

Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) exhibits considerable variability in clinical outcomes. Identifying specific phenotypic profiles within MASLD is essential for developing targeted therapeutic strategies. Here we investigated the heterogeneity of MASLD using partitioning around medoids clustering based on six simple clinical variables in a cohort of 1,389 individuals living with obesity. The identified clusters were applied across three independent MASLD cohorts with liver biopsy (totaling 1,099 participants), and in the UK Biobank to assess the incidence of chronic liver disease, cardiovascular disease and type 2 diabetes. Results unveiled two distinct types of MASLD associated with steatohepatitis on histology and liver imaging. The first cluster, liver-specific, was genetically linked and showed rapid progression of chronic liver disease but limited risk of cardiovascular disease. The second cluster, cardiometabolic, was primarily associated with dysglycemia and high levels of triglycerides, leading to a similar incidence of chronic liver disease but a higher risk of cardiovascular disease and type 2 diabetes. Analyses of samples from 831 individuals with available liver transcriptomics and 1,322 with available plasma metabolomics highlighted that these two types of MASLD exhibited distinct liver transcriptomic profiles and plasma metabolomic signatures, respectively. In conclusion, these data provide preliminary evidence of the existence of two distinct types of clinically relevant MASLD with similar liver phenotypes at baseline, but each with specific underlying biological profiles and different clinical trajectories, suggesting the need for tailored therapeutic strategies.
Subject terms: Medical research, Metabolic disorders
Partitioning clustering based on clinical variables applied to multiple patient cohorts identifies two subtypes of metabolic dysfunction-associated steatotic liver disease with different associations to hepatic and cardiovascular outcomes.
Main
Nonalcoholic fatty liver disease, now referred to as metabolic dysfunction-associated steatotic liver disease (MASLD)1,2, is currently the most common chronic liver disease worldwide, with an estimated global prevalence of approximately 30% (ref. 3).
MASLD comprises a spectrum of disorders ranging from isolated steatosis to metabolic dysfunction-associated steatohepatitis (MASH), ultimately leading to advanced fibrosis, cirrhosis and hepatocellular carcinoma4. However, not every individual diagnosed with MASLD will progress to MASH and later stages of liver disease, indicating the presence of a substantial interindividual variation in the disease progression5. Furthermore, MASLD harbors an increased risk of cardiovascular disease and type 2 diabetes6,7, which also widely varies among individuals. This interindividual variability in the severity and progression of MASLD and its extrahepatic consequences, together with the challenges of finding a specific drug treatment, highlight the need for more personalized approaches8–10. Given this context, advancements in diagnostic strategies for risk stratification and efficient testing of new drugs in at-risk populations are urgently needed11.
Emerging evidence points to the clinical relevance of distinguishing different types of MASLD on the basis of distinct pathophysiological mechanisms and rates of disease progression5. For example, genetic predisposition to hepatic steatosis is associated with increased risk of liver-related events, while offering protection against coronary artery disease12,13. Specifically, PNPLA3 rs738409 (p.I148M), the strongest genetic variant predisposing to MASLD, is associated with a reduction in intrahepatic turnover of lipids droplets but is not causally linked to ischemic heart disease in individuals with MASLD14. In contrast, other mechanisms central to MASLD pathophysiology, such as hepatic de novo lipogenesis or adipose tissue dysfunction, have been associated with insulin resistance and a higher risk for type 2 diabetes and cardiovascular disease, but with only a moderate risk of liver-related events10.
In the present study, we identified two types of MASLD by using a data-driven clustering approach focused on key hepatic and cardiometabolic traits. These two MASLD types have distinct biological profiles and risks for cardiometabolic disease and diabetes, despite having the same severity of MASLD on liver histology. We then clustered four independent cohorts of individuals at-risk for MASLD from Italy, Finland, Belgium and the United Kingdom, with consistent results, supporting the validity of the proposed clustering.
Results
Cluster analysis identifies two distinct types of MASLD
Cluster analysis and identification of MASLD types were performed on the basis of the data of 1,389 French participants from the Atlas Biologique de l’Obésité Sévère (ABOS) cohort (Extended Data Fig. 1). Overall, we identified six clusters with distinctive patterns of the six clustering variables in the ABOS cohort (Fig. 1). We then added patients from three independent cohorts to these clusters, namely, the Universitair Ziekenhuis Antwerpen (UZA) cohort from Belgium (n = 463), the Molecular Architecture of FAtty Liver Disease in individuals with obesity undergoing bAriatric surgery (MAFALDA) cohort from Italy (n = 261) and the Helsinki cohort from Finland (n = 375) (Extended Data Fig. 2). Due to the low number of participants in some individual clusters across cohorts, we pooled the three cohorts for the following analyses, resulting in a consolidated cohort of 1,099 individuals, referred to hereafter as the validation cohort (Fig. 1).
Extended Data Fig. 1.

Participant flowchart for the ABOS cohort.
Fig. 1. Characteristics of the six data-driven clusters in the ABOS cohort and in the validation cohort.

a,b, The distribution of data-driven clusters in the ABOS cohort (a) and the validation cohort (b). c,d, Radar charts representing the median values of age, BMI, HbA1c, LDL, triglycerides and ALT for each cluster in the ABOS cohort (n = 1,389) (c) and the validation cohort (n = 1,099) (d). The dark gray line represents the 95th percentile observed in the ABOS cohort. e,f, Bar plots representing the proportion of patients with MASH at histology in the ABOS cohort (n = 1,325) (e) and the validation cohort (n = 1,099) (f). Statistical tests used include either a chi-squared test or Fisher’s exact test, both two-sided with Bonferroni correction. Significance levels are indicated as follows: ***P < 0.001, $ indicates P = 0.011 (e); $ indicates P = 0.0052, @ indicates P = 0.0046, ***P < 0.001 (f). g,h, Radar charts represent the proportion of patients with NAS ≥4, steatosis grade ≥1, lobular inflammation grade ≥1, ballooning grade ≥1, and fibrosis stage ≥1 and ≥2 for each cluster in the ABOS cohort (g) and the validation cohort (h).
Extended Data Fig. 2. Cluster characteristics in UZA, MAFALDA and HELSINKI cohorts.

Distribution of data-driven clusters: UZA (a), MAFALDA (b), and HELSINKI (c). Radar charts representing the median values of age, BMI, HbA1c, LDL, triglycerides and ALT in UZA (d), MAFALDA (e), and HELSINKI (f). The dark gray line represents the 95th percentile of the ABOS cohort. Bar plots represent the proportion of patients with MASH in: UZA (g), MAFALDA (h), and HELSINKI (i). Radar charts represent the proportion of patients with NAS ≥ 4, steatosis grade ≥1, lobular inflammation grade ≥1, ballooning grade ≥1, fibrosis stage ≥ 1 and ≥2 in: UZA (j), MAFALDA (k), and HELSINKI (l). Statistical tests used include either a Chi-squared test or Fisher’s exact test, both two-sided with Bonferroni correction. Significance levels are indicated as follows: (g) * p = 0,0496; @ p = 0.0253, $ p = 0.0011, *** p < 0.001; (h) $ p = 0.0202; @ p = 0.0016, *** p < 0.001; (i) $ p = 0.0196,*** p < 0.001. Abbreviations: ALT, alanine aminotransferase; BMI, body mass index; HbA1c, hemoglobin A1c; LDL, low-density lipoprotein cholesterol.
In the ABOS cohort, cluster 1 contained 18% of participants and was characterized by older age and hypertension; cluster 2 included 11% of participants and had the highest hemoglobin A1c (HbA1c), high triglycerides and hypertension; cluster 3 had 13% of participants, young age and the highest body mass index (BMI); cluster 4 had 26% of participants and the highest low-density lipoprotein (LDL) cholesterol levels; cluster 5 had 7% of participants and the highest alanine aminotransferase (ALT) levels; and cluster 6 had 24% of participants and a majority of females with a more favorable metabolic profile (Fig. 1 and Extended Data Table 1).
Extended Data Table 1.
Patient characteristics based on cluster allocation in the ABOS cohort (n=1,389)
| Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | Cluster 6 | Adj-p Cluster 1–6 | |
|---|---|---|---|---|---|---|---|
| N | 256 | 158 | 180 | 361 | 99 | 335 | − |
| Clinical data | |||||||
| Age (years) | 53 (10) | 52 (11.75) | 34 (16) | 46 (11) | 37 (15) | 30 (10) | <0.001 |
| Women n (%) | 175 (68.4) | 86 (54.4) | 123 (68.3) | 290 (80.3) | 57 (55.6) | 310 (92.5) | <0.001 |
| BMI (Kg/m2) | 45.5 (7.5) | 44.85 (9.4) | 59.7 (7.93) | 44.4 (7.2) | 43.9 (6.5) | 43.8 (5.9) | <0.001 |
| Waist circumference (cm) | 140 (19.75) | 134 (20.5) | 161 (20.25) | 138 (17) | 137 (15) | 139 (14) | <0.001 |
| Significant alcohol intake1 (n)1 | 9 (6.9) | 7 (8.0) | 6 (6.0) | 15 (8.4) | 3 (6.7) | 12 (6.7) | 1 |
| Glucose profile | |||||||
| HbA1c (%) | 6.2 (1.03) | 9.2 (2.28) | 5.8 (0.8) | 5.8 (0.7) | 5.9 (1.05) | 5.4 (0.5) | <0.001 |
| Fasting glucose (mmol/L) | 6.1 (1.79) | 10.24 (5.3) | 5.49 (1.22) | 5.55 (1.14) | 5.83 (1.72) | 5.11 (0.61) | <0.001 |
| Fasting insulin (UI/L)2 | 13.9 (11.7) | 15.1 (16.05) | 16.75 (10.35) | 13.7 (9.8) | 19.65 (15.23) | 13.7 (9.62) | <0.001 |
| Lipid profile | |||||||
| Total cholesterol (mmol/L) | 4.37 (0.84) | 4.47 (1.33) | 4.7 (0.96) | 5.86 (0.86) | 5.09 (0.89) | 4.6 (0.89) | <0.001 |
| HDL cholesterol (mmol/L) | 1.16 (0.36) | 0.98 (0.29) | 1.11 (0.31) | 1.16 (0.31) | 1.01 (0.31) | 1.14 (0.34) | <0.001 |
| LDL cholesterol (mmol/L) | 2.47 (0.75) | 2.53 (1.05) | 2.97 (0.81) | 3.85 (0.7) | 3.33 (0.9) | 2.9 (0.77) | <0.001 |
| Triglycerides (mmol/L) | 1.4 (0.77) | 2.34 (1.56) | 1.27 (0.68) | 1.49 (0.73) | 1.61 (0.8) | 1.11 (0.6) | <0.001 |
| Liver function tests | |||||||
| AST (UI/L) | 22 (10) | 30 (18) | 22 (11) | 23 (8) | 44 (20.75) | 21 (9) | <0.001 |
| ALT (UI/L) | 25 (15) | 39 (26) | 26 (17) | 26 (14) | 75 (26.5) | 21 (15) | <0.001 |
| GGT (UI/L) | 31 (24.25) | 58 (71.75) | 28.5 (21.25) | 30 (25) | 53.5 (47.75) | 22 (16) | <0.001 |
| Comorbidities | |||||||
| Hypertension n (%) | 201 (78.5) | 138 (87.3) | 109 (60.6) | 200 (55.4) | 55 (55.6) | 107 (31.9) | <0.001 |
| Type 2 diabetes n (%) | 140 (54.7) | 156 (98.7) | 50 (27.8) | 98 (27.1) | 41 (41.4) | 23 (6.9) | <0.001 |
| Dyslipidemia n (%) | 137 (53.5) | 132 (83.5) | 75 (41.7) | 332 (92.0) | 59 (59.6) | 83 (24.8) | <0.001 |
| Medications | |||||||
| Anti-hypertensive drugs n (%) | 180 (70.3) | 125 (79.1) | 62 (34.4) | 139 (38.5) | 34 (34.3) | 37 (11%) | <0.001 |
| Oral glucose-lowering drugs n (%) | 122 (47.8) | 148 (94.3) | 34 (18.9) | 63 (17.5) | 29 (29.3) | 14 (4.2) | <0.001 |
| Insulin n (%) | 30 (11.8) | 83 (52.5) | 5 (2.8) | 9 (2.5) | 3 (3.0%) | 2 (0.6) | <0.001 |
| Lipid-lowering drugs n (%) | 112 (43.8) | 95 (60.1) | 18 (10.0) | 52 (14.4) | 10 (10.1) | 9 (2.7) | <0.001 |
| Statins n (%) | 104 (40.6) | 81 (51.3) | 11 (6.1) | 42 (11.6) | 5 (5.1) | 8 (2.4) | <0.001 |
| Liver histology 3 | |||||||
| Steatosis grade ≥ 1 n (%) | 213 (85.9) | 150 (97.4) | 150 (85.2) | 303 (85.8) | 90 (92.8) | 213 (64.5) | <0.001 |
| Lobular inflammation grade ≥ 1 n (%) | 76 (31.4) | 83 (54.6) | 51 (30.4) | 105 (30.1) | 53 (55.8) | 79 (24.6) | <0.001 |
| Ballooning grade ≥ 1 n (%) | 29 (12.0) | 59 (38.8) | 20 (11.8) | 23 (6.6) | 24 (25.3) | 15 (4.7) | <0.001 |
| MASH n (%) | 16 (6.6) | 51 (33.6) | 14 (8.3) | 16 (4.6) | 23 (24.2) | 8 (2.5) | <0.001 |
| Fibrosis stage ≥ 2 n (%) | 26 (11.3) | 49 (33.3) | 22 (13.3) | 21 (6.3) | 19 (20.0) | 12 (3.9) | <0.001 |
| Fibrosis stage 3-4 n (%) | 15 (6.5) | 32 (21.8) | 7 (4.2) | 9 (2.7) | 15 (15.8) | 4 (1.3) | <0.001 |
| NAS score | 2 (2) | 3 (3) | 1 (2) | 1 (1) | 3 (2.5) | 1 (2) | <0.001 |
| Genetics | |||||||
| PNPLA3 rs738409 n (CC/CG+GG) | 129 (54.9) | 79 (57.7) | 95 (59.0) | 195 (59.1) | 31 (36.0) | 189 (61.0) | 0.009 |
| TM6SF2 rs58542926 n (CC/CT+TT) | 197 (84.9) | 118 (86.8) | 147 (90.7) | 298 (90.0) | 69 (80.2) | 273 (87.2) | 0.42 |
| MBOAT7 rs641738 n (CC/CT+TT) | 74 (32.2) | 38 (27.5) | 48 (29.8) | 109 (32.8) | 21 (24.4) | 104 (33.3) | 1 |
| GCKR rs1260326 n (CC/CT+TT) | 76 (32.9) | 41 (29.7) | 54 (33.5) | 111 (33.6) | 23 (26.7) | 105 (33.5) | 1 |
| PRS-HFC +4 | 0.27 (0.27) | 0.26 (0.27) | 0.19 (0.33) | 0.26 (0.27) | 0.39 (0.41) | 0.19 (0.27) | <0.001 |
| PRS-HFC−5 | 0.13 (0.13) | 0.13 (0.13) | 0.13 (0.13) | 0.13 (0.13) | 0.13 (0.07) | 0.13 (0.13) | 1 |
Data were reported as median (interquartile range) for continuous variables and frequencies (percentages) for categorical variables. Clusters were compared using Kruskal-Wallis test, Chi-squared test, or Fisher’s exact test, as appropriate. Differences were considered statistically significant when p-value(s) adjusted for multiple comparisons using Bonferroni correction, performed separately for clinical data, histological data and genetic data, were less than 0.05.
1 Significant alcohol intake was defined as a daily consumption above 20 g in women and 30 g in men
2 Patients receiving insulin were excluded.
3 Liver histology was available from 1325 participants
4: PRS-HFC + Polygenic Risk Score was calculated with the formula: prs=0.266∗PNPLA3_012 + 0.274∗TMS6F2_012 + 0.065∗GCKR_012 + 0.063∗MBOAT7_012
5: PRS-HFC - Polygenic Risk Score was calculated without PNPLA3 with the formula: prs=0.274∗TMS6F2_012 + 0.065∗GCKR_012 + 0.063∗MBOAT7_012
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; eGFR, estimates of glomerular filtration rate; GCKR, glucokinase regulator; GGT, gamma glutamyltransferase; HbA1c, hemoglobin A1c; HDL, high-density lipoprotein; HOMA2-B, homeostasis model assessment 2 estimates of beta-cell function; HOMA2-IR, homeostasis model assessment 2 estimates of insulin-resistance; LDL, low-density lipoprotein; MBOAT7, membrane-bound O-acyltransferase domain-containing 7; PNPLA3, patatin-like phospholipase domain-containing 3; PRS-HFC, polygenic risk score of hepatic fat content; TM6SF2, transmembrane 6 superfamily member, Adj-p, adjusted-p.
