

Abstract
Next-generation sequencing is widely applied to the investigation of pedigree data for gene discovery. However, identifying plausible disease-causing variants within a robust statistical framework is challenging. Here, we introduce BICEP: a Bayesian inference tool for rare variant causality evaluation in pedigree-based cohorts. BICEP calculates the posterior odds that a genomic variant is causal for a phenotype based on the variant cosegregation as well as a priori evidence such as deleteriousness and functional consequence. BICEP can correctly identify causal variants for phenotypes with both Mendelian and complex genetic architectures, outperforming existing methodologies. Additionally, BICEP can correctly down-weight common variants that are unlikely to be involved in phenotypic liability in the context of a pedigree, even if they have reasonable cosegregation patterns. The output metrics from BICEP allow for the quantitative comparison of variant causality within and across pedigrees, which is not possible with existing approaches.
Keywords: pedigree, next-generation sequencing, Bayesian inference, prior odds of causality, Bayes factor, posterior odds of causality
Introduction
The emergence of next-generation sequencing has prompted renewed interest in family-based study designs for disease–gene prioritization [1–5]. In the absence of other robust approaches [6], linkage analysis remains the de facto standard statistical methodology in implicating genomic regions in disease or trait susceptibility [7]. However, data for many individuals or from multiple independent pedigrees are required to achieve sufficient statistical power [7, 8]. As an alternative, a simple, nonstatistical, identity-by-state (IBS) filtering approach can be used to examine variants present in affected individuals but absent in unaffected individuals within a pedigree [9–12]. This can be augmented with identity-by-descent methods to identify inherited haplotypes [13–16], depending on the pedigree structure and number of individuals sequenced. Downstream approaches to prioritize likely causal variants typically rely on filtering using population-level metrics such as allele frequency or deleteriousness scores [9].
These filtering-based strategies are straightforward to implement but have limitations. Since there is no quantitative measure of cosegregation, there is no means to rank results or compare information across different pedigree structures. For example, we cannot know whether there is more evidence for the cosegregation of a variant in a large sibship or for the same variant in a smaller multigenerational pedigree. The IBS and IBD filtering approaches for rare variants typically assume that all affected individuals must carry a risk variant, which is potentially an oversimplification for complex genetic disorders where there may be multiple risk variants, reduced penetrance, or the presence of phenocopies. There is no obvious way to relax this assumption that would be unbiased and consistent across different pedigree structures and sizes [17]. Furthermore, the population-level filtering strategy used to prioritize variants, even if guided by empirical work, can be subjective and arbitrary. The selection of a strict cut-off value for filtering eliminates much information about that metric and risks removing reasonable candidate variants with slightly subthreshold characteristics [18].
One tool that aims to address some of these issues is pVAAST [19], which provides a unified framework to prioritize likely disease-causing variants from next-generation sequencing data in a pedigree. pVAAST combines gene-based rare-variant association analysis with a novel formulation of linkage analysis. However, the tool requires an ancestry-matched set of control genomes, and allele frequency mismatches between the control and pedigree data sets are known to introduce false positives [20]. The distribution of the pVAAST output scores is not specified, so while the score may be used as a relative metric, it requires a thorough understanding to be correctly interpreted. It is possible that the number of observed genotypes differs across variants in a gene, and so, the p-values from the pVAAST scores may not necessarily be comparable across genes, even within the same pedigree. Similarly, the output metrics of a gene may not be comparable across two independent pedigrees as they depend on the individuals who are sequenced in the pedigrees.
A Bayesian inference approach represents an alternative method to evaluate cosegregating variants in pedigrees that addresses the issues detailed above. To classify variants of unknown clinical significance for pedigree data, Petersen et al. derived a Bayes factor to assess missense variants in BRCA1 and BRCA2 genes with a predicted dominant effect on breast and colorectal cancer [21]. Their methodology forms the basis for another disease-gene prioritization tool PERCH [22]. Mohammadi et al. considered a similar approach, extending the model to account for individuals in the pedigree who were not sequenced [23]. In addition, the authors assumed that the causal variant would be rare in the general population, which greatly reduces the variant search space making the model computationally feasible. While both methods perform comparably [24], they contain information about variant cosegregation only, and no measure is included regarding how likely the variant is to be causal a priori. When variants with reasonable cosegregation metrics have been selected, the standard filtering approaches referenced previously still apply for prioritizing variants.
Here we introduce BICEP, a Bayesian Inference model for Causality Evaluation of rare variants in Pedigrees. This method provides a quantitative framework to facilitate the ranking of rare DNA variants based on their prior evidence of causality and their cosegregation with a binary phenotype within a pedigree. The underlying assumptions of the Bayesian model are that causal variants are population-rare (therefore likely inherited from a common ancestor), and that they cosegregate with high penetrance and low phenocopy rates. Here, we showcase BICEP using pedigree-based whole genome sequencing (WGS) data from two real human disease cohorts and a simulated dataset. For each pedigree, at least one candidate causal variant has been previously identified using traditional approaches or by generating synthetic data (see Methods). In each analysis, we compare the performance of BICEP to pVAAST in identifying the causal variant. For the analyses of the three datasets, the prior information for BICEP was generated using protein-coding single-nucleotide variants (SNVs), although this can be extended to any class of DNA variant.
