

Abstract
Introduction: Extramedullary disease (EMD) is a rare manifestation of Waldenström macroglobulinemia (WM), and its clinical and prognostic implications are poorly understood. Methods: In this single‐center study, we investigated the clinical significance of EMD in a cohort of 469 WM patients. Results: EMD was identified in 30 (6.4%) patients, with the central nervous system, kidneys, and lungs being the most frequently affected sites. The cumulative incidence of EMD was 12.6% at 15 years. Median overall survival rates at 5 and 10 years for patients with EMD were 63% and 37%, respectively. Conclusion: Our findings indicate a persistent risk of EMD throughout the disease course, with no significant impact on long‐term survival.
Keywords: extramedullary, extranodal, lymphoplasmacytic lymphoma, prognosis, Waldenström macroglobulinemia
1. INTRODUCTION
Waldenström macroglobulinemia (WM) is a hematological malignancy defined by bone marrow (BM) infiltration of a lymphoplasmacytic lymphoma (LPL) and secretion of an immunoglobulin M (IgM) monoclonal protein [1]. Although WM predominantly affects the BM, a subset of patients also develops extramedullary disease (EMD), which is considered a rare manifestation of the disease [2]. While several small retrospective case series have detailed specific EMD sites, only two population‐based studies have investigated its clinical significance [3, 4, 5]. In a single‐center study, Banwait et al. [2] identified EMD in 4.4% of patients, with the lungs and central nervous system (CNS) being the most frequently involved sites. The 10‐year survival rate was 79% among patients with EMD, but comparative survival data for patients without EMD were not provided. In contrast, Cao et al. [5] reported that extramedullary involvement at the time of WM diagnosis was associated with a worse prognosis. However, their study included patients with adenopathy and cases without pathological confirmation. As a result, the clinical and prognostic significance of extramedullary involvement remains unclear.
To address these uncertainties, we conducted an observational study to assess the incidence, clinical features, and prognostic implications of EMD in patients with WM.
2. METHODS
Patients with WM, as defined by the European Consortium for Waldenström's macroglobulinemia consensus report criteria, were included [6]. The patients were diagnosed between 2000 and 2022 in the Danish Region Zealand, which covers 15% of the Danish population. The study was approved by the Danish National Research Ethics Committee (ID 2113049) and the Regional Data Protection Agency (REG‐020‐2022).
Only WM patients with LPL in tissue samples or cytological material from one or more extramedullary sites were characterized as EMD. Cases with nodal and splenic involvement alone were not considered EMD, and patients with histological transformation to diffuse large B‐cell lymphoma prior to the occurrence of EMD were excluded. Clinical data were collected from patient records. Data on immunohistochemical profiles, flow cytometry, and real‐time quantitative polymerase chain reaction (qPCR) or next‐generation sequencing (NGS) analysis for MYD88 L265P mutations were retrospectively retrieved from pathology reports. Cases with EMD not initially tested for MYD88 L265P were analyzed with qPCR to determine the mutational status, provided sufficient material was available. All diagnostic EMD specimens were examined by an expert hematopathologist. Response to treatment was evaluated using the criteria from the sixth International Workshop on Waldenstrom's macroglobulinemia [7].
Descriptive statistics were used to summarize clinical characteristics. Comparisons between groups were performed with the chi‐square and the Mann–Whitney tests. Cumulative incidence analyses were estimated using a competing risk model. Overall survival (OS) was calculated from the date of WM or EMD diagnosis to the date of death or follow‐up. Survival curves were generated using the Kaplan–Meier method and differences were compared with the log‐rank test. Univariate and multivariate analyses were performed using a Cox proportional hazards regression model. Statistical analyses were calculated using Stata Statistical Software version 18.0.
