

Abstract
Progression of chronic liver injury to fibrosis, abnormal liver regeneration, and HCC is driven by a dysregulated dialog between epithelial cells and their microenvironment, in particular immune, fibroblasts, and endothelial cells. There is currently no antifibrogenic therapy, and drug treatment of HCC is limited to tyrosine kinase inhibitors and immunotherapy targeting the tumor microenvironment. Metabolic reprogramming of epithelial and nonparenchymal cells is critical at each stage of disease progression, suggesting that targeting specific metabolic pathways could constitute an interesting therapeutic approach. In this review, we discuss how modulating intrinsic metabolism of key effector liver cells might disrupt the pathogenic sequence from chronic liver injury to fibrosis/cirrhosis, regeneration, and HCC.
INTRODUCTION
Progression of chronic liver injury to fibrosis, abnormal liver regeneration, and HCC is driven by a dysregulated dialog between epithelial cells and their microenvironment, in particular immune, fibroblasts, and endothelial cells. Acute liver injury leads to hepatocyte death followed by a wound repair process, with immune cell recruitment, extracellular matrix remodeling driven by HSC, together with hepatocyte proliferation to restore the liver mass (Figure 1). When the liver is chronically injured, sustained wound repair leads to fibrogenesis, characterized by abnormal extracellular matrix accumulation and degradation by HSC, together with inflammatory cell recruitment and activation, as well as sustained hepatocyte proliferation.1,2 This makes fibrosis and regeneration 2 faces of the same “wound repair coin.” At the end-stage cirrhosis, hepatocytes become senescent and stop proliferating, leading to an inhibition of liver regeneration and to the induction of a ductular reaction in periportal areas (Figure 1). Cirrhosis is a prelude to HCC, since this liver primary tumor arises in a large proportion of cases (>80%) in patients with cirrhosis regardless of the causal risk factor. Liver regeneration process acts as a stimulus promoting the growth of precancerous and neoplastic lesions, particularly in a cirrhotic liver, providing growth factors and cytokines favorable for tumorigenesis and HCC progression. HCC is composed of cancer cells with their surrounding tumor stromal cells embedded in a fibrotic matrix. This complex 3 dimensional microenvironment comprises activated HSCs, the precursor of cancer associated fibroblasts,3 endothelial, immune cells, in particular tumor-associated monocytes/macrophages (TAM) and myeloid-derived suppressor cells.4 (Figure 1).
FIGURE 1.
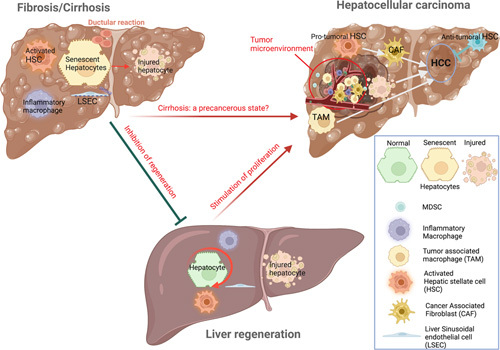
Fibrogenesis, regeneration and cancer: a delicate balance. The same events that promote liver regeneration, particularly the activation of HSC, the induction of endothelial cell dysfunction and the switch of macrophages toward an inflammatory phenotype, drive liver fibrogenesis. During the first steps of fibrogenesis, hepatocytes maintain their proliferative capacity. When disease progress to cirrhosis, hepatocytes become senescent, show impaired proliferative capacity, and a ductular reaction made of cells with progenitor features develops around the portal tract. In this context of cirrhosis, an uncontrolled cell proliferation stimulus may lead to transformation of preneoplastic lesions to HCC. TAM and activated HSC, the precursor of CAF infiltrate HCC microenvironment stimulating tumor progression and invasiveness. A distinct HSC subpopulation with tumor-suppressive functions has also been recently identified.5 Abbreviations: CAF, cancer associated fibroblasts; TAM, tumor associated macrophages.
Accumulating data have demonstrated that metabolic reprogramming of epithelial and nonparenchymal cells is critical at each stage of the sequence fibrosis/cirrhosis-regeneration-HCC and drives disease progression. In this review, we discuss how dysregulation of the intrinsic metabolism in key effector liver cells drives the pathogenic sequence from chronic liver injury to fibrosis/cirrhosis, regeneration, and HCC.
AN OVERVIEW OF INTRINSIC CELLULAR METABOLISM
To face anabolic demands and supply the required energy for homeostasis, cells undergo metabolic reprogramming. Mitochondria play an essential role in this process, by oxidizing glucose, amino acids, and fatty acids and by linking the tricarboxylic acid (TCA) cycle to oxidative phosphorylation (Figure 2).
FIGURE 2.
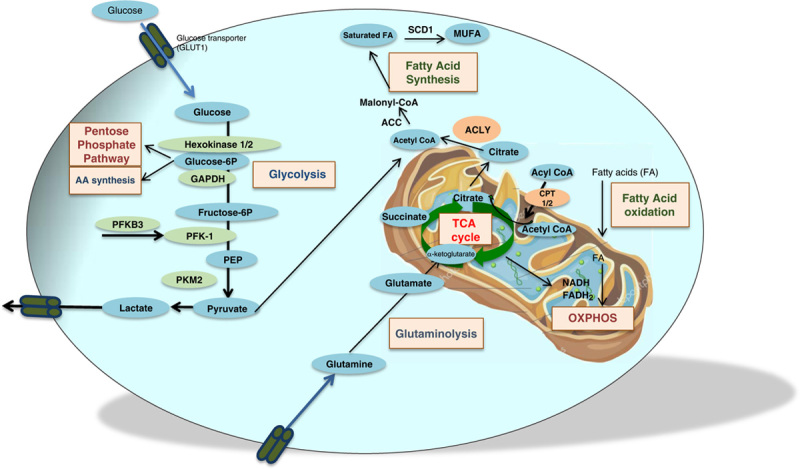
Main intrinsic metabolic pathways. Glucose enters the cell through glucose transporters and is converted into pyruvate through glycolysis. In parallel to glycolysis, the pentose phosphate pathway generates ribose 5 phosphate for the synthesis of nucleotides and amino acids. Pyruvate is either converted into lactate or is oxidized in acetyl coA, fueling the TCA cycle inside the mitochondria and giving rise to NADH and FADH2 that can also be produced by fatty acid oxidation. Fatty acid synthesis produces fatty acids from acetyl coA derived either from glycolysis or from citrate. The TCA cycle can also be fueled by amino acids through glutaminolysis. Abbreviations: ACC, acetyl-coenzyme A carboxylase; ACLY, ATP citrate lyase; CPT, carnitine palmitoyl transferase; GLUT, glucose transporter protein type; MUFA, monounsaturated fatty acid; PFK, phosphofructokinase; PKM2, pyruvate kinase isoform M2; SCD1, stearoyl-coA desaturase; TCA, tricarboxylic acid.
The TCA cycle and oxidative phosphorylation are highly efficient to generate ATP. Pyruvate or fatty acids transferred to the mitochondrial matrix are oxidized into acetyl CoA to enter the TCA cycle through condensation with oxaloacetate to form citrate. Citrate can be exported to the cytosol following oxidation by the TCA cycle or following hydrolysis by ATP citrate lyase (ACLY) to regenerate oxaloacetate and acetyl CoA, needed to synthetize cholesterol and fatty acids. The TCA cycle produces NADH and flavin adenine dinucleotide (FADH2), which provide electrons to the electron transport chain to support oxidative phosphorylation and generate about 30–32 ATP molecules from 1 molecule of glucose converted into pyruvate (Figure 2). Glutamate can also fuel the TCA cycle through its conversion into α-ketoglutarate by glutaminolysis. In contrast to oxidative phosphorylation, glycolysis produces ATP from glucose rapidly but very inefficiently (2 ATP/molecule of glucose). Glycolysis involves 3 rate-limiting enzymes, including hexokinases (HKs), PFK, and the pyruvate kinases. The first step of glycolysis is the uptake of glucose by the cell and its transformation by HK1 into glucose-6-phosphate that can enter the pentose phosphate pathway, providing nucleotides and amino acids precursors. Conversion of phosphoenolpyruvate by pyruvate kinase isoform M2 (PKM2) leads to pyruvate, which is either converted to lactate, the end product of anaerobic glycolysis, or is metabolized in the mitochondria into acetyl CoA to the TCA cycle (Figure 2). The rise in glucose transporter or in glycolytic enzyme (HK2) expressions facilitates glycolysis, which is also used for amino acid and fatty acid synthesis. While normal cells currently use mitochondrial oxidative phosphorylation in aerobic conditions to produce ATP, it has been shown that cancer cells are biased toward glycolysis even in an aerobic environment, a so-called “Warburg effect.”6
TARGETING METABOLISM FOR ANTIFIBROGENIC THERAPY
In response to chronic liver injury, extracellular matrix accumulation, the hallmark of liver fibrogenesis, is mainly driven by HSCs and portal fibroblasts that migrate and accumulate at the site of injury.1,2 Acquisition of the fibrogenic phenotype is controlled by a complex cell-cell network, involving interactions of HSC with hepatocytes, as well as endothelial and immune cells. Sustained hepatic inflammation governs liver fibrosis progression, since professional immune cells, including resident and recruited innate, adaptive immune cells and innate-like lymphocytes, control HSC accumulation.1,7,8 Inflammatory and profibrogenic signals can also arise from liver parenchymal cells, in particular hepatocytes, through release of innate immune proteins with fibrogenic potential, and from nonparenchymal cells including endothelial cells, mainly through their capacity to produce inflammatory mediators and to present antigens.1,7,9 Among nonparenchymal cells, different subsets of liver macrophages control fibrosis progression and regression, the restorative population promoting fibrosis resolution by inducing apoptosis of HSC and/or their reversion to a quiescent-like phenotype.7,10,11
The high energy demand required to promote fibrosis progression is associated with metabolic modifications in the different cellular components of liver fibrosis. Metabolic reprogramming of HSCs driving their transition to a myofibroblastic phenotype has been well documented and includes loss of lipid droplets, inhibition of gluconeogenesis, as well as concomitant activation of aerobic glycolysis and glutaminolysis.12–14 Moreover, the resulting accumulation of lactate sustains and perpetuates the immuno-fibroproliferative phenotype. Although the emblematic function of hepatocytes is to support metabolic homeostasis, the consequences of their metabolic changes on the fibrogenic process are much less characterized. In addition, despite the recognized requirement of metabolic reprogramming for switches in immune cell phenotype, how metabolic changes, particularly in macrophages, may influence fibrosis progression and regression is only emerging.15 The following section describes the recent elements that support the development of antifibrogenic strategies targeting cell-intrinsic metabolism, ie, how intracellular metabolic modification in a given liver cell impacts on its functions.
Inhibiting glycolysis to reduce fibrosis progression
The role of glycolysis in fibrosis progression was initially suggested by the increase in glycolytic gene expression observed in experimental models and in liver human samples with chronic liver injury.12,16 It was reinforced by the reduction of fibrosis promoted by glycolysis inhibitors, such as 2-deoxyglucose (2DG), TEPP-46, or 3PO, which inhibit HK2, PKM2, and PFKFB3, respectively, as well as by pfkfb3 knockdown.16,17 Studies at the cellular level revealed that the profibrogenic functions of glycolysis are related to its impact on HSC activation, proinflammatory functions in macrophages, and reduction of stiffness-induced angiocrine signaling in endothelial cells (Figure 3).
FIGURE 3.
