

Abstract
The assessment of cellular anti-viral immunity is often hampered by the limited availability of adequate samples, especially when attempting simultaneous, high-resolution determination of T cell responses against multiple viral infections. Thus, the development of assay systems, which optimize cell usage, while still allowing for the detailed determination of breadth and magnitude of virus-specific cytotoxic T lymphocyte (CTL) responses, is urgently needed. This study provides an up-to-date listing of currently known, well-defined viral CTL epitopes for HIV, EBV, CMV, HCV and HBV and describes an approach that overcomes some of the above limitations through the use of peptide matrices of optimally defined viral CTL epitopes in combination with anti-CD3 in vitro T cell expansion and re-use of cells from negative ELISpot wells. The data show that, when compared to direct ex vivo cell preparations, antigen-unspecific in vitro T cell expansion maintains the breadth of detectable T cell responses and demonstrates that harvesting cells from negative ELISpot wells for re-use in subsequent ELISpot assays (RecycleSpot), further maximized the use of available cells. Furthermore when combining T cell expansion and RecycleSpot with the use of rationally designed peptide matrices, antiviral immunity against more than 400 different CTL epitopes from five different viruses can be reproducibly assessed from samples of less than 10 milliliters of blood without compromising information on the breadth and magnitude of these responses. Together, these data support an approach that facilitates the assessment of cellular immunity against multiple viral co-infections in settings where sample availability is severely limited.
Keywords: Cytotoxic T Cells, HIV, EBV, CMV, HCV, HBV, CTL, epitope, peptide, cell expansion, anti-CD3, ELISpot, peptide matrix
Introduction
Cell-mediated immunity is considered critical for the prevention and control of many viral infections [1-6]. The approaches developed to detect these responses in vitro have evolved over the years and have provided quantitative and qualitative information on virus-specific T cells for a number of viral infections. These assays include, besides others, lymphoproliferative assays using 3H-thymidine incorporation or CFSE staining, limiting dilution precursor-frequency assays for the enumeration of CTL precursor frequencies, intracellular cytokine staining (ICS) and enzyme-linked immunospot (ELISpot) assays [7-10]. Although these assays differ in their minimal cell requirements, the detailed, simultaneous analysis of anti-viral immunity against multiple viral infections is often limited by cell availability, regardless of the assay employed.
The ELISpot assay has become widely used for rapidly assessing cellular immune responses to extensive numbers of antigens while using relatively few cells. A number of studies have also employed peptide matrix approaches, where every antigenic peptide is tested in two peptide pools, so that responses to reactive pools sharing a specific peptide can help to identify the targeted peptide [9,11]. This has reduced the required cell numbers significantly, so that for instance HIV-specific responses can generally be comprehensively assessed using less than 15 × l06 cells [9]. However, despite such advances, the simultaneous enumeration of virus-specific immunity to multiple viral infections still exceeds the required sample size that can routinely be obtained. Sample size may not be of great concern when assessing CTL mediated immune responses against single, small genome viruses such as HIV and HCV, which can be tested in a comprehensive manner using overlapping peptide sets spanning the entire expressed viral genome [9,12]. Nevertheless, such comprehensive approaches are not feasible for larger viruses, such as DNA-based herpesviruses like EBV, CMV and KSHV [4,13]. Instead, immune analyses need either to be restricted to a selected number of specific viral proteins, or to the use of previously defined, optimal CTL epitopes. Responses against such optimally defined epitopes can account for a significant part of the total virus-specific immune responses, especially when they represent immunodominant epitopes covering the most immunogenic proteins of specific viral genomes. For well-studied viruses such as HIV, HCV, EBV and CMV, large sets of such optimally defined CTL epitopes, restricted by common HLA alleles, have been described in the past [14-17], and provide a valuable alternative to measure pathogen-specific CTL responses without the need to synthesize comprehensive peptide sets spanning the entire viral genomes.
The present study describes an algorithm by which matrices of optimally defined CTL epitopes derived from five different human viral infections are used in the same ELISpot assay. As not all wells of the ELISpot plate contain antigens to which the tested PBMCs will respond, there are consistently some wells with cells that have not been stimulated during this first assay. Theoretically, these cells could be recovered from the ELISpot plate before developing it and re-used in subsequent analyses. Indeed, others have suggested the use of "recycled" cells for DNA isolation[18], however, to our knowledge, no data exist on re-using these cells in functional assays. Since the peptide matrix approach is ideally followed by the subsequent confirmation of single targeted peptides present in two corresponding peptide pools, recycled cells from unstimulated ELISpot wells could be used for these assays. Although this second step could be achieved using in vitro expanded cells, for instance by anti-CD3 monoclonal antibody (mAb) stimulation, expanded cells may lose some of the responses compared directly to cells tested ex vivo [19,20]. In addition, the absolute and relative magnitude of responses may be distorted during cell expansion and assays can only be run after prolonged in vitro culture[21,22] Therefore, as long as functionality of recycled cells in secondary assays can be ensured, they may provide a simple way to complete initial ELISpot screenings, yielding reliable information on the magnitude of specific CTL responses. The feasibility of this approach was tested and it was shown that combined use of optimal epitope matrices, in vitro T cell expansion and RecycleSpot can provide relevant immune data on multiple viral infections even when cell availability is severely limited.
Materials and methods
Isolation of fresh PMBCs from whole blood
Whole blood was collected using Citrate Vacutainer tubes (BD, Franklin Lakes, NJ) and peripheral blood mononuclear cells (PBMC) were isolated by Histopaque (Histopaque® 1077, Sigma, St. Louis, MO) density centrifugation as described [9]. Fresh PBMC were either used directly after isolation, after in vitro expansion or after freezing and thawing with and without subsequent in vitro expansion. For in vitro use, cells were re-suspended in R10 medium (RPMI 1640 containg 10% heat inactivated FCS (both Sigma), 2 mM L-glutamine, 50 U/ml penicillin, 50 μg/ml streptomucin and 10 mM HEPES (all Mediatech, Hemdon, VA)) at a concentration of 1 × 106 cells/ml. Cells were thawed using R10 medium containing 50 U/ml DNAse (Deoxyribnuclease I, RNase-free, Sigma), washed twice in the same medium, re-suspended in R10 and incubated at 37°C with 5% CO2 for 3–4 hours before they were counted and re-suspended in R10 at 1 × 106 cells/ml. The thawed cells were then either used directly in ELISpot assays or expanded.
For in vitro expansion, 1 to 5 × 106 PBMC were added to 25 ml culture flasks in 10 ml R10 supplemented with 1 μl of the anti-CD3 specific monoclonal antibody (mAb) 12F6 [23]. Cells were fed twice a week using R10 supplemented with 50 U/ml of recombinant Interleukin 2 (IL-2) for 2 weeks. Before use in ELISpot assays, cells were washed twice in R10 medium and incubated overnight at 37°C with 5% CO2 in the absence of IL-2. This overnight starving step was necessary to eliminate background in the subseqnet ELISpot assay, which was, in our hands, not an issue, regardless of how long the in vitro culture had been maintained.
Design of Optimal Peptide Matrix
A total of 416 optimal epitopes from five different viruses were assembled in 98 different peptide pools and used in 5 peptide matrices each containing peptides from a single virus. The number of pools and total number of peptides contained in each virus-specific peptide matrix are summarized in Table 1. Each peptide was present at a final concentration of 200 μg/ml in the peptide pools. Detailed lists of all optimal epitopes included in this study, along with their sequence and HLA restriction, are given in Tables 2 through 6.
Table 1.
Virus specific peptide matrix design using previously defined HLA class I restricted CTL epitopes
| Virus | Optimal epitopes | No. of peptide pools | Max. no. of peptides per pools |
| HIV | 173 | 29 | 14 |
| EBV | 91 | 23 | 12 |
| CMV | 38 | 13 | 7 |
| HCV | 77 | 19 | 11 |
| HBV | 37 | 14 | 8 |
Table 2.
Optimal HIV-derived HLA class I restricted CTL epitopes
| Protein | HLA Restriction | Sequence | Position |
| gp120 | A02 | RGPGRAFVTI | 311–320 |
| gp120 | A03 | TVYYGVPVWK | 37–46 |
| gp120 | A11l | SVITQACPK | 199–207 |
| gp120 | A24 | LFCASDAKAY | 53–62 |
| gp120 | A29 | SFEPIPIHY | 209–217 |
| gp120 | A30 | HIGPGRAFY | 310–318 |
| gp120 | A32 | RIKQIINMW | 419–427 |
| gp120 | B07 | RPNNNTRKSI | 303–312 |
| gp120 | B08 | RVKEKYQHL | 2–10 |
| gp120 | B1516/Cw04 | SFNCGGEFF | 379–387 |
| gp120 | B3801 | MHEDIISLW | 104–112 |
| gp120 | B35 | VPVWKEATTTL | 42–52 |
