

Highlights
-
•
The design and analysis of a novel multi-epitope vaccine (MEV) named ZL12138L against Mycobacterium tuberculosis (MTB) using bioinformatics and immunoinformatics approaches.
-
•
The identification of immunodominant epitopes from MTB antigens with broad population coverage, predicted to cover HLA class I and II allele genes in 92.41% and 90.17% of the global population, respectively.
-
•
The demonstration of the ZL12138L vaccine's significant immunogenicity and antigenicity, with no toxicity or allergenicity, through comprehensive bioinformatics analysis.
-
•
The successful molecular docking of the ZL12138L vaccine with Toll-like receptor (TLR) 2 and TLR4, indicating its potential to activate both innate and adaptive immune responses.
-
•
The prediction of robust immune responses, including the activation of NK cells, macrophages, B lymphocytes, and T lymphocytes, following immunization with the ZL12138L vaccine.
Keywords: Tuberculosis, Multi-epitope vaccine, Computer-aided design, Artificial intelligence, Immunogenicity
Abstract
Background
Tuberculosis (TB) remains a global public health challenge. The existing Bacillus Calmette–Guérin vaccine has limited efficacy in preventing adult pulmonary TB, necessitating the development of new vaccines with improved protective effects.
Methods
Computer-aided design and artificial intelligence technologies, combined with bioinformatics and immunoinformatics approaches, were used to design a multi-epitope vaccine (MEV) against TB. Comprehensive bioinformatics analyses were conducted to evaluate the physicochemical properties, spatial structure, immunogenicity, molecular dynamics (MD), and immunological characteristics of the MEV.
Results
We constructed a MEV, designated ZL12138L, containing 13 helper T lymphocyte epitopes, 12 cytotoxic T lymphocyte epitopes, 8 B-cell epitopes, as well as Toll-like receptor (TLR) agonists and helper peptides. Bioinformatics analyses revealed that ZL12138L should exhibit excellent immunogenicity and antigenicity, with no toxicity or allergenicity, and had potential to induce robust immune responses and high solubility, the immunogenicity score was 4.14449, the antigenicity score was 0.8843, and the immunological score was 0.470. Moreover, ZL12138L showed high population coverage for human leukocyte antigen class I and II alleles, reaching 92.41% and 90.17%, respectively, globally. Molecular docking analysis indicated favorable binding affinity of ZL12138L with TLR-2 and TLR-4, with binding energies of −1173.4 and −1360.5 kcal/mol, respectively. Normal mode analysis and MD simulations indicated the stability and dynamic properties of the vaccine construct. Immune simulation predictions suggested that ZL12138L could effectively activate innate and adaptive immune cells, inducing high levels of Type 1 T helper cell cytokines.
Conclusions
This study provides compelling evidence for ZL12138L as a promising TB vaccine candidate. Future research will focus on experimental validation and further optimization of the vaccine design.
Graphical abstract

1. Introduction
Tuberculosis (TB), an ancient disease caused by Mycobacterium tuberculosis (MTB), continues to pose a considerable global public health challenge [[1], [2], [3]]. According to the World Health Organization 2023 report, TB remains one of the leading infectious causes of mortality worldwide, with millions of new cases and hundreds of thousands of deaths reported annually [4]. The Bacillus Calmette–Guérin (BCG) vaccine, which is in widespread use and has shown some efficacy in preventing TB in children, has a limited protective effect in adults and fails to provide long-term immunity [[5], [6], [7]]. Consequently, the development of novel vaccines to prevent TB in adults is highly desirable.
Multi-epitope vaccines (MEVs) are an innovative type of vaccine that can enhance the breadth and intensity of the immune response by combining multiple immunodominant epitopes [[8], [9], [10], [11]]. Compared with traditional vaccines, MEVs can activate a broader range of immune cell populations, including T cells and B cells, thereby providing more comprehensive protection [12]. Furthermore, the design of MEVs allows for customization enabling different antigens of pathogens to be targeted, which provides an effective solution to address pathogen variation and immune evasion.
With the recent considerable advancements in computational power and the continuous optimization of algorithms, computer-aided design (CAD) and artificial intelligence (AI) technologies have enabled breakthrough progress to be made in biomedical applications [[13], [14], [15], [16], [17]]. These technologies have been used in the design and development of TB vaccines, bringing revolutionary changes to MEV research for tuberculosis. In the present study, we employed advanced CAD tools to analyze and predict epitopes with high immunogenic potential from MTB antigens. These epitopes included helper T lymphocyte (HTL) epitopes, cytotoxic T lymphocyte (CTL) epitopes, and B lymphocyte epitopes, which are key factors in activating immune responses [18]. Furthermore, we used machine learning and deep learning algorithms from AI technology to conduct screening and optimization of these immunodominant epitopes, subsequently constructing a novel TB MEV. We predicted the physicochemical properties, antigenicity, immunogenicity, solubility, and spatial structure of this MEV. We further analyzed the interaction characteristics between the candidate MEV and Toll-like receptors (TLRs), as well as the features of the immune responses induced by the MEV.
2. Materials and methods
2.1. Selection of target antigens and acquisition of sequence information
A schematic flowchart of the MEV development and analysis process is shown in Fig. 1. We focused on five primary antigens, three antigens used in proliferative vaccines that have entered clinical trials: Ag85a, Ag85b, and Mtb39A [[19], [20], [21], [22]] and two antigens being investigated in preclinical research: MPT64 (Rv1980c) and PPE68 (Rv3873) [23,24]. These antigens were selected because they are expressed at different stages of the MTB life cycle and have the potential to elicit host immune responses.
Fig. 1.

Schematic flowchart of the ZL12138L vaccine development and analysis process. This process encompasses the selection of candidate antigens, identification of immunodominant HTL, CTL, and B-cell epitopes, construction of the ZL12138L vaccine, analysis of the vaccine's physicochemical properties, analysis of the vaccine's spatial structure, and prediction of the immunological characteristics.
Abbreviations: HTL, helper T lymphocyte; CTL, cytotoxic T lymphocyte; IFN-γ, interferon-γ; IL, interleukin; IEDB, the Immune Epitope Database.
We used the Mycobrowser database (https://mycobrowser.epfl.ch/), a specialized bioinformatics resource for the Mycobacterium genus that provides extensive genomic and proteomic data, to obtain detailed sequence information on these candidate antigens [25]. Specifically, the species MTB H37Rv was selected. The desired antigen gene name (e.g., Rv3873) was entered in the “Gene name or function” field. Subsequently, the full-length amino acid sequence of the antigen in FASAT format was retrieved from the “Protein sequence” section of the results page. Through this platform, we successfully retrieved and documented the full-length amino acid sequences of the selected antigens, laying the foundation for subsequent immunogenic epitope analysis and vaccine design. During the sequence acquisition process, particular attention was paid to the accuracy and completeness of the sequences to ensure the reliability of subsequent bioinformatics analyses. The obtained sequence information will be used for further epitope prediction and vaccine construction to identify key epitopes capable of activating host immune responses.
2.2. Screening of immunodominant T-cell and B-cell epitopes
To construct an effective TB vaccine, we focused on screening T-cell epitopes capable of eliciting strong immune responses. Our aim was to identify epitopes that bind to major histocompatibility complex (MHC) I and MHC II molecules, corresponding to CTL and HTL epitopes, respectively. Using information from previous studies and a literature review, we employed the MHC class servers in the Immune Epitope Database (IEDB I, http://tools.iedb.org/mhci/ and IEDB II, http://tools.iedb.org/mhcii/) for epitope prediction, precisely setting the length range for CTL and HTL epitopes at 9–10 and 12–18 amino acid residues, respectively. Then, we used the VaxiJen v2.0 server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) to evaluate the antigenicity of the predicted epitopes, ensuring the selected epitopes possessed high predicted immunogenicity [26].
We combined multiple online prediction tools, including the Interferon (IFN) Epitope Server (https://webs.iiitd.edu.in/raghava/ifnepitope/predict.php), IL4pred (https://webs.iiitd.edu.in/raghava/il4pred/predict.php), and IL10pred (https://webs.iiitd.edu.in/raghava/il10pred/predict3.php), to assess the ability of the HTL epitopes to induce specific cytokine secretion [27]. We prioritized epitopes capable of inducing IFN-γ-positive responses, while suppressing interleukin-4 (IL-4) and IL-10 secretion, to enhance Th1-type immune responses. To assess the safety of the vaccine candidates, we evaluated the allergenicity and toxicity of the epitopes using AllerTOP v.2.0 (https://www.ddg-pharmfac.net/AllerTOP/) and ToxinPred (https://webs.iiitd.edu.in/raghava/toxinpred/multi_submit.php) servers [28,29]. Finally, we predicted the immunogenicity of the CTL epitopes using the Class I Immunogenicity server (http://tools.iedb.org/immunogenicity/) to determine that the selected epitopes were likely to induce effective immune responses.
Because B lymphocytes are important in anti-infection immunity, we used the ABCpred server (https://webs.iiitd.edu.in/raghava/abcpred/) to predict linear B-cell epitopes [30]. This server employs recurrent neural network technology to identify epitopes that are likely to induce strong immune responses with high accuracy. We restricted the length of the B-cell epitopes, prioritizing the epitopes with high scores and lengths not exceeding 20 amino acid residues.
In addition to linear epitopes, discontinuous B-cell epitopes are also common and play crucial roles in immune responses. Therefore, the accurate prediction of these discontinuous epitopes is critical for refining the spatial structure of vaccine candidates. We used the ElliPro server (http://tools.iedb.org/ellipro/), an advanced non-linear B-cell epitope prediction tool, to predict discontinuous B-cell epitopes for use in the candidate MEV. ElliPro identifies potential epitope regions by analyzing the three-dimensional structure of proteins, providing important structural information. We set a screening threshold of 0.693 based on previous studies and server recommendations. Only discontinuous B-cell epitopes scoring above this threshold were selected for subsequent MEV construction [[31], [32], [33]].
We used the IEDB server (http://tools.iedb.org/bcell/) to predict the surface accessibility, flexibility, β-turns, antigenicity, and hydrophilicity of the epitopes [[34], [35], [36], [37]]. Through these stringent screening criteria, we were able to identify potential epitopes that were likely to enhance the immunogenicity and protective efficacy of a vaccine. This refined epitope prediction and screening process provided a precise structural basis for the MEV design, contributing to the development of an effective and safe TB vaccine.
2.3. Construction of MEV and population coverage analysis of HLA-I and HLA-II alleles
We employed an innovative bioinformatics strategy to predict and screen immunodominant T-cell and B-cell epitopes. We linked the selected epitopes using carefully designed amino acid linkers, including GPGPG, AAY, and KK, aiming to enhance the immunogenicity and improve the vaccine stability of the epitopes. To further boost the immune-stimulating capacity of the MEV, we introduced specific TLR agonists, the TLR2 agonist EAST-6 and the TLR-4 agonist RS-09 [38,39], at both ends of the vaccine sequence. These agonists were chosen because they have known immune-enhancing properties and are capable of effectively activating immune cells. Additionally, we added the PADRE helper peptide, a widely studied immunoenhancing element that helps induce robust immune responses [40], after the RS-09 agonist. To facilitate vaccine purification and functional studies, we added a 6 × His tag at the end of the MEV sequence, which should enable the efficient purification of the vaccine candidate through affinity chromatography.
A crucial factor in the development of a vaccine is its applicability to diverse global populations; thus, MHC restriction should be considered in the design of epitope vaccines. To assess the global coverage potential of our designed MEV candidate, we conducted a comprehensive analysis using the population coverage tool in the IEDB database (http://tools.iedb.org/population/), combined with human leukocyte antigen (HLA)-I and HLA-II allele frequency data from the allele frequency database. These data cover 115 countries and 21 ethnicities across 16 geographic regions worldwide, which should ensure that our designed vaccine can provide protection for the majority of the global population, and thus have the potential for widespread application.
2.4. Biological properties of the candidate MEV
After constructing the candidate MEV, we conducted detailed predictive analyses of key biological properties using a series of advanced bioinformatics tools. These properties, which include the antigenicity, immunogenicity, allergenicity, and toxicity, are crucial for evaluating the efficacy and safety of a vaccine. We first used AntigenPro (https://scratch.proteomics.ics.uci.edu/) and VaxiJen v2.0 servers to assess the antigenicity and immunogenicity of the MEV [26,41]. These tools were used to predict the potential of the MEV to activate immune cells through an in-depth analysis of specific features in the sequence. Additionally, application of the IEDB immunogenicity server allowed further evaluation of the immunogenicity of the MEV to predict its ability to induce immune responses [42]. For the safety assessment, we employed Allergen FP v.1.0 and AllerTOP v.2.0 servers to predict the allergenicity of the MEV. These analyses helped to identify and exclude epitopes that might cause allergic reactions. Simultaneously, to comprehensively assess the vaccine's safety, we also used ToxinPred and ToxinPred2 servers (https://www.ddg-pharmfac.net/AllerTOP/) to predict the toxicity of the MEV [28,43]. These comprehensive bioinformatics analyses indicated that the MEV candidate should possess good immunogenicity while meeting safety requirements, i.e., should be non-toxic and non-allergenic. These analyses provided a solid foundation for experimental research and the potential clinical application of the candidate vaccine, as well as providing an important scientific basis for developing more effective and safer tuberculosis MEV candidate vaccines.
2.5. Physicochemical properties and solubility of the candidate MEV
To comprehensively understand the biological function and immunogenicity of the candidate MEV, we conducted a detailed analysis of the physicochemical properties. These properties included the predicted in vivo and in vitro half-lives, theoretical isoelectric point (pI), instability index, aliphatic index, and grand average of hydropathicity (GRAVY). These parameters are crucial for evaluating the structural stability, the behavior under physiological conditions, and potential interactions of the MEV [44]. Using the Expasy Protparam server (https://www.expasy.org/resources/protparam/) we predicted these key physicochemical parameters. These data, not only helped to understand the stability of the MEV, but also provided important information on the dynamics of the MEV in biological systems. In particular, the solubility of a vaccine is a key factor affecting the immunogenicity. A high solubility facilitates the uniform distribution of a vaccine during injection and thus contributes to the formation of the correct spatial structures and induction of effective immune responses. To assess the solubility of the MEV, we used the protein-sol server (https://protein-sol.manchester.ac.uk/) for prediction [45]. From the results obtained, we set a solubility value >0.45 as the standard for good solubility, providing a scientific basis for vaccine formulation development and injection formulation strategies.
