

Abstract
We studied the distribution of germline and somatic variants in epilepsy surgery patients with (suspected) malformations of cortical development (MCD) who underwent surgery between 2015 and 2020 at University Medical Center Utrecht (the Netherlands) and pooled our data with four previously published cohort studies. Tissue analysis yielded a pathogenic variant in 203 of 663 (31%) combined cases. In 126 of 379 (33%) focal cortical dysplasia (FCD) type II cases and 23 of 37 (62%) hemimegalencephaly cases, a pathogenic variant was identified, mostly involving the mTOR signaling pathway. Pathogenic variants in 10 focal epilepsy genes were found in 48 of 178 (27%) FCDI/mild MCD/mMCD with oligodendroglial hyperplasia and epilepsy cases; 36 of these (75%) were SLC35A2 variants. Six of 69 (9%) patients without a histopathological lesion had a pathogenic variant in SLC35A2 (n = 5) or DEPDC5 (n = 1). A germline variant in blood DNA was confirmed in all cases with a pathogenic variant in tissue, with a variant allele frequency (VAF) of ~50%. In seven of 114 patients (6%) with a somatic variant in tissue, mosaicism in blood was detected. More than half of pathogenic somatic variants had a VAF < 5%. Further analysis of the correlation between genetic variants and surgical outcomes will improve patient counseling and may guide postoperative treatment decisions.
Keywords: epilepsy surgery, genetics, germline variant, MCD, somatic variant
1. INTRODUCTION
Epilepsy surgery is the most effective treatment in epilepsy patients who develop refractory seizures, provided that the epileptogenic zone is focal, well delineated, preferably of presumed structural origin, and outside eloquent areas. 1 Genetic malformations of cortical development (MCD), such as mild MCD (mMCD), mMCD with oligodendroglial hyperplasia and epilepsy (MOGHE), focal cortical dysplasia (FCD; especially FCDIIa and FCDIIb subtypes), and hemimegalencephaly (HMEG) account for more than one third of children who undergo surgery. 2 , 3
Most MCD are found to arise from variants in genes that encode proteins essential for neurodevelopment. These variants can be detected—de novo or inherited—in the patient's germline or arise during embryogenesis (postzygotic), and lead to somatic mosaicism with variant allelic frequencies (VAFs) < 50%. In recent years, genetic testing has been increasingly applied in focal epilepsy patients who are considered for epilepsy surgery, and the outcome may guide the selection of eligible patients for invasive intracranial monitoring and resective surgery. 3 , 4 , 5 Possibilities to comprehensively test epilepsy patients for a genetic etiology with next generation sequencing (NGS) techniques have expanded 6 and have showed that somatic variants in mTOR pathway genes (AKT3, DEPDC5, MTOR, PIK3CA, RHEB, PTEN, TSC1, and TSC2) or in the UDP‐galactose transporter gene SLC35A2 are the major genetic causes of HMEG, FCD, and pathology‐negative epilepsy surgery patients. 7 , 8 Approximately 70% of reported pathogenic somatic variants have a very low‐allelic fraction (i.e., VAF < 5%), which is unlikely to be detected by conventional NGS (whole exome sequencing [WES] or whole genome sequencing [WGS]) with low read depth and Sanger sequencing of tissue. Nevertheless, somatic variants with a low VAF of merely 1% in the affected brain have been shown to be sufficient to cause refractory epilepsy. This suggests that high read depth (deep) sequencing (e.g., ~1000 × read depth), which is mostly cost‐effective when performed using a gene panel, is required to determine how variants in MCD genes affect epilepsy. 9
In the current study, we performed genetic analysis of the resected tissue from patients with (suspected) MCD to identify somatic or germline variants in known focal epilepsy‐related genes, with the use of deep sequencing techniques, and pooled the results from our center with the data from three recent cohort studies. 10 , 11 , 12
2. MATERIALS AND METHODS
2.1. Cohort