Despite marked differences in age and prevalence of type 2 diabetes between clusters 2 and 5, liver histology revealed high prevalence of MASH and advanced fibrosis (F ≥ 3) in these two subgroups, as compared with other clusters combined: 33.6% and 24.2% versus 5.0%, and 21.8% and 15.8% versus 3.4%, respectively (all adjusted P < 0.001 versus other clusters combined). To further examine the potential differences in mechanisms driving MASH, we pooled the clusters with lower severity of MASLD (clusters 1, 3, 4 and 6) in a ‘control’ cluster, which was compared with cluster 2 and cluster 5 (Fig. 2 and Table 1).
Fig. 2. Characteristics of the three clusters across the ABOS cohort, validation cohort and UK Biobank.

a–i, Characteristics of the liver-specific, cardiometabolic and control clusters in the ABOS cohort (a–c), in the validation cohort (d–f) and in the UK Biobank (g–i). In a, d and g, the distribution of data-driven clusters is presented. The radar charts represent the median values of age, BMI, HbA1c, LDL, triglycerides and ALT for each cluster in the ABOS cohort (b), validation cohort (e) and UK Biobank (h). The dark gray line represents the 95th percentile observed in the ABOS cohort. The bar plots represent the proportion of patients with MASH at histology in the ABOS cohort (n = 1325) (c) and the validation cohort (n = 1,099) (f), or at-risk MASH on MRI in the UK Biobank (n = 6,792) (i). Statistical tests used include either a chi-squared test or Fisher’s exact test, both two-sided with Bonferroni correction. Significance levels are indicated as follows: ***P < 0.001 (c); $ P = 0.0011, ***P < 0.001 (f); ***P < 0.001 (i). cT, iron-corrected T1; adj-p, adjusted P value.
Table 1.
Patient characteristics based on cluster allocation in the ABOS cohort (n = 1,389)
| Control | Cardiometabolic | Liver-specific | Adjusted P | Adjusted P cardiometabolic versus liver-specific | Adjusted P cardiometabolic versus control | Adjusted P liver-specific versus control | |
|---|---|---|---|---|---|---|---|
| N | 1,132 | 158 | 99 | − | − | − | |
| Clinical data | |||||||
| Age (years) | 41 (18) | 52 (11.75) | 37 (15) | <0.001 | <0.001 | <0.001 | 0.75 |
| Women (n) | 898 (79.3) | 86 (54.4) | 55 (55.6) | <0.001 | 1 | <0.001 | <0.001 |
| BMI (kg m−2) | 45.75 (9.8) | 44.85 (9.4) | 43.9 (6.5) | 0.007 | 1 | 1 | 0.04 |
| Waist circumference (cm) | 141 (20) | 134 (20.5) | 137 (15) | <0.001 | 1 | <0.001 | 0.03 |
| Significant alcohol intake (n)a | 42 (7.1) | 7 (8.0) | 3 (6.7) | 1 | − | − | − |
| Glucose profile | |||||||
| HbA1c (%) | 5.7 (0.8) | 9.2 (2.28) | 5.9 (1.05) | <0.001 | <0.001 | <0.001 | 0.02 |
| Fasting glucose (mmol l−1) | 5.39 (1.17) | 10.24 (5.3) | 5.83 (1.72) | <0.001 | <0.001 | <0.001 | 0.26 |
| Fasting insulin (IU l−1)b | 14.1 (10.7) | 15.1 (16.05) | 19.65 (15.23) | <0.001 | 1 | 1 | <0.001 |
| Lipid profile | |||||||
| Total cholesterol (mmol l−1) | 4.91 (1.21) | 4.47 (1.33) | 5.09 (0.89) | <0.001 | <0.001 | <0.001 | 1 |
| HDL cholesterol (mmol l−1) | 1.14 (0.34) | 0.98 (0.29) | 1.01 (0.31) | <0.001 | 1 | <0.001 | <0.001 |
| LDL cholesterol (mmol l−1) | 3.1 (1.08) | 2.53 (1.05) | 3.33 (0.9) | <0.001 | <0.001 | <0.001 | 0.17 |
| Triglycerides (mmol l−1) | 1.32 (0.76) | 2.34 (1.56) | 1.61 (0.8) | <0.001 | <0.001 | <0.001 | 0.05 |
| Liver function tests | |||||||
| AST (U l−1) | 22 (9) | 30 (18) | 44 (20.75) | <0.001 | <0.001 | <0.001 | <0.001 |
| ALT (U l−1) | 24 (15) | 39 (26) | 75 (26.5) | <0.001 | <0.001 | <0.001 | <0.001 |
| GGT (U l−1) | 27 (22) | 58 (71.75) | 53.5 (47.75) | <0.001 | 1 | <0.001 | <0.001 |
| Comorbidities | |||||||
| Hypertension n (%) | 617 (54.5) | 138 (87.3) | 55 (55.6) | <0.001 | <0.001 | <0.001 | 1 |
| Type 2 diabetes n (%) | 311 (27.5) | 156 (98.7) | 41 (41.4) | <0.001 | <0.001 | <0.001 | 0.32 |
| Dyslipidemia n (%) | 627 (55.4) | 132 (83.5) | 59 (59.6) | <0.001 | 0.003 | <0.001 | 1 |
| Medications | |||||||
| Antihypertensive drugs n (%) | 418 (36.9) | 125 (79.1) | 34 (34.3) | <0.001 | <0.001 | <0.001 | 1 |
| Oral glucose-lowering drugs n (%) | 233 (20.6) | 148 (94.3) | 29 (29.3) | <0.001 | <0.001 | <0.001 | 1 |
| Insulin n (%) | 46 (4.1) | 83 (52.5) | 3 (3.0) | <0.001 | <0.001 | <0.001 | 1 |
| Lipid-lowering drugs n (%) | 191 (16.9) | 95 (60.1) | 10 (10.1) | <0.001 | <0.001 | <0.001 | 1 |
| Statins n (%) | 165 (14.6) | 81 (51.3) | 5 (5.1) | <0.001 | <0.001 | <0.001 | 0.90 |
| Genetics | |||||||
| PRS-HFC+c | 0.26 (0.27) | 0.26 (0.27) | 0.39 (0.41) | <0.001 | 0.035 | 1 | <0.001 |
| PRS-HFC−d | 0.13 (0.13) | 0.13 (0.13) | 0.13 (0.07) | 0.14 | − | − | − |
| Liver histologye | |||||||
| NAS score | 1 (1) | 3 (2) | 3 (2.5) | <0.001 | 1 | <0.001 | <0.001 |
| Steatosis grade ≥1 (n) | 879 (79.4) | 150 (97.4) | 90 (92.8) | <0.001 | 1 | <0.001 | 0.054 |
| Lobular inflammation grade ≥1 (n) | 311 (28.8) | 83 (54.6) | 53 (55.8) | <0.001 | 1 | <0.001 | <0.001 |
| Ballooning grade ≥1 (n) | 87 (8.0) | 59 (38.8) | 24 (25.3) | <0.001 | 0.96 | <0.001 | <0.001 |
| MASH (n) | 54 (5) | 51 (33.6) | 23 (24.2) | <0.001 | 1 | <0.001 | <0.001 |
| Fibrosis stage ≥2 (n) | 81 (7.8) | 49 (33.3) | 19 (20) | <0.001 | 0.84 | <0.001 | 0.003 |
| Fibrosis stage 3–4 (n) | 35 (3.4) | 32 (21.8) | 15 (15.8) | <0.001 | 1 | <0.001 | <0.001 |
Data were reported as median (interquartile range) for continuous variables and frequencies (percentages) for categorical variables. Clusters were compared using the Kruskal–Wallis test, chi-squared test or Fisher’s exact test, as appropriate. Differences were considered statistically significant when P value(s) adjusted for multiple comparisons using Bonferroni correction, performed separately for clinical data, histological data and genetic data, were less than 0.05. For variables statistically significant, post-hoc analysis was performed comparing pairwise MASH-enriched clusters (2 and 5) and the control cluster (1, 3, 4 and 6) using the Dunn test, chi-squared test or Fisher’s exact test, as appropriate, with Bonferroni adjustment.
aSignificant alcohol intake was defined as a daily consumption above 20 g in women and 30 g in men
bPatients receiving insulin were excluded.
cPRS-HFC+ polygenic risk score was calculated with the following formula: PRS = 0.266 × PNPLA3_012 + 0.274 × TMS6F2_012 + 0.065 × GCKR_012 + 0.063 × MBOAT7_012
dPRS-HFC− polygenic risk score was calculated without PNPLA3 with the following formula: PRS = 0.274 × TMS6F2_012 + 0.065 × GCKR_012 + 0.063 × MBOAT7_012
eLiver histology was available from 1,325 participants
AST, aspartate aminotransferase; GGT, gamma-glutamyltransferase; HDL, high-density lipoprotein.
To replicate these findings, we then assigned the participants of the three validation cohorts with liver histology (UZA, MAFALDA and Helsinki) to the same subgroups, based on which cluster they were most similar to. Results showed similar distributions of clusters across the three cohorts (Figs. 1 and 2, and Extended Data Fig. 2). Like in the ABOS cohort, the potential cardiometabolic cluster (cluster 2), characterized by the highest HbA1c, hypertension and dyslipidemia, and the liver-specific cluster (cluster 5), characterized by the highest ALT, were similarly enriched in participants presenting more severe histological features of MASLD, including MASH and liver fibrosis.
We further confirmed the association of the cardiometabolic and liver-specific clusters with at-risk liver phenotype in a subset of the UK Biobank participants (n = 6,792) who underwent liver magnetic resonance imaging (MRI). Consistent with what was observed with histology in the ABOS cohort, the cardiometabolic and liver-specific clusters in the UK Biobank were similarly enriched in participants presenting typical features of hepatic steatosis (proton density fat fraction (PDFF) >5.5%) and MASH (PDFF >5.5% and iron-corrected T1 (cT1) >800 ms) (Fig. 2 and Extended Data Table 2).
Extended Data Table 2.
Participant characteristics across the clusters in the prospective UK Biobank cohort for liver outcome (n = 213,180) (A), for cardiovascular outcome (n = 195,739) (B), and for type 2 diabetes outcome (n = 196,791) (C)
| Participant characteristics across the clusters in the prospective UK Biobank cohort for liver outcome (n=213,180) (A) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | Cluster 6 | Adj-p | CTRL Cluster | Adj-p 2 vs CTRL | Adj-p 5 vs CTRL | Adj-p 2 vs 5 | |
| N | 53’430 | 5’231 | 20 | 141’570 | 4’366 | 8’563 | − | 203’583 | − | − | − |
| Clinical data | |||||||||||
| Age, years | 61 (6.1) | 59.7 (6.7) | 44 (3.3) | 56.7 (7.6) | 51.8 (7.7) | 43.2 (2.2) | <0.001 | 57.2 (7.9) | <0.001 | <0.001 | <0.001 |
| Women, n (%) | 21,548 (40%) | 1,452 (28%) | 17 (85%) | 73,280 (52%) | 1,101 (25%) | 5,636 (66%) | <0.001 | 100,481 (49%) | <0.001 | <0.001 | 0.016 |
| BMI, kg/m2 | 30 (4.5) | 32.4 (5.4) | 55.4 (1.5) | 29.4 (3.8) | 31.2 (4.2) | 28.2 (3.2) | <0.001 | 29.5 (4) | <0.001 | <0.001 | <0.001 |
| <25 | 1,011 (2%) | 220 (4%) | 0 (0%) | 293 (0%) | 8 (0%) | 61 (1%) | <0.001 | 1,365 (1%) | <0.001 | <0.001 | <0.001 |
| 25–30 | 31,283 (59%) | 1,692 (32%) | 0 (0%) | 94,536 (67%) | 1,983 (45%) | 6,749 (79%) | 132,568 (65%) | ||||
| ≥30 | 21,136 (40%) | 3,319 (63%) | 20 (100%) | 46,741 (33%) | 2,375 (54%) | 1,753 (20%) | 69,650 (34%) | ||||
| Waist circumference, cm | 98.1 (12.2) | 106.7 (13.3) | 136.4 (12.4) | 94.9 (10.8) | 102.4 (11.1) | 88.5 (9.9) | <0.001 | 95.5 (11.3) | <0.001 | <0.001 | <0.001 |
| Significant alcohol intake (n)1 | 10419 (20%) | 928 (18%) | 0 (0%) | 29407 (21%) | 1322 (30%) | 1716 (20%) | <0.001 | 41542 (20%) | < 0.001 | < 0.001 | < 0.001 |
| Liver imaging | |||||||||||
| PDFF, % | 3.8 (2.4–7.1) | 7.7 (4.2–13) | − | 3.9 (2.5–7.1) | 9.6 (4.7–16.3) | 2.6 (1.9–4) | <0.001 | 3.8 (2.5–7) | <0.001 | <0.001 | 0.287 |
| PDFF NA, n (%) | 51,799 (97%) | 5,140 (98%) | 20 (100%) | 136’209 (96%) | 4,199 (96%) | 8,216 (96%) | <0.001 | 196,244 (96%) | <0.001 | 1 | <0.001 |
| Steatosis by PDFF>5.5%, n (%) | 537 (33%) | 58 (64%) | − | 1’819 (34%) | 112 (67%) | 62 (18%) | <0.001 | 2,418 (33%) | <0.001 | <0.001 | 1 |
| cT1, msec | 710 (676–748) | 757.5 (706.5–786) | − | 705 (672–742) | 746 (698–786) | 693 (662–728) | 0.064 | 705 (672–743) | <0.001 | <0.001 | 1 |
| cT1 NA, n (%) | 52,406 (98%) | 5,177 (99%) | 20 (100%) | 138’067 (98%) | 4,261 (98%) | 8,325 (97%) | <0.001 | 198,818 (98%) | <0.001 | 1 | <0.001 |
| MASH by PDFF>5.5% and cT1>800 msec, n (%) | 71 (5%) | 12 (18%) | − | 226 (5%) | 19 (15%) | 13 (4%) | <0.001 | 310 (5%) | <0.001 | <0.001 | 1 |
| Glycolipid profile | |||||||||||
| Glucose, mg/dL | 95.7 (23.4) | 165.8 (75) | 92.5 (22.3) | 90.7 (14.1) | 95.2 (23) | 87.1 (14.4) | <0.001 | 91.9 (17.3) | <0.001 | <0.001 | <0.001 |
| HbA1c, % | 5.5 (5.3–5.8) | 7.7 (6.7–8.7) | 5.5 (5.3–5.7) | 5.4 (5.2–5.6) | 5.4 (5.2–5.7) | 5.1 (4.9–5.3) | <0.001 | 5.4 (5.2–5.6) | <0.001 | <0.001 | <0.001 |
