

Abstract

This Letter details our efforts to develop novel, non-acetylene-containing metabotropic glutamate receptor subtype 5 (mGlu5) negative allosteric modulators (NAMs) with improved pharmacological properties. This endeavor involved replacing the ether-linked pyrimidine moiety, a metabolic liability, with various 5-membered heterocycles. From this exercise, we identified VU6043653, a highly brain penetrant and selective mGlu5 NAM which displayed moderate potency against both human and rat mGlu5. Moreover, VU6043653 has overall improved pharmacological and drug metabolism and pharmacokinetic profiles when compared to its predecessor compounds. Most notably, VU6043653 exhibits low predicted human hepatic clearance, a clean cytochrome P450 profile, and minimal inhibition of the dopamine transporter.
Keywords: Metabotropic Glutamate Receptor Subtype 5, mGlu5, Negative Allosteric Modulator (NAM), Structure−Activity Relationship (SAR), Levodopa-Induced Dyskinesia, Alzheimer’s Disease, Pain, VU6043653
The metabotropic glutamate (mGlu) receptors comprise a family of eight G protein-coupled receptors (GPCRs) that are activated by l-glutamic acid, the major excitatory neurotransmitter of the mammalian central nervous system (CNS). Once activated, the mGlu receptors modulate the strength of synaptic transmission. The eight mGlu receptors are divided into three groups based on structure and sequence homology, downstream signaling partners/pathways, as well as pharmacology. The mGlu5 receptor is widely expressed throughout the CNS and, alongside mGlu1, belongs to group I mGlu receptors, which are predominantly found postsynaptically and couple via Gq to the activation of phospholipase C (PLC).1,2 While designing selective orthostatic ligands that preferentially target one mGlu receptor over another has proven to be extremely challenging, one successful approach to selectively target individual mGlu receptor subtypes is via allosteric modulation. Negative allosteric modulators (NAMs) of mGlu5 are among the most advanced and widely investigated within the field of mGlu receptor allostery.3−8 Preclinical and clinical efficacy has established a multitude of potential therapeutic applications for small molecule mGlu5 NAMs, such as anxiety,9,10 Alzheimer’s disease,11 fragile X syndrome,12−14 autism spectrum disorder,15,16 levodopa-induced dyskinesia experienced by many Parkinson’s disease patients,17−19 gastroesophageal reflux disease,20 addiction disorder,21−23 major depressive disorder,24−26 obsessive-compulsive disorder,27 migraine, and pain.28−31 Early mGlu5 NAMs (e.g., 1 and 2) were based on a key aryl/heterobiaryl acetylene pharmacophore, and this moiety has been carried throughout several subsequent medicinal chemistry optimization efforts (highlighted in Figure 1); however, alkynes, particularly those conjugated to an α-heteroatom, are potentially reactive functional groups.32,33 In fact, acetylene-based mGlu5 NAMs have been linked to hepatotoxicity and glutathione conjugation, as observed in both preclinical and clinical studies.34 AZD9272 (7) utilized an acetylene bioisostere, while fenobam (3) completely lacked the acetylene moiety. Both were advanced to clinical studies; however, their development was halted due to psychosis-like symptoms. Most importantly, further investigation into fenobam and AZD9272 attributed these symptoms to monoamine oxidase-B (MAO-B)-mediated mechanisms rather than mGlu5-mediated mechanisms.35 To date, no mGlu5 NAM has advanced to the market due, in part, to dose-limiting adverse events (such as hallucinations or psychotomimetic effects) observed in some clinical trials.36 Currently, TMP-301 (9) is the only clinical mGlu5 NAM devoid of the acetylene moiety and is undergoing Phase I clinical trials for substance abuse disorders.37 Therefore, endeavors in the field have shifted to identifying novel, non-acetylene-containing mGlu5 NAMs to avoid the pharmacophore-mediated adverse liabilities while exploiting the broad therapeutic utility of a selective mGlu5 NAM.
Figure 1.

Prototypical mGlu5 NAM chemotypes. NAMs 1 and 2 were crucial early tool compounds, and NAMs 4–9 entered human clinical testing.
A major focus of our group has been the development of small molecule mGlu5 NAMs, which ultimately resulted in the identification of clinical candidate 10 (auglurant, VU0424238) (Figure 2).38 Unfortunately, 10 failed in development due to species-specific toxicities observed during a 28-day toxicologic assessment in cynomolgus monkeys, which were not previously observed in rats. Accumulation of a cyno-unique aldehyde oxidase (AO) metabolite was observed after 14 days and resulted in pronounced anemia (non-mechanism-based). Metabolism studies revealed the oxidation of the pyrimidine ring to a 6-oxopyrimidine metabolite, followed by the subsequent formation of a 2,6-oxopyrimidine metabolite. In humans, monkeys, and rats, it was determined that the formation of the 6-oxopyrimide metabolite was mediated by AO; however, there were apparent species differences between monkeys and rats in the enzyme involved in the formation of the 2,6-oxopyrimidine metabolite. While the second metabolite was mediated by AO metabolism in monkeys, it was determined that this process was mediated by xanthine oxidase (XO) metabolism rats.39,40 Therefore, it is possible that species differences in the involvement of AO/XO metabolism may play a role in the observed monkey-specific toxicity.
Figure 2.

Previously published compounds that emerged from optimization of high-throughput screening hits: clinical candidate VU0424238 (auglurant, 10) and backup scaffold 11. Further optimization led to potent mGlu5 NAMs 12.
Attention was shifted to the development of backup analogs 11 in an effort to identify a compound devoid of AO metabolism. While this strategy allowed us to mitigate the role of AO, it did not allow us to fully eliminate this route of metabolism. Additionally, analogs 11 typically suffered from high predicted human hepatic clearance, high plasma protein binding, inhibition of cytochrome P450s (CYPs; in particular 1A2 but also 3A4 and 2C9), and/or inhibition of dopamine transporters (DAT). Thus, further optimization was required. This Letter describes the structure–activity relationship (SAR) development of novel mGlu5 NAMs (12) with various 5-membered heteroaryl groups as replacements for the pyrimidine moiety responsible for the AO-mediated metabolism observed in 10.