It is also essential to consider the potential post-translational modification (PTM) sites of proteins because these are crucial for vaccine development and protein functions. Common PTMs include phosphorylation, acetylation, ubiquitination, and methylation. Notably, phosphorylation is a key mechanism for regulating protein activity and cellular signaling pathways. Analyzing phosphorylation sites can provide valuable insights into protein functions, activation states, and potential interactions within cellular networks. To predict potential phosphorylation sites in the protein of interest, ZL12138L, we employed the NetPhos-3.1 server (https://services.healthtech.dtu.dk/services/NetPhos-3.1/), a widely recognized tool in the field of computational biology. This server uses advanced machine learning algorithms to identify potential phosphorylation sites based on the primary sequence of a protein.
2.6. Bioinformatics prediction of secondary and tertiary structures of the candidate MEV
The spatial structure of a vaccine considerably affects the biological function and immunogenicity. To gain an understanding of the structural characteristics of the candidate MEV, we employed various bioinformatics tools for predictive analysis. First, we used PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) to predict the secondary structure of the MEV. PSIPRED is a protein secondary structure analysis tool that predicts structures from the amino acid sequences or PDB structure files, and is capable of predicting transmembrane topology, helices, folds, and domains. Additionally, we used the SOPMA server (https://npsa.lyon.inserm.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html), which has an accuracy of approximately 69.5%. This server can predict protein secondary structures, including α-helices, β-sheets, and random coils [[46], [47], [48]].
For predicting protein interactions, which is crucial for understanding cellular functions and rational vaccine design, we employed the latest AlphaFold-3 server (https://golgi.sandbox.google.com/). This upgraded version of AlphaFold-2 can predict the structures of complexes including proteins, nucleic acids, small molecules, ions, and modified residues [15,49]. AlphaFold-3 has improved accuracy compared with many specialized tools, particularly in predicting protein–ligand, protein–nucleic acid, and antibody–antigen interactions [50]. After obtaining the tertiary structure prediction, we used the Galaxy Refine web server (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) to optimize the model, a crucial step in ensuring the reliability of the 3D structure prediction of the MEV [[51], [52], [53], [54]]. Through these comprehensive structural prediction and optimization analyses, we were able to obtain a solid structural foundation for the design and optimization of a MEV with good performance as a vaccine.
2.7. 3D structure model validation of the candidate MEV
After completing the 3D structure prediction and optimization of the MEV, we implemented validation procedures to ensure the accuracy and reliability of the obtained model. This process is crucial for refining vaccine design as it helps to identify and correct potential structural defects. First, we used the ProSA-web server (https://prosa.services.came.sbg.ac.at/prosa.php) to conduct a detailed validation of the 3D structure models before and after optimization. ProSA-web is a tool that uses PDB format files to detect errors in protein models by calculating Z-scores to evaluate model quality [55,56]. A comparison of the Z-scores with local quality scores of proteins provided an indication of possible errors in the model [57]. Subsequently, to further assess the structural quality of the MEV, we employed the ERRAT server (https://saves.mbi.ucla.edu/). ERRAT evaluates the quality of protein spatial structures based on non-covalent interactions between atoms, identifying potentially erroneous regions in the structure and providing important information for structural optimization. In addition, to comprehensively assess the stability of the protein structure, we generated Ramachandran plots using UCLA-DOE LAB-SAVES v6.0 (https://saves.mbi.ucla.edu/Jobs/1564711/pc/saves_01.png) [10,[58], [59], [60], [61]]. Ramachandran plots are a visualization tool for evaluating protein spatial structures. By displaying the distribution of amino acid residues in the core, allowed, and disallowed regions, the stability of a protein structure can be assessed. A higher number of residues in the core region indicate a more stable structure, while a higher number in the disallowed region may suggest structural instability. Through these comprehensive validation steps, the 3D structural model of the candidate MEV was shown to possess high accuracy and reliability for use in vaccine design.
2.8. Molecular docking analysis of the candidate MEV with TLRs
TLRs, especially TLR-2 and TLR-4, play crucial roles in the innate immune system by recognizing MTB and activating signaling pathways for pro-inflammatory cytokines. To enhance the immunogenicity of the candidate MEV, TLR-2 and TLR-4 agonists were incorporated into the vaccine design to boost the immune responses by targeting TLR-2 and TLR-4. We obtained the three-dimensional structural information of TLR-2 (PDB ID: 2Z7X) and TLR-4 (PDB ID: 3FXI) from the NCBI molecular modeling database (MMDB; https://www.ncbi.nlm.nih.gov/structure/). These detailed structural data provided the foundation for conducting precise molecular docking analyses. Using the AlphaFold-3 server, we performed ligand–receptor docking analyses to predict the interactions between the MEV and both TLR-2 and TLR-4. To visually demonstrate the interactions between the MEV and the TLRs, we generated 3D visualization models using PyMOL software and created DIMPLOT diagrams showing cross-interface interaction effects using LigPlot software. These images not only provided an intuitive perspective of the binding modes between the MEV and the TLRs but also revealed key interaction sites. This information is crucial for understanding how a MEV enhances immunogenicity through its interactions with TLRs [58,59]. The molecular docking analysis provided valuable insights into (1) the binding affinity between the MEV and the TLRs; (2) the specific amino acid residues involved in the interactions; (3) the types of interactions (e.g., hydrogen bonds and hydrophobic interactions); and (4) potential conformational changes upon binding. This detailed analysis of the MEV–TLR interactions contributed to our understanding of the vaccine's mechanism of action and its potential efficacy in stimulating the immune response against tuberculosisbr [62,63].
2.9. NMA, MD simulations, and MM/GBSA analysis of the candidate MEV complex
To understand the dynamic properties and stability of the candidate MEV, we implemented the following steps:
2.9.1. Normal mode analysis (NMA)
We used the iMODS web server (https://imods.iqf.csic.es/) to perform NMA, a computational method for exploring collective motions of macromolecules at biologically relevant scales. This analysis is crucial for understanding the biological function of molecules [64]. We conducted detailed studies of the dynamic behavior of the MEV complex using covariance matrices, deformation plots, eigenvalues, elastic network models, and B-factor values [65]. These analyses revealed the primary motion modes and potential function-related changes of the MEV complex, providing important insights into its dynamic properties under biological conditions.
2.9.2. MD simulations
To further understand the interactions between the MEV and its target proteins, we performed 150 ns MD simulations using Desmond software from Schrödinger LLC. We simulated the stability of protein–ligand complexes under near-physiological conditions. Prior to simulation, we optimized the protein and ligand using Maestro's Protein Preparation Wizard to eliminate potential steric hindrances and unfavorable contacts. We constructed the simulation system using the System Builder tool, employing the TIP3P solvent model and OPLS_2005 force field. Counter ions were added for neutralization, and 0.15 M sodium chloride was included to mimic physiological conditions. A temperature of 300K and pressure of 1 atm were maintained throughout the simulation. Through these simulations, we were able to evaluate the stability of the MEV in a physiological environment and analyze the dynamics of protein–ligand complexes using root mean square deviation (RMSD) and principal component analysis (PCA) [66,67].
2.9.3. Mechanics/generalized Born surface area (MM/GBSA) analysis
We used the prime module of Schrödinger software to perform MM/GBSA analysis and calculate the binding free energies. Specific components included the total binding free energy (ΔGbind), molecular mechanics gas-phase energy (ΔEgas), solvation free energy (ΔGsol), and entropy (TΔS). The specific equations used for these calculations can be found in previously published papers [[68], [69], [70]].
These analyses provided insights into the interactions between the MEV and its targets, contributing to further optimization of the vaccine design. The combination of NMA, MD simulations, and MM/GBSA analysis enabled a comprehensive view of the behavior, stability, and binding characteristics of the MEV, which are crucial for predicting its efficacy as a vaccine candidate.
2.10. C-ImmSim immune simulation of the candidate MEV
To evaluate the immunogenicity of the candidate MEV, particularly its effectiveness against tuberculosis infection, we used the C-ImmSim server (https://150.146.2.1/CIMMSIM/index.php) to simulate and predict immune responses. This simulation included (1) innate immune cells: natural killer (NK) cells, macrophages, and dendritic cells (DCs); (2) adaptive immune cells: B cells, HTLs, and CTLs; and (3) antibody and cytokine levels induced by the MEV in these immune cells. This comprehensive simulation provided insights into how the MEV might stimulate various components of the immune system, offering a prediction of its potential efficacy as a vaccine candidate.
2.11. Codon optimization and in silico cloning
Because codon usage has an impact on protein antigen expression in vitro, we optimized the codons of the candidate MEV to enhance its expression success rate. We used the Optimizer server (http://genomes.urv.es/OPTIMIZER/) for this optimization, following the method used in previous reports [71,72]. Specifically, (1) Expression Host: we selected the widely used Escherichia coli 536 as the expression host; (2) Output Parameters: the server provided several important parameters, including the codon adaptation index (CAI; the ideal value is 1) and GC content percentage (the recommended range is 30%–70%); (3) Cloning Simulation: using SnapGene software, we simulated the insertion of the optimized MEV gene sequence into the pET28a(+) plasmid through XhoI and BamHI restriction sites to construct a recombinant plasmid; (4) Electrophoresis Simulation: we simulated 1% agarose gel electrophoresis to visualize the cloning results. This in silico optimization and cloning process provided a theoretical foundation for the subsequent experimental validation and expression of the MEV. This process helps to predict potential issues in protein expression and allows for adjustments before actual wet-laboratory experiments, potentially saving time and resources in the vaccine development process. For a summary of the scripts of the key bioinformatics tools used in the design of the ZL12138L vaccine, as well as the key parameters and specified parameter values used in the design and execution, the reasons for selecting these parameters, and any potential impact they may have on the results, please refer to the Supplementary Material Table S1 for detailed information on the key parameters of the servers and software used in the design and execution of the ZL12138L vaccine model.
3. Results
3.1. Identification of immunodominant epitopes, MEV construction, stability, solubility, and population coverage analysis
We screened and identified 13 HTL epitopes, 12 CTL epitopes, and 8 B-cell epitopes from five potential MTB antigens (Fig. 2A). These epitopes exhibited favorable adjusted rank, antigenicity, and IFN-γ scores, and were non-toxic and non-allergenic (Table 1). To enhance the immunogenicity of the MEV, we linked these epitopes using amino acid linkers (GPGPG, AAY, and KK) and incorporated the TLR-4 agonist RS-09, the TLR-2 agonist EAST-6, and the PADRE helper peptide using EAAAK linkers (Fig. 2B). The final MEV construct was designated ZL12138L.
Fig. 2.

Comprehensive characterization of the ZL12138L vaccine construct: design strategy, amino acid composition, solubility profile, and global population coverage. (A) The ZL12138L vaccine construct integrates multiple immunostimulatory components, including the TLR-2 agonist EAST-6, the pan-DR T helper epitope PADRE, 12 HTL epitopes, 13 CTL epitopes, 8 B-cell epitopes, and the TLR-4 agonist RS-09, to enhance immunogenicity. (B) The primary sequence of ZL12138L is depicted with the distinct immunological elements highlighted in different colors to facilitate the visual identification of each component. (C) Solubility assessment using the Protein-Sol server revealed a calculated solubility score exceeding 0.45, indicative of favorable solubility properties. (D) The secondary structure of ZL12138L was predicted using the PSIPRED server. The structural elements, α-helices (pink), β-strands (yellow), and coils (gray), provide a detailed view of the conformational landscape. (E) Global demographic analysis of HLA allele coverage by the ZL12138L vaccine.
Abbreviations: HLA, human leukocyte antigen.
Table 1.
Detailed information on the HTL, CTL, and B-cell epitopes selected to construct the ZL12138L vaccine.
| Protein | Peptide sequence | Length (aa) | Alleles | Percentile ranka | Antigenicity scoreb | IFN-γ scorec | Immunogenicity scored | ABC pred scoree | AllergenFP v.2.0f | Toxin Predg | IL-4h | IL-10i |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HTL epitopes | ||||||||||||
| Ag85a | VSGLVGAVGGTATAGA | 16 | HLA-DQA1*05:01/DQB1*03:01/ DQB1*06:02 |
0.39 | 1.184 | 0.80257189 | Non | Non | Non | Non | ||
| GLVGAVGGTATAGAF | 15 | HLA-DQA1*05:01/DQB1*03:01 | 0.27 | 0.840 | 0.47981667 | Non | Non | Non | Non | |||
| SGLVGAVGGTATAGA | 15 | HLA-DQA1*05:01/DQB1*03:01 | 0.21 | 1.245 | 0.59277922 | Non | Non | Non | Non | |||
| VSGLVGAVGGTATAG | 15 | HLA-DQA1*05:01/DQB1*03:01 /DPB1*04:02/DRB1*01:01/DRB1*09:01 |
0.31 | 1.203 | 0.49398202 | |||||||
| Ag85b | GLAGGAATAGAFSRPGL | 17 | HLA-DQA1*05:01/DQB1*03:01 | 0.37 | 0.542 | 1.5668744 | Non | Non | Non | Non | ||
| LPGLVGLAGGAATAGA | 16 | HLA-DQA1*05:01/DQB1*03:01 | 0.45 | 0.786 | 1.0183619 | Non | Non | Non | Non | |||
| VGLAGGAATAGA | 12 | HLA-DQA1*05:01/DQB1*03:01 | 0.27 | 1.282 | 0.52408286 | |||||||
| AAIGLSMAGSSAMILAA | 17 | HLA-DRB1*09:01/DQA1*01:02 HLA-DQB1*06:02 |
0.26 | 0.689 | 1.1091804 | |||||||
| Mtb39A | ARMYAGPGSASLVAA | 15 | HLA-DQA1*05:01/DQB1*03:01 | 0.35 | 0.564 | 0.015333722 | Non | Non | Non | Non | ||
| Rv1980 | SDPAYNINISLPSYYPDQ | 18 | HLA-DRB3*02:02/DRB1*09:01 HLA-DRB1*14:01 |
0.41 | 1.047 | 0.8399143 | Non | Non | Non | Non | ||
| Rv3873 | GSDKALAAATPMVVWLQ | 17 | HLA-DQA1*06:01/DQB1*03:03 | 0.36 | 0.533 | 0.55646741 | Non | Non | Non | Non | ||
| GSDKALAAATPMVVW | 15 | HLA-DQA1*06:01/DQB1*03:03 | 0.22 | 0.744 | 0.13794349 | |||||||
| CTL epitopes | ||||||||||||
| Ag85a | RVWVYCGNGK | 10 | HLA-A*11:01,HLA-A*30:01 HLA-A*68:01 |
0.42 | 0.715 | 0.10358 | Non | Non | ||||
| GLPVEYLQV | 9 | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*02:06 | 0.27 | 0.706 | 0.05492 | Non | Non | |||||
| MGPTLIGLAM | 10 | HLA-B*35:01/ HLA-B*07:02 | 0.47 | 0.524 | 0.19966 | |||||||
| Ag85b | NAAGGHNAVF | 10 | HLA-B*35:01,HLA-B*53:01 HLA-A*26:01 |
0.33 | 1.475 | 0.16235 | Non | Non | ||||
| NTPAFEWYY | 9 | HLA-A*26:01,HLA-C*03:04, HLA-B*15:01 |
0.1 | 1.945 | 0.4288 | Non | Non | |||||
| NAAGGHNAV | 9 | HLA-A*68:02 | 0.2 | 1.995 | 0.12765 | |||||||
| Mtb39A | ATATATATL | 9 | HLA-A*68:02/*32:01 | 0.47 | 1.001 | 0.1821 | Non | Non | ||||
| LPPEINSARM | 10 | HLA-B*35:01,HLA-B*51:01 | 0.4 | 1.064 | 0.12798 | |||||||
| Rv1980 | GGTHPTTTYK | 10 | HLA-A*11:01,HLA-B*15:02, HLA-C*07:02 |
0.02 | 1.627 | 0.13929 | Non | Non | ||||
| VSIAPNAGL | 9 | HLA-C*03:04, HLA-C*01:02 | 0.16 | 0.764 | 0.1185 | |||||||
| Rv3873 | DAQAVELTAR | 10 | HLA-A*33:03 | 0.36 | 1.389 | 0.18465 | Non | Non | ||||
| SLPEIAANHI | 9 | HLA-C*01:02 | 0.4 | 0.608 | 0.31512 | Non | Non | |||||
| GINTIPIAL | 9 | HLA-A*02:01 | 0.49 | 0.861 | 0.2913 | |||||||
| B cellular epitopes | ||||||||||||
| Ag85a | AGGYKASDMWGPKEDP | 16 | 0.84 | Non | Non | |||||||
| QDAYNAGGGHNGVFDF | 16 | 0.73 | Non | Non | ||||||||
| Ag85b | LRAQDDYNGWDINTPA | 16 | 0.85 | Non | Non | |||||||
| YCGNGTPNELGGANIP | 16 | 0.79 | Non | Non | ||||||||
| Rv1980 | YELNITSATYQSAIPP | 16 | 0.86 | Non | Non | |||||||
| RKPITYDTLWQADTDP | 17 | 0.88 | Non | Non | ||||||||
| Rv3873 | PGASQSTTNPIFGMPS | 16 | 0.85 | Non | Non | |||||||
| VNTLFEKLEPMASILD | 0.8 | Non | Non | |||||||||
Abbreviations: HTL, helper T lymphocyte; CTL, cytotoxic T lymphocyte; IFN-γ, interferon-γ; IL, interleukin.