Patients who underwent epilepsy surgery between 2015 and 2020 at University Medical Center Utrecht (the Netherlands [NL]) were enrolled in this study. Patients were eligible if they had an established histopathological diagnosis consistent with mMCD, MOGHE, FCDI or FCDII, or HMEG, or if they were suspected to have an MCD but histopathological examination of resected tissue demonstrated no abnormalities or merely nonspecific reactive gliosis. Neuropathological diagnoses were classified according to the International League Against Epilepsy guidelines. 3 FCDI referred to isolated lesions with cortical dyslamination, which were classified as FCDIa when an excess of microcolumns was observed; FCDII referred to cortical dyslamination and the presence of dysmorphic neurons, with (FCDIIb) or without (FCDIIa) balloon cells. mMCD referred to surgical cases with blurred gray–white matter boundaries and excess of heterotopic neurons in cortical layer 1 (mMCD type I) or in the deep white matter (mMCD type II). MOGHE referred to surgical cases with an increase in oligodendroglia and heterotopic neurons in the white matter. DNA for genetic analysis was derived from specimens that were formalin‐fixed and paraffin‐embedded (FFPE) by the neuropathology department, or immediately after surgery frozen in liquid nitrogen (fresh frozen [FFZ]). Matched blood DNA samples from the included patients, when available from regular clinical care, were used to investigate the presence of postzygotic mosaicism and germline variants in both brain and peripheral sequencing data. For unmatched samples, variant callers were employed to mitigate the risk of overestimating pathogenicity. All patients consented to participate in this study, as approved by the medical ethics review committee. For the pooled data analysis, we included published data from the other four cohorts according to the methods previously described. 10 , 11 , 12 , 13
2.2. DNA extraction from samples and sequencing
Genomic DNA was extracted from FFPE brain samples using QIAamp DNA FFPE kits (Qiagen). From FFZ brain samples, genomic DNA was extracted using QIAamp DNA Mini Kits (Qiagen). For peripheral blood DNA samples, QIAamp DNA Blood Mini Kits (Qiagen) were used. Patients were analyzed with use of two different panels. Panel 1 concerned a hybrid capture‐based NGS with a coverage of ~1000 × read depth that required a minimum input of >200 ng DNA, and consisted of the following genes: BRAF, FGFR1, DEPDC5, NPRL2, NPRL3, AKT1, AKT3, MTOR, PIK3CA, PIK3R2, SLC35A2, TSC1, TSC2, PTEN, RYR3, and TBC1D7. Panel 2 involved amplicon sequencing with a mean coverage of ~1500 × per amplicon, and consisted of the following genes: BRAF, FGFR1, DEPDC5, NPRL2, NPRL3, AKT1, AKT3, MTOR, PIK3CA, PIK3R2, SLC35A2, TSC1, TSC2, PTEN, TBC1D7, RHEB, PIK3R1, and RPS6. Panel 2 was utilized for cases where only FFPE tissue or tissue with limited DNA content (2–10 ng) was available and for FCDII patients with negative results from panel 1 (after isolating pS6‐positive cells with laser capture). For all candidate variants, validation sequencing with use of targeted amplicon sequencing (Miseq Dx sequencer; Illumina) was performed. Variants were reported as (likely) pathogenic according to the American College of Medical Genetics and Genomics (ACMG) guide adapted for classification of somatic missense variants. 14 , 15 Based on these criteria, we excluded germline variants with uncertain pathogenicity. We classified variants as pathogenic only if they were somatic missense variants in mTOR pathway activators previously reported, shown to cause hyperactivation of the mTOR pathway in vitro, or germline/somatic loss‐of‐function variants in mTOR pathway repressors or in SLC35A2. Missense variants in TSC1/2 or SLC35A2 were classified as pathogenic only if already reported. Unreported variants were considered likely pathogenic if confirmed as somatic and absent in gnomAD. For PTEN and DEPDC5, single‐nucleotide variants resulting in protein truncation or loss of function were considered to be pathogenic because their pathogenic mechanism is loss of function. Germline mutations in TSC1/2 reported as benign or with conflicting interpretation according to the ACMG guidelines were excluded. A full list of genes included in each panel version is available in the supplemental data (Table S2).
2.3. Analysis
Differences in proportion of patients with a pathogenic variant between the centers (NL vs. pooled cohort) were examined by use of χ 2 tests for categorical variables. Categorical data are presented as percentages. Data analysis was performed with IBM SPSS 23.