The synthesis of analogs 22 was straightforward and began by reacting commercially available nitrile 13 with various commercially available 5-membered heteroaryl alcohols under basic conditions to afford the SNAr products 14 (Scheme 1). Basic hydrolysis of nitriles 14 to the carboxylic acids 18 proceeded smoothly in 32–98% yield. Finally, conversion to the acid chloride and reaction with various heterocyclic amines in situ afforded analogs 22. We next turned our attention to exploring further modifications to the central pyridine core with the synthesis of intermediates 15–17. To prepare intermediate 15, we utilized standard SNAr protocols to react commercially available bromide 26 with alcohol 27 to provide intermediate 28, which could then undergo a palladium-catalyzed cross-coupling with zinc cyanide to afford nitrile 15 (Scheme 2). Similar to intermediate 14, nitrile 15 underwent basic hydrolysis to yield carboxylic acid 19. Subsequent conversion to the acid chloride and reaction with various heterocyclic amines in situ afforded analogs 23. The heterocyclic amines (R4) highlighted in Table 1 were select for evaluation based on prior endeavors in which these amines provided potent compounds with promising plasma protein binding and plasma clearance profiles.38
Scheme 1. Synthesis of mGlu5 NAM Analogs 18–25.

Reagents and conditions: (a) R3 = OH, K2CO3, DMF, μW 150 °C, 74–98%; (b) NaOH, EtOH/H2O, 100 °C, 32–98%; (c) NaOH, 1,4-dioxane/H2O, 98%; (d) POCl3, R4 = NH2, pyridine, 0 °C to r.t., 8–89%.
Scheme 2. Synthesis of mGlu5 NAM Intermediate 15.

Reagents and conditions: (a) Cs2CO3, DMSO, 79%; (b) Zn(CN)2, Pd(PPh3)4, DMF, μW 140 °C, 68%.
Table 1. Structures and Activities for Analogs 22–25a.
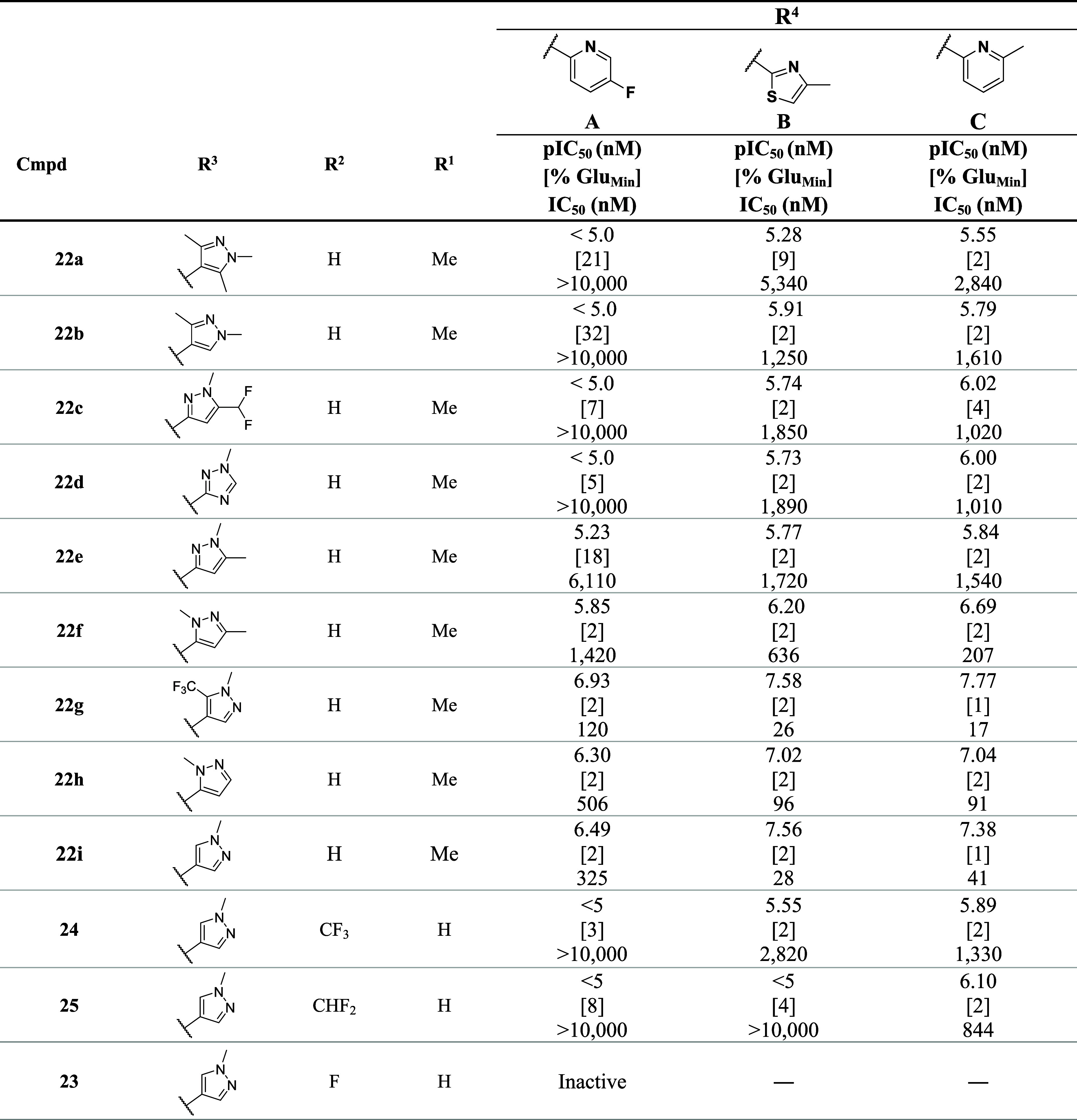
Calcium mobilization assays in human mGlu5-HEK293A cells were performed in the presence of an EC80 fixed concentration of glutamate, n = 2 independent experiments in triplicate. The % GluMin is the measure of efficacy of the NAM to reduce an EC80 response of glutamate.