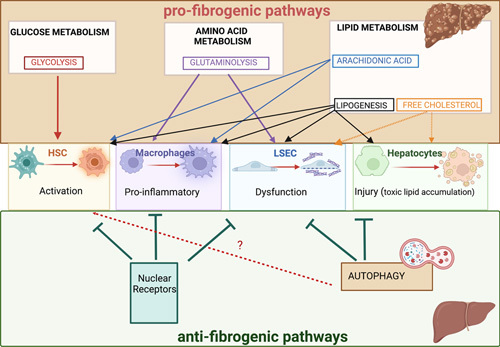
Role of intracellular metabolic pathways in liver fibrogenesis depending on the targeted cell type. The impact of the main intrinsic metabolic pathways (glycolysis, glutaminolysis, lipogenesis, autophagy, nuclear receptors and YAP/TAZ) on HSC activation, macrophage inflammatory phenotype, endothelial dysfunction, and hepatocyte injury and their consequences on liver fibrogenesis are depicted. Abbreviation: YAP/TAZ, yes-associated protein and the transcriptional coactivator with PDZ-binding motif.
In cultured HSC, acquisition of a myofibroblastic phenotype is associated with a rise in the expression of glucose transporters, particularly GLUT1, and of glycolytic enzymes, including HK2, PKM2, or PFKFB3, with concomitant lactate accumulation and downregulation of gluconeogenic genes.12,16,17 Accordingly, pharmacological inhibition of HK2, PKM2, or PKF3 blocks the transition of HSC toward a myofibroblastic phenotype.12,16,17 A link between glycolysis in macrophages and progression of liver fibrosis has also been reported. Thus, phosphorylation and nuclear translocation of PKM2 promoted by follistatin-like protein 1, a TGF-β -stimulated secreted glycoprotein of the SPAR family, results in enhanced glycolysis and glycolysis-dependent immune responses.18 Moreover, myeloid-specific FSTL1 deficiency dampens glycolysis and the proinflammatory properties of liver macrophages, attenuating the progression of liver fibrosis in mouse models.18 Finally, recent data also indicate that mice with endothelial cell-specific deletion of HK2 or inhibition of glycolysis by 3PO attenuates neutrophil infiltration, development of portal hypertension, and early fibrosis.19
Interfering with lipid metabolism as an antifibrogenic strategy
Lipogenesis is profibrogenic
Accumulating data indicate that interfering with key lipogenic pathways limits fibrosis progression, most likely by reducing hepatocyte steatosis and injury, as well as by directly inhibiting HSC activation (Figure 3). Thus, in experimental mouse models, global pharmacological inhibition of lipogenesis through blockade of either stearoyl-coA desaturase (SCD1) by Amarchol, acetyl-coenzyme A carboxylase (ACC) with Fircostat or Compound 1,20–22 FASN by Denifanstat,23 or the upstream target ATP citrate lyase by bempedoic acid24 blunts liver fibrosis. Conversely, mice with constitutively active mutation in ACC show steatosis and fibrosis.25 At the cellular level, inhibiting lipogenesis limits hepatocyte injury and suppresses the activation of HSC, as shown by the reduction of the fibrogenic functions of HSC when exposed to ACC inhibitors, following selective targeting of SCD-1 or exposure to the ACLY inhibitor bempedoic acid. However, ACC inhibition leads to increases in circulating triglycerides in mice and humans,21,26 thereby limiting their use in patients with NASH. Pharmacological inhibitors of ACLY could provide novel opportunities in the treatment of NASH-associated fibrosis. Indeed, although genetic knockout of ACLY in hepatocytes reduces liver steatosis and hepatocyte ballooning but not fibrosis, bempedoic acid has additional direct effects on inflammation and fibrosis in vivo, independent of its effects on hepatocytes, through decreased fatty acid and cholesterol synthesis in HSC, and reduction of HSC activation and proliferation in vitro.24 Data on the role of lipogenesis in immune cells definitely need to be investigated in the context of liver fibrosis, since antifibrogenic properties of lipogenesis inhibitors are anticipated, related to their anti-inflammatory effects in macrophages and attenuation of Th17 responses reported in other pathophysiological situations.27–29
Targeting the arachidonic acid pathway
Fatty acid synthesis is central for the generation of lipids, either phospholipids, which are key components of membrane structures, or triacylglycerols, which can be further converted to di-acylglycerols and monoacylglycerols. In the liver, hydrolysis of monoacylglycerols into glycerol and fatty acids is ensured by monoacylglycerol lipase (MAGL), which constitutes the main source of arachidonic acid and proinflammatory eicosanoids in the liver.30 Although the role of arachidonic-derived metabolites in liver fibrosis is still controversial, inhibiting the upstream target MAGL shows promising results. Indeed, pharmacological inhibition or genetic deletion of MAGL results in a decrease of hepatocyte injury, anti-inflammatory, and antifibrogenic effects and promotes fibrosis regression, most likely through direct lipid reprograming in hepatocytes and macrophages.31–33 In addition, specific knockout of magl in hepatocytes protects against liver injury, whereas knockdown in myeloid cells results in anti-inflammatory and antifibrogenic effects in the liver, through a shift of macrophage phenotype from an arachidonic acid/prostaglandin proinflammatory profile toward an anti-inflammatory 2-arachydonoyl-glycerol-producing phenotype31 (Figure 3).
Targeting cholesterol metabolism
Cholesterol has a global profibrogenic impact, and its metabolism is dysregulated in NAFLD. All strategies aiming at decreasing free cholesterol accumulation (inhibition of lipoprotein lipase or sterol regulatory-element binding protein 2, anti-mir33a-bearing vitamin A-coupled liposomes, etc.) have antifibrogenic effects in mice.34 Conversely, increasing free cholesterol concentrations, either by feeding mice with a high-cholesterol diet or following inactivation of acetyl CoA acetyltransferase 1, an enzyme that converts free cholesterol into cholesterol esters, leads to exacerbation of liver fibrosis.35,36 In vitro, free cholesterol activates HSC through induction of a TLR4 signaling pathway that sensitizes them to TGF-β production.35,36 Moreover, cholesterol crystals formed by the hydrolysis of cholesterol in excess into free cholesterol induce hepatocyte lipotoxicity, activate the inflammasome, and trigger endoplasmic reticulum stress leading to hepatocyte injury and liver fibrosis.37 Studies have also suggested that cholesterol overload or free cholesterol accumulation could play a role in KC activation and LSEC dysfunction and capillarization, but further studies are required to better understand the mechanisms involved.38
Modulating mitochondrial metabolism to limit fibrosis
Mitochondrial dysfunction is a characteristic feature of chronic liver diseases that results in metabolic disturbances in parenchymal and nonparenchymal cells and triggers fibrogenesis. Moreover, mitochondrial carriers that support transport of metabolic molecules across the mitochondrial matrix have emerged as interesting targets.
Fatty acid oxidation is profibrogenic
Fatty acids are oxidized in mitochondria to provide ATP and NADH to facilitate gluconeogenesis and generate acetyl CoA (Figure 2). Long-chain fatty acid β-oxidation is governed by the regulated translocation of activated fatty acids (acyl-CoAs) from the cytoplasm to the matrix of the mitochondria. This is mediated by successive acyltransferases carnitine palmitoyl transferases 1 and 2 (CPT1 and CPT2) (Figure 2). Both pharmacological and global genetic inhibition of CPT1A blunt HSC activation by reducing mitochondrial activity. In isolated HSC, it has been shown that fatty acid oxidation (FAO) provides energy demand to decrease lipid droplets during activation. In keeping, mice with specific deletion of cpt1a in HSCs are protected against liver fibrosis.39 Interestingly, increasing FAO in hepatocytes by either overexpressing cpt1 or exposure with oleic acid leads to ROS-induced activation of fibrogenic genes in HSC, suggesting that FAO in hepatocytes may also contribute to fibrogenesis during chronic liver injury.39
TCA cycle
Several TCA metabolic intermediates have been involved in the regulation of liver fibrosis, including succinate or acetoacetate (Figure 2).
Succinate. Succinate is as a classical mitochondrial TCA intermediate metabolite formed from succinyl-CoA by succinyl-CoA synthetase and subsequently oxidized to fumarate by succinate dehydrogenase, which is part of the respiratory electron transport chain (Figure 2). However, the fibrogenic properties of succinate mainly rely on succinate secretion in the extracellular space, where it functions as a metabolic stress signal sensed by a Gi-coupled receptor succinate receptor 1 or GRP91. In vitro studies indicate that the succinate/GRP91 pathway is fibrogenic by direct or indirect activation of HSC. In HSC exposed to succinate, or in which intracellular succinate levels are increased by either palmitate or an inhibitor of succinate dehydrogenase, fibrogenesis is increased through a GRP91-dependent pathway.40 In addition, succinate released by stressed hepatocytes enhances the profibrogenic properties of HSC. Interestingly, inhibition of succinate/GPR91 pathway has also shown beneficial antifibrogenic effects in NASH experimental models.41,42 However, further studies should evaluate its impact on inflammation. Indeed, although the succinate/GRP91 pathway is generally considered as a proinflammatory driver as it potentiates T-cell activation and switches macrophage phenotype toward a proinflammatory profile,43,44 conflicting results have reported anti-inflammatory effects on macrophages.45,46
Acetoacetate. Acetoacetate is a ketone body produced by the liver that attenuates liver fibrogenesis on systemically administration. Succinyl-CoA-oxoacid transferase, which mediates terminal acetoacetate oxidation within the TCA cycle, is highly expressed in macrophages but not in hepatocytes. Consequently, hepatocytes cannot metabolize ketones, and the acetoacetate released by the hepatocytes is oxidized in alternatively polarized macrophages.47 This hepatocyte-macrophage shuttle seems detrimental to protect the liver against fibrogenesis since mice lacking scot specifically in macrophages are prone to excessive extracellular matrix production and fibrosis development under a high-fat diet.47 Moreover, acetoacetate preserves macrophage mitochondrial function from proinflammatory lactic acidosis.48
Mitochondrial carriers
Mitochondrial pyruvate carrier (MPC). Transport of pyruvate generated in the cytosol into the mitochondrial matrix is critical in the TCA cycle to produce acetyl CoA or oxaloacetate. Pyruvate transport is achieved by an inner MPC, which recently emerged as a potential antifibrogenic target. Indeed, specific deletion of mpc2 in hepatocytes, one of the MPC protein component, limits hepatocyte injury in a NASH model and reduces liver fibrosis, through a paracrine inhibition of HSC fibrogenic features.49
Solute mitochondrial carriers. Members of the solute mitochondrial carrier family facilitate the transport of molecules involved in a variety of metabolic processes across the inner membrane of mitochondria. Among solute carrier 25 family members, SLC25A47 is specifically expressed in the liver, within hepatocytes, and its deletion leads to hepatic metabolic dysregulation, impaired mitochondrial respiration, and increased stress response. Recent data indicate that specific deletion of slc25 in hepatocytes leads to enhanced collagen accumulation and macrophage infiltration.50 Whether the fibrogenic consequences of hepatocyte mitochondrial metabolism are mediated through a direct cross talk with HSC and/or macrophages remains to be elucidated, but these data further argue for a central role of hepatocyte mitochondrial dysfunction in liver fibrogenesis.
Targeting amino acid metabolism: glutaminolysis is profibrogenic
The role of amino acid metabolism in the control of liver fibrogenesis is still poorly characterized and mainly restricted to the glutaminolytic pathway. Glutaminolysis is a source of ATP in a 2-step reaction: glutamate is generated from glutamine by glutaminases and then metabolized into α-ketoglutarate that will fuel the TCA cycle (Figure 2). Enhanced hepatic glutaminolytic activity has been observed in livers from patients with NASH,51 and increase in glutaminolysis byproducts such as a-ketoglutarate, fumarate, or citrate correlates with fibrosis severity.51 Interestingly, pharmacological blockade of glutaminase, ie, inhibition of glutaminolysis, reduces fibrosis progression through its impact on HSC and hepatocytes13,52 (Figure 3). Thus, inhibition of glutaminolysis blocks HSC activation, and mirroring this observation, glutaminase 1 increases during transdifferentiation of HSC into myofibroblasts.53 Along the same line, alcohol exposure stimulates glutamate production by hepatocytes, leading to the release of steatogenic/fibrogenic mediators by neighboring HSC.54,55 The impact of glutaminolysis inhibition on macrophage phenotype remains to be evaluated in the context of liver fibrosis, in light of studies in bone marrow-derived macrophages showing that the production of alpha-ketoglutarate through glutaminolysis shifts macrophages toward a wound healing phenotype.56
Targeting connecting metabolic hubs
YAP/TAZ as a connecting fibrogenic hub between metabolic pathways?
Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) modulate glycolysis by upregulating glucose transporter protein type 1 and genes involved in glucose metabolism. They also promote glutaminolysis and are activated by monounsaturated fatty acids, therefore providing a link to lipid metabolism. The profibrogenic role of the YAP/TAZ pathway is increasingly recognized (Figure 3), but whether it is mediated by metabolic modifications in liver cells has yet to be further explored. Verteporfin or acid ceramidase inhibitors reduce fibrosis development in toxic liver injury mouse models through YAP/TAZ signaling inhibition.57,58 In activated HSC, inhibitors of YAP revert their phenotype by blocking glutaminolysis.13,59 In hepatocytes, the profibrogenic role of YAP has been demonstrated in mouse models of hepatocyte-specific yap overexpression or downregulation.60,61 In keeping, in a mouse model of NASH-induced fibrosis, increase and internalization of cholesterol in hepatocytes leads to upregulation of taz and promotes fibrosis.62 A macrophage-specific deficiency in yap attenuates liver fibrosis by dampening activation of NLRP3 inflammasome in myeloid cells.63,64 Finally, YAP activation in LSECs governs HIF-1α and VEGF-A expressions and promotes angiogenesis, a process exacerbating liver injury and fibrosis.65 Interestingly, blocking the cross talk between YAP-deficient macrophages and endothelial cells attenuates liver fibrosis in rodent and minipig NASH models and promotes liver regeneration.63
Autophagy as an antifibrogenic pathway?
Autophagy plays a central role in the control of fibrogenesis66,67 (Figure 3). Indeed, autophagy regulates cellular metabolism and energy, degrades proteins involved in lipid or glucose metabolism, refueling amino acid pool or removing damaged organelles and lipid droplets, and controls immune cell phenotype, in particular macrophages. In humans, loss-of-function mutations in ATG7 (P426L variant) increase the risk of developing severe NAFLD and predispose to cirrhosis and HCC.68 Pharmacological activation of autophagy by the mammalian target of rapamycin inhibitor rapamycin or by a neutralizing monoclonal antibody to the autophagy inhibitory protein acyl-CoA–binding protein/diazepam-binding inhibitor) reduces liver fibrosis,69–71 demonstrating that global targeting of autophagy in the liver may be considered for antifibrogenic therapy.66,67 In keeping, data from mice with specific deletion of autophagic genes in hepatocytes, myeloid cells, and endothelial cells show that autophagy deficiency promotes or exacerbates liver fibrosis, whereas conflicting results have been obtained in HSC. Indeed, invalidation of Atg5 Atg7 or vmp1 specifically in hepatocytes spontaneously promotes liver fibrosis, whereas Atg5 or Atg7 deficiency in endothelial cells potentiates chronic toxic liver injury.72–74 In addition, deletion of Atg5 in myeloid cells polarizes macrophages toward an inflammatory phenotype and exacerbates inflammation-driven liver fibrosis.75 This phenotype could result from activation of a noncanonical autophagic pathway (ie, LC3-associated phagocytosis), since the deleterious Atg5 KO phenotype is also reproduced on inactivation of the LAP protein rubicon in myeloid cells.76 Regarding HSC, recent findings demonstrate that autophagy in HSC inhibits the release of fibrogenic extracellular vesicles with antifibrogenic consequences.77 However, the beneficial role of autophagy in these cells merits further investigation, since lipophagy-induced β-oxidation of free fatty acids is required for lipid droplet disappearance associated with HSC activation.78,79
Nuclear receptors
Nuclear receptors are at the crossroad between cellular metabolism and inflammation, binding directly to DNA to regulate the transcription of a large variety of genes. Their natural ligands are small lipophilic molecules and include bile acids, oxysterols, fatty acids, and cholesterol.80
Liver X receptors (LXR) regulate the metabolism of cholesterol and bile acids through 2 isoforms, LXRα, which is expressed in hepatocytes, KCs, and endothelial cells, and LXRβ, mostly expressed in HSC. LXR deficiency favors fibrogenesis in mice,81 whereas LXR activation displays antifibrogenic properties, decreasing activation of HSC, suppressing the release of inflammatory mediators in liver macrophages, and reducing endothelial cell capillarization (Figure 3).81–83 However, contradictory results have been obtained with LXR inverse synthetic agonists that have shown antifibrotic effects in various NASH and alcohol-associated liver disease mouse models.84–86 Moreover, global LXR activators also induce side effects assumed to be due to the steatogenic properties of LXRα.87 Therefore, the respective contribution of LXRα and LXRβ in steatosis and fibrogenesis needs to be further explored before designing selective or pan-agonists.
Farnesoid X receptors (FXR) regulate bile acids, lipids, and glucose metabolism. Mice with global FXR knockout show spontaneous steatosis, insulin resistance, and hypertriglyceridemia and are more susceptible to inflammation. FXR is mainly expressed in hepatocytes and poorly in HSCs. FXR agonists have anti-inflammatory and antifibrotic effects in animal and clinical studies.88–93 The beneficial effects of FXR agonists rely on their combined impact on hepatocytes, endothelial cells, and macrophages. In hepatocytes, FXR agonists increase cholesterol but repress genes involved in triglycerides synthesis, as well as inhibit lipogenesis and neoglucogenesis94 FXR agonists also improve endothelial dysfunction and reduce proinflammatory gene expression in macrophages.91,95
Peroxisome proliferator-activated receptors (PPARs) include 3 isoforms (α, β/δ, and γ) differentially expressed in liver cells: PPARβ/δ is ubiquitously expressed, PPARα is mainly found in hepatocytes and not in HSCs, and PPARγ is mainly expressed in macrophages and HSCs.96 The respective role of each PPAR isoforms was initially evaluated in animal models. Regarding PPARα agonists or ligands, decrease or even reversal of liver fibrosis was reported97,98 (Figure 3). This beneficial effect resulted from combined effects on hepatocytes and nonparenchymal cells. Indeed, hepatocyte-specific deletion of pparα exacerbates NAFLD and liver inflammation in mice under a high-fat regimen99 activation of PPARα in KCs limits cytokine release and prevents accumulation of lipids in hepatocytes through blockade of IL1β release100 and leads to improvement of endothelial functions.101,102 Results are more contrasted with PPARβ/δ agonists; some data show reduced hepatotoxicity, anti-inflammatory, and antifibrotic impact in experimental mouse models of toxic liver injury103 while others showed HSC proliferation and enhanced liver fibrosis (Figure 3).104,105 Finally, PPARγ displays antifibrotic properties targeting both HSC, through maintenance of their quiescence or even reversal of their phenotype toward quiescence106,107 and macrophages, in which PPARγ decreases their inflammatory response in vitro and in vivo107–109 (Figure 3). The description of beneficial antifibrogenic effects of drugs selectively targeting each PPAR isoforms led to the development of pan-PPAR agonists that demonstrated preventive and curative antifibrotic effects in various mouse models of liver fibrosis (high-fat/high-sucrose diet, choline-deficient, amino acid-defined high-fat diet) and chronic carbon tetrachloride administration96,110 and are currently evaluated in phase III clinical trials (see below).
Clinical trials targeting metabolism for antifibrogenic therapy: where are we?
There is still no current approved pharmacotherapy for liver fibrosis, although a sustained effort has been made in drug development targeting metabolism for patients with NASH. The main classes of compounds that target intrinsic cellular metabolism have been investigated and include agonists for PPAR, FXR, glucagon like peptide 1, and fibroblast growth factor (FGF) and inhibitors for SCD1, ACC1, and sodium glucose transporter 1/2 as reviewed in Mantovani et al.111 While drugs used in monotherapy often gave disappointed results in clinical trials for fibrosis in patients with NASH, using combination of drugs targeting different metabolic pathways is now considered as a more promising option.112,113
Among drugs used in monotherapy, the PPARα/δ agonist elafibranor showed no decrease in liver stiffness and the PPARγ agonist rosiglitazone showed no amelioration in liver fibrosis following 1- or 2-year treatment.114–116 However, treatment with pioglitazone, another PPARγ agonist, for up to 24 months was associated with reversal of advanced fibrosis stage independently of the presence of diabetes.117 The pan-PPAR agonist lanifibranor seems also promising, since in phase II studies, improvement of not only NASH markers but also fibrosis was observed, allowing to go through a phase III clinical trial, of which results are expected in 2024.118 Although encouraging results were initially obtained with the FXR agonist obeticholic acid with antifibrotic effects in the interim analysis in the intention to treat for 22% of patients versus 12% in the placebo group,119 the clinical trial assay was halted since the other end point (NASH reduction with no worsening of fibrosis) was not reached.
Finally, drugs targeting FGF19, which inhibits the de novo synthesis of bile acids and improves energy homeostasis, or FGF21, which regulates glucose and lipid homeostasis, are currently under investigation in phase II studies, with preliminary data indicating improvement of fibrosis for a long-acting Fc-FGF21 fusion protein efruxifermin but not for the FGF agonist aldafermin.112,120,121
Combination therapy seems a more promising approach. Indeed, the FXR agonist cilofexor combined with the ACC inhibitor firsocostat reduces liver stiffness assessed by transient elastrography.122 In the same line, a combination of these 2 compounds with the antidiabetic glucagon-like peptide 1 agonist semaglutide indicated greater improvement of fibrosis in noninvasive tests than monotherapies.123 Moreover, based on the promising efficacy on multiple noninvasive markers of NASH, an ongoing study will assess the effects of the sodium glucose cotransporter 2 inhibitor licogliflozin, in combination with the FXR agonist tropifexor (ELIVATE, ClinicalTrials.gov identifier NCT04065841). Finally, future studies evaluating combination therapy with drugs targeting immune cells should also be considered.112,113
METABOLIC REGULATION OF LIVER REGENERATION
Liver regeneration is required to restore liver mass and maintain liver functions during wound healing. During this process, proliferation of hepatocytes is associated with HSC activation and macrophage reprogramming toward an inflammatory phenotype. However, on chronic liver injury, the wound healing response is exaggerated and leads to dysfunction of liver regeneration (Figure 1).
Although the hepatocytes are the first liver cells to enter the cell cycle during liver regeneration, they rely on numerous other cell types to replicate. Indeed, within a few minutes after a two-third partial hepatectomy, liver macrophages produce cytokines, such as IL-6 and TNF-α, required for the hepatocytes to acquire a replicative competence and become receptive to growth factors. Accordingly, all metabolic modifications inducing a controlled proinflammatory program in macrophages in the context of fibrosis are supposed to positively impact liver regeneration. Within hepatic microenvironment, HSCs are also important mediators of liver regeneration,124,125 not only through their production of growth factors particularly HGF, and cytokines such as IL-6, but also because they are the main source of metalloproteinases and inhibitors involved in remodeling the extracellular matrix.1,2 LSEC are the paradigmatic cells of the liver regeneration/fibrosis balance.126,127 While involved in the initiation of the regenerative response through the secretion of growth factors such as HGF or Wnt 2, their aberrant activation may shift their response toward fibrosis.126–128
Because they represent 80% of liver cells and are essential for liver function, hepatocytes have been the focus of most studies that try to decipher the role of cell metabolic reprogramming in liver regeneration. The vast majority of hepatocytes is able to enter the cell cycle in response to liver injury in normal conditions. However, growing body of evidence unveiled the heterogeneous proliferative capacity of hepatocytes (for review, see Pu and Zhou129). Interestingly, zonated hepatocytes also differ in their metabolism according to their position in the liver lobule. Periportal hepatocytes display an oxidative metabolism (β-oxidation and amino acid metabolism) while pericentral hepatocytes have essentially a glycolytic metabolism and perform lipogenesis. Of note, proliferating hepatocytes transiently downregulate the expression of metabolic genes.130,131 However, the hepatocyte proliferative capacity is not correlated to their metabolic zonation, cells having the highest proliferative capacity being rather located in the midlobular region under homeostatic conditions.132,133 Liver regeneration is associated with a huge metabolic remodeling of nonparenchymal and parenchymal cells, requiring large amounts of lipids, proteins, and nucleic acids required to reconstitute the liver. Yet, the modulation of liver cell metabolism has been proposed as a strategy to control or modulate hepatocyte proliferation.