3. RESULTS
3.1. Patient characteristics
We included 123 epilepsy surgery patients at our center. In the pooled data analysis (n = 663), we further included 80 patients from France, 10 107 patients from the USA, 11 170 patients from the Korean study (which reported the results of 177 patients, of whom seven were already included in our cohort), 12 and 180 patients from the USA (Cleveland) and European Epilepsy Brain Bank (EEBB) consortium study. 13 Matched peripheral DNA samples were available for sequencing in 44 (36%) NL patients, and 342 of the 663 patients (51%) from all centers combined. Genetic findings of the Dutch cohort and the pooled patient population are listed in Table 1.
TABLE 1.
Findings (proportional) of genetic testing in brain tissue according to histopathology in the NL cohort (n = 123) and in all four cohorts combined (n = 663).
| Histopathology | Genetic + MRI findings | MRI‐negative | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | No pathogenic variant | (Likely) pathogenic variant, n = 166 | |||||||||
| All | SLC35A2 | DEPDC5 | NPRL2/3 | MTOR | PTEN, PIK3CA, AKT3, RHEB | TSC1/2 | Other | ||||
| NL, n (%) | |||||||||||
| All | 123 | 84 (68) | 42 (34) | 7 (6) | 7 (6) | 2 (2) | 16 (13) | 3 (2) | 7 (6) | 0 | 34 (28) |
| No lesion | 26 | 25 (96) | 1 (4) | 1 (4) | 0 | 0 | 0 | 0 | 0 | 0 | 15 (58) |
| FCDI | 1 | 1 (100) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| mMCD | 29 | 19 (66) | 10 (34) | 5 (17) | 1 (3) | 0 | 2 (7) | 0 | 2 (7) | 0 | 7 (25) |
| MOGHE | 1 | 0 | 1 (100) | 1 (100) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| FCDII | 65 | 33 (51) | 32 (49) | 0 | 6 (12) | 2 (3) | 14 (21) | 3 (5) | 5 (78) | 0 | 11 (17) |
| HMEG | 1 | 1 (100) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Pooled cohorts, n (%) | |||||||||||
| All | 663 | 460 (69) | 203 (31) | 41 (6) | 23 (3) | 7 (1) | 78 (12) | 23 (3) | 23 (3) | 8 (1) | 74 (27) a |
| No lesion | 69 | 63 (91) | 6 (9) | 5 (8) | 1 (1) | 0 | 0 | 0 | 0 | 0 | 52 (76) |
| FCDI | 69 | 59 (86) | 10 (14) | 4 (6) | 1 (2) | 0 | 0 | 0 | 0 | 5 (8) | 0 |
| mMCD | 77 | 57 (74) | 20 (26) | 14 (18) | 2 (4) | 0 | 2 (4) | 0 | 2 (4) | 0 | 6 (18) |
| MOGHE | 32 | 14 (44) | 18 (56) | 18 (56) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| FCDII | 379 | 253 (67) | 126 (33) | 0 | 18 (5) | 7 (2) | 70 (18) | 8 (2) | 21 (6) | 2 (1) | 16 (15) |
| HMEG | 37 | 14 (38) | 23 (62) | 0 | 1 (3) | 0 | 6 (16) | 15 (40) | 0 | 1 (3) | 0 |
Abbreviations: FCDI, focal cortical dysplasia type I; FCDII, focal cortical dysplasia type II; HMEG, hemimegalencephaly; mMCD, mild malformations of cortical development; MOGHE, mMCD with oligodendroglial hyperplasia and epilepsy; MRI, magnetic resonance imaging; NL, the Netherlands.
No exact information on MRI outcome could be retrieved in 381 patients.
3.2. Tissue analysis
Outcomes (pooled) of genetic analysis of brain tissue samples according to histopathological diagnosis are illustrated in Figure 1.
FIGURE 1.
