

Abstract

Human sapiens caseinolytic protease P (ClpP) is essential for maintaining mitochondrial proteome homeostasis, and its activation is increasingly recognized as a promising cancer therapy strategy. Herein, based on structure-guided drug design, we discovered a series of potent ClpP activators by introducing a methyl group to the imipridone scaffold of the ClpP activator ONC201 in Phase III clinical trials. Through structural optimization of the lead compound, the most optimal compound, CLPP-1071, exhibited exceptionally potent ClpP agonistic activity (EC50 = 23.5 nM, 107.1-fold stronger than ONC201) and inhibited the proliferation of HL60 cells (IC50 = 4.6 nM, 169.2-fold stronger than ONC201). CLPP-1071 possesses good pharmacokinetic properties and effectively prolongs the lifespan in the MOLM13 and HL60 xenograft models in mice through oral administration. CLPP-1071 is the most potent and orally efficacious ClpP activator reported to date.
Introduction
Mitochondria serve as the energy production centers within cells.1 Cancer cells typically exhibit high metabolic activity and consequently have much greater energy demands than normal cells, leading to a heightened dependence on mitochondrial function.2,3Human sapiens caseinolytic protease P (ClpP) is a vital mitochondrial protease responsible for mitochondrial proteostasis, regulates protein quality control,4,5 mitochondrial metabolism,6,7 and the integrity of oxidative phosphorylation,8 plays a critical role in maintaining mitochondrial function.9−12 An increasing number of studies suggest that the abnormalities in ClpP can lead to mitochondrial dysfunction, thereby contributing to various human diseases, including cancer.13−16 Clinically, overexpression of ClpP has been found in patients with multiple myeloma, various lymphomas, chronic myeloid leukemia, and certain solid tumors.17−19 Therefore, modulating the activity of ClpP is a promising therapeutic strategy for solid tumors and hematological malignancies.20
In recent years, research has revealed that ClpP activators can enhance the protease activity of ClpP, leading to the degradation of numerous proteins within the mitochondria and resulting in mitochondrial dysfunction.21−23 When mitochondrial function is impaired, cancer cells cannot sustain their high energy demands and metabolic activities, ultimately leading to apoptosis.24−27 Due to the overexpression of ClpP in cancer cells, ClpP activators can selectively induce cancer cell lethality and the hyperactivation of ClpP to degrade proteins in mitochondria and promote the apoptosis of tumor cells. These features allow ClpP activators to minimize damage to normal cells, thus reducing the side effects of treatment.9 Furthermore, studies have shown that when used in combination with other chemotherapeutic or targeted drugs, ClpP activators can enhance the efficacy of these drugs, further inhibiting the growth and survival of cancer cells.29,30 These features position ClpP activators as a promising potential strategy for cancer treatment.31
In drug discovery, it is challenging to directly activate a protease with a small molecule; fortunately, discovering activators for ClpP is achievable.32 In the past few years, several classes of ClpP activators have been reported (Figure 1). The earliest breakthroughs in ClpP activator research were achieved through the study of the antibiotic ADEP and its derivatives. For example, ADEP-41 (EC50 = 0.42 μM) has shown significant effects in activating ClpP and inducing cytotoxicity.33D9 can selectively bind to human ClpP by recognizing the YYW motif that controls ClpP protein activity, making it a highly species-selective human ClpP activator with an EC50 of 110 μM.34ONC201, a first-generation imipridone scaffold ClpP activator with an EC50 of 1.25 μM, progressed to Phase III clinical trials in 2023 for the treatment of H3 K27 M mutant gliomas (NTC05580562) and other types of cancer.35 In this clinical trial, ONC201 was tolerated at various dose levels (125–750 mg), resulting in a 12 month survival rate of 57% and a 24 month survival rate of 35%. At a dosage of 625 mg taken twice weekly, 51.4% of patients experienced adverse events such as fatigue, nausea, and vomiting. So far, it has spurred further research in this field, leading to the subsequent development of several imipridone derivatives with enhanced enzyme activity (EC50 of ONC206 = 0.26 μM, EC50 of ONC212 = 0.13 μM, and EC50 of TR57 = 0.12 μM).9,36−38 Over the past three years, medicinal chemists have made significant progress, developing molecules with different scaffolds and properties. Yang et al. have reported two distinct types of ClpP activators, ZG111(39,40) and ZK53.41 Notably, ZK53 exhibits high selectivity for human ClpP and shows no activity against bacterial ClpP proteins. Luo and colleagues developed four types of ClpP activators, including compound 16z(EC50 = 0.20 μM),42 which showed antitumor activity against HCT116 cells. Subsequent developments NCA029 (EC50 = 0.15 μM),437k (EC50 = 0.79 μM),44 and SL44 (EC50 = 1.30 μM)45 were applied to colorectal cancer, acute myeloid leukemia, and hepatocellular carcinoma, respectively. Very recently, Sun et al. reported novel tricyclic scaffold ClpP activators XT6 (EC50 = 0.08 μM)46 and ZYZ-17 (EC50 = 0.26 μM),47 which also exhibited good antitumor potency. Although several different types of activators have been reported, existing activators still exhibit low enzymatic activity or unfavorable pharmacokinetic properties, restricting the therapeutic strategy’s development. For example, when orally administered to rats at a dose of 15 mg/kg, ONC201 showed a lower peak concentration (Cmax = 565.84 ng/mL) and AUC0–t (894.21 h·ng/mL).42 Therefore, it is crucial to find more small-molecule activators that are more potent and efficient and possess superior pharmacokinetics.
Figure 1.