Glycolysis and glutaminolysis
Hypoglycemia is the first metabolic manifestation of liver regeneration.134 However, liver regeneration is associated with a reduction in liver glycolytic activity and an induction of gluconeogenesis, potentially compensating hypoglycemia at the expense of ATP production.135,136 Glycolysis is required at the early stages of liver regeneration (Figure 4), with an increased glucose transport through Glut4 and a switch from the glycolytic pyruvate kinase isoform from L to PKM2 nuclear accumulation, which induces hepatocyte proliferation.136,137 It is likely that the proregenerative capacities of glycolysis also rely on macrophage production of proinflammatory cytokines required for liver regeneration.
FIGURE 4.
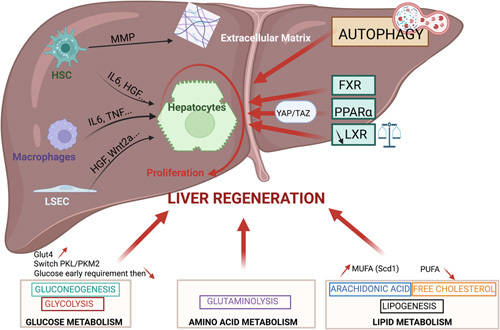
Involvement of intrinsic metabolic pathways in liver regeneration. The consequence of the activation of various metabolic pathways (glycolysis, glutaminolysis, lipogenesis, autophagy, nuclear receptors, and YAP/TAZ) on hepatocyte proliferation either directly or indirectly through the secretion of cytokines, growth factors, or metalloproteinase by nonparenchymal liver cells is shown. Abbreviations: FXR, farnesoid X receptors; HK, hexokinase; MUFA, monounsaturated fatty acids; PPAR, peroxisome proliferator-activated receptors; YAP/TAZ, yes-associated protein and the transcriptional coactivator with PDZ-binding motif.
Proliferating cells have also an increased need for glucogenic amino acids such as glutamine to supply nitrogen and carbon and produce ATP through the TCA cycle. Indeed, glutaminolysis is a hallmark of proliferating cells and enhances the regenerative capacity of the liver138 (Figure 4).
Lipid metabolism
The early regenerating liver transiently accumulates lipids. This transient accumulation of lipids in hepatocytes favors liver regeneration, supposedly in providing phospholipids required for the formation of new cell membranes. However, contradictory results led to the conclusion that a threshold of hepatocyte triglycerides content may probably be required, below which regeneration cannot occur.139 In a NAFLD context, however, a high level of hepatocyte triglyceride content will delay liver regeneration, emphasizing the need for a balanced level of hepatocyte lipids.140,141 This probably also applies to cholesterol, a structural component of biological membranes, whose hepatic levels increase after partial hepatectomy, concomitantly with a LXR transcriptional pathway downregulation required to ensure the required cholesterol levels for hepatocyte proliferation.142 Interestingly, a recent study has reported a change in lipid metabolic pathways engaged when hepatocytes proliferate as compared with resting conditions, with an increase in monounsaturated fatty acids–containing phosphatidyl choline and free cholesterol that provide metabolic energy required for liver regeneration and decreased polyunsaturated fatty acid-containing phosphatidyl choline.143 Accordingly, the expression of SCD1, involved in de novo synthesis of monounsaturated fatty acids, is increased more than 3 times in regenerating liver after major resection and may contribute to liver regeneration by modulating the Wnt pathway. Importantly, lipid reprogramming of hepatocytes during liver regeneration is also a hallmark of liver cancer cells during HCC development.143
Regarding polyunsaturated fatty acid, most of the arachidonic acid metabolites such as prostaglandins, prostacyclin, or thromboxane support hepatocyte proliferation.144–149 In keeping, MAGL displays proregenerative properties in the liver with a direct contribution of MAGL from hepatocytes through eicosanoid production and indirect effects through the release of mitogenic cytokines from macrophages.150
Modulating mitochondrial metabolism
To face the massive energy demand required to promote liver regeneration, several mechanisms providing ATP, including mitochondrial metabolism and autophagy, have been shown to facilitate the regenerative process.
Increased mitochondrial respiration induced by the depletion in its inhibitor methylation-controlled J protein increases ATP production, induces succinate dehydrogenase activity, enhances antioxidant defences, and activates macrophage cytokine production. All these factors contribute to the acceleration of liver regeneration and the reduction of liver damage during repair.151
Activation of mitochondrial β-oxidation also participates to sustained intracellular hepatocyte ATP after partial hepatectomy. Activation of mitochondrial β-oxidation is consecutive to enhanced autophagy, as reflected by an increased number of autophagosomes and enhanced autophagic flux, that allows the removal of damaged mitochondria. In keeping, autophagy-deficient mice (atg5−/−) show impaired liver regeneration with reduced hepatocellular proliferation, increased hepatocyte apoptosis, and senescence (Figure 4). This defect is associated with an altered metabolic response combining reduced ATP production, reduced liver free fatty acid levels, and increased glycemia.152,153
Nuclear receptors PPAR and FXR
PPARα and FXR control liver energy balance.154 FXR promotes liver repair through both direct hepatic-autonomous effects and intestinal expression of FGF15155 (Figure 4). PPARα plays a role in liver regeneration mainly by activating hepatocyte proliferation. Indeed, pharmacological activation of PPARα promotes hepatomegaly and accelerates liver regeneration, while this effect is abolished in hepatocyte-specific PPARα-deficient mice156,157 (Figure 4). This impact of PPARα activation is zonated, increasing hepatocyte size around the central vein and promoting hepatocyte proliferation in the periportal area. Interestingly, it has been recently demonstrated that PPARα interacts with YAP to promote its nuclear translocation. The cell-specific role of the Hippo pathway and its effectors YAP and TAZ in liver regeneration is controversial. Indeed, recent data challenged the commonly accepted idea that YAP/TAZ hepatocyte expression is required for regeneration.158 In contrast, YAP/TAZ biliary expression allows macrophage recruitment and polarization and hampers the activation of pregnane X receptors in hepatocytes.158 However, the fact that verteporfin, a YAP inhibitor, suppresses the hepatomegaly induced by a synthetic PPARα activator suggests that YAP drives the regenerative answer to PPARα agonists157 (Figure 4). It might thus be interesting to test whether PPARα agonists such as bezafibrate, besides its impact on liver fibrosis, could also promote liver regeneration in case of large liver resections.
TARGETING CELL METABOLISM IN HCC
Primary liver cancers define a spectrum of malignancies including HCC, intrahepatic cholangiocarcinomas, and combined tumors (or hepatocholangiocarcinomas), the latter being characterized by the presence of both HCC and intrahepatic cholangiocarcinomas components within the same nodule.159 In this chapter, we focus on HCC as it represents one of the main complications of end-stage chronic liver diseases, ranking as the third-leading cause of cancer-related mortality worldwide with an increasing global prevalence, poor outcome, and innate resistance to chemotherapy.160 HCC results from a sequential process, from fibrosis/cirrhosis to the development of preneoplastic lesions (ie, dysplastic nodules).161 In the context of cirrhosis, normal hepatocyte proliferation is impaired, at least in part because of hepatocyte senescence, in correlation with telomerase inactivation.162 In most HCC, telomerase reactivation is required to ensure uncontrolled cell proliferation. HCC is characterized by a wide heterogeneity, with distinct molecular classes reflecting different biological backgrounds that impact prognosis and response to therapy.163 A multiomics approach also stratified these heterogeneous tumors in distinct subtypes, according to their differences in their metabolic network expression, which might help to identify new targets for drug development.164 However, in addition to this intertumor heterogeneity, the intratumor heterogeneity of HCC complicates the opportunity to find a unique therapeutic approach.
Interactions between liver microenvironmental cells and cancer cells are determinant to stimulate tumor progression, angiogenesis, and invasiveness (Figure 1). Interestingly, the mechanisms by which HSCs impact hepatocarcinogenesis have been recently investigated. However, there are far fewer reports focusing on metabolic reprogramming of cancer-associated fibroblast in HCC than in cholangiocarcinomas or pancreatic cancers, which are characterized by a more prominent desmoplastic fibrous stroma. Recent studies have led to the identification of distinct HSC subpopulations with protumor and tumor-suppressive functions, defined respectively as collagen I-expressing and cytokine (HGF)-expressing HSC.5 The balance between these 2 HSC subpopulations seems to determine the fate of HCC from tumor growth restriction to progression.5
TAM, which mainly originate from circulating monocytes, are among the most abundant cell type in the HCC microenvironment.165 They promote HCC progression through their immunosuppressive activity and by secreting inflammatory mediators that interact with cancer and microenvironmental cells to enhance matrix remodeling, angiogenesis, metastasis, and therapeutic resistance.166
Since cancer cells need to adapt their metabolic activity to generate the energy required for tumor growth, targeting their metabolism emerges as an attractive therapeutic option.167 Indeed, alterations in metabolic pathways and their consequences on HCC progression have been initiated through genome-scale metabolic modeling and further extensively studied in mouse models (reviewed in Satriano et al167). Importantly, modulating metabolic activity of cancer cells may also potentialize sensitivity of tumor cells toward other antineoplastic drugs. Moreover, blocking the dialog between HCC cells and microenvironmental cells and/or modulating their metabolism may provide an additional therapeutic approach for HCC.
Targeting lipid metabolism: far from the goal
The interest to modulate lipid metabolism in liver oncogenesis comes from studies that characterized the modifications of the lipidome in HCC.168,169 Indeed, Li et al169 identified a set of 53 lipids deregulated between human HCC and nontumoral liver, with a specific upregulation of saturated triacylglycerols correlated with the severity of the disease. Levels of monounsaturated palmitic acid, the product of SCD1 activity, were increased in aggressive HCC.170 Moreover, palmitic acid (C16:0) treatment significantly reduced cell proliferation, migration, and invasion in in vitro and in vivo experimental models through a decrease in cell membrane fluidity and inhibition of glucose uptake.168
However, up to now, targeting lipid metabolism in liver cancer cells gave conflicting results, for example, opposite effects of cholesterol depending on the stage of the disease, the targeted cell type, and the HCC etiology.171 Along this line, drugs that lower cholesterol levels such as statins gave controversial results, most of the studies showing a beneficial effect on HCC development and/or recurrence,172–177 while others reported no impact (eg, in NAFLD-associated HCC).178
De novo lipogenesis is also a characteristic feature of cancer cells that supports their proliferation.179 HCC is characterized by upregulation of sterol regulatory element-binding protein 1 and its target lipogenic genes ACLY, ACC, and FASN.179 Again, both beneficial and deleterious results were obtained on HCC development when lipogenesis is restrained with Fasn or ACC inhibitors.180–183 This might reflect the metabolic heterogeneity of HCC164 when stratified based on their metabolic profile, particularly on their levels of FAO. Indeed, targeting FAO may represent a particularly relevant approach in a subset of HCC tumors that harbor β-catenin (ctnnb1) mutations and that use FAO rather than the classical Warburg effect to generate the required energy for tumor growth. In keeping, using a mouse model recapitulating ctnnb1-mutated HCC, Senni et al demonstrated that pharmacological inhibition of FAO with etomoxir was sufficient to abolish tumor initiation and slow down tumor progression. However, the consequences of FAO inhibition might be opposite in other subsets of HCC, since downregulation of CPT2 and the resulting decrease in FAO have been observed in obesity-driven HCC and poorly differentiated HCC. Importantly, CPT2 downregulation participates in liver carcinogenesis and promotes chemoresistance.185 All these data need to be further confirmed in patients but put forward the potential to adapt these metabolic therapeutic approaches to the HCC subtype.
