

Abstract
Hepatorenal tyrosinaemia (HT1) is an autosomal recessive disorder of tyrosine degradation resulting in hepatic and renal dysfunction, neurological sequelae may occur in some patients. The use of nitisinone (NTBC) has revolutionised treatment and outcome of this disorder. NTBC has to be combined with a low protein diet. While NTBC modulates the disease course in HT1 patients, several issues are open. Optimal dosage, doses per day, therapeutic range of NTBC concentration, mode of protein restriction and biomarkers are not well defined. HCC and neurocognitive deficits are long‐term sequelae. Early diagnosis and treatment are essential to minimise the risk for these complications. Clinical guidance for management of HT1‐patients is required. Randomised clinical studies are difficult in the presence of therapeutic options. We discussed these issues in a consensus group of 10 paediatricians, 1 adult hepatologist, 1 geneticist, 2 dieticians, 2 newborn screening specialists with experience in HT1, 1 psychologist and 2 representatives of a patient group from the German‐speaking countries (DACH). Recommendations were based on scientific literature and expert opinion, also taking into account recent experience with newborn screening. There was strong consensus that newborn screening using succinylacetone (SA) and early treatment are essential for a good outcome. The dose of NTBC should be as low as possible without losing metabolic control. This has to be accompanied by a low protein diet, in some patients a simplified diet without calculation of protein intake. Specific education and psychosocial support are recommended. Indications for liver transplantation were defined. Monitoring shall include clinical findings, levels of SA, tyrosine, phenylalanine and NTBC in (dried) blood.
Keywords: hepatorenal tyrosinaemia, newborn screening, nitisinone, succinylacetone, tyrosine
1. INTRODUCTION
Hepatorenal tyrosinaemia (HT1; OMIM 276700; Orpha Code 882) is a rare inborn error of tyrosine catabolism due to deficiency of the enzyme fumarylacetoacetase/fumarylacetoacetatehydratase (FAH; MIM 613871; E.C. 3.7.1.2). This disorder is inherited as an autosomal recessive trait and is due to biallelic pathogenic variants of FAH. The incidence in Central Europe is estimated at 1:100000–1: 125000 1 , 2 but HT1 is more frequent in other regions based on founder mutations like c.1062+5G>A (old nomenclature IVS12+5G>A) in patients from the Saguenay‐ Lac‐St Jean region in Canada. 3
Before the introduction of nitisinone, HT1 was lethal in most of the children affected. Ninety percent of HT1 patients died within the first 2 years of life. 4 Probably, there was an ascertainment bias as milder phenotypes are difficult to diagnose clinically.
Most newborns and infants with HT1 are born after an uneventful pregnancy and show normal development in the first few months of life. Without newborn screening, the presenting symptom is mostly liver dysfunction with hepato(spleno)megaly, jaundice, coagulation disorders, hypoglycaemia, edema due to hypoproteinaemia, liver cirrhosis and acute liver failure. 5 Tubular dysfunction with hypophosphataemic, vitamin D‐resistant rickets, hyperaminoaciduria, renal tubular acidosis resulting from renal bicarbonate wasting, proteinuria and growth failure are additional renal features. 4 , 6 , 7 , 8 Furthermore, ‘porphyric crises’ with peripheral neuropathy, painful dysaesthesia, paralysis and muscular hypertonia due to inhibition of delta‐aminolaevulinic acid dehydrogenase by succinylacetone (SA) were observed. In the long run, there was an increased risk for the development of hepatocellular carcinoma (HCC) 4 , 9 , 10 , 11 , 12 in the clinically diagnosed cases. The natural course of the disease was variable based on the residual activity of FAH. 13 , 14 , 15 , 16 , 17
Until the early 1990ies, the disease was often life‐threatening and lethal. Life expectancy and quality of life were significantly reduced. The introduction of nitisinone (NTBC; 2‐(2‐nitro‐4‐trifluoro‐methylbenzoyl)‐1,3 cyclohexandione) into clinical medicine revolutionised the course of the disease. Nitisinone was originally developed as a herbicide and subsequently repurposed as a drug for treatment of HT1 after it became clear that it inhibited an enzyme in the degradative pathway of tyrosine (for a review see References 18, 19). The enzyme 4‐hydroxy‐phenylpyruvate dioxygenase is inhibited by nitisinone leading to a reduction of toxic compounds found in body fluids of untreated patients suffering from HT1. Based on its disease‐modulating effect, nitisinone was first successfully initiated as a ‘compassionate use’ treatment in the early 1990s 20 , 21 and it took until 2005 for nitisinone to be approved as an ‘orphan drug’ by the European Medicines Agency under ‘exceptional circumstances’ accompanied by a post‐marketing study (OPAL [Orfadin Post Authorization Long‐term Safety]‐study). Nitisinone was approved by the US Food and Drug Administration in 2002. Preclinical data for nitisinone were scarce and the post‐marketing study included very basic data only.
Nitisinone blocks the degradative pathway of tyrosine by inhibiting the enzyme 4‐hydroxy‐phenylpyruvate dioxygenase and thus blocks the synthesis of toxic compounds. 19 SA is used as a surrogate parameter of metabolic control. Blocking the degradative pathway of tyrosine leads to hypertyrosinaemia. To avoid tyrosine toxicity, a protein reduced diet supplemented with a phenylalanine‐ and tyrosine‐free amino acid mixture has to be introduced. 14 , 15 , 22 Nitisinone modifies the clinical course of the disease significantly not only with respect to HCC development but also regarding liver and kidney dysfunction as well as porphyria‐like crises. 8 , 23 , 24
Despite this treatment, neurocognitive delay may occur. 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 The pathophysiology of this neurocognitive compromise is yet unclear.
In an international cross‐sectional study, 8 we collected data from HT1 patients and identified a considerable unmet need to define diagnostic, therapeutic and monitoring procedures for the clinical management of HT1‐patients. Early diagnosis and treatment with nitisinone are essential to prevent chronic liver disease and HCC. Importantly, the odds‐ratio to develop HCC is 13 if treatment with nitisinone is started after the first birthday compared to treatment initiation in the neonatal period. 8
Recommendations regarding the management of HT1‐patients were previously published. 34 , 35 , 36 Chinsky and coauthors recommended newborn screening, nitisinone dosing and target values for tyrosine concentrations in blood. However, the long‐term impact of newborn screening, dosing of nitisinone, optimal therapeutic concentrations of nitisinone in blood, modalities of protein‐reduced diet and optimal therapeutic concentrations of phenylalanine and tyrosine in blood are still unclear in the absence of appropriate randomised clinical studies. These trials are difficult in rare disorders, especially if an efficient therapy is available, due to ethical considerations.
This prompted us to develop clinical practice recommendations based on real world data and experience of metabolic clinicians, dieticians, clinical chemists and psychologists including first experience with newborn screening.
2. METHODOLOGY AND OBJECTIVES
2.1. Guideline development
Methods for the development of these guidelines followed rules and regulations of the German AWMF (Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften; Working group of scientific medical societies), 2nd edition 2020 (http://www.awmf.org/leitlinien/awmf-regelwerk.html). Members of the guideline development group were volunteers recruited from the members of the German speaking ‘working group for Inborn Disorders of Metabolism’ (APS, Arbeitsgemeinschaft für Pädiatrische Stoffwechselstörungen) and of the German speaking ‘working group for inborn disorders of metabolism in internal medicine’ (ASIM, Arbeitsgemeinschaft Stoffwechselerkrankungen in der Inneren Medizin). The guideline development group consisted of 10 paediatricians with expertise in treating HT1‐patients (AMD as chairman, DB, DH, JH, PF, DK, EM, SSB, NW, JZ), 1 adult hepatologist with expertise in treating HT1‐patients (SD), 2 dietitians with expertise in treating HT1‐patients (UM, SR), 2 newborn screening specialists (NJ, JS), 1 geneticist (SM), 2 patient advocacy group representatives of the German Interest Group‐PKU (DIG‐PKU: TH, CJ) and an independent moderator and medical psychologist (KL). During the kick‐off meeting members were advised on AWMF rules for developing a guideline at S2k‐level (consensus‐based guideline, graduated recommendations, limited number of evidence‐based recommendations), and working groups were formed. These working groups wrote different draft chapters, respectively. In a follow‐up meeting, the drafts were discussed and modified; consensus statements for recommendations were presented in another meeting and discussed. Tentative recommendations were sent to all participants and statements were registered. Recommendations and statements were summarised by the independent moderator. Finally, all recommendations were graded based on voting of the members.
2.2. Systematic literature search, review and grading
A systematic literature search in the PubMed database was performed based on the following keywords: Tyrosinaemia, hepatorenal, succinyl acetonuria, newborn screening. Literature dealing with tyrosinaemia type 2 and tyrosinaemia type 3 was excluded. References up to September 2022 were included.
Grading of recommendations followed the AWMF‐methodology: >95% strong consensus, >75% consensus, >50% consensus by the majority of participants, <50% no consensus. The final grading recommendations were classified as ‘strong recommendation’ (terminology: ‘shall’ or ‘shall not’ in the text), ‘recommendation’ (terminology ‘should’ or ‘should not’), ‘recommendation undecided’ (terminology ‘can be considered’ or ‘can be done without’).
2.3. Disclaimer
These practice guidelines/recommendations are supposed to help clinical decision making in HT1 patients. Although based on the best‐available evidence, scientific data on treatment and monitoring are scarce. Hence, some of the recommendations given here only reflect expert opinion. Therefore, clinical practice may deviate from the recommendations given here, if indicated in individual cases. These guidelines cannot guarantee a favourable outcome in all patients.
3. RESULTS
3.1. Diagnosis
3.1.1. Diagnosis by newborn screening
In most individuals with HT1, there is a clinically latent phase, first symptoms occur at the age of several months. 8 This makes early clinical diagnosis difficult but on the other hand allows efficient newborn screening with SA as screening parameter.
For newborn screening, determination of tyrosine concentration in dried blood shows low sensitivity and specificity. 37 , 38 , 39 , 40 , 41 , 42 SA has been shown to be a reliable screening parameter in terms of specificity and sensitivity. 37 , 38 , 39 , 40 , 41 , 43 , 44 , 45 A SA concentration above a certain laboratory‐specific cut‐off (i.e., 99.9th percentile) is regarded as a positive screening result. In mild forms of HT1 temporarily slightly elevated SA concentrations have been described in asymptomatic children which do not require treatment. 39 , 46 , 47 Such ‘pseudodeficient’ individuals reduce the sensitivity of testing.