| Total cholesterol, mmol/L | 4.5 (0.6) | 4.6 (0.9) | 4.7 (0.6) | 6.3 (0.9) | 5.8 (1.1) | 4.7 (0.5) | <0.001 | 5.8 (1.2) | <0.001 | 0.089 | <0.001 |
| LDL cholesterol, mmol/L | 2.6 (0.4) | 2.8 (0.7) | 2.9 (0.4) | 4.1 (0.7) | 3.7 (0.8) | 2.8 (0.3) | <0.001 | 3.7 (0.9) | <0.001 | <0.001 | <0.001 |
| HDL cholesterol, mmol/L | 1.3 (0.4) | 1.1 (0.3) | 1.1 (0.2) | 1.4 (0.3) | 1.2 (0.3) | 1.4 (0.3) | <0.001 | 1.4 (0.3) | <0.001 | <0.001 | <0.001 |
| Triglycerides, mmol/L | 1.5 (1.1–2.1) | 3.1 (2.1–4.5) | 1.6 (1.2–2) | 1.8 (1.3–2.5) | 2.2 (1.6–3.2) | 1 (0.8–1.4) | <0.001 | 1.7 (1.2–2.3) | <0.001 | <0.001 | <0.001 |
| Liver function tests | |||||||||||
| ALT, U/L | 22.1 (17.2–28.9) | 34.9 (25.5–47.3) | 22.5 (16.4–26.9) | 21.7 (16.7–29) | 73.7 (65.2–86.7) | 16.6 (12.9–22.1) | <0.001 | 21.6 (16.6–28.8) | <0.001 | <0.001 | <0.001 |
| AST, U/L | 25.2 (21.7–29.6) | 29.3 (23.4–37.7) | 23.3 (20.9–25.8) | 24.6 (21.2–28.7) | 48 (39.8–59.8) | 21.2 (18.4–25.1) | <0.001 | 24.6 (21.2–28.9) | <0.001 | <0.001 | <0.001 |
| ALP, U/L | 82.1 (69.1–97.5) | 88.8 (73.7–106.1) | 86.2 (76.7–107.1) | 82.4 (69.6–97.5) | 87.2 (72.4–106.1) | 70.9 (60–83.7) | <0.001 | 81.8 (69–97) | <0.001 | <0.001 | 0.246 |
| GGT, U/L | 29.4 (21–44) | 48.2 (32.8–76) | 25.8 (21.8–46.3) | 29.1 (20.6–44.1) | 71 (47.1–116.3) | 19 (14.5–27) | <0.001 | 28.7 (20.3–43.4) | <0.001 | <0.001 | <0.001 |
| Bilirubin, mg/dL | 0.5 (0.3) | 0.5 (0.2) | 0.4 (0.1) | 0.5 (0.2) | 0.6 (0.3) | 0.5 (0.3) | <0.001 | 0.5 (0.2) | 0.015 | <0.001 | <0.001 |
| Albumin, g/dL | 4.5 (0.3) | 4.5 (0.3) | 4.2 (0.2) | 4.5 (0.3) | 4.6 (0.3) | 4.5 (0.3) | <0.001 | 4.5 (0.3) | 1 | <0.001 | <0.001 |
| Platelets, 10e3/uL | 244 (59.5) | 246.7 (63.2) | 289.1 (67.4) | 257.8 (58.2) | 245.1 (56.7) | 256.3 (57.7) | <0.001 | 254.1 (58.8) | <0.001 | <0.001 | 1 |
| Comorbidities | |||||||||||
| Hypertension, n (%) | 30,851 (58%) | 3,797 (73%) | 10 (50%) | 38,678 (27%) | 1,545 (35%) | 893 (10%) | <0.001 | 70,432 (35%) | <0.001 | 0.844 | <0.001 |
| Dyslipidemia, n (%) | 31,124 (58%) | 3,943 (75%) | 6 (30%) | 16,948 (12%) | 864 (20%) | 403 (5%) | <0.001 | 48,481 (24%) | <0.001 | <0.001 | <0.001 |
| Type 2 diabetes, n (%) | 8,007 (15%) | 3,948 (75%) | 5 (25%) | 2,369 (2%) | 243 (6%) | 130 (2%) | <0.001 | 10,511 (5%) | <0.001 | 0.72 | <0.001 |
| Genetic variants | |||||||||||
| PNPLA3 rs738409 C>G, n (%) | 33,068 (62%) | 3,156 (60%) | 8 (40%) | 88,103 (62%) | 2,081 (48%) | 5,332 (62%) | <0.001 | 126,511 (62%) / 67,779 (33%) / 9,111 (4%) | 0.005 | <0.001 | <0.001 |
| CC | 17,836 (33%) | 1,792 (34%) | 11 (55%) | 47,093 (33%) | 1,811 (41%) | 2,839 (33%) | |||||
| CG | 2,482 (5%) | 280 (5%) | 1 (5%) | 6,243 (4%) | 472 (11%) | 385 (4%) | |||||
| GG | |||||||||||
| TM6SF2 rs58542926 C>T, n (%) | 44,769 (84%) | 4,426 (85%) | 19 (95%) | 121,870 (86%) | 3474 (80%) | 7133 (83%) | <0.001 | 173791 (86%) | 0.622 | <0.001 | <0.001 |
| CC | 8,047 (15%) | 758 (15%) | 1 (5%) | 18,881 (13%) | 837 (19%) | 1331 (16%) | 28260 (14%) | ||||
| CT | 487 (1%) | 33 (1%) | 0 (0%) | 473 (0%) | 47 (1%) | 79 (1%) | 1039 (1%) | ||||
| TT | |||||||||||
| MBOAT7 rs641738 C>T, n (%) | 16,639 (31%) | 1,664 (32%) | 6 (30%) | 43,966 (31%) | 1,221 (28%) | 2,675 (31%) | <0.001 | 63286 (31%) | 0.908 | <0.001 | <0.001 |
| CC | 26,255 (50%) | 2,505 (48%) | 10 (50%) | 69,222 (49%) | 2,177 (50%) | 4,223 (50%) | 99710 (49%) | ||||
| CT | 10,093 (19%) | 1,017 (20%) | 4 (20%) | 27,205 (19%) | 924 (21%) | 1,605 (19%) | 38907 (19%) | ||||
| TT | |||||||||||
| GCKR rs1260326 C>T, n (%) | 20,307 (38%) | 1,905 (37%) | 8 (40%) | 51,475 (37%) | 1,493 (34%) | 3,341 (39%) | 0.010 | 75,131 (37%) | 0.649 | 0.002 | 0.217 |
| CC | 25,140 (47%) | 2,466 (47%) | 9 (45%) | 67,510 (48%) | 2,143 (49%) | 3,983 (47%) | 96,642 (48%) | ||||
| CT | 7,775 (15%) | 842 (16%) | 3 (15%) | 21,985 (16%) | 712 (16%) | 1,205 (14%) | 30,968 (15%) | ||||
| TT | |||||||||||
| PRS–HFC | 0.193 (0.126–0.394) | 0.256 (0.126–0.394) | 0.329 (0.128–0.394) | 0.193 (0.126–0.394) | 0.331 (0.128–0.459) | 0.193 (0.126–0.394) | <0.001 | 0.193 (0.126–0.394) | 0.01 | <0.001 | <0.001 |
| PRS-HFC cut-offs, n (%) | <0.001 | 12,474 (6%) | 0.004 | <0.001 | <0.001 | ||||||
| <10° percentile | 3,326 (6%) | 300 (6%) | 2 (10%) | 8,622 (6%) | 163 (4%) | 524 (6%) | 171,816 (84%) | ||||
| 10°–90° percentile | 44,515 (83%) | 4,360 (83%) | 16 (80%) | 120,155 (85%) | 3,334 (76%) | 7,130 (83%) | 19,293 (9%) | ||||
| >90° percentile | 5,589 (10%) | 571 (11%) | 2 (10%) | 12,793 (9%) | 869 (20%) | 909 (11%) | |||||
| Liver events | |||||||||||
| CLD, n (%) | 787 (1.473%) | 210 (4.015%) | 1 (5%) | 1,417 (1.001%) | 194 (4.443%) | 67 (0.782%) | <0.001 | 2,272 (1.116%) | <0.001 | <0.001 | 0.922 |
| Follow-up, years | 13.3 (12.4–14) | 13.2 (12.3–14) | 13.9 (12.9–14.3) | 13.4 (12.7–14.1) | 13.5 (12.7–14.2) | 13.5 (12.8–14.2) | <0.001 | 13.4 (12.6–14.1) | <0.001 | <0.001 | <0.001 |
| Participant characteristics across the clusters in the prospective UK Biobank cohort for cardiovascular outcome (n=195,739) (B) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | Cluster 6 | Adj-p | CTRL Cluster | Adj-p 2 vs CTRL | Adj-p 5 vs CTRL | Adj-p 2 vs 5 | |
| N | 42,093 | 3,999 | 19 | 137,039 | 4,137 | 8,452 | − | 187603 | − | − | − |
| Clinical data | |||||||||||
| Age, years | 60.5 (6.3) | 59.1 (6.9) | 43.5 (2.7) | 56.5 (7.6) | 51.6 (7.6) | 43.2 (2.2) | <0.001 | 56.8 (7.9) | <0.001 | <0.001 | <0.001 |
| Women, n (%) | 18,394 (44%) | 1,197 (30%) | 16 (84%) | 71,263 (52%) | 1042 (25%) | 5,579 (66%) | <0.001 | 95,252 (51%) | <0.001 | <0.001 | <0.001 |
| BMI, kg/m2 | 29.9 (4.5) | 32.3 (5.4) | 55.3 (1.5) | 29.3 (3.8) | 31.2 (4.1) | 28.2 (3.2) | <0.001 | 29.4 (3.9) | <0.001 | <0.001 | <0.001 |
| <25 | 852 (2%) | 185 (5%) | 0 (0%) | 262 (0%) | 7 (0%) | 60 (1%) | <0.001 | 1,174 (1%) | <0.001 | <0.001 | <0.001 |
| 25–30 | 24,979 (59%) | 1,345 (34%) | 0 (0%) | 92,030 (67%) | 1,885 (46%) | 6,670 (79%) | 123,679 (66%) | ||||
| ≥30 | 16,262 (39%) | 2,469 (62%) | 19 (100%) | 44,747 (33%) | 2,245 (54%) | 1,722 (20%) | 62,750 (33%) | ||||
| Waist circumference, cm | 97.4 (12.2) | 106.1 (13.4) | 135.2 (11.4) | 94.8 (10.7) | 102.3 (11.1) | 88.5 (9.9) | <0.001 | 95.1 (11.2) | <0.001 | <0.001 | <0.001 |
| Significant alcohol intake (n)1 | 8446 (20%) | 753 (19%) | 0 (0%) | 28560 (21%) | 1255 (30%) | 1693 (20%) | <0.001 | 38699 (21%) | <0.001 | <0.001 | <0.001 |
| Liver imaging | |||||||||||
| PDFF, % | 3.8 (2.4–7.1) | 7.7 (3.9–13.5) | − | 3.9 (2.5–7.1) | 9.7 (4.7–16.4) | 2.6 (1.9–4) | <0.001 | 3.8 (2.4–6.9) | <0.001 | <0.001 | 0.316 |
| PDFF NA, n (%) | 40,706 (97%) | 3,918 (98%) | 19 (100%) | 131,782 (96%) | 3,975 (96%) | 8,109 (96%) | <0.001 | 180,616 (96%) | <0.001 | 1 | <0.001 |
| Steatosis by PDFF>5.5%, n (%) | 453 (33%) | 53 (65%) | − | 1,773 (34%) | 109 (67%) | 61 (18%) | <0.001 | 2,287 (33%) | <0.001 | <0.001 | 1 |
| cT1, msec | 709 (675–746) | 757 (704–786) | − | 705 (672–742) | 745 (697.5–785.2) | 691 (662–728) | <0.001 | 705 (672–742) | <0.001 | <0.001 | 1 |
| cT1 NA, n (%) | 41,215 (98%) | 3,950 (99%) | 19 (100%) | 133,606 (97%) | 4,037 (98%) | 8,217 (97%) | <0.001 | 183,057 (98%) | <0.001 | 1 | <0.001 |
| MASH by PDFF>5.5% and cT1>800 msec, n (%) | 58 (5%) | 11 (19%) | − | 220 (5%) | 18 (15%) | 13 (4%) | <0.001 | 291 (5%) | <0.001 | <0.001 | 1 |
| Glycolipid profile | |||||||||||
| Glucose, mg/dL | 95.7 (23.7) | 166.6 (76.1) | 93.2 (22.8) | 90.6 (14) | 95.1 (22.8) | 87.1 (14.4) | <0.001 | 91.6 (16.9) | <0.001 | <0.001 | <0.001 |
| HbA1c, % | 5.5 (5.2–5.8) | 7.7 (6.6–8.7) | 5.6 (5.4–5.7) | 5.4 (5.2–5.6) | 5.4 (5.2–5.7) | 5.1 (4.9–5.3) | <0.001 | 5.4 (5.2–5.6) | <0.001 | <0.001 | <0.001 |
| Total cholesterol, mmol/L | 4.6 (0.6) | 4.7 (1) | 4.6 (0.5) | 6.3 (0.9) | 5.9 (1) | 4.7 (0.5) | <0.001 | 5.9 (1.1) | <0.001 | 1 | <0.001 |
| LDL cholesterol, mmol/L | 2.7 (0.4) | 2.8 (0.7) | 2.9 (0.4) | 4.1 (0.7) | 3.8 (0.8) | 2.8 (0.3) | <0.001 | 3.7 (0.9) | <0.001 | 0.004 | <0.001 |
| HDL cholesterol, mmol/L | 1.4 (0.4) | 1.1 (0.3) | 1.1 (0.2) | 1.4 (0.3) | 1.2 (0.3) | 1.4 (0.3) | <0.001 | 1.4 (0.3) | <0.001 | <0.001 | <0.001 |
| Triglycerides, mmol/L | 1.5 (1.1–2) | 3.1 (2.1–4.5) | 1.6 (1.2–1.9) | 1.8 (1.3–2.5) | 2.2 (1.6–3.2) | 1 (0.8–1.4) | <0.001 | 1.7 (1.2–2.3) | <0.001 | <0.001 | <0.001 |
| Liver function tests | |||||||||||
| ALT, U/L | 21.9 (17–28.7) | 35.4 (26–47.8) | 23 (16.9–27.4) | 21.7 (16.7–29) | 73.6 (65.1–86.3) | 16.5 (12.9–22.1) | <0.001 | 21.5 (16.5–28.7) | <0.001 | <0.001 | <0.001 |
| AST, U/L | 25 (21.5–29.3) | 29.4 (23.6–37.8) | 23.3 (20.8–25.9) | 24.5 (21.2–28.7) | 47.6 (39.6–59.2) | 21.2 (18.4–25.1) | <0.001 | 24.5 (21.1–28.7) | <0.001 | <0.001 | <0.001 |
| ALP, U/L | 81.9 (69.1–97.2) | 88.5 (73.7–105.4) | 85.6 (76.5–106.3) | 82.3 (69.5–97.4) | 87.2 (72.5–105.9) | 70.8 (60–83.6) | <0.001 | 81.7 (68.9–96.8) | <0.001 | <0.001 | 0.59 |
| GGT, U/L | 28.6 (20.4–42.5) | 47.7 (32.6–74.5) | 26.6 (22.1–46.5) | 29 (20.5–43.8) | 70.7 (47–116.7) | 19 (14.5–26.9) | <0.001 | 28.3 (20.1–42.8) | <0.001 | <0.001 | <0.001 |
| Bilirubin, mg/dL | 0.5 (0.3) | 0.5 (0.2) | 0.4 (0.1) | 0.5 (0.2) | 0.6 (0.3) | 0.5 (0.3) | <0.001 | 0.5 (0.2) | 0.016 | <0.001 | <0.001 |
| Albumin, g/dL | 4.5 (0.3) | 4.5 (0.3) | 4.2 (0.2) | 4.5 (0.3) | 4.6 (0.3) | 4.5 (0.3) | <0.001 | 4.5 (0.3) | 1 | <0.001 | <0.001 |
| Platelets, 10e3/uL | 246 (59.3) | 248.4 (62.9) | 288.6 (69.2) | 257.8 (58.1) | 245.6 (56.6) | 256.4 (57.6) | <0.001 | 255.1 (58.5) | <0.001 | <0.001 | 0.728 |
| Comorbidities | |||||||||||
| Hypertension, n (%) | 21,060 (50%) | 2,642 (66%) | 9 (47%) | 35,734 (26%) | 1,360 (33%) | 833 (10%) | <0.001 | 57,636 (31%) | <0.001 | 0.01 | <0.001 |
| Dyslipidemia, n (%) | 20,783 (49%) | 2,816 (70%) | 5 (26%) | 14,784 (11%) | 698 (17%) | 351 (4%) | <0.001 | 35,923 (19%) | <0.001 | <0.001 | <0.001 |
| Type 2 diabetes, n (%) | 6,165 (15%) | 2,974 (74%) | 5 (26%) | 2,048 (1%) | 215 (5%) | 127 (2%) | <0.001 | 8,345 (4%) | <0.001 | 0.074 | <0.001 |
| Genetic variants | |||||||||||
| PNPLA3 rs738409 C>G, n (%) | 25,995 (62%) | 2,371 (59%) | 7 (37%) | 85,196 (62%) | 1,981 (48%) | 5,258 (62%) | <0.001 | 116,456 (62%) | <0.001 | <0.001 | <0.001 |
| CC | 14,104 (34%) | 1,389 (35%) | 11 (58%) | 45,655 (33%) | 1,708 (41%) | 2,806 (33%) | 62,576 (33%) | ||||
| CG | 1,959 (5%) | 237 (6%) | 1 (5%) | 6,060 (4%) | 446 (11%) | 381 (5%) | 8,401 (4%) | ||||
| GG | |||||||||||
| TM6SF2 rs58542926 C>T, n (%) | 35,098 (84%) | 3,370 (84%) | 18 (95%) | 117,961 (86%) | 3,285 (80%) | 7,039 (83%) | <0.001 | 160,116 (86%) | 0.191 | <0.001 | <0.001 |
| CC | 6,465 (15%) | 591 (15%) | 1 (5%) | 18,292 (13%) | 798 (19%) | 1,315 (16%) | 26,073 (14%) | ||||
| CT | 424 (1%) | 28 (1%) | 0 (0%) | 453 (0%) | 46 (1%) | 78 (1%) | 955 (1%) | ||||
| TT | |||||||||||
| MBOAT7 rs641738 C>T, n (%) | 13,130 (31%) | 1,274 (32%) | 6 (32%) | 42,546 (31%) | 1,163 (28%) | 2,638 (31%) | 0.002 | 58,320 (31%) | 1 | <0.001 | 0.003 |
