

Summary
Inflammation, acinar atrophy, and ductal hyperplasia drive pancreatic remodeling in newborn cystic fibrosis (CF) ferrets lacking a functional cystic fibrosis conductance regulator (CFTR) channel. These changes are associated with a transient phase of glucose intolerance that involves islet destruction and subsequent regeneration near hyperplastic ducts. The phenotypic changes in CF ductal epithelium and their impact on islet function are unknown. Using bulk RNA sequencing (RNA-seq), single-cell RNA sequencing (scRNA-seq), and assay for transposase-accessible chromatin using sequencing (ATAC-seq) on CF ferret models, we demonstrate that ductal CFTR protein constrains PDX1 expression by maintaining PTEN and GSK3β activation. In the absence of CFTR protein, centroacinar cells adopted a bipotent progenitor-like state associated with enhanced WNT/β-Catenin, transforming growth factor β (TGF-β), and AKT signaling. We show that the level of CFTR protein, not its channel function, regulates PDX1 expression. Thus, this study has discovered a cell-autonomous CFTR-dependent mechanism by which CFTR mutations that produced little to no protein could impact pancreatic exocrine/endocrine remodeling in people with CF.
Subject areas: Physiology, Molecular biology, Cell biology, Transcriptomics
Graphical abstract
Highlights
-
•
The lack of CFTR leads to activation of PDX1 in pancreatic ductal epithelium
-
•
CFTR-KO duct epithelium has open chromatin at endocrine regulatory loci
-
•
Centroacinar cell expansion is observed in CFTR-KO epithelium
-
•
The absence of CFTR protein inhibits PTEN and activates WNT and PDX1 expression
Physiology; Molecular biology; Cell biology; Transcriptomics
Introduction
Cystic fibrosis (CF) is caused by mutations in the cystic fibrosis conductance regulator (CFTR), a chloride and bicarbonate channel that plays important roles in regulating the hydration and pH of epithelial secretions in the lung, pancreas, gallbladder, liver, and intestine.1 The pancreas is one of the earliest affected organs in CF, due to the lack of CFTR-mediated bicarbonate secretion into pancreatic ducts, which maintains an alkaline pH required to inhibit premature activation of pancreatic enzyme prior to exit into the intestine.1,2 In the absence of a functional CFTR channel, pancreatic acinar cell loss, severe inflammation, fibrosis, and adipogenesis alter the microenvironment of the pancreatic islets and ductal epithelium leading to pancreatic insufficiency and diabetes.3,4 At a histologic level, initial stages of CF pancreatic remodeling share similarities with developing pancreatic adenocarcinoma (PDAC), where hyperproliferative ducts form budding structures through a process thought to involve acinar to ductal metaplasia (ADM).5,6 However, little is known about the phenotypic alterations that occur in CF ductal cells and their impact on the function of islets within the pathologically altered pancreatic environment.3,7
CF pancreatic disease and its associated diabetes have been particularly difficult to study in mice lacking the CFTR gene. While certain genotypes of CF mice appear to exhibit some endocrine dysfunction and have reduced islet mass,8,9,10 they fail to develop major exocrine disease in the pancreas like humans.7 However, CF ferret models have proven useful to study pancreatitis and CF-related diabetes (CFRD) due to greater organ level conservation with human.2,7,11 We have previously categorized pancreatic tissue remodeling and associated abnormalities in glucose tolerance broadly into 4 phases using a CFTR-KO (CF) ferret model.12 A period of normoglycemia (phase I) is followed by a period of spontaneous glycemic instability at ∼1- to 2-months of age (phase II) with accompanied loss of endocrine hormone producing islets, severe fibrosis, and inflammation. This is followed by a transient recovery at ∼3-month of age (phase III) with continued tissue remodeling (adipogenesis) and temporary normalization of glucose tolerance during a period of islet resurgence.12 As CF ferrets age (phase IV), they then go on to develop the more classically studied CFRD. Similar to PDAC, CF pancreatic ducts proliferate and form budding structures that contain endocrine hormone-expressing cells during these transitions.6 Furthermore, similar phases of transient glucose intolerance followed by recovery are observed in young children with CF.13,14
Previous studies have implicated phenotypic alterations to CFTR-KO ferret ductal epithelium that can alter islet function through paracrine signaling of proinflammatory factors such as interleukin (IL)-615 and trophic factors such as IGFBP7.16 Further support for altered exocrine to endocrine signaling in the CFTR-KO ferret pancreas comes from analyses of the pancreatic ductal secretome and whole cell proteome.16 These studies have implicated CF-associated alterations in transforming growth factor β (TGF-β), bone morphogenetic protein (BMP), phosphatase and tensin homolog (PTEN), AKT, wingeless-related integration site (WNT/β-catenin), and pancreatic duaodenal homeobox-1 (PDX1) signaling pathways and changes in the expression of proteins that directly bind with CFTR.16 Lastly, analysis of autocrine-paracrine circuits between exocrine/endocrine cell type, derived from 7 different human pancreatic islet single-cell RNA sequencing (scRNA-seq) datasets (7,603 cells), led to the discovery that BMP and WNT signaling is altered in the CF pancreas devoid of acinar cells.17 Despite the growing evidence for phenotypic changes in exocrine and endocrine cell types in the CF pancreas, the molecular mechanisms for these changes have remained elusive.
The PDX1 transcription factor is considered a master regulator of pancreatic development serving critical roles in the initiation of pancreas formation, the formation of multipotent progenitors in ductal epithelium, and the specification and maintenance of beta cells.18,19,20 Expression of both PDX1 and SOX9 in the duct epithelium facilitates an endocrine differentiation program via NGN3.21 However, ectopic expression of PDX1 in acinar cells has been shown to induce trans-differentiation to endocrine lineages.22 PDX1 also appears to play a critical role in the initiation of PDAC and progression during epithelial-to-mesenchymal transition (EMT).23 The mechanistic basis of aberrant PDX1 expression and its role in progression of pancreatic malignancies are still largely unknown. Integral to EMT in the progression of malignancies is the loss of apical-basolateral polarity maintained by apical anchoring proteins such as PTEN.24,25 In this context, PTEN has been shown to constrain centroacinar cell identity and loss of PTEN leads to the expansion of PDX1-expressing ductal cell, acinar cell loss, and PDAC.26
Here we characterized the transcriptomic and epigenomic landscape of wild-type (WT) and CF ferret pancreatic ductal epithelia to investigate whether mechanisms of ADM could explain genotypic changes previously observed in the ductal epithelial proteome and secretome.16 These studies discovered that high-level PDX1 expression in CFTR-knockout (KO) ductal epithelia likely originates from expansion of centroacinar cells with an altered epigenetic landscape that confers EMT-like features with differentially open chromatin at endocrine lineage associated transcription factor loci. Notably, these altered properties of CF pancreatic ductal epithelia were dependent on the loss of the CFTR protein, but not its channel function, and were driven by inhibition of the PTEN/GSK3β pathway leading to enhanced Wnt/β-catenin signaling. These findings suggest that CFTR residence on the apical membrane of ductal progenitors regulates their cell fate and may have implications for disease phenotypes in people with CF that harbor CFTR mutations that produce no or little protein.
Results
Pancreatic ductal epithelium activates PDX1 expression in CFTR-KO ferrets
Given the shared features in pancreatic histopathology between CF and PDAC, we hypothesized that adaptive mechanisms that drive ADM may be similar. PDAC is frequently associated with the aberrant expression of pancreatic development genes PDX15,23 and SOX9,21,27,28 which are required transcription factor for pancreas formation and PDAC progression. To this end, we evaluated expression of PDX1 and SOX9 in the CFTR-KO ferret pancreas at the stage of disease (∼2-month-old) associated with peak inflammation, fibrosis, and glycemic instability.12,13 Consistent with ADM-like features, expression of these master regulators of the pancreatic development program was elevated in a subset of CFTR-KO (CF) ductal epithelial cells, as compared to age-matched WT controls (Figures 1A–1E, 1H). Similar to previous findings,6 sporadic insulin-expressing cells within CF pancreatic ducts were seen but were not present in WT pancreas (Figures 1F and 1G). Additionally, a subset of CF ductal cells expressed higher levels of acinar markers AMY2B29 and RNASE130 (Figures S1A–S1D, S1G), but had reduced expression of the ductal specific marker HNF631 (Figures S1E–S1G), as compared to WT controls. These findings support a change in CF ductal cell phenotype that appears similar to those observed in ADM and PDACs.
Figure 1.

Aberrant gene expression in CF pancreatic ducts
(A–E) WT and CF 2-month-old ferret pancreas immunostained for (A and B) PDX1 and INS or (C–E) SOX9. Insets are single-channel images of the regions marked by the dotted boxes.
(F and G) Immunofluorescent images of 2-month-old WT and CF ferret pancreas stained for INS. Islets are identified by the expression of insulin and are marked by arrows. Ducts are identified by the presence of a lumen and marked by dotted lines. All images were acquired on a confocal microscope (Zeiss 880) at 20X magnification and processed for maximum intensity projection. Scale bars are 50 μm.
(H) Mean intensity of nuclear PDX1 or SOX9 immunoreactivity within ductal cells and frequency of PDX1- or SOX9-positive cells from WT (n = 4 donors) and CF (n = 4 donors) 2-month-old pancreata. Four ducts were quantified from each donor and averaged. Data show the mean ± SEM. Nonparametric Mann-Whitney t test was used to evaluate significance (∗p < 0.05).
(I) Schematic of in vitro approached used to establish ferret pancreatic duct epithelium (PDE) from passaged pancreatic ductal cells (PDCs).
(J) Relative expression of pancreatic duct enriched genes in WT and CF PDC and PDE quantified using RT-qPCR. Nonparametric Mann-Whitney t test was used to evaluate significance (∗p < 0.05).
To delineate cell-autonomous alterations to the CF ductal cell phenotype, we used an approach previously developed for the expansion and polarization of WT and CF ferret pancreatic ductal cells (PDCs) in culture16 (Figure 1I). To obtain a uniform population of pan-cytokeratin-expressing ductal epithelial cells, primary cells were cultured for 10 passages prior to polarization at an air-liquid interface (ALI). We observed genotypic differences in mRNA expression for several ductal markers in proliferating PDCs and polarized pancreatic ductal epithelia (henceforth called PDE when speaking about ALI cultures), with KRT7, CDH1, and SOX9 expression being elevated in CF and HNF6 being lower in CF (Figure 1J). Additionally, PDX1 expression was uniformly elevated in CF in PDC and PDE cultures, as compared to WT controls (Figure 1J). Thus, in vivo changes in PDX1, SOX9, and HNF6 expression in CF pancreatic ducts were also observed at the mRNA level in PDC and PDE cultures.
CFTR-KO PDE cultures adopt mesenchymal features following polarization
To further clarify genotype-specific changes in ductal cell phenotype, we performed bulk RNA sequencing (RNA-seq) on WT and CF PDE cultures. Differential gene expression analysis followed by gene ontology (GO) and upstream regulator analysis inferred alterations to signaling pathways in CF PDE cultures (Figures 2A–2C and S2). Of the 16,574 expressed genes, 923 genes were differentially expressed after benjamini hoschberg (BH) correction (corrected p value <0.05) in CF PDE cultures (Figures S2A, 2A, and Table S1A). Of the differentially expressed genes (DEGs), 27 genes were upregulated (Log2FC > 2) in CF PDEs and 9 genes were downregulated (Log2FC < −2) (Table S1A). GO term analysis of DEGs revealed pathways involved in Mesenchyme Differentiation and Development, Positive regulation of Cell Migration, and Cell-Cell Junction Organization, and Protein Localization in Membrane (Figure S2B and Table S2A), suggesting CF PDEs retain features associated with a loss in epithelial characteristics. In support of this hypothesis, the upregulation of collagen genes plays key role during EMT32 and 10 collagen genes were upregulated (Log2FC > 2) in CF PDEs (Table S1A). However, genes known to repress or activate EMT, such as TMEM45B, ECRG4, CHRDL1, and PCDH10, were also among the most highly upregulated genes in CF PDE, suggesting a transitioning cellular state (Table S1A). PDX1 expression was significantly (p = 4.44E−04) increased 6.6-fold in CF PDEs (Table S1A). Notably, BMP signaling inhibitor (CHRDL1) and WNT signaling activator (TRIM14) were upregulated 5.7-fold and 5.6-fold in CF PDEs (p < 0.05), respectively (Table S1A), indicating potential repression of BMP and activation of WNT signaling.33,34 This was supported by GO term and upstream regulator analysis of DEGs where cell differentiation, mesenchyme differentiation, cell migration, and EMT pathways including WNT and TGF-β were activated and BMP signaling was inhibited (Figures S2B and S2C, Figures 2B and 2C, Tables S1B and S1C, Tables S2A–S2F).
Figure 2.

Transcriptional changes in CF PDE implicate WNT activation and BMP repression
(A) The bulk transcriptomes of newborn WT and CF PDE cultures were sequenced. Heatmap shows differentially expressed genes following Benjamini-Hochberg correction (p < 0.05).
(B) Upstream regulators of observed DEGs were obtained from IPA analysis. The Z scores and p values of the top regulators are shown.
(C) Overlap between putative upstream regulators of DEGs found in PDE cultures and known regulators of pancreatic endocrine progenitors specific genes. Activation Z scores are displayed in the heatmap. Association of the candidate upstream regulators with WNT or BMP signaling is indicated on the right.
(D) Whole-mount localization of pSMAD5 (BMP regulator) and nuclear CTNNB1 (β-catenin, WNT regulator) with insulin in WT and CF PDE cultures. Images were obtained on confocal microscope Zeiss 880 at 20X magnification and processed for maximum intensity projection.
(E) Quantification of nuclear pSMAD5 in WT (n = 5 donors) and CF (n = 5 donors) PDE cultures.
(F) Quantification of cytoplasmic and nuclear CTNNB1 in WT (n = 6 donors) and CF (n = 5 donors) PDE cultures.
(G) Quantification of insulin expression in WT (N = 6 donors) and CF (N = 7 donors) PDE cultures. Bar plots in (E–G) show mean intensity of expression from 3 transwells per donor +/− SEM. Significance was calculated using nonparametric Mann-Whitney t test (∗∗p < 0.01).
The putative upstream regulators of the DEGs in CF PDEs that were associated with mesenchymal transition were found to include WNT and BMP signaling regulators (Figure 2C and Table S1B). For example, activation of upstream regulator ID2 (p = 4.7E−6; Z score = 3.0) has been correlated with increased WNT signaling cancer stem cells.35 Upstream WNT regulator MYCBP36,37 was also activated (p = 3.8E−7; Z score = 2.0), while inhibition of BMP receptor 2 (BMPR2) was observed (p = 2.54E−6; Z score = −4.0) (Figure 2B and Table S1B). Similarly, we observed activation of UBE2K (p = 1.01E−4; Z score = 3.973), a target of WNT signaling that aids the progression of EMT (Figure 2C, Tables S1B and S1C),38 whereas upstream regulated NFATC3 was inhibited (p = 2.22E−7; Z score = −3.0)—a transcription factor activated by BMP signaling that is known to prematurely disrupt mesenchymal transition (Figures 2C, Tables S1B and S1C).39,40 Similarly, BMP signaling activator SMARCA4, a factor known for maintenance of epithelial-like gene signatures,41 was inhibited (p = 3.05E−6; Z score = −3.5) (Figures 2C and Table S1C). Overall, the enhanced mesenchymal signatures in the bulk transcriptome of CF PDEs implicated changes to signaling pathways (BMP, TGF-β, and WNT) that also play important roles in cell fate decisions during pancreatic development and disease (Figures S2B, S2C, 2B, and 2C).
Despite in vitro culture for 10 passages prior to polarization at an ALI, genes associated with inflammation remained upregulated in CF PDEs, including the inflammasome-associated PYCARD (Log2FC = 5.62, p = 2.28E−08)42 and activator of nuclear factor κB (NF-κB)/tumor necrosis factor alpha (TNF-α) signaling in pancreatic cancer VRK2 (Log2FC = 2.25, p = 8.6E−04)43 (Table S1A). Furthermore, many chromatin remodeling genes were lower in CF PDEs (Figure S2E and Table S1A) including CBX2,44 KDM6A,45 and H1-0,46 suggestive of an altered chromatin state (Table S2G).