Materials and Methods
Overview and assumptions of the Bayesian inference model
A full derivation of the statistical model underlying BICEP is given in Supplementary Methods, which we summarize here. For a given input variant, our causal model (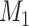 ) states that the variant is the sole cause of the binary phenotype. The neutral model (
) states that the variant is the sole cause of the binary phenotype. The neutral model (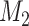 ) states that the variant does not contribute to the phenotype. Given the pedigree data (
) states that the variant does not contribute to the phenotype. Given the pedigree data (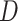 ), the posterior odds for causality (PostOC) are given by:
), the posterior odds for causality (PostOC) are given by:
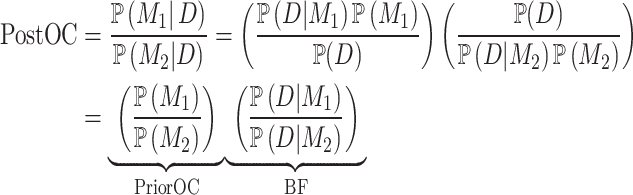 |
(1) |
Following Mohammadi et al., we assume that the variant cosegregates with the phenotype and is rare, therefore likely originating from a founder within the pedigree (whether sequenced or not) and unlikely to be carried by marry-in individuals [23]. We note that the term ‘marry-in’ used here is antiquated as we consider biological relationships, not sociological ones, but we retain the term here in the absence of an established alternative. We further assume that a causal variant has a dominant effect on the phenotype and that, for a neutral variant, the phenotypes are independent of all genotypes and are determined by the population incidence rate. The data in our model consist of the observed genotypes (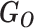 ) and the known binary phenotypes of the pedigree members (
) and the known binary phenotypes of the pedigree members (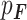 ). Where the phenotype status of an individual is unknown, they are assumed to be unaffected. The parameters of our model are the unobserved genotypes (
). Where the phenotype status of an individual is unknown, they are assumed to be unaffected. The parameters of our model are the unobserved genotypes (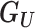 ), the in-pedigree penetrance (β), the in-pedigree phenocopy rate (φ), the population incidence rate (α), and the proband (p). Here, the proband refers to an affected variant carrier in the pedigree (see Supplementary Methods).
), the in-pedigree penetrance (β), the in-pedigree phenocopy rate (φ), the population incidence rate (α), and the proband (p). Here, the proband refers to an affected variant carrier in the pedigree (see Supplementary Methods).
Prior odds
We used a logistic regression model to construct the prior odds that a protein-coding variant is causal (for full details, see Supplementary Methods). As prior regression data, pathogenic and benign variants were extracted from the ClinVar database [25]. We fitted two regression models separately based on variant type: missense variants, and nonmissense SNVs. As predictors for the regression models, we used the variant functional consequence and the maximal allele frequency across all genomic ancestry clusters from gnomAD [26], using v2.1.1 (exome) for data aligned to GRCh37 and v4.1 (joint) for data aligned to GRCh38. For the missense model, we also used the following deleteriousness metrics: Missense badness, PolyPhen2, Constraint (MPC) [27], Sorting Intolerant From Tolerant (SIFT) [28], Polymorphism Phenotyping v2 (PolyPhen2) [29], Rare Exome Variant Ensemble Learning (REVEL) [30], and Functional Analysis Through Hidden Markov Models (FATHMM) [31]. Combined Annotation Dependent Depletion (CADD) deleteriousness scores [32] were included for the nonmissense SNV model.
For the schizophrenia pedigrees, we aimed to generate a prior that reflected the prioritization strategy implemented in the SCHEMA analysis and our own previous analyses of these pedigrees [33, 34]. To this end, in the ‘nonmissense SNV’ regression model, we used allele frequency and functional consequence as predictors, and, for the ‘missense’ model, we used allele frequency and the MPC score. The SCHEMA analysis focused on constrained genes, as determined by the pLI scores [35]. Therefore, the output of BICEP and pVAAST was manually filtered to retain variants in genes with pLI > 0.9.
Bayes factor
To calculate the likelihood of the causal model, we marginalize over the parameters, which results in the following:
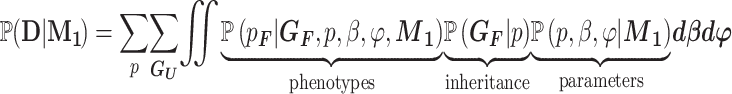 |
(2) |
The assumption of a dominant effect means that an individual’s phenotype is determined solely by whether they carry the variant. Therefore, the ‘phenotypes’ term in Equation (2) becomes a product of the penetrance values and phenocopy rates. If 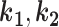 are the number of affected and unaffected variant carriers and
are the number of affected and unaffected variant carriers and 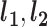 are the number of affected and unaffected variant noncarriers, then:
are the number of affected and unaffected variant noncarriers, then:
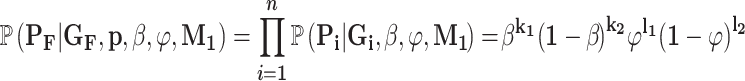 |
The calculation of the likelihood of the neutral model is similar to that of the causal model and results in the following:
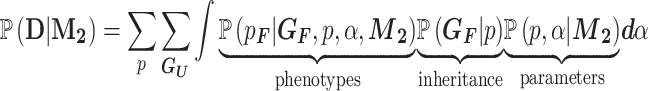 |
(3) |
The main difference from the causal model in Equation (2) is that instead of the penetrance and phenocopy terms, this model depends on the population incidence α. Also, the ‘phenotypes’ term in Equation (3) is simplified since the genotypes are independent of the phenotypes. We can calculate this term as:
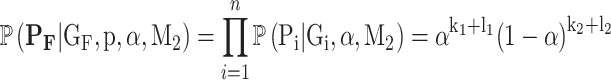 |
Evaluation data
We selected three WGS datasets to evaluate BICEP (details in Supplementary Methods). The first is a large pedigree presenting with an autosomal dominant congenital heart defects phenotype, for which WGS data have been generated [19, 36]. The second is a collection of three multiplex schizophrenia-affected pedigrees that had been previously analysed using an IBS filtering approach [33]. The third is a collection of synthetic phenotypes based on the three-generational CEPH1463 pedigree, for which WGS data have been generated [37]. While BICEP can account for variant missingness, this can often be an indicator of sequencing artefacts or low-quality data (see Supplementary Methods). Since the sample sizes in some of the pedigrees were modest, we removed variants with any missingness.