Preparation of intermediate 16 began with commercially available iodide 29, which underwent an Ullmann biaryl ether formation in the presence of alcohol 27 to afford ether 30 (Scheme 3). A subsequent palladium-catalyzed carbonylation provided ethyl ester 16. Next, the synthesis of intermediate 17 began with a Wohl–Ziegler bromination of commercially available ester 31 to yield gem-dibromide 32 (Scheme 4). Geminal halide hydrolysis of intermediate 32 using AgNO3 as the oxidizing agent provided aldehyde 33, which could undergo further transformation with diethylaminosulfur trifluoride (DAST) to give the difluoro intermediate 34. Utilizing standard SNAr conditions to react intermediate 34 with alcohol 27 afforded intermediate 17. Saponification of esters 16 and 17 to carboxylic acids 20 and 21, respectively, proceeded smoothly in near quantitative yields. Finally, conversion to the acid chloride and reaction with various heterocyclic amines in situ afforded analogs 24 and 25.
Scheme 3. Synthesis of mGlu5 NAM Intermediate 16.

Reagents and conditions: (a) CuI, Cs2CO3, DMF, μW 150 °C, 40%; (b) CO(g), NaOAc, Pd(dppf)Cl2·CH2Cl2, EtOH/H2O (5:1), 70 °C, 99%.
Scheme 4. Synthesis of mGlu5 NAM Intermediate 17.

Reagents and conditions: (a) NBS, AIBN, CCl4, 90 °C, 63%; (b) AgNO3, EtOH/H2O (10:1), 50 °C, 99%; (c) DAST, DCM, 53%; (d) Cs2CO3, DMF, μW 150 °C, 22%.
Select analogs 22–25 were screened against human mGlu5 (hmGlu5) to determine potency, with results highlighted in Table 1. These results emphasize the importance of the amide tail (R4). For instance, when the 5-fluoropyridine amide tail was installed (22aA–22dA), the hmGlu5 IC50’s were >10 μM; however, when the amide tail was exchanged for a 4-methylthiazole amide tail (22aB–22dB) or 6-methylpyridine (22aC–22dC), we observed hmGlu5 IC50’s = 1–5 μM. Moreover, it became evident with further SAR development that the combination of amide tail (R4) and 5-membered heteroaryl ether (R3) was crucial for activity. For example, while the 5-fluoropyridine amide tail provided several analogs with hmGlu5 IC50’s > 10 μM (22a–dA, 24, and 25), several analogs containing alternate heteroaryl ethers had IC50’s ≤ 500 nM (22hA, hmGlu5 IC50 = 506 nM; 22iA, hmGlu5 IC50 = 325 nM; and 22gA, hmGlu5 IC50 = 120 nM). This phenomenon was also observed in the 4-methylthaizole series (22aB, hmGlu5 IC50 = 5.3 μM vs 22gB, hmGlu5 IC50 = 26 nM) as well as the 6-methylpyridine series (22aC, hmGlu5 IC50 = 2.8 μM vs 22hC, hmGlu5 IC50 = 91 nM).
With the exceptions of 22f and 22g, di- or trisubstituted 5-membered heteroaryl analogs (22a–e) only afforded compounds with hmGlu5 IC50’s ≥ 1 μM. Interestingly, comparing 22bC (hmGlu5 IC50 = 1.6 μM) with a constitutional isomer 22fC (hmGlu5 IC50 = 207 nM) gave a 7.8-fold increase in potency. Introduction of a trifluoromethyl electron-withdrawing group to the 1-methyl-1H-pyrazole (22gA, hmGlu5 IC50 = 120 nM) resulted in a ∼3-fold increase in potency in the context of the 5-fluoropyridine amide tail when compared to 22iA (hmGlu5 IC50 = 325 nM); however, this modification had no effect on potency when comparing analogs with the 4-methylthaizole amide tail (22gB, hmGlu5 IC50 = 26 nM vs 22iB, hmGlu5 IC50 = 28 nM). It was also noted that analogs 22iA-C were generally more potent than regioisomers 22hA-C; however, the changes in potency varied with the amine tail (22iA vs 22hA, 1.6-fold increase; 22iB vs 22hB, 3.4-fold increase).
Finally, we evaluated alternative picolinamide cores (23–25). Exchanging the 6-methylpicolinamide core (22iA; hmGlu5 IC50 = 325 nM) to a 5-fluoropicolinamide core (23A) resulted in a complete loss of activity. While the 5-(trifluoromethyl)picolinamide core was tolerated, only micromolar potencies could be achieved (24B, hmGlu5 IC50 = 2.8 μM and 24C, hmGlu5 IC50 = 1.3 μM). Additionally, the 5-(difluoromethyl)picolinomide core was tolerated only with the 6-methylpyrdine tail (25C, hmGlu5 IC50 = 844 nM). These results highlight the significance of the 6-methylpicolinamide core.
Of these compounds, 22f-C, 22gA-B, 22hB-C, and 22iA-C were advanced into a battery of in vitro DMPK assays and our standard rat plasma:brain level (PBL) cassette paradigm (Table 2).41,42 Regarding physicochemical properties, these analogs all possessed molecular weights less than 450 Da, with 22gA, 22gB, 22hB, and 22iB having the most attractive CNS xLogP values (2.07–3.01). Analogs 22fC, 22gB, 22hC, and 22iC displayed high human and rat predicted hepatic clearance (CLhep) based on microsomal CLint data (human CLhep > 15 mL/min/kg; rat CLhep > 46 mL/min/kg); however, analogs 22gA and 22iB were predicted to have moderate human and rat hepatic clearance (human CLhep of 7 and 14 mL/min/kg, rat CLhep of 38 and 27 mL/min/kg, respectively). Interestingly, 22hB was predicted to have moderate rat hepatic clearance (CLhep = 27 mL/min/kg) but high human hepatic clearance (CLhep = 19 mL/min/kg). Analog 22iA provided the best predicted hepatic clearance profile, with low human (CLhep = 6 mL/min/kg) and moderate rat (CLhep = 28 mL/min/kg) clearances.