The percentile ranking of the selected epitopes; the inclusion criterion was a ranking score <0.5.
The antigenicity score; epitopes with antigenicity score >0.5 were selected.
IFN-γ score; the epitopes with the highest positive scores were selected.
The immunogenicity score; epitopes were selected in order of score.
The linear B-cell epitope prediction score; epitopes were selected in order of score.
The result of the sensitization or toxicity test, “Non” indicates that the epitope was non-toxic or non-sensitizing.
The result of the Toxin; “Non” indicates that the epitope was non-toxic.
Stimulated cytokine IL-4 secretion. HTL epitopes were selected that do not secrete cytokine IL-4.
Stimulated cytokine IL-10 secretion. HTL epitopes were selected that do not secrete cytokine IL-10.
The ZL12138L vaccine antigen molecule comprised 691 amino acids with a molecular weight of 67331.45 Da. We further analyzed the physicochemical properties of ZL12138L (Table S2), revealing the molecule had a theoretical pI value of 7.24, instability index of 28.42, aliphatic index of 71.36, and GRAVY value of −0.025. These parameters collectively indicated the stability of ZL12138L and its behavior under physiological conditions. The predicted half-life of ZL12138L was determined to be 30 h in mammalian reticulocytes, 20 h in yeast, and 10 h in E. coli. These results demonstrated the favorable stability of ZL12138L in various host cells, contributing to its role in effecting an immune response.
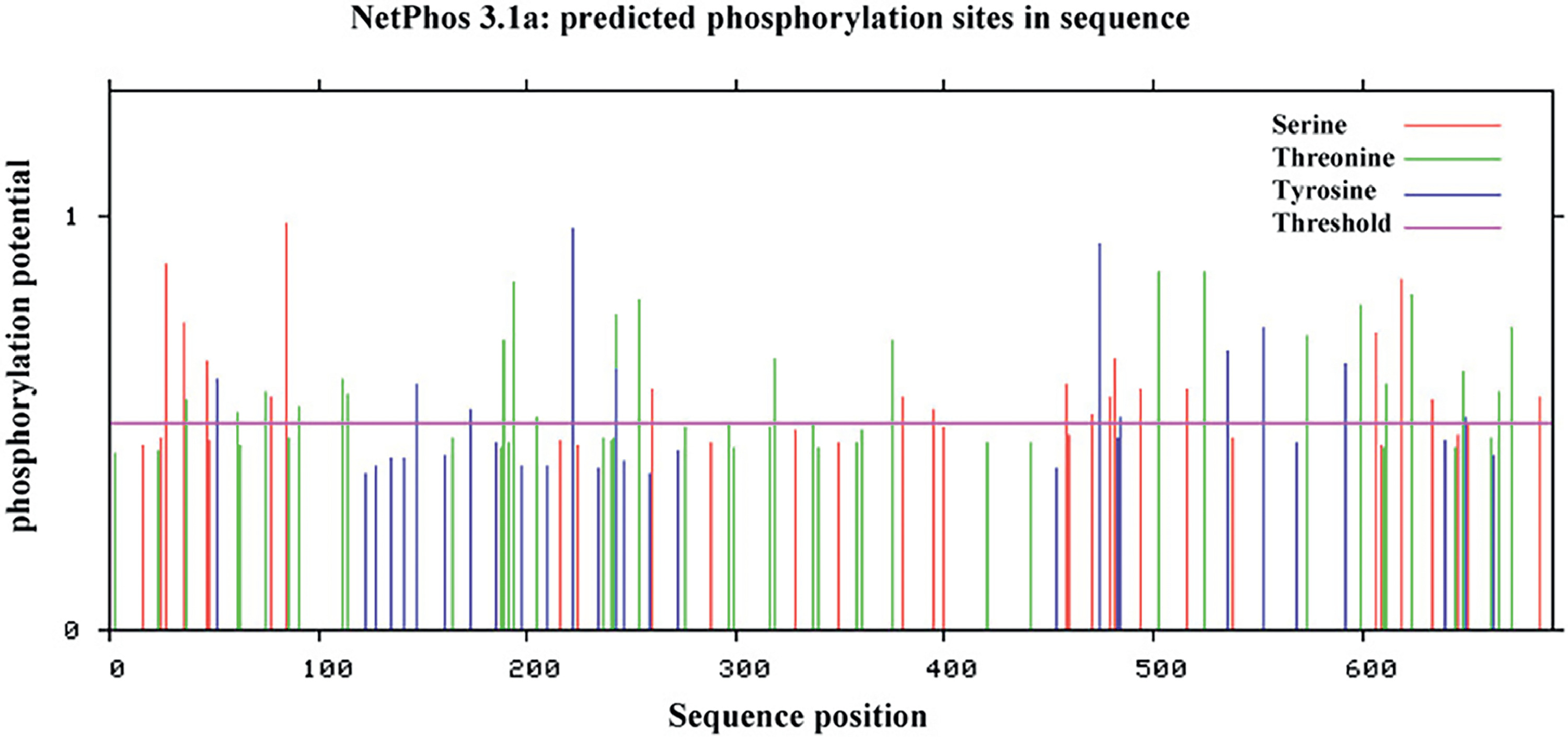
Additionally, ZL12138L exhibited a predicted solubility value of 0.47, surpassing the acceptable threshold of 0.45 (Fig. 2C). As previously mentioned, PTM sites play a crucial role in vaccine design. Our comprehensive analysis of the ZL12138L protein sequence revealed several potential phosphorylation sites, primarily on serine, threonine, and tyrosine residues (Fig. S1). Phosphorylation, a key PTM, can appreciably alter protein function, stability, and interactions, thereby influencing the immune recognition and vaccine efficacy. The identified phosphorylation sites were predominantly clustered in specific regions of the protein, suggesting functional importance. Serine residues showed the highest propensity for phosphorylation, followed by threonine and tyrosine. This pattern was consistent with the general distribution of phosphorylation sites in eukaryotic proteins. These findings provided valuable insights into the complex regulatory mechanisms governing protein activity and the pivotal roles of proteins in signaling pathways.
Secondary structure analysis of the ZL12138L antigen molecule revealed proportions of 32.71% α-helix, 17.08% β-sheet, and 41.53% random coil conformation (Fig. 2D). We conducted a population coverage analysis of the immunodominant HTL and CTL epitopes within the MEV using the IEDB database (Fig. 2E). This analysis demonstrated that the designed ZL12138L vaccine achieved a global coverage of 90.17% for HLA-II alleles (HTL epitopes) and 92.41% for HLA-I alleles (CTL epitopes). These data suggested that the designed MEV had broad applicability and the potential to provide protection for the majority of the global population.
3.2. Analysis of the antigenicity, immunogenicity, and safety of the ZL12138L vaccine
To comprehensively evaluate the immunological characteristics of the ZL12138L vaccine, we employed a series of advanced bioinformatics tools for predictive analysis; the results are summarized in Table S2. Initially, assessment through the IEDB immunogenicity server yielded a high score of 4.14449 for ZL12138L, indicating its potential to elicit robust immune responses. Further antigenicity predictions using VaxiJen v2.0 and AntigenPro servers resulted in scores of 0.763582 and 0.8843, respectively, suggesting that ZL12138L had the capacity to induce effective antibody responses and was a potential vaccine candidate.
In a safety evaluation, ZL12138L demonstrated non-allergenic and non-toxic properties, which are crucial factors for the clinical application of a vaccine. These safety characteristics were investigated through predictive analyses using Allergen FP v.1.0, AllerTOP v.2.0, ToxinPred, and ToxinPred2 servers. Collectively, these bioinformatics analyses revealed that ZL12138L should not only exhibit excellent immunogenicity and antigenicity but also predicted a good safety profile, establishing a solid scientific foundation for further vaccine development and evaluation.
3.3. 3D structure prediction of the ZL12138L vaccine
To investigate the 3D spatial configuration of the ZL12138L vaccine, we employed the AlphaFold-3 server, successfully predicting five 3D models. After careful evaluation, we selected model 0 as the basis for further analysis. To enhance the accuracy and reliability of the model, we used the Galaxy Refine web server for optimization. During this process, we considered multiple evaluation metrics, including the global distance test-high accuracy (GDT-HA), RMSD, MolProbity, Clash score, poor rotamers, and Ramachandran score. Ultimately, we selected model 1, which exhibited the highest GDT-HA value, lowest MolProbity score, and highest Ramachandran score, as the final 3D model for the ZL12138L vaccine (Fig. 3A).
Fig. 3.

Structural analysis and optimization of the ZL12138L vaccine. (A) Representation of the ZL12138L vaccine structure before and after structural optimization. The left panel shows the vaccine's initial structure, while the right panel illustrates the optimized structure, highlighting improvements in secondary and tertiary structural features. (B) Z-score distribution of residues in the initial ZL12138L vaccine structure, compared with X-ray and NMR reference datasets. The plot displays the Z-scores indicating structural deviations of individual residues relative to high-resolution structures. (C) Z-score distribution of residues in the optimized ZL12138L vaccine structure, compared with X-ray and NMR reference datasets. Post-optimization Z-scores demonstrated reduced structural deviations and improved stability. (D) Knowledge-based energy profile of the initial ZL12138L vaccine structure, calculated using different window sizes (10 and 40). Peaks in the energy profile indicate regions with higher energy and potential instability. (E) Knowledge-based energy profile of the optimized ZL12138L vaccine structure, highlighting energy minimization and increased stability across the sequence positions. (F) Ramachandran plot of the initial ZL12138L vaccine structure, showing the distribution of dihedral angles (phi and psi) for all residues. The plot identifies energetically favorable (dark red), allowed (yellow), and disallowed (white) regions. (G) Ramachandran plot of the optimized ZL12138L vaccine structure. Post-optimization, a greater proportion of residues fall within the most favored and allowed regions, reflecting improved structural quality and reduced steric clashes.
Further validation was conducted using ProSA-web and UCLA-DOE LAB SAVES v6.0 servers. The Z-scores before and after optimization were −3.2 (Fig. 3B) and −3.55 (Fig. 3C), respectively, indicating an improvement in statistical energy of the optimized model, bringing the model closer to known high-quality protein structures. Figs. 3D and 3E illustrates the energy changes in the ZL12138L vaccine 3D model before and after optimization. In Fig. 3D, some non-uniformity was observed in the energy distribution of the pre-optimized model, potentially indicating local instabilities in certain regions. Post-optimization, as shown in Fig. 3E, the energy distribution became more uniform, suggesting a considerable improvement in the overall model stability.
Additionally, we performed Ramachandran plot analysis to assess the stereochemical quality of the conformations of the amino acid residues in the model. Prior to optimization, 85% of the amino acid residues were located in the core region, 12.7% in the allowed region, 1.4% in the generously allowed region, and 1.0% in the disallowed region (Fig. 3F). After optimization, these proportions improved to 97.3% in the core region, 2.1% in the allowed region, 0.4% in the generously allowed region, and 0.2% in the disallowed region (Fig. 3G), further indicating the effectiveness of the model optimization. Through this series of analyses and optimizations, the accuracy of the ZL12138L vaccine 3D model was improved and a solid structural foundation was established for further research and development of the vaccine.