Newborn screening for HT1 as a target disease using SA as screening parameter has been started in Germany in 2018 48 , 49 and 2022 in Austria, while it is not available in Switzerland. A positive screening should be confirmed via gas chromatography/mass spectrometry as a second, independent analytical procedure showing elevated levels of SA in urine and blood. 41 Parameters of liver and kidney function in blood should be checked 34 as these organs are primarily affected by the disease. Genetic testing for variants in FAH is recommended as confirmatory testing to confirm that SA‐elevation is due to a FAH‐variant. If no variant in FAH can be found, GSTZ1 encoding zeta class glutathione transferase (also known as maleylacetoacetate isomerase) shall be tested, 46 , 47 preferably using whole exome sequencing. If not available, Sanger sequencing can be done. The diagnostic work‐up is shown in Figure 1 (Recommendation 1).
FIGURE 1.
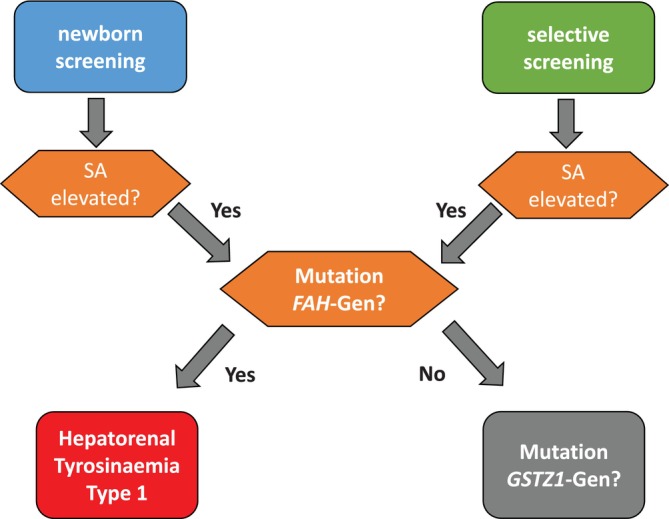
Diagnostic work‐up of a positive newborn screening result via newborn screening (left) or selective screening (right).
Recommendation 1.
Newborn screening for HT1 shall be performed using SA as the screening parameter. In case of a positive screening result, confirmatory tests shall be performed. These include quantitation of SA in urine and/or blood, analysis of amino acids in blood, liver function tests and molecular analysis of FAH. If no variant can be found in FAH, GSTZ1 shall be analysed.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
3.1.2. Diagnosis in HT1 patients with clinical symptoms
Newborn screening for HT1 is not performed in all countries across the world. Immigrants from countries where a newborn screening programme for HT1 is not implemented may not have undergone newborn testing and may only be diagnosed based on clinical symptoms at the age of a few months. Testing via tyrosine concentration may have been done in the home country which is not sensitive/specific enough. If a child is suspected to suffer from HT1 based on clinical symptoms or an index case in the family, SA shall be determined in dried blood and/or urine. If this is positive, molecular genetic analysis of FAH is recommended, together with liver and kidney function tests. If inconclusive, GSTZ1 should be analysed (Recommendation 2).
Recommendation 2.
If a child is clinically suspected to suffer from HT1, SA in dried blood and/or urine shall be quantified. Furthermore, amino acids in plasma and urine, parameters of liver and kidney function and alpha fetoprotein shall be determined. If SA is elevated, sequencing of FAH, and GSTZ1 shall be performed.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
3.1.3. Genetic basis of HT1
HT1 is due to biallelic pathogenic variants in FAH on the long arm of chromosome 15 (15q25.1). Both parents are asymptomatic heterozygotes (‘carriers’) of one variant in FAH in most cases. We recommend genetic analysis of both parents to prove that variants in the patient are biallelic. If variants are not biallelic, we recommend analysis of the GSZT1. If variants of unclear significance (VUS) are found, GSZT1 should be analysed. If no biallelic pathogenic variants of GSZT1 can be identified, functional tests and symptoms of patients should be assessed (clinical symptoms, SA, tyrosine, liver and kidney function etc.) which may lead to a re‐classification of the VUS.
Compound heterozygosity of the rare ‘pseudodeficiency’ variant of FAH, c.1021C>T (p.Arg341Trp) may result in a benign course with mildly elevated SA‐levels without characteristic clinical symptoms. 46 , 47
Fertility of carriers is normal. 50 , 51 The risk to suffer from HT1 in siblings of an index case is 25%, following autosomal recessive inheritance, if both parents are carriers. Genetic counselling of relatives of the parents of an affected child should be offered, particularly in consanguineous families.
Prenatal/preimplantation diagnosis by genetic means can be offered if an index case exists in a family. 52 If the foetus is affected, termination of pregnancy may be discussed. As HT1 is a treatable disorder with a favourable outcome if treatment is initiated early in life, termination of pregnancy is highly controversial and has to be discussed interdisciplinary (paediatrician, geneticist, obstetrician) with the parents on an individual basis. A clinical ethics committee may be involved if necessary.
There is weak genotype–phenotype correlation, 53 even in the same family. Nevertheless, molecular genetic testing shall be offered to all patients who are suspected to suffer from HT1. This test is especially helpful in children with an atypical course of the disease, for example, absence of SA elevation, 54 ‘pseudodeficiency’ variant 46 , 47 or an attenuated phenotype 55 (Recommendation 3).
Recommendation 3a.
Molecular genetic testing of FAH shall be performed as a confirmatory test after a positive result in newborn screening, selective screening or strong clinical suspicion, even if SA is not elevated. If no variants can be detected in FAH, GSTZ1 shall be analysed.
Molecular genetic testing shall not delay pharmacological and dietary treatment.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
Recommendation 3b
To confirm that variants of FAH are bi‐allelic, testing of both parents should be performed.
Expert consensus: 18 yes; 0 no; Abstention 1 – consensus.
Recommendation 3c
Results of molecular genetic analysis should be communicated to the patient and parents in the context of genetic counselling. The option of prenatal diagnosis should be mentioned. If the foetus is affected this is not an obligatory indication for termination of pregnancy, termination of pregnancy can be considered in individual cases.
Expert consensus: 18 yes; 0 no; Abstention 1 – consensus.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
3.2. Therapy of HT1
Therapy of HT1 consists of pharmacological treatment with nitisinone, accompanied by a low protein diet supplemented with an amino acid mixture (AAM) devoid of tyrosine and its precursor phenylalanine.
3.2.1. Pharmacological therapy using nitisinone
Nitisinone has revolutionised the outcome of patients suffering from HT1. 56 It is essential to start therapy early in life, if possible, already in the neonatal period. 8 Nitisinone inhibits the enzyme 4‐hydroxyphenylpyruvate dioxygenase (Figure 2) and thus leads to a reduction in the concentrations of toxic compounds such as 4‐maleyl‐ and 4‐fumarylacetoacetate.
FIGURE 2.
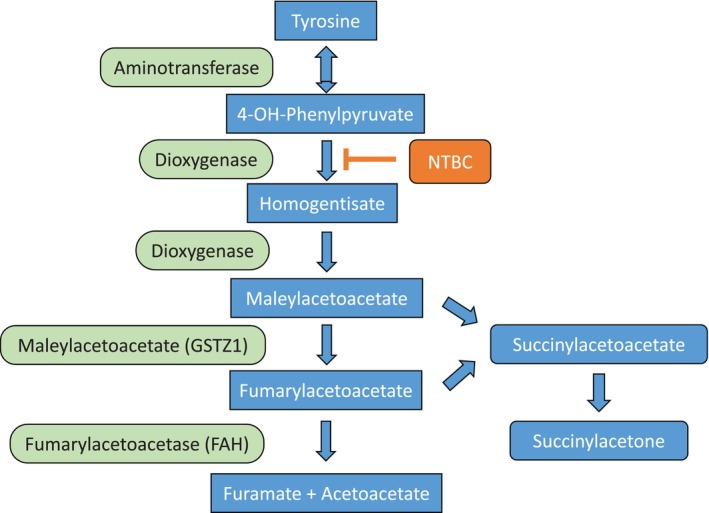
Degradation of tyrosine to fumarate and acetoacetate. FAH is deficient in HT1, nitisinone results in reduction of toxic metabolites, succinylacetone serves as a surrogate marker for toxicity.
Acute liver and kidney dysfunction, predominantly in infants, require symptomatic therapy, like intravenous fluid with glucose and electrolytes, vitamin K (A, D, E) supplementation, albumin and fresh frozen plasma.
The initial dose of nitisinone is 1–2 mg/kg and day. 8 , 34 , 35 , 57 Nitisinone acts fast and can prevent long‐term dysfunction of kidney and liver. 6 , 7 , 58 , 59 , 60 In our experience, the daily dose of nitisinone can be titrated down without hampering metabolic control measured in terms of SA concentration in blood and/or urine which should be monitored regularly. This has also been reported by others. 8 , 34 , 61 For younger patients, nitisinone can be given as a suspension, which facilitates application.
It is generally recommended to give nitisinone in two daily doses. Based on the long half‐life of 54 h in healthy adults 62 it was suggested to give nitisinone in a single daily dose, in order to increase adherence. 63 Nitisinone is unstable in an acidic environment. If given in a single dose, some of the drug may be destroyed due to gastric acidity. 64 Results of different observational studies are controversial.
Nitisinone is well tolerated, rare side effects are for example ocular symptoms (caused by highly elevated tyrosine concentrations) and low platelet counts (Recommendation 4). 8 , 34 , 56 , 57
Recommendation 4a.
Children suspected to suffer from HT1 shall be treated with nitisinone at a dose of 1–2 mg/kg per day. The dose should be titrated down in the further course of disease, metabolic control is monitored based on SA in blood and urine (target: below detection limit). Adapting the dose to increasing weight is rarely necessary.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
Recommendation 4b
Furthermore, symptomatic treatment shall be provided in patients with acute liver and/or kidney dysfunction.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
Recommendation 4c
Nitisinone should be given twice a day. In children weighing more than 20 kg, nitisinone can be given once daily in an attempt to increase adherence.
Treatment shall not at all be interrupted as this may cause acute ‘porphyria‐like crises’.