Table 2. In Vitro DMPK and Rat PBL Data for Select Analogs 22fC, 22gA–C, 22hB–C, and 22iA–C.
| 22fC | 22gA | 22gB | 22gC | 22hB | 22hC | 22iA | 22iB | 22iC | |
|---|---|---|---|---|---|---|---|---|---|
| Property | VU6043937 | VU6044946 | VU6045093 | VU6073906 | VU6043657 | VU6043658 | VU6043653 | VU6043654 | VU6043655 |
| MW | 337.38 | 395.31 | 397.37 | 391.35 | 329.38 | 323.35 | 327.31 | 329.38 | 323.35 |
| xLogPa | 1.86 | 2.07 | 3.01 | 2.17 | 2.5 | 1.66 | 1.16 | 2.1 | 1.26 |
| TPSAa | 81.9 | 81.9 | 81.9 | 81.9 | 81.9 | 81.9 | 81.9 | 81.9 | 81.9 |
| hmGlu5 IC50 (nM) | 207 | 120 | 26 | 17 | 96 | 91 | 325 | 28 | 41 |
| In Vitro PK Parametersb | |||||||||
| CLint (mL/min/kg), rat | 436 | 82 | 817 | 600 | 45 | 320 | 48 | 44 | 234 |
| CLhep (mL/min/kg), rat | 60 | 38 | 65 | 63 | 27 | 57 | 28 | 27 | 54 |
| CLint (mL/min/kg), human | 71 | 11 | 70 | 68 | 216 | 241 | 9 | 46 | 77 |
| CLhep (mL/min/kg), human | 16 | 7 | 16 | 16 | 19 | 19 | 6 | 14 | 17 |
| Rat fu,plasma | NDd | NDd | NDd | NDd | 0.219 | NDd | 0.059 | 0.089 | 0.059 |
| Human fu,plasma | 0.037 | 0.012 | 0.004 | 0.011 | 0.062 | 0.034 | 0.059 | 0.063 | 0.041 |
| Rat fu,brain | 0.008 | 0.002 | 0.003 | 0.005 | 0.029 | 0.021 | 0.012 | 0.014 | 0.013 |
| Brain Distribution (0.25 h) (SD Rat; 0.2 mg/kg IV)c | |||||||||
| Kp, brain:plasma | 1.02 | 3.08 | 5.57 | 2.98 | 1.13 | 2.42 | 1.68 | 1.37 | 1.04 |
| Kp,uu, brain:plasma | NDd | NDd | NDd | NDd | 0.15 | NDd | 0.34 | 0.22 | 0.23 |
TPSA and xLogP were calculated using Dotmatics platform.
fu = fraction unbound; equilibrium dialysis assay; brain = rat brain homogenates;
Kp = total brain-to-plasma partition ratio; Kp,uu = unbound brain-to-plasma partition ratio [(brain fu × total brain)/(plasma fu × total plasma)].
ND = not determined; samples had low analyte peaks, possibly unstable in rat plasma.
Of the compounds tested, only 22gB displayed high protein binding to human plasma with unbound fraction (fu,plasma) < 0.01. Conversely, the best human plasma binding profiles belonged to compounds 22hB and 22iA-C (fu,plasma > 0.04). Analogs 22fC, 22gA, and 22gB were highly bound to rat brain homogenates (fu,brain < 0.01) and were determined to possibly be unstable in rat plasma. By contrast, compounds 22hB (fu,brain = 0.029), 22hC (fu,brain = 0.021), and 22iA-C (fu,brain = 0.012–0.014) were moderately bound to rat brain homogenates. Although 22hC was determined to potentially be unstable in rat plasma, analogs 22hB and 22iA-C displayed a high free fraction in rat plasma (fu,plasma’s > 0.04). All analogs tested were determined to have excellent CNS penetration (rat brain:plasma Kp ≥ 1.0); however, compound 22iA displayed the best CNS distribution of unbound drug (Kp,uu = 0.34). The moderate CNS distribution of unbound drug of VU6043653 is likely due to moderate binding to brain homogenate (fu,brain = 0.012). VU6043653 (22iA) gave the best overall DMPK profile and was selected for further characterization.
When evaluated for a full mGlu selectivity profile in functional assays, VU6043653 (22iA) displayed high subtype selectivity across the mGlu receptors (mGlu1, mGlu2, mGlu4, mGlu7, and Glu8 = inactive; mGlu3 > 10 μM) (Table 3). Additionally, VU6043653 displayed an excellent cytochrome (CYP) P450 inhibition profile, with IC50’s ≥ 30 μM across all isoforms tested (1A2, 2D6, 2C9, and 3A4). Highlighted in Table 4 are the in vivo rat PK parameters. VU6043653 displayed 40% oral bioavailability at a 10 mg/kg dose and moderate plasma clearance (41 mL/min/kg) in rats. The volume of distribution was moderate (2.0 L/kg), indicating minimal tissue binding, and elimination t1/2 was ∼45 min. With promising rat PK in hand, VU6043653 was progressed into higher species in vivo PK studies (Table 4). VU6043653 displayed moderate oral bioavailability (20% at a 3 mg/kg dose) in dogs; however, suprahepatic plasma clearance (38 mL/min/kg) halted further progress toward clinical candidate status.
