

Abstract
Background
Primary central nervous system lymphoma (PCNSL) is a rare lymphoid malignancy. Systemic profiling of the PCNSL tumor microenvironment (TME) was previously conducted through gene expression analysis. We investigated the prognostic impact of TME on survival to establish novel prognostic biomarkers in PCNSL patients.
Methods
We analyzed expression levels of 770 neuroinflammation‐related (NFR) genes via NanoString nCounter technology in tumor samples from 30 PCNSL patients. Genes related to the “recurrence group (RG)” or “non‐recurrence group (NRG)” were identified and validated using whole transcriptomic analysis of an independent PCNSL cohort (n = 30).
Results
Forty‐five of 770 NFR genes were highly expressed in the RG (3‐year overall survival (OS, 22.2%), compared with the NRG group (3‐year OS 66.7%). Signatures related to glial cells were enriched in the RG‐associated gene set. Multivariate analysis revealed that high expressions of TUBB4A (p = 0.028, HR: 3.88), S100B (p = 0.046, HR: 3.093), and SLC6A1 (p = 0.034, HR: 3.765) were significantly related to death. Expression levels of these three genes were also significantly associated with poor OS in the validation cohort. Immunohistochemical staining against TUBB4A, S100B, and proteins specific to glial cells (GFAP, OLIG2, and CD68) revealed significantly higher positivity in RG glial cells.
Conclusion
These data suggest that TME‐related genes play a crucial role in the pathogenesis of PCNSL, complementing the well‐known involvement of the NF‐kB signaling pathway. TME targeting, especially glial cell‐specific proteins, may thus open new and complementary avenues of therapy for all stages of PCNSL.
Keywords: Gliosis, Primary central nervous system lymphoma, Tumor microenvironment in central nervous system
1. INTRODUCTION
Non‐Hodgkin, primary central nervous system lymphoma (PCNSL) is defined as a malignancy that arises within the brain, commonly presenting as diffuse large B‐cell lymphoma (DLBCL) representing 90–95% of cases [1]. While rare, accounting for approximately 2–3% of all brain tumors, the advanced peak onset age in the 60 s (median age of 68 in our department) complicates treatment [1, 2]. Untreated disease is rapidly fatal and 5‐year survival rates are around 20%, but even treatment cannot prevent early mortality (<60 days) in approximately 10% of the population [3].
Fortunately, the advent of next‐generation sequencing has facilitated a number of comprehensive genetic analysis studies on PCNSL [2, 4–6], and novel, molecular genetic classifications of DLBCL based on gene mutation profiles have been proposed. In spite of this, profiling efforts remain complicated by the genetically heterogeneous nature of systemic DLBCL, with more than 150 genetic abnormalities currently reported, while only a small number are detected with a frequency of more than 10% [7, 8, 9]. Such variability and progressive mutations over time make specific targeting difficult from a therapeutic standpoint. However, in 2018, Schmitz et al. [9] published the results of a sequencing analysis of systemic DLBCL, which revealed four persistent subtypes (MCD, BN2, N1, and EZB). Chapuy et al. [8] reported a similar sequencing analysis, which showed that DLBCL can be subdivided into five subtypes (C1‐5). In a recent molecular classification proposed for DLBCL, most PCNSL has been grouped with the MCD/C5 subtype in terms of genetic abnormalities [10]. Meanwhile, comprehensive gene mutation profiling of PCNSL revealed genomic abnormalities in targets, such as MYD88 and CD79B, to be associated with the B‐cell receptor (BCR) signaling pathway and immune escape [2, 5, 6, 11]. Although this has led to advances in targeted therapies, including tirabrutninb and ibrutinib (a Bruton's tyrosine kinase [BTK] inhibitor targeting downstream of BCR signaling), candidate biomarkers for diagnosis and prediction of survival in PCNSL patients remain scarce [12]. Despite the introduction of multiagent methotrexate‐based induction therapies, recurrence still occurs at rates of over 60% and therapeutic resistance is a significant challenge in the management of relapsed PCNSL patients [13]. Additionally, 10–15% demonstrate a primary refractory disease, increasing early mortality and highlighting an unmet need for alternative therapeutic options [14].
Notably, recent reports related to the genetic activity of PCNSL revealed that gene mutations in MYD88 L265P, plus gene expression levels of MYC, telomerase activity, and telomerase reverse transcriptase (TERT), impact clinical factors and response in PCNSL [2, 15, 16]. Additionally, genes related to the systemic profiling of tumor microenvironment (TME) cells were also investigated in several study groups [17, 18, 19, 20, 21]. The TME of PCNSL, which contributes significantly to tumor activity and chemotherapy resistance, consists of immune cells (e.g., neutrophils, dendric cells, and lymphocytes) and glial cells (e.g., astrocytes, oligodendrocytes, and microglial cells) [22]. Systemic TME profiling for PCNSL is usually performed through gene expression analysis by whole transcriptome analysis and single‐cell RNA sequencing analysis, with stratification by spatial transcriptome analysis [19, 21]. The status of infiltrating T cells (TILs) and the expression of PD‐L1 in microglial cells/macrophages (tumor‐associated macrophages) are particularly important in this regard, as PD‐L1 is closely linked to tumor status [23, 24]. However, the lack of a validation cohort is the primary limitation of these analyses and the impact of TME on PCNSL prognosis remains uncertain.
Here, we investigated the roles of TME cells by linking a transcriptomic data analysis of 770 RNA targets related to neuroinflammation with clinical information and histopathology.
2. MATERIALS AND METHODS
2.1. Patients and cohorts
Two cohorts were included in this study (Figure 1A): a discovery cohort from our institute paired with a validation cohort from a controlled access database. We extracted tumor tissues from PCNSL cases at the University of Tsukuba Hospital from 2005 to 2019. Pathological diagnoses were made in accordance with the revised 4th WHO classification [25]. Formalin‐fixed paraffin‐embedded (FFPE) tumor tissues from 30 PCNSL patients were analyzed by the nCounter system (NanoString Technologies) with 770 neuroinflammation‐related (NFR) genes as the discovery cohort.
FIGURE 1.