3.4. Analysis of discontinuous B-cell epitopes in the ZL12138L vaccine
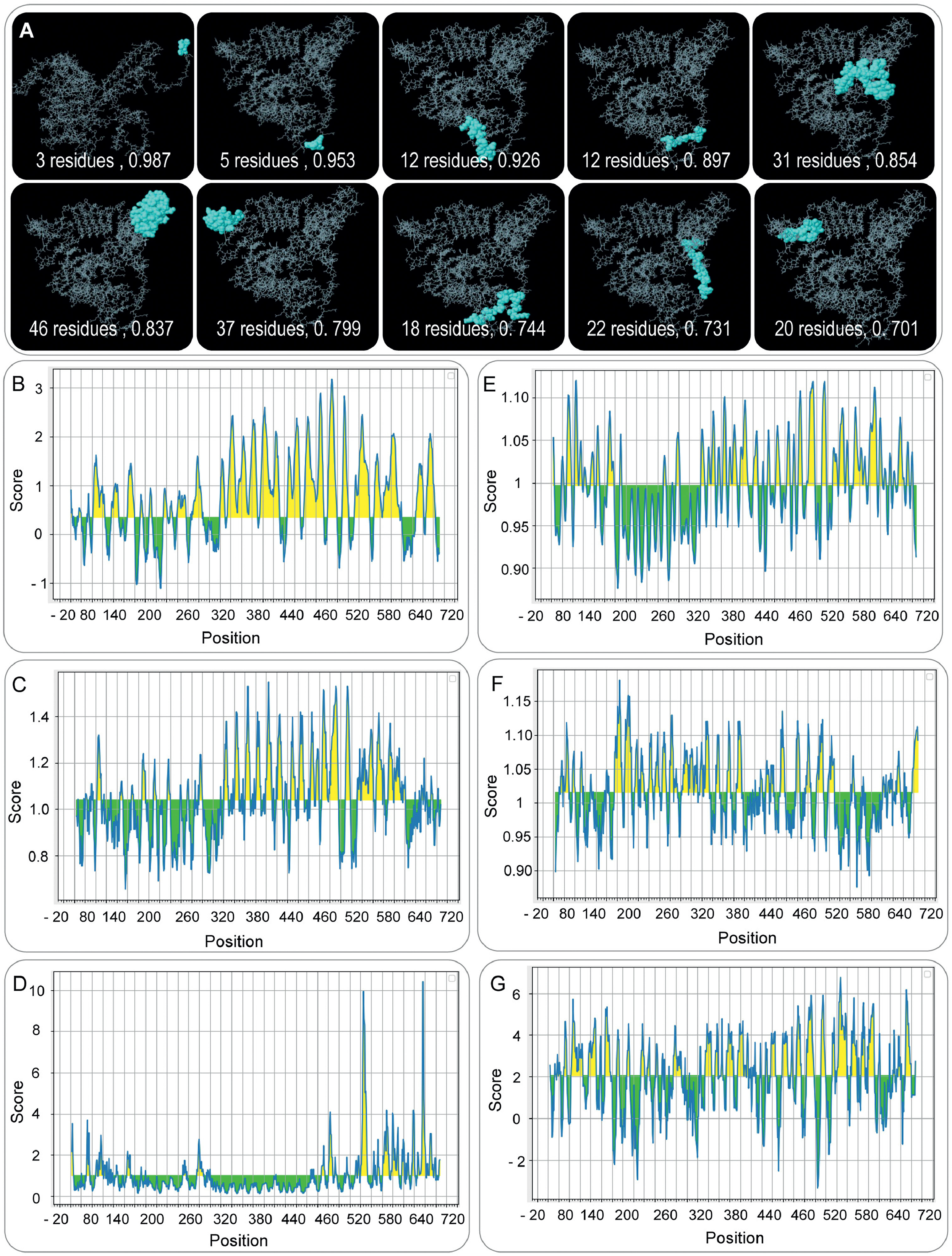
To further enhance the immunogenicity of the ZL12138L vaccine, we used the ElliPro server to predict and analyze the discontinuous B-cell epitopes. These epitopes play a crucial role in vaccine design because of their ability to activate B-cell responses and induce protective antibodies. In our prediction process, we typically selected models with scores >0.69 for in-depth analysis. Using this criterion, we successfully predicted 10 discontinuous B-cell epitopes in the ZL12138L vaccine (Fig. S2A). These epitopes contained 3, 5, 12, 12, 31, 46, 37, 18, 22, and 20 residues, with corresponding core scores of 0.987, 0.953, 0.926, 0.897, 0.854, 0.837, 0.799, 0.744, 0.731, and 0.701, respectively (Table S3). These results indicated that the ZL12138L vaccine possessed multiple potential antigenic epitopes capable of activating B-cell responses and potentially inducing protective antibodies against MTB.
Using the IEDB online server, we systematically analyzed various properties of the ZL12138L vaccine antigen sequence, revealing potential epitope regions. The Bepipred linear epitope prediction (Fig. S2B) showed high scores for many residues in the 340–640 position range, particularly in the yellow regions, suggesting a high probability of these residues being part of epitopes. The β-turn prediction (Fig. S2C) similarly displayed high scores in the same region, reflecting favorable characteristics for β-turn formation in these residues. Surface accessibility analysis (Fig. S2D) revealed high scores in the 520–640 region, indicating these residues may be easily recognized by antibodies. In contrast, flexibility analysis (Fig. S2E) showed high flexibility scores for residues in the 380–580 range, which are crucial for epitope dynamics and accessibility. Antigenicity prediction (Fig. S2F) highlighted the 200–520 region as having high antigenicity scores, suggesting these residues may play important roles in inducing immune responses. Hydrophilicity analysis (Fig. S2G) showed high hydrophilicity scores in specific regions. The presence of hydrophilic regions aids in exposing epitopes to the aqueous environment for immune system recognition. In conclusion, the data indicated that certain residue regions, particularly between positions 340 and 640, combine multiple favorable epitope characteristics. These results provided strong theoretical information for designing vaccines and diagnostic tools aimed at targeted immune responses.
3.5. The ZL12138L vaccine binds tightly to TLR-2 and TLR-4
We employed computational simulation methods to analyze in detail the interactions between the ZL13128L vaccine and TLR-2 (Fig. 4A) and TLR-4 (Fig. 4B). The results demonstrated that ZL13128L formed complex interaction networks when bound to both TLR-2 and TLR-4. The binding regions were magnified to showcase the interactions between key amino acid residues. Detailed analysis revealed that multiple forces contributed to the stable binding of ZL13128L with TLRs, including ligand bonds, hydrogen bonds, and hydrophobic contacts.
Fig. 4.

Molecular docking and interaction analysis of the ZL12138L vaccine with TLRs. 3D structural representation of the docking configuration between the ZL12138L vaccine (teal) and the TLR-2 molecule (pink, A upper panel) or TLR-4 molecule (pink, B upper panel), predicted using AlphaFold-3 and visualized with PyMOL. The inset shows a magnification of the key interface region, displaying detailed interactions between the vaccine and TLR-2 at the atomic level, highlighting potential binding sites and interaction residues. Then, LIGPLOT was used to analyze the key interacting residues between the ZL12138L vaccine and TLR-2 (A lower panel) or TLR-4 (B lower panel). This visualization identified critical residues involved in the interactions, including hydrogen bonds, hydrophobic contacts, and ligand bonds.
Specifically, 27 hydrogen bonds were observed between the ZL13128L vaccine and TLR-2, with lengths ranging from 2.7 to 3.9 Å. For example, the hydrogen bond between Arg587 and Glu455 had a length of 2.7 Å, while the bond between Arg460 and Trg592 was 3.3 Å (Fig. 4A). Similarly, 41 hydrogen bonds were observed between the ZL13128L vaccine and TLR-4, with lengths varying from 2.3 to 3.5 Å (Fig. 4B). Numerous amino acid residues participated in hydrophobic contacts and ligand bond formation, further enhancing the stability of the complexes. The binding sites involved, not only ligand bonds and hydrogen bonds, but also the hydrophobic contacts of many non-ligand residues, demonstrating the complexity and diversity of the binding interfaces.
Overall, the ZL13128L vaccine formed stable bonds with TLR-2 and TLR-4 receptors through various intermolecular forces, including hydrophobic interactions, ligand bonds, and multiple hydrogen bonds involving key amino acids. These findings, not only revealed the potential of the ZL13128L vaccine to stimulate immune responses through molecular interactions, but also provided a valuable structural biology basis for future vaccine design and optimization.
3.6. Normal mode analysis of ZL12138L vaccine complexes with TLR-2 and TLR-4
To investigate the dynamic interactions between the ZL12138L vaccine and TLR-2 and TLR-4 in depth, we employed the iMOD server for NMA. This analysis provided crucial information regarding the dynamic characteristics of both complexes. For the TLR-2-ZL12138L complex (Fig. S3): (1) The deformability plot (Fig. S3A) showed appreciable peaks, especially near the first 200 and around the 800 positions, indicating high flexibility in these regions; (2) the eigenvalue plot (Fig. S3B) displayed the distribution of eigenvalues for the first 20 modes, with the first mode having a particularly small eigenvalue (1.527547e−5), suggesting its importance in the overall dynamics; (3) the variance plot (Fig. S3C) showed that the first seven modes explained over 80% of the variance, indicating that most dynamic information was concentrated in a few modes; (4) the B-factor comparison (Fig. S3D) demonstrated good agreement between NMA-calculated and PDB-derived B-factors, validating the reliability of the dynamic model; (5) the covariance matrix (Fig. S3E) revealed positive and negative covariances between residues, suggesting coordinated or opposing motions; and (6) the correlation plot (Fig. S3F) showed the strength of motion coupling between atoms, with high-density areas indicating important correlated motions.
For the TLR-4-ZL12138L complex (Fig. S4): (1) the deformability plot (Fig. S4A) showed appreciable fluctuations, with notable peaks around indices 200, 800, and 1300, indicating high flexibility in these regions; (2) the eigenvalue plot (Fig. S4B) showed a very small eigenvalue for the first mode (3.275133e−5), emphasizing its importance in overall dynamics; (3) the variance plot (Fig. S4C) indicated that the first 10 modes explained approximately 80% of the variance; (4) the B-factor comparison (Fig. S4D) showed good agreement between NMA-calculated and PDB-derived B-factors; (5) the covariance matrix (Fig. S4E) revealed appreciable coordinated or opposing motions, especially in regions around indices 200, 800, and 1300; and (6) the correlation plot (Fig. S4F) highlighted the areas of strong motion coupling, particularly around indices 500 and 1500. These analyses provided valuable insights into the dynamic behavior of the ZL12138L vaccine in complex with TLR-2 and TLR-4, highlighting key regions of flexibility and correlated motions that may be crucial for the function and stability of the vaccine.
3.7. MD simulation and principal component analysis of ZL12138L vaccine-TLR-4 complex
Further analysis was conducted of the MD simulation of the ZL12138L vaccine-TLR-4 complex. Fig. 5A illustrates the RMSD values of the Cα atoms in the ZL12138L vaccine over the course of the MD simulation. RMSD, a widely used metric for assessing protein structural stability, provides insight into structural changes over time. The graph shows that there were three distinct phases: an initial phase (0−20 ns) characterized by a rapid increase in the RMSD value from approximately 3 to 6 Å, indicating considerable conformational changes and adjustments; a middle phase (20–60 ns) showing overall stability with fluctuations between 6 and 7 Å, suggesting a more balanced conformation; and a late phase (60−100 ns) where the RMSD value was stabilized between 7 and 8 Å with minimal fluctuations, indicating structural equilibrium. Fig. 5B presents the RMSD values for each residue of the ZL12138L vaccine, providing a detailed analysis of the distribution of the flexibility and stability during the simulation. Regions near residue indices 0−300 and 1000 exhibited higher flexibility, potentially indicating areas of instability or considerable conformational changes. In contrast, the central region (residue indices 300−1000) demonstrated greater stability. The secondary structure analysis depicted in Fig. 5C shows the percentage of secondary structure elements (α-helices and β-sheets) for each residue. The region spanning residue indices 0−400 was predominantly α-helical, while the area between 200−500 contained some β-sheet structures. The middle region exhibited lower secondary structure content, potentially indicating unstructured or dynamic areas. The region from indices 800−1100 displayed rich structural diversity. Fig. 5D provides comprehensive structural dynamics information, illustrating the proportion of secondary structure elements over time and the changes in secondary structure for each residue during the MD simulations. The upper portion of the figure shows the stability of the overall secondary structure proportions throughout the 100 ns simulation, indicating overall structural stability of the protein. The lower heatmap in the figure shows local structural stability information, with some regions, such as residue indices 100−400 and 900−1100, showing relative stability in the secondary structure, while the region from 500−700 appears more dynamic.
Fig. 5.

MD simulations and principal component analysis (PCA) of the ZL12138L vaccine-TLR-4 complex. (A) Root Mean Square Deviation (RMSD) plot of the ZL12138L vaccine-TLR-4 complex over 100 ns of MD simulation. The RMSD values indicate structural deviations from the initial configuration, providing insights into the stability of the complex over time. (B) RMSD fluctuation per residue index over the simulation time. This plot highlights specific regions within the ZL12138L vaccine-TLR-4 complex that exhibit appreciable conformational changes, suggesting potential flexible and stable regions. (C) Secondary structure element (SSE) distribution along the residue index. The plot indicates the percentage of time each residue spends in specific secondary structures (e.g., α-helices and β-strands) throughout the simulation, providing insights into the structural dynamics of the complex. (D) Time evolution of the SSE. The upper part of the plot shows the percentage of each SSE over time, and the heatmap below illustrates the temporal changes in secondary structure along the residue index, highlighting regions of structural transitions and stability during the simulation. (E) PCA of the ZL12138L vaccine-TLR-4 complex. The first panel shows a projection of the motion along the first two principal components (PC1 and PC2), capturing 42.68% and 12.79% of the variance, respectively. The clusters in the plot indicate distinct conformational states visited during the simulation. The second panel shows the projection of the motion along PC2 and PC3, capturing 12.79% and 7.86% of the variance, respectively. The third panel shows the projection of the motion along PC1 and PC3. The last panel shows the plot of the proportion of variance explained by each principal component, with an inset showing the cumulative variance. This analysis quantifies the contribution of each mode to the overall motion, demonstrating the dominant dynamics captured by the first few principal components.
PCA of the protein dynamics elucidated the trajectory motions of the ZL12138L vaccine and TLR-4 in the MD simulations. Fig. 5E presents multiple graphs illustrating the distribution of these trajectories in different principal component spaces and the variation in eigenvalues. Scatter plots (PC1 vs. PC2, PC2 vs. PC3, and PC1 vs. PC3) demonstrate the spatial distribution of ZL12138L and TLR-4 across the first three principal components, with PC1, PC2, and PC3 explaining 42.68%, 12.79%, and 7.86% of the variance, respectively. The scree plot displays the relationship between eigenvalues and eigenvector indices for the initial 20 principal components, explaining the contribution of the principal components to the total variance. The first five eigenvectors are particularly prominent, with higher eigenvalues indicating stronger fluidity regulation, suggesting the considerable stability and explanatory power of these principal components in hyperspace fluctuations. These results revealed the primary changes and intrinsic structural relationships of ZL12138L and TLR-4 trajectories in low-dimensional space, providing insights into the dynamic behavior of these proteins in MD simulations.