Expert consensus: 17 yes; 0 no; Abstention 2 – consensus.
3.2.2. Dietary therapy in HT1
Based on its effect on tyrosine catabolism (see Figure 2: inhibition of 4‐hydroxy‐phenylpyruvate dioxygenase), nitisinone therapy has to be combined with a low protein diet to avoid toxicity of tyrosine. 8 , 34 , 35 , 65 This diet has to be supplemented with amino acid mixtures (AAM) devoid of tyrosine and its precursor phenylalanine containing minerals, trace elements and vitamins. 66 This secures a normal physical development. Palatability of AAM hampers adherence to the diet. In the last years, new AAM formulations have been developed, in which amino acids are replaced by glycomacropeptides, 67 which only contain small amounts of phenylalanine and tyrosine. Glycomacropeptides are a side‐product of whey production. As an intact polypeptide they are more palatable, have fewer gastrointestinal side‐effects and lead to long‐lasting satiety compared to AAM. 68 In order to avoid catabolism associated with elevated tyrosine concentrations, caloric needs of the HT1 patients have to be met. The diet has to be followed for life and should regularly be adjusted to the individual needs of the patient, ideally by a dietician with expertise in the field of inborn disorders of metabolism.
For many years, HT1 patients and their families have been advised to calculate their protein intake. In recent years, it has been shown that a simplified diet using categorisation of food according to protein content (three categories are used: ‘harmless’, ‘can be occasionally consumed’, ‘should be avoided’) is possible without hampering metabolic control. 69 A retrospective longitudinal study has shown that larger amounts of natural food can be consumed after school‐age without hampering metabolic control and adherence (Recommendation 5). 70
Recommendation 5.
Nutritional therapy is necessary in all HT1 patients treated with nitisinone and shall be adapted to the individual needs of the patient. These depend on age, growth, energy requirement, health state, the psychosocial situation and other factors. Life‐long adaptation and evaluation of the nutrition is necessary. Relaxation and simplification of the diet (e.g., by categorisation of food instead of protein calculation) may improve adherence, metabolic control and quality of life.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
Regular assessment and evaluation of the diet is necessary to ensure normal growth and development. 66 Nutritional advise should be given to families and patients by a specialised and qualified dietician with expertise in the treatment of patients with inborn errors of metabolism. Nutritional protocols should regularly be assessed and the diet should be adapted accordingly.
Feeding difficulties may be addressed during dietary consultation. In older children, self‐empowerment may be an issue to be addressed by dieticians. Dieticians should be part of a multi‐professional team. Avoidance of catabolism should be a topic during consultations (Recommendation 6).
Recommendation 6.
Process‐oriented nutritional consultations and training shall be regularly performed by a qualified dietician with expertise in inborn disorders of metabolism. A qualified dietician shall be part of the multi‐professional team taking care of HT1 patients.
Expert consensus: 18 yes; 0 no; Abstention 1 – consensus.
3.2.3. Monitoring therapeutic success
Metabolic control is judged by SA concentration as a surrogate parameter of toxicity in dried blood and/or urine. Concentrations below detection limit are to be achieved 8 , 34 , 35 which is specific for each laboratory. 71 , 72 As SA in blood is bound to proteins, blood values reflect a longer time interval compared to concentrations in urine. When comparing plasma and dried blood concentrations, a laboratory‐specific correction factor has to be used. In an international quality control study, average plasma concentrations were 2.34 times higher than dried blood levels. 72
Nitisinone can be reliably measured in dried blood 73 and plasma 74 using the tandem‐mass spectrometry technique. Measurement of nitisinone can be combined with the determination of SA, phenylalanine and tyrosine in dried blood allowing comprehensive, easy sampling at home. 73 , 75 When comparing plasma and dried blood concentrations, a laboratory‐specific correction factor has to be applied as mentioned above. The presumed therapeutic range for nitisinone in blood (trough level) is 20–60 μM. 8 , 34 , 35 , 57 In our experience, even lower concentrations can be tolerated without risking metabolic control. The therapeutic range for nitisinone in blood is still not well defined. The minimal nitisinone dose ensuring metabolic control has to be individually determined for each patient based on SA levels. 8
The therapeutic range for tyrosine concentration in plasma is presumably 200–800 μM, 8 , 26 , 34 , 57 however this is not based on controlled randomised trials. These values are far above the tyrosine concentration in healthy individuals. Tyrosine concentrations should be as low as possible without having a strong impact on quality of life. Tyrosine levels below 600 μM are feasible in our experience. 8 Tyrosine can be measured both in dried blood and plasma, again a correction factor, which is laboratory specific, is necessary to compare values.
Phenylalanine concentrations in blood should be in the lower range of normal. If phenylalanine levels in plasma are too low (<30 μM) natural protein intake should be increased or phenylalanine supplemented as a single amino acid. 57 If phenylalanine levels are low, higher amounts of natural food shall be consumed as long as tyrosine levels are within the low or medium therapeutic range. If phenylalanine levels are low and the tyrosine concentration is in the upper therapeutic range or above, phenylalanine shall be selectively supplemented as a single amino acid (Recommendation 7).
Recommendation 7a.
Patients with HT1 shall be treated for life with the lowest dose of nitisinone possible which ensures metabolic control based on SA levels in blood and/or urine (below detection limit). SA levels in blood reflect a longer time‐interval than excretion of SA in urine. Nitisinone treatment shall be accompanied by a low‐protein diet supplemented with AAM.
The therapeutic range of nitisinone concentration in plasma is supposed to be 20–60 μM, however lower concentrations can be tolerated as long as SA production is suppressed.
Expert consensus: 18 yes; 0 no; Abstention 1 – consensus.
Recommendation 7b
Tyrosine levels in plasma shall be in the target range of 200–600 μM.
Phenylalanine levels in plasma shall be in the lower reference range. If phenylalanine levels in plasma are decreased (< 30 μM) the intake of natural protein shall be increased or phenylalanine shall be supplemented to reach low normal plasma concentrations.
Blood should be drawn before breakfast or at least 4 hours after a meal.
Expert consensus: 18 yes; 0 no; Abstention 1 – consensus.
Recommendation 7c
Analysis in dried blood allows the simultaneous determination of SA, tyrosine, phenylalanine and nitisinone. This enables comprehensive home sampling of monitoring parameters.
For the comparison of concentrations in dried blood and plasma a laboratory‐specific correction factor shall be used.
Expert consensus: 17 yes; 0 no; Abstention 2 – consensus.
3.3. Liver
3.3.1. Liver transplantation
Since treatment with nitisinone in combination with a protein reduced diet became available in the early 1990ies, the number of liver transplantations in HT1 patients has significantly declined and the age at transplantation increased. In countries with a screening programme for HT1, the number of transplantations has further decreased. However, some patients do still require liver transplantation. A liver transplantation shall be performed under the following circumstances:
Acute liver failure refractory to adequate nitisinone therapy (as indicated by suppression of SA production)
Strong suspicion of hepatocellular carcinoma (HCC) or signs for malignancy in liver parenchyma (inhomogeneous structure, nodules discovered by imaging and/or significant increase of alpha‐fetoprotein concentration in blood)
Chronic liver failure that does not respond to nitisinone treatment in combination with diet
Advanced liver cirrhosis
Alternatively, stereotactic radio‐frequency ablation has been suggested in cases where hepatic malignancies are suspected, 76 however there are no randomised controlled studies comparing this modality with liver transplantation.
The outcome of liver transplantation in patients suffering from HT1 is better than in those with other underlying conditions. 30 , 31 , 32 , 57 , 77
In exceptional cases, liver transplantation may also be considered in patients with poor adherence to therapy, but this has to be carefully discussed with the parents/patients as adherence to immunosuppression is essential after transplantation.
Preemptive transplantation is not recommended in patients diagnosed and treated pre‐symptomatically. However, regular monitoring is required in these patients.
After liver transplantation, SA levels remain slightly elevated in both blood and urine. This is due to SA production by the kidneys. These SA elevations do not result in liver or kidney damage. 21 , 34 , 78 Therefore, nitisinone‐treatment is not required after liver transplantation in HT1 patients (Recommendation 8).
Recommendation 8a.
Liver transplantation shall be performed in HT1 patients with therapy‐refractory liver failure, localised HCC, chronic liver failure despite adequate therapy with nitisinone and diet and/or liver cirrhosis.
If adherence to pharmacological and nutritional therapy is poor, the indication for liver transplantation shall be individually discussed (adherence to immunosuppressive therapy required after transplantation).
Expert consensus: 18 yes; 0 no; Abstention 1 – consensus.
Recommendation 8b
After liver transplantation, nitisinone therapy shall not be continued despite slight elevation of SA.
Expert consensus: 13 yes; 0 no; Abstention 6 – consensus by majority of participants.
3.3.2. Training and education of children, adolescents with HT1 and their parents/caregivers
Once diagnosed, treatment needs to be started immediately. As with other inborn errors of metabolism, parents and other caregivers of newborns need to be educated and trained first. 79 , 80 , 81 Later, children, adolescents and adult individuals with HT1 themselves need to be educated and supported. A multiprofessional team is required to care for individuals with HT1 and their families (paediatrician with expertise in the treatment of patients with HT1 as clinical lead, dietician, psychologist, social worker). The principles and goals of the therapy have to be communicated in a consistent way. 82 , 83
If the patient is diagnosed by newborn screening, a structured training and education procedure should be followed. 84 , 85 Due to the rarity of HT1 individual training and education are mostly offered. Standardised training may be supported by brochures and videos (Recommendation 9).
Recommendation 9a.
Structured training and education of children, adolescents and their parents and/or other individuals primarily caring for the patient are integral parts of treatment and shall be offered based on individual needs. Life‐long therapy and adherence shall be addressed.
Expert consensus: 18 yes; 0 no; Abstention 1 – consensus.
Recommendation 9b
Quality controlled age‐adapted training and education shall be offered repeatedly to patients with HT1 and their parents/caregivers as part of the long‐term management.
Expert consensus: 18 yes; 0 no; Abstention 1 – consensus.
Recommendation 9c
Regarding timing, training and education for children shall be offered at least before the start of primary school, at start of puberty and during adolescence (‘emerging adulthood’).
If possible, transition to a qualified metabolic centre for adults shall be encouraged.
Expert consensus: 18 yes; 0 no; Abstention 1 – consensus.