As a comparison to BICEP, we examined the output of pVAAST v2.2.0 for each pedigree (details in Supplementary Methods). We also considered PERCH [22] as a comparison tool but were unable to resolve issues with the implementation during a preliminary evaluation (see Supplementary Note 2). Since the schizophrenia data were aligned to GRCh38 and pVAAST cannot process data on this build, we removed variants at unstable positions [38] and converted the quality-controlled data to GRCh37 using liftOver [39] prior to applying pVAAST. All other datasets were generated on build GRCh37. Note that BICEP ranks each individual variant, whereas pVAAST aggregates the variant-level information to a gene-based score that is used for ranking.
Implementation
BICEP was implemented in Python 3. The software was evaluated on a Scientific Linux server with 20 Intel Xeon 2.20GHz central processing units (CPUs) and 32GB of memory. As input, BICEP requires a variant call format (VCF) file with the jointly genotyped genomic data of individuals sequenced in the pedigree and a FAM file describing the pedigree structure. To generate the prior, we used a VCF file of ClinVar variants that was annotated with the variant effect predictor from Ensembl [40]. Pre-annotated ClinVar VCF files are supplied with the BICEP source code for convenience.
Results
Bayesian Inference model for Causality Evaluation of rare variants in Pedigrees overview
For each input variant, BICEP uses a Bayesian inference model to evaluate if the variant is causal for a binary phenotype in the pedigree (Fig. 1). The posterior odds for causality (PostOC), which is used to rank the variants, is calculated as the product of the prior odds for causality (PriorOC) and the Bayes factor (BF) (see Methods, Equation (1), and Fig. 1). The PriorOC represents the odds of the variant being causal a priori and is generated using a logistic regression model fitted on data independent of the pedigree variants. Here, we used benign and pathogenic protein-coding SNVs from the ClinVar database [25] for our prior regression data, but the regression models can be fitted on data from a variety of sources/analyses (see Discussion). Variant allele frequency, functional consequence, and deleteriousness metrics were used as predictors for this logistic regression model. Indels and noncoding SNVs were not evaluated due to insufficient data in ClinVar.
Figure 1.

Schematic representation of BICEP. The prior odds of causality (PriorOC) are generated from genomic annotation information, independent of the pedigree data. The BF is generated based on the sequencing data, the pedigree structure, and the phenotypes. These are then considered on the base 10 logarithmic scale (logPriorOC and logBF) and summed to give the posterior odds of causality on the base 10 logarithmic scale (logPostOC). The dashed lines in the BF and logBF plots indicate the maximum achievable value for the pedigree (i.e. perfect cosegregation with the phenotype with complete penetrance and no phenocopies).
We evaluated the predictive ability of the prior by splitting the prior regression data into training and hold-out test subsets (see Supplementary Methods). We found that the prior had strong predictive performance on the hold-out test data, although this varied depending on the predictors chosen for the regression models (see Supplementary Fig. 1). We also generated logPriorOC values for variants of unknown significance (VUS) from ClinVar to further investigate the prior model. The benign variants from the hold-out test data had broadly negative logPriorOC values, and the pathogenic variants had mostly positive logPriorOC scores (Supplementary Fig. 2). The VUS had mixed logPriorOC values, lying between the benign and pathogenic values.
The BF compares the probability of the cosegregation pattern of a variant if it were causal versus if it had no effect on the phenotype. The underlying model for the BF is adapted from Mohammadi et al. and assumes that the variant has a dominant effect on the phenotype and is rare in the general population [23]. As rare variants are less likely to be carried by marry-in individuals compared to common variants, a rare causal variant is expected to be inherited identically by descent. Note, however, that a variant’s allele frequency is not explicitly used in the BF calculation but is rather incorporated through the prior. Using the BF, we can now directly compare evidence of cosegregation for variants in the same pedigree or across multiple pedigrees. We can also compare the BF of a variant to the maximum achievable BF based on the samples sequenced, which corresponds to perfect cosegregation with complete penetrance and no phenocopies (represented by the dashed horizontal line in the BF plots, see Fig. 1).
For convenience, we consider the three BICEP metrics (PriorOC, BF, and PostOC) on the base 10 logarithmic scale (denoted logPriorOC, logBF, and logPostOC respectively). The logPostOC is easily interpretable, with positive values indicating evidence for causality and negative values indicating evidence against it.
Mendelian congenital heart defects pedigree
Garg et al. described a large pedigree harbouring an autosomal dominant congenital heart defects phenotype (see Supplementary Fig. 3) and identified the G296S missense variant in GATA4 as the causal variant [36]. We applied BICEP to WGS data for this pedigree (see Methods), and the same GATA4 variant ranked first out of the 32 010 protein-coding variants investigated (Fig. 2). The logPostOC score of 8.05 for the GATA4 variant indicates that it is 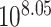 times more likely to be causal than neutral for the congenital heart defects in this pedigree. Of note, the GATA4 variant was the only variant that perfectly cosegregated with the congenital heart defects phenotype and also had a positive logPriorOC (Fig. 2). The variant that ranked second had a logPostOC of 4.59, indicating that it had almost 3000 times less evidence for causality than the GATA4 variant (
times more likely to be causal than neutral for the congenital heart defects in this pedigree. Of note, the GATA4 variant was the only variant that perfectly cosegregated with the congenital heart defects phenotype and also had a positive logPriorOC (Fig. 2). The variant that ranked second had a logPostOC of 4.59, indicating that it had almost 3000 times less evidence for causality than the GATA4 variant (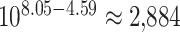 ). Hu et al. analysed this pedigree using pVAAST and found that GATA4 ranked first among all genes evaluated, with the G296S missense variant listed by pVAAST as the likely disease-causing variant [19].