3.8. MM/GBSA analysis of the ZL12138L-TLR-4 complex
The MD simulations results revealed strong interactions between the newly designed multi-epitope tuberculosis vaccine Z12138L and TLR-4. MM/GBSA calculations (Table S4) indicated the Z12138L-TLR-4 complex had a total binding free energy (ΔGbind) of −522.96 kcal/mol, indicating exceptionally high binding affinity. This robust binding was primarily driven by van der Waals forces (−519.26 kcal/mol) and hydrophobic interactions (−306.52 kcal/mol), with additional contributions from hydrogen bonding (−20.20 kcal/mol), which are all crucial factors in vaccine–receptor recognition. Despite some unfavorable electrostatic (30.41 kcal/mol) and solvation (166.90 kcal/mol) effects, the overall binding was highly favorable. The conformational adaptability of Z12138L, reflected in the strain energy of −79.22 kcal/mol, further enabled the binding to TLR-4. On average, each non-hydrogen atom contributed −0.13 kcal/mol to the binding energy, highlighting the efficiency of Z12138L as a multi-epitope vaccine. These results strongly supported the potential of Z12138L to effectively activate TLR-4, likely inducing a robust immune response. This analysis provided compelling evidence for Z12138L as a promising candidate for tuberculosis vaccine development.
3.9. Capacity of the ZL12138L vaccine to activate innate and adaptive immune responses
Both innate and adaptive immune responses play critical roles in a host's elimination of MTB. We employed the C-IMMSIM server to predict the capacity of the ZL12138L vaccine to induce the activation of NK cells, macrophages, B cells, CD4+ T cells (Th1 and Th2 cells), and CD8+ T cells. The simulation results indicated the vaccine should have the ability to activate multiple types of immune cells, providing robust evidence for its immunogenicity.
In the innate immune response, NK cell numbers were predicted to be rapidly increased following initial immunization with ZL12138L, peaking at day 80 and maintaining elevated levels thereafter (Fig. 6A). The predicted levels of presenting-2 and active macrophages peaked and then rapidly decreased 90 days post-immunization, while resting macrophage numbers increased correspondingly (Fig. 6B). DC (Fig. 6C) and epithelial cell (Fig. 6D) numbers exhibited minor fluctuations but generally remained at high levels post-vaccination. ZL12138L was predicted to induce changes in B lymphocytes, memory B cells, T helper (TH) lymphocytes, and CTLs, indicating the potential of ZL12138L to activate adaptive immune responses. Post-vaccination simulations showed increased B lymphocyte and memory B cell numbers, suggesting the vaccine may have the ability to induce robust adaptive immune responses (Fig. 6E and 6F). The increase in TH cell numbers and decrease in memory TH cells emphasized that booster vaccinations would be necessary to maintain long-term immunity (Fig. 6G and 6H). Furthermore, ZL12138L-induced CTL responses and Th1-type immune response activation were crucial mechanisms for MTB clearance (Fig. 6I, 6J, and 6K). Additionally, ZL12138L was predicted to induce the secretion of high levels of cytokines by immune cells, including IFN-γ, TGF-β, IL-2, IL-10, and IL-12, which play key roles in the immune response (Fig. 6L). Concurrently, the vaccine stimulated B lymphocytes to produce high levels of antibodies, further enhancing the immune defense (Fig. S5). In conclusion, ZL12138L was predicted to have potential as an effective vaccine through the activation of both innate and adaptive immune cells and promoting cytokine and antibody production, exhibiting promise for further development.
Fig. 6.

Prediction of innate and adaptive immune responses induced by the ZL12138L vaccine simulated by the C-ImmSim server. We conducted a simulation of three immunizations with the ZL12138L vaccine to predict the innate and adaptive immune responses. (A) NK cell population (cells per mm³). This plot shows the total number of NK cells over time post-vaccination, indicating the fluctuations in NK cell activation. (B) Macrophage population per state (cells per mm³). The plot categorizes macrophages into internalized, presenting (two states), and resting states, along with the total population, demonstrating the role of macrophages in antigen processing and presentation. (C) Dendritic cell (DC) population per state (cells per mm³). DCs are shown in internalized, presenting (two states), and resting states, along with the total population, highlighting their involvement in immune activation. (D) Epithelial cell population per state (cells per mm³). This plot illustrates the total, active, presenting, and actively infected states of epithelial cells, providing insights into the epithelial response post-vaccination. (E) B cell population per state (cells per mm³). The distribution of B cells in internalized, presenting, active, duplicating, and anergic states, depicting the dynamics of B cell activation and antibody production. (F) Total B cell population (cells per mm³). The total B cell count over time, including B memory cells and immunoglobulin levels (IgM, IgG1, and IgG2), reflecting the humoral immune response to the vaccine. (G) T helper (TH) cell population (cells per mm³). The total TH cell count, including non-memory and memory TH cells, showing the evolution of helper T cells post-immunization. (H) TH cell population per state (cells per mm³). Distribution of TH cells in active, resting, duplicating, and anergic states, providing insights into different stages of TH cell activation and function. (I) Cytotoxic T (TC) cell population (cells per mm³). Total TC cell count, including non-memory and memory TC cells, indicating the cytotoxic response elicited by the vaccine. (J) TC cell population per state (cells per mm³). The state-wise distribution of TC cells into active, resting, duplicating, and anergic states, reflecting different functional phases of cytotoxic T cells. (K) Changes in each subtype of helper T cells (cells per mm³). Dynamics of various TH cell subtypes (TH0, TH1, TH2, TH17, and Treg) over time, highlighting their respective roles in orchestrating the immune response. (L) Cytokine concentration (ng/mL). Changes in cytokine levels induced by the ZL12138L vaccine, showcasing the cytokine environment and immune signaling pathways activated during the immune response.
Abbreviations: DC, dendritic cell; NK, natural killer; TH, T helper.
3.10. Codon optimization, recombinant plasmid construction, and simulated gel electrophoresis of the ZL12138L vaccine
A detailed analysis of codon optimization, recombinant plasmid construction, and simulated results for the ZL12138L vaccine was conducted. The codon optimization results revealed a CAI value of 0.81 for the ZL12138L vaccine gene sequence, indicating high compatibility with the host cell's preferred codon usage pattern. This optimization is expected to enhance the mRNA translation efficiency and protein yield. Fig. 7A illustrates the plasmid construction circular map. The ZL13128L-pET28a plasmid, with a total length of 7408 bp, incorporates several crucial elements including the T7 promoter, lacI promoter, 6 × His tag, and multiple cloning sites (MCS). The XhoI and BamHI restriction sites denote the specific insertion location of the ZL13128L gene, represented by the pink region in Fig. 7A. Fig. 7B displays the predicted agarose gel electrophoresis (1% agarose) results, which validated the accuracy of the recombinant plasmid construction. The observed band sizes aligned with the expected plasmid construction, indicating successful plasmid assembly and correct target fragment insertion. Fig. 7C depicts the GC content distribution of the ZL12138L gene within the plasmid, with an average GC content of 60%. The optimized gene coding region, spanning from the XhoI site at 158 bp to the BamHI site at 2237 bp, exhibited a high GC content. This characteristic is conducive to enhancing gene expression efficiency and stability in host cells. These findings collectively demonstrated the successful optimization and construction of the theoretical ZL12138L vaccine plasmid, providing a solid foundation for subsequent expression and functional studies.
Fig. 7.

Codon optimization and recombinant plasmid construction of the ZL12138L vaccine. (A) Codon optimization and plasmid construction. Computational methods were used to optimize the codons of the ZL12138L vaccine gene for increased expression efficiency in E. coli. The optimized gene sequence was subsequently inserted into the pET28a plasmid using XhoI and BamHI restriction sites. The resulting recombinant expression plasmid, named ZL12138L-pET28a(+), encompasses vital elements, including the T7 promoter, lac operator, ribosome binding site (RBS), 6 × His tag, and the multiple cloning site (MCS). (B) Agarose gel electrophoresis simulation. The SnapGene software was employed to simulate agarose gel electrophoresis to validate the construction of the recombinant plasmid. Lane 1 represents the molecular weight marker (ZL12138L), lane 2 shows the uncut pET-28a(+) plasmid, and lane 3 displays the ZL12138L-pET28a(+) recombinant plasmid, indicating the successful insertion and expected molecular sizes. (C) GC content analysis. The GC content of the codon-optimized ZL12138L vaccine gene was assessed using SnapGene software, revealing a GC percentage of 60%. The plot illustrates the distribution of GC content across the length of the optimized gene sequence, with regions of varying GC proportions indicated by different colors: blue (low GC), green to yellow (medium GC), and red (high GC).
4. Discussion
Recent advances in computational power and algorithm optimization have established CAD and AI as pivotal forces driving biomedical research. CAD technology, through precise 3D modeling and simulation, enables the prediction and optimization of the structural features of vaccine candidates at the atomic level. AI techniques, particularly machine learning and deep learning algorithms, have markedly improved the ability to screen and optimize immunodominant epitopes from vast datasets by recognizing complex patterns and associations. The integration of these technologies has not only accelerated the vaccine design process but also enhanced the precision and success rate [73,74].
In the present study, we employed advanced CAD tools to analyze and predict immunodominant epitopes of MTB antigens, including HTL, CTL, and B lymphocyte epitopes, which are crucial for immune response activation. We further refined these epitopes using machine learning and deep learning algorithms, which can identify patterns and predict epitope immunogenicity, guiding the selection of the most effective epitope combinations [74,75].
Our vaccine design incorporated amino acid linkers, including GPGPG, AAY, and KK, as well as TLR agonists, to enhance the immunogenicity and stability [76], consistent with strategies employed in our previous studies [11,31,32,61,[77], [78], [79]]. Physicochemical analysis of the ZL12138L vaccine revealed a molecular weight of 67331.45 Da and a predicted solubility value of 0.47, indicating an acceptable size and solubility [80]. The predicted instability index of 28.42, well below the threshold of 40, suggested good stability [81]. Bioinformatics analyses indicated excellent immunogenicity and antigenicity, with an immunogenicity score of 4.14449 and high global population coverage for HLA class I and II alleles (92.41% and 90.17%, respectively), suggesting the potential for broad applicability and efficacy.
The structural analysis of ZL12138L revealed rich secondary and tertiary structural features crucial for biological function. The vaccine comprised 32.71% α-helices, 17.08% extended strands, and 41.53% random coils, facilitating antigen-specific antibody recognition [9,27,82]. Tertiary structure prediction showed 97.3% of the amino acid residues were in core regions, indicating an acceptable spatial model quality. Molecular docking analyses demonstrated good binding capacity with TLR-2 and TLR-4, with binding energies of −1173.4 and −1360.5 kcal/mol, respectively. Previous studies have shown that TLR-2 induces the secretion of pro-inflammatory cytokines, limiting MTB replication [83], while TLR-4 plays a crucial role in anti-tuberculosis immune responses [84]. NMA and MD simulations provided insights into the dynamic properties and stability of the vaccine under biological conditions. NMA revealed the major motion modes and potential function-related changes, particularly in the flexible regions near the TLR-2 and TLR-4 binding sites. MD simulations showed that ZL12138L could reach equilibrium in physiological environments, which is crucial for immunogenicity. MM/GBSA analysis calculated the binding free energy between ZL12138L and TLR-4, indicating high binding affinity primarily because of van der Waals forces, hydrophobic interactions, and hydrogen bonding.
Innate and adaptive immune responses both play crucial roles in the elimination of MTB from a host [5,[85], [86], [87], [88]]. The immune simulation results demonstrated the ability of ZL12138L to effectively activate various immune cells, including NK cells, macrophages, DCs, B lymphocytes, and T lymphocytes. The vaccine was predicted to induce an increase in the numbers of B lymphocytes and memory B cells, and high levels of IFN-γ and IL-2 production by T lymphocytes, which are crucial Th1-type cytokines for resisting MTB infections [58,[89], [90], [91]].
This method for designing vaccines based on bioinformatics tools offers considerable advantages over conventional techniques, which are primarily reflected in the following five aspects: (1) Efficient screening and analysis. Bioinformatics tools can rapidly screen for key antigen sequences of pathogens. This process can considerably shorten the time and cost required by traditional experimental methods, enabling the rapid identification of the targets of candidate vaccines. (2) Personalized vaccine design. Bioinformatics methods allow the analysis of genetic variations in pathogens across different populations, as well as analysis of the genetic diversity of hosts. This analysis facilitates the design of personalized vaccines for specific populations, thereby improving the vaccine efficacy and reducing adverse reactions. (3) Predicting vaccine immunogenicity and safety. Bioinformatics technologies can predict the immunogenicity (such as T-cell and B-cell epitopes) and safety (such as avoiding the risk of autoimmune reactions) of candidate vaccines. This early-stage elimination of unsuitable vaccine candidates reduces the risks associated with subsequent clinical trials. (4) Accelerating the development of new vaccines. During outbreaks of emerging pathogens (such as COVID-19), bioinformatics methods can quickly identify potential vaccine targets by analyzing the genetic sequences of the pathogens. For instance, in the development of COVID-19 vaccines, bioinformatics played a crucial role in identifying the spike protein of the SARS-CoV-2 virus as a vaccine target. (5) Reducing the number of experiments and animal use. Bioinformatics tools can simulate and predict experimental results, thereby, reducing the number of experiments and the use of animals in the vaccine design process. This approach not only improves the ethical standards of research but also considerably reduces the costs.
However, while the bioinformatics analyses provided strong evidence supporting the potential of ZL12138L, we acknowledge the limitations of the present study. Firstly, the effectiveness of the predicted immunodominant epitopes in actual immune responses requires experimental validation. The diversity of MHC molecules among individuals increases the complexity of epitope selection, necessitating extensive experimental verification in future work. Secondly, experimental research is required on the vaccine stability and immunogenicity in real biological systems, an essential step toward clinical application.
Future research directions will focus on continuously optimizing the design of MEVs while conducting appropriate experiments to evaluate their safety and effectiveness in real-world environments. To this end, we propose the following steps for future in vitro and in vivo validation of the vaccine designed in the present study: (1) Protein expression and purification. The use of recombinant DNA technology to express and purify vaccine proteins or peptides in E. coli expression systems. Identification and analysis of the molecular weight and purity of the vaccine antigens using mass spectrometry to verify the correct expression of the designed antigen sequence. (2) Analysis of the spatial structure of the ZL12138L vaccine. X-ray crystallography analysis of the 3D structure of vaccine antigens, ensuring that their folded state conforms to their natural configuration, which is crucial for eliciting effective immune responses. (3) Immunogenicity evaluation. Detection of the levels of antigen-specific antibodies and cytokines induced by the ZL12138L vaccine in mice using ELISA, ELISPOT, and other methods to investigate the reliability of the immune simulation results obtained in the present study. (4) Safety and protective efficiency evaluation: Evaluation of the safety of the vaccine and the protective efficiency against infection with the H37Rv strain using a ZL12138L vaccine-immunized mouse model. By following these steps, we aim to further substantiate the potential of ZL12138L as a highly effective and stable vaccine candidate, paving the way for its clinical application.