Physical and cognitive development of the patients requires continuous monitoring and adaptation of diet and pharmacological treatment. Hence, repeated training of patients and parents or caregivers is required. Apart from coping with the disease, parents/caregivers need support regarding therapy, expected outcome of disease management, social integration and disease‐specific educational needs. 86
Beginning at kindergarten‐age, children and adolescents need age‐specific support to better cope with age‐specific challenges. Training and education should be individualised based on cultural and family background. Self‐empowerment is an important aspect in older children and adolescents. A training programme for amino acid disorders has been suggested. 87 For the German‐speaking countries, a modular training programme has been developed for the amino acid disorder phenylketonuria (PKU). 80 , 88 This could serve as a blueprint for a modular training programme for patients with HT1s. (Recommendation 9).
3.3.3. Psychosocial support
HT1 is a severe chronic disorder, getting the unexpected positive result of newborn screening is a severe psychological burden for the parents/caregivers. They are often overwhelmed when they are faced with the diagnosis. This may result in shock‐like symptoms, anxiety and even guilt and may hamper their ability to cope with the challenges of HT1 treatment.
Therapeutic interventions including psychological support may reduce posttraumatic stress as observed in other metabolic diseases. 89 , 90 , 91 , 92
Having a child with a severe chronic disease may be a financial challenge, therefore advice on existing social laws and support may be helpful.
A psychosocial screening could reveal other burdens. These burdens may occur at any age. If such psychological challenges exist, specific support and if necessary, therapy should be offered (Recommendation 10).
Recommendation 10a.
Individualised psychological support and advice about social rights shall be offered to parents/caregivers when revealing the diagnosis and to children, adolescents and adults with HT1 during long‐term disease management.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
Recommendation 10b
A regular screening for an exceptional psychosocial burden should be offered to prevent poor adherence to therapy.
Expert consensus: 16 yes; 0 no; Abstention 3 – consensus.
3.4. Long‐term therapy and monitoring
Long‐term management includes regular history taking and physical examination. Dietary advice should be given twice per year, the frequency of visits in the outpatient clinics depends on metabolic stability.
3.4.1. Laboratory parameters
After start of treatment, laboratory parameters shall be monitored every week or every other week, after initial stabilisation every 3 months, in infancy and adolescence every 6 months, in adulthood every 6–12 months.
Regular laboratory parameters shall include: blood count, electrolytes including calcium and phosphate, liver function, total protein, alpha‐fetoprotein, urea, creatinine, cystatin C, coagulation parameters, vitamin D‐state, parathyroid hormone, ferritin. Special metabolic laboratory parameters include: amino acids in plasma and urine, SA in dried blood and urine, nitisinone concentration in (dried) blood.
While tyrosine concentrations in plasma are quite stable, phenylalanine levels show fluctuations throughout the day. 93 Reproducibility of the levels of both amino acids is best if blood is drawn before breakfast, 94 however this may be a logistical challenge and catabolism may alter some of the amino acids. Elevation of branched chain amino acids (valine, leucine and isoleucine) may indicate catabolism (Recommendation 11). 95
Recommendation 11a.
After the start of treatment, frequent follow‐up visits shall take place in a metabolic centre.
Frequency of life‐long monitoring visits shall be tailored according to individual needs. Monitoring shall take place every 3 months up to the age of 1 year, in childhood and adolescence every 6 months, in adults every 6–12 months based on metabolic stability.
Expert consensus: 18 yes; 0 no; Abstention 1 – consensus.
Recommendation 11b
Monitoring of therapy shall be performed by a multiprofessional team (paediatrician and dietician with expertise in inborn errors of metabolism, psychologist, social worker).
Expert consensus: 19 yes; 0 no; Abstention 0– strong consensus.
Recommendation 11c
Physical examination, including weight and height and developmental state shall be accompanied by monitoring of parameters of organ function and metabolic tests like amino acids in plasma (tyrosine, phenylalanine, branched chain amino acids), SA in blood and/or urine, nitisinone in (dried) blood.
Expert consensus: 18 yes; 0 no; Abstention 1– consensus.
Recommendation 11d
Home blood sampling should be done in‐between outpatient visits using dried blood. A correction factor shall be used when comparing plasma and dried blood.
Expert consensus: 18 yes; 0 no; Abstention 1 – consensus.
Monitoring of SA, tyrosine, phenylalanine and nitisinone concentrations is possible by home sampling using dried blood. 73 , 75 , 96 These measurements shall be performed once in‐between visits of outpatient clinics.
3.5. Hepatocellular carcinoma (HCC) in HT1
Alpha‐Fetoprotein (AFP) is regarded as the best biomarker for HCC in terms of specificity and sensitivity, we recommend measuring this parameter every 6–12 months. In the newborn period as well as at diagnosis of HT1 before start of treatment, this parameter is usually elevated. After the start of nitisinone‐treatment, AFP may remain elevated for some time (several months) which makes the diagnosis of HCC difficult during this period. However, a secondary rise in AFP is suspicious for HCC. 9 , 30 , 31 , 32 , 97 In exceptional cases, initially elevated AFP levels do not normalise. 98
Abdominal ultrasound is recommended every 6–12 months to assess liver and kidney morphology. The sensitivity of ultrasound imaging to detect HCC is 60%–80%, specificity 45%–94%, depending on the experience of the operator to perform ultrasound and the quality of the technical equipment. 98 If HCC is suspected, an MRI of the liver is indicated, sensitivity to detect HCC is 89%–100%, even small lesions can be detected. Specificity is 95%. 5 , 30 , 31 , 32 , 99 , 100 , 101 , 102
Liver biopsy is discouraged to avoid dissemination of tumour cells in the body (Recommendation 12). 103
Recommendation 12a.
AFP levels in blood shall be regularly determined during outpatient clinic visits as a biomarker for HCC. After initiation of nitisinone treatment, AFP may remain elevated for some time. Absence of normalisation or a secondary rise shall prompt imaging.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
Recommendation 12b
Abdominal ultrasound shall be performed as the primary imaging technique if sufficient expertise exists, as this technique can be used in infants and children without sedation/anaesthesia. However, MRI has a higher sensitivity and specificity. Due to the risk of disseminating tumour cells, liver biopsy shall not be performed.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
3.6. Ophthalmological examination in HT1
Ophthalmological symptoms are related to highly elevated tyrosine levels in blood (mostly >800 μM) on nitisinone treatment with insufficient protein restriction. 8 , 34 Routine ophthalmological follow‐up exams are not recommended. In case of symptoms like conjunctivitis, pain in the eyes, corneal clouding 104 and visual disturbances, eye examinations shall be performed. Poor dietary control may be another indication for consulting an ophthalmologist (Recommendation 13).
Recommendation 13.
There is no indication for routine follow‐up ophthalmological examinations in HT1‐patients and should not be routinely performed. In case of eye symptoms or poor dietary control ophthalmological examinations shall be performed.
Expert consensus: 17 yes; 0 no; Abstention 2 – consensus.
3.7. Cardiac examination in HT1
Controversy exists regarding the occurrence of clinically relevant cardiac symptoms in HT1. 8 , 34 , 105 If cardiac anomalies were observed, they were quickly reversible upon nitisinone treatment 106 , 107 . Routine monitoring of cardiac function is not recommended, tests shall be performed when clinical symptoms occur (Recommendation 14).
Recommendation 14.
The occurrence of cardiac symptoms directly related to HT1 is controversially discussed. If observed, they are of temporary nature or clinically irrelevant. Therefore, cardiac examinations shall not be performed routinely. If clinical symptoms are observed cardiac examinations shall be initiated.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
3.8. Neurocognitive examination
Cross‐sectional studies in HT1 patients have shown deficiencies of executive functions like working memory, cognitive flexibility, reduced IQ‐scores 25 , 26 , 29 , 108 behaviour and attention 28 as well as motor function, 108 even under treatment with nitisinone. Some studies have found progression of neuropsychological disabilities. 25 , 108 Therefore, some authors recommend examining cognitive function of HT1 patients regularly, starting at school age in order to specifically support school performance (Recommendation 15). Low phenylalanine levels were associated with neurocognitive deficits. 25 , 108 Other authors could not find an association with phenylalanine or tyrosine concentrations. 28
Recommendation 15.
The neurocognitive development of children and adolescents shall be monitored when entering school and at regular intervals thereafter, to secure adequate schooling and support when learning difficulties occur.
Expert consensus: 17 yes; 0 no; Abstention 2 – consensus.
Several hypotheses regarding the pathophysiology of neurocognitive deficits in HT1 patients have been proposed) 25 , 29 , 108 :
The disease itself with production of toxic side products and liver dysfunction (natural course of the disease)
Treatment with nitisinone
High tyrosine concentrations in blood interfering with the uptake of amino acids by the brain and leading to reduced intracerebral synthesis of proteins and neurotransmitters
Low phenylalanine concentrations in plasma and brain 109 , 110
Low protein intake and inadequate dietary adherence
3.9. Vaccination in HT1
There are no restrictions regarding vaccinations recommended by the national regulatory bodies (Recommendation 16).
Recommendation 16.
All vaccinations recommended by the local regulatory bodies shall be given according to the national vaccination scheme.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
4. ADULTS WITH HT1
4.1. Long‐term outpatient care
As in other inborn errors of amino acid metabolism, therapy for life including medication and diet is recommended. Adherence to therapy seems to be poorer in adolescents and adults compared to children and infants. 111 , 112 This has previously been observed in other disorders of amino acid metabolism, for instance in phenylketonuria (PKU). 113 On the other hand, protein tolerance seems to increase with age. 70
Ideally, a transition process should be initiated during adolescence (‘emerging adults’) with a structured transitioning to adult medicine. However, not all centers of adult medicine offer care for patients with HT1. Therefore, even adults with HT1 are often treated by paediatricians.
4.2. Pregnancy in HT1
As treated females with HT1 reach adulthood, pregnancies of HT1‐patients are an issue. Women with HT1 are fertile. The manufacturers of nitisinone discourage patients from using nitisinone during pregnancy due to lack of experience. While teratogenicity of nitisinone has been observed in animal models, several successful pregnancies of females with HT1 on nitisinone treatment have been reported in the literature. 114 , 115 , 116 , 117 , 118 Nitisinone could be detected in cord blood, SA was below detection limit. In one case, the foetus was affected by HT1 as well with a good outcome for mother and child. 115
Breast feeding is possible in a number of inborn errors of metabolism, not only in PKU. 119 It is discouraged by the manufacturers of nitisinone as there are no data regarding drug concentration in breast milk. Single cases of breast feeding have been reported without sequelae. If the mother strongly wishes to breast feed this may be discussed on an individual basis (Recommendation 17).