). Hu et al. analysed this pedigree using pVAAST and found that GATA4 ranked first among all genes evaluated, with the G296S missense variant listed by pVAAST as the likely disease-causing variant [19].
Figure 2.

BICEP applied to the cardiac pedigree. The top 50 variants out of 32 010 ranked by the logPostOC (top panel) from BICEP in the congenital heart defect pedigree. The causal G296S missense variant in GATA4 is shaded and ranked first overall. Also shown for each variant are the logPriorOC (bottom panel) and the logBF (middle panel). The horizontal dashed line in the logBF plot represents the maximum achievable logBF in the pedigree.
Schizophrenia-associated pedigrees
To examine the performance of BICEP on a complex genetic disorder, we considered three pedigrees enriched for schizophrenia (see Supplementary Fig. 4). These pedigrees have been examined previously using a strict IBS-filtering approach, where ultrarare, deleterious SNVs with strong cosegregation patterns were prioritized [33]. This method identified three such variants, one private to each of the pedigrees. Here, we aimed to identify plausible variants that would have been missed by the previous analysis as well as those identified by the IBS-filtering approach. To tailor our analysis for this phenotype (see Methods), we generated a prior based on the prioritization strategy used by the schizophrenia exome meta-analysis (SCHEMA) consortium analysis of rare, protein-coding variants [34]. For consistency with the SCHEMA analysis, we considered variants in constrained genes only [35]. The output metrics from BICEP for the top five variants in each pedigree are shown in Fig. 3 (see also Supplementary Fig. 5) and are detailed in Table 1. In contrast to the congenital heart defects pedigree, the top results do not necessarily perfectly cosegregate with illness, which is consistent with the complex genetic architecture of schizophrenia.
Figure 3.

BICEP applied to the three schizophrenia pedigrees. The output metrics for the top five variants ranked by the logPostOC from BICEP in each of the three schizophrenia pedigrees (K1494, K1524, K1546). The three pedigree-private variants previously prioritized using the IBS-filtering approach [33] are shaded in each pedigree. The horizontal dashed line represents the maximum achievable logBF in each pedigree.
Table 1.
Results for BICEP applied to the three schizophrenia pedigrees.
| Ped | BICEP rank | CHR:POS:REF:ALT | Gene | Functional consequence |
gnomAD AF |
MPC | log PriorOC |
AFF | UN | log BF |
log PostOC |
pVAAST rank |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K1494 | 1 | chr14:71724684:A:G | SIPA1L1 | Missense |
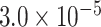
|
1.26 | 0.813 | 4 / 4 | 0 / 1 | 1.026* | 1.839 | 1 / 5 |
| 2 | chr19:42232651:A:G | GSK3A | Missense |
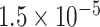
|
2.39 | 1.480 | 3 / 4 | 0 / 1 | 0.313 | 1.793 | N/A | |
| 3 | chr16:30669556:G:A | FBRS | Missense |
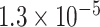
|
0.34 | 0.294 | 4 / 4 | 0 / 1 | 1.026* | 1.320 | 3 / 5 | |
| 4 | chr15:87929240:A:G | NTRK3 | Missense |
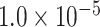
|
1.49 | 0.964 | 3 / 4 | 0 / 1 | 0.313 | 1.277 | N/A | |
| 5 | chr22:18083561:C:T | PEX26 | Missense |
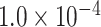
|
0.68 | 0.410 | 3 / 4 | 0 / 1 | 0.708 | 1.118 | N/A | |
| K1524 | 1 | chr10:99610923:T:C | SLC25A28 | Missense |
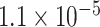
|
2.11 | 1.324 | 3 / 4 | N/A | 0.206 | 1.530 | N/A |
| 2 | chr6:43257902:C:T | TTBK1 | Missense |
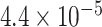
|
1.04 | 0.672 | 4 / 4 | N/A | 0.781* | 1.453 | 4 / 9 | |
| 3 | chr20:51674280:T:C | ATP9A | Missense |
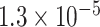
|
0.93 | 0.637 | 4 / 4 | N/A | 0.781* | 1.418 | 6 / 9 | |
| 4 | chr7:5313814:A:T | TNRC18 | Missense |
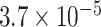
|
0.87 | 0.580 | 4 / 4 | N/A | 0.781* | 1.361 | 7 / 9 | |
| 5 | chr10:75398805:G:A | ZNF503 | Missense |
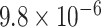
|
1.51 | 0.972 | 3 / 4 | N/A | 0.206 | 1.178 | N/A | |
| K1546 | 1 | chr1:35189207:G:A | SFPQ | Stop gain |
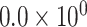
|
- | 2.573 | 1 / 5 | 0 / 3 | 0.153 | 2.726 | N/A |
| 2 | chr3:10360021:G:A | ATP2B2 | Missense |
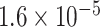
|
2.23 | 1.387 | 4 / 5 | 0 / 3 | 0.798 | 2.185 | 4 / 13 | |
| 3 | chr19:41986225:T:G | ATP1A3 | Missense |
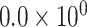
|
3.09 | 1.904 | 1 / 5 | 0 / 3 | 0.010 | 1.914 | N/A | |
| 4 | chr19:41986226:A:G | ATP1A3 | Missense |
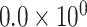
|
2.91 | 1.797 | 1 / 5 | 0 / 3 | 0.010 | 1.807 | N/A | |
| 5 | chr15:77614763:G:A | LINGO1 | Missense |
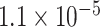
|
1.92 | 1.210 | 2 / 5 | 0 / 3 | 0.431 | 1.641 | N/A |
Shown are the output metrics for the top five variants ranked by BICEP in each of the three schizophrenia pedigrees: K1494, K1524, and K1546. Note that the MPC deleteriousness metric is only defined for missense variants [27]. Genes harbouring previously prioritised variants [36] are shown in bold (see also Figure 3). AFF represents the number of affected carriers out of all affected samples. UN represents the number of unaffected carriers out of all unaffected samples (N/A here indicates that there are no unaffected samples in this pedigree). The maximum logBF per pedigree is denoted with an asterisk (corresponding to dashed lines in logBF plots in Figure 3). The pVAAST rankings of the genes out of all constrained genes passing Bonferroni correction are also shown (N/A here indicates that the gene was not ranked by pVAAST). CHR: chromosome; POS: position (GRCh38); REF: reference allele; ALT: alternate allele; AF: allele frequency.