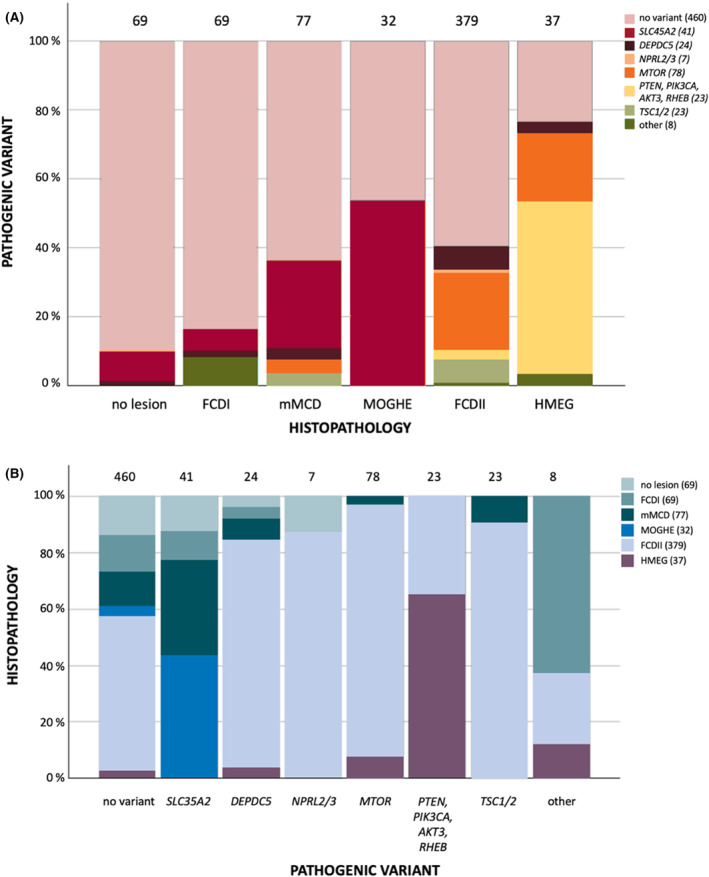
Distribution (proportional) of outcome of genetic testing (somatic and germline variants) in brain tissue according to histopathology (A) and of histopathology according to outcome of genetic testing in brain tissue (B) in all four cohorts combined (n = 663). FCDI, focal cortical dysplasia type I; FCDII, focal cortical dysplasia type II; HMEG, hemimegalencephaly; mMCD, mild malformations of cortical development; MOGHE, mMCD with oligodendroglial hyperplasia and epilepsy.
There was a similar rate of pathogenic variants (somatic and/or germline) in the NL cohort (42 of 123 cases, 34%) and the pooled cohorts (203 of 663 cases, 31%; p‐value of difference = .51), comprising of pathogenic variants in 10 of 69 FCDI cases (14%), 20 of 77 mMCD cases (26%), 18 of 32 MOGHE cases (56%), 126 of 379 FCDII cases (33%), 23 of 37 HMEG cases (62%), and six of 69 patients without a lesion (9%). In 146 of the 149 FCDII/HMEG cases with an identified variant (98%), a pathogenic variant in mTOR pathway genes was identified; 36 of 48 FCDI/mMCD/MOGHE cases with a variant (75%) had an SLC35A2 variant. Six of 69 patients (9%) without a histopathological lesion also had a pathogenic variant, in either SLC35A2 (n = 5) or DEPDC5 (n = 1). The "other" variants category contained five FCDI patients with detected CASK, KRAS, NF1, or NIBPL variants, or a 1q amplification. Two FCDII patients had an SCN1A variant or a 1q amplification/PTEN variant, and one HMEG case had a somatic uniparental disomy of the p‐arm of chromosome 16. Genetic findings of individual patients with a (likely) pathogenic variant are listed in Table S1. In 20 of 34 NL cases (59%) and 89 of 170 patients from the pooled cohorts (52%; p‐value of difference = .64) with a somatic pathogenic variant, a low‐level variant with a VAF < 5% was identified. Somatic variants and germline variants accounted for 29 (24%) and eight (7%) patients in our cohort, and 162 (24%) and 33 (5%) among all patients combined, respectively. In five patients (4%) of our cohort and eight patients (2%) of all cohorts combined, a germline and (second hit) somatic variant in the same gene was identified in the resected tissue.