| gp120 | B35 | DPNPQEVVL | 77–85 |
| gp120 | B44 | AENLWVTVY | 30–38 |
| gp120 | B51 | LPCRIKQII | 416–424 |
| gp120 | B55 | VPVWKEATTT | 42–51 |
| gp120 | A33 | VFAVLSIVNR | 187–196 |
| gp120 | A33 | EVAQRAYR | 320–327 |
| gp41 | A01 | RRGWEVLKY | 787–795 |
| gp41 | A02 | SLLNATDIAV | 818–827 |
| gp41 | A0205 | RIRQGLERA | 335–343 |
| gp41 | A03/A30 | RLRDLLLIVTR | 775–785 |
| gp41 | A23/A24 | RYLKDQQLL | 591–598 |
| gp41 | A30 | IVNRNRQGY | 704–712 |
| gp41 | A30 | KYCWNLLQY | 794–802 |
| gp41 | A6802 | IVTRIVELL | 782–790 |
| gp41 | B07 | IPRRIRQGL | 843–851 |
| gp41 | B08 | YLKDQQLL | 591–598 |
| gp41 | B08 | RQGLERALL | 848–856 |
| gp41 | B14 | ERYLKDQQL | 589–597 |
| gp41 | B2705 | GRRGWEALKY | 791–799 |
| gp41 | B35 | TAVPWNASW | 611–619 |
| gp41 | B4001 | QELKNSAVSL | 810–819 |
| gp41 | Cw3/Cw15 | RAIEAQQHL | 46–54 |
| p17 | A02 | SLYNTVATL | 77–85 |
| p17 | A03 | KIRLRPGGK | 18–26 |
| p17 | A03 | RLRPGGKKK | 20–28 |
| p17 | A03 | RLRPGGKKKY | 20–29 |
| p17 | A11 | TLYCVHQRI | 84–92 |
| p17 | A24 | KYKLKHIVW | 28–36 |
| p17 | A30 | RSLYNTVATLY | 74–86 |
| p17 | B08 | GGKKKYKL | 24–31 |
| p17 | B08 | ELRSLYNTV | 74–82 |
| p17 | B2705 | IRLRPGGKK | 19–27 |
| p17 | B35 | WASRELERF | 36–44 |
| p17 | B35 | NSSKVSQNY | 124–132 |
| p17 | B4001 | IEIKDTKEAL | 92–101 |
| p17 | B4002 | GELDRWEKI | 11–19 |
| p24 | A0207 | YVDRFYKTL | 164–172 |
| p24 | A11 | ACQGVGGPGHK | 349–359 |
| p24 | A24/B44 | RDYVDRFFKTL | 296–306 |
| p24 | A25 | QAISPRTLNAW | 145–155 |
| p24 | B07 | SPRTLNAWV | 148–156 |
| p24 | B07/B42/B81/Cw8 | TPQDLNTML | 48–56 |
| p24 | B07 | GPGHKARVL | 223–231 |
| p24 | B07 | HPVHAGPIA | 84–92 |
| p24 | B08 | EIYKRWII | 260–267 |
| p24 | B08 | DCKTILKAL | 329–337 |
| p24 | B14 | DRFYKTLRA | 298–306 |
| p24 | B1501 | GLNKIVRMY | 267–277 |
| p24 | B18 | FRDYVDRFYK | 293–302 |
| p24 | B2703 | RRWIQLGLQK | 260–269 |
| p24 | B2705 | KRWIILGLNK | 265–274 |
| p24 | B35 | NPVPVGNIY | 245–253 |
| p24 | B35 | PPIPVGDIY | 254–262 |
| p24 | B39 | GHQAAMQML | 193–201 |
| p24 | B4001 | SEGATPQDL | 176–184 |
| p24 | B4002 | KETINEEAA | 70–78 |
| p24 | B4002 | AEWDRVHPV | 78–86 |
| p24 | B44 | AEQASQDVKNW | 174–184 |
| p24 | B44 | EEKAFSPEV | 28–36 |
| p24 | B52 | RMYSPTSI | 143–150 |
| p24 | B53 | TPYDINQML | 48–56 |
| p24 | B53/B57 | QASQEVKNW | 176–184 |
| p24 | B57 | ISPRTLNAW | 15–23 |
| p24 | B57 | KAFSPEVIPMF | 30–40 |
| p24 | B57 | TSTLQEQIGW | 108–118 |
| p24 | B57 | KAFSPEVI | 30–37 |
| p24 | B58 | TSTLQEQIGW | 108–117 |
| p24 | B58 | TSTVEEQIQW | 108–117 |
| p24 | Cw0I | VIPMFSAL | 36–43 |
| p24 | A25 | ETINEEAAEW | 71–80 |
| p24 | A26 | EVIPMFSAL | 35–43 |
| p15 | A02 | FLGKIWPSYK | 1–10 |
| p15 | B14 | CRAPRKKGC | 42–50 |
| p15 | B4001 | KELYPLTSL | 33–41 |
| p15 | B4002 | TERQANFL | 64–71 |
| Protease | A6802/A74 | ITLWQRPLV | 3–11 |
| Protease | A6802 | DTVLEEMNL | 30–38 |
| Integrase | A30 | KIQNFRVYY | 219–227 |
| Integrase | A03/A11 | AVFIHNFKRK | 179–188 |
| Integrase | B1503 | RKAKIIRDY | 263–271 |
| Integrase | B42 | VPRRKAKII | 260–268 |
| Integrase | B57 | KTAVQMAVF | 173–181 |
| RT | A26 | ETKLGKAGY | 604–612 |
| RT | A02 | ALVEICTEM | 33–41 |
| RT | A02 | VIYQYMDDL | 179–187 |
| RT | A02 | ILKEPVHGV | 309–317 |
| RT | A03 | ALVEICTEMEK | 33–43 |
| RT | A03 | GIPHPAGLK | 93–101 |
| RT | A03/A1 1 | AIFQSSMTK | 158–166 |
| RT | A03 | QIYPGIKVR | 269–277 |
| RT | A03 | KLVDFRELNK | 73–82 |
| RT | A03 | RMRGAHTNDVK | 356–366 |
| RT | A11 | IYQEPFKNLK | 341–350 |
| RT | A11 | QIIEQLIKK | 80–88 |
| RT | B51 | TAFTIPSI | 128–135 |
| RT | B57 | IVLPEKDSW | 244–252 |
| RT | B58 | IAMESIVIW | 375–383 |
| RT | B81 | LFLDGIDKA | 715–723 |
| RT | B1503 | VTDSQYALGI | 651–660 |
| RT | A30 | KQNPDIVIY | 173–181 |
| RT | A30 | KLNWASQIY | 263–271 |
| RT | A30 | RMRGAHTNDV | 356–365 |
| RT | A32 | PIQKETWETW | 392–401 |
| RT | B08 | GPKVKQWPL | 18–26 |
| RT | B1501 | LVGKLNWASQIY | 260–271 |
| RT | B1501 | IKLEPVHGVY | 309–318 |
| RT | B35 | TVLDVGDAY | 107–115 |
| RT | B35 | VPLDEDFRKY | 118–127 |
| RT | B35 | NPDIVIYQY | 175–183 |
| RT | B35 | HPDIVIYQY | 175–183 |
| RT | B4001 | IEELRQHLL | 202–210 |
| RT | B42 | YPGIKVRQL | 271–279 |
| RT | B51 | EKEGKISKI | 42–50 |
| Vpr | A02 | AIIRILQQL | 59–67 |
| Vpr | B07/B81 | FPRIWLHGL | 34–42 |
| Vpr | B51 | EAVRHFPRI | 29–37 |
| Vpr | B57 | AVRHFPRIW | 30–38 |
| Tat | A6801 | ITKGLGISYGR | 39–49 |
| Tat | B1503 | FQTKGLGISY | 38–47 |
| Tat | B53 | EPVDPRLEPW | 2–11 |
| Tat | Cw12 | CCFHCQVC | 30–37 |
| Vif | A03 | RIRTWKSLVK | 17–26 |
| Vif | A03 | HMYISKKAK | 28–36 |
| Vif | A03 | KTKPPLPSVKK | 158–168 |
| Vif | B07 | HPRVSSEVHI | 48–57 |
| Vif | B18 | LADQLIHLHY | 102–111 |
| Vif | B57 | ISKKAKGWF | 31–39 |
| Nef | A02 | PLTFGWCYKL | 136–145 |
| Nef | A02 | VLEWRFDSRL | 180–189 |
| Nef | A03/A11 | QVPLRPMTYK | 73–82 |
| Nef | A03/A11 | AVDLSHFLK | 84–92 |
| Nef | A11 | PLRPMTYK | 75–82 |
| Nef | A24 | RYPLTFGW | 134–141 |
| Nef | A33 | TRYPLTFGW | 133–141 |
| Nef | B07 | FPVTPQVPLR | 68–77 |
| Nef | B07 | FPVTPQVPL | 68–76 |
| Nef | B07 | TPQVPLRPM | 71–79 |
| Nef | B07 | RPMTYKAAL | 77–85 |
| Nef | B07 | TPGPGVRYPL | 128–137 |
| Nef | B07 | RQDILDLWIY | 106–115 |
| Nef | B08 | WPTVRERM | 13–20 |
| Nef | B08 | FLKEKGGL | 90–97 |
| Nef | B1501 | TQGYFPDWQNY | 117–127 |
| Nef | B1501 | RMRRAEPAA | 19–27 |
| Nef | B1503 | WRFDSRLAF | 183–191 |
| Nef | B18/B53 | YPLTFGWCY | 135–143 |
| Nef | B2705 | RRQDILDLWI | 105–114 |
| Nef | B35 | VPLRPMTY | 74–81 |
| Nef | A01/A29/837/857 | YFPDWQNYT | 120–128 |
| Nef | B40 | KEKGGLEGL | 92–100 |
| Nef | B42 | TPGPGVRYPL | 128–137 |
| Nef | B53 | YPLTFGWCF | 135–143 |
| Nef | B57 | HTQGYFPDWQ | 116–125 |
| Nef | B57 | HTQGYFPDW | 116–124 |
| Nef | Cw07 | RRQDILDLWIY | 105–115 |
| Nef | Cw7 | KRQEILDLWVY | 105–115 |
| Nef | Cw8 | AAVDLSHFL | 83–91 |
| Rev | A03 | ERILSTYLGR | 57–66 |
| Rev | B57/B58 | KAVRLIKFLY | 14–23 |
| Rev | Cw05 | SAEPVPLQL | 67–75 |
| Vpu | A33 | EYRKILRQR | 29–37 |
All epitopes were referred from the Los Alamos HIV Immunology Database 2004 [24].
Table 6.
Optimal HBV-derived HLA class I restricted CTL epitopes
| Protein | HLA Restriction | Sequence | Position | Reference |
| Core | A2 | FLPSDFFPSV | 18–27 | [84] |
| Core | A2 | CLTFGRETV | 107–115 | [85] |
| Core | A2 | VLEYLVSFGV | 115–124 | [85] |
| Core | A2/A24 | EYLVSFGVW | 117–125 | [86, 87] |
| Core | A2 | ILSTLPETTV | 139–148 | [86] |
| Core | A33/A68 | STLPETTVVRR | 141–151 | [88] |
| Core | A2 | AILSKTGDPV | 152–161 | [89] |
| Env | A2 | LLDPRVRGL | 131–139 | [85] |
| Env | A2 | VLQAGFFLL | 177–185 | [90] |
| Env | A2 | FLLTRILTI | 183–191 | [91] |
| Env | A2 | SLNFLGGTTV | 201–210 | [92] |
| Env | A2 | FLGGTPVCL | 204–212 | [89] |
| Env | A2 | LLLCLIFLL | 250–258 | [86] |
| Env | A2 | LLCLIFLLV | 251–259 | [92] |
| Env | A2 | LLDYQGMLPV | 260–269 | [92] |
| Env | A2 | LVLLDYQGML | 269–278 | [85] |
| Env | A2 | VLLDYQGML | 270–278 | [85] |
| Env | A2 | LLDYQGMLPV | 271–280 | [85] |
| Env | A2 | WLSLLVPFV | 335–343 | [92] |
| Env | A2 | LLVPFVQWFV | 338–347 | [92] |
| Env | A2 | GLSPTVWLSV | 348–357 | [92] |
| Env | A2 | SIVSPFIPLL | 370–379 | [89] |
| Env | A2 | LLPIFFCLWV | 378–387 | [92] |
| Env | A2 | ILSPFFFLPLL | 382–390 | [85] |
| x-Protein | A2 | VLCLRPVGA | 15–23 | [93] |
| x-Protein | A2 | TLPSPSSSA | 36–44 | [93] |
| x-Protein | A2 | HLSLRGLFV | 52–60 | [93] |
| x-Protein | A2 | VLHKRTLGL | 92–100 | [93] |
| x-Protein | A2 | AMSTTDLEA | 102–110 | [93] |
| x-Protein | A2 | CLFKDWEEL | 115–123 | [93] |
| Pol | A24 | LYSSTVPVF | 62–70 | [90] |
| Pol | A2 | GLSRYVARL | 455–463 | [90] |
| Pol | A2 | YMDDVVLGA | 551–559 | [91] |
| Pol | A2 | FLLSLGIHL | 575–583 | [90] |
| Pol | A24 | KYTSFPWLL | 756–764 | [87] |
| Pol | A2 | ILRGTSFVYV | 773–782 | [91] |
| Pol | A2 | SLYADSPSV | 816–824 | [91] |
ELISpot assay
96-well polyvinylidene plates (Millipore, Bedford, MA), pre-coated overnight with 2 μg/ml of anti-interferon gamma (IFN-γ) mAb 1-D1K (Mabtech, Stockholm, Sweden), were washed six times with sterile phosphate buffered saline (DPBS, no Ca & Mg, Mediatech) containing 1% fetal calf serum (FCS) before use. After washing, 30 μl of R10 were added to each well to avoid drying of the membrane, and 100,000 to 200,00 cells per well were added in 100 μl R10. 100,00 cells/well were used to detect responses to HIV, CMV and EBV, whereas responses to HCV and HBV were tested using 200,000 cells/well. Each peptide was added at a final concentration of 14 μg/ml (both single peptides as well as pools). As a negative control, cells were incubated in medium alone, and PHA was added at a concentration of 1.8 μg/ml to serve as a positive control. Plates were incubated for 16 h at 37°C with 5% CO2 before being developed. After washing six times with PBS, 100 μl of biotinylated anti-IFN-γ mAB 7-B6-1 (0.5 μg/ml, Mabtech) were added and plates were incubated for 1 hour at room temperature (RT). The plates were washed again and incubated with a 1:2000 dilution of streptavidin-coupled alkaline phosphatase (Streptavidin-ALP-PQ Mabtech) for 1 hour at RT in the dark. After washing the plates again, IFN-γ production was detected as dark spots after a short incubation of 10–20 minutes with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate (BioRad, Hercules, CA). The color reaction was stopped by washing plates with tap water and the plates were air-dried before counting using a AID ELISPOT Reader Unit (Autoimmun Diagnostika GmbH, Strassberg, Germany). Results were expressed as spot forming cells (SFC) per million input cells. Thresholds for positive responses were determined as either 5 spots (50 SFC/106 input cells) or as the mean plus 3 standard deviations of negative control wells, whichever was higher.