The top two ranked variants in pedigree K1494 have similar logPostOC scores but for different reasons. Although the highest-ranking variant, a SIPA1L1 missense variant, perfectly cosegregates with schizophrenia, it is only moderately deleterious. In contrast, the variant that ranks second, a GSK3A missense variant, is highly deleterious but has a reduced cosegregation pattern with schizophrenia. The third-ranked variant, in FBRS, also perfectly cosegregates but is much less deleterious than the variant in SIPA1L1. Given this, the top two ranked variants are the most plausible contributors to schizophrenia susceptibility in this pedigree and are potential candidates for further experimental validation.
In pedigree K1524, the top-ranked variant is a highly deleterious missense variant in SLC25A28, which has a reduced cosegregation pattern with schizophrenia. However, variants ranked second to fourth had perfect cosegregation and reasonable logPriorOC scores. Of note, the second-ranked variant is an ultra-rare missense variant in TTBK1, with a logPostOC score similar to the first-ranked variant in SLC25A28 (see Table 1). An ultrarare, pathogenic, de novo missense variant in TTBK1 listed in ClinVar has been reported in childhood-onset schizophrenia [41]. The variant in TTBK1 identified by BICEP was missed by the previous IBS-filtering approach as it was not considered sufficiently deleterious [33].
In pedigree K1546, the highest-ranked variant is an ultrarare stop-gain variant in SFPQ and, as such, has a large logPriorOC score (see Table 1). However, the evidence for cosegregation (i.e. logBF score) is not compelling, as the variant is carried by only one of the five affected members of the pedigree. In contrast, the second highest–ranked variant in this pedigree is a highly deleterious missense variant in ATP2B2, carried by four of the five affected members of the pedigree and absent from unaffected members (see Table 1). This result is supported by the previous IBS filtering analysis on the same data [33] and the SCHEMA analysis, which reported a nominal excess of ultrarare, deleterious, missense variants in ATP2B2 [34]. No variant ranked in the top 50 achieved the maximum logBF (see Supplementary Fig. 5c). The highest-ranking variant with perfect cosegregation has a negative logPriorOC, a negative logPostOC, and ranks 591st overall (not shown). Therefore, the ATP2B2 missense variant is the most plausible variant to contribute to the increased liability for schizophrenia in this pedigree.
We applied pVAAST to each of the three pedigrees separately after preprocessing the WGS data (see Methods and Supplementary Methods). pVAAST prioritized 5, 9, and 13 constrained genes in pedigrees K1494, K1524, and K1546 respectively (see Supplementary Table 1). However, not all variants could be evaluated (9 of the 15 genes harbouring variants in Table 1). From the previous IBS-filtering approach [33], both SLC25A28 and GSK3A also could not be scored. ATP2B2 received a nonzero pVAAST score and ranked fourth across the 13 constrained genes that had a significant output score (Table 1 and Supplementary Table 1).
In our previous analysis of these pedigrees [33], we were unable to quantify the evidence for causality with schizophrenia. A major advantage of BICEP is that it can be used to compare results across pedigrees. Of the variants identified by BICEP above that merit further investigation (which include the three variants from the previous analysis), the ATP2B2 variant has the most evidence for causality (see Table 1). Among the top five ranked variants in pedigree K1494, variants in GSK3A, NTRK3, and PEX26 were carried by three of the four affected samples and absent from the marry-in sample (see Table 1). The previous filtering analysis could not differentiate between these based on cosegregation alone, as it did not take the pedigree structure into account. However, the logBF indicates that the PEX26 variant has better cosegregation with schizophrenia than the other two variants.
Synthetic Mendelian phenotype
In any pedigree analysis, by random chance, we expect to observe common variants perfectly cosegregating with the phenotype. For phenotypes driven by rare, deleterious variants, these common variants should not rank highly. To this end, we used the three-generational CEPH1463 pedigree (see Supplementary Fig. 6) to generate synthetic phenotypes to investigate BICEP’s ability to correctly discriminate between rare and common variants with perfect cosegregation. We used a simple filtering approach to identify 21 deleterious, protein-altering variants that had a Mendelian dominant inheritance pattern (see Supplementary Methods). These included 6 rare variants (allele frequency < 1%), 2 low-frequency variants (1% ≤ allele frequency < 5%), and 13 common variants (allele frequency ≥ 5%). We created a collection of synthetic phenotypes by assuming that carriers are affected, and noncarriers are unaffected. We applied BICEP and pVAAST to each of the synthetic phenotypes individually, and the respective output metrics from both tools on the independent pseudo-causal variants are shown in Fig. 4 and detailed in Supplementary Table 2.
Figure 4.

BICEP and pVAAST applied to the CEPH 1463 pedigree with the synthetic phenotypes. Output scores for the pseudo-causal variants, including (A) the logPostOC from BICEP and (B) the gene-based composite likelihood ratio test score from pVAAST. For ease of presentation, the logPostOC scores were capped below at −2, indicated with an asterisk. Variants are coloured by their allele frequency (AF): Common (AF ≥ 5%), low frequency (1% ≤ AF < 5%), and rare (AF < 1%). Genes are ordered according to genomic position.