TGF-β1, BMP, and WNT signaling are altered in CFTR-KO PDCs in vivo
We assessed whether the inferred transcriptional changes in BMP and WNT signaling of CF PDE cultures reflected those observed in vivo. Active BMP signaling is associated with increases in nuclear phosphorylated suppressor of mothers against decapentaplegic (pSMAD), and thus we hypothesized that pSMAD levels would be lower in CF ductal epithelia. Indeed, nuclear pSMAD5 levels in CF PDEs (Figures 2D and 2E) and CF pancreas (Figures S3G, S3H, and S3L) were ∼2-fold lower than WT counterparts, supporting the hypothesis that BMP signaling is suppressed in CF ductal cells.
Accumulation of nuclear β-catenin is associated with activation of canonical WNT signaling, and nuclear β-catenin is elevated in CF human pancreatic ducts.17 Similarly, CF PDEs demonstrated an increase in nuclear β-catenin (∼2-fold) and decrease (∼3-fold) in cytoplasmic β-catenin (Figures 2D–2F). To substantiate that WNT signaling is activated in the CF ferret pancreas, we evaluated localization of WNT7A,47 which was previously shown to be upregulated in human CF PDCs by scRNA-seq.17 Indeed, clusters of WNT7A-expressing cells were observed in ∼2-month-old CF pancreatic ducts but were largely absent in WT controls (Figures S3C, S3D, and S3J). Similarly, AXIN2 expression, a marker of WNT activation,47 was increased (∼18-fold) in CF ductal pancreatic epithelium (Figures S3A, S3B, and S3I). These findings support the hypothesis that WNT signaling is activated in CF ductal cells in vitro and in vivo.
TGF-β1 plays major roles in EMT during development and disease and similarly promotes endocrine differentiation during pancreatic development.48,49 Consistent with CF PDE gene signatures, TGFB1 mRNA expression was significantly (p < 0.05; ∼100-fold) higher in 2-months-old CF ferret ductal epithelium as compared to WT controls (Figures S3E, S3F, and S3K). Given that TGF-β1 and EMT are involved in endocrine progenitor fate initiation,48 we stained WT and CF PDEs for insulin following polarization. CF PDEs retained greater numbers of insulin-expressing cells compared to WT (p < 0.01) (Figures 2D and 2G). As previously shown,6 CF ferret pancreatic ductal epithelium also contained sporadic insulin-expressing cells in vivo, which was not observed in WT pancreas (Figures 1F and 1G). Collectively, the phenotypic changes observed in CF PDEs in vitro and pancreatic ducts in vivo appeared similar to those typically associated with multipotent ductal progenitors during pancreatic development.
The epigenome of CF PDEs is reprogrammed with multipotent progenitor signatures
The reduced expression of chromatin regulatory genes in CF PDEs suggested epigenomic modifications could be responsible for altering the CF ductal cell phenotype. To this end, we used bulk assay for transposase-accessible chromatin using sequencing (ATAC-seq) to evaluate differentially accessible chromatin in WT and CF PDEs following 14-day of polarization. Approximately 13,000 regions were significantly (p < 0.05) differentially accessible as determined by DiffBind analysis (Figure 3A) (Table S3A). Genomic loci of endocrine progenitor transcription factors PDX1 (Log2FC = 4.3, p = 2.94E−27) and PAX6 (Log2FC = 2.25, p = 1.9E−05) among others were significantly more open in CF as compared to WT PDE cultures (Figure 3B and Table S3A), consistent with enhanced PDX1 expression in CF PDE cultures (Figure 1J and Table S1A). Histone-modifying genes described to suppress endocrine progenitor specification,50,51,52 including histone methyltransferase EZH2 (Log2FC = −2.00, p = 0.000901), histone deacetylases HDAC4 (Log2FC = −3.29, p = 8.46E−16), and HDAC9 (Log2FC = −2.18, p = 1.35E−5), were differentially closed in CF PDE cultures (Table S3A). Disruption of these genes in mice51,52 or treatment with HDAC inhibitors53 promotes endocrine specification, whereas overexpression of HDACs inhibits specification of beta and delta cells.52 With the exception of SOX9, which was more open in CF PDEs and has roles in maintaining both bipotent pancreatic progenitors and pancreatic ductal identity,21,54 the chromatin of exocrine fate-related factors were relatively unchanged in CF vs. WT PDE cultures (Figure 3B).
Figure 3.

Altered epigenome in CF PDE cultures leads to greater accessibility of endocrine lineage-related genes
(A) Differentially open regions (DORs) in the genome of CF relative to WT PDE cultures are shown in the volcano plot. Regions in the genome with −2< or >2 Log2FC read alignment were considered differentially open between genotypes.
(B) Histograms of the number of reads aligned to genomic loci of endocrine fate-related factors and exocrine fate-related factors.
(C) Analysis of motif enrichment (AME) for TF binding site motifs in the open regions of the CF PDE genome. Enriched motifs of endocrine fate-related TFs are shown. The percentage of open regions with the shown motifs is given in parentheses.
(D) RT-qPCR quantification of endocrine fate-associated gene expression in WT (n = 11 cultures from 6 donors) and CF (n = 13 cultures from 6 donors) PDE cultures. Boxplots indicate mean relative expression +/− SEM. Significance was calculated using nonparametric Mann-Whitney t test (∗p < 0.05).
While SOX9 is a marker of ductal cell lineages in the adult pancreas, during pancreatic development SOX9 induces expression of NEUROG3—the transcription factor required for specification of endocrine cell lineages.54 SOX9 has also been described to initiate acinar to ductal reprogramming and maintain the pancreatic multipotent and bipotent progenitor pool.21 Thus, open chromatin at the SOX9 locus (Figure 3B) and enhanced SOX9 expression in CF PDCs and PDE (Figure 1J) are also consistent with expansion of ductal-derived progenitors in CF ductal epithelium. Additionally, NR5A2 (Log2FC = 3.9, p = 3.2E−08), a trunk-specific transcription factor known for its role in maintaining the progenitor population55 and generation of acinar cells during development,56 and PROM1 (Log2FC = 2.94, p = 0.00474), a duct epithelial progenitor marker,57 were both differentially open in CF PDE (Table S3A). These findings suggest the presence of a mixture of progenitor cell phenotypes in CF PDE capable of exocrine and endocrine lineage specification.
To gain a better understanding of the altered transcriptional landscape in CF PDE, the differentially open genomic regions were evaluated for enrichment of transcription factor binding motifs using analysis of motif enrichment (Figure 3C and Table S3B). This analysis revealed significant enrichment of binding sites within open chromatin for several transcription factors involved in regulating pancreatic endocrine and exocrine fate including FOXA2 (enriched in ∼62% of the sequences),58 NEUROD1 (enriched in ∼75% of the sequences),59 and NR5A2 (enriched in ∼35% of sequences)55 among others (Table S3B). GO term analysis of enriched motifs indicated activation of endocrine developmental program, with increased expression of endocrine lineage-related factors found in CF PDEs (Figure S2D and Table S2H). As expected, pancreatic progenitor and endocrine specification transcription factors PDX1 (100-fold, p < 0.001), PAX6 (4-fold, p < 0.05), and NKX6.1 (5-fold, p < 0.05) were expressed at significantly higher levels in CF PDE cultures (Figure 3D). These results further substantiate the observed transcriptional changes in CF PDCs that implicate altered potential for differentiating toward exocrine and endocrine lineages.
WT and CF PDCs contain divergent and distinct lineages during polarization and differentiation
To better understand the genotype-specific differences in fate potential of PDCs, we performed scRNA-seq on neonatal WT and CF PDCs during polarization and differentiation at an ALI. The WT and CF PDEs were sequenced on 2, 5, 7, and 9 days of polarization (Figure S4A). Three WT and three CF donors with an average difference in PDX1 expression of ∼1,600-fold in PDC proliferating culture, and ∼100-fold following polarization, were pooled and used for scRNA-seq (Figures S4B and S4C). An average of ∼6,000 cells per sample were sequenced. Only cells with mitochondrial RNA less than 20% of the total RNA were included in the subsequent analyses. Principal-component analysis followed by clustering using Seurat identified prominent pancreatic cell states at all stages of differentiation.60 Clustifyr was used to assign cell states to the predicted clusters61 by correlating average expression of each gene to previously published single-cell expression datasets.62,63,64,65,66 The cell state with the highest correlation was assigned to the cluster.
The predicted identities of the cell clusters were acinar, ductal, proliferating acinar, proliferating ductal, centroacinar progenitor, and centroacinar (Figures S4E, S4F, Figure 4A, and Tables S4A–S4F). Clusters with differentially upregulated expression of ADIRF,67 CAPG, S100A1063, and keratins (KRT5, KRT15, KRT19) were assigned ductal identity (Figures S4E, S4F, and Table S4B).65 Enrichment of IPA pathway analysis terms for this ductal cluster included branching morphogenesis of epithelial tube, negative regulation of cell migration, and columnar/cuboidal epithelial cell differentiation. Together with ductal expressed markers ADIRF,67 ADIRF, and S100A10,63 differential expression of cell-cycle markers like CCNA2, CCNB1/2, CDC20, and CDCA2 was used to classify proliferating ductal cells (Figures S4E, S4F, and Table S4D). Acinar cells were marked by LDHA, ASNS, PRDX2, PRDX4, FKBP4, FKBP11, and NUPR1, which were also found as acinar markers in previous single-cell studies63 (Figure S4F and Table S4A). The lack of expression of prominent acinar markers like AMY2B is believed to be due to the ductal origin of these acinar-like cells (Figures S4E, S4F, and Table S4A). Clusters differentially expressing the acinar genes PRDX2, PRDX4, and FKBP11,63 as well as cell-cycle regulatory genes like CCNB1/2, CDC20, and CDCA2, were assigned as proliferating acinar cells (Figures S4D–S4F and Table S4C). Clusters called centroacinar cells had a combination of ductal (KRT7, KRT16) and the centroacinar-enriched genes (WSB1, PROM1, S100A6, S100A4) previously reported by scRNA-seq65 (Figures S4F and Table S4F). Finally, clusters assigned centroacinar progenitor cell identity was characterized by ALDH1A1 and BCL2A1 enriched expression68,69 together with expression of multipotency regulator proteins like KLF4 and TFF3,65,70 progenitor-like pancreatic duct epithelial marker TSPAN8,65 mucins (MUC1, MUC13, MUC16, MUC4, MUC5AC, MUC5B, MUC20), and exocrine markers CEACAM (Figures S4E, S4F, and Table S4E).
Figure 4.

Single-cell transcriptomes of actively differentiating PDCs reveal acquisition of an epithelial-to-mesenchymal transitional phenotype in CF PDE cultures
(A) WT and CF PDE on day 2 (D2), day 5 (D5), day 7 (D7), and day 9 (D9) of differentiation at ALI underwent 10× single-cell RNA sequencing (scRNA-seq). Shown are uniform manifold approximation and projections (UMAPs) of cell types at each time point of differentiation; cell markers for classification used a combined mouse and human scRNA-seq pancreatic datasets.
(B) Heatmaps of DEGs in CF relative to WT PDE cultures at each time point. Genes relevant to epithelial-to-mesenchymal transition (EMT), diabetes, and cystic fibrosis are highlighted. Time point of differentiation is shown in the color-coded panel on top of each heatmap (using the legend shown in C).
(C) Violin plots of DEGs found in WT vs. CF PDE cultures depicting Log2(TPM) expression level at each time point of differentiation.
(D) Putative upstream regulators of the DEGs in CF PDE cultures. Activation Z score of candidate regulators is shown at each time point of differentiation. Z scores >2 indicate activation (dotted lines) and <−2 indicates inhibition.
(E) Activation Z scores of IPA pathways altered in CF relative to WT PDE cultures at each time point of differentiation.
The WT and CF PDEs had variable differentiation trajectories following polarization with significant genotype-specific differences in the derived cell states. WT PDEs were composed of predominantly ductal cells on day 2 and differentiated to a mixture of ductal and centroacinar cells by day 9 (Figures 4A and 5D). CF PDEs were also predominantly composed of duct cells on day 2 and by day 9 were predominantly composed of centroacinar cells (Figures 4A and 5D). Differential gene expression analysis between the two genotypes at each time point showed presence of signatures that altered during the course of differentiation. For example, PAX6 was differentially enriched in CF PDEs on day 2 and higher in CF PDEs relative to WT PDEs at all time points (Figures 4B, 4C and Tables S5A–S5D), suggestive of endocrine differentiation71 and/or a phenotype similar to early duct/endocrine progenitors.72,73 Increased HES1 in day-5 CF PDE was associated with the expansion of centroacinar cells74 and centroacinar progenitor cells on day 7 for which HES1 has known roles75,76 (Tables S6A–S6F). SPP1 is a marker for undifferentiated pancreatic progenitors65 and was elevated in CF PDE at day 7 to day 9 (Figures 4B and 4C, Tables S5C–S5D, and Tables S6A–S6F).
Figure 5.

CF PDE cultures predominantly differentiate to centroacinar cells
(A and B) Lineage trajectories observed in differentiating (A) WT and (B) CF PDE cultures using slingshot pseudotime ordering.
(C) Variable expression profile of genes associated with centroacinar progenitors and centroacinar cell development in CF and WT PDE lineages.
(D) Relative proportions of cell types in WT and CF PDE cultures at different time points of differentiation. A higher relative proportion of centroacinar cells is observed in CF PDE cultures.
(E) DEGs in centroacinar progenitor and centroacinar cells generated from WT and CF PDE cultures at day 9 of differentiation.
CF PDEs acquire cell migration-associated markers during differentiation
Putative upstream regulators of the scRNA-seq DEGs in CF PDEs indicated the activation of AKT and PI3K and the repression of PTEN at day 5–9 (Figure 4D; Tables S7A–S7D). Epithelial identity is closely associated with cellular polarity with defined apical and basolateral domains. Cytoskeletal remodeling alters the distribution of epithelial polarity complex proteins and can change cellular identity.76 A known cell migration-inducing factor integrin subunit beta (ITGB) was upregulated in CF PDEs (Tables S5C, S5D, S6C, and S6D).77,78 Migration is associated with loss of adherence junctions, which eliminates apical-basal polarity and initiates front-rear polarization.79 Consistent with this, GAS6, a protein that associates with AXL and induces migration through ERK signaling, was upregulated in CF PDE (Tables S5C, S5D, Tables S6C, and S6D).80 Similarly, LAMB1 important for basement membrane formation and for induction of cell migration by the ERK pathway,81 was higher in CF PDEs (Tables S5C, S5D, S6C, and S6D). Substantiating this, PTEN (p = 2.30E−10) and twist related protein (TWIST) (p = 1.65E−13) were shown to be upstream regulators of the observed DEGs at each time point (Tables S7A–S7D and Figure 4D).
PTEN dephosphorylates PIP3 only when transiently bound to cell membrane. This restricts PTEN activity spatially and ensures increased density of PIP3 on the apical membrane, which is important for apical-basal polarity.25,82 Likewise, TWIST is necessary for cytoskeletal rearrangement when cells lose apical-basal polarity and acquire invasiveness during EMT.83 Activated β-catenin signaling was identified as an upstream regulator of CF ductal cell phenotype (Figure 4D and Tables S7A–S7D) and was supported by enhanced nuclear accumulation of β-catenin in vivo (Figures 2D and 2F). Furthermore, β-catenin depletion from adherence junctions and cytoplasm is a prominent mechanism for loss of epithelial polarity.84 Pathway analysis of CF PDE DEGs also included integrin signaling and actin cytoskeleton initiating on day 7 (Figure 4E and Tables S7E–S7H). GP6 signaling is also necessary for cellular migration85 and was also an upstream regulator activated in CF relative to WT on day 7 and day 9 (Figure 4E and Tables S7E–S7H). Taken together, upregulation of pathways that contribute to loss in epithelial polarity and cell fate transitions was observed in CF PDE cultures.
Multiple ductal subpopulations were identified in both WT and CF PDE (Figure S6A and Tables S8A–S8F) each of which had unique changes in gene expression (Figure S6B and Tables S8A–S8F); GO term analysis of the differential gene expression patterns from WT and CF PDE ductal cells (Figures S6C and S6D) indicated inhibition of BMP signaling and activation of PI3K-AKT signaling, which was similar to the analysis on bulk RNA-seq of WT and CF PDE.
