.
MACHETE circumvents the inefficiency of retrieving cells with large deletions that limits previous methods.
Acknowledgements
We thank all members of the Lowe laboratory for advice and helpful discussions. F.M.B. was supported by a Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center (GMTEC) Postdoctoral Fellowship, a Memorial Sloan Kettering Cancer Center (MSKCC) Translational Research Oncology Training Fellowship (5T32CA160001-08) and a Young Investigator Award by the Edward P. Evans Foundation. This work was also supported by MSKCC’s David Rubenstein Center for Pancreatic Research Pilot Project (to S.W.L.), the Agilent Thought Leader Program (to S.W.L.) and the GMTEC Classic Individual Funding (to S.W.L.). S.W.L. is an investigator in the Howard Hughes Medical Institute and the Geoffrey Beene Chair for Cancer Biology. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Related links
Key reference using this protocol
- Barriga FM et al. Nat. Cancer (2022): 10.1038/s43018-022-00443-5 [DOI] [Google Scholar]
Footnotes
Competing interests
S.W.L. is a consultant and holds equity in Blueprint Medicines, ORIC Pharmaceuticals, Mirimus, PMV Pharmaceuticals, Faeth Therapeutics, and Senescea Therapeutics and is a consultant for Fate Therapeutics. F.M.B. declares no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41596-024-00953-9.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data related to this protocol are based on our previously published work14.
References
- 1.Kim YG, Cha J & Chandrasegaran S Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl Acad. Sci. USA 93, 1156–1160 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cermak T. et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39, e82–e82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jinek M. et al. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wood AJ et al. Targeted genome editing across species using ZFNs and TALENs. Science 333, 307–307 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller JC et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol 25, 778–785 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Doudna JA The promise and challenge of therapeutic genome editing. Nature 578, 229–236 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joung J. et al. Genome-scale CRISPR–Cas9 knockout and transcriptional activation screening. Nat. Protoc 12, 828–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fauser F, Schiml S & Puchta H Both CRISPR/Cas-based nucleases and nickases can be used efficiently for genome engineering in Arabidopsis thaliana. Plant J. 79, 348–359 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Friedland AE et al. Heritable genome editing in C. elegans via a CRISPR–Cas9 system. Nat. Methods 10, 741–743 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shalem O, Sanjana NE & Zhang F High-throughput functional genomics using CRISPR–Cas9. Nat. Rev. Genet 16, 299–311 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng S. et al. Strategies for high-efficiency mutation using the CRISPR/Cas system. Front. Cell Dev. Biol 9, 803252 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Varshney GK et al. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 25, 1030–1042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canver MC et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J. Biol. Chem 289, 21312–21324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barriga FM et al. MACHETE identifies interferon-encompassing chromosome 9p21.3 deletions as mediators of immune evasion and metastasis. Nat. Cancer 3, 1367–1385 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spiliopoulou P. et al. All is not lost: learning from 9p21 loss in cancer. Trends Immunol. 43, 379–390 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Sherr CJ The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol 2, 731–737 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Cai Y. et al. Loss of chromosome 8p governs tumor progression and drug response by altering lipid metabolism. Cancer Cell 29, 751–766 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Carroll D. Genome engineering with targetable nucleases. Annu. Rev. Biochem 83, 409–439 (2014). [DOI] [PubMed] [Google Scholar]
- 19.He Z. et al. Highly efficient targeted chromosome deletions using CRISPR/Cas9: highly efficient targeted chromosome deletions. Biotechnol. Bioeng 112, 1060–1064 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Boroviak K, Doe B, Banerjee R, Yang F & Bradley A Chromosome engineering in zygotes with CRISPR/Cas9. Genesis 54, 78–85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shalem O. et al. Genome-scale CRISPR–Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y. et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 531, 471–475 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor AM et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell 33, 676–689.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosco N. et al. KaryoCreate: a CRISPR-based technology to study chromosome-specific aneuploidy by targeting human centromeres. Cell 186, 1985–2001.e19 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Truong MA et al. A kinesin-based approach for inducing chromosome-specific mis-segregation in human cells. EMBO J. 42, e111559 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fu Y. et al. High-frequency off-target mutagenesis induced by CRISPR–Cas nucleases in human cells. Nat. Biotechnol 31, 822–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Höijer I. et al. CRISPR–Cas9 induces large structural variants at on-target and off-target sites in vivo that segregate across generations. Nat. Commun 13, 627 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez AR et al. GuideScan software for improved single and paired CRISPR guide RNA design. Nat. Biotechnol 35, 347–349 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt H. et al. Genome-wide CRISPR guide RNA design and specificity analysis with GuideScan2. Preprint at bioRxiv 10.1101/2022.05.02.490368 (2022). [DOI] [Google Scholar]
- 30.Gundry MC et al. Highly efficient genome editing of murine and human hematopoietic progenitor cells by CRISPR/Cas9. Cell Rep. 17, 1453–1461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stewart-Ornstein J & Lahav G Dynamics of CDKN1A in single cells defined by an endogenous fluorescent tagging toolkit. Cell Rep. 14, 1800–1811 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data related to this protocol are based on our previously published work14.