For BICEP, each pseudo-causal variant achieved the maximum logBF, as expected. In Fig. 4A, we see that the rare pseudo-causal variants all show evidence for causality with a positive logPostOC. The rare pseudo-causal variants (with the exception of the SMYD1 variant) were ranked in the top two variants scored by BICEP for their respective synthetic phenotype. Although the SMYD1 variant is rare and predicted deleterious by CADD, the other deleteriousness metrics used in the regression model for the logPriorOC are low (see Supplementary Table 2). These metrics indicate that the variant is not considered deleterious, which results in the negative logPriorOC and a modest logPostOC. As expected, all common pseudo-causal variants showed evidence against causality, despite their perfect cosegregation. The two low-frequency pseudo-causal variants showed mixed evidence for causality. The OR1J1 variant was a deleterious stop-gain variant that received a positive logPostOC, whereas the TSACC missense variant was modestly deleterious and so received a negative logPostOC.
For pVAAST, the gene harbouring each pseudo-causal variant was ranked at least fifth across all genes evaluated for the respective synthetic phenotype (see Supplementary Table 2). For each gene, the pseudo-causal variant was listed as one of the likely disease-causing variants, having achieved the maximum LOD score. However, pVAAST was unable to differentiate between rare and common pseudo-causal variants (Fig. 4B). For 9 of the 14 common pseudo-causal variants, the gene carrying the variant was ranked first overall.
Discussion
Here, we introduce BICEP, a novel Bayesian framework to evaluate rare variant causality in pedigree-based cohorts (see Fig. 1). We have extended previous Bayesian models to measure variant cosegregation with a binary phenotype [23] by incorporating prior information derived from genomic annotation data. The logPostOC output score is straightforward to interpret and can be used as a stand-alone measure for variant causality or as a relative measure to compare variants within or across pedigrees. The logBF explicitly incorporates penetrance and phenocopy rates in its derivation. This is useful for complex genetic traits where we may not always expect variants with perfect cosegregation, as with the schizophrenia-affected pedigrees. Users can interpret the output in the context of the genetic architecture underlying the selected phenotype. For example, if a user wishes to identify a variant likely responsible for the phenotype throughout the pedigree, they might ignore variants that ranked highly due to a large logPriorOC but with a modest logBF.
BICEP has several advantages over non-statistical methods such as filtering approaches. The output metrics provide quantitative measures that can be used to compare information across pedigrees. For example, using the logPostOC for the three previously prioritized variants in the schizophrenia pedigrees (see Table 1), we can definitively say that the ATP2B2 variant has the most evidence of causality for schizophrenia. Additionally, BICEP identified a rare missense variant in TTBK1 that perfectly cosegregated with schizophrenia in pedigree K1524 (see Table 1). An ultrarare pathogenic variant in TTBK1 has been reported in the ClinVar database for childhood-onset schizophrenia [25, 41], indicating that this gene is relevant for follow-up investigation. This variant identified by BICEP was missed by the previous analysis due to the use of strict cut-off thresholds, highlighting the benefits of the more sophisticated prioritization strategy proposed here.
This analysis focused on protein-coding SNVs due to limitations of the prior regression data from ClinVar used to calculate the logPriorOC. BICEP can easily be extended to other classes of genomic variants such as indels, noncoding variants, or copy number variants through the use of appropriate prior regression data. We used deleteriousness scores and functional consequence as predictors for the prior regression models, but BICEP allows for the selection of any genomic annotation to tailor the prior for the selected phenotype and its expected genomic architecture. As an example of this, we selected specific metrics used by the SCHEMA analysis to prioritize rare, damaging variants likely implicated in schizophrenia (see Fig. 3). Alternatively, the user can provide other annotated prior regression data where appropriate if it better suits the phenotype. The logPriorOC accurately distinguishes between hold-out benign and pathogenic variants, although the performance depends on the predictors selected (Supplementary Fig. 1). The logPriorOC gives a mixed distribution of values for VUS, which is consistent with their lack of sufficient evidence for or against pathogenicity (Supplementary Fig. 2). We showed that by requiring allele frequency as a predictor in the prior, BICEP will naturally prioritize rare variants over common variants, even those with strong cosegregation patterns (see Fig. 4A). However, rare variants must also be deleterious to receive a large logPriorOC score (see Supplementary Table 2 and Supplementary Fig. 7).
We compared the performance of BICEP to pVAAST, a tool that aims to perform disease–gene prioritization for similar pedigree-based data. pVAAST correctly identified some of the previously prioritized variants but was unable to score others, although the reason behind this is unclear. Additionally, pVAAST was unable to distinguish between common and rare variants that perfectly cosegregated with the synthetic phenotypes (see Fig. 4B). This is a major issue for phenotypes where there is selection against rare, deleterious variants, as common variants can perfectly cosegregate by random chance. For pVAAST, users can remove variants based on their allele frequencies [20], although this process suffers from the usual limitation of selecting a strict cut-off threshold for allele frequency filtering.
While BICEP performs well in the cohorts described here, there are some scenarios for which further development is required. As is often the case with standard genome-wide analytical methods, sex and mitochondrial chromosomes were not included since generating a prior for such variants is challenging. We expect that as appropriate prior regression data become available, the current implementation of BICEP could easily be adapted. The BF presented here does not account for the age of the pedigree individuals, which may be limiting for age-dependent phenotypes [21, 23]. BICEP has a reasonable runtime on the pedigrees described here (see Supplementary Table 3). However, increasing the number of nonsequenced individuals in the pedigree is expected to exponentially increase the runtime since BICEP iterates over all unknown genotypes. Genotype missingness decreases the logBF values on average, and the position of the individuals with missing data in the pedigree structure can result in more pronounced decreases (see Supplementary Section 2.1 and Supplementary Figs 8 and 9). The logBF calculation is simplified by assuming no consanguinity in the pedigree, and, therefore, BICEP cannot be used to investigate such complex pedigree structures. This work focuses on binary traits only, but the BICEP framework could be extended to continuous or multiclass traits by modifying the BF and obtaining appropriate prior regression data. Finally, BICEP currently evaluates variants independently for causality, but aggregating variant evidence at the gene level or across gene networks could be beneficial for a two-hit or multihit model. This would also facilitate comparing evidence across pedigrees since we may not always expect independent pedigrees to share the same rare causal variant, especially for complex genetic traits.