WT and CF PDE cultures contain progenitors with unique trajectories during polarization and differentiation
In order to determine the temporal dynamics of genotypic changes in PDE phenotype following polarization, the sequenced transcriptomes were ordered on pseudotime using Slingshot. Slingshot first determines the number of lineages and branching points for each trajectory and then estimates cell-level pseudotime variable for each lineage.86 Using this method, three principal lineages were predicted for both WT and CF PDE cultures, with the ductal cells as the starting point for WT and centroacinar cells for the starting point of CF for all predicted lineages (Figures 5A and 5B). In the WT PDEs, the ductal cells transitioned through a centroacinar-like state and differentiated mostly to acinar or ductal cells, whereas the CF centroacinar cells differentiated primarily to centroacinar progenitors and ductal-like cells (Figures 5A and 5B). Given the genotype-dependent discrete clustering observed for the derived cell states, we sought to understand the genetic differences between WT and CF-derived centroacinar, centroacinar progenitors, acinar, and ductal cells. Particularly, we characterized the difference in WT and CF-derived centroacinar and centroacinar progenitor cells in WT and CF lineage 3 (Figures 5A and 5B).
Genes differentially expressed in the WT and CF pseudotime lineage 3 trajectory are shown in Figure 5C. Retinoic acid signaling has been shown to be important for centroacinar cell differentiation and maintenance,87,88 and cellular retinoic acid binding protein 2 (CRABP2) gene expression was upregulated over time in the CF trajectory during differentiation (Figure 5C). Similarly, AGR2, which is known to induce translocation of epidermal growth factor receptor (EGFR) to the membrane during pancreatic regeneration and for proliferation of progenitors in vitro,89,90 also increased with time of CF PDE differentiation (Figure 5C). Genes associated with fibrosis (FABP5) and activation of stellate cells (ISG20)88,91,92 were also higher in the CF lineage (Figure 5C). ARL4C, which is activated by WNT signaling,93,94 was higher in CF at all stages of differentiation (Figure 5C). Notably, MUC16 and ICAM1 were lower in CF and are genes shown to be downregulated during EMT (MUC16).95 Likewise, cancer biomarker (ICAM1) was downregulated in CF PDEs96,97 (Figure 5C). Quantification of relative proportions of predicted cell types at each time point of differentiation showed an increase in centroacinar cells in CF PDEs from ∼25% (day 2) to ∼80% (day 9) (Figure 5D). By contrast, centroacinar cells were absent from WT PDE cultures on day 2 but rose to ∼40% of the culture by day 9.
PTEN-associated signaling pathways are upregulated in CF PDE centroacinar cells
During late phases of pancreas development, Notch and its target gene Hes1 promote ductal differentiation from exocrine-restricted progenitors and then maintenance of centroacinar cell fate in the mature pancreas.74,98 Notch/HES1 also repress cell fate commitment by multipotent and bipotent pancreatic progenitors during development,99 and thus lower HES1 expression in CF-derived centroacinar progenitors is consistent with a more pliable progenitor cell state (Table S6E and Figure 5E). Furthermore, higher expression of PAX6, ID1, and ID3 in CF centroacinar progenitors supports known functions in ID protein maintenance of a stem cell state and PAX6 involvement in endocrine cell specification73,100,101 (Tables S6E and Figure 5E). Additionally, CF centroacinar progenitors appeared more proliferative given the upregulation of cell-cycle genes CCND1 and CCND2 (Tables S6E and Figure 5E).
Like CF centroacinar progenitors, DEGs in CF centroacinar cells included upregulation of endocrine lineage genes ID2 and ID3 (Table S6E and S6F), but also enhanced expression of WNT mediator CTNNB1102 (Table S6F) and PI3K signaling subunit PIK3R1, and downregulation of ductal genes like MUC1 and KRT8 (Tables S6E and S6F and Figure 5E). Upstream regulator analysis on DEGs between WT and CF-derived centroacinar cells and centroacinar progenitors showed upregulation of TGF-β (p = 2.67E−55), WNT (p = 1.37E−37), and AKT (p = 4.41E−18) signaling (activation Z score >2) and inhibition of PTEN (p = 6.34E−21) only in CF centroacinar cells (activation Z score < −2) (Tables S9A and B) (Figures S5A and S5B). AKT signaling-associated genes TSC2 and EIF4E, both regulators of cell cycle and mTOR-related tissue regenerative mechanisms,103,104 were higher in CF centroacinar cells (Table S9C). Enhanced expression of the PTEN inhibitor CREB3L2 was observed in CF centroacinar cells (Table S9D). PTEN signaling contributes to epithelial polarity,25 and loss of PTEN is thought to promote PDAC from centroacinar cells and EMT.105 Furthermore, PDPK1 (PDK1), a gene required for expansion of exocrine and endocrine pancreatic progenitors during development,106 was also elevated in CF centroacinar cells (Table S9D). TGF-β signaling is known for its roles in EMT during the duct to endocrine transition of pancreatic development,49 and THBS1, a TGF-β signaling target gene and EMT activator,107 was upregulated in CF centroacinar cells (Table S9F). Upstream activators associated with WNT signaling in CF centroacinar cells (Figures S5A and S5B) included enhanced expression of LEF1 and TCF7L2 relative to WT centroacinar cells (Table S9E), which are known to be associated with enhanced proliferation in pancreatic cancer.108 Taken together, the altered phenotype in CF PDEs centered around TGF-β-mediated inhibition of PTEN signaling, activation of PTEN downstream target AKT, and WNT activation likely through AKT-mediated inhibition of GSK3B. These scRNA-seq studies thus confirmed many of the findings in bulk RNA-seq and ATAC-seq and implicate these pathways as mediators of cellular programs governed by PDX1 and global changes in the epigenetic landscape of CF PDEs.
Cell-autonomous reprogramming of pancreatic ductal epithelium occurs in the absence of CFTR protein not function
Pancreatic pathology in CF is hypothesized to be caused by the lack of CFTR-mediated bicarbonate secretion, which lowers pH and leads to premature activation of pancreatic enzymes and the inflammatory destruction of acinar cells.2 Based on the transcriptional signatures of CF ductal epithelium, we hypothesized that ductal cell reprogramming was secondary to inflammatory-dependent expansion of a unique progenitor and/or CFTR-dependent channel functions (i.e., luminal pH regulation) that impact epithelial phenotype. To approach the later of these hypotheses, we asked whether inhibition of CFTR function in WT PDEs would impact PDX1 expression (Figures 6A and 6B). Notably, continuous treatment of differentiating WT PDEs for 14-day with CFTR inhibitor GlyH101 inhibition did not significantly alter PDX1 expression (Figure 6B), despite effective inhibition of CFTR-mediated chloride currents (Figure 6A). We have previously shown that CFTR-G551D ferret PDEs are responsive to the CFTR modulator VX-770, which restores channel gating.109 Thus, we next sought to evaluate whether VX-770 rescue of CFTR-G551D function in CF PDEs would inhibit PDX1 expression. Contrary to our hypothesis, PDX1 expression in CFTR-G551D PDEs was 1000-fold lower than CFTR-KO PDEs regardless of whether they were differentiated in the presence of VX-770 (Figure 6C). Given that CFTR-G551D PDC cultures were derived from ferrets with similar pancreatic pathology to CFTR-KO PDC cultures (i.e., untreated with VX-770), these findings suggested that the altered PDX1 phenotype of CFTR-KO PDCs and PDEs was not a direct consequence of the inflamed state from which the cells were derived or CFTR channel function.
Figure 6.

CFTR protein presence, but not function, regulates PDX1 expression
(A) Short circuit current measurement in WT PDE cultures differentiated in the presence of CFTR inhibitor GlyH101 or vehicle (DMSO). Responses to sequential addition of amiloride, DIDS, IBMX/Forskolin (IF), and GlyH101 are shown (n = 4 donors with 2 cultures averaged per donor). Inset is a representative current trace for each condition.
(B) RT-qPCR quantification of PDX1 expression under the conditions show in (A). PDX1 expression in CFTR-KO PDE cultures without GlyH101 is shown for comparison (n = 4 donors with 3–4 cultures combined for RNA).
(C) PDX1 expression in PDE cultures derived from ferret PDCs with homozygous CFTR-G551D germ line and differentiated in the presence of VX-770 (+) or DMSO (−) (n = 3 donors with 3–4 cultures combined for RNA). PDX1 expression in CFTR-KO PDEs is shown for comparison (n = 3 donors with 3–4 transwells combined for RNA). PDX1 expression is normalized to that of (B) WT (−) or (C) G551D (−).
(D) Schematic of human CFTR (hCFTR) complementation in CFTR-KO PDCs using a lentiviral vector that also expresses tdTomato. A vector expressing just tdTomato was used as a control (cont). FACS-enriched tdTomato-positive cells were used to generate PDE cultures. Representative images of PDC cultures before and after FACS enrichment are shown below the schematic.
(E) Short circuit current measurements of PDE cultures-derived lentiviral transduced CFTR-KO PDCs expressing hCFTR/tdTomato or tdTomato alone. Responses to sequential addition of amiloride, DIDS, IBMX/Forskolin (IF), and GlyH101 are shown (n = 3 donors). Inset is a representative trace of current for each condition.
(F) RT-qPCR quantification of ductal (SOX9, HNF6) and endocrine (PDX1, PAX6, NKX6.1) genes expression from the conditions in (E) (n = 3 donors with 2 cultures combined for RNA). Expression levels are normalized to CFTR-KO tdTomato PDE cultures.
(G) Immunofluorescence images of PDX1 and INS expression in 2-month-old WT ferrets and CFTR mutants ferrets with variable expression of CFTR protein (percent CFTR expression is shown on top of each image). Images were obtained on confocal microscope Zeiss 880 at 20X magnification and processed for maximum intensity projection. Scale bars, 50 μm.
(H) Quantification of PDX1 expression in ductal epithelium (n = 3 donors for each genotype). All graphs show the mean ± SEM. Significance was calculated using nonparametric Mann-Whitney t test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Since CFTR-KO PDEs lack both CFTR function and protein presence on the membrane, we asked whether reconstituting the CFTR protein in CFTR-KO PDEs would restore PDX1 expression to WT levels. CFTR-KO PDCs were genetically modified using lentiviral vectors to express hCFTR/tdTomato or tdTomato alone and then enriched for tdTomato-expressing cells by fluorescence-activated cell sorting (FACS) prior to plating at an ALI (Figure 6D). As expected, PDEs cultures generated from hCFTR/tdTomato-expressing PDCs generated CFTR currents that were significantly greater (p < 0.0001) than tdTomato-expressing controls (Figure 6E). Notably, complementation of CFTR expression led to reduced PDX1, PAX6, and SOX9 expression toward that of WT but had no effect on NKX6.1 expression (Figure 6F). Collectively, these findings implicated the cell-intrinsic presence of the CFTR protein, not its function, as the root cause of altered CFTR-KO ductal cell phenotype.
We next sought to obtain in vivo data to support a correlation between CFTR presence in PDCs and PDX1 expression. To this end, we evaluated PDX1 expression in pancreata derived from a series of CF ferret models with CFTR genotypic variants that alter the abundance of CFTR protein. These models included ferrets harboring (1) a biallelic CFTR-G551D mutation that expresses 100% of WT CFTR (GKGK),110 (2) a hypomorphic biallelic G551D mutation with only 50% CFTR expression (GG) due to a neomycin selection cassette in neighboring CFTR intron,109 and (3) a compound heterozygote harboring one CFTR-KO allele111 and one hypomorphic G551D mutation109 with only 25% CFTR expression (GKO). Finding from these pancreata demonstrated that PDX1 expression in the ductal epithelium of GKGK animals was similar to WT, while in GG and GKO pancreata PDX1 expression increased in concert with the extent of hypomorphic CFTR expression (Figures 6G and 6H). These findings support the in vitro observations implicating a cell-autonomous process by which CFTR protein presence, not function, alters the observed PDX1 phenotype in CFTR-KO ductal cells.
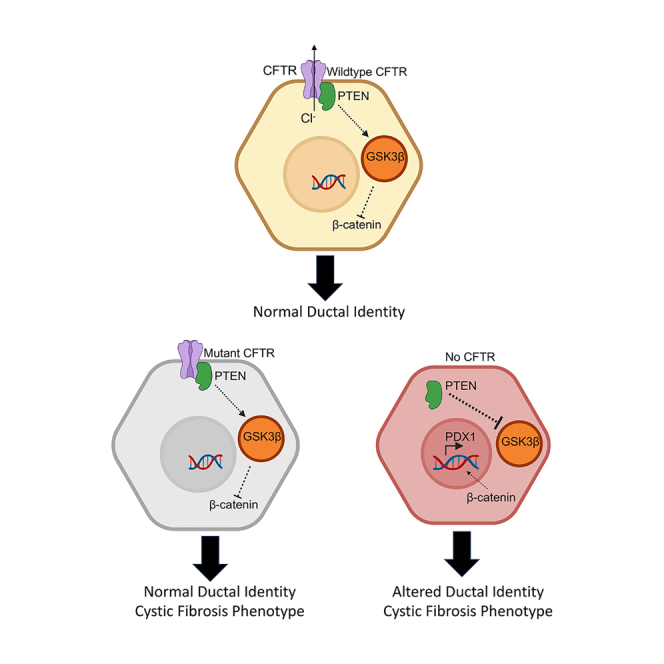
PTEN inhibition and WNT activation alter ductal cell phenotype
Our results indicate that PTEN signaling in CF PDEs is perturbed and is a major upstream regulator of their altered transcriptional signature. Apical CFTR has been previously shown to be associated with multiple membrane proteins, including PTEN, where CFTR serves as a membrane anchor and regulator of PTEN activity, and this regulation does not require a functional CFTR channel.112,113 This is postulated to be necessary to restrict PTEN activity to the apical membrane and establish the apical-basal axis for epithelial polarization.25,114 PTEN inhibits PIP2 to PIP3 conversion by PI3K, necessary for AKT phosphorylation and activation. Activated AKT inhibits GSK3B to stimulate WNT signaling by repressing GSK3B-mediated phosphorylation and degradation of beta-catenin. Hence, active PTEN inhibits downstream WNT signaling by de-repression of GSK3B activity.115 However, the loss of CFTR-PTEN complex impairs PTEN activity112 and thus could activate WNT signaling.
Since the lack of CFTR protein leads to enhanced PDX1 expression in CFTR-KO PDCs and PDEs, we hypothesized that a similar mechanism of CFTR-mediated PTEN inhibition was responsible for alterations in CFTR-KO ductal phenotype. To formally test this, we perturbed the PTEN pathway at two important nodes—PTEN and GSK3B. We polarized WT PDEs in the presence of PTENi or GSK3Bi and evaluated their effect of PDX1 and related progenitor genes SOX9 and NKX6.1 (Figure 7). Repression of PTEN activity resulted in significant increases in PDX1 (p < 0.05) and SOX9 (p < 0.05), but not NKX6.1 (Figure 7A). While pancreas-specific knockdown of PTEN has been previously reported to elevate PDX1 expression in ducts,26 its effect on SOX9 expression has not been shown. However, increased SOX9 expression is consistent with EMT initiation and also associated with PTEN inhibition.116,117
Figure 7.

PTEN inhibition and WNT activation induce PDX1 expression in WT PDE
(A and B) RT-qPCR quantification of PDX1, SOX9, and NKX6.1 mRNA in WT PDEs exposed to PTEN inhibitor (A) (PTENi) and WNT agonist (B) (CHIR). Boxplots show the mean relative expression +/− SEM for n = 4 donors per condition with ≥3 PDE cultures analyzed per donor and averaged. Significance was calculated using nonparametric Mann-Whitney t test (∗p < 0.05, ∗∗p < 0.01).
(C) Schematic of proposed model for CFTR/PTEN-mediated PDX1 regulation. The reactions that are proposed to be active in the presence or absence of CFTR are shown by green arrows (activating) and lines with caps (inhibitory), whereas reactions that are suppressed are indicated in red arrows and lines with caps. Dotted arrows indicate presence of intermediate reactions that are not shown in the schematic.
Activation of WNT by inhibiting GSK3B led to significantly higher levels of PDX1 (p < 0.001), SOX9 (p < 0.05), and NKX6.1 (p < 0.05) in WT PDEs, again mirroring the CFTR-KO PDE phenotype (Figure 7B). Non-canonical WNT signaling has been shown to induce PDX1 expression and prime the foregut cells for pancreatic lineage and prevent liver differentiation.118 WNT signaling also increases SOX9 expression during ductal branching morphogenesis. Additionally, PDX1 and SOX9 have been described to act cooperatively for pancreatic lineage specification and pancreatic progenitor maintenance.50,54,119 Collectively, these data support a structural role for CFTR in the maintenance of ductal cell phenotype though the control of PTEN/GSK3B axis.