Table 3. Further In Vitro Characterization of VU6043653 (21iA).
| Metabotropic Glutamate Selectivity | ||
|---|---|---|
| IC50 (nM) | [%GluMin] | |
| human mGlu1a | inactive | |
| human mGlu2b | inactive | |
| human mGlu3b | >10,000 | [58] |
| human mGlu4a | inactive | |
| human mGlu7a | inactive | |
| human mGlu8a | inactive | |
| P450 Inhibition IC50 (μM)c | |||
|---|---|---|---|
| 1A2 | 2D6 | 2C9 | 3A4 |
| >30 | >30 | >30 | >30 |
Calcium mobilization assay.
G-protein-gated inwardly rectifying potassium channel (GIRK) assay.
Assay performed in pooled human liver microsomes (HLM) in the presence of NADPH with CYP-specific probe substrates.
Table 4. In Vivo Rat and Dog Pharmacokinetics of VU6043653.

| IV PK | |||||
|---|---|---|---|---|---|
| Species | Dose (mg/kg) | t1/2 (h)b | MRT (h)b | CLp (mL/min/kg)b | Vss (L/kg)b |
| Rat (SD)a | 1.0 | 0.74 | 0.84 | 41 | 2.0 |
| Dog (beagle)c | 0.5 | 1.55 | 1.41 | 38 | 3.2 |
| PO PK | |||||
|---|---|---|---|---|---|
| Species | Dose (mg/kg) | Tmax (h)e | Cmax (ng/mL)e | AUC0-∞ (h·ng/mL)e | %Fe |
| Rat (SD)d | 10.0 | 1.95 | 402 | 1320 | 43 |
| Dog (beagle)f | 3.0 | 0.67 | 70.3 | 264 | 20 |
Male Sprague–Dawley rats (n = 3); vehicle = 10% ethanol, 70% PEG400, 20% saline.
t1/2 = terminal phase plasma half-life; MRT = mean residence time; Vss = volume of distribution at steady-state; CLp = plasma clearance.
Male beagle dogs (n = 3); vehicle = 10% ethanol, 70% PEG400, 20% saline.
Male Sprague–Dawley rats (n = 3); vehicle = 0.5% aqueous methylcellulose with 0.1% Tween 80.
Tmax = time at which Cmax occurs; Cmax = maximum concentration; AUC = area under the curve; %F = oral bioavailability.
Male beagle dogs (n = 3); 0.5% aqueous methylcellulose with 0.1% Tween 80 in saline.
Nonetheless, as a non-aryl/heterobiaryl acetylene mGlu5 NAM with an encouraging in vivo rodent PK profile, we wished to further assess VU6043653 as a novel chemotype. Therefore, we compared metabolites in multiple species to better understand species differences in clearance and metabolism. These metabolism experiments, utilizing cryopreserved hepatocytes, identified amide hydrolysis as a major metabolite across all species tested (rats, dogs cynomolgus monkeys, and humans). Consistent with the high plasma clearance observed in dogs, high turnover was observed more so in dog hepatocytes than any other species tested (see the Supporting Information for additional details and results). To further evaluate our novel chemotype, the off-target and safety/toxicity profiles for this compound were further investigated. An ancillary pharmacology screen (Eurofins Panlabs)38 revealed both Adenosine A3 and Androgen receptors as potential off-target liabilities (≥70% inhibition at 10 μM) (see the Supporting Information for the full ancillary pharmacology profile).
In conclusion, we have established that 5-membered heterocycles are able to serve as competent isosteres for the metabolically labile pyrimidine of clinical candidate VU0424238 (10) and predecessor compounds 11. Of analogs assessed, VU6043653 (22iA) displayed the best overall PK profile, with low human predicted hepatic clearance (CLhep = 6 mL/min/kg), favorable rat and human plasma protein binding (fu,plasma = 0.059), and high brain penetration (Kp = 1.68; Kp,uu = 0.34). VU6043653 displayed high selectivity for mGlu5 over all other mGlu receptors evaluated (mGlu1–4 and mGlu7–8) and provided an improved CYP inhibition profile (CYP 2C9, 2D6, 3A4 IC50’s ≥ 30 μM) when compared to predecessor compounds 11. In fact, VU6043653 addressed many other challenges associated with compounds 11, such as high predicted human CLhep, poor fu, and DAT inhibition. However, VU6043653 did not progress forward due to its moderate potency in inhibiting human mGlu5 as well as poor higher species PK. Although this exercise did not provide mGlu5 NAMs with suitable DMPK profiles to warrant further advancement, it did highlight SAR insights for future scaffold designs. These refinements will be reported in due course.
Acknowledgments
We thank the NIH for funding via the Molecular Libraries Probe Center Network (U54MH084659 to C.W.L.), Vanderbilt NCDDG (U01MH087965), and the HEAL Initiative (1UG3NS116218-01 to J.M.R and C.W.L). We also thank William K. Warren, Jr., and the William K. Warren Foundation, which funded the William K. Warren, Jr., Chair in Medicine (to C.W.L.).
Glossary
Abbreviations
- AO
Aldehyde oxidase
- CLhep
Hepatic clearance
- CLint
Intrinsic clearance
- CNS
Central nervous system
- CYP
Cytochrome P450
- DAT
Dopamine transporter
- DMPK
Drug metabolism and pharmacokinetics
- GIRK
G-protein-gated inwardly rectifying potassium channel
- MAO-B
Monoamine oxidase-B
- hmGlu5
Human metabotropic glutamate receptor subtype 5
- mGluR
Metabotropic glutamate receptor
- NAM
Negative allosteric modulator
- SAR
Structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00481.