| CC | 20,675 (50%) | 1,911 (48%) | 10 (53%) | 66,996 (49%) | 2,059 (50%) | 4,171 (50%) | 91,852 (49%) | ||||
| CT | 7,927 (19%) | 781 (20%) | 3 (16%) | 26,357 (19%) | 875 (21%) | 1,585 (19%) | 35,872 (19%) | ||||
| TT | |||||||||||
| GCKR rs1260326 C>T, n (%) | 16,111 (38%) | 1,468 (37%) | 8 (42%) | 49,948 (37%) | 1,412 (34%) | 3,294 (39%) | <0.001 | 69,361 (37%) | 1 | 0.002 | 0.162 |
| CC | 19,764 (47%) | 1,898 (48%) | 8 (42%) | 65,287 (48%) | 2,034 (49%) | 3,933 (47%) | 88,992 (48%) | ||||
| CT | 6,045 (14%) | 619 (16%) | 3 (16%) | 21,221 (16%) | 673 (16%) | 1,191 (14%) | 28,460 (15%) | ||||
| TT | |||||||||||
| PRS–HFC | 0.193 (0.126–0.394) | 0.256 (0.126–0.394) | 0.329 (0.128–0.394) | 0.193 (0.126–0.394) | 0.331 (0.128–0.459) | 0.193 (0.126–0.394) | <0.001 | 0.193 (0.126–0.394) | <0.001 | <0.001 | <0.001 |
| PRS–HFC cut–offs, n (%) | 2,633 (6%) | 228 (6%) | 2 (11%) | 8,355 (6%) | 156 (4%) | 514 (6%) | <0.001 | 11,504 (6%) | <0.001 | <0.001 | <0.001 |
| <10° percentile | 34,948 (83%) | 3,301 (83%) | 15 (79%) | 116,290 (85%) | 3,158 (76%) | 7,040 (83%) | 158,293 (84%) | ||||
| 10°–90° percentile | 4,512 (11%) | 470 (12%) | 2 (11%) | 12,394 (9%) | 823 (20%) | 898 (11%) | 17,806 (9%) | ||||
| >90° percentile | |||||||||||
| Cardiovascular events | |||||||||||
| CLD, n (%) | 5,647 (13.416%) | 875 (21.88%) | 1 (5.263%) | 13,642 (9.955%) | 394 (9.524%) | 162 (1.917%) | <0.001 | 19,452 (10.369%) | <0.001 | 0.238 | <0.001 |
| Follow–up, years | 13.1 (12.2–14) | 13 (11–13.9) | 14 (13.2–14.3) | 13.3 (12.4–14) | 13.4 (12.5–14.2) | 13.5 (12.7–14.1) | <0.001 | 13.4 (12.7–14.1) | <0.001 | <0.001 | <0.001 |
| Participant characteristics across the clusters in the prospective UK Biobank cohort for type 2 diabetes outcome (n=196,791) (C) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | Cluster 6 | Adj-p | CTRL Cluster | Adj-p 2 vs CTRL | Adj-p 5 vs CTRL | Adj-p 2 vs 5 | |
| N | 45,021 | 942 | 15 | 138,365 | 4,017 | 8,431 | − | 191,832 | − | − | − |
| Clinical data | |||||||||||
| Age, years | 60.9 (6.1) | 59.5 (6.8) | 44.9 (3.3) | 56.6 (7.6) | 51.6 (7.7) | 43.2 (2.2) | <0.001 | 57 (7.9) | <0.001 | <0.001 | <0.001 |
| Women, n (%) | 18,476 (41%) | 151 (16%) | 13 (87%) | 71,711 (52%) | 1,004 (25%) | 5,555 (66%) | <0.001 | 95,755 (50%) | <0.001 | <0.001 | <0.001 |
| BMI, kg/m2 | 29.8 (4.1) | 31.3 (4.4) | 55.5 (1.6) | 29.3 (3.7) | 31 (4) | 28.2 (3.2) | <0.001 | 29.4 (3.8) | <0.001 | <0.001 | 0.274 |
| <25 | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | <0.001 | 0 (0%) | <0.001 | <0.001 | 0.895 |
| 25−30 | 28,290 (63%) | 427 (45%) | 0 (0%) | 93,366 (67%) | 1,899 (47%) | 6,708 (80%) | 128,364 (67%) | ||||
| ≥30 | 16,731 (37%) | 515 (55%) | 15 (100%) | 44,999 (33%) | 2,118 (53%) | 1,723 (20%) | 63,468 (33%) | ||||
| Waist circumference, cm | 97.3 (11.5) | 104.9 (11.2) | 136.2 (14.2) | 94.8 (10.7) | 101.9 (10.9) | 88.6 (9.8) | <0.001 | 95.1 (11) | <0.001 | <0.001 | <0.001 |
| Significant alcohol intake (n)1 | 9126 (20%) | 295 (31%) | 0 (0%) | 28884 (21%) | 1224 (30%) | 1695 (20%) | < 0.001 | 39705 (21%) | < 0.001 | < 0.001 | 1 |
| Liver imaging | |||||||||||
| PDFF, % | 3.7 (2.4−6.4) | 7.6 (5.9−11.9) | − | 3.9 (2.5−7.1) | 9.5 (4.8−16.5) | 2.6 (1.9−4) | 1 | 3.8 (2.4−6.8) | 0.003 | <0.001 | 1 |
| PDFF NA, n (%) | 43,594 (97%) | 923 (98%) | 15 (100%) | 133,080 (96%) | 3,857 (96%) | 8,089 (96%) | <0.001 | 184,778 (96%) | 0.016 | 0.929 | 0.008 |
| Steatosis by PDFF>5.5%, n (%) | 433 (30%) | 15 (79%) | − | 1,772 (34%) | 108 (68%) | 61 (18%) | <0.001 | 2,266 (32%) | <0.001 | <0.001 | 1 |
| cT1, msec | 708 (675−746) | 762.3 (758−803.5) | − | 705 (672−742) | 746 (699−786) | 693 (662−728.5) | 1 | 705 (672−742) | 0.002 | <0.001 | 0.367 |
| cT1 NA, n (%) | 44,118 (98%) | 932 (99%) | 15 (100%) | 134,916 (98%) | 3916 (97%) | 8,196 (97%) | <0.001 | 187,245 (98%) | 0.015 | 1 | 0.014 |
| MASH by PDFF>5.5% and cT1>800 msec, n (%) | 58 (5%) | 3 (27%) | − | 217 (5%) | 18 (15%) | 13 (4%) | <0.001 | 288 (5%) | 0.035 | <0.001 | 1 |
| Glycolipid profile | |||||||||||
| Glucose, mg/dL | 91.1 (12.6) | 97.4 (18.9) | 86 (10.6) | 89.9 (11.5) | 91.9 (14.8) | 86.6 (10.2) | <0.001 | 90.1 (11.7) | <0.001 | <0.001 | <0.001 |
| HbA1c, % | 5.4 (5.2−5.7) | 5.7 (5.5−6) | 5.4 (5.2−5.6) | 5.4 (5.2−5.6) | 5.4 (5.2−5.6) | 5.1 (4.9−5.3) | <0.001 | 5.4 (5.2−5.6) | <0.001 | <0.001 | <0.001 |
| Total cholesterol, mmol/L | 4.6 (0.6) | 5 (0.7) | 4.7 (0.5) | 6.3 (0.9) | 5.9 (1) | 4.7 (0.5) | <0.001 | 5.8 (1.1) | <0.001 | 1 | <0.001 |
| LDL cholesterol, mmol/L | 2.7 (0.4) | 2.9 (0.5) | 3 (0.3) | 4.1 (0.7) | 3.8 (0.8) | 2.8 (0.3) | <0.001 | 3.7 (0.9) | <0.001 | <0.001 | <0.001 |
| HDL cholesterol, mmol/L | 1.3 (0.4) | 1 (0.2) | 1.2 (0.2) | 1.4 (0.3) | 1.2 (0.3) | 1.4 (0.3) | <0.001 | 1.4 (0.3) | <0.001 | <0.001 | <0.001 |
| Triglycerides, mmol/L | 1.5 (1.1−2) | 5.4 (4.5−6.1) | 1.4 (1.2−1.9) | 1.8 (1.3−2.5) | 2.2 (1.6−3.2) | 1 (0.8−1.4) | <0.001 | 1.7 (1.2−2.3) | <0.001 | <0.001 | <0.001 |
| Liver function tests | |||||||||||
| ALT, U/L | 22 (17.1−28.8) | 41.1 (31.7−51.7) | 22 (16.9−24.1) | 21.7 (16.6−29) | 73.2 (65−85.5) | 16.5 (12.9−22.1) | <0.001 | 21.5 (16.5−28.7) | <0.001 | <0.001 | <0.001 |
| AST, U/L | 25.4 (21.9−29.8) | 33.8 (28.3−40.6) | 23.3 (20.6−24.9) | 24.6 (21.2−28.7) | 47.4 (39.6−58.9) | 21.2 (18.4−25.1) | <0.001 | 24.6 (21.2−28.9) | <0.001 | <0.001 | <0.001 |
| ALP, U/L | 82 (69.2−97.3) | 86.2 (72.5−101.5) | 99.1 (77.7−107.7) | 82.3 (69.5−97.3) | 87.1 (72.4−105.7) | 70.9 (60−83.7) | <0.001 | 81.7 (68.9−96.8) | <0.001 | <0.001 | 0.141 |
| GGT, U/L | 29.1 (20.8−43.6) | 55.9 (39.2−85.8) | 24.2 (20.2−34.1) | 29 (20.5−43.8) | 69.8 (46.4−115.1) | 19 (14.5−27.1) | <0.001 | 28.5 (20.2−43.1) | <0.001 | <0.001 | <0.001 |
| Bilirubin, mg/dL | 0.5 (0.3) | 0.5 (0.3) | 0.4 (0.1) | 0.5 (0.2) | 0.6 (0.3) | 0.5 (0.3) | <0.001 | 0.5 (0.2) | 1 | <0.001 | <0.001 |
| Albumin, g/dL | 4.5 (0.3) | 4.6 (0.3) | 4.1 (0.2) | 4.5 (0.3) | 4.6 (0.3) | 4.5 (0.3) | <0.001 | 4.5 (0.3) | <0.001 | <0.001 | <0.001 |
| Platelets, 10e3/uL | 243.6 (58.9) | 238.5 (56.9) | 290.2 (67.4) | 257.7 (58) | 245.6 (56.5) | 256.4 (57.6) | <0.001 | 254.4 (58.5) | <0.001 | <0.001 | 0.005 |
| Comorbidities | |||||||||||
| Hypertension, n (%) | 24,433 (54%) | 578 (61%) | 8 (53%) | 36,937 (27%) | 1,343 (33%) | 855 (10%) | <0.001 | 62233 (32%) | <0.001 | 0.553 | <0.001 |
| Dyslipidemia, n (%) | 23,912 (53%) | 553 (59%) | 3 (20%) | 15,744 (11%) | 701 (17%) | 363 (4%) | <0.001 | 40022 (21%) | <0.001 | <0.001 | <0.001 |
| Genetic variants | |||||||||||
| PNPLA3 rs738409 C>G, n (%) | 27,892 (62%) | 586 (62%) | 6 (40%) | 86,045 (62%) | 1,923 (48%) | 5,255 (62%) | <0.001 | 119,198 (62%) | 1 | <0.001 | <0.001 |
| CC | 15,001 (33%) | 307 (33%) | 8 (53%) | 46,075 (33%) | 1,655 (41%) | 2,790 (33%) | 63,874 (33%) | ||||
| CG | 2,088 (5%) | 48 (5%) | 1 (7%) | 6,115 (4%) | 437 (11%) | 380 (5%) | 8,584 (4%) | ||||
| GG | |||||||||||
| TM6SF2 rs58542926 C>T, n (%) | 37,710 (84%) | 820 (88%) | 14 (93%) | 119,152 (86%) | 3,190 (80%) | 7,024 (83%) | <0.001 | 163,900 (86%) | 0.706 | <0.001 | <0.001 |
| CC | 6,795 (15%) | 114 (12%) | 1 (7%) | 18,414 (13%) | 774 (19%) | 1,310 (16%) | 26,520 (14%) | ||||
| CT | 414 (1%) | 3 (0%) | 0 (0%) | 459 (0%) | 46 (1%) | 78 (1%) | 951 (0%) | ||||
| TT | |||||||||||
| MBOAT7 rs641738 C>T, n (%) | 14,088 (32%) | 300 (32%) | 5 (33%) | 43,002 (31%) | 1,101 (28%) | 2,639 (32%) | <0.001 | 59,734 (31%) | 1 | <0.001 | 0.073 |
| CC | 22,080 (49%) | 459 (49%) | 7 (47%) | 67,621 (49%) | 2,021 (51%) | 4,164 (50%) | 93,872 (49%) | ||||
| CT | 8,467 (19%) | 179 (19%) | 3 (20%) | 26,588 (19%) | 852 (21%) | 1,569 (19%) | 36,627 (19%) | ||||
| TT | |||||||||||
| GCKR rs1260326 C>T, n (%) | 16,955 (38%) | 270 (29%) | 6 (40%) | 50,225 (36%) | 1,363 (34%) | 3,290 (39%) | <0.001 | 70,476 (37%) | <0.001 | 0.003 | 0.004 |
| CC | 21,242 (47%) | 480 (51%) | 6 (40%) | 66,043 (48%) | 1,981 (50%) | 3,926 (47%) | 91,217 (48%) | ||||
| CT | 6,655 (15%) | 189 (20%) | 3 (20%) | 21,508 (16%) | 655 (16%) | 1,181 (14%) | 29,347 (15%) | ||||
| TT | |||||||||||
| PRS−HFC2 | 0.193 (0.126−0.394) | 0.193 (0.128−0.394) | 0.329 (0.128−0.426) | 0.193 (0.126−0.394) | 0.331 (0.128−0.459) | 0.193 (0.126−0.394) | <0.001 | 0.193 (0.126−0.394) | 0.949 | <0.001 | <0.001 |
| PRS−HFC cut−offs, n (%) | 2,809 (6%) | 39 (4%) | 2 (13%) | 8,428 (6%) | 146 (4%) | 513 (6%) | <0.001 | 11,752 (6%) | 0.097 | <0.001 | <0.001 |
| <10° percentile | 37,507 (83%) | 818 (87%) | 11 (73%) | 117,435 (85%) | 3,066 (76%) | 7,023 (83%) | 161,976 (84%) | ||||
| 10–90° percentile | 4,705 (10%) | 85 (9%) | 2 (13%) | 12,502 (9%) | 805 (20%) | 895 (11%) | 18,104 (9%) | ||||
| >90° percentile | |||||||||||
| Diabetes events | |||||||||||
| Type 2 diabetes, n (%) | 2,934 (6.517%) | 257 (27.282%) | 1 (6.667%) | 4,939 (3.57%) | 375 (9.335%) | 57 (0.676%) | <0.001 | 7,931 (4.134%) | <0.001 | <0.001 | <0.001 |
| Follow-up, years | 13.2 (12.3–14) | 12.8 (9.5–13.9) | 13.9 (12.8–14.4) | 13.4 (12.6–14.1) | 13.4 (12.5–14.2) | 13.5 (12.8–14.2) | <0.001 | 13.3 (12.6–14.1) | <0.001 | 0.339 | <0.001 |
Continuous variables are shown as mean (SD) or median (IQR) as appropriate. Categorical variables are shown as frequency (percentage).
Cluster control group is defined as cluster 1 + 3 + 4 + 6. Clusters were compared using Kruskal-Wallis test, Chi-squared test, or Fisher’s exact test, as appropriate.The adjusted P value is reported for comparisons across the three clusters, as well as for post hoc comparisons between cluster 2 versus CTRL, cluster 5 versus CTRL, and cluster 2 versus cluster 5. Differences were considered statistically significant when p-value(s) adjusted for multiple comparisons using Bonferroni correction were less than 0.05. For variables statistically significant, post-hoc analysis was performed comparing pairwise MASH-enriched clusters (2 and 5) and the combined non-enriched MASH clusters (1, 3, 4, and 6) using the Dunn test, Chi-squared test, or Fisher’s exact test, as appropriate, with Bonferroni adjustment.
1: Significant alcohol intake was defined as a daily consumption above 20 g in women and 30 g in men.
2:PRS + Polygenic Risk Score was calculated with the formula: prs=0.266∗PNPLA3_012 + 0.274∗TMS6F2_012 + 0.065∗GCKR_012 + 0.063∗MBOAT7_012
ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; eGFR, estimates of glomerular filtration rate; GCKR, glucokinase regulator; GGT, gamma-glutamyltransferase; HbA1c, hemoglobin A1c; HDL, high-density lipoprotein; HOMA2-B, homeostasis model assessment 2 estimates of beta-cell function; HOMA2-IR, homeostasis model assessment 2 estimates of insulin-resistance; LDL, low-density lipoprotein; MBOAT7, membrane-bound O-acyltransferase domain-containing 7; PNPLA3, patatin-like phospholipase domain-containing 3; PRS-HFC, polygenic risk score of hepatic fat content; TM6SF2, transmembrane 6 superfamily member, Adj-p, adjusted-p.