3. RESULTS
The total cohort consisted of 469 WM patients and EMD was detected in 30 (6.4%) of patients. Baseline characteristics are shown in Table 1. Nine (30%) patients had EMD at the time of WM diagnosis. The remaining 21 (70%) patients developed EMD at a later stage of their disease with a median timespan from WM diagnosis to the occurrence of EMD being 4.6 years (range 0.3–14.9). The cumulative incidence of EMD, with death as a competing event, was 3.5% at 5 years, 7.2% at 10 years, and 12.6% at 15 years from diagnosis of WM (Supporting Information Figure 1). CNS, pleural/pulmonary, and kidney involvement were the most common extramedullary manifestations, representing 70% of EMD cases (Supporting Information Table 1). The MYD88 L265P mutational status was available in 262 out of 469 (56%) patients from the overall cohort and 26 out of 30 (87%) patients with EMD. Mutated MYD88 L265P was found in 96% of the total cohort, and no cases of MYD88 WT were detected in patients with EMD. Paired biopsies, including samples from the extramedullary component, were analyzed for the MYD88 L265P mutation in 21 patients, and the mutation was found in both components in all cases.
TABLE 1.
Baseline characteristics of patients with Waldenström macroglobulinemia with or without extramedullary disease.
| Characteristics | Patients with EMD (n = 30) | Patients without EMD (n = 439) | p |
|---|---|---|---|
| Age (years) | |||
| Median (range) | 68 (38–84) | 73 (41–96) | 0.02 |
| Sex | |||
| M/F ratio | 1.5 | 1.5 | 0.99 |
| IgM (g/L) | |||
| Median (range) | 15.6 (1.2–78.1) | 16.6 (1.1–109) | 0.95 |
| Missing data (n) | 1 | 3 | |
| Hemoglobin (g/dL) | |||
| Median (range) | 11.6 (5.8–15.5) | 12.4 (5.1–19.2) | 0.03 |
| Missing data (n) | 1 | 2 | |
| Platelet count (109/L) | |||
| Median (range) | 255 (56–574) | 257 (1–1207) | 0.72 |
| Missing data (n) | 1 | 3 | |
| β2M (g/dL) | |||
| Median (range) | 4.5 (1.6–7.7) | 3.0 (1.3–16.1) | 0.02 |
| Missing data (n) | 8 | 53 | |
| LDH | |||
| Median (range) | 161 (106–600) | 164 (60–1600) | 0.44 |
| Missing data (n) | 1 | 8 | |
| Albumin (g/L) | |||
| Median (range) | 35 (20–46) | 36 (12–47) | 0.44 |
| Missing data (n) | 1 | 11 | |
| Symptomatic disease | |||
| n (%) | 22 (73) | 260 (59) | 0.13 |
Abbreviations: β2M, beta‐2 microglobulin; EMD, extramedullary disease; IgM, immunoglobulin M; LDH, lactate dehydrogenase.
At the time of EMD diagnosis, 17 (57%) patients were treatment‐naïve. The remaining 13 (43%) patients received a median of 2 (range 1–5) prior lines of therapy. Treatment was initiated in 23 (77%) patients at the time of EMD diagnosis. First‐line treatments included bendamustine–rituximab (39%), Bruton tyrosine kinase (BTK) inhibitors (22%), rituximab, and alkylator‐based combinations (13%). The remaining treatments (26%) were rituximab and nucleoside analogue combinations, high‐dose methotrexate‐based combinations, rituximab monotherapy, and radiation therapy. Response evaluation indicated that 39% of patients achieved either a complete response (CR) or a very good partial response (VGPR), 39% attained a partial response (PR), while 22% did not respond to treatment.
The median follow‐up time was 5.9 years (range 0.2–21.9). The survival estimates of patients with and without EMD were comparable, with 5‐year and 10‐year survival rates after primary WM diagnosis at 63% and 37% in patients with EMD, and 66% and 44% in patients without EMD (Figure 1). The median OS after the occurrence of EMD was 5.4 years (95% confidence interval [CI], 1.5–6.4) for patients diagnosed with EMD at the time of primary WM diagnosis, and 4.2 years (95% CI, 2.8–8.1) for those diagnosed with EMD at progression (p = 0.8). The cumulative incidence of death was comparable between patients with and without EMD when calculating EMD as a competing risk (Supporting Information Table 2). In the Cox regression analysis including other clinical risk factors, EMD had no significant impact on survival (Supporting Information Table 3).
FIGURE 1.
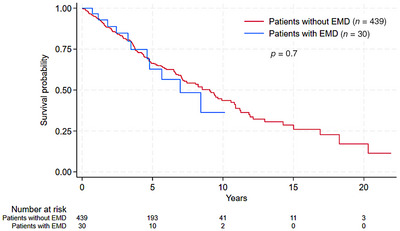
Overall survival from diagnosis of Waldenström macroglobulinemia in patients with and without extramedullary disease (EMD).