HCC and amino acid metabolism rewiring
In most HCC, the levels of nonessential amino acids, such as glutamine and aspartate levels, are increased while glycine and branched-chain are decreased. Glutamine provides a carbon source for de novo lipogenesis and support tumor cell growth. The inhibition of a rate-limiting component of the mitochondrial oxoglutarate deshydrogenase complex promotes HCC by reprogramming glutamine metabolism.186 Amino acid metabolism also impacts the immune cell function. In this line, it has been shown that enhanced glutamine metabolism facilitates HCC and hampers immunotherapy efficacy by PD-L1 exosome activity.187 Thus, the HCC amino acid metabolic profile may help in defining new therapeutic approaches and treatment sensitivity. Two different molecular HCC subtypes have been recently identified based on their amino acid–related gene expression,188 but this classification needs to be refined in the future.
Targeting glycolysis in liver cancer cells
While aerobic glycolysis is less efficient energetically compared with oxidative phosphorylation, it is a faster process to support rapid growth of cancer cells. In addition, lactate produced by tumor cells will enhance the immunosuppressive functions of myeloid-derived suppressor cell and shift macrophages toward a protumoral TAM phenotype, resulting in sustained tumor growth. Aerobic glycolysis also contributes to tumor aggressiveness, inducing angiogenesis and immune evasion and drug resistance.167 Among the main rate-limiting enzymes of aerobic glycolysis, HKs, and pyruvate kinase are the most promising targets, whereas the contribution of phosphofructokinase is less well characterized.189
Inhibiting HKs as an antitumoral strategy
Among HK, the HK2 isoform is highly upregulated in many fetal tissues and cancers, including HCC, while HK4 (glucokinase), highly expressed in the normal liver, is repressed.190 Immuno-analysis of HK2 expression showed a stepwise increase from normal tissue to cirrhosis, cirrhosis to cirrhosis with dysplasia, and cirrhosis with dysplasia to HCC. These data underlined the potential role of HK2 in the transition between dysplastic nodules and HCC, by providing energy and promoting liver cancer cell survival.190 Interestingly, silencing expression of hk2 using in vitro and in vivo models of liver carcinogenesis through pharmacological (3-bromopyruvate) or genetic approaches demonstrated significant antitumoral effect.191 In addition, hk2 silencing is also able to enhance sensitivity of HCC tumor cells to drugs such as metformin and sorafenib.191 Altogether, these data support the interest to use HK2 inhibitors in patients with HCC. An additional protumoral function for glycolysis in the tumor microenvironment was recently uncovered with the identification of the protumoral role of the HK1 isoform in HSC. Indeed, HK1 is mainly expressed by HSC compared with macrophages or cancer cells. HK1 is secreted by HSC into large extracellular vesicles, which are taken up by HCC cells to promote glycolysis and proliferation, leading to HCC growth192 (Figure 5). Whether HK1 is primarily secreted by a specific subpopulation of HSC, ie, the profibrogenic collagen I-expressing HSC,1 remains to be investigated.
FIGURE 5.
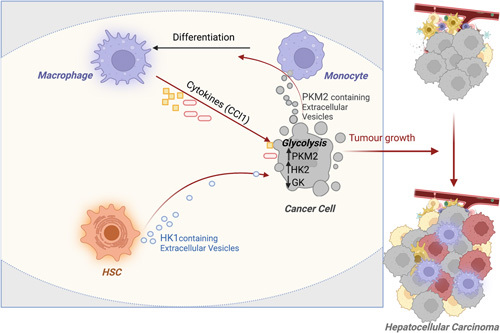
Targeting glycolysis for HCC therapy? Hexokinases (HK1 in HSC and HK2 in tumor cells) and PKM2 facilitate HCC progression through increased glycolysis in tumor cells and by promoting the dialog between tumor and microenvironmental cells. Abbreviations: FXR, farnesoid X receptors; HK, hexokinase; PKM2, pyruvate kinase M2.
Targeting PKM2 for HCC therapy?
In tumor development, a shift in the pyruvate kinase isoform composition is observed in favor of the embryonic M2 isoform, which is expressed in most adult tissues.193 PKM2 overexpression is associated with aggressive clinicopathological features (vascular invasion and presence of satellite nodules) and poor prognosis, with higher 1-year recurrence rate as well as shorter overall survival.194 Preclinical data have highlighted the potential of targeting PKM2 for HCC therapy. Indeed, knockdown of PKM2 leads to reversal of the Warburg effect, with reduction in human HCC cell line proliferation and in HCC tumor growth.194 In addition, the reported nonglycolytic functions of PKM2 in tumor growth, such as binding to histone H3 or regulation of β-catenin expression195,196 could also be considered in the context of HCC. An additional mechanism of the protumoral functions of PKM2 arises from data showing that PKM2 contained in microvesicles secreted from liver cancer cells interacts with monocytes to promote their differentiation into TAM, thereby triggering tumor microenvironment remodeling. In turn, chemokine production by TAM further sustains tumor growth through enhanced release of PKM2 microvesicles from HCC cells197 (Figure 5).
Taken together, these data highlight that targeting glycolysis, through combined inhibition of HK1 and HK2 and/or of PKM2, could provide an interesting antitumoral approach.
CONCLUSIONS
Intrinsic metabolic reprogramming is undoubtedly a characteristic feature of the fibrosis/regeneration/HCC sequence. Inhibition of glycolysis seems the more promising common approach to limit fibrogenesis, abnormal regeneration, and HCC. Nevertheless, given the encouraging antifibrogenic effects obtained in preclinical studies when inhibiting other metabolic pathways, including autophagy, lipogenesis, or glutaminolysis, better delineation of their impact on liver regeneration and HCC at different stages remains to be explored. In addition, whether synergistic effects could be achieved with existing multikinase inhibitors or immunotherapy will be the key challenge of future studies.
Acknowledgments
FUNDING INFORMATION
This work was supported by grants from INSERM (France), the Université Paris Cité, Agence Nationale pour la Recherche (ANR-20-CE14-0038 to Sophie Lotersztajn), and Fondation pour la Recherche Médicale (Equipe FRM EQU202203014642 to Sophie Lotersztajn).
CONFLICTS OF INTEREST
The authors have no conflicts to report.
Footnotes
Abbreviations: ACC, acetyl-coenzyme A carboxylase; ACLY, ATP citrate lyase; CAF, cancer associated fibroblast; CPT, carnitine palmitoyl transferase; FAO, fatty acid oxidation; FGF, fibroblast growth factor; FXR, farnesoid X receptors; GLUT, glucose transporter protein type; HK, hexokinase; LXR, liver X receptors; MAGL, monoacylglycerol lipase; MPC, mitochondrial pyruvate carrier; MUFA, monounsaturated fatty acids; PFK, phosphofructokinase; PKM2, pyruvate kinase isoform M2; PPAR, peroxisome proliferator-activated receptor; SCD1, stearoyl-coA desaturase; TAM, tumor-associated macrophages; TAZ, transcriptional coactivator with PDZ-binding motif; TCA, tricarboxylic acid; YAP, Yes-associated protein.
Contributor Information
Hélène Gilgenkrantz, Email: helene.gilgenkrantz@inserm.fr.
Valérie Paradis, Email: valerie.paradis@aphp.fr.
Sophie Lotersztajn, Email: sophie.lotersztajn@inserm.fr.
REFERENCES
- 1.Lotersztajn S, Julien B, Teixeira-Clerc F, Grenard P, Mallat A. Hepatic fibrosis: molecular mechanisms and drug targets. Annu Rev Pharmacol Toxicol. 2005;45:605–28. [DOI] [PubMed] [Google Scholar]
- 2.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397–411. [DOI] [PubMed] [Google Scholar]
- 3.Sun L, Wang Y, Wang X, Navarro-Corcuera A, Ilyas S, Jalan-Sakrikar N, et al. PD-L1 promotes myofibroblastic activation of hepatic stellate cells by distinct mechanisms selective for TGF-beta receptor I versus II. Cell Rep. 2022;38:110349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capece D, Fischietti M, Verzella D, Gaggiano A, Cicciarelli G, Tessitore A, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int. 2013;2013:187204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filliol A, Saito Y, Nair A, Dapito DH, Yu LX, Ravichandra A, et al. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature. 2022;610:356–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peiseler M, Schwabe R, Hampe J, Kubes P, Heikenwalder M, Tacke F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease —novel insights into cellular communication circuits. J Hepatol. 2022;77:1136–60. [DOI] [PubMed] [Google Scholar]
- 8.Hegde P, Weiss E, Paradis V, Wan J, Mabire M, Sukriti S, et al. Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat Commun. 2018;9:2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14:181–94. [DOI] [PubMed] [Google Scholar]
- 10.Papachristoforou E, Ramachandran P. Macrophages as key regulators of liver health and disease. Int Rev Cell Mol Biol. 2022;368:143–212. [DOI] [PubMed] [Google Scholar]
- 11.Mabire M, Hegde P, Hammoutene A, Wan J, Caer C, Sayegh RA, et al. MAIT cell inhibition promotes liver fibrosis regression via macrophage phenotype reprogramming. Nat Commun. 2023;14:1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Choi SS, Michelotti GA, Chan IS, Swiderska-Syn M, Karaca GF, et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology. 2012;143:1319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, et al. Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology. 2018;154:1465–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilgenkrantz H, Mallat A, Moreau R, Lotersztajn S. Targeting cell-intrinsic metabolism for antifibrotic therapy. J Hepatol. 2021;74:1442–54. [DOI] [PubMed] [Google Scholar]
- 16.Mejias M, Gallego J, Naranjo-Suarez S, Ramirez M, Pell N, Manzano A, et al. CPEB4 increases expression of PFKFB3 to induce glycolysis and activate mouse and human hepatic stellate cells, promoting liver fibrosis. Gastroenterology. 2020;159:273–88. [DOI] [PubMed] [Google Scholar]
- 17.Zheng D, Jiang Y, Qu C, Yuan H, Hu K, He L, et al. Pyruvate kinase M2 tetramerization protects against hepatic stellate cell activation and liver fibrosis. Am J Pathol. 2020;190:2267–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao J, Wang H, Ni M, Wang Z, Wang Z, Wei S, et al. FSTL1 promotes liver fibrosis by reprogramming macrophage function through modulating the intracellular function of PKM2. Gut. 2022;71:2539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greuter T, Yaqoob U, Gan C, Jalan-Sakrikar N, Kostallari E, Lu J, et al. Mechanotransduction-induced glycolysis epigenetically regulates a CXCL1-dominant angiocrine signaling program in liver sinusoidal endothelial cells in vitro and in vivo. J Hepatol. 2022;77:723–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamura YO, Sugama J, Iwasaki S, Sasaki M, Yasuno H, Aoyama K, et al. Selective acetyl-CoA carboxylase 1 inhibitor improves hepatic steatosis and hepatic fibrosis in a preclinical nonalcoholic steatohepatitis model. J Pharmacol Exp Ther. 2021;379:280–9. [DOI] [PubMed] [Google Scholar]
- 21.Kim CW, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X, et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: A bedside to bench investigation. Cell Metab. 2017;26:394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goedeke L, Bates J, Vatner DF, Perry RJ, Wang T, Ramirez R, et al. Acetyl-CoA carboxylase inhibition reverses NAFLD and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology. 2018;68:2197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Farrell M, Duke G, Crowley R, Buckley D, Martins EB, Bhattacharya D, et al. FASN inhibition targets multiple drivers of NASH by reducing steatosis, inflammation and fibrosis in preclinical models. Sci Rep. 2022;12:15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrow MR, Batchuluun B, Wu J, Ahmadi E, Leroux JM, Mohammadi-Shemirani P, et al. Inhibition of ATP-citrate lyase improves NASH, liver fibrosis, and dyslipidemia. Cell Metab. 2022;34:919–36 e918. [DOI] [PubMed] [Google Scholar]