RecycleSpot
After overnight incubation in a primary ELISpot assay, cells from all wells of the ELISpot plate were transferred to a 96-well round-bottom plate and incubated at 37°C with 5% CO2 while developing the ELISpot assay. Cells from wells without any spots (including negative control wells) were then pooled, counted and used for secondary ELISpot assays. In control experiments, cells corresponding to wells with positive responses were also pooled, washed extensively (>5 times) and re-used in subsequent, secondary ELISpot assays as well. Cells from positive control wells (PHA stimulated) were not used for subsequent assays.
Results
Design of the optimal epitope matrix for five viral infections
To simultaneously test CTL responses against five different viruses with a limited number of PBMCs, a peptide matrix approach was used that included all previously published, well-defined CTL epitopes in HIV, HCV, HBV, EBV and CMV. The total number of described CTL epitopes for these viruses varied from 37 described optimal epitopes in HBV to more than 170 optimal epitopes in HIV. A list of all the optimal epitopes included in the present study is given in Tables 2 through 6, totaling 416 well-defined, HLA class I-restricted CTL epitopes. The included HIV epitopes were derived from the annually updated list of HIV CTL epitopes at the Los Alamos National Laboratory HIV immunology database[24]. For all the other pathogens, the epitopes listed were those for which, to the best of our knowledge, at least one publication existed showing CTL activity against this epitope in at least one infected individual. While the optimal epitopes in HIV, HCV and HBV cover large parts of their respective viral genomes, the epitopes defined in EBV and CMV represent only a portion of the proteins expressed by these viruses. Given the approximately 100 open reading frames in these large-genome viruses, complete representation of all viral proteins can hardly be achieved and most studies on these pathogens have thus focused on a relatively small number of viral proteins, especially concentrating on those containing serological determinants and those characterized by specific viral gene expression patterns. Thus, described EBV and CMV encoded CTL epitopes are derived from eleven and four different viral proteins respectively, whereas the known HIV, HCV and HBV epitopes cover all the viral proteins in these small-genome pathogens.
As the number of described optimal CTL epitopes varies between pathogens, separate peptide matrices were designed for each virus (Table 1). Importantly, the first set of pools ("protein pools") was designed so that the pools contained all the epitopes derived from the same viral protein, whereas the second half of matrix peptide pools contained the epitopes in a non-protein specific composition ("random pools"). This matrix design allowed assessment of the virus specific immune response at different levels of resolution including i) a "total virus" specific response by adding up all the protein pool specific or random peptide pool specific responses, ii) a "protein" specific responses by focusing on single pools containing all the epitopes of a given protein; and iii) upon single peptide confirmation, on a single epitope level, by comparing responses in pools containing the same epitope. Together, the epitope matrix design facilitated the assessment of T cell responses to more than 400 CTL epitopes from five different viruses simultaneously, using less than 10 × 106 PBMCs while still allowing determination of breadth and magnitude of virus-, protein-, and epitope-specific responses for each virus separately.
Moreover, since each epitope is tested twice in different pools, it should reflect the same magnitude of response in each pool, thus the matrix approach provides its own internal control. Additionally, "protein pools" and "random pools" should theoretically yield the same total magnitude of responses since they, as a whole, contain the same set of peptides. To test this, and rule out the possibility that peptide compositions in the different pools interfered with the detection of specific responses, the magnitude of all "protein pool" and "random pool" specific responses were compared in 19 subjects infected with EBV (n = 19) and co-infected with CMV (n = 14), HIV (12), and HCV (9). These analyses showed a statistically highly significant correlation between total magnitudes of responses detected by either set of peptide pools, indicating that the peptide mixtures in the pools sharing a specific response did not significantly impact the detection of the targeted epitope (Figure 1). Of note, for all four viruses analyzed, the "random pools" detected a slightly higher, statistically not significant total virus-specific response than the "protein pools". This is likely due to the presence of highly reactive epitopes which, when tested in the same peptide pool, can exceed the upper detection limit of the ELISpot assay and may thus underestimate the total virus-specific magnitude of responses. This may be more likely for epitopes in "peptide pools" than "random pools" if some proteins elicit generally stronger immune responses than others. A protein pool accumulating strongly reactive epitopes would result in fewer spots than the total of the respective "random pools" containing these epitopes equally distributed and fully quantitative.
Figure 1.
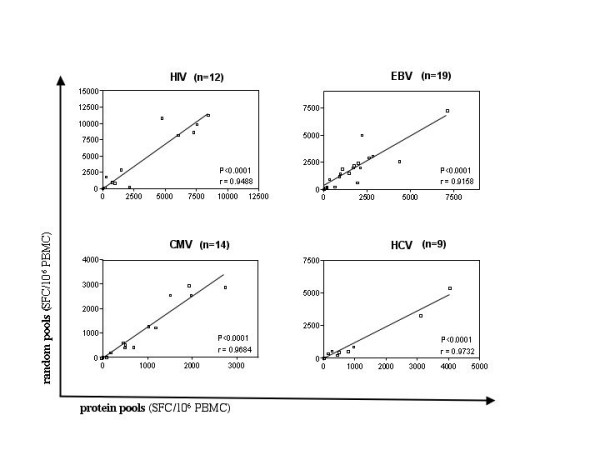
Comparable magnitude of responses detected by "protein" and "random" peptide pools: The magnitude of CTL responses was determined by adding magnitudes for all "protein" or "random" pools for each virus. Responses on the Y-axes represent the total of all virus specific "random pools", the X-axes indicate total responses detected using the "protein pools". Data from 12 HIV-, 19 EBV-, 14 CMV-, 9 HCV-infected individuals were tested against either set of peptide pools for A) HIV, B) HCV, C) EB V, and D) CMV and compared using the non-parametric Wilcoxon matched pairs test.
Cells from negative ELISpot wells can be used in secondary ELISpot assays (RecycleSpot)
In order to maximize cell use in samples with limited cell availability, we investigated whether cells from initial ELISpot matrix screens could be re-used in subsequent functional assays. Specifically, cells from wells that did not respond to peptides added in the first assay as well as the cells in the negative control wells may be used for secondary ELISpot assays. To assess the feasibility of this strategy, all wells from the initial ELISpot plate were transferred to a 96-well plate and incubated at 37°C with 5% CO2 while the ELISpot plate was developed. Cells from negative ELISpot wells were then used to confirm the identity of the epitope(s) targeted in the matrix peptide pools. In separate experiments, cells from initially positive wells were also tested in subsequent assays to determine if continuing IFN-γ production in these cells would prevent them from being used in further ELISpot assays. The analyses also compared ELISpot results in plates that were either undisturbed, or from which cells were transferred for later use.
Representative RecyleSpot assays using PBMC and recovered cells from initial ELISpot assays from three individuals are shown in Figure 2. In all cases, negative wells from initial peptide matrix ELISpot assays were re-used to reconfirm the identity of the presumed, single targeted epitope shared by the two pools. Further, initially positive pools were re-tested to assess whether recycled cells responded with a different magnitude compared to the initial assay. The data show that sufficient cells were recovered from initial assays to perform reconfirmations of single targeted epitopes in the RecycleSpot, and that background activity and magnitude of responses were not significantly different between the first and the subsequent assays. RecycleSpot assays that used initially positive wells, or mixtures of initially positive and negative wells, showed high background in the secondary assay, indicating ongoing IFN-γ production and thus precluding these cells from use in the RecycleSpot (data not shown). No effects on the quality and the number of spots between the manipulated and non-manipulated wells were observed, indicating that harvesting cells from the ELISpot plate did not negatively interfere with the quality of the assay, at least when cells are removed by careful pipetting using a 12-channel pipetor Furthermore, RecycleSpot assays were performed using both fresh and frozen/thawed cells and showed that HIV-and EBV-specific responses were maintained in recycled cells in both cases (data not shown). Together, the data indicate that RecycleSpot can provide sufficient numbers of cells from initial assays and that these cells maintain functional capacity for use in subsequent assays, without raising background activity. Also, the data show that re-using the cells form negative wells after an overnight incubation did not reduce the magnitude of responses to a statistically significant level.
Figure 2.

RecycleSpot using recycled cells for the de-convolution of positive peptide pools: Wells of primary ELISpot and secondary RecycleSpot are shown. Line A shows the data from the initial ELISpot assay, including two positive wells indicating cellular response to EBV peptide pools, three negative and one positive control wells. Line B shows the same outline as in A, this time with recycled cells in a secondary ELISpot analysis and, one separate well, using the predicted targeted epitopes from the matrix analysis. The numbers indicate the spot forming cells per million PBMC.
In vitro expanded T cells mount responses detected in fresh ex vivo PBMC samples
Even though rational optimal epitope matrix design and RecycleSpots may help in reducing the required cell numbers for in vitro analyses, cell availability may still be limiting in settings where only very small biological samples can be obtained. In such instances, investigators have resorted to the use of in vitro expanded cells [19,20,25]. However, despite its potential usefulness in situations of small sample size (e.g. tissue biopsies or small volume peripheral blood samples), relatively little is known on how in vitro expansion impacts magnitude and breadth of detectable responses [20,25]. Furthermore, CTL responses to pathogens like HIV, for which a defect in their proliferative capacity has been shown, may be severely distorted by in vitro expansion, even when stimulated unspecifically [7]. To address this issue and to investigate whether stimulation of PBMC with an anti-CD3 mAb (12F6) expands CTL of different specificity equally well, we tested cells either directly or after expansion against peptide sets of described HIV- and EBV-specific epitopes restricted by the individual's HLA alleles.
These analyses included twelve subjects, of which seven were tested for responses to HIV and EBV epitopes, while the remaining five were tested for EBV-specific responses only (Figure 3).
Figure 3.
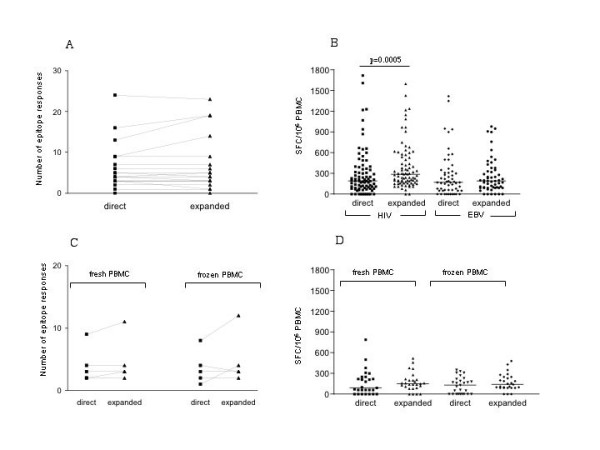
In vitro expansion of thawed cells increases the magnitude and breadth of HIV and EBV specific responses: Thawed PBMC from 12 individuals were tested against HIV and EBV peptide pools (n = 7 subjects) or against EBV peptide pools only (n = 5). Cells were used either directly after thawing or after thawing and a subsequent two-week in vitro expansion using the anti-CD3 mAb 12F6. A) The breadth of the detected responses (number of peptide pools reacting) and B) the total magnitude (sum of all positive peptide pools) is compared between the two cell preparation using the non-parametric Wilcoxon matched pairs test. C) PBMC from 5 EBV infected individuals were used either directly after isolation of after a two-week in vitro expansion or as frozen/thawed cells with and without in vitro expansion, and compared for the breadth (number of pools recognized) and D) total magnitude of the EBV specific responses.