4. DISCUSSION
In the present study, we examined the incidence, clinicopathological features, and outcomes of patients with extramedullary involvement in a cohort of WM patients spanning two decades. EMD was detected in 6.4% of patients across the total study cohort. Roughly one‐third of patients with EMD presented with extramedullary involvement at the time of WM diagnosis, confirming the findings of a previous report that EMD primarily develops at progression [2]. The cumulative risk of EMD was evenly distributed and persistent throughout the disease course, with a cumulative incidence of 12.6% at 15 years, accounting for death without EMD as a competing risk. Banwait et al. [2] reported a slightly lower rate of EMD, but all cases were diagnosed prior to 2013, which may have affected the accuracy of the diagnosis.
EMD has mainly been evaluated in various case series that focus on specific extramedullary sites [3, 4, 8, 9]. In our study, the most common sites were the CNS (also known as Bing–Neel syndrome), kidneys, and lungs/pleura, which is consistent with a previous study suggesting a predisposition toward these sites [2]. Notably, monoclonal gammopathy‐related lesions or deposition of amyloid are important competing causes of extramedullary complications, emphasizing the significance of a thorough evaluation of WM patients presenting with unexplained symptoms or extramedullary manifestations [10].
Approximately half of the patients in our study were treatment‐naïve at the time of extramedullary involvement. Treatment was initiated in 76% of patients directly following the EMD diagnosis and the majority achieved a response equivalent to at least a PR. We observed no difference in survival rates between patients with and without EMD. Due to a small number of cases, potential differences in survival between specific EMD sites could not be reliably compared. Banwait et al. [2] reported higher survival rates in EMD patients, but their findings were limited by the lack of direct comparison to patients without EMD. Conversely, Cao et al. [5] found a significantly shorter median OS in patients with EMD at the time of WM diagnosis. However, their analysis was primarily focused on patients with nodal involvement, making their data not directly comparable to ours. As the first population‐based comparative analysis, our data suggest that WM patients with and without EMD generally have similar long‐term outcomes. Nonetheless, because EMD encompasses a diverse range of clinical complications, there is a need for further studies to explore the potential prognostic implications of specific EMD sites.
Somatic mutations in MYD88 and CXCR4 have been established as genetic hallmarks of WM [11, 12]. In our study, the MYD88 L265P mutation was highly recurrent in patients with EMD and was present in both primary diagnostic samples and the extramedullary component in all cases assessed. However, detecting subtle differences may be challenging given the infrequency of MYD88 WT in WM; therefore, our findings require validation in larger studies. The chemokine receptor CXCR4 is involved in regulating cell trafficking and homing of malignant B cells in WM [13]. Moreover, CXCR4 mutations have been shown to enhance tumor dissemination to extramedullary sites in mouse models [14]. Unfortunately, data on CXCR4 mutations were not available in our cohort. Further studies investigating the role of CXCR4 mutations and other novel molecular markers in patients with EMD are warranted.
Our study is a retrospective analysis with a limited sample size, reflecting the rarity of extramedullary involvement in WM. The rate of detection also depends on the extent of examination, entailing a potential risk of underestimating the incidence. Despite these limitations, our study is the first to demonstrate a continuous risk of EMD throughout the entire course of the disease. In addition, our findings indicate that EMD has limited impact on survival in patients with WM.
AUTHOR CONTRIBUTIONS
Simon Østergaard wrote the first draft of the manuscript; Mette Ølgod Pedersen and Lise Mette Rahbek Gjerdrum provided molecular data and performed pathology reviews; Simon Østergaard and Lars Munksgaard collected clinical data. All the authors contributed to the conceptual design of the study, the data analysis, and to the editing of the manuscript. All the authors read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
ETHICS STATEMENT
This study was approved by the Danish National Research Ethics Committee (ID: 2113049) and the Region Zealand Data Protection Agency (ID: REG‐020‐2022).
PATIENT CONSENT STATEMENT
The authors have confirmed patient consent statement is not needed for this submission.
CLINICAL TRIAL REGISTRATION
The authors have confirmed clinical trial registration is not needed for this submission.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors have nothing to report.