Discussion
Here we demonstrate that PDX1-positive ductal progenitors expand within the CF pancreas in the setting of acinar cell loss. In vitro expansion and differentiation of these progenitors suggest they are centroacinar cell derived and harbor bipotent progenitor cell transcriptional signatures that are typically associated with the specification of exocrine and endocrine fates during pancreatic development. Notably, these altered properties of CF PDCs were dependent on the loss of the CFTR protein, but not function, and are phenotypically distinct from WT ductal cells. Inhibition of PTEN or GSK3β led to the activation of PDX1 expression in WT ductal epithelium, giving rise to a similar phenotypic state in the CFTR-KO pancreas with enhanced Wnt/β-catenin activation and PDX1 expression. These findings suggest that CFTR residence on the apical membrane of ductal progenitors regulates their cell fate and may have implications for disease phenotypes in people with CF that harbor CFTR mutations that produce no or little protein.
Changes in CF pancreatic ductal phenotype have not been carefully studied, largely because CF mouse models lack the pancreatic phenotype observed in people with CF. In contrast to mice, the CF ferret pancreas undergoes significant remodeling within the first two months of life involving islet destruction, inflammation, fibrosis, adipogenesis, and ductal hyperplasia.6,11,12,109,120 The observed increase in PDX1 expression of CFTR-KO pancreatic ducts was postulated to be either due to the altered signals from the remodeled pancreatic environment or due to a cell-autonomous change associated with ADM, as often observed in PDX1-positive PDAC caused by chronic pancreatitis.23 Our in vitro studies with CFTR-KO proliferating PDCs and differentiated PDE, which remove extrinsic signals from the CF pancreas, support cell-autonomous changes in ductal cell phenotypes that enhance PDX1 expression. However, we appreciate that the loss of cellular microenvironment in in vitro systems can diminish the impact of environmental factors found in vivo and understand that in vitro differentiation can mask physiological lineage dynamics. Hence, we have attempted to verify our in vitro findings in vivo but emphasize that our results warrant further cell-specific validation.
PDX1 has a well-characterized developmental role in initiating pancreatic and beta cell differentiation.18 Given that PDX1 marks multipotent progenitors in the developing pancreas, higher PDX1 and SOX9 expression in CFTR-KO PDEs suggested a similar cellular state might exist in CF pancreatic ducts.28,119,121 Furthermore, ∼2% of the differentially open regions in the CFTR-KO ductal genome have been previously shown to bind PDX1 in pancreatic progenitors,122 highlighting PDX1 prominence in establishing the CF phenotype. While PDX1 binds to a Swi/Snf chromatin-modifying complex to initiate pancreatic progenitor differentiation,123 the cooperative role of both PDX1 and SOX9 in establishing and maintaining the progenitor pool is well established.50 In the adult pancreas, centroacinar cells are progenitors marked by SOX9 and HES1 but express little to no PDX1.21,75,105,119 However, PDX1 expression is observed in centroacinar cells and ductal cells of PDAC,23 and the ductal pattern in early stage cancers is strikingly similar to that in CFTR-KO ducts. Lineage tracing aberrant PDX1 expression in the CF pancreas could provide a specific cellular context to changes in signaling.
Inactivation of pancreatic developmental marker loci like PBX1 (Table S3A) in CF PDE, previously known to induce islet malformations and aberrant ductal morphogenesis during development,124 suggested potential loss in normal differentiation potential. However, increased accessibility of islet differentiation factors like ISL1 and PAX6 (Table S2A) infers a disposition to endocrine lineages. Taken together, CF PDE displayed a mix of epigenetic signatures of a primordial state with abnormal regenerative capability. Given that ATAC-seq was a bulk assay, multiple differentially accessible genes were not differentially expressed. However, in addition to PDX1, one of the genes that was differentially open and differentially expressed in CF PDEs was CACNA2D1 (Tables S1A and S3A). Notably this gene encodes a calcium channel complex that enables influx of Ca2+ and when defective leads to impaired islet insulin secretion and diabetes.125
PTEN is known to play an important role in maintenance of epithelial polarity, EMT, and cancer progression,24,82,117 and the transcription signatures of these pathways were altered in CFTR-KO PDEs (Figure S2). The localization of PTEN at the apical membrane of epithelial cells maintains PIP2 density important for polarity maintenance, and the loss of PTEN promotes EMT and malignant transformation.26,117 Pancreas-specific PI3K activation via PTEN KO increases expression of pancreatic progenitor genes PDX1 and HES1 in pancreatic ducts of mice via centroacinar cell metaplasia.26 Our bulk RNA-seq (Figure S2) and scRNA-seq (Figure S5) on CFTR-KO PDEs demonstrated transcriptional changes consistent with EMT, suppression of the PTEN pathway, and centroacinar cell expansion. Inhibition of PTEN would be expected to increase nuclear β-catenin and Wnt signaling, and this is consistent with enhancement of WNT7A, AXIN2, and nuclear β-catenin in CF pancreatic ducts and/or PDEs (Figures S3, S2, and 2C). The physical association of PTEN with CFTR at the plasma membrane maintains PTEN activity in airway epithelia, and CFTR absence inhibits PTEN activity leading to the activation of NF-κB though PI3K/AKT activation of inhibitor of nuclear factor k (IKK).112,113 These findings are strikingly similar to ours studying CFTR-KO ductal epithelium, in that CFTR mutants with stable membrane expression failed to inhibit the PTEN pathway and subsequent signaling alterations. Furthermore, the level of CFTR protein expression from hypomorphic mutants correlated with the ductal cell activation of PDX1 expression in vivo.
This study comes with limitations. Further analysis is needed to validate the CFTR/PTEN interaction in PDE cultures and determine the cell types in vivo for which this occurs. Utilization of transgenic ferrets with varying abundance of CFTR protein on the membrane (including newly reported CFTRF508del)126 would enable these studies and provide a better understanding of the PDX1-CFTR/PTEN axis in vivo using molecular markers of functional changes obtained in this study. These additional in vivo studies would provide greater physiological context to this research, given that our in vitro studies exclude a significant number of cell-cell interactions (duct-adipocyte, duct-fibroblast, etc) that could impact cell fate in vivo. Lastly, it is now possible to fate map progenitors in vivo using multi-transgenic ferret models127; thus, with the appropriate cell-specific Cre driver it will be possible in the future to fate map these phenotypic changes and better understand their in vivo implications to CF disease progression.
Our findings have potential implications for people with CF harboring CFTR mutation that produces no or little mutant protein like F508del. One previous clinical study in a relatively small cohort of people with CF (average age of 26–27 years), found that 23% of F508del/F508del patients developed impaired glucose tolerance or CFRD, as compared to 8% of patients with one G551D allele and a second severe mutation.128 In comparison to our studies in ferrets, these findings would suggest that reduced CFTR protein at the membrane in ductal cells has a negative impact on progression of CFRD. Whether this altered human phenotype relates to altered phenotype of ductal cells in the CF pancreas remains to be determined.
We propose that loss of CFTR at the membrane of centroacinar cells represses PTEN leading to the activation of AKT, inhibition of GSK3β, nuclear accumulation of β-catenin, and activation of WNT signaling (Figure 7C). Supporting this model are CFTR complementation experiments in CF PDEs and PTEN and GSK3β chemical inhibition experiments in WT PDEs, which either reserve or promote CF-associated changes in PDX1, SOX9, PAX6, and/or NKX6.1 expression. Coupled with scRNA-seq experiments in actively differentiating PDE cultures, we conclude that CFTR plays a structural role in maintaining pancreatic ductal epithelial phenotype, the loss of which leads to expansion of pancreatic progenitor cell state that is closely related to centroacinar cells.
Limitations of the study
In this study we show that CFTR presence on the membrane regulates PDX1 expression in PDCs via GSK3β activity. This involves contact-based inactivation of PTEN due to lack of/reduced CFTR presence on the cell membrane. We believe mechanistic validation of CFTR-PTEN interaction regulating PDX1 in genotypes with varying abundance of CFTR protein on the membrane would provide better understanding of the PDX1-CFTR/PTEN axis. Additionally, cell type specificity of the PDX1-CFTR/PTEN axis in vivo would have provided more physiological context to the study. Furthermore, in regards to the in vitro ductal cell polarization system, we appreciate that it removes cellular environment-related changes that might contribute to PDX1 regulation in CF duct epithelium. We understand that the in vitro system described in this study can also mask differentiation phenotypic data found in vivo.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, John F. Engelhardt (john-engelhardt@uiowa.edu).
Materials availability
This study did not generate new unique materials.
Data and code availability
-
•
All data generated for this paper will be shared by the lead contact upon request. The data are publicly available at GEO database. The accession numbers are listed in the key resources table.
-
•
All original code is deposited on GitHub. The URL to the code is listed in key resources table.
-
•
Any other information required will be provided by the lead author.
Acknowledgments
This work was supported by NIH grants (P30 DK054759, RC2 DK124207, and NHLBI Federal Contract 75N92024C00008 to J.F.E.); Cystic Fibrosis Foundation grant (ENGELH21XX0 to JFE) the Carver Chair in Molecular Medicine (to J.F.E.). Biorender was used for figure and graphical abstract schematics.
Author contributions
Conceptualization, P.G.R. and J.F.E.; methodology, P.G.R., X.S., B.L., M.W., and Y.Z.; software, K.L.W. and N.C.; validation, P.G.R., G.G., F.Y., I.A.-E., and Y.Y.; formal analysis, P.G.R., Y.Y., G.G., F.Y., I.A.-E., P.W., G.L., S.C., R.R., A.E.S., and K.L.W.; investigation, P.G.R., G.G., F.Y., I.A.-E., and Y.Y.; formal analysis, P.G.R., Y.Y., G.G., F.Y., I.A.-E., P.W., G.L., S.C., R.R., A.E.S., and K.L.W.; resources, J.F.E., A.W.N., and L.S.; data curation, K.L.W.; writing – original draft, P.G.R. and J.F.E.; writing – review and editing, P.G.R., J.F.E., A.W.N., L.S., and K.L.W.; supervision, J.F.E., A.W.N., L.S., and A.U.; project administration, P.G.R., J.F.E., and A.W.N.; funding acquisition. J.F.E.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti PDX1 | Abcam | RRID: AB_777179 |
| Mouse anti SOX9 | Abcam | RRID: AB_2194156 |
| Guinea Pig anti INS | Invitrogen | RRID: AB_794668 |
| Rabbit anti CTNNB1 | Abcam | RRID: AB_305407 |
| Rabbit anti pSMAD5 | Abcam | RRID: AB_10561456 |
| Rabbit anti Axin2 | Abcam | RRID: AB_2290204 |
| Bacterial and Virus Strains | ||
| pLenti6/V5-GW/LacZ | Thermo Fisher | Cat#K495510 |
| One Shot Stbl3 Chemically Competent E.coli | Thermo Fisher | Cat#C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Hoechst 33258 | Molecular Probes | Cat#H3569 |
| Accutase | Stem Cell Technologies | Cat#07920 |
| Pneumacult Ex+ | Stem Cell Technologies | Cat#05040 |
| Pneumacult ALI | Stem Cell Technologies | Cat#05050 |
| PTEN inhibitor VO-OHpic trihydrate | Sigma | Cat#8639 |
| CHIR 99021 (GSK3b inhibitor/Wnt agonist) | Tocris | Cat#4423 |
| Aqua-Mount | Thermo Scientific | Cat#13800 |
| 3-Isobutyl-1-methylxanthine (IBMX) | Sigma | Cat# 28822-58-4 |
| Forskolin | Sigma | Cat# 66575-29-9 |
| Critical Commercial Assays | ||
| KAPA2G Robust PCR kit with dNTP 250 U | Roche Applied Science | Cat#07960743001 |
| High-Capacity cDNA reverse transcription kit | Thermo Fisher | Cat#4368814 |
| RNeasy plus mini kit | QIAGEN | Cat#74134 |
| TaqMan Universal Master Mix | IDT | 10007067 |
| View RNA ISH kit | ThermoFisher Scientific | Cat#QVT0400 |
| Illumina Tagment DNA Enzyme and Buffer Small Kit | Illumina | 20034197 |
| NEBNext High-Fidelity 2x PCR Master Mix | New England Biolabs | Cat#M0541S |
| Nextera DNA CD Indexes | Illumina | H503 |
| AmpureXP Bead based Reagen | Beckman Coulter | A63881 |
| MACS Dead Cell Removal Kit | Miltenyi Biotech | Cat#130-090-101 |
| Lenti-X Concentrator | Takara | Cat#631232 |
| Experimental Models: Organisms/Strains | ||
| Ferret: CFTRWT/WT | Sun et al., 2010 | N/A |
| Ferret: CFTRG551D−KI/G551D−KI | Yan et al., 2022 | N/A |
| Ferret: CFTRG551D/G551D | Sun et al., 2019 | N/A |
| Ferret: CFTRG551D/KO | Sun et al., 2019 | N/A |
| Ferret: CFTRKO/KO | Sun et al., 2010 | N/A |
| Oligonucleotides | ||
| PDX1 primer 1: CCTCCCTTTGTCTTCCTTTTCC | This paper | N/A |
| PDX1 primer 2: ACCCTCGCAAGATGTTCTC | This paper | N/A |
| ACTB1 primer 1: TGAAGGTCTCGAACATGATCTG | This paper | N/A |
| ACTB1 primer 2: ACCACACCTTCTACAATGAGC | This paper | N/A |
| SOX9 primer 1: ACCTACACGGGCAGCTA | This paper | N/A |
| SOX9 primer 2: TGTAGTGGCTGGGACTCA | This paper | N/A |
| NKX6.1 primer 1: CAAACGAAATACTTGGCGGG | This paper | N/A |
| NKX6.1 primer 2: CGTGCTTCTTCCTCCACTTG | This paper | N/A |
| PAX6 primer 1: GAGTTATGACACCTACACCCC | This paper | N/A |
| PAX6 primer 2: ACATATCAGGTTCACTTCCAGG | This paper | N/A |
| CDH1 primer 1: GAGTGTGCCCCATTACCTAC | This paper | N/A |
| CDH1 primer 2: TCCCTTCATAGTCAAACACCAG | This paper | N/A |
| HNF6 primer 1: GAGGATGTGGAAGTGGCTG | This paper | N/A |
| HNF6 primer 2: ACATCTGTGAAGACCAACCTG | This paper | N/A |
| Software and Algorithms | ||
| Analysis Code | This paper | https://github.com/kwells4/sussel_ferret_sc_220429 |
| PyMiner | Tyler et al., 2019 | N/A |
| Deposited Data | ||
| Bulk RNA-seq of WT and CF PDE | This paper | GSE264019 |
| Bulk ATACseq of WT and CF PDE | This paper | GSE264021 |
| Single cell RNAseq of D2, D5, D7, D9 WT and CF PDE | This paper | GSE264022 |
Experimental models and study participant details
Animal models
All procedures involving animals were performed in compliance with a protocol approved by the Institutional Animal Care and Use Committee of the University of Iowa and under institutional assurances including: AAALAC accreditation (#000833, since November 1994), USDA research facility registration (USDA No. 42-R-0004), and PHS Animal Welfare Assurance approval (D16–00009, A3021-01). Three ferret models of cystic fibrosis were used in these studies and express differing amounts of CFTR. The CFTR knockout ferret model (CFTRKO) was generated by somatic cell nuclear transfer using gene targeted fibroblasts.111 Homozygous CFTRKO/KO ferrets (abbreviated as KO/KO) expresses no CFTR mRNA or protein. CFTRKO/KO kits were reared as previously described.129 The CFTRG551D ferret model was also generated by somatic cell nuclear transfer using gene targeted fibroblasts.109 This model is a hypomorph due to a selection cassette in the reverse orientation within the intron adjacent to the targeted exon. As a result, CFTRG551D/G551D homozygous ferrets (abbreviated as G/G) expresses 50% CFTR mRNA and protein as compared to wildtype (WT) ferrets.109 CFTRG551D/G551D kits were reared as previously described on the CFTR modulator (VX-770) from embryonic day 28.109 The CFTRG551D−KI ferret model was generated by CRISPR/Cas9 targeting in zygotes.110 Unlike the CFTRG551D/G551D ferret model, CFTRG551D−KI/G551D−KI homozygous ferrets (abbreviated as GK/GK) have a ‘clean’ knock-in (KI) of the G551D mutation and express WT levels of CFTR mRNA. CFTRG551D−KI/G551D−KI kits were reared as previously described on the CFTR modulator (VX-770) from embryonic day 28.109 CF ferrets harboring the G551D mutation are typically weaned off VX-770 at 1–2 months after birth, allowing for pancreatic remodeling to initiate in the absence of CFTR function.2 The equivalent pathophysiologic state for a 2-months-old CFTR knockout ferret would be a 3-months-old CFTRG551D−KI/G551D−KI ferret harboring removed from VX-770 at 1 month of age.2 We used animals with a CF age around 1.5-month, which is the peak of inflammation and fibrosis. WT ferrets of sable coat color were obtained from Marshall Farms (North Rose, NY). Various allelic CFTR mutation combinations were generated through cross-breeding of the CF ferret lines. Offspring from this cross-breeding gave rise to ferrets with various levels of CFTR expression (Table 1).