Recommendation 17a.
Pregnancy can be considered in females with HT1, though clinical experience is limited. Nitisinone shall be continued during pregnancy. Metabolic control should be stable preconceptionally.
Expert consensus: 17 yes; 0 no; Abstention 2 – consensus.
Recommendation 17b
Breast feeding on nitisinone treatment of the mother with HT1 (‘maternal’ tyrosinaemia) has been described in a few case reports with successful outcome. There is not enough scientific evidence to make a general recommendation. Therefore, breast feeding in HT1‐mothers shall not be encouraged, however this shall be discussed on an individual basis.
Expert consensus: 16 yes; 0 no; Abstention 3 – consensus.
4.3. Psychosocial counselling in HT1
HT1 requires a challenging, life‐long therapy, especially at the end of the second decade and in the third decade of live coinciding with classical developmental steps (leaving the parents' home, starting an independent life, finishing vocational training, starting a professional career, partnership, own family). During this period, regular counselling of (‘emerging’) adults with HT1 is necessary and helpful. Integration of therapy in their own daily life‐routine and considering the outcome of HT1 are common issues.
External support is required for those adult patients with HT1 suffering from (progressive) neurocognitive deficits who cannot meet therapeutic requirements. Individually tailored social support should be instituted. This concerns vocational integration, rehabilitation and support of daily life. Similarly, patients with psychological issues may not be able to follow a demanding therapeutic regimen (medication and diet). If problems with adherence are suspected, a psychological screening should be performed and individual therapeutic support offered (Recommendation 18).
Recommendation 18a.
Adults with HT1 shall be offered individually tailored psychosocial counselling to enable integration of the metabolic disorder in vocational and private life.
Expert consensus: 18 yes; 0 no; Abstention 1 – consensus.
Recommendation 18b
Psychosocial screening shall be performed if a specific burden or problems with adherence are suspected.
Expert consensus: 19 yes; 0 no; Abstention 0 – strong consensus.
5. CONCLUSION
Recommendations for dealing with individuals with HT1 have been previously published. 34 , 35 , 36 Only the study by Chinsky et al. followed a structured consensus procedure. Our recommendations, which originally focused on the German‐speaking scientific community, are based on a generic, standardised and structured consensus approach within a multi‐professional group. The statements in the present guideline are in line with those of the US and Canadian consensus groups. 34 However, our data are more robust because we were able to gain experience and collect data over a longer period of time. Both groups recommend neonatal screening with succinylacetone as a screening parameter; treatment includes nitisinone and diet. We point out that tyrosine intake calculation is no longer required. Categorisation of foods can facilitate diet adherence without compromising metabolic control. Between outpatient clinic visits, home monitoring of dried blood samples is possible after discussion within our group. More experience is available regarding the pregnancy of women suffering from HT1. We are therefore more confident that pregnancies are possible for women with HT1 without harming mother or child.
We hope that our current recommendations are useful to physicians and dieticians even beyond the German speaking countries.
AUTHOR CONTRIBUTIONS
Anibh M. Das (guarantor) and Karin Lange conceptualised and organised the consensus process, Karin Lange acted as the independent monitor. All authors participated in the consensus discussions, drafting, critical review and revision of the manuscript before submission.
FUNDING INFORMATION
No external funding was provided.
CONFLICT OF INTEREST STATEMENT
Anibh M. Das, Diana Ballhausen, Dorothea Haas, Johannes Häberle, Tobias Hagedorn, Cecilia Janson‐Mutsaerts, Nils Janzen, Johannes Sander, Peter Freisinger, Daniela Karall, Uta Meyer, Eberhard Mönch, Susanne Morlot, Stefanie Rosenbaum‐Fabian, Sabine Scholl‐Bürgi, Stephan vom Dahl, Natalie Weinhold, Jiri Zeman, and Karin Lange declare that they have no conflict of interest.
ETHICS STATEMENT
As no patient data were involved an ethics approval was not required.
INFORMED CONSENT STATEMENT
This article does not contain any studies with human or animal subjects performed by any of the authors.
PATIENT CONSENT STATEMENT
No individual patient data were used in this study.
ACKNOWLEDGEMENTS
We did not get financial support for our consensus meetings and writing up the results. The consensus was approved by the members of the German working group for inborn errors of metabolism (Arbeitsgemeinschaft für Pädiatrische Stoffwechselstörungen, APS, now Gesellschaft für angeborene Stoffwechselerkrankungen, GfAS) and the working group for inborn errors of metabolism in internal medicine (Arbeitsgemeinschaft Stoffwechselerkrankungen in der Inneren Medizin, ASIM).
Das AM, Ballhausen D, Haas D, et al. Diagnosis, treatment, management and monitoring of patients with tyrosinaemia type 1: Consensus group recommendations from the German‐speaking countries. J Inherit Metab Dis. 2025;48(1):e12824. doi: 10.1002/jimd.12824
†Eberhard Mönch is recently deceased.
Communicating Editor: Jirair Krikor Bedoyan MD, PhD
DATA AVAILABILITY STATEMENT
Data supporting the results of this manuscript can be requested from Anibh M. Das or Karin Lange.
REFERENCES
- 1. Angileri F, Bergeron A, Morrow G, et al. Geographical and ethnic distribution of mutations of the fumarylacetoacetate hydrolase gene in hereditary tyrosinemia type 1. JIMD Rep. 2015;19:43‐58. doi: 10.1007/8904_2014_363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arranz JA, Piñol F, Kozak L, et al. Splicing mutations, mainly IVS6‐1(G>T), account for 70% of fumarylacetoacetate hydrolase (FAH) gene alterations, including 7 novel mutations, in a survey of 29 tyrosinemia type I patients. Hum Mutat. 2002;20(3):180‐188. doi: 10.1002/humu.10084 [DOI] [PubMed] [Google Scholar]
- 3. Poudrier J, St‐Louis M, Lettre F, et al. Frequency of the IVS12 + 5G‐‐>A splice mutation of the fumarylacetoacetate hydrolase gene in carriers of hereditary tyrosinaemia in the French Canadian population of Saguenay‐Lac‐St‐Jean. Prenat Diagn. 1996;16(1):59‐64. doi: [DOI] [PubMed] [Google Scholar]
- 4. Russo PA, Mitchell GA, Tanguay RM. Tyrosinemia: a review. Pediatr Dev Pathol. 2001;4(3):212‐221. doi: 10.1007/s100240010146 [DOI] [PubMed] [Google Scholar]
- 5. Halac U, Dubois J, Mitchell GA. The liver in tyrosinemia type I: clinical management and course in Quebec. Adv Exp Med Biol. 2017;959:75‐83. doi: 10.1007/978-3-319-55780-9_6 [DOI] [PubMed] [Google Scholar]
- 6. Maiorana A, Dionisi‐Vici C. NTBC and correction of renal dysfunction. Adv Exp Med Biol. 2017;959:93‐100. doi: 10.1007/978-3-319-55780-9_8 [DOI] [PubMed] [Google Scholar]
- 7. Maiorana A, Malamisura M, Emma F, et al. Early effect of NTBC on renal tubular dysfunction in hereditary tyrosinemia type 1. Mol Genet Metab. 2014;113(3):188‐193. doi: 10.1016/j.ymgme.2014.07.021 [DOI] [PubMed] [Google Scholar]
- 8. Mayorandan S, Meyer U, Gokcay G, et al. Cross‐sectional study of 168 patients with hepatorenal tyrosinaemia and implications for clinical practice. Orphanet J Rare Dis. 2014;9:107. doi: 10.1186/s13023-014-0107-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koelink CJ, van Hasselt P, van der Ploeg A, et al. Tyrosinemia type I treated by NTBC: how does AFP predict liver cancer? Mol Genet Metab. 2006;89(4):310‐315. doi: 10.1016/j.ymgme.2006.07.009 [DOI] [PubMed] [Google Scholar]
- 10. Paradis K, Weber A, Seidman EG, et al. Liver transplantation for hereditary tyrosinemia: the Quebec experience. Am J Hum Genet. 1990;47(2):338‐342. [PMC free article] [PubMed] [Google Scholar]
- 11. Schady DA, Roy A, Finegold MJ. Liver tumors in children with metabolic disorders. Transl Pediatr. 2015;4(4):290‐303. doi: 10.3978/j.issn.2224-4336.2015.10.08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Spronsen FJ, Bijleveld CM, van Maldegem BT, Wijburg FA. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2‐(2 nitro‐4‐3 trifluoro‐ methylbenzoyl)‐1, 3‐cyclohexanedione treatment. J Pediatr Gastroenterol Nutr. 2005;40(1):90‐93. doi: 10.1097/00005176-200501000-00017 [DOI] [PubMed] [Google Scholar]
- 13. Demers SI, Russo P, Lettre F, Tanguay RM. Frequent mutation reversion inversely correlates with clinical severity in a genetic liver disease, hereditary tyrosinemia. Hum Pathol. 2003;34(12):1313‐1320. doi: 10.1016/s0046-8177(03)00406-4 [DOI] [PubMed] [Google Scholar]
- 14. Morrow G, Angileri F, Tanguay RM. Molecular aspects of the FAH mutations involved in HT1 disease. Adv Exp Med Biol. 2017;959:25‐48. doi: 10.1007/978-3-319-55780-9_3 [DOI] [PubMed] [Google Scholar]
- 15. Morrow G, Tanguay RM. Biochemical and clinical aspects of hereditary tyrosinemia type 1. Adv Exp Med Biol. 2017;959:9‐21. doi: 10.1007/978-3-319-55780-9_2 [DOI] [PubMed] [Google Scholar]