5. Conclusions
A novel MEV, ZL12138L, was successfully designed in the present study using CAD techniques and AI algorithms. The designed vaccine incorporated precisely screened HTL, CTL, and B-cell epitopes, and integrated TLR agonists and PADRE helper peptides to enhance the immunostimulatory capacity. Bioinformatics analyses revealed the predicted MEV had excellent immunogenicity and antigenicity, as well as being non-toxic and non-allergenic, with broad applicability across global populations. Furthermore, molecular docking and dynamics simulations indicated that ZL12138L had strong binding capacity toward TLR-2 and TLR-4 receptors, providing crucial evidence for its ability to activate innate immune responses. The immune simulation results suggested that the ZL12138L vaccine could induce high levels of Th1-type immune responses. Although further experimental validation is needed to support these findings, these analyses of ZL12138L have already provided new strategies toward tuberculosis prevention.
We anticipate that through continued research and development, the ZL12138L vaccine could become an effective tool to prevent tuberculosis infections, making a considerable contribution to global public health efforts. The present study demonstrated the potential of integrating advanced computational methods with immunological insights in the design of next-generation vaccines, paving the way for more efficient and targeted approaches in vaccine development against challenging pathogens such as MTB.
Funding
This work was sponsored by the Beijing Nova Program (20240484526). The funding body did not participate in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
CRediT authorship contribution statement
Li Zhuang: Writing – original draft, Visualization, Methodology, Formal analysis, Data curation. Awais Ali: Methodology. Ling Yang: Methodology, Formal analysis. Zhaoyang Ye: Methodology, Formal analysis. Linsheng Li: Methodology, Formal analysis. Ruizi Ni: Methodology, Formal analysis. Yajing An: Methodology, Formal analysis. Syed Luqman Ali: Writing – review & editing, Visualization, Conceptualization. Wenping Gong: Writing – review & editing, Visualization, Supervision, Project administration, Conceptualization.
Acknowledgments
None.
Declaration of competing interest
The authors declare no conflict of interest.
Data available statement
All data generated or analyzed during this study are included in the published article.
Ethics statement
Not applicable.
Informed consent
Not applicable.
Footnotes
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.imj.2024.100148.
Contributor Information
Syed Luqman Ali, Email: luqmanali@awkum.edu.pk.
Wenping Gong, Email: gwp891015@whu.edu.cn.
Appendix. Supplementary materials
Fig. S1. Phosphorylation sites predicted in the ZL12138L vaccine sequence. The green line represents possible phosphorylation sites of serine (S). The red line represents the possible phosphorylation sites of threonine (T). The blue line represents possible phosphorylation sites of tyrosine (Y). The purple horizontal line represents the threshold, usually 0.5 or 0.6, used to distinguish between high and low likelihood phosphorylation sites.
{kind=link}
Fig. S2. Discontinuous B-cell epitope prediction of the ZL12138L vaccine. (A) 3D structures of the 12 predicted discontinuous B-cell epitopes of the ZL12138L vaccine. Each panel represents a cluster of residues forming an epitope (yellow sphere), annotated with the number of residues involved and their respective clustering scores. (B–G) Predicted scores of discontinuous B-cell epitopes using the IEDB online server analyses. Each plot displays a different scoring metric along the sequence positions of the ZL12138L vaccine, identifying potential B-cell epitopes based on various properties. (B) Scores reflecting the antigenicity of residues, identifying most antigenic regions. (C) Scores based on residue solvent accessibility, highlighting areas that are likely to be extracellular and accessible to immune cells. (D) Scores indicating flexibility and likelihood of epitope formation, identifying highly flexible regions. (E) Scores representing hydrophilicity, marking regions likely to interact with water molecules and thus be more immunogenic. (F) Scores based on the presence of beta-turns, which are commonly found in B-cell epitopes. (G) Scores summarizing all attributes, providing a comprehensive overview of the predicted epitope regions. The highlighted regions in each plot (in yellow) indicate the specific positions within the ZL12138L sequence that are predicted to serve as potential discontinuous B-cell epitopes. These regions are key targets for inducing a strong immune response.
{kind=link}
Fig. S3. Normal mode analysis (NMA) of the interaction between the ZL12138L vaccine and TLR-2. (A) Deformability plot of the ZL12138L vaccine and TLR-2 complex, highlighting the flexibility of each atom index within the protein structure. Peaks indicate regions with higher flexibility, signifying potential conformational changes within the complex. (B) Eigenvalue plot displaying the relationship between eigenvalues and their corresponding mode indices for the first 20 modes. Lower eigenvalues represent more flexible modes, indicating intrinsic motions within the complex. (C) Cumulative variance explained by the first 20 normal modes. This plot quantifies the percentage of total motion captured by these modes, providing insights into the dominant dynamic behaviors of the ZL12138L and TLR-2 interaction. (D) B-factor comparison between the theoretical values obtained from NMA (red) and experimental values from the PDB structure (gray). This comparison helps in validating the normal mode calculations, highlighting consistent and divergent flexibility regions within the complex. (E) Covariance matrix of the residue indices, showcasing correlated motions between pairs of residues in the ZL12138L and TLR-2 complex. Red regions represent positive correlations (residues moving together), while blue regions denote negative correlations (residues moving inversely). (F) Elastic network model representing the inter-residue contact map of the complex. Darker shades indicate stronger interactions between residue pairs, which are essential for maintaining structural stability within the ZL12138L and TLR-2 complex.
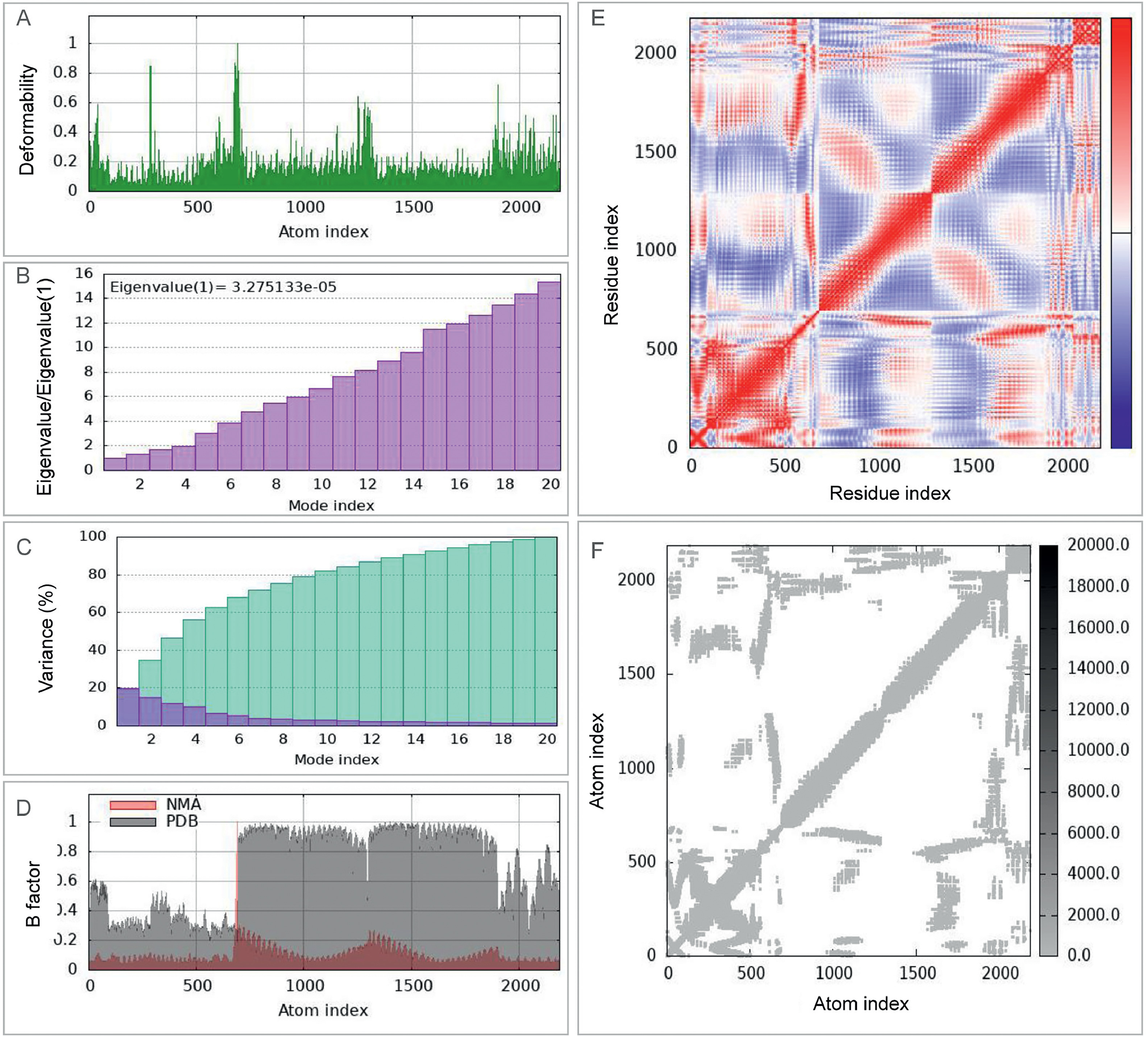{kind=link}
Fig.S4. Normal mode analysis (NMA) of the interaction between the ZL12138L vaccine and TLR-4. (A) Deformability plot of the ZL12138L vaccine and TLR-4 complex, highlighting the flexibility of each atom index within the protein structure. Peaks indicate regions with higher flexibility, signifying potential conformational changes within the complex. (B) Eigenvalue plot displaying the relationship between eigenvalues and their corresponding mode indices for the first 20 modes. Lower eigenvalues represent more flexible modes, indicating intrinsic motions within the complex. (C) Cumulative variance explained by the first 20 normal modes. This plot quantifies the percentage of total motion captured by these modes, providing insights into the dominant dynamic behaviors of the ZL12138L and TLR-4 interaction. (D) B-factor comparison between the theoretical values obtained from NMA (red) and experimental values from the PDB structure (gray). This comparison helps in validating the normal mode calculations, highlighting consistent and divergent flexibility regions within the complex. (E) Covariance matrix of the residue indices, showcasing correlated motions between pairs of residues in the ZL12138L and TLR-4 complex. Red regions represent positive correlations (residues moving together), while blue regions denote negative correlations (residues moving inversely). (F) Elastic network model representing the inter-residue contact map of the complex. Darker shades indicate stronger interactions between residue pairs, which are essential for maintaining structural stability within the ZL12138L and TLR-4 complex.
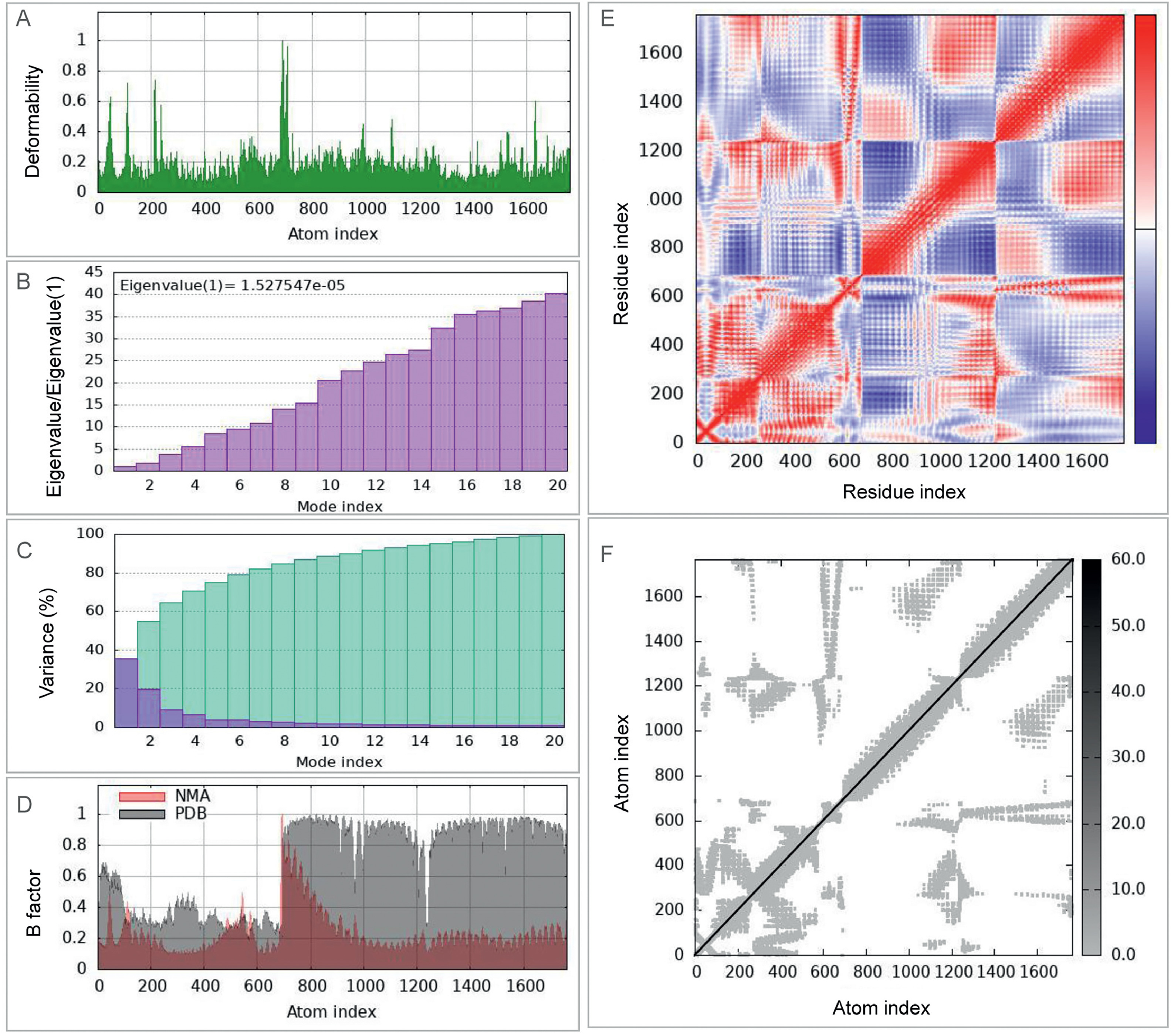{kind=link}
Fig. S5. The ability of the ZL12138L vaccine to induce B lymphocytes to secrete ZL12138L-specific antibodies. The simulation results of the immune response induced by the ZL12138L vaccine, focusing on the production of immunoglobulins and the formation of immunocomplexes over time.