The liver-specific cluster is enriched in at-risk genetic variants
MASLD has a strong genetic component with variants in PNPLA3, TM6SF2, MBOAT7 and GCKR accounting for a large fraction of its heritability and accelerating liver disease progression to MASH, cirrhosis and hepatocellular carcinoma15–17. We hypothesized that the liver-specific cluster could be enriched in these genetic variants. Therefore, we examined the difference of polygenic risk score of hepatic fat content (PRS-HFC) distribution in the liver-specific cluster 5 compared with the cardiometabolic and control clusters in ABOS, finding an enrichment of PRS-HFC in this cluster (adjusted P = 0.034 and adjusted P < 0.001 versus the cardiometabolic and control clusters, respectively) (Table 1). Results were similar when we considered only the PNPLA3 rs738409 variant (P < 0.01 and P < 0.001 versus the cardiometabolic and control clusters, respectively) (Fig. 3). These results were confirmed in UK Biobank participants (Extended Data Table 2).
Fig. 3. Genotype distribution of the PNPLA3 rs738409 C > G stratified by clusters in the ABOS cohort and UK Biobank and differential hepatic gene expression and plasma metabolomics across clusters in the ABOS cohort.

a, Genotype distribution of the PNPLA3 rs738409 C > G stratified by clusters in the ABOS cohort. The bar graph shows the percentages of homozygotes (GG) and heterozygotes (CG) patients at risk across liver-specific (LS), cardiometabolic (CM) and control clusters. Statistical tests were chi-squared test or Fisher exact test as appropriate, two-sided with Bonferroni correction. Significance levels are indicated as follows: $ indicates P = 0.0079, ***P < 0.001. b,c, Differential hepatic gene expression and plasma metabolomics across clusters. The Euler diagrams illustrate the differential gene expression in liver tissue (b) and plasma metabolomics (c), across the three clusters: cardiometabolic (CM), liver-specific (LS) and control (CTRL). The sizes of the areas in the Euler diagram are proportional to the number of differentially expressed features they represent.
Risk of liver and cardiovascular outcomes, and type 2 diabetes
In the UK Biobank, individuals allocated in the six clusters exhibited similar characteristics to those observed in the ABOS cohort (Extended Data Table 2 and Extended Data Fig. 4).
Extended Data Fig. 4. Distribution of clustering variables in the UK Biobank cohort.

Distribution of clustering variables in the various subpopulations used for analyzing cumulative incidences of (a) liver outcome (n = 213,180), (b) cardiovascular disease (n = 195,739), and (c) type 2 diabetes (n = 196,791), in the UK Biobank cohort. Radar charts represent the median values of age, BMI, HbA1c, LDL, triglycerides, and ALT for each cluster. The dark gray line represents the 95th percentile observed in the ABOS cohort. ALT, alanine aminotransferase; BMI, body mass index; HbA1c, hemoglobin A1c; LDL, low-density lipoprotein cholesterol.
During a median (interquartile range) follow-up of 13.4 (12.6–14.1) years, there were 2,676 (1.12%) individuals who developed chronic liver disease, with the liver-specific and cardiometabolic clusters being the ones with the highest cumulative incidence (both P < 0.001 versus control cluster) (Fig. 4 and Extended Data Table 2). Following adjustment for age, sex and alcohol intake, the liver-specific and cardiometabolic clusters had a more than fourfold increased risk of chronic liver disease compared with the control cluster (adjusted hazard ratio (HR) 4.52, 95% confidence interval (CI) 3.88–5.26, P < 0.001, and adjusted HR 4.04, 95% CI 3.50–4.66, P < 0.001, respectively) (Fig. 4).
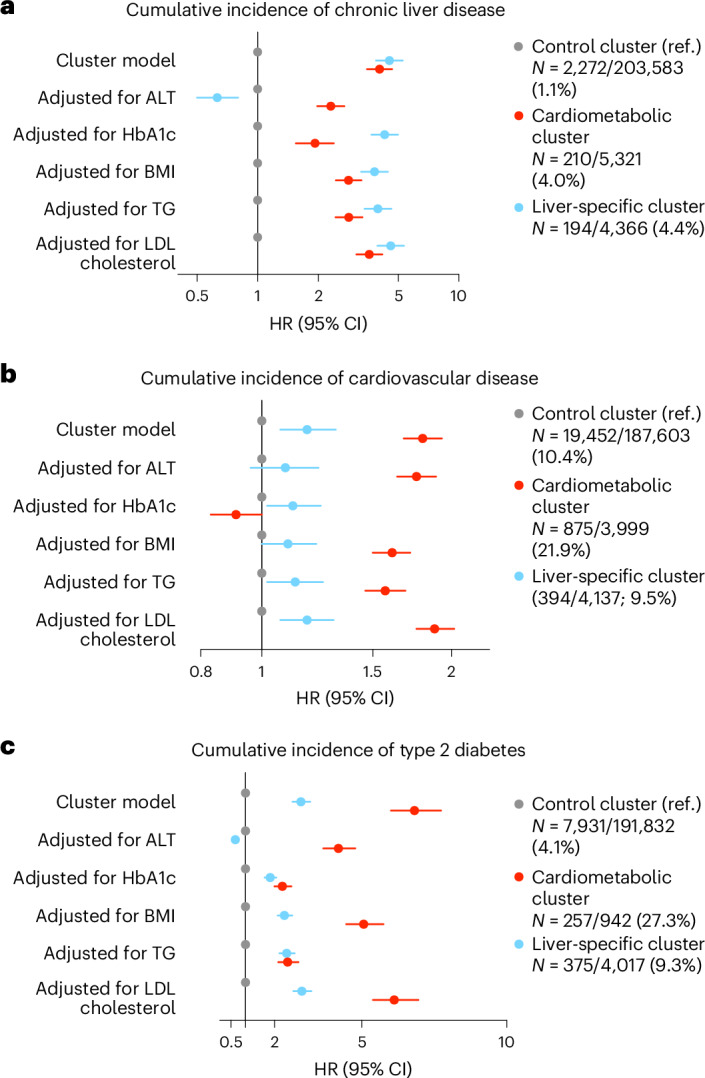
Fig. 4. Cumulative incidence of chronic liver disease, cardiovascular disease and type 2 diabetes across clusters in the prospective UK Biobank.

a–c, Cumulative incidence of chronic liver disease (a), cardiovascular disease (b) and type 2 diabetes (c) across clusters in the prospective UK Biobank. In each panel, the lines represent the cumulative incidence in the different clusters (cardiometabolic (CM) in red, liver-specific (LS) in blue and control in gray), with the shaded area representing 95% CI. HRs with 95% CIs and corresponding P value were calculated by Cox proportional hazards models for cardiometabolic (in red) and liver-specific (in blue) clusters versus control cluster (in gray), adjusted for age, sex and alcohol intake (g per day). Survival curves were compared using the pairwise log-rank test, with Holm correction.
During a median (interquartile range) follow-up of 13.4 (12.7–14.1) years, there were 20,721 (10.59%) individuals who developed cardiovascular disease, with the cardiometabolic cluster being the one with the highest cumulative incidence: 21.88% in the cardiometabolic cluster versus 10.37% in the control cluster (HR 2.31, 95% CI 2.16–2.47; P < 0.001 versus control), and 9.52% in the liver-specific cluster (HR 0.91, 95% CI 0.82–1.00; P = 0.054 versus control) (Fig. 4 and Extended Data Table 2). When the analysis was adjusted for age, sex and alcohol intake, the cardiometabolic cluster had a significantly increased risk of experiencing cardiovascular disease compared with the control cluster (adjusted HR 1.80, 95% CI 1.68–1.93; P < 0.001), which was also significantly higher than the increase in risk of the liver-specific cluster compared with the control cluster (adjusted HR 1.18, 95% CI 1.07–1.31; P = 0.001) (Fig. 4).
During a median (interquartile range) follow-up of 13.3 (12.6–14.1) years, there were 8,563 (4.35%) individuals who developed type 2 diabetes, with the cardiometabolic cluster being the one with the highest cumulative incidence (P < 0.001 versus both liver-specific and control clusters) (Fig. 4 and Extended Data Table 2). Following adjustment for age, sex and alcohol intake, the cardiometabolic cluster had a nearly sevenfold increased risk of developing type 2 diabetes compared with the control cluster (adjusted HR 6.82, 95% CI 6.01–7.73; P < 0.001), which was higher than the increase in risk of the liver-specific cluster compared with the control cluster (adjusted HR 2.91, 95% CI 2.62–3.23; P < 0.001) (Fig. 4).
Of note, a majority of participants from the cardiometabolic cluster also presented with type 2 diabetes, which may explain the higher risk of cardiovascular disease observed in this cluster. Likewise, the mean HbA1c level remained superior in the cardiometabolic cluster after excluding patients with preexisting type 2 diabetes for analyzing incident diabetes (Extended Data Table 2). However, adjusting for HbA1c did not fully remove the association of the cardiometabolic cluster with type 2 diabetes risk.
Sensitivity analyses excluding individuals with BMI <27 kg m−2 or those with excessive alcohol consumption (>50/60 g per day for women/men) showed similar results to the main analysis (Extended Data Table 3).
Extended Data Table 3.
Risk of incident chronic liver disease, cardiovascular disease, and type 2 diabetes, across the clusters in the prospective UK Biobank cohort including only those with BMI ≥ 27 kg/m2 (A). Risk of incident chronic liver disease, cardiovascular disease, and type 2 diabetes, across the clusters in the prospective UK Biobank cohort excluding those with harmful alcohol consumption (>50/60 g/day for women/men) (B)
| Risk of incident chronic liver disease, cardiovascular disease, and type 2 diabetes, across the clusters in the prospective UK Biobank cohort including only those with BMI≥27 kg/m2 (A). | |||
|---|---|---|---|
| Control | Cardiometabolic | Liver-specific | |
| Chronic liver disease | |||
| N | 140,872 | 4,569 | 3,747 |
| Events, n (%) | 1928 (1.369%) | 199 (4.355%) | 183 (4.884%) |
| Follow-up, years | 13.4 (12.6–14.1) | 13.2 (12.3–14) | 13.5 (12.7–14.2) |
| Adjusted model | Reference | 3.59 (3.09–4.16) | 4.04 (3.45–4.73) |
| P value | - | <0.001 | <0.001 |
| Cardiovascular disease | |||
| N | 128632 | 3449 | 3546 |
| Events, n (%) | 14153 (11.003%) | 756 (21.919%) | 344 (9.701%) |
| Follow-up, years | 13.4 (12.6–14.1) | 13.3 (12.4–14.1) | 13.5 (12.7–14.2) |
| Adjusted HR (95% CI) | Reference | 2.17 (2.02–2.33) | 0.87 (0.78–0.96) |
| P value | − | <0.001 | 0.009 |
| Type 2 diabetes | |||
| N | 131978 | 819 | 3427 |
| Events, n (%) | 6845 (5.186%) | 242 (29.548%) | 343 (10.009%) |
| Follow-up, years | 13.3 (12.5–14.1) | 12.7 (9.1–13.9) | 13.4 (12.4–14.2) |
| Adjusted HR (95% CI) | Reference | 6.07 (5.33–6.91) | 2.41 (2.15–2.69) |
| P value | − | <0.001 | <0.001 |
| Risk of incident chronic liver disease, cardiovascular disease, and type 2 diabetes, across the clusters in the prospective UK Biobank cohort excluding those with harmful alcohol consumption (>50/60 g/day for women/men) (B). | |||
|---|---|---|---|
| Control | Cardiometabolic | Liver-specific | |
| Chronic liver disease | |||
| N | 197,729 | 5,026 | 4,05 |
| Events, n (%) | 2,185 (1.105%) | 202 (4.019%) | 179 (4.42%) |
| Follow-up, years | 13.4 (12.6–14.1) | 13.2 (12.3–14) | 13.5 (12.7–14.2) |
| Adjusted model | Reference | 3.99 (3.44–4.62) | 4.55 (3.88–5.32) |
| P value | − | <0.001 | <0.001 |
| Cardiovascular disease | |||
| N | 182186 | 3826 | 3836 |
| Events, n (%) | 18785 (10.311%) | 842 (22.007%) | 358 (9.333%) |
| Follow-up, years | 13.4 (12.7–14.1) | 13.3 (12.4–14.1) | 13.5 (12.7–14.2) |
| Adjusted HR (95% CI) | Reference | 1.81 (1.68–1.94) | 1.17 (1.05–1.3) |
| P value | - | <0.001 | 0.004 |
| Type 2 diabetes | |||
| N | 186269 | 850 | 3723 |
| Events, n (%) | 7689 (4.128%) | 239 (28.118%) | 345 (9.267%) |
| Follow-up, years | 13.3 (12.6–14.1) | 12.8 (9.5–13.9) | 13.4 (12.5–14.2) |
| Adjusted HR (95% CI) | Reference | 6.87 (6.03–7.82) | 2.87 (2.57–3.21) |
| P value | - | <0.001 | <0.001 |
Cluster control group is defined as cluster 1 + 3 + 4 + 6.
HRs with 95% CIs were calculated by Cox proportional hazards models adjusted for age, sex and alcohol intake (g/day).
Abbreviations: CI, confidence interval; CLD, chronic liver disease; CVD, cardiovascular disease; HR, hazard ratio.
In summary, the cardiometabolic cluster had a higher risk of developing cardiovascular disease and type 2 diabetes, and a similar risk of developing chronic liver disease, as compared with the liver-specific cluster.
The added value of clustering beyond individual variables
We then explored the added value of the proposed clustering, beyond each of its individual components, to predict the various clinical outcomes. For that purpose, for each outcome, we first examined the overall predictive power of each variable of interest compared with clustering alone. No individual variable performed better than clustering at predicting simultaneously the three clinical outcomes (Extended Data Table 4). For example, ALT alone predicted incident chronic liver disease better than clustering, but clustering was superior at predicting cardiovascular disease. In contrast, HbA1c predicted incident cardiovascular disease better than clustering, but clustering performed better in the prediction of chronic liver disease. Likewise, among patients without diabetes at the time of inclusion, age, BMI, HbA1c, ALT and triglycerides performed better in predicting the risk of incident diabetes better than clustering alone. In contrast, clustering did better than LDL cholesterol alone at predicting all outcomes.
Extended Data Table 4.
Association between each individual variable included in clustering and the cumulative incidence of the three clinical outcomes among UK Biobank participants
| Chronic liver disease | AIC | ||
|---|---|---|---|
| Hazard ratio | LogLik, | ||
| (95%CI), p | p variable vs clustering | ||
| Clustering | − | −29826 | 59656 |
| ALT | 1.03 (1.03–1.03), <0.001 | −29699, <0.001 | 59399 |
| HbA1c | 1.48 (1.43–1.53), <0.001 | −29873, <0.001 | 59748 |
| Triglycerides | 1.32 (1.29–1.37), <0.001 | −29883, <0.001 | 59768 |
| Body mass index | 1.11 (1.10–1.12), <0.001 | −29657, <0.001 | 59316 |
| Age | 1.00 (1.00–1.01), 0.044 | −30016, <0.001 | 60033 |
| LDL cholesterol | 0.84 (0.80–0.87), <0.001 | −29990, <0.001 | 59982 |
| Cardiovascular disease | |||
| Hazard ratio | LogLik, | AIC | |
| (95%CI), p | p variable vs clustering | ||
| Clustering | − | −250222 | 500448 |
| ALT | 1.00 (1.00–1.01), <0.001 | −250419, <0.001 | 500839 |
| HbA1c | 1.40 (1.38–1.42), <0.001 | −249694, <0.001 | 499390 |
| Triglycerides | 1.16 (1.15−1.18),<0.001 | −250378, <0.001 | 500557 |
| Body mass index | 1.03 (1.02–1.03),<0.001 | −250312, <0.001 | 500627 |
| Age | 1.07 (1.07–1.08),<0.001 | −247706, <0.001 | 495413 |
| LDL cholesterol | 0.97 (0.96–0.99), 0.001 | −250450, <0.001 | 500902 |
| Type 2 diabetes | |||
| Hazard ratio | LogLik, | AIC | |
| (95%CI), p | p variable vs clustering | ||
| Clustering | − | −103310 | 206624 |
| ALT | 1.02 (1.02–1.02), <0.001 | −103179, <0.001 | 206361 |
| HbA1c | 33.72 (31.74–35.82), <0.001 | −97390, <0.001 | 194781 |
| Triglycerides | 1.43 (1.41–1.46), <0.001 | −102906, <0.001 | 205814 |
| Body mass index | 1.13 (1.12–1.13), <0.001 | −102129, <0.001 | 204259 |
| Age | 1.05 (1.04–1.05), <0.001 | −103205, <0.001 | 206413 |
| LDL cholesterol | 0.80 (0.78–0.82), <0.001 | −103556, <0.001 | 207113 |
Univariate analysis of the association between each individual variable included in clustering and the cumulative incidence of the three clinical outcomes (chronic liver disease, cardiovascular disease and Type 2 diabetes) among UK Biobank participants.