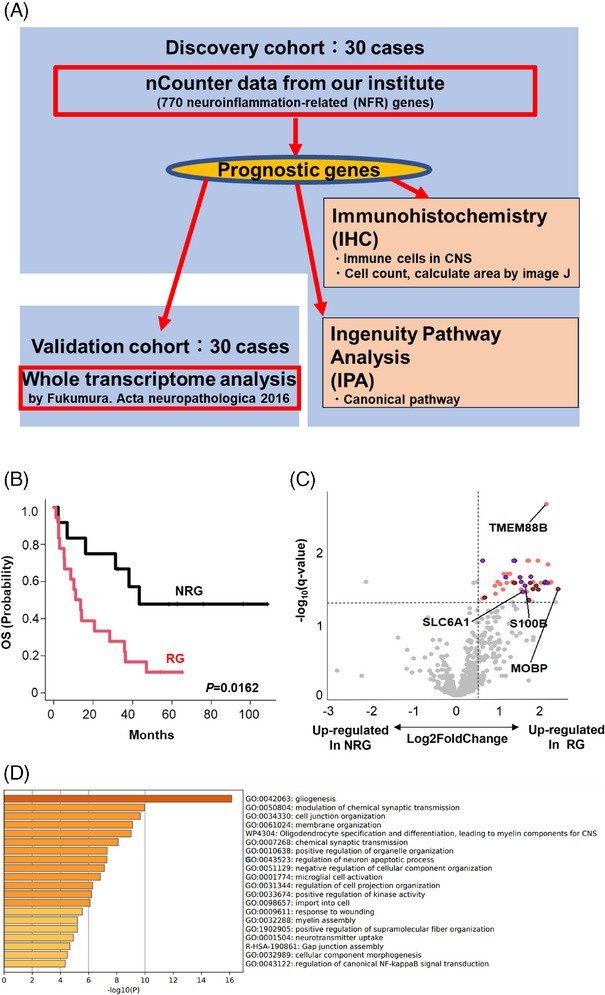
Identification of predicting factors for primary central nervous system lymphoma (PCNSL) outcomes. (A) Schematic representation of overall study design. Thirty cases of newly diagnosed PCNSL were recruited for a discovery cohort using nCounter methods, followed by analysis of the validation cohort. Whole transcriptome analysis data (previously published) was included as a validation cohort. (B) The discovery cohort was divided into two groups according to prognoses. Relapsed PCNSL patients were classified into either the “recurrence group (RG)” or the “non‐recurrence group (NRG)”. (C) A volcano plot indicates differentially expressed genes highly expressed in RG (right) and NRG (left) of the discovery cohort. p‐value adjustment was performed using the Benjamini–Hochberg method. Forty‐five genes were significantly upregulated in RG (q‐value ≤ 0.05, log2 fold change > 0.5). Among these 45 genes, firebrick dots represent genes linked to gliogenesis (GO:0042063) (D), purple dots represent genes linked to modulation of chemical synaptic transmission (GO:0050804) (D) and orange dots represent other genes. (D) Gene ontology (GO) analysis of 45 prognostic genes from the differential expression analysis (C) was performed using the Metascape web application (http://metascape.org). Enriched GO terms are shown by ID and category. GO, gene ontology; NRG, nonrecurrence group; OS, overall survival; PCNSL, primary central nervous system lymphoma; RG, recurrence group; WTA, whole transcriptome analysis.
The controlled access datasets of whole transcriptomic analyses from 30 PCNSL patients (previously published by Fukumura et al. [6]) in the NBDC Human Database (https://humandbs.biosciencedbc.jp/) were chosen as the validation cohort.
2.2. Gene‐expression profiling using the nCounter system in the discovery cohort
We used the Human Neuroinflammation panel (NanoString Technologies), which consists of 770 NFR genes, for nCounter‐based gene‐expression measurements to extract candidate prognostic genes in the discovery cohort (Table S1). Detailed information about sample preparation and assay procedure is provided in the Supporting Information Methods. For differential expression analyses, p‐values were adjusted using the Benjamini–Hochberg correction algorithm [26].
Gene Ontology (GO) enrichment analysis was performed using the Metascape web application (http://metascape.org) and ingenuity pathway analysis (IPA) (Qiagen). Candidate prognostic genes were applied to find top upstream regulator pathways enriched from these genes.
2.3. Gene‐mutation analysis in the discovery cohort
Targeted deep sequencing (TDS) was performed in genomic DNA extracted from tumors using our in‐house PCNSL panel for 12 candidate genes (Table S2).
2.4. Clinical data collection and prognostic analysis in the discovery cohort
Clinical data, including age, Karnofsky performance status (KPS), lactate dehydrogenase (LDH) levels, and altered mental status, were available in 30 patients. Overall survival (OS) was defined as the time from diagnosis to the last follow‐up or death.
2.5. Immunohistochemical analysis
We used immunohistochemical (IHC) analyses to probe the expression levels of proteins of interest as described in the Supplementary Methods. ImageJ (National Institutes of Health) was used for densitometric analyses and quantification of protein expression.
2.6. Statistical analyses
OS probability was estimated using the Kaplan–Meier method. A Cox proportional hazards model was used to assess predictive values of the expression level of each gene and clinical risk factors in univariate and multivariate analyses. Gene expression data related to RG were subjected to multivariate analysis with clinical variables with p‐values less than 0.15 in the univariate analysis. The results of analyses were validated using the whole transcriptome analysis data in an independent PCNSL cohort. A Student's t‐test was used to analyze the differences in marker‐positive areas/cells in IHC. Statistical analyses were performed using R ver. 4.3.1 (http://CRAN.R‐project.org/). Detailed protocols are available in the Supporting Information Methods.
2.7. Ethical considerations
Diagnosed patients with PCNSL in the discovery cohort provided written, informed consent for tissue collection and subsequent research purposes. The use of data from the validation cohort was approved by the University of Tokyo, Kyorin University, Saitama Medical University, National Cancer Center Research Institute, and National Cancer Center Hospital. This study conformed to the principles of the Declaration of Helsinki and was approved by the Institutional Review Board of the University of Tsukuba Institute of Medicine (approval # R02‐84).
3. RESULT
3.1. Differences in gene regulation are significantly associated with poor prognosis
Data regarding the observation period and patient survival status were available for all 30 PCNSL patients in the discovery cohort (Table 1; Table S3). The median age at diagnosis was 69 years old (range, 32–83), with a median follow‐up period of 25 months (range, 1–110 months), an OS at 3 years of 39.7%, and progression‐free survival at 3 years of 25.4%.
TABLE 1.
Summary of patient characteristics.
| Characteristic | Discovery cohort | Validation cohort |
|---|---|---|
| No. of patients (%) | ||
| Age > 70 years | 14 (47) | 11 (36) |
| Male sex | 19 (63) | 22 (73) |
| Karnofsky performance status >60 | 7 (23) | 17 (56) |
| Elevated lactate dehydrogenase | 14 (47) | 20/29 (69) |
| Deep brain lesions | 22 (73) | 14/30 (47) |
| Altered mentation | 19 (63) | 9/30 (30) |
| Frontline treatment | ||
| HDMTX‐based chemotherapy | 28 (93) | 28 (93) |
| Others | 2 (7) | 2 (7) |
| Non‐GCB (Hans algorithm) | 24 (80) | 25 (83) |
We identified genes correlated with patient outcomes by analyzing FFPE samples obtained at initial diagnosis from 30 PCNSL patients. These patients were divided into two groups according to their clinical course (Figure 1B). Similar to previous reports, all relapses occurred within 2 years in PCNSL in our institute [27]. Next, 18 patients with relapsed or refractory PCNSL were defined as the “recurrence group (RG)” while the remaining 12 patients were classified into the “non‐recurrence group (NRG)” group. The OS of RG was significantly poorer than NRG (3‐year OS 22.2% vs. 66.7%).