In a first analysis, frozen PBMC were either tested directly or after a 2-week stimulation using 12F6 and the number of targeted HIV or EBV epitopes were compared, resulting in 19 data points (seven individuals tested for HIV and EBV responses and five subjects tested for EBV-specific responses). Flow cytometry in nine individuals showed preferential expansion of CD8 T cells, as CD4 expressing T cells ranged between 0.5% and 14% only, independent of HIV infection and starting CD4 T cell counts (data not shown). The Elispot results revealed no difference in the breadth of responses (number of targeted epitopes) between the directly tested and the expanded cells, as a median of 6.4 and 6.9 positive responses were detected for HIV and EBV, respectively (Figure 3A). The recognition of HIV- and EBV-derived epitopes was equally frequent by the two different cell preparations (data not shown). When the magnitude of responses was compared between directly used and expanded cells, expanded cells responded with a slightly higher magnitude than unexpanded cells. This trend was more prominent when HIV and EBV responses were analyzed separately. The HIV responses in directly tested cells showed a median of 185 SFC/106 PBMC, as compared to 285 SFC/106 PBMC in expanded cells (p = 0.0005); whereas the median EBV-specific responses had a magnitude of 170 SFC/106 in unexpanded PBMC compared to 190 SFC/106PBMC in expanded cells (p > NS).
Moreover, to determine whether freshly isolated cells could also be expanded without drastic changes in their response patterns, PBMC from five EBV-infected subjects were tested directly after isolation, or after freezing, and with or without in vitro expansion. In agreement with the data from frozen samples, no significant difference in the number of pools targeted or the median magnitude of these responses was observed (Figure 3 C and 3 D). Despite concordance among the response patterns between the different cell preparations that was as low as 80%, the overall breadth and magnitude of these responses did not change. In addition, when comparing the magnitudes of the responses between each other, the relative magnitude of the responses was maintained between the four different cell preparations (data not shown). Combined, the data demonstrate that anti-CD3 expanded cells maintain their specificity and relative magnitudes when compared to unexpanded cells (both when used fresh or after thawing) indicating that in vitro expansion could be employed when the breadth, but not the absolute magnitude of responses, is being assessed. This was the case for the assessment of HIV- as well as the EBV-specific responses, suggesting that cells specific for HIV do not significantly differ from EBV specific cells in their ability to undergo in vitro expansion using a non-antigenic stimulus.
Discussion
Cell availability can severely hamper in vitro analyses of antigen specific immune responses, hence approaches which optimize cell use are urgently needed. This is especially true for assays requiring extensive sets of antigens to be tested while only a limited number of cells can be obtained. However, logistic considerations may prevent repetitive sample collection for larger trials, and re-use of fresh or frozen samples could provide more effective ways to perform necessary analyses. The present study introduces a novel approach by which some of the sample limitations can be overcome, and may prove helpful in routine laboratory tests that currently do not make optimal use of available cells. This may not only facilitate currently performed assays, but may open possibilities to expand analyses to simultaneous assessment of even larger sets of antigens and additional functional aspects.
In the present study, we have designed and tested an approach that allows the assessment of the CTL mediated immunity against five different viral infections, including HIV, HCV, HBV, EBV and CMV. We provide an up-to-date listing of currently determined viral epitopes for which the minimal length and HLA restriction have been established. In the case of the small genome viruses HIV and HCV, these optimal epitopes represent a large portion of the respective immune targets [26]. Although they do not include all responses detected in OLP screenings, our comparative analyses of HIV-specific responses from 100 individuals detected by either overlapping peptide (OLP) sets or optimal epitopes show that on average 68% of the observed OLP responses are covered by previously established HIV optimal epitopes (data not shown).
The present data also show that PBMC recycled from negative wells from an ELISpot assay can be re-used for subsequent functional assays. Depending on the analyses performed in the subsequent assay, such as reconfirmation of single epitope responses predicted in the initial matrix analyses, relatively small numbers of cells maybe required. Thus, although individuals with broad responses in the initial ELISpot assay will not yield many negative wells from which to recycle cells, the wells with non-targeted peptide in addition to the negative control wells often provide sufficient quantities of recycled cells to complete the matrix based analyses. Since the responses in the RecycleSpot are not significantly diminished as compared to the initial assay (Figure 2), the magnitude of responses in the subsequent assay can still provide adequate data at the single epitope level.
In vitro expanded cells have been used in a number of studies where cell availability has been the limiting factor[21,22]. However, no study has directly compared for instance biopsy and PBMC-derived responses in a systematic manner and on a single epitope level, and it is unclear whether the in vitro expansion provides identical data. In the present report, we have compared the response patterns to EBV- and HIV-derived antigens in directly ex vivo and in vitro expanded PBMC preparations. No significant differences were observed, although some responses are lost or gained upon expansion. As no difference in the concordance between EBV- and HIV-specific responses was observed, the data indicate that responses to both viruses are equally well expandable in vitro using an antigen-unspecific stimulus, despite the ongoing viral replication in most HIV infected subjects tested here.
Thus, optimal epitope matrices, RecycleSpot and in vitro expansion of cells can be combined to achieve maximal information on an extensive set of antigens, even if sample availability is limited. As a practical approach, expanded cells from frozen PBMC aliquots can be used initially to screen a large number of antigens to determine the approximate breadth of responses within the set of antigens used. Subsequent studies using unexpanded cells and antigen matrices in conjunction with RecycleSpot would then allow determination of the true breadth and, more importantly, the true magnitude of these responses while requiring minimal cell numbers. Furthermore, cells can be successfully recovered from the RecycleSpot once more to be used for genetic analyses such as HLA typing. This combined approach should facilitate future work in settings in which cell availability is of constant concern.
Table 3.
Optimal EBV-derived HLA class I restricted CTL epitopes
| Protein | HLA Restriction | Sequence | Position | Reference |
| BMLF1 | A1 | LVSDYCNVLNKEFT | 25–39 | [27] |
| BMLF1 | A2 | GLCTLVAML | 280–288 | [28] |
| BMLF1 | B18 | DEVEFLGHY | 397–405 | [28] |
| BMLF1 | n.d.* | KDTWLDARM | 265–273 | [29] |
| BMLF1 | A24 | DYNFVKQLF | 320–328 | [30] |
| BHRF | A2 | LLWAARPRL | 204–212 | [31] |
| BZLF1 | B7 | LPCVLWPVL | 44–52 | [13] |
| BZLF1 | B8 | RAKFQLL | 190–197 | [32] |
| BZLF1 | Cw6 | RKCCRAKFKQLLQH | 186–201 | [1] |
| BMRF1 | Cw6 | YRSGIIAW | 268–276 | [16] |
| BMRF1 | Cw3 | FRNLAYGRTCVLGK | 86–100 | [16] |
| BRLF1 | A2 | YVLDHLIVV | 109–117 | [33] |
| BRLF1 | A2 | RALIKTLPRASYSSH | 225–239 | [27] |
| BRLF1 | A3 | RVRAYTYSK | 148–156 | [1] |
| BRLF1 | A11 | ATIGTAMYK | 134–142 | [16] |
| BRLF1 | A24 | DYCNVLNKEF | 28–37 | [27] |
| BRLF1 | A24 | TYPVLEEMF | 198–206 | [30] |
| BRLF1 | B61 | QKEEAAICGQMDLS | 529–543 | [1] |
| BRLF1 | Cw4 | ERPIFPHPSKPTFLP | 393–407 | [1] |
| gp110 | A2 | ILIYNGWYA | 106–114 | [1] |
| gp110 | B35 | VPGSETMCY | 544–552 | [1] |
| gp110 | B35 | APGWLIWTY | 190–198 | [1] |
| gp85 | A2 | TLFIGSHVV | 420–428 | [1] |
| gp85 | A2 | LMPIIPLINV | 542–550 | [1] |
| gp85 | A2 | SLVIVTTFV | 225–233 | [1] |
| gp350 | A2 | VLQWASLAV | 863–871 | [1] |
| gp350 | A2 | VLTLLLLLV | 871–879 | [34] |
| gp350 | A2 | LIPETVPYI | 152–160 | [34] |
| gp350 | A2 | QLTPHTKAV | 67–75 | [34] |
| EBNA1 | A2 | FMVFLQTHI | 562–570 | [13] |
| EBNA1 | B7 | RPQKRPSCI | 72–80 | [35] |
| EBNA1 | B7 | IPQCRLTPL | 528–536 | [35] |
| EBNA1 | B53 | HPVGEADYF | 407–415 | [35] |
| EBNA2 | A2/B51 | DTPLIPLTIF | 42–50 | [36] |
| EBNA3A | A2 | SVRDRLARL | 596–604 | [37] |
| EBNA3A | A3 | RLRAEAQVK | 603–611 | [38] |
| EBNA3A | A24 | RYSIFFDY | 246–253 | [37] |
| EBNA3A | A29 | VFSDGRVAC | 491–499 | [16] |
| EBNA3A | A30 | AYSSWMYSY | 176–184 | [1] |
| EBNA3A | B7 | RPPIFIRRL | 379–387 | [39] |
| EBNA3A | B7 | VPAPAGPIV | 502–510 | [16] |
| EBNA3A | B8 | QAKWRLQTL | 158–166 | [37] |
| EBNA3A | B8 | FLRGRAYGL | 325–333 | [40] |
| EBNA3A | B35 | YPLHEQYGM | 458–466 | [37] |
| EBNA3A | B46 | VQPPQLTLQV | 617–625 | [41] |
| EBNA3A | B62 | LEKARGSTY | 406–414 | [16] |
| EBNA3A | n.d.* | HLAAQGMAY | 318–326 | [16] |
| EBNA3B | A1l | NPTQAPVIQLHAVY | 101–115 | [40] |
| EBNA3B | A1l | AVFDRKSDAK | 399–408 | [16] |
| EBNA3B | A1l | LPGPQVTAVLLHEES | 481–495 | [40] |
| EBNA3B | A1l | DEPASTEPVHDQLL | 551–563 | [40] |
| EBNA3B | Al1 | IVTDFSVIK | 416–424 | [40] |
| EBNA3B | A24 | TYSAGIVQI | 217–225 | [16] |
| EBNA3B | A27 | RRARSLSAERY | 243–253 | [42] |
| EBNA3B | B35 | AVLLHEESM | 488–496 | [1] |
| EBNA3B | B44 | VEITPYKPTW | 657–666 | [16] |
| EBNA3B | B58 | VSFIEFVGW | 279–287 | [43] |
| EBNA3B | B62 | GQGGSPTAM | 831–839 | [16] |
| EBNA3C | B7 | QPRAPIRPI | 881–889 | [39] |
| EBNA3C | B27 | RRIYDLIEL | 258–266 | [44] |
| EBNA3C | B27 | HRCQAIRK | 149–157 | [16] |
| EBNA3C | B27 | FRKAQIQGL | 343–351 | [16] |
| EBNA3C | B27 | RKIYDLIEL | 258–266 | [45] |
| EBNA3C | B27 | RRIFDLIEL | 258–266 | [45] |
| EBNA3C | B27 | LRGKWQRRYR | 249–258 | [44] |
| EBNA3C | B37 | LDFVRFMGV | 285–293 | [46] |
| EBNA3C | B39 | HHIWQNLL | 271–278 | [16] |
| EBNA3C | B44 | KEHVIQNAF | 335–343 | [47] |
| EBNA3C | B44 | EENLLDFVRF | 281–290 | [40] |
| EBNA3C | B44 | EGGVGWRHW | 163–171 | [48] |
| EBNA3C | B62 | QNGALAINTF | 213–222 | [49] |
| EBNALP | A2 | SLREWLLRI | 284–292 | [43] |
| LMP1 | A2 | YLQQNWWTL | 159–167 | [50] |
| LMP1 | A2 | YLLEMLWRL | 125–133 | [50] |
| LMP1 | A2 | LLVDLLWLL | 167–175 | [50] |
| LMP1 | A2 | TLLVDLLWL | 166–174 | [50] |
| LMP1 | A2 | LLLIALWNL | 92–100 | [50] |
| LMP1 | B51 | DPHGPVQLSYYD | 393–404 | [51] |
| LMP2 | A2 | FLYALALLL | 356–364 | [52] |
| LMP2 | A2 | LLWTLWLL | 329–337 | [53] |
| LMP2 | A2 | CLGGLLTMV | 426–434 | [54] |
| LMP2 | A2 | LTAGFLIFL | 453–461 | [53] |
| LMP2 | A11 | SSCSSCPLSKI | 340–350 | [53] |
| LMP2 | A23 | PYLFWLAAI | 131–139 | [55] |
| LMP2 | A2 | LLSAWILTA | 447–455 | [43] |
| LMP2 | A24 | TYGPVFMCL | 419–427 | [53] |
| LMP2 | A24 | IYVLVMLVL | 222–230 | [30] |
| LMP2 | A25 | VMNSNTLLSAW | 442–451 | [16] |
| LMP2 | A27 | RRRWRRLTV | 236–244 | [44] |
| LMP2 | B40 | IEDPPFNSL | 200–208 | [53] |
| LMP2 | B63 | WTLWLLI | 331–338 | [1] |
*not determined
Table 4.