General methods for the synthesis and characterization for key compounds and experimental details for calcium mobilization assays, in vitro and in vivo DMPK protocols, multispecies hepatocyte metabolism studies, and off-target assessment (PDF)
Author Contributions
E.S.C, R.A.C, K.E.C, M.L.M. A.M.B, A.S.F, and K.J.T. performed synthetic chemistry. E.S.C. and K.J.T. provided compound characterization. M.A.M, N.B.B., H.P.C., A.L.R., and C.M.N. performed and analyzed molecular pharmacology data. W.P., J.M.R., A.T.G, and C.K.J. performed and analyzed in vivo pharmacology experiments. S.C., A.L.B., and O.B. performed and analyzed DMPK experiments. P.J.C, C.M.N., H.C.P., C.K.J, J.M.R., and C.W.L. and oversaw experimental design, and K.J.T. wrote the manuscript with input from all authors.
The authors declare the following competing financial interest(s): R.A.C., A.S.F., C.W.L, P.J.C., and K.J.T are inventors on applications for composition of matter patents that protect several series of mGlu5 negative allosteric modulators.
Supplementary Material
References
- Golubeva A. V.; Moloney R. D.; O’Connor R. M.; Dinan T. G.; Cryan J. F. Metabotropic glutamate receptors in central nervous system disease. Curr. Drug Targets 2016, 17, 538–616. 10.2174/1389450116666150316224011. [DOI] [PubMed] [Google Scholar]
- Conn P. J.; Pin J. P. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 205–237. 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Niswender C. M.; Conn P. J. Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295. 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett K. A.; Doré A. S.; Christopher J. A.; Weiss D. R.; Marshall F. H. Structures of mGluRs shed light on the challenges of drug development of allosteric modulators. Curr. Opin. Pharmacol. 2015, 20, 1–7. 10.1016/j.coph.2014.09.022. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Emmitte K. A.; Hopkins C. R.; Bridges T. M.; Gregory K. J.; Niswender C. M.; Conn P. J. Practical strategies and concepts in GPCR allosteric modulator discovery: Recent advances with metabotropic glutamate receptors. Chem. Rev. 2016, 116, 6707. 10.1021/acs.chemrev.5b00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmitte K. A. mGlu5 negative allosteric modulators: a patent review (2013–2016). Expert Opin. Ther. Pat. 2017, 27, 691. 10.1080/13543776.2017.1280466. [DOI] [PubMed] [Google Scholar]
- Emmitte K. A. Recent Advances in the Design and Development of Novel Negative Allosteric Modulators of mGlu5. ACS Chem. Neurosci. 2011, 2, 411–432. 10.1021/cn2000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J.; Xiong H. SAR Studies on mGlu5 Receptor Positive Allosteric Modulators (2003- 2013). Curr. Top. Med. Chem. 2014, 14, 1789–1841. 10.2174/1568026614666140826120419. [DOI] [PubMed] [Google Scholar]
- Jaeschke G.; Kolczewski S.; Spooren W.; Vieira E.; Bitter-Stoll N.; Boissin P.; Borroni E.; Büttelmann B.; Ceccarelli S.; Clemann N.; David B.; Funk C.; Guba W.; Harrison A.; Hartung T.; Honer M.; Huwyler J.; Kuratli M.; Niederhauser U.; Pähler A.; Peters J. U.; Petersen A.; Prinssen E.; Ricci A.; Rueher D.; Rueher M.; Schneider M.; Spurr P.; Stoll T.; Tännler D.; Wichmann J.; Porter R. H.; Wettstein J. G.; Lindemann L. Metabotropic glutamate receptor 5 negative allosteric modulators: discovery of 2-chloro-4-[1-(4-fluorophenyl)-2,5-dimethyl-1H-imidazol-4-ylethynyl]pyridine (basimglurant, RO4917523), a promising novel medicine for psychiatric diseases. J. Med. Chem. 2015, 58, 1358. 10.1021/jm501642c. [DOI] [PubMed] [Google Scholar]
- Carcache D.; Vranesic I.; Blanz J.; Desrayaud S.; Fendt M.; Glatthar R. Benzimidazoles as potent and orally active mGlu5 receptor antagonists with and improved PK profile. ACS Med. Chem. Lett. 2011, 2, 58. 10.1021/ml100215b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro F. M.; Vieira L. B.; Pires R. G.; Olmo R. P.; Ferguson S. S. Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol. Res. 2017, 115, 179–191. 10.1016/j.phrs.2016.11.013. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E.; Des Portes V.; Hagerman R.; Jacquemont S.; Charles P.; Visootsak J.; Brinkman M.; Rerat K.; Koumaras B.; Zhu L.; Barth G. M.; Jaecklin T.; Apostol G.; von Raison F. Mavoglurant in fragile X syndrome: Results of two randomized, double-blind, placebo-controlled trials. Sci.Transl. Med. 2016, 8, 321ra5. 10.1126/scitranslmed.aab4109. [DOI] [PubMed] [Google Scholar]
- Bailey D. B. Jr; Berry-Kravis E.; Wheeler A.; Raspa M.; Merrien F.; Ricart J.; Koumaras B.; Rosenkranz G.; Tomlinson M.; von Raison F.; Apostol G. Mavoglurant in adolescents with fragile X syndrome: analysis of Clinical Global Impression-Improvement source data from a double-blind therapeutic study followed by an open-label, long-term extension study. J. Neurodev. Disord. 2016, 8, 1. 10.1186/s11689-015-9134-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop A. S.; Gomez-Mancilla B.; Neri G.; Willemsen R.; Gasparini F. Fragile X syndrome: a preclinical review on metabotropic glutamate receptor 5 (mGluR5) antagonists and drug development. Psychopharmacology. 2014, 231, 1217. 10.1007/s00213-013-3330-3. [DOI] [PubMed] [Google Scholar]