We then used commercially available probe sets that include 770 NFR genes (Table S1). This screen identified 45 genes with statistically significant upregulation in the RG (q‐value < 0.05, log2 fold change > 0.5, Figure 1C; Table S4). GO analysis then further revealed that most prognostic genes were associated with gliogenesis, with oligodendrocyte and astrocyte‐related signatures also enriched in the RG‐associated gene set (Figure 1D). Furthermore, IPA identified the neuroinflammation signaling pathway as the most significant among the up‐regulated pathways in the RG (Figure S1). Therefore, it is logical that the pathway is induced by the activation of microglial cells and leads to the regulation of NF‐κB activity [28].
3.2. Identification of genes leading to poor prognosis independent of clinical factors
A univariate Cox proportional hazards model was conducted to identify genes significantly associated with OS of PCNSL patients in our cohort. Cut‐off expression values of each gene were determined using maximally selected rank statistics [29]. Among these 45 genes, high expression of 19 genes were significantly associated with poor prognoses (Table 2).
TABLE 2.
Univariate analysis of clinical factors and neuroinflammation‐related gene expressions in the discovery cohort.
| Variable | Group | Univariate analysis | |||
|---|---|---|---|---|---|
| n | 2 years OS, % | Median, years | p‐value | ||
| Clinical factors | |||||
| Age, years | ≤70 | 16 | 50 | 1.86 | 0.965 |
| >70 | 14 | 50 | 2.36 | ||
| KPS | <70 | 23 | 34.8 | 1.14 | 0.0362 |
| ≥70 | 7 | 100 | NA | ||
| Sex | Male | 19 | 52.6 | 2.62 | 0.518 |
| Female | 11 | 45.5 | 1.73 | ||
| LDH > upper limit | Low | 16 | 62.5 | 3.04 | 0.431 |
| High | 14 | 35.7 | 1.16 | ||
| Consciousness levels at diagnosis | No | 11 | 72.7 | 3.19 | 0.127 |
| Yes | 19 | 36.8 | 0.93 | ||
| Involvement of deep brain | No | 8 | 62.5 | 2.5 | 0.902 |
| Yes | 22 | 45.5 | 1.53 | ||
| Cell of origin | Non‐GCB | 24 | 50 | 1.86 | 0.353 |
| GCB | 6 | 50 | 1.73 | ||
| Gene expressions | |||||
| SOX10 | Low | 8 | 75 | 3.03 | 0.0438 |
| High | 22 | 40.9 | 0.89 | ||
| MOG | Low | 5 | 100 | NA | 0.0396 |
| High | 25 | 40 | 1.17 | ||
| MAG | Low | 7 | 85.7 | NA | 0.0248 |
| High | 23 | 39.1 | 1.17 | ||
| GJB1 | Low | 5 | 80 | NA | 0.0448 |
| High | 25 | 44 | 1.35 | ||
| MAL | Low | 8 | 75 | NA | 0.0438 |
| High | 22 | 40.9 | 1.26 | ||
| PLXNB3 | Low | 7 | 71.4 | NA | 0.0444 |
| High | 23 | 43.5 | 1.35 | ||
| S100B | Low | 8 | 68.2 | 3.03 | 0.00569 |
| High | 22 | 0 | 0.89 | ||
| PMP22 | Low | 8 | 75 | NA | 0.0462 |
| High | 22 | 40.9 | 1.26 | ||
| MYRF | Low | 13 | 69.2 | NA | 0.0311 |
| High | 17 | 35.2 | 1.17 | ||
| TMC7 | Low | 6 | 83.3 | NA | 0.0326 |
| High | 24 | 41.7 | 1.26 | ||
| PLEKHB1 | Low | 8 | 75 | NA | 0.0478 |
| High | 22 | 40.9 | 1.26 | ||
| SLC6A1 | Low | 5 | 60 | 2.99 | 0.0042 |
| High | 25 | 0 | 0.73 | ||
| MAPT | Low | 12 | 66.7 | 3.19 | 0.0112 |
| High | 18 | 25 | 1.03 | ||
| TUBB4A | Low | 8 | 63.6 | 3.03 | 0.00663 |
| High | 22 | 11.7 | 0.89 | ||
| GRM3 | Low | 8 | 59.1 | 2.83 | 0.0411 |
| High | 22 | 25 | 0.99 | ||
| ERBB3 | Low | 10 | 70 | NA | 0.0287 |
| High | 20 | 40 | 1.26 | ||
| NLGN2 | Low | 11 | 63.2 | 3.19 | 0.0183 |
| High | 19 | 27.3 | 1.14 | ||
| DST | Low | 12 | 66.7 | 3.03 | 0.0273 |
| High | 18 | 25 | 0.89 | ||
| GJA1 | Low | 11 | 63.2 | 2.99 | 0.0231 |
| High | 19 | 27.3 | 0.93 | ||
Abbreviation: KPS, Karnofsky performance status.
We identified several specific clinical parameters reported previously as prognostic factors of PCNSL in our discovery cohort. These include consciousness levels at diagnosis and elevated creatinine clearance (CrCl); age > 50 years and KPS < 70; age > 75 years; increased serum LDH level; cerebrospinal fluid protein concentration; and lymphoma involvement of deep brain structures such as the basal ganglia, corpus callosum, brainstem, periventricular region, and cerebellum [13, 30]. As expected, KPS > 70 (p = 0.0362), and consciousness levels at diagnosis (other than JCS 0–1) (p = 0.127) were associated with poor OS (p < 0.15) after univariate analysis in the discovery cohort (Table 2).
We next used multivariate analysis to measure the effect of clinical factors such as KPS and consciousness level on transcript levels of each of the 19 genes among 30 patients (Table S3). The analysis revealed that high expressions of TUBB4A (p = 0.028, HR:3.88), S100B (p = 0.046, HR:3.093), and SLC6A1 (p = 0.034, HR:3.765) exhibited a prognostic value independent of KPS and consciousness level (Table 3).
TABLE 3.
Multivariate analysis of clinical factors and neuroinflammation‐related gene expressions in the discovery cohort.
| Multivariate analysis | ||||
|---|---|---|---|---|
| Variable | Hazard ratio | Lower 95%CI | Upper 95%CI | p‐value |
| TUBB4A | 3.88 | 1.155 | 13.04 | 0.028 |
| KPS | 3.421 | 0.978 | 11.96 | 0.054 |
| Consciousness levels at diagnosis | 1.134 | 0.398 | 3.279 | 0.8171 |
| S100B | 3.093 | 1.02 | 9.385 | 0.046 |
| KPS | 2.53 | 0.697 | 9.184 | 0.158 |
| Consciousness levels at diagnosis | 1.552 | 0.602 | 3.999 | 0.362 |
| SLC6A1 | 3.765 | 1.106 | 12.81 | 0.034 |
| KPS | 2.865 | 0.81 | 10.13 | 0.102 |
| Consciousness levels at diagnosis | 1.423 | 0.539 | 3.792 | 0.481 |
Abbreviation: KPS, kernofsky performance status.