Optimal CMV-derived HLA class I restricted CTL epitopes
| Protein | HLA Restriction | Sequence | Position | Reference |
| pp65 | B35 | IPSINVHHY | 123–131 | [56] |
| pp65 | B35 | DDVWTSGSDSDEELV | 397–411 | [57] |
| pp65 | B35 | VFPTKDVAL | 187–195 | [17] |
| pp65 | B38 | PTFTSQYRIQGKL | 367–379 | [17] |
| pp65 | B7 | TPRVTGGGAM | 417–426 | [57] |
| pp65 | B7 | RPHERNGFTVL | 265–275 | [17] |
| pp65 | A1 | YSEHPTFTSQY | 363–373 | [17] |
| pp65 | A1101 | SVLGPISGHVLK | 13–24 | [17] |
| pp65 | A2402 | FTSQYRIQGKL | 369–379 | [17] |
| pp65 | A68 | FVFPTKDVALP | 186–196 | [17] |
| pp65 | A2 | NLVPMVATV | 495–503 | [57] |
| pp65 | A2 | VLGPISGHV | 14–22 | [58] |
| pp65 | A2 | MLNIPSINV | 120–128 | [58] |
| pp65 | B44 | EFFWDANDIY | 512–521 | [57] |
| pp65 | A2402 | VYALPLKML | 113–121 | [59] |
| pp65 | A2402/Cw0401 | QYDPVAALF | 341–349 | [60, 61] |
| pp65 | B5201 | QMWQARLTV | 155–163 | [62] |
| pp65 | A0207 | RIFAELEGV | 522–530 | [61] |
| pp65 | A1101 | ATVQGQNLK | 501–509 | [61] |
| pp65 | B1501 | KMQVIGDQY | 215–223 | [61] |
| pp65 | B4001 | CEDVPSGKL | 232–240 | [61] |
| pp65 | B40 | HERNGFTVL | 267–275 | [61] |
| pp65 | B4006 | AELEGVWQPA | 525–534 | [61] |
| pp65 | B4403 | SEHPTFTSQY | 364–373 | [61] |
| pp65 | B5101 | DALPGPCI | 545–552 | [61] |
| pp65 | Cw0102 | RCPEMISVL | 7–15 | [61] |
| pp65 | Cw0801 | VVCAHELVC | 198–206 | [61] |
| pp65 | Cw1202 | VAFTSHEHF | 294–302 | [61] |
| pp65 | A33 | SVNVHNPTGR | 91–100 | [63] |
| pp150 | A0301 | TTVYPPSSTAK | 945–955 | [17] |
| pp150 | A68 | QTVTSTPVQGR | 792–802 | [17] |
| IE | B7 | CRVLCCYVL | 309–317 | [64] |
| IE | A2 | YILEETSVM | 315–323 | [65] |
| IE | B18 | ELKRKMIYM | 199–207 | [65] |
| IE | B18 | CVETMCNEY | 279–287 | [65] |
| IE | B18 | DEEDAIVAY | 379–387 | [65] |
| IE | B18 | SDEEEAIVAYTL | 378–389 | [56] |
| GB | A2 | FIAGNSAYEYV | 618–628 | [66] |
Table 5.
Optimal HCV-derived HLA class I restricted CTL epitopes
| Protein | HLA Restriction | Sequence | Position | Reference |
| Core | B60 | GQIVGGVYLL | 28–37 | [67] |
| Core | A0201 | YLLPRRGPRL | 35–44 | [68] |
| Core | B7 | GPRLGVRAT | 41–49 | [69] |
| Core | B44 | NEGCGWAGW | 88–96 | [70] |
| Core | A0201 | DLMGYIPLV | 132–140 | [71] |
| Core | A0201 | ALAHGVRAL | 150–158 | [15] |
| Core | A0201 | LLALLSCLTV | 178–187 | [72] |
| Core | A11 | MSTNPKPQK | 1–9 | [73] |
| P7 | A29 | FYGMWPLLL | 790–798 | [15] |
| P7 | Cw7 | FYGMWPLL | 790–797 | [15] |
| E1 | A0201 | ILHTPGCV | 220–227 | [74] |
| E1 | B35 | NASRCWVAM | 234–242 | [25] |
| E1 | A0201 | QLRRHIDLLV | 257–266 | [74] |
| E1 | A23 | FLVGQLFTF | 285–293 | [15] |
| E1 | A0201 | MMMNWSPTT | 322–330 | [15] |
| E1 | A0201 | SMVGNWAKV | 363–371 | [74] |
| E1 | B35 | CPNSSIVY | 207–214 | [15] |
| E2 | A0201 | SLLAPGAKQNV | 401–411 | [74] |
| E2 | B53 | CRPLTDFDQGW | 460–469 | [69] |
| E2 | B51 | YPPKPCGI | 489–496 | [73] |
| E2 | B60 | GENDTDVFVL | 530–539 | [75] |
| E2 | B50 | CVIGGAGNNT | 569–578 | [73] |
| E2 | A0201 | RLWHYPCTV | 614–622 | [76] |
| E2 | A11 | TINYTIFK | 621–628 | [69] |
| E2 | B60 | LEDRDRSEL | 654–662 | [75] |
| E2 | A2402 | EYVLLLFLL | 717–725 | [77] |
| E2 | B57 | NTRPPLGNWF | 541–550 | [15] |
| NS2 | A29 | MALTLSPY | 827–834 | [25] |
| NS2 | A25 | SPYYKRYISW | 832–841 | [78] |
| NS2 | A23 | YISWCLWWL | 838–845 | [69] |
| NS3 | A24 | AYSQQTRGL | 1031–1039 | [79] |
| NS3 | A0201 | CINGVCWTV | 1073–1081 | [68] |
| NS3 | A0201 | LLCPAGHAV | 1169–1177 | [68] |
| NS3 | A0201 | LLCPSGHAV | 1169–1177 | [68] |
| NS3 | A11 | TLGFGAYMSK | 1261–1270 | [80] |
| NS3 | A0201 | ATLGFGAYM | 1260–1268 | [81] |
| NS3 | A0201 | TLHGPTPLL | 1617–1625 | [81] |
| NS3 | A0201 | TGAPVTYSTY | 1287–1296 | [79] |
| NS3 | A2402 | TYSTYGKFL | 1292–1300 | [77] |
| NS3 | B35 | HPNIEEVAL | 1359–1367 | [82] |
| NS3 | B8 | HSKKKCDEL | 1395–1403 | [69] |
| NS3 | A0201 | KLVALGINAV | 1406–1415 | [68] |
| NS3 | B8 | LIRLKPTL | 1611–1618 | [75] |
| NS3 | A11 | TLTHPVTK | 1636–1643 | [80] |
| NS3 | A68 | HAVGLFRAA | 1175–1184 | [15] |
| NS3 | A0201 | GLLGCIITSL | 1038–1047 | [81] |
| NS4 | A2402 | FWAKHMWNF | 1760–1768 | [77] |
| NS4 | B35 | IPDREVLY | 1695–1712 | [15] |
| NS4 | A24 | VIAPAVQTNW | 1745–1754 | [15] |
| NS4 | B57 | LTTSQTLLF | 1801–1809 | [15] |
| NS4B | A25 | EVIAPAVQTNW | 1744–1754 | [78] |
| NS4B | A25 | ETFWAKHMW | 1758–1766 | [78] |
| NS4B | A0201 | SLMAFTAAV | 1789–1797 | [68] |
| NS4B | A0201 | LLFNILGGWV | 1807–1816 | [72] |
| NS4B | A0201 | ILAGYGAGV | 1851–1859 | [72] |
| NS4B | B37 | SECTTPCSGSW | 1966–1976 | [78] |
| NS4B | B38 | AARVTAIL | 1941–1948 | [75] |
| NS5 | A2 | VLSDFKTWL | 1987–1995 | [83] |
| NS5 | B35 | EPEPDVAVL | 2162–2170 | [15] |
| NS5 | B57 | LGVPPLRAWR | 2912–2921 | [15] |
| NS5A | B60 | HEYPVGSQL | 2152–2160 | [75] |
| NS5A | B35 | PCEPEPDVAVL | 2161–2171 | [75] |
| NS5A | B38 | NHDSPDAEL | 2218–2226 | [75] |
| NS5A | A2 | SPDAELIEANL | 2221–2231 | [75] |
| NS5A | A25 | ELIEANLLW | 2225–2233 | [78] |
| NS5A | A0201 | ILDSFDPLV | 2252–2260 | [68] |
| NS5A | B60 | REISVPAEIL | 2267–2275 | [80] |
| NS5B | A3 | SLTPPHSAK | 2510–2518 | [80] |
| NS5B | A3 | RVCEKMALY | 2588–2596 | [69] |
| NS5B | A2 | ALYDWTKL | 2594–2602 | [78] |
| NS5B | B57 | KSKKTPMGF | 2629–2637 | [80] |
| NS5B | A0201 | GLQDCTMLV | 2727–2735 | [72] |
| NS5B | B38 | HDGAGKRVYL | 2794–2804 | [80] |
| NS5B | A25 | TARHTPVNSW | 2819–2828 | [78] |
| NS5B | A2402 | RMILMTHFF | 2841–2849 | [77] |
| NS5B | A2402 | CYSIEPLDL | 2870–2878 | [77] |
| NS5B | A31 | VGIYLLPNR | 3003–3011 | [80] |
Acknowledgments
Acknowledgements
This work was supported by a grant of the Swiss National Science Foundation to FKB (SNF-PBSKB-102686) and by the Solid Organ Transplantation in HIV: Multi-Side Study (AI052748) funded by the National Institute of Allergy and Infectious Diseases.