Second, we performed multivariable analyses, in which the clustering model was first adjusted for sex, age and alcohol use, and second, one by one, ALT, HbA1c, triglycerides, BMI or LDL cholesterol (Fig. 5). Although in most cases the HR estimates of at-risk clusters were reduced after further adjustment for one other clustering variable, all values remained statistically significant compared with the control cluster in at least one at-risk cluster for each outcome. Collectively, these data show that clustering was superior to each individual variable in predicting simultaneously all three clinical trajectories.
Fig. 5. Added value of the clustering model to predict cumulative incidence of chronic liver disease, cardiovascular disease and type 2 diabetes, among UK Biobank participants.
a–c, Added value of the clustering model to predict cumulative incidence of chronic liver disease (a), cardiovascular disease (b) and type 2 diabetes (c), among UK Biobank participants. Multivariable analyses evaluating the predictive value of the clustering model adjusted for age/sex/alcohol, independently of each additional individual variable, are included in the clustering. TG, triglycerides. The dots represent the HR estimates, and the error bars represent the 95% CIs.
Differential liver transcriptomic analysis across clusters
To gain insights into the biological differences between the cardiometabolic and liver-specific clusters, we performed differential gene expression analysis in the liver in a subset of the ABOS cohort participants, including 97 individuals from the cardiometabolic cluster, 63 from the liver-specific cluster and 671 from the control cluster.
The comparison of the cardiometabolic and the liver-specific clusters showed upregulation of genes involved in cholesterol metabolism and biosynthesis (for example, HMGCS1, MVD, CYP51A1, LSS, SC5D and LDLR) and glycolysis (for example, ALDOC) in the cardiometabolic cluster (Fig. 3 and Supplementary Table 1), which were identified as enriched pathways also by Gene Ontology biological processes (GO-BP) analysis, together with alcohol metabolic processes (Extended Data Fig. 3). The chitinase 3-like 1 (CHI3L1) gene, linked to liver fibrogenesis18, was the most highly differentially expressed, possibly reflecting a slightly higher albeit not significantly different fibrosis stage in the individuals in this cluster as well as an older age (Table 1). Similar results were obtained when comparing the cardiometabolic and the control clusters, confirming the upregulation of genes involved in cholesterol metabolism and synthesis in the cardiometabolic cluster (Extended Data Fig. 3), mirroring the higher metabolic dysfunction, type 2 diabetes and cardiovascular risk observed in this cluster.
Extended Data Fig. 3. Differential Liver Gene Expression, Pathway Enrichment, and Plasma Metabolomics Across Cardiometabolic and Liver-Specific Clusters.

Upper volcano plots illustrate the comparative liver gene expression in (a) the cardiometabolic cluster (CM; n = 97) vs control cluster (CTRL; n = 671), (b) the liver-specific cluster (LS; n = 63) vs control cluster, and (c) the cardiometabolic cluster vs liver-specific cluster. Horizontal and vertical lines represent adjusted p-value with Benjamin Hochberg correction (0.05) and log2FoldChange (0.26) significance thresholds respectively. Bar plots representing Gene Ontology Biological Processes (GO-BP) enrichment analysis for differentially expressed genes in the liver between (d) the cardiometabolic cluster (CM; n = 97) vs the control cluster (CTRL; n = 671), (e) the liver-specific cluster (LS; n = 63) vs the control cluster, and (f) the cardiometabolic cluster vs the liver-specific cluster. Graphs were generated using the GSEA enrichment method from the R package clusterProfiler. Lower volcano plots illustrate the comparative metabolomics in (g) cardiometabolic cluster (CM; n = 151) vs control cluster (CTRL; n = 1076), (h) liver-specific cluster (LS; n = 95) vs control cluster, and (i) cardiometabolic cluster vs liver-specific cluster. Horizontal and vertical lines represent adjusted p-value of 0.05 with Benjamini-Hochberg correction and log2FoldChange (0.26) significance thresholds respectively. The statistical tests are based on Limma’s linear model, performing a two-sided moderated t-test for each variable. P-values are adjusted for multiple comparisons using the Benjamini-Hochberg method to control the False Discovery Rate (FDR).
When comparing the liver-specific and the control clusters, we observed upregulation of genes involved in lipid droplet homeostasis and intrahepatic lipid transport, including FABP4 and FABP5, in the liver-specific cluster. This cluster also showed upregulation of genes implicated in inflammation, including CXCL9 and SPP1, and liver carcinogenesis, including ANXA2P1 and HULC (Extended Data Fig. 3 and Supplementary Table 1). GO-BP analysis confirmed these results, showing an upregulation of lipid localization, immunoregulatory, inflammatory and wound healing processes19 and mirroring the elevated liver enzymes observed in this cluster as well as a higher risk of progressive liver disease in UK Biobank (Extended Data Fig. 3).
Differential metabolomic analysis across clusters
To further elucidate biological differences between the cardiometabolic and liver-specific clusters, we analyzed the metabolomics data available in ABOS (Fig. 3). When comparing the cardiometabolic and liver-specific clusters, we observed increased concentrations of carbohydrates in the cardiometabolic cluster (Extended Data Fig. 3), reflecting the dysglycemic state (Table 1). However, most differences concerned amino acid and lipid metabolites, and particularly the amino acid metabolites tyramine O-sulfate, homocitrulline, p-cresol glucuronide, phenylacetylglutamine, phenylacetylglutamate, 4-hydroxyphenylacetylglutamine, 4-hydroxyphenylacetate and imidazole propionate, previously associated with the gut microbiota20–22, had the highest and most significant increase in the cardiometabolic cluster. Deoxycholate, a secondary bile acid, was also elevated, suggesting changes in lipid metabolism and liver function. These metabolites were also differentially abundant between the cardiometabolic and control clusters (Extended Data Fig. 3 and Supplementary Table 1) and, therefore, probably linked to the dysmetabolic state.
Differences were also observed in the comparison between the liver-specific and control clusters, with elevated levels of 5α-androstan-3α,17β-diol monosulfate, its disulfate form, glycoursodeoxycholic acid sulfate, and taurochenodeoxycholic acid 3-sulfate suggesting changes in steroid processing. Furthermore, higher levels of ursodeoxycholate, glycochenodeoxycholate glucuronide and glycochenodeoxycholate 3-sulfate and decreased levels of cysteine-glutathione disulfide were observed in both the liver-specific and cardiometabolic clusters compared with the control cluster (Extended Data Fig. 3 and Supplementary Table 1). Possibly linked to oxidative stress and liver function, we observed decreased levels of cysteine-glutathione disulfide both in the liver-specific and in the cardiometabolic cluster compared with the control cluster, thus indicating that reduced antioxidant capacity might be a common feature in the two MASH subtypes or a consequence of the severe phenotype.
Taken together, these transcriptomics and metabolomics analyses support the existence of two biologically distinct types of severe MASLD.
Molecular features of the cardiometabolic cluster versus dysglycemia
Since a majority of individuals in the cardiometabolic cluster have type 2 diabetes, we also investigated if the molecular features of that cluster differ from those merely associated with dysglycemia. For that purpose, we analyzed liver gene transcripts and metabolites that were differentially abundant between the cardiometabolic cluster versus the control cluster, as compared with those that were differentially abundant between individuals with type 2 diabetes versus nondiabetic controls. We found that the cardiometabolic cluster differentially exhibited a set of 199 unique liver transcripts that were not overexpressed in the type 2 diabetes group, indicating a distinctive transcriptional signature corresponding to 58 pathways expressed in the cardiometabolic cluster but not present in the type 2 diabetes group. Specifically, the cardiometabolic cluster shows distinct molecular pathways that involve unique aspects of lipid transport and metabolism, immune response modulation, oxidative stress and extracellular matrix remodeling, suggesting a heightened state of metabolic activity and cellular defense, as well as active involvement in managing inflammation (Supplementary Table 1). Regarding metabolites, our analyses also revealed a significant overlap between type 2 diabetes and cardiometabolic cluster, with 151 metabolites that were differentially abundant in both subgroups, many being directly linked to dysglycemia, such as monosaccharides and disaccharides (for example, glucose and sucrose). However, we identified a distinctive subset of 88 metabolites unique to the cardiometabolic cluster. These ‘cardiometabolic-specific’ metabolites include glycerophospholipids, sphingolipids, amino acid derivatives, protein metabolism and metabolites of bile acids unveiling a metabolic signature particular to this cluster at risk for MASH. These metabolites highlight disturbances in lipid processing, protein and energy metabolism, inflammatory profile and potential gut microbiome interactions that are not present in the type 2 diabetes profile (Supplementary Table 1).
Discussion
In the present study, using unsupervised hard clustering, we identified two distinct endotypes of at-risk MASLD, namely, cardiometabolic MASLD and liver-specific MASLD. Both types were characterized by a severe liver phenotype at baseline; however, they showed different underlying biological profiles and distinct clinical progression patterns.
These two newly defined types of MASLD could be robustly identified in several independent and well-characterized cohorts, using a simple algorithm based on six widely available traits: age, BMI, HbA1c, ALT, LDL cholesterol and triglycerides (https://ulr-metrics.univ-lille.fr/masldclusters/). The two types of at-risk MASLD could not be distinguished by their liver phenotype assessed by histology nor by MRI, and they were both associated with an increased risk of incident chronic liver disease. The cardiometabolic MASLD was, however, specifically characterized by a higher prevalence of dyslipidemia, hypertension and dysglycemia, resulting in a high risk of incident cardiovascular disease and type 2 diabetes. In contrast, the liver-specific MASLD was characterized by a more pronounced elevation of liver enzymes at a younger age and showed limited risk of diabetes progression and incident cardiovascular disease. The liver-specific MASLD was also characterized by a specific genetic background with a higher frequency of the minor allele of PNPLA3 rs738409 and a higher polygenic risk score for hepatic fat content.
Importantly, the proposed clustering outperformed its individual components in simultaneously predicting liver phenotype and future risk of the different clinical outcomes.
As expected, several individual continuous variables also showed a good predictive value for predicting specific clinical outcomes in the overall UK Biobank population, namely, ALT for chronic liver disease and HbA1c for cardiovascular disease and incident diabetes. In contrast, the clustering approach surpassed all individual variables for simultaneously predicting the three outcomes. Of note, after adjustment for ALT in multivariable analysis, the risk of chronic liver disease became lower in the liver-specific cluster than in the control cluster, while it remained increased in the cardiometabolic cluster. Confirming the strong association between the risk of liver disease and ALT in the liver-specific cluster, this result also indicates that ALT may overestimate the risk of chronic liver disease when other clustering variables are not considered. Similarly, the positive association between the cardiometabolic cluster and cardiovascular risk became negative after adjustment for HbA1c, suggesting that HbA1c alone may overestimate the risk of cardiovascular disease, in which other clustering variables such as triglycerides or age may favor cardiovascular disease, independently of dysglycemia. Finally, in the liver-specific cluster, the elevated risk of incident diabetes was eliminated after adjustment for ALT, underlying the specific role played by the liver in the physiopathology of dysglycemia23. Taken together, our findings highlight the potential of clustering to provide a more comprehensive risk assessment, identifying patients at risk for a range of liver and cardiometabolic diseases rather than focusing on a single condition.
In addition, the resulting assignment of individuals into two clearly labeled clusters of at risk MASLD facilitated the exploration of their biological nature. Specifically, the cardiometabolic cluster exhibited unique liver gene transcripts and pathways not present in type 2 diabetes, involving lipid transport, immune response and inflammation and vascular function-related pathways. In addition, metabolomic analyses identified numerous metabolites common to both type 2 diabetes and the cardiometabolic cluster, mostly linked to dysglycemia but also some metabolites uniquely associated with the cardiometabolic cluster. These unique metabolites, including glycerophospholipids, sphingolipids and bile acid metabolites, indicate specific disturbances in lipid processing, protein and energy metabolism, and inflammation.
The cardiometabolic cluster was also characterized by an increase of several gut microbiota metabolites previously linked to insulin resistance and diabetes pathogenesis, such as imidazole propionate, p-cresol glucuronide, phenylacetylglutamine, 4-hydroxyphenylacetylglutamine and phenylacetylglutamate20–22. Similarly, higher levels of p-cresol glucuronide and 4-hydroxyphenylacetylglutamine have been linked to cardiovascular toxicity and mortality22,24,25. These metabolites, which are produced by the gut microbiota from aromatic amino acids, might explain at least in part the increased cardiovascular risk observed in this cluster. In contrast, the liver-specific MASLD was more related to changes in lipid metabolism confined to the hepatocyte, in line with its specific genetic background.
In this study we identify distinctive endotypes of at-risk MASLD with a similar baseline liver phenotype, but different biological mechanisms, ultimately resulting in distinct clinical trajectories. Two studies have previously employed data-driven clustering in MASLD26,27. However, none of these studies examined liver histology across proposed clusters, assessed the risk of liver-related outcomes nor explored the underlying molecular biology.
Overall, our results demonstrate the heterogeneity of MASLD and underscore the distinct pathophysiological profile of the newly identified clusters, highlighting the need for more targeted therapeutic approaches. Likewise, the thyroid hormone receptor agonist Resmetirom, recently approved for the treatment of MASH, was found ineffective in a large fraction of individuals, potentially due to disease heterogeneity28. According to the present study, liver-specific MASLD, characterized by abnormal lipid droplet homeostasis and intrahepatic lipid transport genes, may respond more favorably to this drug that specifically reduces hepatic lipid content and inflammation. In contrast, cardiometabolic MASLD may respond better to drugs regulating lipid and glucose metabolism such as the fibroblast growth factor 21 analog pegozafermin29 and the pan-peroxisome proliferator-activated receptor agonist lanifibranor30, or to drugs favoring weight loss and cardiovascular risk reduction, namely, the glucagon-like peptide-1 (GLP1) receptor agonist semaglutide31, the GLP1–glucose-dependent insulinotropic polypeptide receptor dual agonist tirzepatide32 or the GLP1–glucagon receptor dual agonist survodutide33. Taken together with existing evidence, the newly proposed stratification could help refine emerging therapeutic strategies based on specific molecular pathomechanisms underlying each MASLD endotype.
These findings align with partitioned polygenic risk score analyses based on genetic associations with MASLD, including intrahepatic lipoprotein retention, which identify two distinct subtypes: one primarily liver-confined with more aggressive liver disease and another systemic with a higher risk of cardiometabolic disease34.
Some limitations of our study must be acknowledged. First, unsupervised clustering largely depends on the traits used in the analysis. We therefore selected six biomarkers embedded in the pathological mechanisms of MASLD, with high biological plausibility. It is noteworthy that we focused the present analysis on the two clusters associated with at-risk MASLD. The other clusters may, however, also represent distinct and potentially clinically relevant subgroups of MASLD, warranting further exploration in future studies. Second, the absence of lean or overweight individuals in the validation cohort could limit the generalizability of the proposed stratification across the full spectrum of steatotic liver disease. Moreover, ABOS participants were not screened on the basis of additional clinical or biochemical markers, unlike most studies where biopsies are performed only on at-risk individuals. Of note, the robustness of the new stratification was confirmed in independent cohorts with a higher incidence of MASH or more diverse BMI categories. In addition, an independent parallel study based on partitioned polygenic risk score associated with MASLD identified two similar subtypes: one primarily liver-confined with more aggressive liver disease and another systemic with a higher risk of cardiometabolic disease34. Another debatable aspect of the present study is the use of hard clustering, which assigns each patient to a single cluster. While this method facilitates the interpretation, it also ignores uncertainties within clusters, particularly for individuals at cluster boundaries. Alternative statistical approaches that provide probabilities for cluster membership, for example, model-based clustering35, could capture within-cluster differences more effectively and influence the clinical decision. Reversed graph embedding approaches such as discriminative dimensionality reduction via learning a tree (DDRTree) could also offer a more nuanced understanding of patient profiles36. Finally, all the study cohorts comprised primarily Europeans, and our findings remain to be confirmed in other ethnic groups, with other genetic backgrounds.
In conclusion, this study unveiled the existence of at least two distinct types of at-risk MASLD, displaying a similar liver phenotype at baseline, but different biological mechanisms and specific outcomes, ultimately resulting in distinct clinical trajectories, with regard to cardiovascular disease and diabetes. Therefore, it is reasonable to state that the search for drug treatment should reflect and selectively target these different biological pathways. Future prospective studies are needed to assess the clinical value of these two MASLD types for guiding prevention and treatment.