3.3. The independent cohorts validated the prognostic impact of three candidate genes
We next validated the prognostic value of three candidate genes using an independent cohort of 30 PCNSL cases (Figure 1A). As expected, the Kaplan–Meier survival curve successfully delineated clinical outcomes of validation cohort PCNSL patients (Figure 2B) [29].
FIGURE 2.
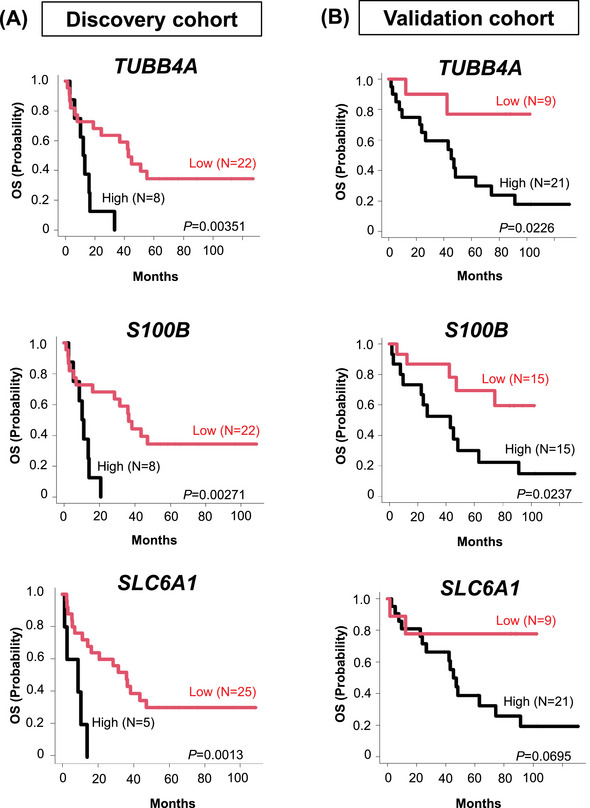
Clinical impact of extracted three prognostic genes in the discovery and validation cohort. Kaplan–Meier curves show the duration of OS based on expression levels of unfavorable candidate genes in the discovery (left) and the validation (right) cohorts. Statistical analysis was performed using the two‐sided log‐rank test. OS, overall survival.
Validation cohort patients with PCNSL whose specimens showed high expression of TUBB4A (p = 0.0226), S100B (p = 0.0237), and SLC6A1 (p = 0.0695) exhibited poor prognoses (Figure 2B).
3.4. Unfavorable prognostic genes derived from specific glial cells
We next performed IHC to identify the origin of unfavorable prognostic genes in the discovery cohort, evaluating the protein expression of TUBB4A and S100B by IHC in 30 PCNSL samples (Table S5).
In the RG, IHC staining for TUBB4A and S100B showed diffuse infiltration of strongly stained cells into tumors (samples A and B in Figure 3). Conversely, in the NRG, expression levels of TUBB4A and S100B were significantly lower (samples C and D in Figure 3). IHC of these prognostic proteins suggest TME‐resident immune cells together with lymphoma cells were actively infiltrating into tumors of RG.
FIGURE 3.
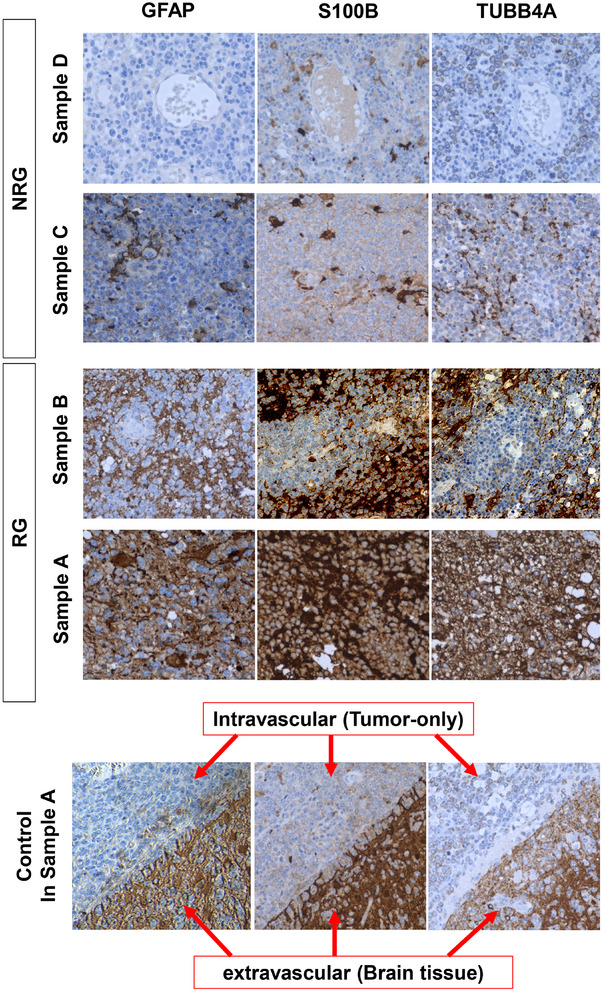
Immunostaining of PCNSL for prognostic proteins. TUBB4A and S100B expression levels were examined by immunohistochemical (IHC) staining in the RG (samples A and B) and NRG (samples C and D) groups, respectively. In control in IHC staining of sample A, expression levels were compared between intravascular (only tumor cells inside the vessel) and extravascular (brain tissue infiltrated by tumor cells) space. Original magnification 400×. NRG, nonrecurrence group; PCNSL, primary central nervous system lymphoma; RG, recurrence group.
In order to characterize the details of TME cells in the RG, IHC analysis of proteins specific to glial cell subclasses such as oligodendrocytes, astrocytes, and microglial cells was additionally performed (Table S5). Staining against GFAP, OLIG2, and CD68 indicated more aggressive infiltration of astrocytes, oligodendrocytes, and microglia/macrophages in tumors of the RG versus the NRG (Figure 4A,C,E).
FIGURE 4.