Contributor Information
Florian K Bihl, Email: fbihl@partners.org.
Elisabetta Loggi, Email: epalab@med.unibo.it.
John V Chisholm, III, Email: jchisholmiii@partners.org.
Hannah S Hewitt, Email: hshewitt@partners.org.
Leah M Henry, Email: lmhenry@partners.org.
Caitlyn Linde, Email: clinde1@partners.org.
Todd J Suscovich, Email: tsuscovich@partners.org.
Johnson T Wong, Email: jwong1@partners.org.
Nicole Frahm, Email: nfrahm@partners.org.
Pietro Andreone, Email: andreone@med.unibo.it.
Christian Brander, Email: brander@helix.mgh.harvard.edu.
References
- Khanna R, Burrows SR. Role of cytotoxic T lymphocytes in Epstein-Barr virus- associated diseases. Annu Rev Microbiol. 2000;54:19–48. doi: 10.1146/annurev.micro.54.1.19. [DOI] [PubMed] [Google Scholar]
- Brander C, Walker BD. T lymphocyte responses in HIV-1 infection. Implications for vaccine development. Curr Opin Immunol. 1999;11:451–459. doi: 10.1016/S0952-7915(99)80076-4. [DOI] [PubMed] [Google Scholar]
- Cerny A, Chisari FV. Pathogenesis of chronic hepatitis C: immunological features of hepatic injury and viral persistence. Hepatology. 1999;30:595–601. doi: 10.1002/hep.510300312. [DOI] [PubMed] [Google Scholar]
- Brander C, O'Connor P, Suscovich T, Jones NG, Lee Y, Kedes D, Ganem D, Martin J, Osmond D, Southwood S, Sette A, Walker BD, Scadden DT. Definition of an optimal cytotoxic T lymphocyte epitope in the latently expressed Kaposi's sarcoma- associated herpesvirus kaposin protein. J Infect Dis. 2001;184:119–126. doi: 10.1086/322003. [DOI] [PubMed] [Google Scholar]
- Riddell SR, Rabin M, Geballe AP, Britt WJ, Greenberg PD. Class IMHC-restricted cytotoxic T lymphocyte recognition of cells infected with human cytomegalovirus does not require endogenous viral gene expression. J Immunol. 1991;146:2795–2804. [PubMed] [Google Scholar]
- Bertoletti A, Maini M, Williams R. Role of hepatitis B virus specific cytotoxic T cells in liver damage and viral control. Antiviral Res. 2003;60:61–66. doi: 10.1016/j.antiviral.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Lichterfeld M, Yu XG, Waring MT, Mui SK, Johnston MN, Cohen D, Addo MM, Zaunders J, Alter G, Pae E, Strick D, Allen TM, Rosenberg ES, Walker BD, Altfeld M. HIV-1-specific cytotoxicity is preferentially mediated by a subset of CD8(+) T cells producing both interferon-gamma and tumor necrosis factor-alpha. Blood. 2004;104:487–494. doi: 10.1182/blood-2003-12-4341. [DOI] [PubMed] [Google Scholar]
- Brander C, Hartman KE, Trocha AK, Jones NG, Johnson RP, Korber B, Wentworth P, Buchbinder SP, Wolinsky S, Walker BD, Kalams SA. Lack of strong immune selection pressure by the immunodominant, HLA-A*0201 restricted CTL response in chronic HIV-1 infection. j Clin Invest. 1998;101:2559–2566. doi: 10.1172/JCI2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahm N, Korber BT, Adams CA, Szinger JJ, Draenert R, Addo MM, Feeney ME, Yusim K, Sango K, Brown NV, SenGupta D, Piechocka-Trocha A, Simonis T, Marincola FM, Wurcel A, Stone DR, Russell CJ, Adolf P, Cohen D, Roach T, StJohn A, Khatri A, Davis K, Mullins J, Goulder PJ, Walker BD, Brander C. Consistent cytotoxic-T- lymphocyte targeting of immunodominant regions in Human Immunodeficiency Virus across multiple ethnicities. J Virol. 2004;78:2187–2200. doi: 10.1128/JVI.78.5.2187-2200.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brander C, Goulder P. HIV Molecular immunology database. Los Alamos National Laboratory: Theoretical Biology and Biophysics. Los Alamos, NM, USA; 1999. Recent advances in HIV-1 CTL epitope charactrization. [Google Scholar]
- Addo MM, Yu XG, Rathod A, Cohen D, Eldridge RL, Strick D, Johnston MN, Corcoran C, Wurcel AG, Fitzpatrick CA, Feeney ME, Rodriguez WR, Basgoz N, Draenert R, Stone DR, Brander C, Goulder PJ, Rosenberg ES, Altfeld M, Walker BD. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)- specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J Virol. 2003;77:2081–2092. doi: 10.1128/JVI.77.3.2081-2092.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer GM, Ouchi K, Chung RT, Nguyen TN, Day CL, Purkis DR, Reiser M, Kim AY, Lucas M, Klenerman P, Walker BD. Comprehensive analysis of CD8(+)-T-cell responses against hepatitis C virus reveals multiple unpredicted specificities. J Virol. 2002;76:6104–6113. doi: 10.1128/JVI.76.12.6104-6113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodberry T, Henry LM, Suscovich TJ, Walker BD, Scadden DT, Wang F, Brander C. Differential targeting and shifts in immunodominance in EBV- specific CD8 and CD4 T-cell responses from acute to persistent infection. Journal of Infections Disease. 2005. [DOI] [PubMed]
- Frahm N, Goulder P, Brander C. HIV Molecular immunology database. Los Alamos National Laboratory: Theoretical Biology and Biophysics. Los Alamos, NM, USA; 2002. Total assessment of HIV specific CTL responses: Epitope clustering, processing preferences and the impact of HIV sequence heterogeneity. [Google Scholar]
- Lauer GM, Barnes E, Lucas M, Timm J, Ouchi K, Kim AY, Day CL, Robbins GK, Casson DR, Reiser M, Dusheiko G, Allen TM, Chung RT, Walker BD, Klenerman P. High resolution analysis of cellular immune responses in resolved and persistent hepatitis C virus infection. Gastroenterology. 2004;127:924–936. doi: 10.1053/j.gastro.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Rickinson AB, Moss DJ. Human cytotoxic T lymphocyte responses to Epstein- Barr virus infection. Annu Rev Immunol. 1997;15:405–431. doi: 10.1146/annurev.immunol.15.1.405. [DOI] [PubMed] [Google Scholar]
- Longmate J, York J, La Rosa C, Krishnan R, Zhang MY, Senitzer D, Diamond D. Population coverage by HLA class-I restricted cytotoxic T-lymphocyte epitopes. Immunogenetics. 2001;52:165–173. doi: 10.1007/s002510000271. [DOI] [PubMed] [Google Scholar]
- Meiklejohn DA, Karlsson RK, Karlsson AC, Chapman JM, Nixon DF, Schweighardt B. ELISPOT cell rescue. J Immunol Methods. 2004;288:135–147. doi: 10.1016/j.jim.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Wilson CC, Wong JT, Girard DD, Merrill DP, Dynan M, An DD, Kalams SA, Johnson RP, Hirsch MS, D'Aquila RT, et al. Ex vivo expansion of CD4 lymphocytes from human immunodeficiency virus type 1-infected persons in the presence of combination antiretroviral agents. Journal of Infectious Diseases. 1995;172:88–96. doi: 10.1093/infdis/172.1.88. [DOI] [PubMed] [Google Scholar]
- Jones N, Agrawal D, Elrefaei M, Hanson A, Novitsky V, Wong JT, Cao H. Evaluation of antigen-specific responses using in vitro enriched T cells. J Immunol Methods. 2003;274:139–147. doi: 10.1016/S0022-1759(02)00510-0. [DOI] [PubMed] [Google Scholar]
- Ibarrondo FJ, Anton PA, Fuerst M, Ng HL, Wong JT, Matud J, Elliott J, Shih R, Hausner MA, Price C, Hultin LE, Hultin PM, Jamieson BD, Yang OO. Parallel human immunodeficiency virus type 1-specific CD8+ T-lymphocyte responses in blood and mucosa during chronic infection. J Virol. 2005;79:4289–4297. doi: 10.1128/JVI.79.7.4289-4297.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacklett BL, Yang O, Hausner MA, Elliott J, Hultin L, Price C, Fuerst M, Matud J, Hultin P, Cox C, Ibarrondo J, Wong JT, Nixon DF, Anton PA, Jamieson BD. Optimization of methods to assess human mucosal T-cell responses to HIV infection. J Immunol Methods. 2003;279:17–31. doi: 10.1016/S0022-1759(03)00255-2. [DOI] [PubMed] [Google Scholar]
- Wong JT, Eylath AA, Ghobrial I, Colvin RB. The mechanism of anti-CD3 monoclonal antibodies. Mediation of cytolysis by inter-T cell bridging. Transplantation. 1990;50:683–689. doi: 10.1097/00007890-199010000-00030. [DOI] [PubMed] [Google Scholar]
- Korber B, Brander C, Haynes B, Koup R, Kuiken C, Moore J, BD W, Watkins D. Theoretical Biology and Biophysics Group. Los Alamas National Laboratory. Los Alamos, NM, USA; 2004. HIV Molecular Immunology Database 2004. [Google Scholar]
- Koziel MJ, Dudley D, Wong JT, Dienstag J, Houghton M, Ralston R, Walker BD. Intrahepatic cytotoxic T lymphocytes specific for hepatitis C virus in persons with chronic hepatitis. J Immunol. 1992;149:3339–3344. [PubMed] [Google Scholar]
- Frahm N, Goulder P, Brander C. HIV Molecular immunology database. Los Alamos National Laboratory: Theoretical Biology and Biophysics. Los Alamos, NM, USA; 2003. Broad HIV-1 specific CTL responses reveal extensive HLA class I binding promiscuity of HIV-derived, optimally defined CTL epitopes. [Google Scholar]
- Pepperl S, Benninger-Doring G, Modrow S, Wolf H, Jilg W. Immediate-early transactivator Rta of Epstein-Barr virus (EBV) shows multiple epitopes recognized by EBV-specific cytotoxic T lymphocytes. J Virol. 1998;72:8644–8649. doi: 10.1128/jvi.72.11.8644-8649.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven NM, Leese AM, Annels NE, Lee SP, Rickinson AB. Epitope focusing in the primary cytotoxic T cell response to Epstein-Barr virus and its relationship to T cell memory. J Exp Med. 1996;184:1801–1813. doi: 10.1084/jem.184.5.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven NM, Annels NE, Kumar A, Leese AM, Kurilla MG, Rickinson AB. Immediate early and early lytic cycle proteins are frequent targets of the Epstein- Barr virus-induced cytotoxic T cell response. J Exp Med. 1997;185:1605–1617. doi: 10.1084/jem.185.9.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzushima K, Hayashi N, Kudoh A, Akatsuka Y, Tsujimura K, Morishima Y, Tsurumi T. Tetramer-assisted identification and characterization of epitopes recognized by HLA A*2402-restricted Epstein-Barr virus-specific CD8+ T cells. Blood. 2003;101:1460–1468. doi: 10.1182/blood-2002-04-1240. [DOI] [PubMed] [Google Scholar]