- 25.Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013;19:1649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergman A, Carvajal-Gonzalez S, Tarabar S, Saxena AR, Esler WP, Amin NB. Safety, tolerability, pharmacokinetics, and pharmacodynamics of a liver-targeting acetyl-CoA carboxylase inhibitor (PF-05221304): A three-part randomized phase 1 study. Clin Pharmacol Drug Dev. 2020;9:514–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. 2014;20:1327–33. [DOI] [PubMed] [Google Scholar]
- 28.Bechara R, McGeachy MJ, Gaffen SL. The metabolism-modulating activity of IL-17 signaling in health and disease. J Exp Med. 2021;218:e20202191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Remmerie A, Scott CL. Macrophages and lipid metabolism. Cell Immunol. 2018;330:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Habib A, Chokr D, Wan J, Hegde P, Mabire M, Siebert M, et al. Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver. Gut. 2019;68:522–32. [DOI] [PubMed] [Google Scholar]
- 32.Tardelli M, Bruschi FV, Claudel T, Fuchs CD, Auer N, Kunczer V, et al. Lack of monoacylglycerol lipase prevents hepatic steatosis by favoring lipid storage in adipose tissue and intestinal malabsorption. J Lipid Res. 2019;60:1284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tardelli M, Bruschi FV, Fuchs CD, Claudel T, Auer N, Kunczer V, et al. Monoacylglycerol lipase inhibition protects from liver injury in mouse models of sclerosing cholangitis. Hepatology. 2020;71:1750–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arab JP, Arrese M, Trauner M. Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annu Rev Pathol. 2018;13:321–50. [DOI] [PubMed] [Google Scholar]
- 35.Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, et al. Acyl-CoA:cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J Hepatol. 2014;61:98–106. [DOI] [PubMed] [Google Scholar]
- 36.Teratani T, Tomita K, Suzuki T, Oshikawa T, Yokoyama H, Shimamura K, et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology. 2012;142:152–64. [DOI] [PubMed] [Google Scholar]
- 37.Gan LT, Van Rooyen DM, Koina ME, McCuskey RS, Teoh NC, Farrell GC. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J Hepatol. 2014;61:1376–84. [DOI] [PubMed] [Google Scholar]
- 38.Horn CL, Morales AL, Savard C, Farrell GC, Ioannou GN. Role of cholesterol-associated steatohepatitis in the development of NASH. Hepatol Commun. 2022;6:12–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fondevila MF, Fernandez U, Heras V, Parracho T, Gonzalez-Rellan MJ, Novoa E, et al. Inhibition of carnitine palmitoyltransferase 1A in hepatic stellate cells protects against fibrosis. J Hepatol. 2022;77:15–28. [DOI] [PubMed] [Google Scholar]
- 40.Le CT, Nguyen G, Park SY, Choi DH, Cho EH. LY2405319, an analog of fibroblast growth factor 21 ameliorates alpha-smooth muscle actin production through inhibition of the succinate-G-protein couple receptor 91 (GPR91) pathway in mice. PLoS One. 2018;13:e0192146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li YH, Woo SH, Choi DH, Cho EH. Succinate causes alpha-SMA production through GPR91 activation in hepatic stellate cells. Biochem Biophys Res Commun. 2015;463:853–858. [DOI] [PubMed] [Google Scholar]
- 42.Li YH, Choi DH, Lee EH, Seo SR, Lee S, Cho EH. Sirtuin 3 (SIRT3) regulates alpha-smooth muscle actin (alpha-SMA) production through the succinate dehydrogenase-G protein-coupled receptor 91 (GPR91) pathway in hepatic stellate cells. J Biol Chem. 2016;291:10277–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol. 2008;9:1261–9. [DOI] [PubMed] [Google Scholar]
- 44.Saraiva AL, Veras FP, Peres RS, Talbot J, de Lima KA, Luiz JP, et al. Succinate receptor deficiency attenuates arthritis by reducing dendritic cell traffic and expansion of Th17 cells in the lymph nodes. FASEB J. 2018;32:6550–8; fj201800285. [DOI] [PubMed] [Google Scholar]
- 45.Trauelsen M, Rexen Ulven E, Hjorth SA, Brvar M, Monaco C, Frimurer TM, et al. Receptor structure-based discovery of non-metabolite agonists for the succinate receptor GPR91. Mol Metab. 2017;6:1585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keiran N, Ceperuelo-Mallafre V, Calvo E, Hernandez-Alvarez MI, Ejarque M, Nunez-Roa C, et al. SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat Immunol. 2019;20:581–92. [DOI] [PubMed] [Google Scholar]
- 47.Puchalska P, Martin SE, Huang X, Lengfeld JE, Daniel B, Graham MJ, et al. Hepatocyte-macrophage acetoacetate shuttle protects against tissue fibrosis. Cell Metab. 2019;29:383–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adam C, Paolini L, Gueguen N, Mabilleau G, Preisser L, Blanchard S, et al. Acetoacetate protects macrophages from lactic acidosis-induced mitochondrial dysfunction by metabolic reprograming. Nat Commun. 2021;12:7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCommis KS, Hodges WT, Brunt EM, Nalbantoglu I, McDonald WG, Holley C, et al. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology. 2017;65:1543–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bresciani N, Demagny H, Lemos V, Pontanari F, Li X, Sun Y, et al. The Slc25a47 locus is a novel determinant of hepatic mitochondrial function implicated in liver fibrosis. J Hepatol. 2022;77:1071–82. [DOI] [PubMed] [Google Scholar]
- 51.Du K, Chitneni SK, Suzuki A, Wang Y, Henao R, Hyun J, et al. Increased glutaminolysis marks active scarring in nonalcoholic steatohepatitis progression. Cell Mol Gastroenterol Hepatol. 2020;10:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J, Ghazwani M, Liu K, Huang Y, Chang N, Fan J, et al. Regulation of hepatic stellate cell proliferation and activation by glutamine metabolism. PLoS One. 2017;12:e0182679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bode JG, Peters-Regehr T, Gressner AM, Haussinger D. De novo expression of glutamine synthetase during transformation of hepatic stellate cells into myofibroblast-like cells. Biochem J. 1998;335:697–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi WM, Kim HH, Kim MH, Cinar R, Yi HS, Eun HS, et al. Glutamate signaling in hepatic stellate cells drives alcoholic steatosis. Cell Metab. 2019;30:877–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mallat A, Lotersztajn S. Glutamate signaling in alcohol-associated fatty liver: “Pas de Deux”. Hepatology. 2020;72:350–2. [DOI] [PubMed] [Google Scholar]
- 56.Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18:985–94. [DOI] [PubMed] [Google Scholar]
- 57.Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LF, et al. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol. 2015;63:679–88. [DOI] [PubMed] [Google Scholar]
- 58.Alsamman S, Christenson SA, Yu A, Ayad NME, Mooring MS, Segal JM, et al. Targeting acid ceramidase inhibits YAP/TAZ signaling to reduce fibrosis in mice. Sci Transl Med. 2020;12:eaay8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu HX, Yao Y, Bu FT, Chen Y, Wu YT, Yang Y, et al. Blockade of YAP alleviates hepatic fibrosis through accelerating apoptosis and reversion of activated hepatic stellate cells. Mol Immunol. 2019;107:29–40. [DOI] [PubMed] [Google Scholar]
- 60.Mooring M, Fowl BH, Lum SZC, Liu Y, Yao K, Softic S, et al. Hepatocyte stress increases expression of Yes-associated protein and transcriptional coactivator with PDZ-binding motif in hepatocytes to promote parenchymal inflammation and fibrosis. Hepatology. 2020;71:1813–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salloum S, Jeyarajan AJ, Kruger AJ, Holmes JA, Shao T, Sojoodi M, et al. Fatty acids activate the transcriptional coactivator YAP1 to promote liver fibrosis via p38 mitogen-activated protein kinase. Cell Mol Gastroenterol Hepatol. 2021;12:1297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang X, Cai B, Yang X, Sonubi OO, Zheng Z, Ramakrishnan R, et al. Cholesterol stabilizes TAZ in hepatocytes to promote experimental non-alcoholic steatohepatitis. Cell Metab. 2020;31:969–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qing J, Ren Y, Zhang Y, Yan M, Zhang H, Wu D, et al. Dopamine receptor D2 antagonism normalizes profibrotic macrophage-endothelial crosstalk in non-alcoholic steatohepatitis. J Hepatol. 2022;76:394–406. [DOI] [PubMed] [Google Scholar]
- 64.Wang D, Zhang Y, Xu X, Wu J, Peng Y, Li J, et al. YAP promotes the activation of NLRP3 inflammasome via blocking K27-linked polyubiquitination of NLRP3. Nat Commun. 2021;12:2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang C, Bian M, Chen X, Jin H, Zhao S, Yang X, et al. Oroxylin A prevents angiogenesis of LSECs in liver fibrosis via inhibition of YAP/HIF-1alpha signaling. J Cell Biochem. 2018;119:2258–68. [DOI] [PubMed] [Google Scholar]
- 66.Allaire M, Rautou PE, Codogno P, Lotersztajn S. Autophagy in liver diseases: Time for translation? J Hepatol. 2019;70:985–98. [DOI] [PubMed] [Google Scholar]
- 67.Gual P, Gilgenkrantz H, Lotersztajn S. Autophagy in chronic liver diseases: the two faces of Janus. Am J Physiol Cell Physiol. 2017;312:C263–73. [DOI] [PubMed] [Google Scholar]
- 68.Baselli GA, Jamialahmadi O, Pelusi S, Ciociola E, Malvestiti F, Saracino M, et al. Rare ATG7 genetic variants predispose patients to severe fatty liver disease. J Hepatol. 2022;77:596–606. [DOI] [PubMed] [Google Scholar]
- 69.Zhu J, Wu J, Frizell E, Liu SL, Bashey R, Rubin R, et al. Rapamycin inhibits hepatic stellate cell proliferation in vitro and limits fibrogenesis in an in vivo model of liver fibrosis. Gastroenterology. 1999;117:1198–204. [DOI] [PubMed] [Google Scholar]
- 70.Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–32. [DOI] [PubMed] [Google Scholar]
- 71.Motino O, Lambertucci F, Anagnostopoulos G, Li S, Nah J, Castoldi F, et al. ACBP/DBI protein neutralization confers autophagy-dependent organ protection through inhibition of cell loss, inflammation, and fibrosis. Proc Natl Acad Sci U S A. 2022;119:e2207344119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ni HM, Woolbright BL, Williams J, Copple B, Cui W, Luyendyk JP, et al. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol. 2014;61:617–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hammoutene A, Biquard L, Lasselin J, Kheloufi M, Tanguy M, Vion AC, et al. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J Hepatol. 2020;72:528–38. [DOI] [PubMed] [Google Scholar]
- 74.Jiang X, Fulte S, Deng F, Chen S, Xie Y, Chao X, et al. Lack of VMP1 impairs hepatic lipoprotein secretion and promotes non-alcoholic steatohepatitis. J Hepatol. 2022;77:619–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lodder J, Denaes T, Chobert MN, Wan J, El-Benna J, Pawlotsky JM, et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. 2015;11:1280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wan J, Weiss E, Ben Mkaddem S, Mabire M, Choinier PM, Picq O, et al. LC3-associated phagocytosis protects against inflammation and liver fibrosis via immunoreceptor inhibitory signaling. Sci Transl Med. 2020;12:eaaw8523. [DOI] [PubMed] [Google Scholar]
- 77.Gao J, Wei B, de Assuncao TM, Liu Z, Hu X, Ibrahim S, et al. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J Hepatol. 2020;73:1144–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thoen LF, Guimaraes EL, Dolle L, Mannaerts I, Najimi M, Sokal E, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55:1353–60. [DOI] [PubMed] [Google Scholar]
- 80.Konigshofer P, Brusilovskaya K, Petrenko O, Hofer BS, Schwabl P, Trauner M, et al. Nuclear receptors in liver fibrosis. Biochim Biophys Acta Mol Basis Dis. 2021;1867:166235. [DOI] [PubMed] [Google Scholar]