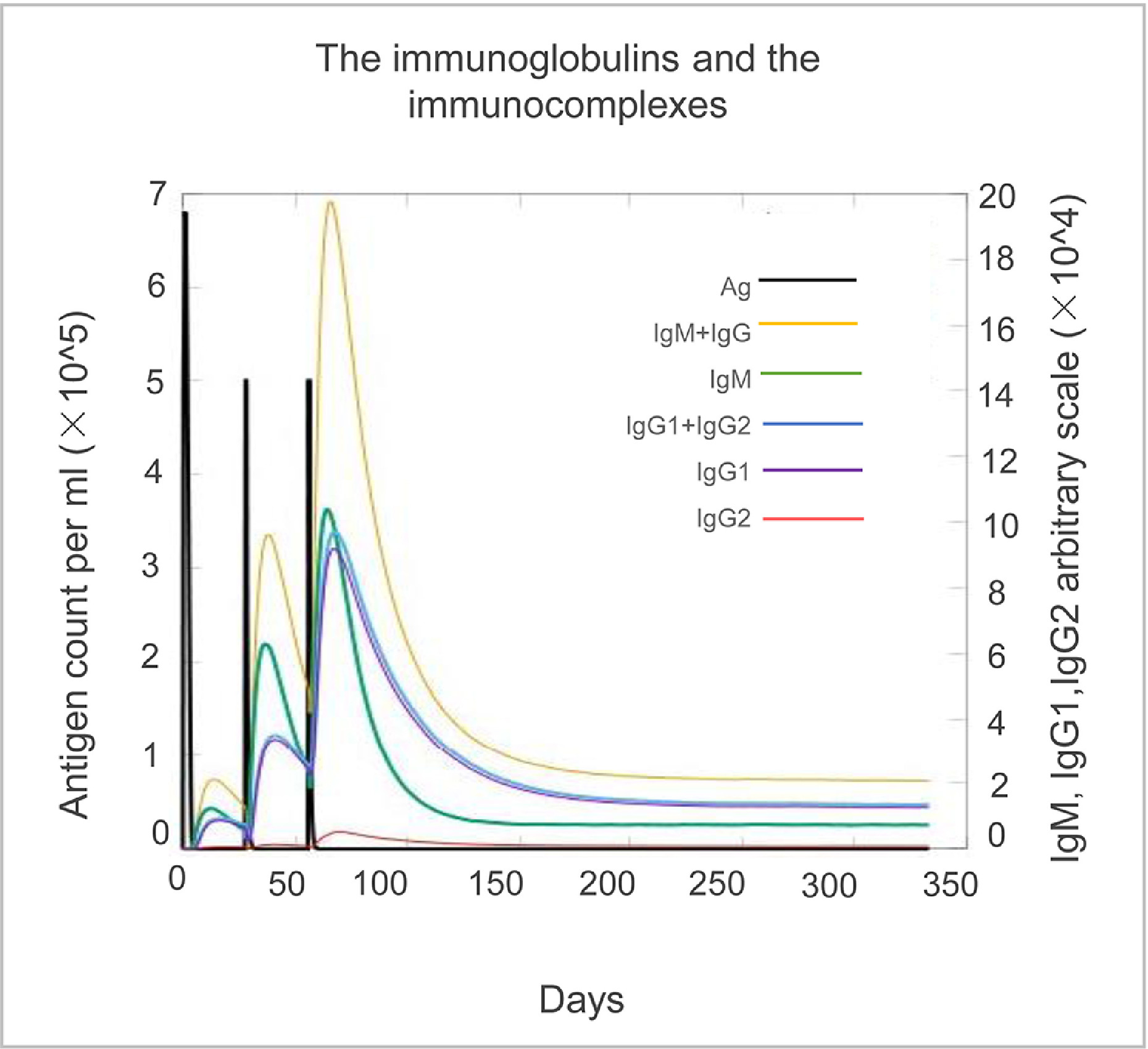{kind=link}
Table S1. Key parameters of servers and software used in the design and execution of the ZL12138L vaccine model.
Table S2. Predicted ZL12138L parameters.
Table S3. The discontinuous B-cell epitope residues of ZL12138L predicted by ElliPro.
Table S4. MM/GBSA analysis of the ZL12138L-TLR-4 complex.
References
- 1.Zhuang L., Yang L., Li L., et al. Mycobacterium tuberculosis: immune response, biomarkers, and therapeutic intervention. MedComm. 2024;5(1):e419. doi: 10.1002/mco2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang L., Ye Z., Li L., et al. Visualization analysis of research progress and trends in coexistence of lung cancer and pulmonary tuberculosis using bibliometrics. Med. Adv. 2024;2(2):144–164. doi: 10.1002/med4.58. [DOI] [Google Scholar]
- 3.Lin Z., Sun L., Wang C., et al. Bottlenecks and recent advancements in detecting Mycobacterium tuberculosis in patients with HIV. iLABMED. 2023;1(1):44–57. doi: 10.1002/ila2.11. [DOI] [Google Scholar]
- 4.WHO . World Health Organization; Geneva: 2023. Global tuberculosis report 2023; pp. 1–75. [Google Scholar]
- 5.Gong W., Du J. Optimising the vaccine strategy of BCG, ChAdOx1 85A, and MVA85A for tuberculosis control. Lancet Infect. Dis. 2024;24(3):224–226. doi: 10.1016/S1473-3099(23)00514-5. [DOI] [PubMed] [Google Scholar]
- 6.Dos Santos P.C.P., Messina N.L., de Oliveira R.D., et al. Effect of BCG vaccination against Mycobacterium tuberculosis infection in adult Brazilian health-care workers: a nested clinical trial. Lancet Infect. Dis. 2024;24(6):594–601. doi: 10.1016/s1473-3099(23)00818-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gong W., Pan C., Cheng P., et al. Peptide-based vaccines for tuberculosis. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.830497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng C., Jiang F., Liu Y., et al. Development and evaluation of a promising biomarker for diagnosis of latent and active tuberculosis infection. Infect. Dis. Immun. 2024;4(1):10–24. doi: 10.1097/id9.0000000000000104. [DOI] [Google Scholar]
- 9.Jiang F., Wang L., Wang J., et al. Design and development of a multi-epitope vaccine for the prevention of latent tuberculosis infection. Med. Adv. 2023;1(4):361–382. doi: 10.1002/med4.40. [DOI] [Google Scholar]
- 10.Jiang F., Peng C., Cheng P., et al. PP19128R, a multiepitope vaccine designed to prevent latent tuberculosis infection, induced immune responses in silico and in vitro assays. Vaccines. 2023;11(4):856. doi: 10.3390/vaccines11040856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang F., Liu Y., Xue Y., et al. Developing a multiepitope vaccine for the prevention of SARS-CoV-2 and monkeypox virus co-infection: a reverse vaccinology analysis. Int. Immunopharmacol. 2023;115 doi: 10.1016/j.intimp.2023.109728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhuang L., Ye Z., Li L., et al. Next-generation TB vaccines: progress, challenges, and prospects. Vaccines. 2023;11(8):1304. doi: 10.3390/vaccines11081304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng F., Liu Y., Yang Y., et al. Assessing computational tools for predicting protein stability changes upon missense mutations using a new dataset. Protein Sci. 2024;33(1):e4861. doi: 10.1002/pro.4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Notin P., Rollins N., Gal Y., et al. Machine learning for functional protein design. Nat. Biotechnol. 2024;42(2):216–228. doi: 10.1038/s41587-024-02127-0. [DOI] [PubMed] [Google Scholar]
- 15.Yang Z., Zeng X., Zhao Y., et al. AlphaFold2 and its applications in the fields of biology and medicine. Signal Transduct. Target. Ther. 2023;8(1):115. doi: 10.1038/s41392-023-01381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang P., Ning K. How much metagenome data is needed for protein structure prediction: the advantages of targeted approach from the ecological and evolutionary perspectives. iMeta. 2022;1(1):e9. doi: 10.1002/imt2.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu Z., Zhang L., Li S., et al. S–6P exhibits better immunogenicity than S–2P at lower doses of COVID-19 mRNA vaccines. Decod. Infect. Transm. 2024;2 doi: 10.1016/j.dcit.2024.100017. [DOI] [Google Scholar]
- 18.Ye Z., Li L., Yang L., et al. Impact of diabetes mellitus on tuberculosis prevention, diagnosis, and treatment from an immunologic perspective. Exploration. 2024;4(5) doi: 10.1002/exp.20230138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawkridge T., Scriba T.J., Gelderbloem S., et al. Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in healthy adults in South Africa. J. Infect. Dis. 2008;198(4):544–552. doi: 10.1086/590185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nemes E., Hesseling A.C., Tameris M., et al. Safety and immunogenicity of newborn MVA85A vaccination and selective, delayed bacille calmette-guerin for infants of human immunodeficiency virus-infected mothers: a phase 2 randomized, controlled trial. Clin. Infect. Dis. 2018;66(4):554–563. doi: 10.1093/cid/cix834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luabeya A.K., Kagina B.M., Tameris M.D., et al. First-in-human trial of the post-exposure tuberculosis vaccine H56: IC31 in Mycobacterium tuberculosis infected and non-infected healthy adults. Vaccine. 2015;33(33):4130–4140. doi: 10.1016/j.vaccine.2015.06.051. [DOI] [PubMed] [Google Scholar]
- 22.Tait D.R., Hatherill M., Van Der Meeren O., et al. Final analysis of a trial of M72/AS01E vaccine to prevent tuberculosis. N. Engl. J. Med. 2019;381(25):2429–2439. doi: 10.1056/nejmoa1909953. [DOI] [PubMed] [Google Scholar]
- 23.Rodríguez D., Vizcaíno C., Ocampo M., et al. Peptides from the Mycobacterium tuberculosis Rv1980c protein involved in human cell infection: insights into new synthetic subunit vaccine candidates. Biol. Chem. 2010;391(2–3):207–217. doi: 10.1515/bc.2010.019. [DOI] [PubMed] [Google Scholar]
- 24.Cockle P.J., Gordon S.V., Lalvani A., et al. Identification of novel Mycobacterium tuberculosis antigens with potential as diagnostic reagents or subunit vaccine candidates by comparative genomics. Infect. Immun. 2002;70(12):6996–7003. doi: 10.1128/IAI.70.12.6996-7003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kapopoulou A., Lew J.M., Cole S.T. The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis. 2011;91(1):8–13. doi: 10.1016/j.tube.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Doytchinova I.A., Flower D.R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J., Jiang F., Cheng P., et al. Construction of novel multi-epitope-based diagnostic biomarker HP16118P and its application in the differential diagnosis of Mycobacterium tuberculosis latent infection. Mol. Biomed. 2024;5(1):15. doi: 10.1186/s43556-024-00177-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dimitrov I., Bangov I., Flower D.R., et al. AllerTOP v.2: a server for in silico prediction of allergens. J. Mol. Model. 2014;20(6):2278. doi: 10.1007/s00894-014-2278-5. [DOI] [PubMed] [Google Scholar]
- 29.Nagpal G., Usmani S.S., Dhanda S.K., et al. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci. Rep. 2017;7:42851. doi: 10.1038/srep42851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saha S., Raghava G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins. 2006;65(1):40–48. doi: 10.1002/prot.21078. [DOI] [PubMed] [Google Scholar]
- 31.Cheng P., Xue Y., Wang J., et al. Evaluation of the consistence between the results of immunoinformatics predictions and real-world animal experiments of a new tuberculosis vaccine MP3RT. Front. Cell. Infect. Microbiol. 2022;12 doi: 10.3389/fcimb.2022.1047306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng C., Tang F., Wang J., et al. Immunoinformatic-based multi-epitope vaccine design for co-infection of Mycobacterium tuberculosis and SARS-CoV-2. J. Pers. Med. 2023;13(1):116. doi: 10.3390/jpm13010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ponomarenko J., Bui H.H., Li W., et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics. 2008;9:514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barkowsky E.V. Prediction of the secondary structure of globular proteins by their amino acid sequence. Acta Biol. Med. Ger. 1982;41(9):751–758. [PubMed] [Google Scholar]
- 35.Emini E.A., Hughes J.V., Perlow D.S., et al. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985;55(3):836–839. doi: 10.1128/JVI.55.3.836-839.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li C.H., Ma X.H., Chen W.Z., et al. A soft docking algorithm for predicting the structure of antibody-antigen complexes. Proteins. 2003;52(1):47–50. doi: 10.1002/prot.10382. [DOI] [PubMed] [Google Scholar]
- 37.Larsen J.E., Lund O., Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome. Res. 2006;2:2. doi: 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuen R., Kuniholm J., Lisk C., et al. Neisserial PorB immune enhancing activity and use as a vaccine adjuvant. Hum. Vaccin. Immunother. 2019;15(11):2778–2781. doi: 10.1080/21645515.2019.1609852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Office F.P. Erratum: Lactobacillus plantarum RS-09 induces M1-type macrophage immunity against Salmonella typhimurium challenge via the TLR2/NF-κB signalling pathway. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.937618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soltan M.A., Elbassiouny N., Gamal H., et al. In silico prediction of a multitope vaccine against Moraxella catarrhalis: reverse vaccinology and immunoinformatics. Vaccines. 2021;9(6):669. doi: 10.3390/vaccines9060669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magnan C.N., Zeller M., Kayala M.A., et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26(23):2936–2943. doi: 10.1093/bioinformatics/btq551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calis J.J., Maybeno M., Greenbaum J.A., et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013;9(10) doi: 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dimitrov I., Naneva L., Doytchinova I., et al. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30(6):846–851. doi: 10.1093/bioinformatics/btt619. [DOI] [PubMed] [Google Scholar]
- 44.Wilkins M.R., Gasteiger E., Bairoch A., et al. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999;112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 45.Hebditch M., Carballo-Amador M.A., Charonis S., et al. Protein-Sol: a web tool for predicting protein solubility from sequence. Bioinformatics. 2017;33(19):3098–3100. doi: 10.1093/bioinformatics/btx345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geourjon C., Deléage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 1995;11(6):681–684. doi: 10.1093/bioinformatics/11.6.681. [DOI] [PubMed] [Google Scholar]
- 47.Buchan D.W.A., Jones D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019;47(W1):W402–W407. doi: 10.1093/nar/gkz297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McGuffin L.J., Bryson K., Jones D.T. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16(4):404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 49.Varga J.K., Schueler-Furman O. Who binds better? let Alphafold2 decide! Angew. Chem. Int. Ed. Engl. 2023;62(28) doi: 10.1002/anie.202303526. [DOI] [PubMed] [Google Scholar]
- 50.Abramson J., Adler J., Dunger J., et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630(8016):493–500. doi: 10.1038/s41586-024-07487-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haron F.N., Azazi A., Chua K.H., et al. In silico structural modeling and quality assessment of Plasmodium knowlesi apical membrane antigen 1 using comparative protein models. Trop. Biomed. 2022;39(3):394–401. doi: 10.47665/tb.39.3.009. [DOI] [PubMed] [Google Scholar]
- 52.Muccee F., Ghazanfar S., Ajmal W., et al. In-silico characterization of estrogen reactivating β-glucuronidase enzyme in GIT associated microbiota of normal human and breast cancer patients. Genes. 2022;13(9):1545. doi: 10.3390/genes13091545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Setlur A.S., Chandrashekar K., Pandey S., et al. Comprehensive molecular interaction studies to construe the repellent/kill activity of geraniol during binding event against Aedes aegypti proteins. Mol. Biotechnol. 2023;65(5):726–740. doi: 10.1007/s12033-022-00560-7. [DOI] [PubMed] [Google Scholar]
- 54.Heo L., Park H., Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41(Web Server issue):384–W388. doi: 10.1093/nar/gkt458. W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gong W., Liang Y., Mi J., et al. Peptides-based vaccine MP3RT induced protective immunity against Mycobacterium tuberculosis infection in a humanized mouse model. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.666290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng P., Wang L., Gong W. In silico analysis of peptide-based biomarkers for the diagnosis and prevention of latent tuberculosis infection. Front. Microbiol. 2022;13 doi: 10.3389/fmicb.2022.947852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wiederstein M., Sippl M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(Web Server issue):W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gong W., Liang Y., Mi J., et al. A peptide-based vaccine ACP derived from antigens of Mycobacterium tuberculosis induced Th1 response but failed to enhance the protective efficacy of BCG in mice. Indian J. Tuberc. 2022;69(4):482–495. doi: 10.1016/j.ijtb.2021.08.016. [DOI] [PubMed] [Google Scholar]