Østergaard S, Munksgaard L, Nielsen TH, Hammer T, Pedersen LM, Ølgod Pedersen M, et al. Extramedullary disease in Waldenström macroglobulinemia: A population‐based observational study. eJHaem. 2024;5:1269–1273. 10.1002/jha2.1037
DATA AVAILABILITY STATEMENT
The data from this study can be made available upon reasonable request from the corresponding author and with the appropriate approvals from the Region Zealand Data Protection Agency (ID: REG‐020‐2022).
REFERENCES
- 1. García‐Sanz R, Hunter ZR, Poulain S, Varettoni M, Owen RG. New developments in the diagnosis and characterization of Waldenström's macroglobulinemia. Expert Rev Hematol. 2023;16(11):835–847. [DOI] [PubMed] [Google Scholar]
- 2. Banwait R, Aljawai Y, Cappuccio J, McDiarmid S, Morgan EA, Leblebjian H, et al. Extramedullary Waldenström macroglobulinemia. Am J Hematol. 2015;90(2):100–104. [DOI] [PubMed] [Google Scholar]
- 3. Vos JM, Gustine J, Rennke HG, Hunter Z, Manning RJ, Dubeau TE, et al. Renal disease related to Waldenstrom macroglobulinaemia: incidence, pathology and clinical outcomes. Br J Haematol. 2016;175:623–630. [DOI] [PubMed] [Google Scholar]
- 4. Simon L, Fitsiori A, Lemal R, Dupuis J, Carpentier B, Boudin L, et al. Bing‐Neel syndrome, a rare complication of Waldenström macroglobulinemia: analysis of 44 cases and review of the literature. A study on behalf of the French Innovative Leukemia Organization (FILO). Haematologica. 2015;100(12):1587–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cao X, Ye Q, Orlowski RZ, Wang X, Loghavi S, Tu M, et al. Waldenström macroglobulinemia with extramedullary involvement at initial diagnosis portends a poorer prognosis. J Hematol Oncol. 2015;8:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dogliotti I, Jiménez C, Varettoni M, Talaulikar D, Bagratuni T, Ferrante M, et al. Diagnostics in Waldenström's macroglobulinemia: a consensus statement of the European Consortium for Waldenström's macroglobulinemia. Leukemia. 2023;37:388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Owen RG, Kyle RA, Stone MJ, Rawstron AC, Leblond V, Merlini G, et al. Response assessment in Waldenström macroglobulinaemia: update from the VIth international workshop. Br J Haematol. 2013;160(2):171–176. [DOI] [PubMed] [Google Scholar]
- 8. Stien S, Durot E, Durlach A, Beylot‐Barry M, Adamski H, Beltraminelli H, et al. Cutaneous involvement in Waldenström's macroglobulinaemia. Acta Derm Venereol. 2020;100:adv00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bhatti K, Nazir A, Ostergaard S, Schejbel L, Norgaard P, Gjerdrum LMR, et al. Bone involvement as a primary rare manifestation of Waldenstrom macroglobulinemia: a case report and prevalence in a nationwide population‐based cohort study. J Hematol. 2022;11(6):233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Higgins L, Nasr SH, Said SM, Kapoor P, Dingli D, King RL, et al. Kidney involvement of patients with waldenström macroglobulinemia and other IgM‐producing B cell lymphoproliferative disorders. Clin J Am Soc Nephrol. 2018;13(7):1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hunter ZR, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM‐like CXCR4 mutations, and small somatic deletions associated with B‐cell lymphomagenesis. Blood. 2014;123:1637–1646. [DOI] [PubMed] [Google Scholar]
- 12. Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenström macroglobulinemia. Blood. 2014;123(18):2791–2796. [DOI] [PubMed] [Google Scholar]
- 13. Ngo HT, Leleu X, Lee J, Jia X, Melhem M, Runnels J, et al. SDF‐1/CXCR4 and VLA‐4 interaction regulates homing in Waldenstrom macroglobulinemia. Blood. 2008;112(1):150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roccaro AM, Sacco A, Jimenez C, Maiso P, Moschetta M, Mishima Y, et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014;123(26):4120–4131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Data Availability Statement
The data from this study can be made available upon reasonable request from the corresponding author and with the appropriate approvals from the Region Zealand Data Protection Agency (ID: REG‐020‐2022).