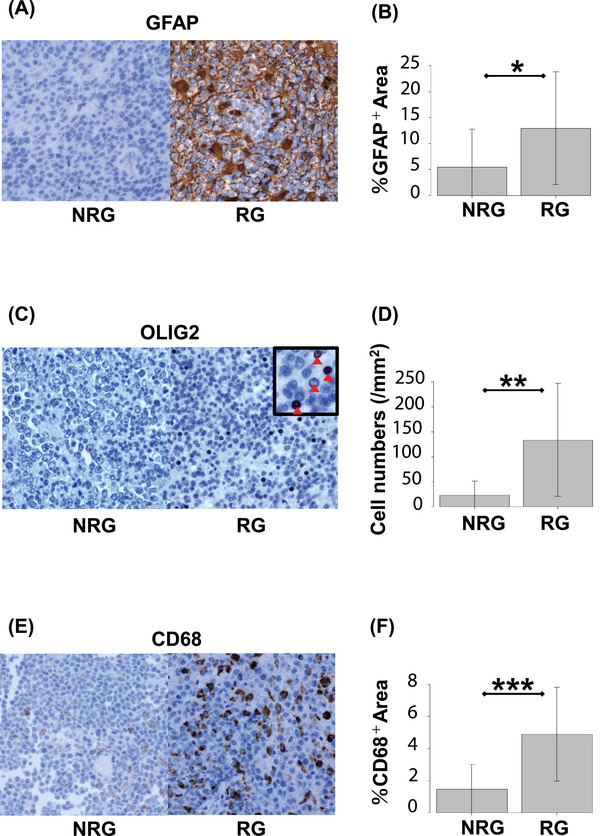
PCNSL tissues harbor tumor microenvironment immune cells. Tumor sections were stained with GFAP (A), OLIG2 (B), and CD68 (C). Representative data from NRG (left) and RG (right) are shown (panels A, C, and E). Quantifications of the immunoreactivity of immunohistochemical (IHC) markers shown in RG compared with NRG (panels B, D, and F). Numbers of OLIG2‐positive cells and percentage of GFAP‐ and CD68‐positive area were counted across sections using scanning software (see “Methods”; panels B, D, and F) *p < 0.05, **p < 0.01, ***p < 0.001. NRG, nonrecurrence group; PCNSL, primary central nervous system lymphoma; RG, recurrence group.
To quantify the immunoreactivity of IHC markers, we used ImageJ to quantitatively measure IHC results comprehensively by calculating the percentage of positive areas and positive cell numbers. GFAP‐ and CD68‐positive areas and OLIG2‐positive cell numbers were significantly higher in the RG than the NRG (GFAP p = 0.0456, OLIG2 p < 0.01, CD68 p < 0.001, Figure 4B,D,F; Table S6). IHC results revealed that, among TME cells, the proportions of these three kinds of glial cells were higher in RG compared with NRG.
Our transcriptomic and histopathological data suggest that the activation of glial cells may play a crucial role and have a prognostic effect on the pathogenesis of human PCNSL (Figures 1D and 4).
3.5. Gene mutation status is not associated with OS status
We identified recurrent mutations in MYD88 L265P (22/30, 73%), CD79B Y196 (13/30, 43%), PIM1 (22/30, 73%), BTG2 (11/30, 37%), TBL1XR1 (5/30, 17%), TOX (4/30, 13%), B2 M (3/30, 10%), CARD11 (5/30, 17%), PRDM1 (3/30, 10%), TNFAIP3 (3/30, 10%), TMEM30A (2/30, 7%), and PRKCD (1/30, 3%) via our in‐house PCNSL panel that includes 12 genes (Tables S2 and S7). According to Fisher's exact test, gene mutation status was not associated with OS status (NRG vs. RG; Table S8).
Next, we integrated gene expression data of 770 NFR genes with the gene mutation data from the 12 genes in our in‐house PCNSL panel (Figure 5, Tables S4 and S7). These data suggest that expression of almost all NFR genes were not significantly up‐ or downregulated in samples harboring somatic mutations in MYD88, CD79B, PIM1, BTG2, TBL1XR1, TOX, and CARD11 within the discovery cohort (Figure S2). Genes such as B2 M, PRDM1, TNFAIP2, TMEM30A, and PRKCD harbored somatic mutations in only 1–3 patients; therefore, these genes were excluded from this combination analysis.
FIGURE 5.
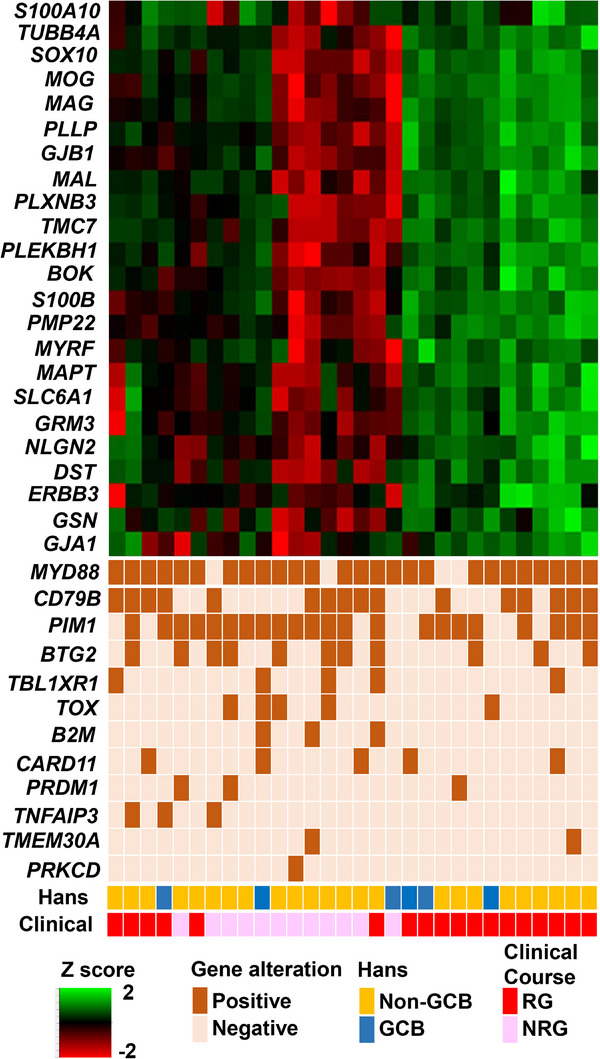
Integrated visualization of transcriptome data. The transcriptome data from nCounter analysis is shown as a heat map, with the 23 informative genes shown as rows and the 30 patient biopsies shown as columns. These were combined with genetic alterations, cell of origin (COO) classification, and prognosis. Gene expression levels are shown as a percentile of the normalized expression values for each gene. GCB, germinal center B; Hans, Hans criteria; NRG, nonrecurrence group; RG, recurrence group.
4. DISCUSSION
To our knowledge, this is the first study that reveals the prognostic effect of genes related to glial cells in the brain. In general, infiltration of immune regulatory cells into the tumor mass from the TME is associated with poor outcomes through induction of chemotherapy resistance and immune evasion. Gliosis within the normally immune‐privileged brain occurs from chronic inflammation and tumor infiltration may thus occur through these reactive glial cells which accumulate in the CNS. Our observations demonstrate that high expression levels of 3 NFR genes (S100B, TUBB4A, and SLC6A1) were associated with poor prognoses in both the discovery and the validation cohorts.
S100B encodes a member of the multigene family of Ca2+‐binding proteins which are thought to be involved in the regulation of cellular activities such as metabolism, motility, and proliferation [31]. S100B protein is constitutively released by astrocytes and reactive microglial cells [32]. In the nervous system, expression of S100B is also induced by various stimuli, such as inflammation or trauma, and it induces the expression of inducible nitric oxide synthase and NF‐κB, which in turn activates microglia cells and astrocytes in an autocrine manner, upregulating the expression of proinflammatory cytokines [33]. Previous studies revealed that S100B was significantly upregulated in PCNSL compared with non‐CNS nodal and extranodal DLBCL [34, 35, 36]. This suggests that S100B is also induced by stimuli from lymphoma cells within the CNS.