3.3. Blood DNA sample analysis
In all 10 patients with a tissue VAF of ~50% whose blood DNA samples were available for analysis (eight DEPDC5 cases, one TSC2 case, and one SCN1A case), the germline variant was confirmed. In addition, in seven of 114 patients (6%) with a somatic variant in tissue and available blood DNA, mosaicism for the variant was shown in blood, with VAFs (tissue/blood %) of 24.0/2.0% and 15.2/2.0% in two MTOR cases, 4.6/2.0%, 3.7/2.2%, 7.0/1.0%, and 4.4/1.0% in four TSC1 cases, and 9/1.4% in one PTEN case.
4. DISCUSSION
Similar to what was previously reported, 8 this study shows that a high rate of pathogenic or likely pathogenic genetic findings in HMEG (62%) as well as FCDII (33%) could be identified. Almost all variants in HMEG and FCDII involved genes comprising the mTOR signaling pathway, which is not surprising given the shared pathological features between these malformations. As reported before, genes mutated in FCDI/mMCD/MOGHE patients were related to glycosylation, the mTOR signaling pathway, or synapse or gene expression pathways, pointing toward a higher genetic heterogeneity in FCDI/mMCD/MOGHE as compared to FCDII. 13 The majority of all somatic pathogenic variants were low‐level (VAF < 5%) variants. Three quarters of mMCD/FCDI/MOGHE cases with a pathogenic variant had SLC35A2 somatic variants. In nonlesional patients, the resected tissue infrequently revealed somatic, mainly SLC35A2 variants. Blood‐derived DNA analysis infrequently showed postzygotic mosaicism.
It is likely that some of the patients without an established genetic pathogenic variant are falsely negative, because a recent study reported evidence that in a substantial part of variant‐negative FCDII cases, ultra‐low‐level somatic variant in mTOR pathway genes can only be detected in pS6‐enriched cells by deep WGS, 16 and because two of the pooled cohorts (USA, USA‐EEBB) used WES, which is a less sensitive approach to identify low‐level somatic variants. 11 , 13 Also, the gene panels that were used in the other cohorts (NL, Korea, France) did not include all 75 currently known MCD‐related genes, and unknown genes might also play a role, although the MCD genes with the highest prevalence were analyzed. 10 , 12 , 17 Lastly, it is probable that there is an underestimation of the histopathological entity MOGHE in this study, because for most of the included previously published cohorts, MOGHE was not yet well recognized as a distinct pathological entity during the study period. As a result, because somatic brain mosaic variants in SCL35A2 are a major etiological factor in MOGHE, it is possible that part of the previously reported SLC35A2 cases with a histopathological diagnosis of FCDI or normal/aspecific histopathological findings should be classified as MOGHE under the current guidelines. 3 , 18
Recent studies emphasize that genetic testing of tissue adherent to stereoelectroencephalographic electrodes and cell‐free DNA derived from cerebrospinal fluid collected during epilepsy surgery show future opportunities for detecting pathogenic variants in epilepsy surgery patients. 19 , 20 Further analysis of the correlation between genetic variants and surgical outcomes in cohorts such as these will improve patient counseling and may guide postoperative treatment decisions.
AUTHOR CONTRIBUTIONS
Study design, data acquisition, manuscript drafting/revision: Maurits W. C. B. Sanders, Bobby P. C. Koeleman, Eva H. Brilstra, and Floor E. Jansen. Data acquisition, manuscript revision: Sara Baldassari, Mathilde Chipaux, Nam Suk Sim, Ara Ko, Hoon‐Chul Kang, Ingmar Blümcke, Dennis Lal, Stéphanie Baulac, and Jeong Ho Lee. Study design, data acquisition, manuscript drafting/revision: Eleonora Aronica and Kees P. J. Braun.
CONFLICT OF INTEREST STATEMENT
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Data S1.
ACKNOWLEDGMENTS
None.
Sanders MWCB, Koeleman BPC, Brilstra EH, Jansen FE, Baldassari S, Chipaux M, et al. Somatic variant analysis of resected brain tissue in epilepsy surgery patients. Epilepsia. 2024;65:e209–e215. 10.1111/epi.18148
Eleonora Aronica and Kees P. J. Braun contributed equally to this study.