- Kienzle N, Sculley TB, Poulsen L, Buck M, Cross S, Raab-Traub N, Khanna R. Identification of a cytotoxic T-lymphocyte response to the novel BARFO protein of Epstein-Barr virus: a critical role for antigen expression. J Virol. 1998;72:6614–6620. doi: 10.1128/jvi.72.8.6614-6620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Precopio ML, Sullivan JL, Willard C, Somasundaran M, Luzuriaga K. Differential kinetics and specificity of EBV-specific CD4+ and CD8+ T cells during primary infection. J Immunol. 2003;170:2590–2598. doi: 10.4049/jimmunol.170.5.2590. [DOI] [PubMed] [Google Scholar]
- Hislop AD, Gudgeon NH, Callan MF, Fazou C, Hasegawa H, Salmon M, Rickinson AB. EBV-specific CD8+ T cell memory: relationships between epitope specificity, cell phenotype, and immediate effector function. J Immunol. 2001;167:2019–2029. doi: 10.4049/jimmunol.167.4.2019. [DOI] [PubMed] [Google Scholar]
- Bharadwaj M, Sherritt M, Khanna R, Moss DJ. Contrasting Epstein-Barr virus- specific cytotoxic T cell responses to HLA A2-restricted epitopes in humans and HLA transgenic mice: implications for vaccine design. Vaccine. 2001;19:3769–3777. doi: 10.1016/S0264-410X(01)00085-8. [DOI] [PubMed] [Google Scholar]
- Blake N, Haigh T, Shaka'a G, Croom-Carter D, Rickinson A. The importance of exogenous antigen in priming the human CD8+ T cell response: lessons from the EBV nuclear antigen EBNA1. J Immunol. 2000;165:7078–7087. doi: 10.4049/jimmunol.165.12.7078. [DOI] [PubMed] [Google Scholar]
- Schmidt C, Burrows SR, Sculley TB, Moss DJ, Misko IS. Nonresponsiveness to an immunodominant Epstein-Barr virus-encoded cytotoxic T-lymphocyte epitope in nuclear antigen 3A: implications for vaccine strategies. Proc Natl Acad Sci USA. 1991;88:9478–9482. doi: 10.1073/pnas.88.21.9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows SR, Gardner J, Khanna R, Steward T, Moss DJ, Rodda S, Suhrbier A. Five new cytotoxic T cell epitopes identified within Epstein-Barr virus nuclear antigen. Journal of General Virology. 1994;75:2489–2493. doi: 10.1099/0022-1317-75-9-2489. [DOI] [PubMed] [Google Scholar]
- Hill AB, Lee SP, Haurum JS, Murray N, Yao QY, Rowe M, Signoret N, Rickinson AB, McMichael AJ. Class I major histocompatibility complex-restricted cytotoxic T lymphocytes specific for Epstein-Barr virus (EBV)-transformed B lymphoblastoid cell lines against which they were raised. Journal of Experimental Medicine. 1995;181:2221–2228. doi: 10.1084/jem.181.6.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A, Worth A, Elliott T, Rowland-Jones S, Brooks J, Rickinson A, McMichael A. Characterization of two Epstein-Barr virus epitopes restricted by HLA-B7. European Journal of Immunology. 1995;25:18–24. doi: 10.1002/eji.1830250105. [DOI] [PubMed] [Google Scholar]
- Burrows SR, Misko IS, Sculley TB, Schmidt C, Moss DJ. An Epstein-Barr virus-specific cytotoxic T-cell epitope present on A- and B-type transformants. J Virol. 1990;64:3974–3976. doi: 10.1128/jvi.64.8.3974-3976.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney BM, Chan AT, Rickinson AB, Lee SP, Lin CK, Johnson PJ. Frequency of Epstein-Barr virus-specific cytotoxic T lymphocytes in the blood of Southern Chinese blood donors and nasopharyngeal carcinoma patients. J Med Virol. 2002;67:359–363. doi: 10.1002/jmv.10073. [DOI] [PubMed] [Google Scholar]
- Brooks JM, Colbert RA, Mear JP, Leese AM, Rickinson AB. HLA-B27 subtype polymorphism and CTL epitope choice: studies with EBV peptides link immunogenicity with stability of the B27: peptide complex. J Immunol. 1998;161:5252–5259. [PubMed] [Google Scholar]
- Lee SP, Chan AT, Cheung ST, Thomas WA, CroomCarter D, Dawson CW, Tsai CH, Leung SF, Johnson PJ, Huang DP. CTL control of EBV in nasopharyngeal carcinoma (NPC): EBV-specific CTL responses in the blood and tumors of NPC patients and the antigen-processing function of the tumor cells. J Immunol. 2000;165:573–582. doi: 10.4049/jimmunol.165.1.573. [DOI] [PubMed] [Google Scholar]
- Brooks JM, Murray RJ, Thomas WA, Kurilla MG, Rickinson AB. Different HLA-B27 subtypes present the same immunodominant Epstein-Barr virus peptide. Journal of Experimental Medicine. 1993;178:879–887. doi: 10.1084/jem.178.3.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks JM, Croom-Carter DS, Leese AM, Tierney RJ, Habeshaw G, Rickinson AB. Cytotoxic T-lymphocyte responses to a polymorphic Epstein-Barr virus epitope identify healthy carriers with coresident viral strains. J Virol. 2000;74:1801–1809. doi: 10.1128/JVI.74.4.1801-1809.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Smith KD, Kurilla MG, Lutz CT. Cytotoxic CD8+ T cells recognize EBV antigen but poorly kill autologous EBV-infected B lymphoblasts: immunodominance is elicited by a peptide epitope that is presented at low levels in vitro. J Immunol. 1997;159:1844–1852. [PubMed] [Google Scholar]
- Khanna R, Burrows SR, Kurilla MG, Jacob CA, Misko IS, Sculley TB, Kieff E, Moss DJ. Localization of Epstein-Barr virus cytotoxic T cell epitopes using recombinant vaccinia: implications for vaccine development. Journal of Experimental Medicine. 1992;176:169–176. doi: 10.1084/jem.176.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan SM, Wilkinson GW, Floettmann E, Blake N, Rickinson AB. A recombinant adenovirus expressing an Epstein-Barr virus (EBV) target antigen can selectively reactivate rare components of EBV cytotoxic T-lymphocyte memory in vitro. J Virol. 1996;70:2394–2402. doi: 10.1128/jvi.70.4.2394-2402.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr BM, Kienzle N, Burrows JM, Cross S, Silins SL, Buck M, Benson EM, Coupar B, Moss DJ, Sculley TB. Identification of type B-specific and cross-reactive cytotoxic T-lymphocyte responses to Epstein-Barr virus. J Virol. 1996;70:8858–8864. doi: 10.1128/jvi.70.12.8858-8864.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R, Burrows SR, Nicholls J, Poulsen LM. Identification of cytotoxic T cell epitopes within Epstein-Barr virus (EBV) oncogene latent membrane protein 1 (LMP1): evidence for HLA A2 supertype-restricted immune recognition of EBV- infected cells by LMPl-specific cytotoxic T lymphocytes. Eur J Immunol. 1998;28:451–458. doi: 10.1002/(SICI)1521-4141(199802)28:02<451::AID-IMMU451>3.3.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Meij P, Leen A, Rickinson AB, Verkoeijen S, Vervoort MB, Bloemena E, Middeldorp JM. Identification and prevalence of CD8(+) T-cell responses directed against Epstein-Barr virus-encoded latent membrane protein 1 and latent membrane protein 2. Int J Cancer. 2002;99:93–99. doi: 10.1002/ijc.10309. [DOI] [PubMed] [Google Scholar]
- Lautscham G, Haigh T, Mayrhofer S, Taylor G, Groom-Carter D, Leese A, Gadola S, Cerundolo V, Rickinson A, Blake N. Identification of a TAP-Independent, Immunoproteasome-Dependent CD8(+) T-Cell Epitope in Epstein-Barr Virus Latent Membrane Protein 2. J Virol. 2003;77:2757–2761. doi: 10.1128/JVI.77.4.2757-2761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SP, Tierney RJ, Thomas WA, Brooks JM, Rickinson AB. Conserved CTL epitopes within EBV latent membrane protein 2: a potential target for CTL-based tumor therapy. J Immunol. 1997;158:3325–3334. [PubMed] [Google Scholar]
- Lee SP, Thomas WA, Murray RJ, Khanim F, Kaur S, Young LS, Rowe M, Kurilla M, Rickinson AB. HLA A2.1-restricted cytotoxic T cells recognizing a range of Epstein-Barr virus isolates through a defined epitope in latent membrane protein LMP2. Journal of Virology. 1993;67:7428–7435. doi: 10.1128/jvi.67.12.7428-7435.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R, Burrows SR, Moss DJ, Silins SL. Peptide transporter (TAP-1 and TAP-2)-independent endogenous processing of Epstein-Barr virus (EBV) latent membrane protein 2A: implications for cytotoxic T-lymphocyte control of EBV- associated malignancies. J Virol. 1996;70:5357–5362. doi: 10.1128/jvi.70.8.5357-5362.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin MA, Gilbert MJ, Riddell SR, Greenberg PD, Bevan MJ. Alkali hydrolysis of recombinant proteins allows for the rapid identification of class I MHC-restricted CTL epitopes. J Immunol. 1993;151:3971–3980. [PubMed] [Google Scholar]
- Wills MR, Carmichael AJ, Mynard K, Jin X, Weekes MP, Plachter B, Sissons JG. The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J Virol. 1996;70:7569–7579. doi: 10.1128/jvi.70.11.7569-7579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solache A, Morgan CL, Dodi AI, Morte C, Scott I, Baboonian C, Zal B, Goldman J, Grundy JE, Madrigal JA. Identification of three HLA-A*0201-restricted cytotoxic T cell epitopes in the cytomegalovirus protein pp65 that are conserved between eight strains of the virus. J Immunol. 1999;163:5512–5518. [PubMed] [Google Scholar]
- Masuoka M, Yoshimuta T, Hamada M, Okamoto M, Fumimori T, Honda J, Oizumi K, Itoh K. Identification of the HLA-A24 peptide epitope within cytomegalovirus protein pp65 recognized by CMV-specific cytotoxic T lymphocytes. Viral Immunol. 2001;14:369–377. doi: 10.1089/08828240152716619. [DOI] [PubMed] [Google Scholar]
- Kuzushima K, Hayashi N, Kimura H, Tsurumi T. Efficient identification of HLA-A*2402-restricted cytomegalovirus-specific CD8(+) T-cell epitopes by a computer algorithm and an enzyme-linked immunospot assay. Blood. 2001;98:1872–1881. doi: 10.1182/blood.V98.6.1872. [DOI] [PubMed] [Google Scholar]
- Kondo E, Akatsuka Y, Kuzushima K, Tsujimura K, Asakura S, Tajima K, Kagami Y, Kodera Y, Tanimoto M, Morishima Y, Takahashi T. Identification of novel CTL epitopes of CMV-pp65 presented by a variety of HLA alleles. Blood. 2004;103:630–638. doi: 10.1182/blood-2003-03-0824. [DOI] [PubMed] [Google Scholar]
- Kern F, Bunde T, Faulhaber N, Kiecker F, Khatamzas E, Rudawski IM, Pruss A, Gratama JW, Volkmer-Engert R, Ewert R, Reinke P, Volk HD, Picker LJ. Cytomegalovirus (CMV) phosphoprotein 65 makes a large contribution to shaping the T cell repertoire in CMV-exposed individuals. J Infect Dis. 2002;185:1709–1716. doi: 10.1086/340637. [DOI] [PubMed] [Google Scholar]