Conclusions
BICEP can be used to discover plausible disease-causing variants for phenotypes with both Mendelian and complex genetic architectures, including variants that would have been missed by traditional approaches. The output metrics can be used to compare variant causality both within and across pedigree datasets. Furthermore, as these metrics are based on Bayesian inference, they provide a quantitative method to rank variants in terms of causality. While we showcase BICEP using protein-coding SNVs here, the model can easily be extended to other classes of rare genomic variation. Given the increased interest in the analysis of pedigree-based genomic data, BICEP addresses the need for a rigorous statistical approach to prioritizing variants using this type of data.
Key Points
This work details a novel Bayesian tool to quantify the evidence that a DNA variant is causal for a genomic trait from pedigree-based data.
BICEP correctly identifies established causal variants found by traditional methods as well as identifying plausible causal variants missed by overly conservative filtering strategies.
The output metrics from BICEP can be used to compare the evidence for causality within and across pedigree cohorts, which is not possible using traditional prioritization approaches or existing tools.
Supplementary Material
Acknowledgements
The authors acknowledge the support of the Trinity Centre for High Performance Computing (ResearchIT). For the congenital heart defects pedigree data, we acknowledge data generation supported by NIH grants R01 GM104390, R01 DK091374, and R01 CA164138 as well as the University of Luxembourg Institute for Systems Biology Program.
Contributor Information
Cathal Ormond, Neuropsychiatric Genetics Research Group, Department of Psychiatry, Trinity Centre for Health Sciences, Trinity College Dublin, St James’s Hospital, Dublin 8, Ireland.
Niamh M Ryan, Neuropsychiatric Genetics Research Group, Department of Psychiatry, Trinity Centre for Health Sciences, Trinity College Dublin, St James’s Hospital, Dublin 8, Ireland.
Mathieu Cap, Neuropsychiatric Genetics Research Group, Department of Psychiatry, Trinity Centre for Health Sciences, Trinity College Dublin, St James’s Hospital, Dublin 8, Ireland.
William Byerley, Department of Psychiatry and Behavioral Sciences, University of California, 1550 Fourth Street, San Francisco, CA 94158, United States.
Aiden Corvin, Neuropsychiatric Genetics Research Group, Department of Psychiatry, Trinity Centre for Health Sciences, Trinity College Dublin, St James’s Hospital, Dublin 8, Ireland.
Elizabeth A Heron, Neuropsychiatric Genetics Research Group, Department of Psychiatry, Trinity Centre for Health Sciences, Trinity College Dublin, St James’s Hospital, Dublin 8, Ireland.
Author contributions
Conceptualisation: C.O., E.A.H., N.M.R., and A.C. Data curation: C.O. Formal analysis: C.O., and E.A.H. Funding acquisition: A.C. Investigation: C.O., E.A.H., N.M.R., and A.C. Methodology: C.O., and E.A.H. Resources: W.B., E.A.H., and A.C. Software: C.O., N.M.R., M.C., and E.A.H. Supervision: A.C., and E.A.H. Validation: N.M.R., M.C., and E.A.H. Visualisation: C.O., and E.A.H. Writing (original draft): C.O., E.A.H, and A.C. Writing (review and editing): C.O., E.A.H, N.M.R., and A.C.
Funding
This work was supported by the National Institutes of Mental Health [U01MH109499-04 and R01MH124875 to A.C.] and Science Foundation Ireland [16/SPP/3324 to A.C.].
Data availability
The source code for BICEP is publicly available at: https://github.com/cathaloruaidh/BICEP. BICEP is available under a dual licence that grants use for academic purposes. WGS data for the congenital heart defects pedigree are available from dbGaP (accession number phs000758.v1.p1). WGS data for the CEP1463 pedigree are available from dbGaP (accession number phs001224.v1.p1). WGS data for the schizophrenia pedigrees are available from the corresponding author upon reasonable request.