- 16. Tanguay RM, Valet JP, Lescault A, et al. Different molecular basis for fumarylacetoacetate hydrolase deficiency in the two clinical forms of hereditary tyrosinemia (type I). Am J Hum Genet. 1990;47(2):308‐316. [PMC free article] [PubMed] [Google Scholar]
- 17. van Spronsen FJ, Thomasse Y, Smit GP, et al. Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology. 1994;20(5):1187‐1191. [PubMed] [Google Scholar]
- 18. Das AM, Mayorandan S, Janzen N. Diagnosing hepatorenal tyrosinaemia in Europe: newborn mass screening versus selective screening. Adv Exp Med Biol. 2017;959:125‐132. doi: 10.1007/978-3-319-55780-9_11 [DOI] [PubMed] [Google Scholar]
- 19. Lock EA. From weed killer to wonder drug. Adv Exp Med Biol. 2017;959:175‐185. doi: 10.1007/978-3-319-55780-9_16 [DOI] [PubMed] [Google Scholar]
- 20. Holme E, Lindstedt S. Diagnosis and management of tyrosinemia type I. Curr Opin Pediatr. 1995;7(6):726‐732. doi: 10.1097/00008480-199512000-00017 [DOI] [PubMed] [Google Scholar]
- 21. Lindstedt S, Holme E, Lock EA, Hjalmarson O, Strandvik B. Treatment of hereditary tyrosinaemia type I by inhibition of 4‐hydroxyphenylpyruvate dioxygenase. Lancet. 1992;340(8823):813‐817. doi: 10.1016/0140-6736(92)92685-9 [DOI] [PubMed] [Google Scholar]
- 22. van Spronsen FJ, van Rijn M, Meyer U, Das AM. Dietary considerations in tyrosinemia type I. Adv Exp Med Biol. 2017;959:197‐204. doi: 10.1007/978-3-319-55780-9_18 [DOI] [PubMed] [Google Scholar]
- 23. Bartlett DC, Lloyd C, McKiernan PJ, Newsome PN. Early nitisinone treatment reduces the need for liver transplantation in children with tyrosinaemia type 1 and improves post‐transplant renal function. J Inherit Metab Dis. 2014;37(5):745‐752. doi: 10.1007/s10545-014-9683-x [DOI] [PubMed] [Google Scholar]
- 24. McKiernan PJ, Preece MA, Chakrapani A. Outcome of children with hereditary tyrosinaemia following newborn screening. Arch Dis Child. 2015;100(8):738‐741. doi: 10.1136/archdischild-2014-306886 [DOI] [PubMed] [Google Scholar]
- 25. Bendadi F, de Koning TJ, Visser G, et al. Impaired cognitive functioning in patients with tyrosinemia type I receiving nitisinone. J Pediatr. 2014;164(2):398‐401. doi: 10.1016/j.jpeds.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 26. de Laet C, Munoz VT, Jaeken J, et al. Neuropsychological outcome of NTBC‐treated patients with tyrosinaemia type 1. Dev Med Child Neurol. 2011;53(10):962‐964. doi: 10.1111/j.1469-8749.2011.04048.x [DOI] [PubMed] [Google Scholar]
- 27. Masurel‐Paulet A, Poggi‐Bach J, Rolland MO, et al. NTBC treatment in tyrosinaemia type I: long‐term outcome in French patients. J Inherit Metab Dis. 2008;31(1):81‐87. doi: 10.1007/s10545-008-0793-1 [DOI] [PubMed] [Google Scholar]
- 28. Pohorecka M, Biernacka M, Jakubowska‐Winecka A, et al. Behavioral and intellectual functioning in patients with tyrosinemia type I. Pediatr Endocrinol Diabetes Metab. 2012;18(3):96‐100. [PubMed] [Google Scholar]
- 29. Thimm E, Richter‐Werkle R, Kamp G, et al. Neurocognitive outcome in patients with hypertyrosinemia type I after long‐term treatment with NTBC. J Inherit Metab Dis. 2012;35(2):263‐268. doi: 10.1007/s10545-011-9394-5 [DOI] [PubMed] [Google Scholar]
- 30. van Ginkel WG, Jahja R, Huijbregts SCJ, van Spronsen FJ. Neurological and neuropsychological problems in tyrosinemia type I patients. Adv Exp Med Biol. 2017a;959:111‐122. doi: 10.1007/978-3-319-55780-9_10 [DOI] [PubMed] [Google Scholar]
- 31. van Ginkel WG, Pennings JP, van Spronsen FJ. Liver cancer in tyrosinemia type 1. Adv Exp Med Biol. 2017b;959:101‐109. doi: 10.1007/978-3-319-55780-9_9 [DOI] [PubMed] [Google Scholar]
- 32. van Ginkel WG, van Vliet D, Burgerhof JGM, et al. Presumptive brain influx of large neutral amino acids and the effect of phenylalanine supplementation in patients with tyrosinemia type 1. PLoS One. 2017c;12(9):e0185342. doi: 10.1371/journal.pone.0185342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Ginkel WG, Jahja R, Huijbregts SC, et al. Neurocognitive outcome in tyrosinemia type 1 patients compared to healthy controls. Orphanet J Rare Dis. 2016;11(1):87. doi: 10.1186/s13023-016-0472-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chinsky JM, Singh R, Ficicioglu C, et al. Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet Med. 2017;19(12):1380‐1395. doi: 10.1038/gim.2017.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de Laet C, Dionisi‐Vici C, Leonard JV, et al. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis. 2013;8. doi: 10.1186/1750-1172-8-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schiff M, Broue P, Chabrol B, et al. Heterogeneity of follow‐up procedures in French and Belgian patients with treated hereditary tyrosinemia type 1: results of a questionnaire and proposed guidelines. J Inherit Metab Dis. 2012;35(5):823‐829. doi: 10.1007/s10545-011-9429-y [DOI] [PubMed] [Google Scholar]
- 37. de Jesús VR, Adam BW, Mandel D, Cuthbert CD, Matern D. Succinylacetone as primary marker to detect tyrosinemia type I in newborns and its measurement by newborn screening programs. Mol Genet Metab. 2014;113(1–2):67‐75. doi: 10.1016/j.ymgme.2014.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. la Marca G, Malvagia S, Pasquini E, et al. Newborn screening for tyrosinemia type I: further evidence that Succinylacetone determination on blood spot is essential. JIMD Rep. 2011;1:107‐109. doi: 10.1007/8904_2011_24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morrissey MA, Sunny S, Fahim A, Lubowski C, Caggana M. Newborn screening for Tyr‐I: two years' experience of the New York State program. Mol Genet Metab. 2011;103(2):191‐192. doi: 10.1016/j.ymgme.2011.02.017 [DOI] [PubMed] [Google Scholar]
- 40. Sander J, Janzen N, Peter M, et al. Newborn screening for hepatorenal tyrosinemia: tandem mass spectrometric quantification of succinylacetone. Clin Chem. 2006;52(3):482‐487. doi: 10.1373/clinchem.2005.059790 [DOI] [PubMed] [Google Scholar]
- 41. Turgeon C, Magera MJ, Allard P, et al. Combined newborn screening for succinylacetone, amino acids, and acylcarnitines in dried blood spots. Clin Chem. 2008;54(4):657‐664. doi: 10.1373/clinchem.2007.101949 [DOI] [PubMed] [Google Scholar]
- 42. Weigel JF, Janzen N, Pfäffle RW, Thiery J, Kiess W, Ceglarek U. Tandem mass spectrometric determination of succinylacetone in dried blood spots enables presymptomatic detection in a case of hepatorenal tyrosinaemia. J Inherit Metab Dis. 2007;30(4):610. doi: 10.1007/s10545-007-0608-9 [DOI] [PubMed] [Google Scholar]
- 43. Allard P, Grenier A, Korson MS, Zytkovicz TH. Newborn screening for hepatorenal tyrosinemia by tandem mass spectrometry: analysis of succinylacetone extracted from dried blood spots. Clin Biochem. 2004;37(11):1010‐1015. doi: 10.1016/j.clinbiochem.2004.07.006 [DOI] [PubMed] [Google Scholar]
- 44. Lund AM, Hougaard DM, Simonsen H, et al. Biochemical screening of 504,049 newborns in Denmark, The Faroe Islands and Greenland‐‐experience and development of a routine program for expanded newborn screening. Mol Genet Metab. 2012;107(3):281‐293. doi: 10.1016/j.ymgme.2012.06.006 [DOI] [PubMed] [Google Scholar]
- 45. Metz TF, Mechtler TP, Merk M, et al. Evaluation of a novel, commercially available mass spectrometry kit for newborn screening including succinylacetone without hydrazine. Clin Chim Acta. 2012;413(15–16):1259‐1264. doi: 10.1016/j.cca.2012.04.007 [DOI] [PubMed] [Google Scholar]
- 46. Yang H, Al‐Hertani W, Cyr D, et al. Hypersuccinylacetonaemia and normal liver function in maleylacetoacetate isomerase deficiency. J Med Genet. 2017a;54(4):241‐247. doi: 10.1136/jmedgenet-2016-104289 [DOI] [PubMed] [Google Scholar]
- 47. Yang H, Rossignol F, Cyr D, et al. Mildly elevated succinylacetone and normal liver function in compound heterozygotes with pathogenic and pseudodeficient FAH alleles. Mol Genet Metab Rep. 2017b;14:55‐58. doi: 10.1016/j.ymgmr.2017.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Spiekerkoetter U, Krude H. Target diseases for neonatal screening in Germany. Dtsch Arztebl Int. 2022;119(17):306‐316. doi: 10.3238/arztebl.m2022.0075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bundesministerium für Gesundheit . Beschluss über eine Änderung der Richtlinie des Bundesausschusses der Ärzte und Krankenkassen über die Früherkennung von Krankheiten bei Kindern bis zur Vollendung des 6. Lebensjahres (Kinder‐Richtlinie) zur Einführung des erweiterten Neugeborenen‐Screenings vom. 2016. https://www.g‐ba.de/downloads/62‐492‐1333/RL_Kinder_2017‐11‐24_iK‐2017‐01‐28.pdf accessed 1.8,2924
- 50. de Braekeleer M, Lamarre V, Scriver CR, Larochelle J, Bouchard G. Fertility in couples heterozygous for the tyrosinemia gene in Saguenay Lac‐St‐Jean. Genet Couns. 1990;1(3–4):259‐264. [PubMed] [Google Scholar]
- 51. de Braekeleer M, Larochelle J. Genetic epidemiology of hereditary tyrosinemia in Quebec and in Saguenay‐Lac‐St‐Jean. Am J Hum Genet. 1990;47(2):302‐307. [PMC free article] [PubMed] [Google Scholar]
- 52. Rafati M, Mohamadhashem F, Hoseini A, Ramandi SD, Ghaffari SR. Prenatal diagnosis of tyrosinemia type 1 using next generation sequencing. Fetal Pediatr Pathol. 2016;35(4):282‐285. doi: 10.3109/15513815.2016.1167149 [DOI] [PubMed] [Google Scholar]