- Lim JB, Kwon OH, Kim HS, Kim HO, Choi JR, Provenzano M, Stroncek D. Adoptive immunotherapy for cytomegalovirus (CMV) disease in immunocompromised patients. Yonsei Med J. 2004:18–22. doi: 10.3349/ymj.2004.45.Suppl.18. [DOI] [PubMed] [Google Scholar]
- Kern F, Khatamzas E, Surel I, Frommel C, Reinke P, Waldrop SL, Picker LJ, Volk HD. Distribution of human CMV-specific memory T cells among the CD8pos. subsets defined by CD57, CD27, and CD45 isoforms. Eur J Immunol. 1999;29:2908–2915. doi: 10.1002/(SICI)1521-4141(199909)29:09<2908::AID-IMMU2908>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Retiere C, Prod'homme V, Imbert-Marcille BM, Bonneville M, Vie H, Hallet MM. Generation of cytomegalovirus-specific human T-lymphocyte clones by using autologous B-lymphoblastoid cells with stable expression of pp65 or IE1 proteins: a tool to study the fine specificity of the antiviral response. J Virol. 2000;74:3948–3952. doi: 10.1128/JVI.74.9.3948-3952.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utz U, Biddison WE. Presentation of three different viral peptides is determined by common structural features of the human lymphocyte antigen-A2.1 molecule. Journal of Immunotherapy. 1992;12:180–182. doi: 10.1097/00002371-199210000-00007. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Nakamura I, Kita H, Hiroishi K, Moriyama T, Imawari M. Three new cytotoxic T cell epitopes identified within the hepatitis C virus nucleoprotein. J Gen Virol. 1996;77:1305–1309. doi: 10.1099/0022-1317-77-6-1305. [DOI] [PubMed] [Google Scholar]
- Cerny A, McHutchison JG, Pasquinelli C, Brown ME, Brothers MA, Grabscheid B, Fowler P, Houghton M, Chisari FV. Cytotoxic T lymphocytes response to Hepatitis C Virus-derived peptides containing the HLA A2.1 binding motif. J Clin Invest. 1995;95:521–530. doi: 10.1172/JCI117694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziel MJ, Dudley D, Afdhal N, Grakoui A, Rice CM, Choo QL, Houghton M, Walker BD. HLA class I-restricted cytotoxic T lymphocytes specific for hepatitis C virus. Identification of multiple epitopes and characterization of patterns of cytokine release. J Clin Invest. 1995;96:2311–2321. doi: 10.1172/JCI118287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita H, Moriyama T, Kaneko T, Harase I, Nomura M, Miura H, Nakamura I, Yazaki Y, Imawari M. HLA B44-restricted cytotoxic T lymphocytes recognizing an epitope on hepatitis C virus nucleocapsid protein. Hepatology. 1993;18:1039–1044. doi: 10.1016/0270-9139(93)90454-U. [DOI] [PubMed] [Google Scholar]
- Shirai M, Okada H, Nishioka M, Akatsuka T, Wychowski C, Houghten R, Pendleton CD, Feinstone SM, Berzofsky JA. An epitope in hepatitis C virus core region recognized by cytotoxic T cells in mice and humans. J Virol. 1994;68:3334–3342. doi: 10.1128/jvi.68.5.3334-3342.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battegay M, Fikes J, Di Bisceglie AM, Wentworth PA, Sette A, Celis E, Ching WM, Grakoui A, Rice CM, Kurokohchi K, et al. Patients with chronic hepatitis C have circulating cytotoxic T cells which recognize hepatitis C virus-encoded peptides binding to HLA-A2.1 molecules. Journal of Virology. 1995;69:2462–2470. doi: 10.1128/jvi.69.4.2462-2470.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziel MJ, Dudley D, Afdhal N, Choo QL, Houghton M, Ralston R, Walker BD. Hepatitis C virus (HCV)-specific cytotoxic T lymphocytes recognize epitopes in the core and envelope proteins of HCV. Journal of Virology. 1993;67:7522–7532. doi: 10.1128/jvi.67.12.7522-7532.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai M, Arichi T, Nishioka M, Nomura T, Ikeda K, Kawanishi K, Engelhard VH, Feinstone SM, Berzofsky JA. CTL responses of HLA-A2.1-transgenic mice specific for hepatitis C viral peptides predict epitopes for CTL of humans carrying HLA-A2.1. J Immunol. 1995;154:2733–2742. [PubMed] [Google Scholar]
- Wong DK, Dudley DD, Dohrenwend PB, Lauer GM, Chung RT, Thomas DL, Walker BD. Detection of diverse hepatitis C virus (HCV)-specific cytotoxic T lymphocytes in peripheral blood of infected persons by screening for responses to all translated proteins of HCV. J Virol. 2001;75:1229–1235. doi: 10.1128/JVI.75.3.1229-1235.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarobe P, Huarte E, Lasarte JJ, Lopez-Diaz de Cerio A, Garcia N, Borras-Cuesta F, Prieto J. Characterization of an immunologically conserved epitope from hepatitis C virus E2 glycoprotein recognized by HLA-A2 restricted cytotoxic T lymphocytes. J Hepatol. 2001;34:321–329. doi: 10.1016/S0168-8278(00)00018-0. [DOI] [PubMed] [Google Scholar]
- Nakamoto Y, Kaneko S, Takizawa H, Kikumoto Y, Takano M, Himeda Y, Kobayashi K. Analysis of the CD8-positive T cell response in Japanese patients with chronic hepatitis C using HLA-A*2402 peptide tetramers. J Med Virol. 2003;70:51–61. doi: 10.1002/jmv.10349. [DOI] [PubMed] [Google Scholar]
- Lechner F, Wong DK, Dunbar PR, Chapman R, Chung RT, Dohrenwend P, Robbins G, Phillips R, Klenerman P, Walker BD. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. 2000;191:1499–1512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokohchi K, Akatsuka T, Pendleton CD, Takamizawa A, Nishioka M, Battegay M, Feinstone SM, Berzofsky JA. Use of recombinant protein to identify a motif- negative human cytotoxic T-cell epitope presented by HLA-A2 in the hepatitis C virus NS3 region. Journal of Virology. 1996;70:232–240. doi: 10.1128/jvi.70.1.232-240.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DK, Dudley DD, Afdhal NH, Dienstag J, Rice CM, Wang L, Houghton M, Walker BD, Koziel MJ. Liver-derived CTL in hepatitis C virus infection: breadth and specificity of responses in a cohort of persons with chronic infection. J Immunol. 1998;160:1479–1488. [PubMed] [Google Scholar]
- Martin P, Parroche P, Chatel L, Barretto C, Beck A, Trepo C, Bain C, Lone YC, Inchauspe G, Fournillier A. Genetic immunization and comprehensive screening approaches in HLA-A2 transgenic mice lead to the identification of three novel epitopes in hepatitis C virus NS3 antigen. J Med Virol. 2004;74:397–405. doi: 10.1002/jmv.20189. [DOI] [PubMed] [Google Scholar]
- Ibe M, Sakaguchi T, Tanaka K, Saito S, Yokota S, Tanaka T, Shimotohno K, Chujoh Y, Shiratori Y, Omata M, Miwa K, Takiguchi M. Identification and characterization of a cytotoxic T cell epitope of hepatitis C virus presented by HLA- B*3501 in acute hepatitis. J Gen Virol. 1998;79:1735–1744. doi: 10.1099/0022-1317-79-7-1735. [DOI] [PubMed] [Google Scholar]
- Urbani S, Uggeri J, Matsuura Y, Miyamura T, Penna A, Boni C, Ferrari C. Identification of immunodominant hepatitis G virus (HCV)-specific cytotoxic T-cell epitopes by stimulation with endogenously synthesized HCV antigens. Hepatology. 2001;33:1533–1543. doi: 10.1053/jhep.2001.25091. [DOI] [PubMed] [Google Scholar]
- Bertoletti A, Chisari FV, Penna A, Guilhot S, Galati L, Missale G, Fowler P, Schlicht HJ, Vitiello A, Chesnut RC, et al. Definition of a minimal optimal cytotoxic T-cell epitope within the hepatitis B virus nucleocapsid protein. Journal of Virology. 1993;67:2376–2380. doi: 10.1128/jvi.67.4.2376-2380.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HG, Lim JS, Lee KY, Choi YK, Choe IS, Chung TW, Kim K. Peptide- specific CTL induction in HBV-seropositive PBMC by stimulation with peptides in vitro: novel epitopes identified from chronic carriers. Virus Res. 1997;50:185–194. doi: 10.1016/S0168-1702(97)00068-3. [DOI] [PubMed] [Google Scholar]
- Rehermann B, Pasquinelli C, Mosier SM, Chisari FV. Hepatitis B virus (HBV) sequence variation of cytotoxic T lymphocyte epitopes is not common in patients with chronic HBV infection. J Clin Invest. 1995;96:1527–1534. doi: 10.1172/JCI118191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobao Y, Sugi K, Tomiyama H, Saito S, Fujiyama S, Morimoto M, Hasuike S, Tsubouchi H, Tanaka K, Takiguch M. Identification of hepatitis B virus-specific CTL epitopes presented by HLA-A*2402, the most common HLA class I allele in East Asia. J Hepatol. 2001;34:922–929. doi: 10.1016/S0168-8278(01)00048-4. [DOI] [PubMed] [Google Scholar]
- Missale G, Redeker A, Person J, Fowler P, Guilhot S, Schlicht HJ, Ferrari C, Chisari FV. HLA-A31- and HLA-Aw68-restricted cytotoxic T cell responses to a single hepatitis B virus nucleocapsid epitope during acute viral hepatitis. J Exp Med. 1993;177:751–762. doi: 10.1084/jem.177.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CL, Tsai SL, Lee TH, Chien RN, Liao SK, Liaw YF. High frequency of functional anti-YMDD and -mutant cytotoxic T lymphocytes after in vitro expansion correlates with successful response to lamivudine therapy for chronic hepatitis B. Gut. 2005;54:152–161. doi: 10.1136/gut.2003.032920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sette A, Vitiello A, Reherman B, Fowler P, Nayersina R, Kast WM, Melief CJ, Oseroff C, Yuan L, Ruppert J, et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J Immunol. 1994;153:5586–5592. [PubMed] [Google Scholar]
- Rehermann B, Fowler P, Sidney J, Person J, Redeker A, Brown M, Moss B, Sette A, Chisari FV. The cytotoxic T lymphocyte response to multiple hepatitis B virus polymerase epitopes during and after acute viral hepatitis. Journal of Experimental Medicine. 1995;181:1047–1058. doi: 10.1084/jem.181.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayersina R, Fowler P, Guilhot S, Missale G, Cerny A, Schlicht HJ, Vitiello A, Chesnut R, Person JL, Redeker AG, et al. HLA A2 restricted cytotoxic T lymphocyte responses to multiple hepatitis B surface antigen epitopes during hepatitis B virus infection. J Immunol. 1993;150:4659–4671. [PubMed] [Google Scholar]
- Hwang YK, Kim NK, Park JM, Lee K, Han WK, Kim HI, Cheong HS. HLA-A2 1 restricted peptides from the HBx antigen induce specific CTL responses in vitro and in vivo. Vaccine. 2002;20:3770–3777. doi: 10.1016/S0264-410X(02)00297-9. [DOI] [PubMed] [Google Scholar]