TUBB4A is a brain‐specific β‐tubulin and is selectively expressed by CNS‐resident oligodendrocytes and microglial cells [37, 38]. In a neuropathology report of a patient with TUBB4A‐related severe atrophy of the cerebellum, CD68 staining for microglia/macrophages showed activation of microglia while staining against the astrocyte‐specific GFAP showed moderately reactive astrogliosis (38). This reveals that the expression level of TUBB4A may correlate with the activation of tumor‐infiltrating glial cells.
SLC6A1 (solute carrier 6 member 1) is abundantly expressed in the developing brain even before the CNS is formed and encodes GABA transporter1 (GAT‐1), globally expressed in the brain, in both astrocytes and neurons [39]. Previous reports in other cancers showed associations between low SLC6A1 and extended survival times, putatively through downregulation of SLC6A1 activity by miR‐200c direct binding [40].
According to the IHC results from our study cohorts, these three key prognostic proteins are speculated to be released mainly from infiltrating glial cells (Figure 3), and several previous studies regarding these genes agree with our results (32, 37). Furthermore, we found that glial cell‐specific proteins, such as GFAP, OLIG2, and CD68, were closely associated with the activation of glial cells in the RG. GO analysis confirmed that oligodendrocyte and astrocyte‐related signatures were enriched in the RG‐associated, 45‐gene set, suggesting that gliosis (defined as activation of glial cells) is related to unfavorable prognoses in PCNSL.
GO analysis also revealed that most prognostic genes were associated with gliogenesis, suggesting that gliosis was more activated in the RG and canonical NFκB signal transduction. Additionally, neuroinflammation signaling pathway gene upregulation was enriched in the RG, indicating glial cell upregulation via the NF‐κB pathway (Figure S1) [28]. These analyses suggest a tight correlation of the gliosis and NF‐κB signaling pathways to the pathogenesis and prognosis of PCNSL. Previous studies have shown that, during brain injury (e.g., ischemic stroke, neurodegenerative disease, or brain tumors), immune cell recruitment, plus upregulation of NF‐κB pathway and related cytokines (TGFβ and interleukin [IL]‐6), drive reactive glial cells [41, 42]. The importance of reactive glial cell activity on the TME of PCNSL was also investigated [43], with a previous study that revealed the activation of glial cells by NF‐κB as a driver of PCNSL pathogenesis in a murine model [43]. Briefly, that report detailed the effect of gliosis‐enhanced, cell‐adhesion molecules and astrocyte‐derived Chemokine (C–C motif) ligand (CCL)19 on the increase of lymphoma cells in mouse brains. Clinically, this is relevant as astrocytic CCL19 was also observed in human PCNSL [43]. Therefore, upregulation of the NF‐κB signaling pathway could be an important factor for promoting PCNSL in addition to other known lymphoma risk factors, such as age and immune evasion [43, 44]. The GO and IPA analysis of our discovery cohort supports the notion that gliosis could support lymphoma growth through NF‐κB signaling, as well as become a driving and prognostic factor for poor PCNSL outcomes. Future studies examining synergistic effects of astrocytic chemokines and glial activation factors on PCNSL will be instructive with regard to infiltration profiling.
Notably, expression levels of NFR genes were not associated with gene mutation status in our cohort. These data are incompatible with previous reports that showed some NFR genes, notably MYD88, PIM1, CARD11, and TBL1XR1, were significantly upregulated in samples having somatic mutations [10, 45–48]. Larger sample sizes are needed to further examine the impact of gene mutations on NFR gene expression.
In spite of the NFR panel being inconclusive, our results highlight the potential clinical value of targeting TME in B‐cell lymphoma [49, 50], supporting current efforts by early‐phase clinical trials to test agents targeting TME cells [50]. However, these studies are ongoing, and clinical trials of a PD‐1 blockade with nivolumab and a BTK inhibitor in relapsed/refractory PCNSL have not produced verifiable results suitable for creating clinical treatment guidelines [12, 44]. Further studies will help promote clinical applications of TME‐based, targeted therapy.
Our study presents several limitations. Identification of prognostic genes was carried out using differential expression analyses obtained from nCounter data, so it may not address the cross‐talk between single cells or the heterogeneity within tumors that other approaches such as single‐cell RNA‐seq or spatial transcriptomics would potentially do. Since the activity of NF‐kB and the functions of TME‐related genes could not be directly examined in this study, we aim to investigate these in future research. Another limitation would be the small cohort size because of the rarity of PCNSL. However, this study may well serve as a foundation for future, multi‐center studies that explore associations between gliosis and PCNSL survival parameters, especially in early mortality cohorts.
In conclusion, our data suggest that genes related to TME may play a crucial role in the pathogenesis of PCNSL, complementing the well‐known involvement of the NF‐kB signaling pathway. Thus, multicomponent strategies that simultaneously target TME cells and tumor progression may be useful to overcome the highly refractory nature of PCNSL.
AUTHOR CONTRIBUTIONS
Contribution: Keiichiro Hattori collected the human samples, performed the experiments and computational analyses, and generated all figures and tables; Ryota Ishii, Yoshiaki Abe, Tatsuhiro Sakamoto, and Naoki Kurita supported the statistical analyses; Mamiko Sakata‐Yanagimoto developed the experimental and analytical nCounter and targeted deep seq systems; Masahide Matsuda, Takao Tsurubuchi, Eiichi Ishikawa, and Shota Tanaka contributed to human sample collection; Ryota Matsuoka and Daisuke Matsubara performed IHC staining; Ryo Nishikawa, Shota Tanaka, Akitake Mukasa, Yoshitaka Narita, Koichi Ichimura, and Motoo Nagane provided clinical information of PCNSL patients in the validation cohort; Mamiko Sakata‐Yanagimoto conceived the study; Keiichiro Hattori and Mamiko Sakata‐Yanagimoto designed the project; Keiichiro Hattori, Bryan J. Mathis, and Mamiko Sakata‐Yanagimoto wrote the manuscript; all authors participated in the discussion and interpretation of the data and results.
CONFLICT OF INTEREST STATEMENT
Lecture honoraria: Eiichi Ishikawa (Daiichi Sankyo, Eisai, Otuka Pharma); Yoshitaka Narita (Ono Pharma) Motoo Nagane (Ono Pharma); Research fund: Sakata‐Yanagimoto Mamiko (Eisai, Bristol Myers Squib, Otuska Pharma, Chugai Pharma), Shigeru Chiba (Thyas, Astellas, Kyowa Kirin); Eiichi Ishikawa (Mitsubishi Electric); Ryo Nishikawa (Ono Pharma) Motoo Nagane (Ono Pharma); Koichi Ichimura (Daiichi Sankyo, Riken Genesis, Gold Ribbon Network, Sumitomo Pharma); Scholarship grant: Shigeru Chiba (Bayer, Eisai, Chugai Pharma, Kyowa Kirin); Eiichi Ishikawa (Eisai) Ryo Nishikawa (Chugai Pharma); Yoshitaka Narita (Ono Pharma, Sumitomo Pharma, Eisai, Taiho Pharma, Ohara Pharma); Motoo Nagane (Chugai Pharma, Bayer, Eisai, Daiichi Sankyo); Endowed chair: Koichi Ichimura (Idorsia Pharmaceuticals).