- 81.Beaven SW, Wroblewski K, Wang J, Hong C, Bensinger S, Tsukamoto H, et al. Liver X receptor signaling is a determinant of stellate cell activation and susceptibility to fibrotic liver disease. Gastroenterology. 2011;140:1052–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xing Y, Zhao T, Gao X, Wu Y. Liver X receptor alpha is essential for the capillarization of liver sinusoidal endothelial cells in liver injury. Sci Rep. 2016;6:21309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hamilton JP, Koganti L, Muchenditsi A, Pendyala VS, Huso D, Hankin J, et al. Activation of liver X receptor/retinoid X receptor pathway ameliorates liver disease in Atp7B(-/-) (Wilson disease) mice. Hepatology. 2016;63:1828–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huang P, Kaluba B, Jiang XL, Chang S, Tang XF, Mao LF, et al. Liver X receptor inverse agonist SR9243 suppresses nonalcoholic steatohepatitis intrahepatic inflammation and fibrosis. Biomed Res Int. 2018;2018:8071093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Griffett K, Welch RD, Flaveny CA, Kolar GR, Neuschwander-Tetri BA, Burris TP. The LXR inverse agonist SR9238 suppresses fibrosis in a model of non-alcoholic steatohepatitis. Mol Metab. 2015;4:353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sengupta M, Griffett K, Flaveny CA, Burris TP. Inhibition of hepatotoxicity by a LXR inverse agonist in a model of alcoholic liver disease. ACS Pharmacol Transl Sci. 2018;1:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Becares N, Gage MC, Voisin M, Shrestha E, Martin-Gutierrez L, Liang N, et al. Impaired LXRalpha phosphorylation attenuates progression of fatty liver disease. Cell Rep. 2019;26:984–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang S, Wang J, Liu Q, Harnish DC. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. 2009;51:380–8. [DOI] [PubMed] [Google Scholar]
- 89.Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004;127:1497–512. [DOI] [PubMed] [Google Scholar]
- 90.McMahan RH, Wang XX, Cheng LL, Krisko T, Smith M, El Kasmi K, et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J Biol Chem. 2013;288:11761–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Verbeke L, Mannaerts I, Schierwagen R, Govaere O, Klein S, Vander Elst I, et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep. 2016;6:33453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fickert P, Fuchsbichler A, Moustafa T, Wagner M, Zollner G, Halilbasic E, et al. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am J Pathol. 2009;175:2392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schwabl P, Hambruch E, Seeland BA, Hayden H, Wagner M, Garnys L, et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J Hepatol. 2017;66:724–33. [DOI] [PubMed] [Google Scholar]
- 94.Evans RM, Mangelsdorf DJ. Nuclear receptors, RXR, and the Big Bang. Cell. 2014;157:255–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jin D, Lu T, Ni M, Wang H, Zhang J, Zhong C, et al. Farnesoid X receptor activation protects liver from ischemia/reperfusion injury by up-regulating small heterodimer partner in Kupffer cells. Hepatol Commun. 2020;4:540–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wettstein G, Luccarini JM, Poekes L, Faye P, Kupkowski F, Adarbes V, et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol Commun. 2017;1:524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39:1286–96. [DOI] [PubMed] [Google Scholar]
- 98.Francque S, Szabo G, Abdelmalek MF, Byrne CD, Cusi K, Dufour JF, et al. Nonalcoholic steatohepatitis: the role of peroxisome proliferator-activated receptors. Nat Rev Gastroenterol Hepatol. 2021;18:24–39. [DOI] [PubMed] [Google Scholar]
- 99.Regnier M, Polizzi A, Smati S, Lukowicz C, Fougerat A, Lippi Y, et al. Hepatocyte-specific deletion of Pparalpha promotes NAFLD in the context of obesity. Sci Rep. 2020;10:6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, et al. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology. 2010;51:511–22. [DOI] [PubMed] [Google Scholar]
- 101.Rodriguez-Vilarrupla A, Lavina B, Garcia-Caldero H, Russo L, Rosado E, Roglans N, et al. PPARalpha activation improves endothelial dysfunction and reduces fibrosis and portal pressure in cirrhotic rats. J Hepatol. 2012;56:1033–9. [DOI] [PubMed] [Google Scholar]
- 102.Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2015;62:720–33. [DOI] [PubMed] [Google Scholar]
- 103.Shan W, Palkar PS, Murray IA, McDevitt EI, Kennett MJ, Kang BH, et al. Ligand activation of peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) attenuates carbon tetrachloride hepatotoxicity by downregulating proinflammatory gene expression. Toxicol Sci. 2008;105:418–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hellemans K, Michalik L, Dittie A, Knorr A, Rombouts K, De Jong J, et al. Peroxisome proliferator-activated receptor-beta signaling contributes to enhanced proliferation of hepatic stellate cells. Gastroenterology. 2003;124:184–201. [DOI] [PubMed] [Google Scholar]
- 105.Kostadinova R, Montagner A, Gouranton E, Fleury S, Guillou H, Dombrowicz D, et al. GW501516-activated PPARbeta/delta promotes liver fibrosis via p38-JNK MAPK-induced hepatic stellate cell proliferation. Cell Biosci. 2012;2:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati-Baroni G, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology. 2002;122:1924–40. [DOI] [PubMed] [Google Scholar]
- 107.Ni XX, Ji PX, Chen YX, Li XY, Sheng L, Lian M, et al. Regulation of the macrophage-hepatic stellate cell interaction by targeting macrophage peroxisome proliferator-activated receptor gamma to prevent non-alcoholic steatohepatitis progression in mice. Liver Int. 2022;42:2696–712. [DOI] [PubMed] [Google Scholar]
- 108.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. [DOI] [PubMed] [Google Scholar]
- 109.Moran-Salvador E, Titos E, Rius B, Gonzalez-Periz A, Garcia-Alonso V, Lopez-Vicario C, et al. Cell-specific PPARgamma deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J Hepatol. 2013;59:1045–53. [DOI] [PubMed] [Google Scholar]
- 110.Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages(). J Hepatol. 2020;73:757–770. [DOI] [PubMed] [Google Scholar]
- 111.Mantovani A, Byrne CD, Targher G. Efficacy of peroxisome proliferator-activated receptor agonists, glucagon-like peptide-1 receptor agonists, or sodium-glucose cotransporter-2 inhibitors for treatment of non-alcoholic fatty liver disease: a systematic review. Lancet Gastroenterol Hepatol. 2022;7:367–78. [DOI] [PubMed] [Google Scholar]
- 112.Dufour JF, Anstee QM, Bugianesi E, Harrison S, Loomba R, Paradis V, et al. Current therapies and new developments in NASH. Gut. 2022;71:2123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wiering L, Tacke F. Treating inflammation to combat non-alcoholic fatty liver disease. J Endocrinol. 2023;256:e220194. [DOI] [PubMed] [Google Scholar]
- 114.Ratziu V, Giral P, Jacqueminet S, Charlotte F, Hartemann-Heurtier A, Serfaty L, et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 2008;135:100–110. [DOI] [PubMed] [Google Scholar]
- 115.Ratziu V, Charlotte F, Bernhardt C, Giral P, Halbron M, Lenaour G, et al. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology. 2010;51:445–53. [DOI] [PubMed] [Google Scholar]
- 116.Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150:1147–59. [DOI] [PubMed] [Google Scholar]
- 117.Musso G, Cassader M, Paschetta E, Gambino R. Thiazolidinediones and advanced liver fibrosis in nonalcoholic steatohepatitis: a meta-analysis. JAMA Intern Med. 2017;177:633–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A randomized, controlled trial of the Pan-PPAR agonist lanifibranor in NASH. N Engl J Med. 2021;385:1547–58. [DOI] [PubMed] [Google Scholar]
- 119.Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394:2184–96. [DOI] [PubMed] [Google Scholar]
- 120.Harrison SA, Ruane PJ, Freilich BL, Neff G, Patil R, Behling CA, et al. Efruxifermin in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled, phase 2a trial. Nat Med. 2021;27:1262–71. [DOI] [PubMed] [Google Scholar]
- 121.Harrison S, Ruane P, Freilich BL, Neff G, Patil R, Behling C, et al. A randomized, double-blind, placebo-controlled phase IIa trial of efruxifermin for patients with compensated NASH cirrhosis. J Hep Rep. 2022;5:100563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Loomba R, Noureddin M, Kowdley KV, Kohli A, Sheikh A, Neff G, et al. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology. 2021;73:625–43. [DOI] [PubMed] [Google Scholar]
- 123.Alkhouri N, Herring R, Kabler H, Kayali Z, Hassanein T, Kohli A, et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: A randomised, open-label phase II trial. J Hepatol. 2022;77:607–18. [DOI] [PubMed] [Google Scholar]
- 124.Kordes C, Sawitza I, Gotze S, Herebian D, Haussinger D. Hepatic stellate cells contribute to progenitor cells and liver regeneration. J Clin Invest. 2014;124:5503–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kitto LJ, Henderson NC. Hepatic stellate cell regulation of liver regeneration and repair. Hepatol Commun. 2021;5:358–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lafoz E, Ruart M, Anton A, Oncins A, Hernandez-Gea V. The endothelium as a driver of liver fibrosis and regeneration. Cells. 2020;9:929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol. 2019;70:1278–91. [DOI] [PubMed] [Google Scholar]
- 129.Pu W, Zhou B. Hepatocyte generation in liver homeostasis, repair, and regeneration. Cell Regen. 2022;11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Walesky CM, Kolb KE, Winston CL, Henderson J, Kruft B, Fleming I, et al. Functional compensation precedes recovery of tissue mass following acute liver injury. Nat Commun. 2020;11:5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chembazhi UV, Bangru S, Hernaez M, Kalsotra A. Cellular plasticity balances the metabolic and proliferation dynamics of a regenerating liver. Genome Res. 2021;31:576–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen F, Jimenez RJ, Sharma K, Luu HY, Hsu BY, Ravindranathan A, et al. Broad distribution of hepatocyte proliferation in liver homeostasis and regeneration. Cell Stem Cell. 2020;26:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wei Y, Wang YG, Jia Y, Li L, Yoon J, Zhang S, et al. Liver homeostasis is maintained by midlobular zone 2 hepatocytes. Science. 2021;371:6532; eabb1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Solhi R, Lotfinia M, Gramignoli R, Najimi M, Vosough M. Metabolic hallmarks of liver regeneration. Trends Endocrinol Metab. 2021;32:731–45. [DOI] [PubMed] [Google Scholar]
- 135.Huang J, Rudnick DA. Elucidating the metabolic regulation of liver regeneration. Am J Pathol. 2014;184:309–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Caldez MJ, Van Hul N, Koh HWL, Teo XQ, Fan JJ, Tan PY, et al. Metabolic remodeling during liver regeneration. Dev Cell. 2018;47:425–38. [DOI] [PubMed] [Google Scholar]
- 137.Hu K, Xu J, Fan K, Zhou D, Li L, Tang L, et al. Nuclear accumulation of pyruvate kinase M2 promotes liver regeneration via activation of signal transducer and activator of transcription 3. Life Sci. 2020;250:117561. [DOI] [PubMed] [Google Scholar]
- 138.Wang L, Zhu L, Wu K, Chen Y, Lee DY, Gucek M, et al. Mitochondrial general control of amino acid synthesis 5 like 1 regulates glutaminolysis, mammalian target of rapamycin complex 1 activity, and murine liver regeneration. Hepatology. 2020;71:643–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rudnick DA, Davidson NO. Functional relationships between lipid metabolism and liver regeneration. Int J Hepatol. 2012;2012:549241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Collin de l’Hortet A, Zerrad-Saadi A, Prip-Buus C, Fauveau V, Helmy N, Ziol M, et al. GH administration rescues fatty liver regeneration impairment by restoring GH/EGFR pathway deficiency. Endocrinology. 2014;155:2545–54. [DOI] [PubMed] [Google Scholar]