- 53. Couce ML, Dalmau J, del Toro M, Pintos‐Morell G, Aldámiz‐Echevarría L. Spanish working group on tyrosinemia type 1. Tyrosinemia type 1 in Spain: mutational analysis, treatment and long‐term outcome. Pediatr Int. 2011;53(6):985‐989. doi: 10.1111/j.1442-200X.2011.03427.x [DOI] [PubMed] [Google Scholar]
- 54. Cassiman D, Zeevaert R, Holme E, Kvittingen EA, Jaeken J. A novel mutation causing mild, atypical fumarylacetoacetase deficiency (Tyrosinemia type I): a case report. Orphanet J Rare Dis. 2009;4:28. doi: 10.1186/1750-1172-4-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Morrow G, Dreumont N, Bourrelle‐Langlois M, Roy V, Tanguay RM. Presence of three mutations in the fumarylacetoacetate hydrolase gene in a patient with atypical symptoms of hereditary tyrosinemia type I. Mol Genet Metab. 2019;127(1):58‐63. doi: 10.1016/j.ymgme.2019.01.019 [DOI] [PubMed] [Google Scholar]
- 56. Spiekerkoetter U, Couce ML, Das AM, et al. Long‐term safety and outcomes in hereditary tyrosinaemia type 1 with nitisinone treatment: a 15‐year non‐interventional, multicentre study. Lancet Diabetes Endocrinol. 2021;9(7):427‐435. doi: 10.1016/S2213-8587(21)00092-9 [DOI] [PubMed] [Google Scholar]
- 57. Alvarez F, Mitchell GA. Tyrosinemia and liver transplantation: experience at CHU Sainte‐Justine. Adv Exp Med Biol. 2017;959:67‐73. doi: 10.1007/978-3-319-55780-9_5 [DOI] [PubMed] [Google Scholar]
- 58. Larochelle J, Alvarez F, Bussières JF, et al. Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Québec. Mol Genet Metab. 2012;107(1–2):49‐54. doi: 10.1016/j.ymgme.2012.05.022 [DOI] [PubMed] [Google Scholar]
- 59. Santra S, Baumann U. Experience of nitisinone for the pharmacological treatment of hereditary tyrosinaemia type 1. Expert Opin Pharmacother. 2008;9(7):1229‐1236. doi: 10.1517/14656566.9.7.1229 [DOI] [PubMed] [Google Scholar]
- 60. Santra S, Preece MA, Hulton SA, McKiernan PJ. Renal tubular function in children with tyrosinaemia type I treated with nitisinone. J Inherit Metab Dis. 2008;31(3):399‐402. doi: 10.1007/s10545-008-0817-x [DOI] [PubMed] [Google Scholar]
- 61. El‐Karaksy H, Fahmy M, El‐Raziky M, et al. Hereditary tyrosinemia type 1 from a single center in Egypt: clinical study of 22 cases. World J Pediatr. 2011;7(3):224‐231. doi: 10.1007/s12519-011-0287-3 [DOI] [PubMed] [Google Scholar]
- 62. Hall MG, Wilks MF, Provan WM, Eksborg S, Lumholtz B. Pharmacokinetics and pharmacodynamics of NTBC (2‐(2‐nitro‐4‐fluoromethylbenzoyl)‐1,3‐cyclohexanedione) and mesotrione, inhibitors of 4‐hydroxyphenyl pyruvate dioxygenase (HPPD) following a single dose to healthy male volunteers. Br J Clin Pharmacol. 2001;52(2):169‐177. doi: 10.1046/j.0306-5251.2001.01421.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schlune A, Thimm E, Herebian D, Spiekerkoetter U. Single dose NTBC‐treatment of hereditary tyrosinemia type I. J Inherit Metab Dis. 2012;35(5):831‐836. doi: 10.1007/s10545-012-9450-9 [DOI] [PubMed] [Google Scholar]
- 64. Barchanska H, Rola R, Szczepankiewicz W, Mrachacz M. LC‐MS/MS study of the degradation processes of nitisinone and its by‐products. J Pharm Biomed Anal. 2019;171:15‐21. doi: 10.1016/j.jpba.2019.03.046 [DOI] [PubMed] [Google Scholar]
- 65. Chakrapani A, Gissen P, McKiernan P. Disorders of tyrosine metabolism. In: Saudubray J‐M, van den Berghe G, Walter JH, eds. Inborn Metabolic Diseases, Chapter 18. 5th ed. Springer; 2012:275‐276. [Google Scholar]
- 66. DACH Deutsche Gesellschaft für Ernährung . Österreichische Gesellschaft für Ernährung, Schweizerische Gesellschaft für Ernährungsforschung, Schweizerische Vereinigung für Ernährung (Hrsg.): Referenzwerte für die Nährstoffzufuhr. Bonn, 2. Auflage, 5. aktualisierte Ausgabe. 2019.
- 67. Ney DM, Stroup BM, Clayton MK, et al. Glycomacropeptide for nutritional management of phenylketonuria: a randomized, controlled, crossover trial. Am J Clin Nutr. 2016;104(2):334‐345. doi: 10.3945/ajcn.116.135293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Daly A, Evans S, Pinto A, Ashmore C, MacDonald A. Casein glycomacropeptide: an alternative protein substitute in tyrosinemia type I. Nutrients. 2021;13(9):3224. doi: 10.3390/nu13093224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bärhold F, Meyer U, Neugebauer AK, et al. Hepatorenal tyrosinaemia: impact of a simplified diet on metabolic control and clinical outcome. Nutrients. 2020;13(1):134. doi: 10.3390/nu13010134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yilmaz O, Daly A, Pinto A, et al. Natural protein tolerance and metabolic control in patients with hereditary tyrosinaemia type 1. Nutrients. 2020;12(4):1148. doi: 10.3390/nu12041148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Adam BW, Hall EM, Meredith NK, et al. Performance of succinylacetone assays and their associated proficiency testing outcomes. Clin Biochem. 2012;45(18):1658‐1663. doi: 10.1016/j.clinbiochem.2012.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Laeremans H, Turner C, Andersson T, et al. Inter‐laboratory analytical improvement of succinylacetone and nitisinone quantification from dried blood spot samples. JIMD Rep. 2020;53(1):90‐102. doi: 10.1002/jmd2.12112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sander J, Janzen N, Terhardt M, et al. Monitoring tyrosinaemia type I: blood spot test for nitisinone (NTBC). Clin Chim Acta. 2011;412(1–2):134‐138. doi: 10.1016/j.cca.2010.09.027 [DOI] [PubMed] [Google Scholar]
- 74. Sundberg J, Wibrand F, Lund AM, Christensen M. Simultaneous quantification of succinylacetone and nitisinone for therapeutic drug monitoring in the treatment of Tyrosinemia type 1. J Chromatogr B Analyt Technol Biomed Life Sci. 2018;1072:259‐266. doi: 10.1016/j.jchromb.2017.11.031 [DOI] [PubMed] [Google Scholar]
- 75. la Marca G, Malvagia S, Materazzi S, et al. LC‐MS/MS method for simultaneous determination on a dried blood spot of multiple analytes relevant for treatment monitoring in patients with tyrosinemia type I. Anal Chem. 2012;84(2):1184‐1188. doi: 10.1021/ac202695h [DOI] [PubMed] [Google Scholar]
- 76. Karall D, Scholl‐Bürgi S, Widmann G, et al. Stereotactic radiofrequency ablation for liver tumors in inherited metabolic disorders. Cardiovasc Intervent Radiol. 2014;37(4):1027‐1033. doi: 10.1007/s00270-013-0756-2 [DOI] [PubMed] [Google Scholar]
- 77. Arnon R, Annunziato R, Miloh T, et al. Liver transplantation for hereditary tyrosinemia type I: analysis of the UNOS database. Pediatr Transplant. 2011;15(4):400‐405. doi: 10.1111/j.1399-3046.2011.01497.x [DOI] [PubMed] [Google Scholar]
- 78. Pierik LJ, van Spronsen FJ, Bijleveld CM, van Dael CM. Renal function in tyrosinaemia type I after liver transplantation: a long‐term follow‐up. J Inherit Metab Dis. 2005;28(6):871‐876. doi: 10.1007/s10545-005-0059-0 [DOI] [PubMed] [Google Scholar]
- 79. Ernst G, Lange K, Szczepanski R, Staab D, Ehrich J, Zinken K. How to train families to cope with lifelong health problems? J Pediatr. 2016;170:349‐350.e502. doi: 10.1016/j.jpeds.2015.11.057 [DOI] [PubMed] [Google Scholar]
- 80. Meyer U, Das A, Ernst G, Lange K, Mit PKU gut leben . Schulungsprogramm und Curriculum für Eltern und betroffene Jugendliche. 2. überarbeitete Auflage, Pabst, Lengerich. 2017.
- 81. Phelan H, Lange K, Cengiz E, et al. ISPAD Clinical Practice Consensus Guidelines 2018: diabetes education in children and adolescents. Pediatr Diabetes. 2018;19(Suppl 27):75‐83. doi: 10.1111/pedi.12762 [DOI] [PubMed] [Google Scholar]
- 82. Lange K, Kordonouri O. Setting the right course at type 1 diabetes diagnosis. Lancet Child Adolesc Health. 2019;3(3):138‐139. doi: 10.1016/S2352-4642(19)30031-8 [DOI] [PubMed] [Google Scholar]
- 83. Skinner TC, Lange KS, Hoey H, et al. Targets and teamwork: understanding differences in pediatric diabetes centers treatment outcomes. Pediatr Diabetes. 2018;19(3):559‐565. doi: 10.1111/pedi.12606 [DOI] [PubMed] [Google Scholar]
- 84. Gramer G, Haege G, Glahn EM, Hoffmann GF, Lindner M, Burgard P. Living with an inborn error of metabolism detected by newborn screening‐parents' perspectives on child development and impact on family life. J Inherit Metab Dis. 2014;37(2):189‐195. doi: 10.1007/s10545-013-9639-6 [DOI] [PubMed] [Google Scholar]
- 85. Weber SL, Segal S, Packman W. Inborn errors of metabolism: psychosocial challenges and proposed family systems model of intervention. Mol Genet Metab. 2012;105(4):537‐541. doi: 10.1016/j.ymgme.2012.01.014 [DOI] [PubMed] [Google Scholar]
- 86. Ernst G, Menrath I, Lange K, et al. Development and evaluation of a generic education program for chronic diseases in childhood. Patient Educ Couns. 2017;100(6):1153‐1160. doi: 10.1016/j.pec.2017.01.001 [DOI] [PubMed] [Google Scholar]
- 87. Zeltner NA, Huemer M, Baumgartner MR, Landolt MA. Quality of life, psychological adjustment, and adaptive functioning of patients with intoxication‐type inborn errors of metabolism ‐ a systematic review. Orphanet J Rare Dis. 2014;9:159. doi: 10.1186/s13023-014-0159-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ernst G, Szczepanski R, eds. Hrsg) Modulares Schulungsprogramm für chronisch kranke Kinder und Jugendliche sowie deren Familien “ModuS”. 3. Auflage Pabst Publisher. 2020.