ETHICS STATEMENT
The use of data from the validation cohort was approved by the University of Tokyo, Kyorin University, Saitama Medical University, National Cancer Center Research Institute, and National Cancer Center Hospital. This study conformed to the principles of the Declaration of Helsinki and was approved by the Institutional Review Board of the University of Tsukuba Institute of Medicine (approval # R02‐84).
PATIENT CONSENT STATEMENT
Diagnosed patients with PCNSL in the discovery cohort provided written, informed consent for tissue collection and subsequent research purposes.
CLINICAL TRIAL REGISTRATION
The authors have confirmed clinical trial registration is not needed for this submission.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
The authors thank the staff at the associated institutions and departments for helping with human sample collection, as well as Y. Sakashita for technical assistance. This work was supported by Grants‐in‐Aid for Scientific Research (KAKENHI: JP23K15317 [S.S.], JP20J20851 [Y.A.], JP23K15316 [K.H.], JP23K15293 [K. M.], JP21K16261 [T. S.], and JP22K19451 and JP21H02945 [M.S.‐Y.]) from the Ministry of Education, Culture, Sports, and Science of Japan; AMED (JP23ck0106644, JP23ck0106797, the Moonshot Research and Development Program, JP22zf0127009 [M.S.‐Y.]); Japan Leukemia Research Fund, SENSHIN Medical Research Foundation, SGH Foundation, the Yasuda Medical Foundation, and Daiichi Sankyo Foundation of Life Science (Y.A.); Takahashi Industrial and Economic Research Foundation (K.H.); and Kobayashi Foundation for Cancer Research, Kobayashi Foundation, the Chemo‐Sero Therapeutic Research Institute, Takeda Science Foundation, and the Uehara Memorial Foundation (M.S.‐Y.).
Hattori K, Makishima K, Suma S, Abe Y, Suehara Y, Sakamoto T, et al. Association between microenvironment‐related genes and prognosis of primary central nervous system lymphoma. eJHaem. 2024;5:1201–1214. 10.1002/jha2.1046
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
REFERENCES
- 1. Villano JL, Koshy M, Shaikh H, Dolecek TA, McCarthy BJ. Age, gender, and racial differences in incidence and survival in primary CNS lymphoma. Brit J Cancer. 2011;105(9):1414–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hattori K, Sakata‐Yanagimoto M, Okoshi Y, Goshima Y, Yanagimoto S, Nakamoto‐Matsubara R, et al. MYD88 (L265P) mutation is associated with an unfavourable outcome of primary central nervous system lymphoma. Brit J Haematol. 2017;177(3):492–494. [DOI] [PubMed] [Google Scholar]
- 3. Lin CH, Yang CF, Yang HC, Fay LY, Yeh CM, Kuan AS, et al. Risk prediction for early mortality in patients with newly diagnosed primary CNS lymphoma. J Cancer. 2019;10(17):3958–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hattori K, Sakata‐Yanagimoto M, Kusakabe M, Nanmoku T, Suehara Y, Matsuoka R, et al. Genetic evidence implies that primary and relapsed tumors arise from common precursor cells in primary central nervous system lymphoma. Cancer Sci. 2019;110(1):401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakamura T, Tateishi K, Niwa T, Matsushita Y, Tamura K, Kinoshita M, et al. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. 2016;42(3):279–290. [DOI] [PubMed] [Google Scholar]
- 6. Fukumura K, Kawazu M, Kojima S, Ueno T, Sai E, Soda M, et al. Genomic characterization of primary central nervous system lymphoma. Acta Neuropathologica. 2016;131(6):865–875. [DOI] [PubMed] [Google Scholar]
- 7. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell. 2017;171(2):481–494.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24(5):679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B‐cell lymphoma. New Eng J Med. 2018;378(15):1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Radke J, Ishaque N, Koll R, Gu Z, Schumann E, Sieverling L, et al. The genomic and transcriptional landscape of primary central nervous system lymphoma. Nat Commun. 2022;13(1):2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chapuy B, Roemer MG, Stewart C, Tan Y, Abo RP, Zhang L, et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood. 2016;127(7):869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Narita Y, Nagane M, Mishima K, Terui Y, Arakawa Y, Yonezawa H, et al. Phase 1/2 study of tirabrutinib, a second‐generation Bruton's tyrosine kinase inhibitor, in relapsed/refractory primary central nervous system lymphoma. Neuro‐oncol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Taoka K, Okoshi Y, Sakamoto N, Takano S, Matsumura A, Hasegawa Y, et al. A nonradiation‐containing, intermediate‐dose methotrexate regimen for elderly patients with primary central nervous system lymphoma. Int J Hematol. 2010;92(4):617–623. [DOI] [PubMed] [Google Scholar]
- 14. Jahnke K, Thiel E, Martus P, Herrlinger U, Weller M, Fischer L, et al. Relapse of primary central nervous system lymphoma: clinical features, outcome and prognostic factors. J Neuro‐oncol. 2006;80(2):159–165. [DOI] [PubMed] [Google Scholar]
- 15. Son SM, Ha SY, Yoo HY, Oh D, Kim SJ, Kim WS, et al. Prognostic impact of MYC protein expression in central nervous system diffuse large B‐cell lymphoma: comparison with MYC rearrangement and MYC mRNA expression. Modern Pathol. 2017;30(1):4–14. [DOI] [PubMed] [Google Scholar]
- 16.Gomes Candido Reis D, Levy D, Lage L, Culler HF, Rocha V, Bydlowski SP, et al. New genetic prognostic biomarkers in primary central nervous system lymphoma (PCNSL). Brain Behav. 2021;11(4):e02061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ruan H, Wang Z, Zhai Y, Xu Y, Pi L, Zheng J, et al. Single‐cell transcriptome analysis of diffuse large B cells in cerebrospinal fluid of central nervous system lymphoma. iScience. 2021;24(9):102972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wei B, Liu Z, Fan Y, Wang S, Dong C, Rao W, et al. Analysis of cellular heterogeneity in immune microenvironment of primary central nervous system lymphoma by single‐cell sequencing. Front Oncol. 2021;11:683007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heming M, Haessner S, Wolbert J, Lu IN, Li X, Brokinkel B, et al. Intratumor heterogeneity and T cell exhaustion in primary CNS lymphoma. Genome Med. 2022;14(1):109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hernández‐Verdin I, Kirasic E, Wienand K, Mokhtari K, Eimer S, Loiseau H, et al. Molecular and clinical diversity in primary central nervous system lymphoma. Annal Oncol. 2022. [DOI] [PubMed] [Google Scholar]