Methods
Study cohorts
ABOS cohort
ABOS is a prospective study (NCT01129297) aiming to identify the key factors influencing the outcomes of bariatric surgery. A total of 1,545 participants enrolled between 2006 and 2021 at the Lille University Hospital, Lille, France, were included in the present analysis. All individuals provided written informed consent before inclusion. Ethical approval for the study was granted by the Comité de Protection des Personnes Nord Ouest VI (Lille, France). Demographic characteristics, anthropomorphic measurements, medical history, concomitant medication and laboratory tests were collected before surgery as previously described37–40. A 75 g oral glucose tolerance test was performed after overnight fasting at baseline and 1 year after surgery. Type 2 diabetes status was defined at baseline on the basis of a previous history of diabetes, use of antidiabetic medications, fasting plasma glucose ≥126 mg dl−1 (7.0 mmol l−1) and/or 2 h plasma glucose ≥200 mg dl−1 (11.1 mmol l−1) during oral glucose tolerance test, and/or HbA1c ≥6.5% (48 mmol l−1)41. Liver histology was obtained at baseline through a percutaneous liver needle biopsy performed during surgery as previously described42–44. All liver biopsies were analyzed at Lille University Hospital by two expert liver pathologists, according to the NASH Clinical Research Network (NASH CRN) scoring system, as previously described45,46. Briefly, pathologists were blinded to the patient’s clinical and biological data. The reports were drawn up using a standardized template adapted to the recommendations of the NASH CRN group. All biopsies obtained before 2011 were reanalyzed and adapted to NASH CRN recommendations. Liver biopsies from patients with ‘borderline NASH’ histology, or with borderline size or length, were reanalyzed by two expert pathologists. The diagnosis of MASH was made by pathologists in the simultaneous presence of steatosis, inflammation and ballooning. Disease activity was subsequently graded with the nonalcoholic fatty liver disease activity score (NAS) according to specific histological features, as the unweighted sum of the scores for steatosis (0–3), lobular inflammation (0–3) and ballooning (0–2) ranging from 0 to 8. Liver fibrosis was scored from F0 to F4 (ref. 45).
UZA cohort
The UZA cohort included 467 patients referred to the Obesity Clinic at Antwerp University Hospital, Edegem, Belgium, for suspected MASLD based on imaging and biochemistry data. The collection of clinical, anthropometric and histological data has been previously described47,48. A percutaneous or laparoscopic-guided percutaneous liver needle biopsy was performed on participants with overweight/obesity as part of the Hepatic and Adipose Tissue and Functions in Metabolic Syndrome (HEPADIP) study (Belgian registration number B30020071389, Antwerp University Hospital File 6/25/125) as previously described47. Liver histology was assessed according to the NASH CRN45,46. Individuals with alcohol consumption above 30/20 g per day in men/women were excluded from the analysis. Written informed consent was obtained from all patients in both cohorts, and the studies were conducted in conformity with the Declaration of Helsinki.
MAFALDA cohort
A total of 264 participants with liver biopsy data from the MAFALDA cohort were included in the analyses49. Briefly, consecutive individuals with morbid obesity eligible for bariatric surgery were recruited from May 2020 to June 2021 at Fondazione Policlinico Universitario Campus Bio-Medico, Rome, Italy. Preoperative clinical and laboratory data were collected using standardized procedures. An intraoperative liver biopsy was obtained. Liver histology was assessed according to the NASH CRN45,46, as described above. Individuals with alcohol consumption above 30/20 g per day in men/women were excluded from the analysis. The MAFALDA study has been approved by the Local Research Ethics Committee (no. 16/20), and it was conducted in accordance with the principles of the Declaration of Helsinki. All participants gave written informed consent to the study.
Helsinki cohort
The Helsinki cohort enrolled 343 consecutive individuals with morbid obesity eligible for bariatric surgery and 42 consecutive individuals with a BMI ≥25 kg m−2 undergoing liver biopsy for suspected MASH, all recruited between 2006 and 2018 at the Helsinki University Hospital, Helsinki, Finland. A week before the liver biopsy, participants underwent clinical examination and blood sampling as previously described50. Liver histology was assessed according to the NASH CRN45,46, as described above. Individuals with alcohol consumption above 30/20 g per day in men/women were excluded from the analysis. The study was approved by the Local Research Ethics Committee at Helsinki University Hospital. All participants gave written informed consent to the study.
UK Biobank cohort
The UK Biobank is a large prospective cohort study recruiting approximately 500,000 participants (age 40–69 years) between 2006 and 2010 throughout the United Kingdom51. Clinical and laboratory data were collected using highly standardized procedures. Medical diagnoses were obtained through linkage of hospital admissions, death and cancer registers from the National Health Service records (data fields 41270, 40001, 40002 and 40006). The UK Biobank study has been approved by the NorthWest Multicenter Research Ethics Committee (no. 21/NW/0157). All participants gave written informed consent to the study. Data used in this study were obtained under application number 37142.
In the current study, we selected unrelated UK Biobank participants of European ancestry on the basis of our quality control pipeline, which has been described in detail previously15,52,53, and we included individuals with BMI ≥25 kg m−2 and/or with type 2 diabetes as defined elsewhere15. Participants were scanned at the UK Biobank Imaging Centre in Cheadle (United Kingdom) using a Siemens 1.5T MAGNETOM Aera as described in detail elsewhere54,55. Briefly, a shortened modified look locker inversion (ShMOLLI) was used to quantify liver T1, and a multi-echo-spoiled gradient echo was used to quantify liver iron and fat. Data were analyzed using LiverMultiScan Discover 4.0 software. Hepatic steatosis was defined by PDFF >5.5%) (ref. 54), MASH by PDFF >5.5% and iron-corrected T1 mapping (cT1) by >800 ms (refs. 54,56).
Cluster analysis
Six variables associated with MASLD physiopathology and increased risk of MASH were selected for clustering in ABOS, namely, age, BMI, HbA1c, ALT, LDL cholesterol and circulating triglycerides. Cluster analysis and identification of MASLD subtypes were performed on 1,389 ABOS participants (Fig. 1), after the exclusion of 54 patients for self-declaration alcohol consumption above 50/60 g per day for women and men, respectively, at the first visit, to avoid any risk of inclusion of patients with alcohol-related liver disease; 58 participants for a BMI ≤30 kg m−2; 27 participants for missing values in clustering traits (that is, age, BMI, HbA1c, ALT, LDL cholesterol and circulating triglycerides); and 17 participants having absolute standardized values of 5 or higher in at least one of the clustering traits (Extended Data Fig. 1). The analysis was performed using the partitioning around medoids method in R (package ‘cluster’, version 2.1.4)57, which is a more robust version of k-means clustering. Distances were computed as Euclidean distances using standardized variables scaled to a mean of 0 and a standard deviation of 1.
To estimate the optimal number of clusters, we evaluated the silhouette widths58 for each clustering, varying the number of clusters going from three clusters to ten clusters. We determined the optimal number of clusters by choosing the configuration that yielded the highest silhouette coefficients, signifying well-delineated clusters whose members are closely related to one another and distinctly separate from individuals in other clusters. We then assessed the stability of the resulting clusters using the R function clusterboot from the fpc package (v.2.2-12), by resampling 2,000 times the original data and computing the Jaccard similarities of the original clusters to the most similar clusters in the resampled data. The mean (standard deviation) Jaccard-similarity measure was 0.73 (0.07) across all clusters. Data from the UZA, MAFALDA and Helsinki cohorts were normalized using ABOS values for centering and scaling. Then, participants were allocated to the cluster they were most similar to after the exclusion of participants having absolute standardized values of 5 or higher in at least one of the clustering traits, calculated as their Euclidean distance from the nearest cluster medoid derived from ABOS coordinates. Data from the UK biobank cohorts were normalized using ABOS values for centering and scaling. Participants were allocated to the cluster they were most similar to after the exclusion of those with self-reported history or medical diagnosis of other causes of liver disease, with a medical diagnosis of the target longitudinal outcome at baseline, or having absolute standardized values of 5 or higher in at least one of the clustering traits, calculated as their Euclidean distance from the nearest cluster medoid derived from ABOS coordinates.
The Calinski–Harabasz Index was 263 for the ABOS cohort and reached 174 in the validation cohort, indicating well-defined clusters and confirming the transportability of the proposed stratification in diverse populations. In the UK Biobank cohort, encompassing a broader BMI range and less clinically extreme cases, the Calinski–Harabasz Index increases even further to 18,774, probably due to the larger and more diverse sample size.
Visualizing individual risk in relation to their phenotype
As a potential aid for assisting clinicians in defining individual profiles of patients with MASLD, we developed an app (https://ulr-metrics.univ-lille.fr/masldclusters/).
Genotyping
In the ABOS cohort, genotyping was available for 1,259 participants and was performed using the Illumina Infinium assay59. This analysis was conducted at the SNO&SEQ Technology Platform, Molecular Medicine, BMC, Husargatan 3, Uppsala, Sweden. Results were analyzed using the software GenomeStudio 2.0.3. The following variants were assessed: PNPLA3 rs738409 C > G (p.I148M), TM6SF2 rs58542926 C > T (p.E167K), MBOAT7 rs641738 C > T and GCKR rs1260326 C > T (p.P446L).
In the UK Biobank, genotyping was available for approximately 490,000 individuals and was performed using two similar genotyping arrays (that is Affymetrix UK BiLEVE and UK Biobank Axiom arrays) as described elsewhere60. The following variants were assessed: PNPLA3 rs738409 C > G (p.I148M), TM6SF2 rs58542926 C > T (p.E167K), MBOAT7 rs641738 C > T and GCKR rs1260326 C > T (p.P446L).
The PRS-HFC was computed according to the originally reported formula61.
Long-term longitudinal outcomes
We analyzed the risk of developing hepatic and extrahepatic outcomes and overall mortality in the UK Biobank cohort. To estimate the incidence of liver outcomes, we selected 213,180 individuals without self-reported history or medical diagnosis of any liver disease (International Classification of Diseases 10th edition (ICD-10) B18, B19, C22.0, E83.0, E83.1, E88.0, I82.0, I85.0, I85.9, K70, K71, K72.1, K72.9, K74.1, K74.2, K74.3, K74.4, K74.5, K74.6, K75.2, K75.3, K75.4, K75.8, K75.9, K76.5, K76.6, K76.7, K76.8, K76.9, K83.0, R18 and Z94.4) at baseline and identified those who developed chronic liver disease (ICD-10 C22.0, I85.0, I85.9, K70, K72.1, K72.9, K73, K74.0, K74.1, K74.2, K74.6, K76.0, K76.6, K76.7, K76.8, K76.9 and Z94.4) across the clusters. Participants were excluded from the analyses if they received a medical diagnosis of competing liver diseases (ICD-10 B18, B19, E83.0, E83.1, E88.0, I82.0, K71, K74.3, K74.4, K74.5, K75.2, K75.3, K75.4, K75.8, K75.9, K76.5 and K83.0) before the diagnosis of liver outcome.
To estimate the incidence of cardiovascular outcomes, we selected 195,739 individuals without self-reported history or medical diagnosis of chronic viral hepatitis (ICD-10 B18 and B19), other causes of liver disease (ICD-10 E83.0, E83.1, E88.0, I82.0, K70, K71, K74.3, K74.4, K74.5, K75.2, K75.3, K75.4, K75.8, K75.9, K76.5, K76.8, K76.9 and K83.0) and cardiovascular disease (ICD-10 I20–I25, I60–I64, I69 and G45) at baseline, and identified those who developed cardiovascular disease across the clusters.
To estimate the incidence of type 2 diabetes, we selected 196,791 individuals without self-reported history or medical diagnosis of chronic viral hepatitis (ICD-10 B18 and B19), other causes of liver disease (ICD-10 E83.0, E83.1, E88.0, I82.0, K70, K71, K74.3, K74.4, K74.5, K75.2, K75.3, K75.4, K75.8, K75.9, K76.5, K76.8, K76.9 and K83.0) and type 2 diabetes as defined elsewhere53 at baseline, and identified those who developed type 2 diabetes (ICD-10 E11 and E14) across the clusters.
Detailed information about the UK Biobank methods and clinical diagnosis is provided in Supplementary Table 2.
Liver transcriptomic data generation and normalization
Liver transcriptomic data were available for a subset of 831 participants from the ABOS cohort, as previously described62. Total RNA was extracted from 30 mg frozen liver biopsies for Affymetrix microarray analysis using TRIzol reagent (Thermo Fisher Scientific), followed by purification on RNeasy columns (Qiagen). RNA purity and quantity were assessed using a Nanodrop spectrometer (Thermo Fisher Scientific). RNA integrity was quantified using the Agilent RNA6000 Nano assay and an Agilent 2100 BioAnalyzer. Raw data from Affymetrix microarrays were first processed with robust multi-array average (RMA) with GC correction and scale intensities (CG-RMA-scale) as a normalization method.
Metabolomic data generation and normalization
In the ABOS cohort, nontargeted global metabolomic analysis was performed on plasma samples in 1,322 participants by Metabolon, using two independent platforms: ultrahigh performance liquid chromatography/tandem mass spectrometry optimized for basic species or acidic species, and gas chromatography–mass spectrometry. Raw data for metabolomics were transformed using log transformation and imputation with minimum observed values for each compound.
Statistical analysis
Data were reported as median (interquartile range) for continuous variables and frequencies (percentages) for categorical variables. Clusters were compared using the Kruskal–Wallis test, chi-squared test or Fisher’s exact test, as appropriate. Raw P values were adjusted for multiple testing separately for clinical data, histological data and genetic data. To control the family-wise error rate, the Bonferroni method was used. Differences were considered statistically significant when adjusted P value(s) were less than 0.05. For statistically significant variables, post hoc analysis was performed comparing pairwise MASH-enriched MASLD clusters (2 and 5) and the combined nonenriched MASLD clusters (1, 3, 4 and 6) using the Dunn test, chi-squared test or Fisher’s exact test, as appropriate, with Bonferroni adjustment.
Differential analysis of liver transcriptomic across the clusters was performed using moderated t-tests from the R Bioconductor package Limma v.3.60.4. The same methodology was also applied to metabolomic after exclusion of xeniobiotics. Differences were considered statistically significant when P value(s) adjusted for multiple comparisons using the Benjamini–Hochberg correction (to control the false discovery rate) were less than 0.05 and the absolute value of log2 fold change was greater than 0.26. Group comparisons for genes were represented using volcano plots. The number of differentially expressed genes between the various clusters were reported through Euler diagrams.
Pathway enrichment on the transcriptome was performed with the R package ClusterProfiler (v.4.7.1), based on GO-BP pathways. The GSEA method was run with the absolute value of the moderated t-test statistic as ranking metric. The P values of enriched pathways were adjusted using the Benjamini–Hochberg procedure, and an adjusted P value <0.05 was considered significant.
In the UK Biobank, clusters were compared using analysis of variance, Kruskal–Wallis test, chi-square test or Fisher’s test as appropriate, adjusted for multiple testing separately for clinical data and genetic data, using the Bonferroni method. Similarly, post hoc comparisons were carried out with Bonferroni correction. The incidence of chronic liver disease, cardiovascular disease and type 2 diabetes were defined as the composite occurrence of the clinical event or event-related death during follow-up. Then, the cumulative incidence of the clinical outcomes was computed according to the Aalen–Johansen method for chronic liver disease, cardiovascular disease and type 2 diabetes, taking into account the competing occurrence of other-cause death, and of selected liver disease (only in the case of chronic liver disease; see above for ICD-10 codes). Cause-specific HRs were calculated through Cox regressions, adjusted for age, sex and alcohol intake. The proportional hazard assumption was verified through the inspection of the Schoenfeld residuals. Sensitivity analyses were performed (1) including only individuals with BMI ≥27 kg m−2 and (2) excluding those with harmful alcohol consumption (>50/60 g per day for women/men).
Statistical analyses and graphical representations were performed using R statistical software v.4.4.1 (R Foundation for Statistical Computing, Vienna, Austria).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41591-024-03283-1.
Supplementary information
Supplementary Table 1. The liver gene expression and plasma metabolites with significant differences between the cardiometabolic and liver-specific clusters (A), cardiometabolic and control (B) and liver-specific and control (C). Metabolites with significant differences between the cardiometabolic and liver-specific clusters (D), cardiometabolic and control (E) and liver-specific and control (F). Molecular features that were differentially expressed between type 2 diabetes and non-T2D groups, and between cardiometabolic and control clusters (G). Supplementary Table 2. Definition of self-reported history of liver disease, cardiovascular disease and type 2 diabetes (UK Biobank data-field 20002) and ICD-10 codes used to define liver disease, cardiovascular disease and type 2 diabetes.