Table 1.
CF ferret line names and abbreviation associated with various levels of CFTR expression
| Formal line name | Abbreviated name | Level of WT CFTR expression |
|---|---|---|
| CFTRWT/WT | WT | 100% |
| CFTRG551D−KI/G551D−KI | GKGK | 100% |
| CFTRG551D/G551D | GG | 50% |
| CFTRG551D/KO | GKO | 25% |
| CFTRKO/KO | CFTR-KO, CFKO, or KO | 0% |
Pancreatic ductal cell harvest, propagation, and differentiation at an ALI
Pancreata were removed from 1 to 3 day old newborn WT and CF ferrets and digested in 5 mg/mL collagenase for 20 min at 37°C. The digested pancreas was incubated overnight in PneumaCult-Ex Plus medium (STEMCELL Technologies, MA, USA) on 804G-coated culture dishes overnight at 37°C in a 5% CO2 incubator. The 804G coating procedure is as previously described for airway basal cells.130 Ductal structures that adhered to the plate were aspirated on the following day and cultured on fresh 804G-coated culture dishes in PneumaCult-Ex Plus medium until near confluence. These cells were then passaged by incubating with Accutase (STEMCELL Technologies, MA, USA) for 5 min at 37°C. The cells were passaged continuously for 10 passages to eliminate contaminating cells and obtain a morphologically homogeneous population of duct cells. Passage-10 cells were harvested using Accutase and transferred to 804G-coated transwell inserts (Corning, NY, USA) at a density of 100,000 cells per well. Following seeding, transwells were cultured in PneumaCult-Ex Plus medium on both apical and basolateral chambers for 3 days. The medium in both the apical and basolateral chamber was then switched to PneumaCult-ALI (STEMCELL Technologies, MA, USA) for 1 day. Air liquid interface (ALI) was established the following day by aspirating the medium from the apical chamber. Cultures were maintained at an ALI for 2 weeks before use in experiments at which time transepithelial resistance should be greater than 1000 Ω. Cells in culture were tested for mycoplasma contamination regularly (once every two months).
Method details
Immunofluorescence staining of differentiated epithelial cultures
Differentiated cultures of WT and CF pancreatic ductal epithelium (PDE) were fixed in 4% paraformaldehyde (PFA) for 20 min at room temperature (RT). After 3 washes in 1X PBS for 5 min, the cells were incubated in blocking solution containing 10% donkey serum in PBS for 1 h at RT. Primary antibodies were diluted and applied overnight at 4°C at the following dilutions (CTNNB1, 1:100; PSMAD5, 1:100; INS, 1:500). Slides were washed in 1X PBS thrice for 15 min each and incubated with secondary antibodies (anti-rabbit Alexa 594, A21207, Invitrogen, USA; anti-mouse Alexa 594, 715-585-151, Jackson ImmunoResearch, USA) at a 1:1000 dilution for 1 h at RT. Slides were mounted using Aquamount (Thermo Scientific,MA,USA) containing Hoescht diluted at 1:2000 and imaged on a Zeiss 880.
Immunofluorescence staining of pancreatic sections
Paraffin sections from newborn or 1- to 2-month old WT and CF ferrets were deparaffinized and dehydrated by sequential 5 min incubation in xylene (3 times) and 100% ethanol (2 times). The sections were rehydrated by sequential 5 min incubations in 90%, 70%, 50%, 30% ethanol followed by water (2 times). Sections in citrate-based antigen retrieval solution were incubated in a water bath at 95°C for 1 h. The slides were placed at RT for 40 min and then incubated in blocking solution (1X PBS with 10% donkey serum) for 1 h at RT. Primary antibodies were applied overnight at 4°C at the following dilutions (PDX1, 1:100; SOX9, 1:100; AXIN2 1:100; INS, 1; 100). The slides were washed for 15 min in 1X PBS thrice. The secondary antibodies were diluted to 1:2000 and applied for 1 h at RT. The slides were washed for 5 min in 1X PBS thrice and mounted using Aquamont containing Hoescht diluted at 1:2000 and imaged on a Zeiss 880.
RNA fluorescent in situ hybridization (FISH) on pancreatic sections
FISH was used to visualize mRNA expression using ViewRNA ISH kits (Thermo Fisher, MA, USA) on paraffin embedded sections of WT and CFTR-KO ferret pancreata. Prior to deparaffinization the pancreatic sections were baked at 60°C for 1 h. During deparaffinization sections were submerged in xylene three times for 5 min, dehydrated in 100% ethanol twice for 5 min, followed by air-drying. Tissue was permeabilized by incubating the sections in permeabilizing solution (Thermo Fisher, MA, USA) for 10 min at 90-95°C. The sections were then incubated in protease solution (Thermo Fisher, MA, USA) at a 1:100 dilution for 10 min at 40°C. Oligonucleotide probes (Thermo Fisher, MA, USA) for ferret transcripts (INS, RNASE1, AMY2B, HNF6, WNT5A, TGF-β) were diluted to 1:40 (except INS at 1:200) with target probe diluent buffer and incubated with the sections for 5 h at 40°C, followed by three rigorous washes using the wash buffer provided in the kit for 15 min each. Sections were incubated in pre-amplifying buffer at 40°C for 25 min, followed by three rigorous washes for 15 min each. Sections were then incubated with amplifying solutions at 40°C for 15 min, followed by three washes lasting 15 min each. Sections were then incubated in labeling probe buffer mixture diluted to 1:1000 at 40°C for 30 min, washed, and developed using Blue or Red substrate bound to the alkaline phosphatase enzyme. Sections were mounted using Hoescht containing (1:2000) Aquamount (Thermo Scientific, MA,USA) and stored at 4°C until they were imaged on a Zeiss 880 Confocal.
Short-circuit current (isc) measurements on PDE cultures
CFTR dependent chloride current measurements on WT and CF PDE cultures were performed using Ussing chambers as previously described.109 Changes in Isc were assessed after the sequential addition of the following channel agonist and antagonists to the apical chamber: 1) Amiloride (100 μM) to inhibit ENaC, 2) 4,4′-Diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS,100 μM) to inhibit non-CFTR Cl− channels, 4) 8-Methoxymethyl-3-isobutyl-1-methylxanthine (IBMX, 100 μM) and Forskolin (10 μM) to stimulate CFTR, and 4) GlyH101 (3 μM) to inhibit CFTR. Amiloride and DIDS inhibit the sodium channels and non-CFTR chloride channels, respectively. IBMX and Forskolin both stimulate increases in intracellular cAMP and activates the CFTR channel. Isc were recorded using Acquire and Analyze software (Physiologic Instruments, CA, USA).
Quantitative qRT-PCR on proliferating pancreatic ductal cells (PDC) and differentiated PDE cultures
RNA was harvested from PDCs and PDEs (2–4 transwells combined per donor) using Qiagen RNEasy Plus Kits (Qiagen, MD, USA). Equal amounts of RNA were used to generate cDNA using the High-capacity cDNA Reverse Transcription kit (Thermo Fisher, MA, USA). Taqman probes were designed against the ferret transcripts for genes of interest (PDX1, SOX9, PAX6, NKX6.1, HNF6, KRT7, CDH1, ACTB) using the IDT tools website (IDT, IA, USA). The Taqman Real-time PCR Master Mix (IDT, IA, USA) was mixed with 2 μL of cDNA. RT-qPCR was performed on the BioRAD thermocycler (BioRAD, CA, USA). The PCR cycling conditions included a denaturation step at 95°C for 3 min followed by 41 cycles of 95°C for 15 s and 60°C for 45 s. Blanks and negative control samples excluding the reverse transcriptase were used in every experiment. ACTB was used as the internal control housekeeping gene for normalization.
PTEN and GSK3β inhibitory studies on actively polarizing WT PDE cultures
Actively polarizing WT PDEs were used to test whether inhibition of PTEN or GSK3β pathways led to a CFTR-KO phenotype. Approximately 100,000 PDCs were plated on 804G coated trans-wells (Corning, NY, USA). Cells were incubated for three days in PneumaCult-Ex Plus in both the apical and basolateral chambers. The media was then replaced with PneumaCult-ALI in both apical and basolateral chambers for overnight incubation. The next day the apical media was then aspirated and PTENi or GSK3βi was added to the basolateral media at 10 μM concentration; Donor-matched controls received vehicle. The cells were sustained for 2 weeks in the presence of the inhibitors. Subsequently, 2–4 trans-wells were combined to harvest RNA using the RNEasy Plus Kit (Qiagen, MD, USA). Three samples (combining 2–4 trans-wells per sample) per donor per condition were used for subsequent assays.
CFTR complementation studies in CFTR-KO PDEs
Lentivirus PGK-hCFTR-dTomato was previously described as pLV-dt/CFTR-Ø.131 It contains a phosphoglycerate kinase 1 (PGK) promoter driving CFTR and CMV beta-actin promoter driven dTomato cassettes. PGK-dTomato (control) virus was generated from pLV-dt/CFTR-Ø by deleting the CFTR gene. Proviral plasmids were amplified using Stbl3 competent E.Coli (Thermofisher, MA, USA). Lentivirus was generated as previously described.131 PDCs at passage 8–10 from CFTR-KO donors were transduced with Lenti-PGK-hCFTR-dTomato or Lenti-PGK-dTomato at an MOI of 100 particles/cell. Cells were passaged for 7 days and then dTomato positive cells were isolated by FACS and expanded for experiments. PDCs were then plated onto tranwells and polarized for 2 weeks at an ALI in PneumaCult-ALI media. RNA was then generated from 2 to 4 transwells combined.
Bulk RNA-seq sample preparation from differentiated PDE cultures
WT and CF PDE cultures differentiated from 3 donors of each genotype were polarized for 2 weeks at an ALI. Triplicate samples per donor were prepared by combining two transwells per replicate to extract RNA from the cells using RNeasy Plus Mini Kit (Qiagen, MA, USA). Genomic DNA was eliminated using gDNA columns (Qiagen, MA, USA). Extracted RNA was analyzed for integrity by assessing their RNA integrity number. Samples with RIN >9 were used to prepare indexed libraries using Trueseq mRNA stranded preparation. Barcoded samples were pooled in equimolar rations and sequenced for 75 bp paired end reads using a HiSeq4000 (Illumina) in the University of Iowa Genomics Division.
ATAC-seq sample preparation from differentiated PDE cultures
WT and CF PDE cultures differentiated from 3 donors of each genotype were polarized for 2 weeks at an ALI. Samples were processed for ATAC-seq according to previously published protocols.132,133 Cells were dissociated using Accutase and ∼50,000 cells from each PDE culture were lysed in ice-cold lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40: Sigma). Transposition was performed using 25 μL tagmentation reaction mix from Tagment DNA kit (Illumina, CA, USA). Tagged DNA was amplified indexed, using the NEBNext High-Fidelity 2x PCR Master Mix (New England Biolabs, MA, USA) and with Nextera DNA CD Indexes (Illumina, CA, USA), using the following settings: 72°C for 5 min; 98°C for 30 s; 12 cycles of 98°C for 10 s, 63°C for 30 s, and 72°C for 1 min. The indexed library was purified with 1.8 times the volume of Ampure XP beads (Beckman Coulter, CA, USA). Library quality was assessed using a BioAnalyzer 2100 High Sensitivity DNA Chip (Agilent Technologies, MA, USA). All DNA libraries that exhibited the correct nucleosome pattern were pooled and processed for 150bp paired-end sequencing using a HiSeq4000 (Illumina) in the University of Iowa Genomics Division.
Single cell RNA-seq sample preparation
WT and CF ferret PDE cultures grown at ALI for 2, 5, 7, and 9 days were dissociated using Accutase followed by DNase treatment. Cells were filtered through a 20 μM strainer and pelleted in 0.04% BSA PBS at 500 g for 10 min. Nonviable dead cells were removed by using MACS Dead Cell Removal Kit following 10X Genomics recommendations (Document CG00039). Single cells were counted on a Thermo countess cell counter and 0.04% BSA/PBS was added to achieve a targeted concentration of 1000 cells/μl. Sequencing libraries were generated by following 10X Genomics recommendations (Document CG000315). Briefly, single cells and reverse transcription master mix were partitioned into Gel Beads in partitioning oil in the 10X Chromium controller. After reverse transcription, cDNA libraries were amplified and fragmentated, followed by adaptor ligation and sample index PCR reaction. Libraries were sequenced on NovaSeq 6000 platform by the University of Iowa Genomics Division.
Quantification and statistical analysis
Image analysis
Image quantification was performed using FIJI. Maximum intensity projection of Z-stacks with at least 15 sections were generated. Regions of interest were manually drawn around ductal structures for quantification of signal intensity and area. Fluorescent signals overlapping with DAPI stain was quantified for nuclear localization. At least 3 donors per condition were used for quantification and statistical analysis. Statistical analysis of the measurements was done using Graphpad PRISM.
Analysis of WT and CF PDE bulk RNA-seq
FASTQ reads obtained after sequencing were mapped to the ferret reference genome using RSEM. The normalized transcripts per million counts for each gene was generated. Differentially expressed genes (DEG) with BH corrected p-value <0.05 were obtained using PyMiner.17 The list of DEGs were used to perform subsequent pathway analysis and upstream regulator analysis on Ingenuity Pathway Analysis (Qiagen Bioinformatic).
ATAC-seq peak calling and differential analysis
ATAC-seq was performed using 150 bp paired-end sequencing reads. Raw FASTQ reads were trimmed using Trimmomatic on Galaxy tools and aligned to ferret genome assembly (MusPutFur1.0) using Bowtie 2 on Galaxy tools with default parameters. DeepTools version 3.3.0 was used to check the reproducibility of the biological replicates and generate bigWig coverage files for visualization. MACS2 was used to call peaks. DiffBind version 2.10 was used to identify differential peaks between WT and CF PDEs with log2 fold-change threshold of >1 and a false discovery rate (FDR) < 0.05, indicating differential accessibility. Peaks were assigned to genes using BED intersect, to identify differentially accessible genes. IGV was used to visualize differentially accessible regions of the genome.
Single cell RNA-seq analysis of WT and CF PDE
Pre-processing
Sequences from scRNA-seq were processed using 10x Genomics Cellranger v 5.0.1 software134 using the ferret ASM1176430v1.1 genome and gtf file. A recently described improved annotation of the ferret transcriptome was used which achieves a median read assignment in scRNA-seq studies of ∼71%.127 The genome and gtf file were made into a cellranger reference using the `mkref’ command.
Raw data generated by Cellranger were then read into R v4.1.2 using the Seurat135 v4.1.1 R package with at least 200 genes per cell and at least 3 cells. Cells were further filtered based on the number of genes per cell and the percent of mitochondrial reads per cell (maximum 20%). The data were normalized by using `NormalizeData’. For each sample, variable genes were found by using `FindVariableFeatures’ and data was scaled using `ScaleData’.
Doublets were removed using `DoubletFinder’136 using the default values except for pK, nExp, and PCs. The pK was identified using the pK associated with the maximum BCmetric value after running `find.pK’ from doublet finder. All samples used PCs 1:20. After doublet removal, 4499 (CFKO_D2), 2750 (CFKO_D5), 1607 (CFKO_D7), 2489 (CFKO_D9), 6224 (WT_D2), 5326 (WT_D5), 4634 (WT_D7), 5416 (WT_D9) cells were used for downstream analysis.
The 8 samples were merged using the `merge’ function from Seurat. The merged data was then normalized and scaled using `NormalizeData’, `FindVariableFeatures’, and `ScaleData’ as described above. Dimensionality reduction and clustering were performed using `RunPCA’, `FindNeighbors’, `FindClusters’, and `RunUMAP’. `RunPCA’ was run using the default values except all variable features were used for the features argument. `FindNeighbors’ was run with default parameters except for the dims argument (dims = 1:30). `FindClusters’ was run with default parameters except for the resolution argument (resolution = 0.6). `RunUMAP’ was run with default parameters except for the dims and metric arguments (dims = 1:30, metric = “correlation”). Because some clusters had good representation of all development days and we wanted to analyze the WT and KO separately, no batch correction was performed.