References
- 1. Glahn DC, Nimgaonkar VL, Raventos H. et al. Rediscovering the value of families for psychiatric genetics research. Mol Psychiatry 2019;24:523–35. 10.1038/s41380-018-0073-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thomas DC, Yang Z, Yang F. Two-phase and family-based designs for next-generation sequencing studies. Front Genet 2013;4:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jiao X, Ke H, Qin Y. et al. Molecular genetics of premature ovarian insufficiency. Trends Endocrinol Metab 2018;29:795–807. 10.1016/j.tem.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 4. Similuk MN, Yan J, Ghosh R. et al. Clinical exome sequencing of 1000 families with complex immune phenotypes: toward comprehensive genomic evaluations. J Allergy Clin Immunol 2022;150:947–54. 10.1016/j.jaci.2022.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuhlen M, Taeubner J, Brozou T. et al. Family-based germline sequencing in children with cancer. Oncogene 2019;38:1367–80. 10.1038/s41388-018-0520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kanzi AM, San JE, Chimukangara B. et al. Next generation sequencing and bioinformatics analysis of family genetic inheritance. Front Genet 2020;11:544162. 10.3389/fgene.2020.544162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ott J, Wang J, Leal SM. Genetic linkage analysis in the age of whole-genome sequencing. Nat Rev Genet 2015;16:275–84. 10.1038/nrg3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pol-Fuster J, Cañellas F, Ruiz-Guerra L. et al. The conserved ASTN2/BRINP1 locus at 9q33.1-33.2 is associated with major psychiatric disorders in a large pedigree from southern Spain. Sci Rep 2021;11:14529. 10.1038/s41598-021-93555-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet 2010;11:415–25. 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 10. Homann OR, Misura K, Lamas E. et al. Whole-genome sequencing in multiplex families with psychoses reveals mutations in the SHANK2 and SMARCA1 genes segregating with illness. Mol Psychiatry 2016;21:1690–5. 10.1038/mp.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deng Z, Chen M, Zhao Z. et al. Whole genome sequencing identifies genetic variants associated with neurogenic inflammation in rosacea. Nat Commun 2023;14:3958. 10.1038/s41467-023-39761-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Delgado-Vega AM, Martínez-Bueno M, Oparina NY. et al. Whole exome sequencing of patients from multicase families with systemic lupus erythematosus identifies multiple rare variants. Sci Rep 2018;8:8775. 10.1038/s41598-018-26274-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ryan NM, Ormond C, Chang YC. et al. Identity-by-descent analysis of a large Tourette's syndrome pedigree from Costa Rica implicates genes involved in neuronal development and signal transduction. Mol Psychiatry 2022;27:5020–7. 10.1038/s41380-022-01771-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cardoso M, Maia S, Brandão A. et al. Exome sequencing of affected duos and trios uncovers PRUNE2 as a novel prostate cancer predisposition gene. Br J Cancer 2023;128:1077–85. 10.1038/s41416-022-02125-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mitani T, Isikay S, Gezdirici A. et al. High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population. Am J Hum Genet 2021;108:1981–2005. 10.1016/j.ajhg.2021.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao L, He Z, Zhang D. et al. A rare variant nonparametric linkage method for nuclear and extended pedigrees with application to late-onset alzheimer disease via WGS data. Am J Hum Genet 2019;105:822–35. 10.1016/j.ajhg.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bush WS, Haines J. Overview of linkage analysis in complex traits. Curr Protoc Hum Genet 2010Chapter 1;64:Unit 1.9.1–18. 10.1002/0471142905.hg0109s64. [DOI] [PubMed] [Google Scholar]
- 18. Altman DG, Royston P. The cost of dichotomising continuous variables. BMJ 2006;332:1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu H, Roach JC, Coon H. et al. A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat Biotechnol 2014;32:663–9. 10.1038/nbt.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yandell M, Huff C, Hu H. et al. A probabilistic disease-gene finder for personal genomes. Genome Res 2011;21:1529–42. 10.1101/gr.123158.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Petersen GM, Parmigiani G, Thomas D. Missense mutations in disease genes: a Bayesian approach to evaluate causality. Am J Hum Genet 1998;62:1516–24. 10.1086/301871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feng BJ. PERCH: a unified framework for disease gene prioritization. Hum Mutat 2017;38:243–51. 10.1002/humu.23158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mohammadi L, Vreeswijk MP, Oldenburg R. et al. A simple method for co-segregation analysis to evaluate the pathogenicity of unclassified variants; BRCA1 and BRCA2 as an example. BMC Cancer 2009;9:211. 10.1186/1471-2407-9-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rañola JMO, Liu Q, Rosenthal EA. et al. A comparison of cosegregation analysis methods for the clinical setting. Fam Cancer 2018;17:295–302. 10.1007/s10689-017-0017-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Landrum MJ, Lee JM, Benson M. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018;46:D1062–d1067. 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karczewski KJ, Francioli LC, Tiao G. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Samocha KE, Kosmicki JA, Karczewski KJ. et al. Regional missense constraint improves variant deleteriousness prediction. bioRxiv 2017;148353. [Google Scholar]
- 28. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–4. 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013. Chapter 7; Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ioannidis NM, Rothstein JH, Pejaver V. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 2016;99:877–85. 10.1016/j.ajhg.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shihab HA, Gough J, Cooper DN. et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 2013;34:57–65. 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rentzsch P, Witten D, Cooper GM. et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47:D886–94. 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ormond C, Ryan NM, Heron EA. et al. Ultra-rare missense variants implicated in Utah pedigrees multiply affected with schizophrenia. Biol Psychiatry Glob Open Sci 2023;3:797–802. 10.1016/j.bpsgos.2023.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Singh T, Poterba T, Curtis D. et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 2022;604:509–16. 10.1038/s41586-022-04556-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lek M, Karczewski KJ, Minikel EV. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91. 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Garg V, Kathiriya IS, Barnes R. et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003;424:443–7. 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 37. Eberle MA, Fritzilas E, Krusche P. et al. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res 2017;27:157–64. 10.1101/gr.210500.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ormond C, Ryan NM, Corvin A. et al. Converting single nucleotide variants between genome builds: from cautionary tale to solution. Brief Bioinform 2021;22;1–7. 10.1093/bib/bbab069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haeussler M, Zweig AS, Tyner C. et al. The UCSC genome browser database: 2019 update. Nucleic Acids Res 2019;47:D853–d858. 10.1093/nar/gky1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McLaren W, Gil L, Hunt SE. et al. The Ensembl variant effect predictor. Genome Biol 2016;17:122. 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ambalavanan A, Girard SL, Ahn K. et al. De novo variants in sporadic cases of childhood onset schizophrenia. Eur J Hum Genet 2016;24:944–8. 10.1038/ejhg.2015.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code for BICEP is publicly available at: https://github.com/cathaloruaidh/BICEP. BICEP is available under a dual licence that grants use for academic purposes. WGS data for the congenital heart defects pedigree are available from dbGaP (accession number phs000758.v1.p1). WGS data for the CEP1463 pedigree are available from dbGaP (accession number phs001224.v1.p1). WGS data for the schizophrenia pedigrees are available from the corresponding author upon reasonable request.