- Silverman J. L.; Smith D. G.; Rizzo S. J.; Karras M. N.; Turner S. M.; Tolu S. S.; Bryce D. K.; Smith D. L.; Fonseca K.; Ring R. H.; Crawley J. N. Negative Allosteric Modulation of the mGluR5 Receptor Reduces Repetitive Behaviors and Rescues Social Deficits in Mouse Models of Autism. Sci. Transl. Med. 2012, 4, 131ra51. 10.1126/scitranslmed.3003501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W.; Choi S. Y.; Lee E.; Park H.; Kang J.; Park H.; Choi Y.; Lee D.; Park S. G.; Kim R.; Cho Y. S.; Choi J.; Kim M. H.; Lee J. W.; Lee S.; Rhim I.; Jung M. W.; Kim D.; Bae Y. C.; Kim E. Social deficits in IRSp53 mutant mice improved by NMDAR and mGluR5 suppression. Nat. Neurosci. 2015, 18, 435. 10.1038/nn.3927. [DOI] [PubMed] [Google Scholar]
- Rascol O.; Fox S.; Gasparini F.; Kenney C.; Di Paolo T.; Gomez-Mancilla B. Use of metabotropic glutamate 5-receptor antagonists for treatment of levodopa-induced dyskinesias. Parkinsonism Relat. Disord. 2014, 20, 947. 10.1016/j.parkreldis.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Trenkwalder C.; Stocchi F.; Poewe W.; Dronamraju N.; Kenney C.; Shah A.; von Raison F.; Graf A. Mavoglurant in Parkinson’s patients with L-Dopa-induced dyskinesias: Two randomized Phase 2 studies. Mov. Disord. 2016, 31, 1054. 10.1002/mds.26585. [DOI] [PubMed] [Google Scholar]
- Tison F.; Keywood C.; Wakefield M.; Durif F.; Corvol J. C.; Eggert K.; Lew M.; Isaacson S.; Bezard E.; Poli S. M.; Goetz C. G.; Trenkwalder C.; Rascol O. A Phase 2A Trial of the movel mGluR5-negative allosteric modulator Dipraglurant for Levodopa-induced dyskinesia in Parkinson’s Disease. Mov. Disord. 2016, 31, 1373. 10.1002/mds.26659. [DOI] [PubMed] [Google Scholar]
- Zerbib F.; Bruley des Varannes S.; Roman S.; Tutuian R.; Galmiche J. P.; Mion F.; Tack J.; Malfertheiner P.; Keywood C. Randomized clinical trial: effects of monotherapy with ADX10059, a mGluR5 inhibitor, on symptoms and reflux events in patients with gastro-oesophageal reflux disease. Aliment. Pharmacol. Ther. 2011, 33, 911–921. 10.1111/j.1365-2036.2011.04596.x. [DOI] [PubMed] [Google Scholar]
- Mihov Y.; Hasler G. Negative Allosteric Modulators of Metabotropic Glutamate Receptors Subtype 5 in Addiction: A Therapeutic Window. Int. J. Neuropsychopharmacol. 2016, 19, pyw002. 10.1093/ijnp/pyw002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass J. T.; Osborne M. P. H.; Watson N. L.; Brown J. L.; Olive M. F. mGluR5 Antagonism Attenuates Methamphetamine Reinforcement and Prevents Reinstatement of Methamphetamine-Seeking Behavior in Rats. Neuropsychopharmacology. 2009, 34, 820. 10.1038/npp.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Fardon R.; Baptista M. A. S.; Dayas C. V.; Weiss F. Dissociation of the Effects of MTEP [3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]piperidine] on Conditioned Reinstatement and Reinforcement: Comparison between Cocaine and a Conventional Reinforcer. J. Pharmacol. Exp. Ther. 2009, 329, 1084. 10.1124/jpet.109.151357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann L.; Porter R. H.; Scharf S. H.; Kuennecke B.; Bruns A.; von Kienlin M.; Harrison A. C.; Paehler A.; Funk C.; Gloge A.; Schneider M.; Parrott N. J.; Polonchuk L.; Niederhauser U.; Morairty S. R.; Kilduff T. S.; Vieira E.; Kolczewski S.; Wichmann J.; Hartung T.; Honer M.; Borroni E.; Moreau J. L.; Prinssen E.; Spooren W.; Wettstein J. G.; Jaeschke G. Pharmacology of Basimglurant (RO4917523, RG7090), a Unique Metabotropic Glutamate Receptor 5 Negative Allosteric Modulator in Clinical Development for Depression. J. Pharmacol. Exp. Ther. 2015, 353, 213. 10.1124/jpet.114.222463. [DOI] [PubMed] [Google Scholar]
- Quiroz J. A.; Tamburri P.; Deptula D.; Banken L.; Beyer U.; Rabbia M.; Parkar N.; Fontoura P.; Santarelli L. Efficacy and Safety of Basimglurant as Adjunctive Therapy for Major Depression: A Randomized Clinical Trial. JAMA Psychiatry. 2016, 73, 675. 10.1001/jamapsychiatry.2016.0838. [DOI] [PubMed] [Google Scholar]
- Du Y.; Gao F.; Sun H.; Wu C.; Zhu G.; Zhu M. Novel substituted 4-(Arylethynyl)-Pyrrolo[2,3-d]pyrimidines negative allosteric modulators (NAMs) of the metabotropic glutamate receptor subtype 5 (mGlu5) treat depressive disorder in mice. Eur. J. Med. Chem. 2023, 261, 115855. 10.1016/j.ejmech.2023.115855. [DOI] [PubMed] [Google Scholar]
- Rutrick D.; Stein D. J.; Subramanian G.; Smith B.; Fava M.; Hasler G.; Cha J. H.; Gasparini F.; Donchev T.; Ocwieja M.; Johns D.; Gomez-Mancilla B. Mavoglurant Augmentation in OCD Patients Resistant to Selective Serotonin Reuptake Inhibitors: A Proof-of-Concept, Randomized, Placebo-Controlled, Phase 2 Study. Adv. Ther. 2017, 34, 524. 10.1007/s12325-016-0468-5. [DOI] [PubMed] [Google Scholar]