- 21. Xia Y, Sun T, Li G, Li M, Wang D, Su X, et al. Spatial single cell analysis of tumor microenvironment remodeling pattern in primary central nervous system lymphoma. Leukemia. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Quail DF, Joyce JA. the microenvironmental landscape of brain tumors. Cancer Cell. 2017;31(3):326–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alame M, Cornillot E, Cacheux V, Rigau V, Costes‐Martineau V, Lacheretz‐Szablewski V, et al. The immune contexture of primary central nervous system diffuse large B cell lymphoma associates with patient survival and specific cell signaling. Theranostics. 2021;11(8):3565–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takashima Y, Kawaguchi A, Sato R, Yoshida K, Hayano A, Homma J, et al. Differential expression of individual transcript variants of PD‐1 and PD‐L2 genes on Th‐1/Th‐2 status is guaranteed for prognosis prediction in PCNSL. Sci Rep. 2019;9(1):10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Swerdlow SH, International Agency for Research on C. WHO classification of tumours of haematopoietic and lymphoid tissues. rev. 4th ed ed: International Agency for Research on Cancer; 2017; p.p. 585. [Google Scholar]
- 26. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun. 2019;10(1):1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nayak L, Hedvat C, Rosenblum MK, Abrey LE, DeAngelis LM. Late relapse in primary central nervous system lymphoma: clonal persistence. Neuro‐oncol. 2011;13(5):525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shabab T, Khanabdali R, Moghadamtousi SZ, Kadir HA, Mohan G. Neuroinflammation pathways: a general review. Int J Neurosci. 2017;127(7):624–633. [DOI] [PubMed] [Google Scholar]
- 29. Hothorn T, Zeileis A. Generalized maximally selected statistics. Biometrics. 2008;64(4):1263–1269. [DOI] [PubMed] [Google Scholar]
- 30. Abrey LE, Ben‐Porat L, Panageas KS, Yahalom J, Berkey B, Curran W, et al. Primary central nervous system lymphoma: the Memorial Sloan‐Kettering Cancer Center prognostic model. J Clin Oncol. 2006;24(36):5711–5715. [DOI] [PubMed] [Google Scholar]
- 31. Huang H, He W, Tang T, Qiu M. Immunological markers for central nervous system glia. Neurosci Bulletin. 2023;39(3):379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adami C, Sorci G, Blasi E, Agneletti AL, Bistoni F, Donato R. S100B expression in and effects on microglia. Glia. 2001;33(2):131–142. [PubMed] [Google Scholar]
- 33. Donato R, Sorci G, Riuzzi F, Arcuri C, Bianchi R, Brozzi F, et al. S100B's double life: intracellular regulator and extracellular signal. Biochimica et biophysica acta. 2009;1793(6):1008–1022. [DOI] [PubMed] [Google Scholar]
- 34. Tun HW, Personett D, Baskerville KA, Menke DM, Jaeckle KA, Kreinest P, et al. Pathway analysis of primary central nervous system lymphoma. Blood. 2008;111(6):3200–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Montesinos‐Rongen M, Siebert R, Deckert M. Primary lymphoma of the central nervous system: just DLBCL or not? Blood. 2009;113(1):7–10. [DOI] [PubMed] [Google Scholar]
- 36. CO Sung, Kim SC, Karnan S, Karube K, Shin HJ, Nam DH, et al. Genomic profiling combined with gene expression profiling in primary central nervous system lymphoma. Blood. 2011;117(4):1291–1300. [DOI] [PubMed] [Google Scholar]
- 37. Fernández‐Arjona MDM, León‐Rodríguez A, López‐Ávalos MD, Grondona JM. Microglia activated by microbial neuraminidase contributes to ependymal cell death. Fluids Barriers CNS. 2021;18(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duncan ID, Bugiani M, Radcliff AB, Moran JJ, Lopez‐Anido C, Duong P, et al. A mutation in the Tubb4a gene leads to microtubule accumulation with hypomyelination and demyelination. Annal Neurol. 2017;81(5):690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mermer F, Poliquin S, Rigsby K, Rastogi A, Shen W, Romero‐Morales A, et al. Common molecular mechanisms of SLC6A1 variant‐mediated neurodevelopmental disorders in astrocytes and neurons. Brain. 2021;144(8):2499–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao Y, Zhou X, He Y, Liao C. SLC6A1‐miR133a‐CDX2 loop regulates SK‐OV‐3 ovarian cancer cell proliferation, migration and invasion. Oncol Letters. 2018;16(4):4977–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pekny M, Pekna M. Reactive gliosis in the pathogenesis of CNS diseases. Biochimica et biophysica acta. 2016;1862(3):483–491. [DOI] [PubMed] [Google Scholar]
- 42. Vandenbark AA, Offner H, Matejuk S, Matejuk A. Microglia and astrocyte involvement in neurodegeneration and brain cancer. J Neuroinflam. 2021;18(1):298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. O'Connor T, Zhou X, Kosla J, Adili A, Garcia Beccaria M, Kotsiliti E, et al. Age‐related gliosis promotes central nervous system lymphoma through ccl19‐mediated tumor cell retention. Cancer Cell. 2019;36(3):250‐67.e9. [DOI] [PubMed] [Google Scholar]
- 44. Nayak L, Iwamoto FM, LaCasce A, Mukundan S, Roemer MGM, Chapuy B, et al. PD‐1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood. 2017;129(23):3071–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jung H, Yoo HY, Lee SH, Shin S, Kim SC, Lee S, et al. The mutational landscape of ocular marginal zone lymphoma identifies frequent alterations in TNFAIP3 followed by mutations in TBL1XR1 and CREBBP. Oncotarget. 2017;8(10):17038–17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Improgo MR, Tesar B, Klitgaard JL, Magori‐Cohen R, Yu L, Kasar S, et al. MYD88 L265P mutations identify a prognostic gene expression signature and a pathway for targeted inhibition in CLL. Brit J Haematol. 2019;184(6):925–936. [DOI] [PubMed] [Google Scholar]
- 47. Wei Z, Zhang Y, Chen J, Hu Y, Jia P, Wang X, et al. Pathogenic CARD11 mutations affect B cell development and differentiation through a noncanonical pathway. Sci Immunol. 2019;4(41). [DOI] [PubMed] [Google Scholar]
- 48. Zhang H, Lu Y, Zhang T, Guan Q, Wang X, Guo Y, et al. PIM1 genetic alterations associated with distinct molecular profiles, phenotypes and drug responses in diffuse large B‐cell lymphoma. Clin Translat Med. 2022;12(4):e808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leivonen SK, Pollari M, Bruck O, Pellinen T, Autio M, Karjalainen‐Lindsberg ML, et al. T‐cell inflamed tumor microenvironment predicts favorable prognosis in primary testicular lymphoma. Haematologica. 2019;104(2):338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu Y, Zhou X, Wang X. Targeting the tumor microenvironment in B‐cell lymphoma: challenges and opportunities. J Hematol Oncol. 2021;14(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.