- 59.Setlur A.S., Chandrashekar K., Pandey S., et al. Comprehensive molecular interaction studies to construe the repellent/kill activity of geraniol during binding event against Aedes aegypti proteins. Mol. Biotechnol. 2023;65(5):726–740. doi: 10.1007/s12033-022-00560-7. [DOI] [PubMed] [Google Scholar]
- 60.Peng C., Tang F., Wang J., et al. Immunoinformatic-based multi-epitope vaccine design for co-infection of Mycobacterium tuberculosis and SARS-CoV-2. J. Pers. Med. 2023;13(1):116. doi: 10.3390/jpm13010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng P., Jiang F., Wang G., et al. Bioinformatics analysis and consistency verification of a novel tuberculosis vaccine candidate HP13138PB. Front. Immunol. 2023;14 doi: 10.3389/fimmu.2023.1102578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vajda S., Yueh C., Beglov D., et al. New additions to the ClusPro server motivated by CAPRI. Proteins. 2017;85(3):435–444. doi: 10.1002/prot.25219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rapin N., Lund O., Bernaschi M., et al. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One. 2010;5(4):e9862. doi: 10.1371/journal.pone.0009862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y., Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57(4):702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 65.Lopéz-Blanco J.R., Garzón J.I., Chacón P. iMod: multipurpose normal mode analysis in internal coordinates. Bioinformatics. 2011;27(20):2843–2850. doi: 10.1093/bioinformatics/btr497. [DOI] [PubMed] [Google Scholar]
- 66.Kitao A. Principal component analysis and related methods for investigating the dynamics of biological macromolecules. J. 2022;5(2):298–317. doi: 10.3390/j5020021. [DOI] [Google Scholar]
- 67.Palma J., Pierdominici-Sottile G. On the uses of PCA to characterise molecular dynamics simulations of biological macromolecules: basics and tips for an effective use. Chemphyschem. 2023;24(2) doi: 10.1002/cphc.202200491. [DOI] [PubMed] [Google Scholar]
- 68.Godschalk F., Genheden S., Söderhjelm P., et al. Comparison of MM/GBSA calculations based on explicit and implicit solvent simulations. Phys. Chem. Chem. Phys. 2013;15(20):7731–7739. doi: 10.1039/c3cp00116d. [DOI] [PubMed] [Google Scholar]
- 69.Hildebrand P.W., Rose A.S., Tiemann J.K.S. Bringing molecular dynamics simulation data into view. Trends Biochem. Sci. 2019;44(11):902–913. doi: 10.1016/j.tibs.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 70.Faris A., Ibrahim I.M., Al Kamaly O., et al. Computer-aided drug design of novel derivatives of 2-amino-7, 9-dihydro-8H-purin-8-one as potent pan-janus JAK3 inhibitors. Molecules. 2023;28(15):5914. doi: 10.3390/molecules28155914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Puigbò P., Guzmán E., Romeu A., et al. OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. 2007;35(Web Server issue):W126–W131. doi: 10.1093/nar/gkm219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gould N., Hendy O., Papamichail D. Computational tools and algorithms for designing customized synthetic genes. Front. Bioeng. Biotechnol. 2014;2:41. doi: 10.3389/fbioe.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tao Y., Zhang Y., Li Y., et al. Computer-aided designing of a novel multi-epitope DNA vaccine against severe fever with thrombocytopenia syndrome virus. BMC Infect. Dis. 2024;24(1):476. doi: 10.1186/s12879-024-09361-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim E.E. Artificial intelligence and computer-aided diagnosis in medicine. Curr. Med. Imaging Rev. 2020;16(1):1. doi: 10.2174/157340561601200106142451. [DOI] [PubMed] [Google Scholar]
- 75.Ramprasadh S.V., Rajakumar S., Srinivasan S., et al. Computer-aided multi-epitope based vaccine design against monkeypox virus surface protein A30L: an immunoinformatics approach. Protein J. 2023;42(6):645–663. doi: 10.1007/s10930-023-10150-4. [DOI] [PubMed] [Google Scholar]
- 76.Shamriz S., Ofoghi H., Moazami N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput. Biol. Med. 2016;76:24–29. doi: 10.1016/j.compbiomed.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 77.Wang J., Jiang F., Cheng P., et al. Construction of novel multi-epitope-based diagnostic biomarker HP16118P and its application in the differential diagnosis of Mycobacterium tuberculosis latent infection. Mol. Biomed. 2024;5(1):15. doi: 10.1186/s43556-024-00177-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang F., Han Y., Liu Y., et al. A comprehensive approach to developing a multi-epitope vaccine against Mycobacterium tuberculosis: from in silico design to in vitro immunization evaluation. Front. Immunol. 2023;14 doi: 10.3389/fimmu.2023.1280299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang F., Peng C., Cheng P., et al. PP19128R, a multiepitope vaccine designed to prevent latent tuberculosis infection, induced immune responses in silico and in vitro assays. Vaccines. 2023;11(4):856. doi: 10.3390/vaccines11040856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Khatoon N., Pandey R.K., Prajapati V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017;7(1):8285. doi: 10.1038/s41598-017-08842-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kaur A., Pati P.K., Pati A.M., et al. Physico-chemical characterization and topological analysis of pathogenesis-related proteins from Arabidopsis thaliana and Oryza sativa using in-silico approaches. PLoS One. 2020;15(9) doi: 10.1371/journal.pone.0239836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Corradin G., Villard V., Kajava A.V. Protein structure based strategies for antigen discovery and vaccine development against malaria and other pathogens. Endocr. Metab. Immune Disord. Drug Targets. 2007;7(4):259–265. doi: 10.2174/187153007782794371. [DOI] [PubMed] [Google Scholar]
- 83.Gopalakrishnan A., Salgame P. Toll-like receptor 2 in host defense against Mycobacterium tuberculosis: to be or not to be-that is the question. Curr. Opin. Immunol. 2016;42:76–82. doi: 10.1016/j.coi.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sepehri Z., Kiani Z., Kohan F., et al. Toll-like receptor 4 as an immune receptor against Mycobacterium tuberculosis: a systematic review. Lab. Med. 2019;50(2):117–129. doi: 10.1093/labmed/lmy047. [DOI] [PubMed] [Google Scholar]
- 85.Tahir Ul Qamar M., Shokat Z., Muneer I., et al. Multiepitope-based subunit vaccine design and evaluation against respiratory syncytial virus using reverse vaccinology approach. Vaccines. 2020;8(2):288. doi: 10.3390/vaccines8020288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhuang L., Yang L., Li L., et al. Mycobacterium tuberculosis: immune response, biomarkers, and therapeutic intervention. MedComm. 2024;5(1):e419. doi: 10.1002/mco2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li L.S., Yang L., Zhuang L., et al. From immunology to artificial intelligence: revolutionizing latent tuberculosis infection diagnosis with machine learning. Mil. Med. Res. 2023;10(1):58. doi: 10.1186/s40779-023-00490-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gong W., Wu X. Differential diagnosis of latent tuberculosis infection and active tuberculosis: a key to a successful tuberculosis control strategy. Front. Microbiol. 2021;12 doi: 10.3389/fmicb.2021.745592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ravesloot-Chávez M.M., Van Dis E., Stanley SA. The innate immune response to Mycobacterium tuberculosis infection. Annu. Rev. Immunol. 2021;39:611–637. doi: 10.1146/annurev-immunol-093019-010426. [DOI] [PubMed] [Google Scholar]
- 90.Cooper A.M., Dalton D.K., Stewart T.A., et al. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med. 1993;178(6):2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Flynn J.L., Chan J., Triebold K.J., et al. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 1993;178(6):2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Phosphorylation sites predicted in the ZL12138L vaccine sequence. The green line represents possible phosphorylation sites of serine (S). The red line represents the possible phosphorylation sites of threonine (T). The blue line represents possible phosphorylation sites of tyrosine (Y). The purple horizontal line represents the threshold, usually 0.5 or 0.6, used to distinguish between high and low likelihood phosphorylation sites.
Fig. S2. Discontinuous B-cell epitope prediction of the ZL12138L vaccine. (A) 3D structures of the 12 predicted discontinuous B-cell epitopes of the ZL12138L vaccine. Each panel represents a cluster of residues forming an epitope (yellow sphere), annotated with the number of residues involved and their respective clustering scores. (B–G) Predicted scores of discontinuous B-cell epitopes using the IEDB online server analyses. Each plot displays a different scoring metric along the sequence positions of the ZL12138L vaccine, identifying potential B-cell epitopes based on various properties. (B) Scores reflecting the antigenicity of residues, identifying most antigenic regions. (C) Scores based on residue solvent accessibility, highlighting areas that are likely to be extracellular and accessible to immune cells. (D) Scores indicating flexibility and likelihood of epitope formation, identifying highly flexible regions. (E) Scores representing hydrophilicity, marking regions likely to interact with water molecules and thus be more immunogenic. (F) Scores based on the presence of beta-turns, which are commonly found in B-cell epitopes. (G) Scores summarizing all attributes, providing a comprehensive overview of the predicted epitope regions. The highlighted regions in each plot (in yellow) indicate the specific positions within the ZL12138L sequence that are predicted to serve as potential discontinuous B-cell epitopes. These regions are key targets for inducing a strong immune response.
Fig. S3. Normal mode analysis (NMA) of the interaction between the ZL12138L vaccine and TLR-2. (A) Deformability plot of the ZL12138L vaccine and TLR-2 complex, highlighting the flexibility of each atom index within the protein structure. Peaks indicate regions with higher flexibility, signifying potential conformational changes within the complex. (B) Eigenvalue plot displaying the relationship between eigenvalues and their corresponding mode indices for the first 20 modes. Lower eigenvalues represent more flexible modes, indicating intrinsic motions within the complex. (C) Cumulative variance explained by the first 20 normal modes. This plot quantifies the percentage of total motion captured by these modes, providing insights into the dominant dynamic behaviors of the ZL12138L and TLR-2 interaction. (D) B-factor comparison between the theoretical values obtained from NMA (red) and experimental values from the PDB structure (gray). This comparison helps in validating the normal mode calculations, highlighting consistent and divergent flexibility regions within the complex. (E) Covariance matrix of the residue indices, showcasing correlated motions between pairs of residues in the ZL12138L and TLR-2 complex. Red regions represent positive correlations (residues moving together), while blue regions denote negative correlations (residues moving inversely). (F) Elastic network model representing the inter-residue contact map of the complex. Darker shades indicate stronger interactions between residue pairs, which are essential for maintaining structural stability within the ZL12138L and TLR-2 complex.
Fig.S4. Normal mode analysis (NMA) of the interaction between the ZL12138L vaccine and TLR-4. (A) Deformability plot of the ZL12138L vaccine and TLR-4 complex, highlighting the flexibility of each atom index within the protein structure. Peaks indicate regions with higher flexibility, signifying potential conformational changes within the complex. (B) Eigenvalue plot displaying the relationship between eigenvalues and their corresponding mode indices for the first 20 modes. Lower eigenvalues represent more flexible modes, indicating intrinsic motions within the complex. (C) Cumulative variance explained by the first 20 normal modes. This plot quantifies the percentage of total motion captured by these modes, providing insights into the dominant dynamic behaviors of the ZL12138L and TLR-4 interaction. (D) B-factor comparison between the theoretical values obtained from NMA (red) and experimental values from the PDB structure (gray). This comparison helps in validating the normal mode calculations, highlighting consistent and divergent flexibility regions within the complex. (E) Covariance matrix of the residue indices, showcasing correlated motions between pairs of residues in the ZL12138L and TLR-4 complex. Red regions represent positive correlations (residues moving together), while blue regions denote negative correlations (residues moving inversely). (F) Elastic network model representing the inter-residue contact map of the complex. Darker shades indicate stronger interactions between residue pairs, which are essential for maintaining structural stability within the ZL12138L and TLR-4 complex.
Fig. S5. The ability of the ZL12138L vaccine to induce B lymphocytes to secrete ZL12138L-specific antibodies. The simulation results of the immune response induced by the ZL12138L vaccine, focusing on the production of immunoglobulins and the formation of immunocomplexes over time.
Table S1. Key parameters of servers and software used in the design and execution of the ZL12138L vaccine model.
Table S2. Predicted ZL12138L parameters.
Table S3. The discontinuous B-cell epitope residues of ZL12138L predicted by ElliPro.
Table S4. MM/GBSA analysis of the ZL12138L-TLR-4 complex.