REFERENCES
- 1. Oderiz CC, von Ellenrieder N, Dubeau F, Eisenberg A, Gotman J, Hall J, et al. Association of cortical stimulation‐induced seizure with surgical outcome in patients with focal drug‐resistant epilepsy. JAMA Neurol. 2019;76:1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blumcke I, Spreafico R, Haaker G, Coras R, Kobow K, Bien CG, et al. Histopathological findings in brain tissue obtained during epilepsy surgery. N Engl J Med. 2017;377:1648–1656. [DOI] [PubMed] [Google Scholar]
- 3. Najm I, Lal D, Alonso Vanegas M, Cendes F, Lopes‐Cendes I, Palmini A, et al. The ILAE consensus classification of focal cortical dysplasia: an update proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia. 2022;63:1899–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanders MWCB, Lemmens CMC, Jansen FE, Brilstra EH, Koeleman BPC, Braun KPJ, et al. Implications of genetic diagnostics in epilepsy surgery candidates: a single‐center cohort study. Epilepsia Open. 2019;4:609–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oegema R, Barakat TS, Wilke M, Stouffs K, Amrom D, Aronica E, et al. International consensus recommendations on the diagnostic work‐up for malformations of cortical development. Nat Rev Neurol. 2020;16(11):618–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Helbig I, Tayoun ANA. Understanding genotypes and phenotypes in epileptic encephalopathies. Mol Syndromol. 2016;7:172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jamuar SS, Lam A‐TN, Kircher M, Wang J, Barry BJ, Zhang X, et al. Somatic mutations in cerebral cortical malformations. N Engl J Med. 2014;371:733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gerasimenko A, Baldassari S, Baulac S. mTOR pathway: insights into an established pathway for brain mosaicism in epilepsy. Neurobiol Dis. 2023;182:106144. [DOI] [PubMed] [Google Scholar]
- 9. Lim JS, Kim WI, Kang HC, Kim SH, Park AH, Park EK, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. 2015;21:395–400. [DOI] [PubMed] [Google Scholar]
- 10. Baldassari S, Ribierre T, Marsan E, Adle‐Biassette H, Ferrand‐Sorbets S, Bulteau C, et al. Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol. 2019;138:885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lai D, Gade M, Yang E, Koh HY, Lu J, Walley NM, et al. Somatic variants in diverse genes leads to a spectrum of focal cortical malformations. Brain. 2022;145:2704–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sim NS, Ko A, Kim WK, Kim SH, Kim JS, Shim KW, et al. Precise detection of low‐level somatic mutation in resected epilepsy brain tissue. Acta Neuropathol. 2019;138:901–912. [DOI] [PubMed] [Google Scholar]
- 13. López‐Rivera JA, Leu C, Macnee M, Khoury J, Hoffmann L, Coras R, et al. The genomic landscape across 474 surgically accessible epileptogenic human brain lesions. Brain. 2023;146(4):1342–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lai A, Soucy A, El Achkar CM, Barkovich AJ, Cao Y, DiStefano M, et al. The ClinGen brain malformation variant curation expert panel: rules for somatic variants in AKT3, MTOR, PIK3CA, and PIK3R2 . Genet Med. 2022;24:2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim JH, Park JH, Lee J, Park JW, Kim HJ, Chang WS, et al. Ultra‐low level somatic mutations and structural variations in focal cortical dysplasia type II. Ann Neurol. 2023;93:1082–1093. [DOI] [PubMed] [Google Scholar]
- 17. Chung C, Yang X, Bae T, Vong KI, Mittal S, Donkels C, et al. Comprehensive multi‐omic profiling of somatic mutations in malformations of cortical development. Nat Genet. 2023;55:209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bonduelle T, Hartlieb T, Baldassari S, Sim NS, Kim SH, Kang HC, et al. Frequent SLC35A2 brain mosaicism in mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE). Acta Neuropathol Commun. 2021;6(9):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim S, Baldassari S, Sim NS, Chipaux M, Dorfmüller G, Kim DS, et al. Detection of brain somatic mutations in cerebrospinal fluid from refractory epilepsy patients. Ann Neurol. 2021;89:1248–1252. [DOI] [PubMed] [Google Scholar]
- 20. Montier L, Haneef Z, Gavvala J, Yoshor D, North R, Verla T, et al. A somatic mutation in MEN1 gene detected in periventricular nodular heterotopia tissue obtained from depth electrodes. Epilepsia. 2019;60(10):e104–e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.