Acknowledgements
This research was supported by the ‘Programme d’Investissement d’Avenir’ (PRECINASH, ANR-16-RHUS-0006; European Genomic Institute for Diabetes, ANR-10-LABX-0046), Lille University (WILL-CHAlRES-23-001), Fondation de la Recherche Médicale (EQU202303016330 PATTOU), EU Horizon 2020 research and innovation program (Innovative Medicines Initiative 2, project SOPHIA 875534). S.R. was supported by the Swedish Cancerfonden (22 2270 Pj), the Swedish Research Council (Vetenskapsradet (VR), 2023-02079), the Swedish state under the Agreement between the Swedish government and the county councils (the ALF agreement, ALFGBG-965360), the Swedish Heart Lung Foundation (20220334), the Wallenberg Academy Fellows from the Knut and Alice Wallenberg Foundation (KAW 2017.0203), the Novonordisk Distinguished Investigator Grant - Endocrinology and Metabolism (NNF23OC0082114) and the Novonordisk Project grants in Endocrinology and Metabolism (NNF20OC0063883). S.F.Q. was supported by the Orion Research Foundation, the Yrjö Jahnsson Foundation (20207313), the Maud Kuistila Memorial Foundation (2021-0301B), the Emil Aaltonen Foundation (210182), the Finnish Medical Foundation (5843) and the Biomedicum Helsinki Foundation (20230241). We thank the staff and the participants from the different cohorts analyzed in this study and the French Institute for Bioinformatics. We also thank the staff and the participants of the ABOS, UZA, MAFALDA, Helsinki and UK Biobank studies. This research has been conducted using the UK Biobank resource (application no. 37142). We thank M.-B. Abdelouahab for thoroughly reviewing and providing valuable feedback on the R code related to the ABOS AQ19 results.
Extended data
Author contributions
V.R., F.T., G.M., S.R. and F.P. conceived and designed the study. E.C., A.D.V., G.M., S.F.Q., U.V.-G., R.C., H.V., C.M., J.V., J.T.H., H.Y-J., G.B., S.B., M.C., N.O-D., V.G. and E.L. were involved in data preparation and data analysis. V.R., F.T., G.M., S.R., F.P., V.T., G.L., L.O. and R.C. interpreted the results and wrote the manuscript. C.S., J.K-C, P.L., J.T.H., S.F., B.S., C.W.L.R., V.T. and P.M. provided critical inputs to the the manuscript.
Peer review
Peer review information
Nature Medicine thanks Ewan Pearson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Data availability
The individual data analyzed in the current study are not publicly available due to national data protection laws and restrictions imposed by the ethics committee to ensure participant privacy. However, researchers can apply for access through an individual project agreement with the principal investigator at the University Hospital of Lille, France. The study protocol and methods (NCT01129297) have been published and are available without restriction. Data access is conditional upon signing a data use agreement, which ensures data usage for the intended research purposes only. Researchers must submit a detailed request outlining their research objectives and methodology directed to the principal investigator of the ABOS study cohort (francois.pattou@univ-lille.fr). Data will be available only to researchers affiliated with recognized institutions and for research that aligns with the original scope of the ABOS cohort study. Access will be granted approximately one month after the interinstitutional agreement for the individual project is finalized and the study is registered on the Lille University Hospital site, in compliance with General Data Protection Regulation regulations. Data from UZA, MAFALDA and Helsinki cohorts are not publicly available due to governance limitations but are available for research by approval from principal investigators. All other data supporting the findings of this study are available within the article. UK Biobank data are publicly available to researchers through an open application via https://www.ukbiobank.ac.uk/register-apply/. Raw transcriptomic files are available at GEO under the accession number GSE130991. Metabolite abundances file is available at BioStudies under the accession number S-BSST1479. Cluster annotations of transcriptomic and metabolomic samples are available at https://gitlab.com/bilille/2024-raverdy_et_al-masld_clusters/-/tree/main/Data
Code availability
Codes used for implementing the partitioning around medoids (pam) method in R (package ‘cluster’, v.2.1.6) algorithm are available publicly in a GitLab repository for ABOS and validation cohort (https://gitlab.com/bilille/2024-raverdy_et_al-masld_clusters/-/blob/main/Code/maincode.Rmd) and in a GitHub repository for UK Biobank (https://github.com/devanto86/ukbb_cluster/blob/main/code).
Competing interests
The authors declare no conflicts of interest related to this manuscript.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Violeta Raverdy, Federica Tavaglione, Estelle Chatelain, Guillaume Lassailly.
Contributor Information
Stefano Romeo, Email: stefano.romeo@wlab.gu.se.
François Pattou, Email: francois.pattou@univ-lille.fr.
Extended data
is available for this paper at 10.1038/s41591-024-03283-1.
Supplementary information
The online version contains supplementary material available at 10.1038/s41591-024-03283-1.
References
- 1.Rinella, M. E. et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Hepatology78, 1966–1986 (2023). [DOI] [PubMed] [Google Scholar]
- 2.Rinella, M. E. et al. AASLD Practice Guidance on the clinical assessment and management of nonalcoholic fatty liver disease. Hepatology77, 1797–1835 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Younossi, Z. M. et al. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology77, 1335–1347 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romeo, S., Sanyal, A. & Valenti, L. Leveraging human genetics to identify potential new treatments for fatty liver disease. Cell Metab.31, 35–45 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Kantartzis, K. & Stefan, N. Clustering NAFLD: phenotypes of nonalcoholic fatty liver disease and their differing trajectories. Hepatol. Commun.7, e0112 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Targher, G., Day, C. P. & Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med.363, 1341–1350 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Younossi, Z. M. et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis. J. Hepatol.71, 793–801 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Eslam, M. et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J. Hepatol.73, 202–209 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Friedman, S. L., Neuschwander-Tetri, B. A., Rinella, M. & Sanyal, A. J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med.24, 908–922 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stefan, N. & Cusi, K. A global view of the interplay between non-alcoholic fatty liver disease and diabetes. Lancet Diabetes Endocrinol.10, 284–296 (2022). [DOI] [PubMed] [Google Scholar]
- 11.Friedman, S. L. & Sanyal, A. J. The future of hepatology. Hepatology78, 637–648 (2023). [DOI] [PubMed] [Google Scholar]
- 12.Liu, D. J. et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat. Genet.49, 1758–1766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stefan, N., Haring, H. U. & Cusi, K. Non-alcoholic fatty liver disease: causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol.7, 313–324 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Lauridsen, B. K. et al. Liver fat content, non-alcoholic fatty liver disease, and ischaemic heart disease: Mendelian randomization and meta-analysis of 279 013 individuals. Eur. Heart J.39, 385–393 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Tavaglione, F. et al. Inborn and acquired risk factors for severe liver disease in Europeans with type 2 diabetes from the UK Biobank. JHEP Rep.3, 100262 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holmer, M. et al. Effect of common genetic variants on the risk of cirrhosis in non-alcoholic fatty liver disease during 20 years of follow-up. Liver Int.42, 2769–2780 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seko, Y. et al. The greater impact of PNPLA3 polymorphism on liver-related events in Japanese non-alcoholic fatty liver disease patients: a multicentre cohort study. Liver Int.43, 2210–2219 (2023). [DOI] [PubMed] [Google Scholar]
- 18.Nishimura, N. et al. Chitinase 3-like 1 is a profibrogenic factor overexpressed in the aging liver and in patients with liver cirrhosis. Proc. Natl Acad. Sci. USA118, e2019633118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lefebvre, P. et al. Interspecies NASH disease activity whole-genome profiling identifies a fibrogenic role of PPARα-regulated dermatopontin. JCI Insight2, e92264 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bar, N. et al. A reference map of potential determinants for the human serum metabolome. Nature588, 135–140 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Nemet, I. et al. Atlas of gut microbe-derived products from aromatic amino acids and risk of cardiovascular morbidity and mortality. Eur. Heart J.44, 3085–3096 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nemet, I. et al. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell180, 862–877 e822 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mantovani, A. et al. Non-alcoholic fatty liver disease and risk of incident diabetes mellitus: an updated meta-analysis of 501 022 adult individuals. Gut70, 962–969 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Liabeuf, S. et al. Does p-cresylglucuronide have the same impact on mortality as other protein-bound uremic toxins? PLoS ONE8, e67168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei, H. et al. Increased circulating phenylacetylglutamine concentration elevates the predictive value of cardiovascular event risk in heart failure patients. J. Intern. Med.294, 515–530 (2023). [DOI] [PubMed] [Google Scholar]
- 26.Yi, J., Wang, L., Guo, J. & Ren, X. Novel metabolic phenotypes for extrahepatic complication of nonalcoholic fatty liver disease. Hepatol. Commun.7, e0016 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye, J. et al. Novel metabolic classification for extrahepatic complication of metabolic associated fatty liver disease: a data-driven cluster analysis with international validation. Metabolism136, 155294 (2022). [DOI] [PubMed] [Google Scholar]
- 28.Cusi, K. Selective agonists of thyroid hormone receptor beta for the treatment of NASH. N. Engl. J. Med.390, 559–561 (2024). [DOI] [PubMed] [Google Scholar]
- 29.Loomba, R. et al. Randomized, controlled trial of the FGF21 analogue pegozafermin in NASH. N. Engl. J. Med.389, 998–1008 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooreman, M. P. et al. The pan-PPAR agonist lanifibranor improves cardiometabolic health in patients with metabolic dysfunction-associated steatohepatitis. Nat. Commun.15, 3962 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newsome, P. N. et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N. Engl. J. Med.384, 1113–1124 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Loomba, R. et al. Tirzepatide for metabolic dysfunction-associated steatohepatitis with liver fibrosis. N. Engl. J. Med.391, 299–310 (2024). [DOI] [PubMed] [Google Scholar]
- 33.Sanyal, A. J. et al. A phase 2 randomized trial of survodutide in MASH and fibrosis. N. Engl. J. Med.391, 311–319 (2024). [DOI] [PubMed] [Google Scholar]
- 34.Jamialahmadi, O. et al. Partitioned polygenic risk scores identify distinct types of metabolic dysfunction-associated steatotic liver disease. Nat. Med.10.1038/s41591-024-03284-0 (2024). [DOI] [PMC free article] [PubMed]
- 35.Gormley, I. C., Murphy, T. B. & Raftery, A. E. Model-based clustering. Ann. Rev. Stat. Appl.10, 573–595 (2023). [Google Scholar]
- 36.Nair, A. T. N. et al. Heterogeneity in phenotype, disease progression and drug response in type 2 diabetes. Nat. Med.28, 982–988 (2022). [DOI] [PubMed] [Google Scholar]
- 37.Raverdy, V. et al. Data-driven subgroups of type 2 diabetes, metabolic response, and renal risk profile after bariatric surgery: a retrospective cohort study. Lancet Diabetes Endocrinol.10, 167–176 (2022). [DOI] [PubMed] [Google Scholar]
- 38.Raverdy, V. et al. Performance of non-invasive tests for liver fibrosis resolution after bariatric surgery. Metabolism153, 155790 (2024). [DOI] [PubMed] [Google Scholar]
- 39.Raverdy, V. et al. Combining diabetes, sex, and menopause as meaningful clinical features associated with NASH and liver fibrosis in individuals with class II and III obesity: a retrospective cohort study. Obesity31, 3066–3076 (2023). [DOI] [PubMed] [Google Scholar]
- 40.Saux, P. et al. Development and validation of an interpretable machine learning-based calculator for predicting 5-year weight trajectories after bariatric surgery: a multinational retrospective cohort SOPHIA study. Lancet Digit. Health5, e692–e702 (2023). [DOI] [PubMed] [Google Scholar]
- 41.American Diabetes Association Professional Practice. 2. Diagnosis and classification of diabetes: standards of care in diabetes-2024. Diabetes Care47, S20–S42 (2024). [DOI] [PMC free article] [PubMed]
- 42.Mathurin, P. et al. Prospective study of the long-term effects of bariatric surgery on liver injury in patients without advanced disease. Gastroenterology137, 532–540 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Lassailly, G. et al. Bariatric surgery reduces features of nonalcoholic steatohepatitis in morbidly obese patients. Gastroenterology149, 379–388 (2015). [DOI] [PubMed]
- 44.Lassailly, G. et al. Bariatric surgery provides long-term resolution of nonalcoholic steatohepatitis and regression of fibrosis. Gastroenterology159, 1290–1301 e1295 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Kleiner, D. E. et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology41, 1313–1321 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Brunt, E. M. et al. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology53, 810–820 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lalloyer, F. et al. Roux-en-Y gastric bypass induces hepatic transcriptomic signatures and plasma metabolite changes indicative of improved cholesterol homeostasis. J. Hepatol.79, 898–909 (2023). [DOI] [PubMed] [Google Scholar]
- 48.Verrijken, A. et al. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology59, 121–129 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Tavaglione, F. et al. Accuracy of controlled attenuation parameter for assessing liver steatosis in individuals with morbid obesity before bariatric surgery. Liver Int.42, 374–383 (2022). [DOI] [PubMed] [Google Scholar]
- 50.Luukkonen, P. K. et al. The PNPLA3-I148M variant confers an antiatherogenic lipid profile in insulin-resistant patients. J. Clin. Endocrinol. Metab.106, e300–e315 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med.12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jamialahmadi, O. et al. Exome-wide association study on alanine aminotransferase identifies sequence variants in the GPAM and APOE associated with fatty liver disease. Gastroenterology160, 1634–1646 e1637 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Bianco, C. et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol.74, 775–782 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilman, H. R. et al. Characterisation of liver fat in the UK Biobank cohort. PLoS ONE12, e0172921 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mojtahed, A. et al. Reference range of liver corrected T1 values in a population at low risk for fatty liver disease—a UK Biobank sub-study, with an appendix of interesting cases. Abdom. Radiol.44, 72–84 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parisinos, C. A. et al. Genome-wide and Mendelian randomisation studies of liver MRI yield insights into the pathogenesis of steatohepatitis. J. Hepatol.73, 241–251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schubert, E. & Rousseeuw, P. J. Fast and eager k-medoids clustering: 0(k) runtime improvement of the PAM, CLARA and CLARANS algorithms. Inf. Syst.101, 101804 (2021).
- 58.Rousseeuw, P. J. & Rousseeuw, P. J. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math.20, 53–65 (1987). [Google Scholar]
- 59.Gunderson, K. L. et al. A genome-wide scalable SNP genotyping assay using microarray technology. Nat. Genet.37, 549–554 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dongiovanni, P. et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J. Intern. Med.283, 356–370 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Margerie, D. et al. Hepatic transcriptomic signatures of statin treatment are associated with impaired glucose homeostasis in severely obese patients. BMC Med. Genomics12, 80 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. The liver gene expression and plasma metabolites with significant differences between the cardiometabolic and liver-specific clusters (A), cardiometabolic and control (B) and liver-specific and control (C). Metabolites with significant differences between the cardiometabolic and liver-specific clusters (D), cardiometabolic and control (E) and liver-specific and control (F). Molecular features that were differentially expressed between type 2 diabetes and non-T2D groups, and between cardiometabolic and control clusters (G). Supplementary Table 2. Definition of self-reported history of liver disease, cardiovascular disease and type 2 diabetes (UK Biobank data-field 20002) and ICD-10 codes used to define liver disease, cardiovascular disease and type 2 diabetes.
Data Availability Statement
The individual data analyzed in the current study are not publicly available due to national data protection laws and restrictions imposed by the ethics committee to ensure participant privacy. However, researchers can apply for access through an individual project agreement with the principal investigator at the University Hospital of Lille, France. The study protocol and methods (NCT01129297) have been published and are available without restriction. Data access is conditional upon signing a data use agreement, which ensures data usage for the intended research purposes only. Researchers must submit a detailed request outlining their research objectives and methodology directed to the principal investigator of the ABOS study cohort (francois.pattou@univ-lille.fr). Data will be available only to researchers affiliated with recognized institutions and for research that aligns with the original scope of the ABOS cohort study. Access will be granted approximately one month after the interinstitutional agreement for the individual project is finalized and the study is registered on the Lille University Hospital site, in compliance with General Data Protection Regulation regulations. Data from UZA, MAFALDA and Helsinki cohorts are not publicly available due to governance limitations but are available for research by approval from principal investigators. All other data supporting the findings of this study are available within the article. UK Biobank data are publicly available to researchers through an open application via https://www.ukbiobank.ac.uk/register-apply/. Raw transcriptomic files are available at GEO under the accession number GSE130991. Metabolite abundances file is available at BioStudies under the accession number S-BSST1479. Cluster annotations of transcriptomic and metabolomic samples are available at https://gitlab.com/bilille/2024-raverdy_et_al-masld_clusters/-/tree/main/Data
Codes used for implementing the partitioning around medoids (pam) method in R (package ‘cluster’, v.2.1.6) algorithm are available publicly in a GitLab repository for ABOS and validation cohort (https://gitlab.com/bilille/2024-raverdy_et_al-masld_clusters/-/blob/main/Code/maincode.Rmd) and in a GitHub repository for UK Biobank (https://github.com/devanto86/ukbb_cluster/blob/main/code).