- 89. Conijn T, Haverman L, Wijburg FA, De Roos C. Reducing posttraumatic stress in parents of patients with a rare inherited metabolic disorder using eye movement desensitization and reprocessing therapy: a case study. Orphanet J Rare Dis. 2021;16(1):126. doi: 10.1186/s13023-021-01768-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Frankel LA, Pereira S, McGuire AL. Potential psychosocial risks of sequencing newborns. Pediatrics. 2016;137(Suppl 1):S24‐S29. doi: 10.1542/peds.2015-3731F [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kenny T, Bogart K, Freedman A, et al. The importance of psychological support for parents and caregivers of children with a rare disease at diagnosis. Rare Dis Orphan Drugs J. 2022;1:7. doi: 10.20517/rdodj.2022.04 [DOI] [Google Scholar]
- 92. Kißgen R, Carlitscheck J, Rapp C, Franke S. Die psychosoziale Versorgung in der Neonatologie in Deutschland: Eine quantitativ‐empirische Bestandsaufnahme aus ärztlicher Perspektive [Psychosocial care in institutional neonatology in Germany: a quantitative‐empirical inventory from the medical professionals' perspective]. Z Geburtshilfe Neonatol. 2012;216(6):259‐268. doi: 10.1055/s-0032-1323795 [DOI] [PubMed] [Google Scholar]
- 93. Daly A, Gokmen‐Ozel H, MacDonald A, et al. Diurnal variation of phenylalanine concentrations in tyrosinaemia type 1: should we be concerned? J Hum Nutr Diet. 2012;25(2):111‐116. doi: 10.1111/j.1365-277X.2011.01215.x [DOI] [PubMed] [Google Scholar]
- 94. van Dam E, Daly A, Venema‐Liefaard G, et al. What is the best blood sampling time for metabolic control of phenylalanine and tyrosine concentrations in tyrosinemia type 1 patients? JIMD Rep. 2017;36:49‐57. doi: 10.1007/8904_2016_37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Illsinger S, Lücke T, Meyer U, Vaske B, Das AM. Branched chain amino acids as a parameter for catabolism in treated phenylketonuria. Amino Acids. 2005;28(1):45‐50. doi: 10.1007/s00726-004-0150-0 [DOI] [PubMed] [Google Scholar]
- 96. Schultz MJ, Netzel BC, Singh RH, et al. Laboratory monitoring of patients with hereditary tyrosinemia type I. Mol Genet Metab. 2020;130(4):247‐254. doi: 10.1016/j.ymgme.2020.06.001 [DOI] [PubMed] [Google Scholar]
- 97. Nobili V, Jenkner A, Francalanci P, et al. Tyrosinemia type 1: metastatic hepatoblastoma with a favorable outcome. Pediatrics. 2010;126(1):e235‐e238. doi: 10.1542/peds.2009-1639 [DOI] [PubMed] [Google Scholar]
- 98. van Ginkel WG, Gouw AS, van der Jagt EJ, et al. Hepatocellular carcinoma in tyrosinemia type 1 without clear increase of AFP. Pediatrics. 2015;135(3):e749‐e752. doi: 10.1542/peds.2014-1913 [DOI] [PubMed] [Google Scholar]
- 99. Ayuso C, Rimola J, García‐Criado A. Imaging of HCC. Abdom Imaging. 2012;37(2):215‐230. doi: 10.1007/s00261-011-9794-x [DOI] [PubMed] [Google Scholar]
- 100. Bolondi L. Screening for hepatocellular carcinoma in cirrhosis. J Hepatol. 2003a;39(6):1076‐1084. doi: 10.1016/s0168-8278(03)00349-0 [DOI] [PubMed] [Google Scholar]
- 101. Bolondi L. Screening tests for hepatocellular carcinoma. Hepatology. 2003b;37(6):1493. doi: 10.1053/jhep.2003.50215 [DOI] [PubMed] [Google Scholar]
- 102. Burrel M, Llovet JM, Ayuso C, et al. MRI angiography is superior to helical CT for detection of HCC prior to liver transplantation: an explant correlation. Hepatology. 2003;38(4):1034‐1042. doi: 10.1053/jhep.2003.50409 [DOI] [PubMed] [Google Scholar]
- 103. Khanna R, Verma SK. Pediatric hepatocellular carcinoma. World J Gastroenterol. 2018;24(35):3980‐3999. doi: 10.3748/wjg.v24.i35.3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wisse RP, Wittebol‐Post D, Visser G, van der Lelij A. Corneal depositions in tyrosinaemia type I during treatment with Nitisinone. BMJ Case Rep. 2012;bcr2012006301. doi: 10.1136/bcr-2012-006301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Arora N, Stumper O, Wright J, Kelly DA, McKiernan PJ. Cardiomyopathy in tyrosinaemia type I is common but usually benign. J Inherit Metab Dis. 2006;29(1):54‐57. doi: 10.1007/s10545-006-0203-5 [DOI] [PubMed] [Google Scholar]
- 106. André N, Roquelaure B, Jubin V, Ovaert C. Successful treatment of severe cardiomyopathy with NTBC in a child with tyrosinaemia type I. J Inherit Metab Dis. 2005;28(1):103‐106. doi: 10.1007/s10545-005-5085-4 [DOI] [PubMed] [Google Scholar]
- 107. Bergman AJ, van den Berg IE, Brink W, Poll‐The BT, Ploos van Amstel J, Berger R. Spectrum of mutations in the fumarylacetoacetate hydrolase gene of tyrosinemia type 1 patients in northwestern Europe and Mediterranean countries. Hum Mutat. 1998;12(1):19‐26. doi: [DOI] [PubMed] [Google Scholar]
- 108. García MI, de la Parra A, Arias C, Arredondo M, Cabello JF. Long‐term cognitive functioning in individuals with tyrosinemia type 1 treated with nitisinone and protein‐restricted diet. Mol Genet Metab Rep. 2017;11:12‐16. doi: 10.1016/j.ymgmr.2017.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. van Ginkel WG, Rodenburg IL, Harding CO, Hollak CEM, Heiner‐Fokkema MR, van Spronsen FJ. Long‐term outcomes and practical considerations in the pharmacological management of tyrosinemia type 1. Paediatr Drugs. 2019a;21(6):413‐426. doi: 10.1007/s40272-019-00364-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. van Ginkel WG, van Reemst HE, Kienstra NS, et al. The effect of various doses of phenylalanine supplementation on blood phenylalanine and tyrosine concentrations in tyrosinemia type 1 patients. Nutrients. 2019b;11(11):2816. doi: 10.3390/nu11112816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. González‐Lamuño D, Sánchez‐Pintos P, Andrade F, Couce ML, Aldámiz‐Echevarría L. Treatment adherence in tyrosinemia type 1 patients. Orphanet J Rare Dis. 2021;16(1):256. doi: 10.1186/s13023-021-01879-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Malik S, NiMhurchadha S, Jackson C, et al. Treatment adherence in type 1 hereditary Tyrosinaemia (HT1): a mixed‐method investigation into the beliefs, attitudes and behaviour of adolescent patients, their families and their health‐care team. JIMD Rep. 2015;18:13‐22. doi: 10.1007/8904_2014_337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Das AM, Goedecke K, Meyer U, et al. Dietary habits and metabolic control in adolescents and young adults with phenylketonuria: self‐imposed protein restriction may be harmful. JIMD Rep. 2014;13:149‐158. doi: 10.1007/8904_2013_273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Äärelä L, Nevalainen PI, Kurppa K, Hiltunen P. First Scandinavian case of successful pregnancy during nitisinone treatment for type 1 tyrosinemia. J Pediatr Endocrinol Metab. 2020;33(5):661‐664. doi: 10.1515/jpem-2019-0540 [DOI] [PubMed] [Google Scholar]
- 115. Garcia Segarra N, Roche S, Imbard A, et al. Maternal and fetal tyrosinemia type I. J Inherit Metab Dis. 2010;33(suppl 3):S507‐S510. doi: 10.1007/s10545-012-9569-8 [DOI] [PubMed] [Google Scholar]
- 116. Kassel R, Sprietsma L, Rudnick DA. Pregnancy in an NTBC‐treated patient with hereditary tyrosinemia type I. J Pediatr Gastroenterol Nutr. 2015;60(1):e5‐e7. doi: 10.1097/MPG.0b013e3182a27463 [DOI] [PubMed] [Google Scholar]
- 117. Vanclooster A, Devlieger R, Meersseman W, et al. Pregnancy during nitisinone treatment for tyrosinaemia type I: first human experience. JIMD Rep. 2012;5:27‐33. doi: 10.1007/8904_2011_88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Zöggeler T, Ramoser G, Höller A, et al. Nitisinone treatment during two pregnancies and breastfeeding in a woman with tyrosinemia type 1—a case report. J Pediatr Endocrinol Metab. 2021;35(2):259‐265. doi: 10.1515/jpem-2021-0465 [DOI] [PubMed] [Google Scholar]
- 119. Pichler K, Michel M, Zlamy M, et al. Breast milk feeding in infants with inherited metabolic disorders other than phenylketonuria—a 10‐year single‐center experience. J Perinat Med. 2017;45(3):375‐382. doi: 10.1515/jpm-2016-0205 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data supporting the results of this manuscript can be requested from Anibh M. Das or Karin Lange.