Cell type identification
We made a first pass at naming clusters on the merged WT and merged CFKO samples separately using a mix of published single cell studies of mouse and human pancreas62,63,64,65,66,137 as a reference and determining cluster identity using `clustifyr’.61 For each reference, we used the top 2000 variable genes and found either the mouse or human ortholog. These orthologs were used as the `query_genes’ and the clusters identified by `FindClusters’ as the `cluster_col’. The top correlated cell type from any reference to each cluster was used as the first determination of cell type.
Cluster markers and pathways
To identify markers of each cluster, `FindAllMarkers’ was run on each merged dataset using default settings except we set only.pos to TRUE. Genes were called differentially expressed if the adjusted p-value was less than 0.05 and the log fold change was greater than 0.5. To generate a heatmap of gene sets, the average expression of all genes within each cluster was determined using `AverageExpression’ from Seurat. Next, the average expression matrix was subset to only the genes in the gene set of interest. Finally, a Z score was computed, and the plot was generated with `pheatmap’ (https://cran.r-project.org/web/packages/pheatmap/index.html).
Trajectory analysis
To perform trajectory analysis, we used `slingshot’. We identified starting clusters based on the cluster containing the most D2 cells for each WT and CFKO. Slingshot was run on the first 30 PCs of each dataset. We then used `tradeseq’ using the pseudotime scores generated by slingshot to identify genes that correlated with pseudotime. We used the function `evaluateK’ to determine the number of knots (5 for the CFKO, 8 for WT) and the function `fitGAM’ to identify correlated genes. To visualize pseudotime on the UMAP, we used the `EmbedCurves’ function to map our pseudotime data onto the UMAP dimensionality reduction.
Figures
Most figures were created using the `scAnalysisR’ package available on github (https://github.com/CUAnschutzBDC/scAnalysisR). All scripts to replicate this analysis are available on github (https://github.com/kwells4/sussel_ferret_sc_220429).
Published: November 15, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111393.
Contributor Information
Kristen L. Wells, Email: kristen.wells-wrasman@cuanschutz.edu.
John F. Engelhardt, Email: john-engelhardt@uiowa.edu.
Supplemental information
(A) Bulk RNA sequencing analysis results of WT (n = 3) and CF (n = 3) PDEs. (B) Upstream Regulator analysis showing transcriptional regulators of DEGs in CF PDEs using IPA. (C) Upstream regulator analysis results for DEG in CF PDEs.
(A) Results from GO term analysis on DEG in CFTR-KO PDE using Panther. (B) Gene list associated with cell differentiation GO term. (C) Gene set associated with cell migration GO term. (D) Gene set for mesenchyme development GO term. (E) Gene set for response to TGF-β stimulus GO term. (F) Gene set for response to WNT signaling is listed. (G) Gene set for endocrine pancreas development GO term. (H) Gene set for histone modification GO term.
(A) The differentially open regions (DORs) associated with coding regions in the chromatin of CF PDEs relative to WT PDEs are shown in the table. (B) Analysis of motif enrichment in the CF DORs.
(A) DEG in acinar cells derived from WT and CF PDE. (B) DEG in ductal cells found in WT and CF PDE. (C) DEG in proliferating acinar cells derived from WT and CF PDE. (D) DEG in proliferating ductal cells derived from WT and CF PDE. (E) DEG in centroacinar progenitor cells derived from WT and CF PDE. (F) DEG in centroacinar cells derived from WT and CF PDE.
(A) DEGs in CF PDEs relative to WT PDEs at D2 of differentiation. (B) DEGs in CF PDEs relative to WT PDEs at D5 of differentiation. (C) The DEGs in CF PDEs relative to WT PDEs at D7 of differentiation. (D) The DEGs in CF PDEs relative to WT PDEs at D9 of differentiation.
(A) The variability in expression of D2 DEGs. (B) The variability in expression of D5 DEGs. (C) The variability in expression of D7 DEGs. (D) The variability in expression of D9 DEGs. (E) The variability in expression of D9 CAC progenitor DEGs. (F) The variability in expression of D9 CAC DEGs.
(A) Upstream regulator analysis for DEGs at D2 of differentiation using IPA. (B) Upstream regulator analysis for DEGs at D5 of differentiation using IPA. (C) Upstream regulator analysis for DEGs at D7 of differentiation using IPA. (D) Upstream regulator analysis for DEGs at D9 of differentiation using IPA. (E) Pathway analysis for CF PDE DEGs at D2 of differentiation. (F) Pathway analysis for CF PDE DEG at D5 of differentiation. (G) Pathway analysis for CF PDE DEGs at D7 of differentiation. (H) Pathway analysis for CF PDE DEGs at D9 of differentiation.
(A) The DEGs in WT ductal 1 subpopulation. (B) The DEGs in WT ductal 2 subpopulation. (C) The DEGs in WT ductal 3 subpopulation. (D) The DEGs in CF ductal 1 subpopulation. (E) The DEGs in CF ductal 2 subpopulation are shown. (F) The DEGs in CF ductal 3 subpopulation are shown. (G) The Z scores of DEGs in ductal subpopulations identified in WT and CF PDEs. (H) The DEGs in ductal cells of WT and CF PDEs, and their Z scores for expression across all time points of differentiation.
(A) The predicted upstream regulatory signaling pathways active in CF CACs and relative to WT CACs. (B) The predicted upstream regulatory signaling pathways active in CF progenitor-like cells relative to WT progenitor-like cells. (C) The Z score enrichment values for AKT signaling genes in WT and CF PDE cells at D9 differentiation. (D) The Z score enrichment values for PTEN signaling genes in WT and CF PDE cells at D9 differentiation. (E) The Z score enrichment values for WNT signaling genes in WT and CF PDE cells at D9 differentiation.
References
- 1.Grasemann H., Ratjen F. Cystic Fibrosis. N. Engl. J. Med. 2023;389:1693–1707. doi: 10.1056/NEJMra2216474. [DOI] [PubMed] [Google Scholar]
- 2.Gibson-Corley K.N., Engelhardt J.F. Animal Models and Their Role in Understanding the Pathophysiology of Cystic Fibrosis-Associated Gastrointestinal Lesions. Annu. Rev. Pathol. 2021;16:51–67. doi: 10.1146/annurev-pathol-022420-105133. [DOI] [PubMed] [Google Scholar]
- 3.Rickels M.R., Norris A.W., Hull R.L. A tale of two pancreases: exocrine pathology and endocrine dysfunction. Diabetologia. 2020;63:2030–2039. doi: 10.1007/s00125-020-05210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hart N.J., Aramandla R., Poffenberger G., Fayolle C., Thames A.H., Bautista A., Spigelman A.F., Babon J.A.B., DeNicola M.E., Dadi P.K., et al. Cystic fibrosis-related diabetes is caused by islet loss and inflammation. JCI Insight. 2018;3 doi: 10.1172/jci.insight.98240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan F.C., Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev. Dynam. 2011;240:530–565. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- 6.Rotti P.G., Xie W., Poudel A., Yi Y., Sun X., Tyler S.R., Uc A., Norris A.W., Hara M., Engelhardt J.F., Gibson-Corley K.N. Pancreatic and Islet Remodeling in Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Knockout Ferrets. Am. J. Pathol. 2018;188:876–890. doi: 10.1016/j.ajpath.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Putman M.S., Norris A.W., Hull R.L., Rickels M.R., Sussel L., Blackman S.M., Chan C.L., Ode K.L., Daley T., Stecenko A.A., et al. Cystic Fibrosis-Related Diabetes Workshop: Research Priorities Spanning Disease Pathophysiology, Diagnosis, and Outcomes. Diabetes. 2023;72:677–689. doi: 10.2337/db22-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edlund A., Barghouth M., Huhn M., Abels M., Esguerra J., Mollet I., Svedin E., Wendt A., Renstrom E., Zhang E., et al. Defective exocytosis and processing of insulin in a cystic fibrosis mouse model. J. Endocrinol. 2019;241:45–57. doi: 10.1530/JOE-18-0570. [DOI] [PubMed] [Google Scholar]
- 9.Fontes G., Ghislain J., Benterki I., Zarrouki B., Trudel D., Berthiaume Y., Poitout V. The DeltaF508 Mutation in the Cystic Fibrosis Transmembrane Conductance Regulator Is Associated With Progressive Insulin Resistance and Decreased Functional beta-Cell Mass in Mice. Diabetes. 2015;64:4112–4122. doi: 10.2337/db14-0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khan D., Kelsey R., Maheshwari R.R., Stone V.M., Hasib A., Manderson Koivula F.N., Watson A., Harkin S., Irwin N., Shaw J.A., et al. Short-term CFTR inhibition reduces islet area in C57BL/6 mice. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-47745-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olivier A.K., Yi Y., Sun X., Sui H., Liang B., Hu S., Xie W., Fisher J.T., Keiser N.W., Lei D., et al. Abnormal endocrine pancreas function at birth in cystic fibrosis ferrets. J. Clin. Invest. 2012;122:3755–3768. doi: 10.1172/JCI60610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yi Y., Sun X., Gibson-Corley K., Xie W., Liang B., He N., Tyler S.R., Uc A., Philipson L.H., Wang K., et al. A Transient Metabolic Recovery from Early Life Glucose Intolerance in Cystic Fibrosis Ferrets Occurs During Pancreatic Remodeling. Endocrinology. 2016;157:1852–1865. doi: 10.1210/en.2015-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yi Y., Norris A.W., Wang K., Sun X., Uc A., Moran A., Engelhardt J.F., Ode K.L. Abnormal Glucose Tolerance in Infants and Young Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2016;194:974–980. doi: 10.1164/rccm.201512-2518OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanquer M., Clainche L.L., Bismuth E., Storey C., Gerardin M., Houdouin V. Very early glucose tolerance abnormalities in children with cystic fibrosis. J. Cyst. Fibros. 2021;20:792–795. doi: 10.1016/j.jcf.2021.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Sun X., Yi Y., Xie W., Liang B., Winter M.C., He N., Liu X., Luo M., Yang Y., Ode K.L., et al. CFTR Influences Beta Cell Function and Insulin Secretion Through Non-Cell Autonomous Exocrine-Derived Factors. Endocrinology. 2017;158:3325–3338. doi: 10.1210/en.2017-00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rotti P.G., Evans I.A., Zhang Y., Liang B., Cunicelli N., O'Malley Y., Norris A.W., Uc A., Engelhardt J.F. Lack of CFTR alters the ferret pancreatic ductal epithelial secretome and cellular proteome: Implications for exocrine/endocrine signaling. J. Cyst. Fibros. 2022;21:172–180. doi: 10.1016/j.jcf.2021.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tyler S.R., Rotti P.G., Sun X., Yi Y., Xie W., Winter M.C., Flamme-Wiese M.J., Tucker B.A., Mullins R.F., Norris A.W., Engelhardt J.F. PyMINEr Finds Gene and Autocrine-Paracrine Networks from Human Islet scRNA-Seq. Cell Rep. 2019;26:1951–1964.e8. doi: 10.1016/j.celrep.2019.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ebrahim N., Shakirova K., Dashinimaev E. PDX1 is the cornerstone of pancreatic beta-cell functions and identity. Front. Mol. Biosci. 2022;9 doi: 10.3389/fmolb.2022.1091757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reichert M., Rustgi A.K. Pancreatic ductal cells in development, regeneration, and neoplasia. J. Clin. Invest. 2011;121:4572–4578. doi: 10.1172/JCI57131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashizawa S., Brunicardi F.C., Wang X.P. PDX-1 and the pancreas. Pancreas. 2004;28:109–120. doi: 10.1097/00006676-200403000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Seymour P.A. Sox9: a master regulator of the pancreatic program. Rev. Diabet. Stud. 2014;11:51–83. doi: 10.1900/RDS.2014.11.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyatsuka T., Kaneto H., Shiraiwa T., Matsuoka T.A., Yamamoto K., Kato K., Nakamura Y., Akira S., Takeda K., Kajimoto Y., et al. Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes Dev. 2006;20:1435–1440. doi: 10.1101/gad.1412806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy N., Takeuchi K.K., Ruggeri J.M., Bailey P., Chang D., Li J., Leonhardt L., Puri S., Hoffman M.T., Gao S., et al. PDX1 dynamically regulates pancreatic ductal adenocarcinoma initiation and maintenance. Genes Dev. 2016;30:2669–2683. doi: 10.1101/gad.291021.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langlois M.J., Bergeron S., Bernatchez G., Boudreau F., Saucier C., Perreault N., Carrier J.C., Rivard N. The PTEN phosphatase controls intestinal epithelial cell polarity and barrier function: role in colorectal cancer progression. PLoS One. 2010;5 doi: 10.1371/journal.pone.0015742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin-Belmonte F., Gassama A., Datta A., Yu W., Rescher U., Gerke V., Mostov K. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell. 2007;128:383–397. doi: 10.1016/j.cell.2006.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stanger B.Z., Stiles B., Lauwers G.Y., Bardeesy N., Mendoza M., Wang Y., Greenwood A., Cheng K.H., McLaughlin M., Brown D., et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8:185–195. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 27.Edelman H.E., McClymont S.A., Tucker T.R., Pineda S., Beer R.L., McCallion A.S., Parsons M.J. SOX9 modulates cancer biomarker and cilia genes in pancreatic cancer. Hum. Mol. Genet. 2021;30:485–499. doi: 10.1093/hmg/ddab064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kopp J.L., von Figura G., Mayes E., Liu F.F., Dubois C.L., Morris J.P., 4th, Pan F.C., Akiyama H., Wright C.V.E., Jensen K., et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:737–750. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J., Akanuma N., Liu C., Naji A., Halff G.A., Washburn W.K., Sun L., Wang P. TGF-beta1 promotes acinar to ductal metaplasia of human pancreatic acinar cells. Sci. Rep. 2016;6 doi: 10.1038/srep30904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hosein A.N., Huang H., Wang Z., Parmar K., Du W., Huang J., Maitra A., Olson E., Verma U., Brekken R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight. 2019;5 doi: 10.1172/jci.insight.129212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kropp P.A., Zhu X., Gannon M. Regulation of the Pancreatic Exocrine Differentiation Program and Morphogenesis by Onecut 1/Hnf6. Cell. Mol. Gastroenterol. Hepatol. 2019;7:841–856. doi: 10.1016/j.jcmgh.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez D.M., Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014;7 doi: 10.1126/scisignal.2005189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gustafson B., Hammarstedt A., Hedjazifar S., Hoffmann J.M., Svensson P.A., Grimsby J., Rondinone C., Smith U. BMP4 and BMP Antagonists Regulate Human White and Beige Adipogenesis. Diabetes. 2015;64:1670–1681. doi: 10.2337/db14-1127. [DOI] [PubMed] [Google Scholar]
- 34.Tan Z., Song L., Wu W., Zhou Y., Zhu J., Wu G., Cao L., Song J., Li J., Zhang W. TRIM14 promotes chemoresistance in gliomas by activating Wnt/beta-catenin signaling via stabilizing Dvl2. Oncogene. 2018;37:5403–5415. doi: 10.1038/s41388-018-0344-7. [DOI] [PubMed] [Google Scholar]
- 35.Dong H.J., Jang G.B., Lee H.Y., Park S.R., Kim J.Y., Nam J.S., Hong I.S. The Wnt/beta-catenin signaling/Id2 cascade mediates the effects of hypoxia on the hierarchy of colorectal-cancer stem cells. Sci. Rep. 2016;6 doi: 10.1038/srep22966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swarup S., Pradhan-Sundd T., Verheyen E.M. Genome-wide identification of phospho-regulators of Wnt signaling in Drosophila. Development. 2015;142:1502–1515. doi: 10.1242/dev.116715. [DOI] [PubMed] [Google Scholar]