- Pereira V.; Goudet C. Emerging Trends in Pain Modulation by Metabotropic Glutamate Receptors. Front. Mol. Neurosci. 2019, 11, 464. 10.3389/fnmol.2018.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzitelli M.; Presto P.; Antenucci N.; Meltan S.; Neugebauer V. Recent Advances in the Modulation of Pain by the Metabotropic Glutamate Receptors. Cells. 2022, 11, 2608. 10.3390/cells11162608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin J. C.; Goadsby P. J. Glutamatergic fine tuning with ADX-10059: a novel therapeutic approach for migrane?. Expert Opin. Invest. Drugs. 2010, 19, 555–561. 10.1517/13543781003691832. [DOI] [PubMed] [Google Scholar]
- Hoffmann J.; Charles A. Glutamate and Its Receptors as Therapeutic Targets for Migraine. Neurotherapeutics. 2018, 15, 361. 10.1007/s13311-018-0616-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini F.; Lingenhöhl K.; Stoehr N.; Flor P. J.; Heinrich M.; Vranesic I.; Biollaz M.; Allgeier H.; Heckendorn R.; Urwyler S.; Varney M. A.; Johnson E. C.; Hess S. D.; Rao S. P.; Sacaan A. I.; Santori E. M.; Veliçelebi G.; Kuhn R. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 1999, 38, 1493. 10.1016/S0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- Cosford N. D. P.; Tehrani L.; Roppe J.; Schweiger E.; Smith N. D.; Anderson J.; Bristow L.; Brodkin J.; Jiang X.; McDonald I.; Rao S.; Washburn M.; Varney M. A. 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: A potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J. Med. Chem. 2003, 46, 204. 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Balan G.; Barreiro G.; Boscoe B. P.; Chenard L. K.; Cianfrogna J.; Claffey M. M.; Chen L.; Coffman K. J.; Drozda S. E.; Dunetz J. R.; Fonseca K. R.; Galatsis P.; Grimwood S.; Lazzaro J. T.; Mancuso J. Y.; Miller E. L.; Reese M. R.; Rogers B. N.; Sakurada I.; Skaddan M.; Smith D. L.; Stepan A. F.; Trapa P.; Tuttle J. B.; Verhoest P. R.; Walker D. P.; Wright A. S.; Zaleska M. M.; Zasadny K.; Shaffer C. L. Discovery and Preclinical Characterization of 1-Methyl-3-(4-methylpyridin-3-yl)-6-(pyridin-2-ylmethoxy)-1H-pyrazolo-[3,4-b]pyrazine (PF470): A Highly Potent, Selective, and Efficacious Metabotropic Glutamate Receptor 5 (mGluR5) Negative Allosteric Modulator. J. Med. Chem. 2014, 57, 861. 10.1021/jm401622k. [DOI] [PubMed] [Google Scholar]
- Varnäs K.; Cselényi Z.; Arakawa R.; Nag S.; Stepanov V.; Moein M. M.; Johnström P.; Kingston L.; Elmore C. S.; Halldin C.; Farde L. The pro-psychotic metabotropic glutamate receptor compounds fenobam and AZD9272 share binding sites with monoamine oxidase-B inhibitors in humans. Neuropharmacology 2020, 162, 107809. 10.1016/j.neuropharm.2019.107809. [DOI] [PubMed] [Google Scholar]
- Multiple Ascending Dose Study of TMP-301 in Healthy Subjects. https://classic.clinicaltrials.gov/ct2/show/NCT06025396 (accessed: 10/26/2023).
- Bennett K. A.; Christopher J. A.; Tehan B. G. Structure-based discovery and development of metabotropic glutamate receptor 5 negative allosteric modulators. Adv. Pharmacol. 2020, 88, 35. 10.1016/bs.apha.2020.03.001. [DOI] [PubMed] [Google Scholar]
- Felts A. S.; Rodriguez A. L.; Blobaum A. L.; Morrison R. D.; Bates B. S.; Thompson Gray A.; Rook J. M.; Tantawy M. N.; Byers F. W.; Chang S.; Venable D. F.; Luscombe V. B.; Tamagnan G. D.; Niswender C. M.; Daniels J. S.; Jones C. K.; Conn P. J.; Lindsley C. W.; Emmitte K. A. Discovery of N-(5-Fluoropyridin-2-yl)-6-methyl-4-(pyrimidin-5-yloxy)picolinamide (VU0424238): A Novel Negative Allosteric Modulator of Metabotropic Glutamate Receptor Subtype 5 Selected for Clinical Evaluation. J. Med. Chem. 2017, 60, 5072. 10.1021/acs.jmedchem.7b00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch R. D.; Blobaum A. L.; Felts A. S.; Conn P. J.; Lindsley C. W. Species-Specific Involvement of Aldehyde Oxidase and Xanthine Oxidase in the Metabolism of the Pyrimidine-Containing mGlu5-Negative Allosteric Modulator VU0424238 (Auglurant). Drug Metab. Dispos. 2017, 45, 1245–1259. 10.1124/dmd.117.077552. [DOI] [PubMed] [Google Scholar]
- Morrison R. D.; Blobaum A. L.; Byers F. W.; Santomango T. S.; Bridges T. M.; Stec D.; Brewer K. A.; Sanchez-Ponce R.; Corlew M. M.; Rush R.; Felts A. S.; Manka J.; Bates B. S.; Venable D. F.; Rodriguez A. L.; Jones C. K.; Niswender C. M.; Conn P. J.; Lindsley C. W.; Emmitte K. A.; Daniels J. S. The role of aldehyde oxidase and xanthine oxidase in the biotransformation of a novel negative allosteric modulator of metabotropic glutamate receptor subtype 5. Drug Metab. Dispos. 2012, 40, 1834–45. 10.1124/dmd.112.046136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankovic Z. CNS Drug Design: Balancing Physiochemical Properties for Optimal Brain Exposure. J. Med. Chem. 2015, 58, 2584–2608. 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci. 2016, 7, 767–775. 10.1021/acschemneuro.6b00029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.