- 141.Allaire M, Gilgenkrantz H. The impact of steatosis on liver regeneration. Horm Mol Biol Clin Investig. 2018;41. doi: 10.1515/hmbci-2018-0050 [DOI] [PubMed] [Google Scholar]
- 142.Lo Sasso G, Celli N, Caboni M, Murzilli S, Salvatore L, Morgano A, et al. Down-regulation of the LXR transcriptome provides the requisite cholesterol levels to proliferating hepatocytes. Hepatology. 2010;51:1334–44. [DOI] [PubMed] [Google Scholar]
- 143.Hall Z, Chiarugi D, Charidemou E, Leslie J, Scott E, Pellegrinet L, et al. Lipid remodeling in hepatocyte proliferation and hepatocellular carcinoma. Hepatology. 2021;73:1028–44. [DOI] [PubMed] [Google Scholar]
- 144.Miura Y, Fukui N. Prostaglandins as possible triggers for liver regeneration after partial hepatectomy. A review. Cell Mol Biol Incl Cyto Enzymol. 1979;25:179–84. [PubMed] [Google Scholar]
- 145.Wernze H, Tittor W, Goerig M. Release of prostanoids into the portal and hepatic vein in patients with chronic liver disease. Hepatology. 1986;6:911–6. [DOI] [PubMed] [Google Scholar]
- 146.Tsujii H, Okamoto Y, Kikuchi E, Matsumoto M, Nakano H. Prostaglandin E2 and rat liver regeneration. Gastroenterology. 1993;105:495–9. [DOI] [PubMed] [Google Scholar]
- 147.Rudnick DA, Perlmutter DH, Muglia LJ. Prostaglandins are required for CREB activation and cellular proliferation during liver regeneration. Proc Natl Acad Sci U S A. 2001;98:8885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Minamino T, Ito Y, Ohkubo H, Hosono K, Suzuki T, Sato T, et al. Thromboxane A(2) receptor signaling promotes liver tissue repair after toxic injury through the enhancement of macrophage recruitment. Toxicol Appl Pharmacol. 2012;259:104–14. [DOI] [PubMed] [Google Scholar]
- 149.De Lujan Alvarez ML, Lorenzetti F. Role of eicosanoids in liver repair, regeneration and cancer. Biochem Pharmacol. 2021;192:114732. [DOI] [PubMed] [Google Scholar]
- 150.Allaire M, Siebert M, Al Sayegh R, Mabire M, Wan J, Morein B, et al. Monoacylglycerol lipase reprograms lipid metabolism in macrophages and hepatocytes to promote liver regeneration. J Hepatol. 2020;73:S19–20. [Google Scholar]
- 151.Goikoetxea-Usandizaga N, Serrano-Macia M, Delgado TC, Simon J, Fernandez Ramos D, Barriales D, et al. Mitochondrial bioenergetics boost macrophage activation, promoting liver regeneration in metabolically compromised animals. Hepatology. 2022;75:550–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Toshima T, Shirabe K, Fukuhara T, Ikegami T, Yoshizumi T, Soejima Y, et al. Suppression of autophagy during liver regeneration impairs energy charge and hepatocyte senescence in mice. Hepatology. 2014;60:290–300. [DOI] [PubMed] [Google Scholar]
- 153.Chen Y, Xu Z, Zeng Y, Liu J, Wang X, Kang Y. Altered metabolism by autophagy defection affect liver regeneration. PLoS One. 2021;16:e0250578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Preidis GA, Kim KH, Moore DD. Nutrient-sensing nuclear receptors PPARalpha and FXR control liver energy balance. J Clin Invest. 2017;127:1193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhang L, Wang YD, Chen WD, Wang X, Lou G, Liu N, et al. Promotion of liver regeneration/repair by farnesoid X receptor in both liver and intestine in mice. Hepatology. 2012;56:2336–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Xie G, Yin S, Zhang Z, Qi D, Wang X, Kim D, et al. Hepatocyte peroxisome proliferator-activated receptor alpha enhances liver regeneration after partial hepatectomy in mice. Am J Pathol. 2019;189:272–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Fan S, Gao Y, Qu A, Jiang Y, Li H, Xie G, et al. YAP-TEAD mediates PPAR alpha-induced hepatomegaly and liver regeneration in mice. Hepatology. 2022;75:74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Verboven E, Moya IM, Sansores-Garcia L, Xie J, Hillen H, Kowalczyk W, et al. Regeneration defects in Yap and Taz mutant mouse livers are caused by bile duct disruption and cholestasis. Gastroenterology. 2021;160:847–62. [DOI] [PubMed] [Google Scholar]
- 159.Torbenson MS, Ng IOL, Park YN, Roncalli M MS. Hepatocellular Carcinoma WHO Classification of Tumours: Digestive System Tumours, 5th ed. International Agency for Research on Cancer; 2019:229–39. [Google Scholar]
- 160.Llovet JM, De Baere T, Kulik L, Haber PK, Greten TF, Meyer T, et al. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2021;18:293–313. [DOI] [PubMed] [Google Scholar]
- 161.Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16:589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nault JC, Ningarhari M, Rebouissou S, Zucman-Rossi J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat Rev Gastroenterol Hepatol. 2019;16:544–58. [DOI] [PubMed] [Google Scholar]
- 163.Rebouissou S, Nault JC. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J Hepatol. 2020;72:215–29. [DOI] [PubMed] [Google Scholar]
- 164.Bidkhori G, Benfeitas R, Klevstig M, Zhang C, Nielsen J, Uhlen M, et al. Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc Natl Acad Sci U S A. 2018;115:E11874–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8. [DOI] [PubMed] [Google Scholar]
- 166.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Satriano L, Lewinska M, Rodrigues PM, Banales JM, Andersen JB. Metabolic rearrangements in primary liver cancers: cause and consequences. Nat Rev Gastroenterol Hepatol. 2019;16:748–66. [DOI] [PubMed] [Google Scholar]
- 168.Lin L, Ding Y, Wang Y, Wang Z, Yin X, Yan G, et al. Functional lipidomics: Palmitic acid impairs hepatocellular carcinoma development by modulating membrane fluidity and glucose metabolism. Hepatology. 2017;66:432–48. [DOI] [PubMed] [Google Scholar]
- 169.Li Z, Guan M, Lin Y, Cui X, Zhang Y, Zhao Z, et al. Aberrant lipid metabolism in hepatocellular carcinoma revealed by liver lipidomics. Int J Mol Sci. 2017;18:2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Budhu A, Roessler S, Zhao X, Yu Z, Forgues M, Ji J, et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology. 2013;144:1066–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhou F, Sun X. Cholesterol metabolism: a double-edged sword in hepatocellular carcinoma. Front Cell Dev Biol. 2021;9:762828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology. 2013;144:323–332. [DOI] [PubMed] [Google Scholar]
- 173.Kim G, Jang SY, Nam CM, Kang ES. Statin use and the risk of hepatocellular carcinoma in patients at high risk: A nationwide nested case-control study. J Hepatol. 2018;68:476–84. [DOI] [PubMed] [Google Scholar]
- 174.Miura K, Ohnishi H, Morimoto N, Minami S, Ishioka M, Watanabe S, et al. Ezetimibe suppresses development of liver tumors by inhibiting angiogenesis in mice fed a high-fat diet. Cancer Sci. 2019;110:771–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.German MN, Lutz MK, Pickhardt PJ, Bruce RJ, Said A. Statin use is protective against hepatocellular carcinoma in patients with nonalcoholic fatty liver disease: A case-control study. J Clin Gastroenterol. 2020;54:733–40. [DOI] [PubMed] [Google Scholar]
- 176.Tran KT, McMenamin UC, Coleman HG, Cardwell CR, Murchie P, Iversen L, et al. Statin use and risk of liver cancer: Evidence from two population-based studies. Int J Cancer. 2020;146:1250–60. [DOI] [PubMed] [Google Scholar]
- 177.Zhang J, Fu S, Liu D, Wang Y, Tan Y. Statin can reduce the risk of hepatocellular carcinoma among patients with nonalcoholic fatty liver disease: a systematic review and meta-analysis. Eur J Gastroenterol Hepatol. 2023;35:353–8. [DOI] [PubMed] [Google Scholar]
- 178.Yi SW, Kim SH, Han KJ, Yi JJ, Ohrr H. Higher cholesterol levels, not statin use, are associated with a lower risk of hepatocellular carcinoma. Br J Cancer. 2020;122:630–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Alannan M, Fayyad-Kazan H, Trezeguet V, Merched A. Targeting lipid metabolism in liver cancer. Biochemistry. 2020;59:3951–64. [DOI] [PubMed] [Google Scholar]
- 180.Li L, Che L, Tharp KM, Park HM, Pilo MG, Cao D, et al. Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology. 2016;63:1900–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wang H, Zhou Y, Xu H, Wang X, Zhang Y, Shang R, et al. Therapeutic efficacy of FASN inhibition in preclinical models of HCC. Hepatology. 2022;76:951–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, et al. Inhibition of acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab. 2019;29:174–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nelson ME, Lahiri S, Chow JD, Byrne FL, Hargett SR, Breen DS, et al. Inhibition of hepatic lipogenesis enhances liver tumorigenesis by increasing antioxidant defence and promoting cell survival. Nat Commun. 2017;8:14689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Senni N, Savall M, Cabrerizo Granados D, Alves-Guerra MC, Sartor C, Lagoutte I, et al. beta-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut. 2019;68:322–34. [DOI] [PubMed] [Google Scholar]
- 185.Fujiwara N, Nakagawa H, Enooku K, Kudo Y, Hayata Y, Nakatsuka T, et al. CPT2 downregulation adapts HCC to lipid-rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut. 2018;67:1493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Dai W, Xu L, Yu X, Zhang G, Guo H, Liu H, et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J Hepatol. 2020;72:909–23. [DOI] [PubMed] [Google Scholar]
- 187.Wei Y, Tang X, Ren Y, Yang Y, Song F, Fu J, et al. An RNA-RNA crosstalk network involving HMGB1 and RICTOR facilitates hepatocellular carcinoma tumorigenesis by promoting glutamine metabolism and impedes immunotherapy by PD-L1+ exosomes activity. Signal Transduct Target Ther. 2021;6:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Li Y, Mo H, Jia S, Wang J, Ma Y, Liu X, et al. Comprehensive analysis of the amino acid metabolism-related gene signature for prognosis, tumor immune microenvironment, and candidate drugs in hepatocellular carcinoma. Front Immunol. 2022;13:1066773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Li S, Dai W, Mo W, Li J, Feng J, Wu L, et al. By inhibiting PFKFB3, aspirin overcomes sorafenib resistance in hepatocellular carcinoma. Int J Cancer. 2017;141:2571–84. [DOI] [PubMed] [Google Scholar]
- 190.Guzman G, Chennuri R, Chan A, Rea B, Quintana A, Patel R, et al. Evidence for heightened hexokinase II immunoexpression in hepatocyte dysplasia and hepatocellular carcinoma. Dig Dis Sci. 2015;60:420–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.DeWaal D, Nogueira V, Terry AR, Patra KC, Jeon SM, Guzman G, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. 2018;9:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chen QT, Zhang ZY, Huang QL, Chen HZ, Hong WB, Lin T, et al. HK1 from hepatic stellate cell-derived extracellular vesicles promotes progression of hepatocellular carcinoma. Nat Metab. 2022;4:1306–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:969–80. [DOI] [PubMed] [Google Scholar]
- 194.Wong CC, Au SL, Tse AP, Xu IM, Lai RK, Chiu DK, et al. Switching of pyruvate kinase isoform L to M2 promotes metabolic reprogramming in hepatocarcinogenesis. PLoS One. 2014;9:e115036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, et al. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480:118–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hou PP, Luo LJ, Chen HZ, Chen QT, Bian XL, Wu SF, et al. Ectosomal PKM2 promotes HCC by inducing macrophage differentiation and remodeling the tumor microenvironment. Mol Cell. 2020;78:1192–206. [DOI] [PubMed] [Google Scholar]