- 37.Jung H.C., Kim K. Identification of MYCBP as a beta-catenin/LEF-1 target using DNA microarray analysis. Life Sci. 2005;77:1249–1262. doi: 10.1016/j.lfs.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 38.Szymanska K., Boldt K., Logan C.V., Adams M., Robinson P.A., Ueffing M., Zeqiraj E., Wheway G., Johnson C.A. Regulation of canonical Wnt signalling by the ciliopathy protein MKS1 and the E2 ubiquitin-conjugating enzyme UBE2E1. Elife. 2022;11 doi: 10.7554/eLife.57593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mandal C.C., Das F., Ganapathy S., Harris S.E., Choudhury G.G., Ghosh-Choudhury N. Bone Morphogenetic Protein-2 (BMP-2) Activates NFATc1 Transcription Factor via an Autoregulatory Loop Involving Smad/Akt/Ca2+ Signaling. J. Biol. Chem. 2016;291:1148–1161. doi: 10.1074/jbc.M115.668939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subbalakshmi A.R., Kundnani D., Biswas K., Ghosh A., Hanash S.M., Tripathi S.C., Jolly M.K. NFATc Acts as a Non-Canonical Phenotypic Stability Factor for a Hybrid Epithelial/Mesenchymal Phenotype. Front. Oncol. 2020;10 doi: 10.3389/fonc.2020.553342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orlando K.A., Douglas A.K., Abudu A., Wang Y., Tessier-Cloutier B., Su W., Peters A., Sherman L.S., Moore R., Nguyen V., et al. Re-expression of SMARCA4/BRG1 in small cell carcinoma of ovary, hypercalcemic type (SCCOHT) promotes an epithelial-like gene signature through an AP-1-dependent mechanism. Elife. 2020;9 doi: 10.7554/eLife.59073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wen H., Gris D., Lei Y., Jha S., Zhang L., Huang M.T.H., Brickey W.J., Ting J.P.Y. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011;12:408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen J., Qiao K., Zhang C., Zhou X., Du Q., Deng Y., Cao L. VRK2 activates TNFalpha/NF-kappaB signaling by phosphorylating IKKbeta in pancreatic cancer. Int. J. Biol. Sci. 2022;18:1288–1302. doi: 10.7150/ijbs.66313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morey L., Santanach A., Di Croce L. Pluripotency and Epigenetic Factors in Mouse Embryonic Stem Cell Fate Regulation. Mol. Cell Biol. 2015;35:2716–2728. doi: 10.1128/MCB.00266-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taube J.H., Sphyris N., Johnson K.S., Reisenauer K.N., Nesbit T.A., Joseph R., Vijay G.V., Sarkar T.R., Bhangre N.A., Song J.J., et al. The H3K27me3-demethylase KDM6A is suppressed in breast cancer stem-like cells, and enables the resolution of bivalency during the mesenchymal-epithelial transition. Oncotarget. 2017;8:65548–65565. doi: 10.18632/oncotarget.19214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Torres C.M., Biran A., Burney M.J., Patel H., Henser-Brownhill T., Cohen A.H.S., Li Y., Ben-Hamo R., Nye E., Spencer-Dene B., et al. The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science. 2016;353 doi: 10.1126/science.aaf1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qian L., Mahaffey J.P., Alcorn H.L., Anderson K.V. Tissue-specific roles of Axin2 in the inhibition and activation of Wnt signaling in the mouse embryo. Proc. Natl. Acad. Sci. USA. 2011;108:8692–8697. doi: 10.1073/pnas.1100328108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee J.H., Lee J.H., Rane S.G. TGF-beta Signaling in Pancreatic Islet beta Cell Development and Function. Endocrinology. 2021;162 doi: 10.1210/endocr/bqaa233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tulachan S.S., Tei E., Hembree M., Crisera C., Prasadan K., Koizumi M., Shah S., Guo P., Bottinger E., Gittes G.K. TGF-beta isoform signaling regulates secondary transition and mesenchymal-induced endocrine development in the embryonic mouse pancreas. Dev. Biol. 2007;305:508–521. doi: 10.1016/j.ydbio.2007.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shih H.P., Seymour P.A., Patel N.A., Xie R., Wang A., Liu P.P., Yeo G.W., Magnuson M.A., Sander M. A Gene Regulatory Network Cooperatively Controlled by Pdx1 and Sox9 Governs Lineage Allocation of Foregut Progenitor Cells. Cell Rep. 2015;13:326–336. doi: 10.1016/j.celrep.2015.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Golson M.L., Kaestner K.H. Epigenetics in formation, function, and failure of the endocrine pancreas. Mol. Metabol. 2017;6:1066–1076. doi: 10.1016/j.molmet.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lenoir O., Flosseau K., Ma F.X., Blondeau B., Mai A., Bassel-Duby R., Ravassard P., Olson E.N., Haumaitre C., Scharfmann R. Specific control of pancreatic endocrine beta- and delta-cell mass by class IIa histone deacetylases HDAC4, HDAC5, and HDAC9. Diabetes. 2011;60:2861–2871. doi: 10.2337/db11-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haumaitre C., Lenoir O., Scharfmann R. Histone deacetylase inhibitors modify pancreatic cell fate determination and amplify endocrine progenitors. Mol. Cell Biol. 2008;28:6373–6383. doi: 10.1128/MCB.00413-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lynn F.C., Smith S.B., Wilson M.E., Yang K.Y., Nekrep N., German M.S. Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proc. Natl. Acad. Sci. USA. 2007;104:10500–10505. doi: 10.1073/pnas.0704054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hale M.A., Swift G.H., Hoang C.Q., Deering T.G., Masui T., Lee Y.K., Xue J., MacDonald R.J. The nuclear hormone receptor family member NR5A2 controls aspects of multipotent progenitor cell formation and acinar differentiation during pancreatic organogenesis. Development. 2014;141:3123–3133. doi: 10.1242/dev.109405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.von Figura G., Morris J.P., 4th, Wright C.V.E., Hebrok M. Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. Gut. 2014;63:656–664. doi: 10.1136/gutjnl-2012-304287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mameishvili E., Serafimidis I., Iwaszkiewicz S., Lesche M., Reinhardt S., Bölicke N., Büttner M., Stellas D., Papadimitropoulou A., Szabolcs M., et al. Aldh1b1 expression defines progenitor cells in the adult pancreas and is required for Kras-induced pancreatic cancer. Proc. Natl. Acad. Sci. USA. 2019;116:20679–20688. doi: 10.1073/pnas.1901075116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ejarque M., Cervantes S., Pujadas G., Tutusaus A., Sanchez L., Gasa R. Neurogenin3 cooperates with Foxa2 to autoactivate its own expression. J. Biol. Chem. 2013;288:11705–11717. doi: 10.1074/jbc.M112.388173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anderson K.R., Torres C.A., Solomon K., Becker T.C., Newgard C.B., Wright C.V., Hagman J., Sussel L. Cooperative transcriptional regulation of the essential pancreatic islet gene NeuroD1 (beta2) by Nkx2.2 and neurogenin 3. J. Biol. Chem. 2009;284:31236–31248. doi: 10.1074/jbc.M109.048694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu R., Gillen A.E., Sheridan R.M., Tian C., Daya M., Hao Y., Hesselberth J.R., Riemondy K.A. clustifyr: an R package for automated single-cell RNA sequencing cluster classification. F1000Res. 2020;9:223. doi: 10.12688/f1000research.22969.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baron M., Veres A., Wolock S.L., Faust A.L., Gaujoux R., Vetere A., Ryu J.H., Wagner B.K., Shen-Orr S.S., Klein A.M., et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 2016;3:346–360.e4. doi: 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Byrnes L.E., Wong D.M., Subramaniam M., Meyer N.P., Gilchrist C.L., Knox S.M., Tward A.D., Ye C.J., Sneddon J.B. Lineage dynamics of murine pancreatic development at single-cell resolution. Nat. Commun. 2018;9:3922. doi: 10.1038/s41467-018-06176-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tabula Muris Consortium. Overall coordination. Logistical coordination. Organ collection and processing. Library preparation and sequencing. Writing group. Supplemental text writing group. Principal investigators. Computational data analysis. Cell type annotation Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018;562:367–372. doi: 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qadir M.M.F., Álvarez-Cubela S., Klein D., van Dijk J., Muñiz-Anquela R., Moreno-Hernández Y.B., Lanzoni G., Sadiq S., Navarro-Rubio B., García M.T., et al. Single-cell resolution analysis of the human pancreatic ductal progenitor cell niche. Proceedings of the National Academy of Sciences. 2020;117:10876–10887. doi: 10.1073/pnas.1918314117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muraro M.J., Dharmadhikari G., Grun D., Groen N., Dielen T., Jansen E., van Gurp L., Engelse M.A., Carlotti F., de Koning E.J., van Oudenaarden A. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016;3:385–394. doi: 10.1016/j.cels.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elyada E., Bolisetty M., Laise P., Flynn W.F., Courtois E.T., Burkhart R.A., Teinor J.A., Belleau P., Biffi G., Lucito M.S., et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019;9:1102–1123. doi: 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Socorro M., Criscimanna A., Riva P., Tandon M., Prasadan K., Guo P., Humar A., Husain S.Z., Leach S.D., Gittes G.K., Esni F. Identification of Newly Committed Pancreatic Cells in the Adult Mouse Pancreas. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-17884-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ioannou M., Serafimidis I., Arnes L., Sussel L., Singh S., Vasiliou V., Gavalas A. ALDH1B1 is a potential stem/progenitor marker for multiple pancreas progenitor pools. Dev. Biol. 2013;374:153–163. doi: 10.1016/j.ydbio.2012.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Q., Wang H., Sun Y., Li S.W., Donelan W., Chang L.J., Jin S., Terada N., Cheng H., Reeves W.H., Yang L.J. The reprogrammed pancreatic progenitor-like intermediate state of hepatic cells is more susceptible to pancreatic beta cell differentiation. J. Cell Sci. 2013;126:3638–3648. doi: 10.1242/jcs.124925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ashery-Padan R., Zhou X., Marquardt T., Herrera P., Toube L., Berry A., Gruss P. Conditional inactivation of Pax6 in the pancreas causes early onset of diabetes. Dev. Biol. 2004;269:479–488. doi: 10.1016/j.ydbio.2004.01.040. [DOI] [PubMed] [Google Scholar]
- 72.Gribben C., Lambert C., Messal H.A., Hubber E.L., Rackham C., Evans I., Heimberg H., Jones P., Sancho R., Behrens A. Ductal Ngn3-expressing progenitors contribute to adult beta cell neogenesis in the pancreas. Cell Stem Cell. 2021;28:2000–2008.e4. doi: 10.1016/j.stem.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang X., Heaney S., Maas R.L. Cre-loxp fate-mapping of Pax6 enhancer active retinal and pancreatic progenitors. Genesis. 2003;35:22–30. doi: 10.1002/gene.10160. [DOI] [PubMed] [Google Scholar]
- 74.Kopinke D., Brailsford M., Shea J.E., Leavitt R., Scaife C.L., Murtaugh L.C. Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development. 2011;138:431–441. doi: 10.1242/dev.053843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hosokawa S., Furuyama K., Horiguchi M., Aoyama Y., Tsuboi K., Sakikubo M., Goto T., Hirata K., Tanabe W., Nakano Y., et al. Impact of Sox9 dosage and Hes1-mediated Notch signaling in controlling the plasticity of adult pancreatic duct cells in mice. Sci. Rep. 2015;5:8518. doi: 10.1038/srep08518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fukuda A., Kawaguchi Y., Furuyama K., Kodama S., Horiguchi M., Kuhara T., Koizumi M., Boyer D.F., Fujimoto K., Doi R., et al. Ectopic pancreas formation in Hes1 -knockout mice reveals plasticity of endodermal progenitors of the gut, bile duct, and pancreas. J. Clin. Invest. 2006;116:1484–1493. doi: 10.1172/JCI27704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shu C., Han S., Hu C., Chen C., Qu B., He J., Dong S., Xu P. Integrin beta1 regulates proliferation, apoptosis, and migration of trophoblasts through activation of phosphoinositide 3 kinase/protein kinase B signaling. J. Obstet. Gynaecol. Res. 2021;47:2406–2416. doi: 10.1111/jog.14782. [DOI] [PubMed] [Google Scholar]
- 78.Pellinen T., Blom S., Sanchez S., Valimaki K., Mpindi J.P., Azegrouz H., Strippoli R., Nieto R., Viton M., Palacios I., et al. ITGB1-dependent upregulation of Caveolin-1 switches TGFbeta signalling from tumour-suppressive to oncogenic in prostate cancer. Sci. Rep. 2018;8:2338. doi: 10.1038/s41598-018-20161-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nelson W.J. Remodeling epithelial cell organization: transitions between front-rear and apical-basal polarity. Cold Spring Harb. Perspect. Biol. 2009;1 doi: 10.1101/cshperspect.a000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abu-Thuraia A., Goyette M.A., Boulais J., Delliaux C., Apcher C., Schott C., Chidiac R., Bagci H., Thibault M.P., Davidson D., et al. AXL confers cell migration and invasion by hijacking a PEAK1-regulated focal adhesion protein network. Nat. Commun. 2020;11:3586. doi: 10.1038/s41467-020-17415-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Petz M., Them N.C.C., Huber H., Mikulits W. PDGF enhances IRES-mediated translation of Laminin B1 by cytoplasmic accumulation of La during epithelial to mesenchymal transition. Nucleic Acids Res. 2012;40:9738–9749. doi: 10.1093/nar/gks760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gerisch G., Schroth-Diez B., Müller-Taubenberger A., Ecke M. PIP3 waves and PTEN dynamics in the emergence of cell polarity. Biophys. J. 2012;103:1170–1178. doi: 10.1016/j.bpj.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y., Liu J., Ying X., Lin P.C., Zhou B.P. Twist-mediated Epithelial-mesenchymal Transition Promotes Breast Tumor Cell Invasion via Inhibition of Hippo Pathway. Sci. Rep. 2016;6 doi: 10.1038/srep24606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim W.K., Kwon Y., Jang M., Park M., Kim J., Cho S., Jang D.G., Lee W.B., Jung S.H., Choi H.J., et al. beta-catenin activation down-regulates cell-cell junction-related genes and induces epithelial-to-mesenchymal transition in colorectal cancers. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-54890-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yadav V.K., Lee T.Y., Hsu J.B.K., Huang H.D., Yang W.C.V., Chang T.H. Computational analysis for identification of the extracellular matrix molecules involved in endometrial cancer progression. PLoS One. 2020;15 doi: 10.1371/journal.pone.0231594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Street K., Risso D., Fletcher R.B., Das D., Ngai J., Yosef N., Purdom E., Dudoit S. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018;19:477. doi: 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang W., Beer R.L., Delaspre F., Wang G., Edelman H.E., Park H., Azuma M., Parsons M.J. Sox9b is a mediator of retinoic acid signaling restricting endocrine progenitor differentiation. Dev. Biol. 2016;418:28–39. doi: 10.1016/j.ydbio.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hughes C.S., ChinAleong J.A., Kocher H.M. CRABP2 and FABP5 expression levels in diseased and normal pancreas. Ann. Diagn. Pathol. 2020;47 doi: 10.1016/j.anndiagpath.2020.151557. [DOI] [PubMed] [Google Scholar]
- 89.Wodziak D., Dong A., Basin M.F., Lowe A.W. Anterior Gradient 2 (AGR2) Induced Epidermal Growth Factor Receptor (EGFR) Signaling Is Essential for Murine Pancreatitis-Associated Tissue Regeneration. PLoS One. 2016;11 doi: 10.1371/journal.pone.0164968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Goncalves C.A., Larsen M., Jung S., Stratmann J., Nakamura A., Leuschner M., Hersemann L., Keshara R., Perlman S., Lundvall L., et al. A 3D system to model human pancreas development and its reference single-cell transcriptome atlas identify signaling pathways required for progenitor expansion. Nat. Commun. 2021;12:3144. doi: 10.1038/s41467-021-23295-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Duan Y.R., Chen B.P., Chen F., Yang S.X., Zhu C.Y., Ma Y.L., Li Y., Shi J. LncRNA lnc-ISG20 promotes renal fibrosis in diabetic nephropathy by inducing AKT phosphorylation through miR-486-5p/NFAT5. J. Cell Mol. Med. 2021;25:4922–4937. doi: 10.1111/jcmm.16280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang H., Chen F., Fan X., Lin C., Hao Y., Wei H., Lin W., Jiang Y., He F. Quantitative Proteomic analysis on Activated Hepatic Stellate Cells reversion Reveal STAT1 as a key regulator between Liver Fibrosis and recovery. Sci. Rep. 2017;7 doi: 10.1038/srep44910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Matsumoto S., Fujii S., Sato A., Ibuka S., Kagawa Y., Ishii M., Kikuchi A. A combination of Wnt and growth factor signaling induces Arl4c expression to form epithelial tubular structures. EMBO J. 2014;33:702–718. doi: 10.1002/embj.201386942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Harada A., Matsumoto S., Yasumizu Y., Shojima K., Akama T., Eguchi H., Kikuchi A. Localization of KRAS downstream target ARL4C to invasive pseudopods accelerates pancreatic cancer cell invasion. Elife. 2021;10 doi: 10.7554/eLife.66721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Comamala M., Pinard M., Thériault C., Matte I., Albert A., Boivin M., Beaudin J., Piché A., Rancourt C. Downregulation of cell surface CA125/MUC16 induces epithelial-to-mesenchymal transition and restores EGFR signalling in NIH:OVCAR3 ovarian carcinoma cells. Br. J. Cancer. 2011;104:989–999. doi: 10.1038/bjc.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lim E.J., Kang J.H., Kim Y.J., Kim S., Lee S.J. Correction: ICAM-1 promotes cancer progression by regulating SRC activity as an adapter protein in colorectal cancer. Cell Death Dis. 2022;13:968. doi: 10.1038/s41419-022-05425-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu X., Chan A., Tai C.H., Andresson T., Pastan I. Multiple proteases are involved in mesothelin shedding by cancer cells. Commun. Biol. 2020;3:728. doi: 10.1038/s42003-020-01464-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kuriyama K., Kodama Y., Shiokawa M., Nishikawa Y., Marui S., Kuwada T., Sogabe Y., Kakiuchi N., Tomono T., Matsumori T., et al. Essential role of Notch/Hes1 signaling in postnatal pancreatic exocrine development. J. Gastroenterol. 2021;56:673–687. doi: 10.1007/s00535-021-01779-y. [DOI] [PubMed] [Google Scholar]
- 99.Xu X., Seymour P.A., Sneppen K., Trusina A., Egeskov-Madsen A.l.R., Jørgensen M.C., Jensen M.H., Serup P. Jag1-Notch cis-interaction determines cell fate segregation in pancreatic development. Nat. Commun. 2023;14:348. doi: 10.1038/s41467-023-35963-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang X., Rowan S., Yue Y., Heaney S., Pan Y., Brendolan A., Selleri L., Maas R.L. Pax6 is regulated by Meis and Pbx homeoproteins during pancreatic development. Dev. Biol. 2006;300:748–757. doi: 10.1016/j.ydbio.2006.06.030. [DOI] [PubMed] [Google Scholar]
- 101.Lasorella A., Benezra R., Iavarone A. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer. 2014;14:77–91. doi: 10.1038/nrc3638. [DOI] [PubMed] [Google Scholar]
- 102.Dey N., Young B., Abramovitz M., Bouzyk M., Barwick B., De P., Leyland-Jones B. Differential activation of Wnt-beta-catenin pathway in triple negative breast cancer increases MMP7 in a PTEN dependent manner. PLoS One. 2013;8 doi: 10.1371/journal.pone.0077425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tee A.R., Fingar D.C., Manning B.D., Kwiatkowski D.J., Cantley L.C., Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xu X., Ahmed T., Wang L., Cao X., Zhang Z., Wang M., Lv Y., Kanwal S., Tariq M., Lin R., et al. The mTORC1-eIF4F axis controls paused pluripotency. EMBO Rep. 2022;23 doi: 10.15252/embr.202153081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stanger B.Z., Dor Y. Dissecting the cellular origins of pancreatic cancer. Cell Cycle. 2006;5:43–46. doi: 10.4161/cc.5.1.2291. [DOI] [PubMed] [Google Scholar]
- 106.Westmoreland J.J., Wang Q., Bouzaffour M., Baker S.J., Sosa-Pineda B. Pdk1 activity controls proliferation, survival, and growth of developing pancreatic cells. Dev. Biol. 2009;334:285–298. doi: 10.1016/j.ydbio.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jayachandran A., Anaka M., Prithviraj P., Hudson C., McKeown S.J., Lo P.H., Vella L.J., Goding C.R., Cebon J., Behren A. Thrombospondin 1 promotes an aggressive phenotype through epithelial-to-mesenchymal transition in human melanoma. Oncotarget. 2014;5:5782–5797. doi: 10.18632/oncotarget.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mu F., Huang J., Xing T., Jing Y., Cui T., Guo Y., Yan X., Li H., Wang N. The Wnt/beta-Catenin/LEF1 Pathway Promotes Cell Proliferation at Least in Part Through Direct Upregulation of miR-17-92 Cluster. Front. Genet. 2019;10:525. doi: 10.3389/fgene.2019.00525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sun X., Yi Y., Yan Z., Rosen B.H., Liang B., Winter M.C., Evans T.I.A., Rotti P.G., Yang Y., Gray J.S., et al. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aau7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yan Z., Vorhies K., Feng Z., Park S.Y., Choi S.H., Zhang Y., Winter M., Sun X., Engelhardt J.F. Recombinant Adeno-Associated Virus-Mediated Editing of the G551D Cystic Fibrosis Transmembrane Conductance Regulator Mutation in Ferret Airway Basal Cells. Hum. Gene Ther. 2022;33:1023–1036. doi: 10.1089/hum.2022.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sun X., Yan Z., Yi Y., Li Z., Lei D., Rogers C.S., Chen J., Zhang Y., Welsh M.J., Leno G.H., Engelhardt J.F. Adeno-associated virus-targeted disruption of the CFTR gene in cloned ferrets. J. Clin. Invest. 2008;118:1578–1583. doi: 10.1172/JCI34599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Riquelme S.A., Lozano C., Moustafa A.M., Liimatta K., Tomlinson K.L., Britto C., Khanal S., Gill S.K., Narechania A., Azcona-Gutiérrez J.M., et al. CFTR-PTEN-dependent mitochondrial metabolic dysfunction promotes Pseudomonas aeruginosa airway infection. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aav4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Riquelme S.A., Hopkins B.D., Wolfe A.L., DiMango E., Kitur K., Parsons R., Prince A. Cystic Fibrosis Transmembrane Conductance Regulator Attaches Tumor Suppressor PTEN to the Membrane and Promotes Anti Pseudomonas aeruginosa Immunity. Immunity. 2017;47:1169–1181.e7. doi: 10.1016/j.immuni.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bruurs L.J.M., van der Net M.C., Zwakenberg S., Rosendahl Huber A.K.M., Post A., Zwartkruis F.J., Bos J.L. The Phosphatase PTPL1 Is Required for PTEN-Mediated Regulation of Apical Membrane Size. Mol. Cell Biol. 2018;38 doi: 10.1128/MCB.00102-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Carnero A., Paramio J.M. The PTEN/PI3K/AKT Pathway in vivo, Cancer Mouse Models. Front. Oncol. 2014;4:252. doi: 10.3389/fonc.2014.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huang J.Q., Wei F.K., Xu X.L., Ye S.X., Song J.W., Ding P.K., Zhu J., Li H.F., Luo X.P., Gong H., et al. SOX9 drives the epithelial-mesenchymal transition in non-small-cell lung cancer through the Wnt/beta-catenin pathway. J. Transl. Med. 2019;17:143. doi: 10.1186/s12967-019-1895-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Qi Y., Liu J., Chao J., Scheuerman M.P., Rahimi S.A., Lee L.Y., Li S. PTEN suppresses epithelial-mesenchymal transition and cancer stem cell activity by downregulating Abi1. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-69698-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rodriguez-Seguel E., Mah N., Naumann H., Pongrac I.M., Cerda-Esteban N., Fontaine J.F., Wang Y., Chen W., Andrade-Navarro M.A., Spagnoli F.M. Mutually exclusive signaling signatures define the hepatic and pancreatic progenitor cell lineage divergence. Genes Dev. 2013;27:1932–1946. doi: 10.1101/gad.220244.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Seymour P.A., Freude K.K., Tran M.N., Mayes E.E., Jensen J., Kist R., Scherer G., Sander M. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc. Natl. Acad. Sci. USA. 2007;104:1865–1870. doi: 10.1073/pnas.0609217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sun X., Olivier A.K., Yi Y., Pope C.E., Hayden H.S., Liang B., Sui H., Zhou W., Hager K.R., Zhang Y., et al. Gastrointestinal pathology in juvenile and adult CFTR-knockout ferrets. Am. J. Pathol. 2014;184:1309–1322. doi: 10.1016/j.ajpath.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhou Q., Law A.C., Rajagopal J., Anderson W.J., Gray P.A., Melton D.A. A multipotent progenitor domain guides pancreatic organogenesis. Dev. Cell. 2007;13:103–114. doi: 10.1016/j.devcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 122.Wang X., Sterr M., Burtscher I., Chen S., Hieronimus A., Machicao F., Staiger H., Häring H.U., Lederer G., Meitinger T., et al. Genome-wide analysis of PDX1 target genes in human pancreatic progenitors. Mol. Metabol. 2018;9:57–68. doi: 10.1016/j.molmet.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Spaeth J.M., Liu J.H., Peters D., Guo M., Osipovich A.B., Mohammadi F., Roy N., Bhushan A., Magnuson M.A., Hebrok M., et al. The Pdx1-Bound Swi/Snf Chromatin Remodeling Complex Regulates Pancreatic Progenitor Cell Proliferation and Mature Islet beta-Cell Function. Diabetes. 2019;68:1806–1818. doi: 10.2337/db19-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim S.K., Selleri L., Lee J.S., Zhang A.Y., Gu X., Jacobs Y., Cleary M.L. Pbx1 inactivation disrupts pancreas development and in Ipf1-deficient mice promotes diabetes mellitus. Nat. Genet. 2002;30:430–435. doi: 10.1038/ng860. [DOI] [PubMed] [Google Scholar]
- 125.Mastrolia V., Flucher S.M., Obermair G.J., Drach M., Hofer H., Renström E., Schwartz A., Striessnig J., Flucher B.E., Tuluc P. Loss of alpha(2)delta-1 Calcium Channel Subunit Function Increases the Susceptibility for Diabetes. Diabetes. 2017;66:897–907. doi: 10.2337/db16-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Evans I.A., Sun X., Liang B., Vegter A.R., Guo L., Lynch T.J., Zhang Y., Zhang Y., Yi Y., Yang Y., et al. In utero and postnatal ivacaftor/lumacaftor therapy rescues multiorgan disease in CFTR-F508del ferrets. JCI Insight. 2024;9 doi: 10.1172/jci.insight.157229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yuan F., Gasser G.N., Lemire E., Montoro D.T., Jagadeesh K., Zhang Y., Duan Y., Ievlev V., Wells K.L., Rotti P.G., et al. Transgenic ferret models define pulmonary ionocyte diversity and function. Nature. 2023;621:857–867. doi: 10.1038/s41586-023-06549-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Comer D.M., Ennis M., McDowell C., Beattie D., Rendall J., Hall V., Elborn J.S. Clinical phenotype of cystic fibrosis patients with the G551D mutation. QJM. 2009;102:793–798. doi: 10.1093/qjmed/hcp120. [DOI] [PubMed] [Google Scholar]
- 129.Sun X., Olivier A.K., Liang B., Yi Y., Sui H., Evans T.I.A., Zhang Y., Zhou W., Tyler S.R., Fisher J.T., et al. Lung phenotype of juvenile and adult cystic fibrosis transmembrane conductance regulator-knockout ferrets. Am. J. Respir. Cell Mol. Biol. 2014;50:502–512. doi: 10.1165/rcmb.2013-0261OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mou H., Vinarsky V., Tata P.R., Brazauskas K., Choi S.H., Crooke A.K., Zhang B., Solomon G.M., Turner B., Bihler H., et al. Dual SMAD Signaling Inhibition Enables Long-Term Expansion of Diverse Epithelial Basal Cells. Cell Stem Cell. 2016;19:217–231. doi: 10.1016/j.stem.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Choi S.H., Reeves R.E., Romano Ibarra G.S., Lynch T.J., Shahin W.S., Feng Z., Gasser G.N., Winter M.C., Evans T.I.A., Liu X., et al. Detargeting Lentiviral-Mediated CFTR Expression in Airway Basal Cells Using miR-106b. Genes. 2020;11 doi: 10.3390/genes11101169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kenny C., Dilshat R., Seberg H.E., Van Otterloo E., Bonde G., Helverson A., Franke C.M., Steingrímsson E., Cornell R.A. TFAP2 paralogs facilitate chromatin access for MITF at pigmentation and cell proliferation genes. PLoS Genet. 2022;18 doi: 10.1371/journal.pgen.1010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8 doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Krentz N.A.J., Lee M.Y.Y., Xu E.E., Sproul S.L.J., Maslova A., Sasaki S., Lynn F.C. Single-Cell Transcriptome Profiling of Mouse and hESC-Derived Pancreatic Progenitors. Stem Cell Rep. 2018;11:1551–1564. doi: 10.1016/j.stemcr.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Bulk RNA sequencing analysis results of WT (n = 3) and CF (n = 3) PDEs. (B) Upstream Regulator analysis showing transcriptional regulators of DEGs in CF PDEs using IPA. (C) Upstream regulator analysis results for DEG in CF PDEs.
(A) Results from GO term analysis on DEG in CFTR-KO PDE using Panther. (B) Gene list associated with cell differentiation GO term. (C) Gene set associated with cell migration GO term. (D) Gene set for mesenchyme development GO term. (E) Gene set for response to TGF-β stimulus GO term. (F) Gene set for response to WNT signaling is listed. (G) Gene set for endocrine pancreas development GO term. (H) Gene set for histone modification GO term.
(A) The differentially open regions (DORs) associated with coding regions in the chromatin of CF PDEs relative to WT PDEs are shown in the table. (B) Analysis of motif enrichment in the CF DORs.
(A) DEG in acinar cells derived from WT and CF PDE. (B) DEG in ductal cells found in WT and CF PDE. (C) DEG in proliferating acinar cells derived from WT and CF PDE. (D) DEG in proliferating ductal cells derived from WT and CF PDE. (E) DEG in centroacinar progenitor cells derived from WT and CF PDE. (F) DEG in centroacinar cells derived from WT and CF PDE.
(A) DEGs in CF PDEs relative to WT PDEs at D2 of differentiation. (B) DEGs in CF PDEs relative to WT PDEs at D5 of differentiation. (C) The DEGs in CF PDEs relative to WT PDEs at D7 of differentiation. (D) The DEGs in CF PDEs relative to WT PDEs at D9 of differentiation.
(A) The variability in expression of D2 DEGs. (B) The variability in expression of D5 DEGs. (C) The variability in expression of D7 DEGs. (D) The variability in expression of D9 DEGs. (E) The variability in expression of D9 CAC progenitor DEGs. (F) The variability in expression of D9 CAC DEGs.
(A) Upstream regulator analysis for DEGs at D2 of differentiation using IPA. (B) Upstream regulator analysis for DEGs at D5 of differentiation using IPA. (C) Upstream regulator analysis for DEGs at D7 of differentiation using IPA. (D) Upstream regulator analysis for DEGs at D9 of differentiation using IPA. (E) Pathway analysis for CF PDE DEGs at D2 of differentiation. (F) Pathway analysis for CF PDE DEG at D5 of differentiation. (G) Pathway analysis for CF PDE DEGs at D7 of differentiation. (H) Pathway analysis for CF PDE DEGs at D9 of differentiation.
(A) The DEGs in WT ductal 1 subpopulation. (B) The DEGs in WT ductal 2 subpopulation. (C) The DEGs in WT ductal 3 subpopulation. (D) The DEGs in CF ductal 1 subpopulation. (E) The DEGs in CF ductal 2 subpopulation are shown. (F) The DEGs in CF ductal 3 subpopulation are shown. (G) The Z scores of DEGs in ductal subpopulations identified in WT and CF PDEs. (H) The DEGs in ductal cells of WT and CF PDEs, and their Z scores for expression across all time points of differentiation.
(A) The predicted upstream regulatory signaling pathways active in CF CACs and relative to WT CACs. (B) The predicted upstream regulatory signaling pathways active in CF progenitor-like cells relative to WT progenitor-like cells. (C) The Z score enrichment values for AKT signaling genes in WT and CF PDE cells at D9 differentiation. (D) The Z score enrichment values for PTEN signaling genes in WT and CF PDE cells at D9 differentiation. (E) The Z score enrichment values for WNT signaling genes in WT and CF PDE cells at D9 differentiation.
Data Availability Statement
-
•
All data generated for this paper will be shared by the lead contact upon request. The data are publicly available at GEO database. The accession numbers are listed in the key resources table.
-
•
All original code is deposited on GitHub. The URL to the code is listed in key resources table.
-
•
Any other information required will be provided by the lead author.