Representative small-molecule activators of ClpP.
Herein, we report our structure-guided discovery of small molecule activators of ClpP. Our efforts have developed CLPP-1071 as an exceptionally potent, orally efficacious ClpP activator, which could prolong the survival of mice in a dose-dependent manner. CLPP-1071 is the most potent and efficacious ClpP activator reported to date (Figure 2).
Figure 2.

Structure-guided design and optimization of ClpP activators.
Results and Discussion
ClpP is composed of two adjacent cylindrical heptamers, with the active sites typically located between two subunits (Figure 3a). By comparing various reported crystal structures of ClpP, it has been observed that the active site can accommodate larger molecules such as ADEP-28 (Figure 3c). Additionally, the indole side chain of the key residue W146 undergoes conformational flipping across different crystal structures, leading to the formation of additional hydrophobic cavities (Figure 3b). These findings indicated that the active site of ClpP exhibits flexibility, allowing for further modifications based on the scaffold structure of ONC201 (Figure 3d,e).
Figure 3.

Cocrystal structures of the activators ONC201 and ADEP-28 with ClpP. (a) Overview of the structure and orientation of activators bound within the hydrophobic pocket between two subunits of ClpP. (b) W146 was observed to undergo conformational flipping in the cocrystal structures of ONC201 (PDB ID:6DL7, in yellow) and ADEP-28 (PDB ID: 6BBA, in cyan). Surface characteristics of the active pocket in crystal structures 6BBA (c) and 6DL7 (d,e), with ONC201 represented as yellow sticks.
The introduction of methyl can significantly enhance the physical and chemical properties of small molecules, such as balancing the lipid–water partition coefficient, which is essential for drug design and development. In addition, the introduction of a methyl group may limit the conformation of the molecule, allowing it to bind more tightly to the protein in a suitable conformation.48−50 We used compound ONC212, a second-generation imipridone with more potent antitumor effects than ONC201, as the starting point for our design efforts. We initiated the synthesis of compounds 2–9 (Table 1) based on the synthetic pathway of ONC212. Methyl groups were initially added at positions 1, 3, 4, and 5 of ONC212 (Figure 4a). Compound 2 and compound 3 each introduced a methyl group onto the methylene group on either side, but this modification resulted in a decrease in enzyme activity (EC50 of 2 = 193.40 ± 18.90 nM and EC50 of 3 = 2659.10 ± 11.70 nM). The binding mode analysis reveals that the methyl group introduced at the 1-position of compound 2 will generate steric clashes with the indole side chain of W146 (Figure 4b). The introduction of a methyl group in 3-position in ONC212 displaces the water molecule that mediates a “water bridge”, thereby disrupting the hydrogen bonding interactions between the ligand and the key residue Q107 (Figure 4c). Introducing one or two methyl groups at the 4-position of the piperidine ring in ONC212 would induce severe steric clashes with W146 and H116 (Figure 4d), leading to a decrease in activity. (EC50 of 4 = 1411.50 ± 20.50 nM, EC50 of 5 = 1648.50 ± 84.50 nM). 6–9 are mixtures because their synthesis involved Dieckmann cyclization reactions, which involved position isomers with similar polarities (Scheme 1).51 Despite trying various separation methods, including normal-phase column chromatography, C18 reversed-phase column chromatography, and recrystallization, we were unable to obtain a single-isomer compound. A similar situation occurred in the subsequent SAR optimization process. We observed a higher proportion of methyl group isomers at 5-position in these mixtures, for example, compound 6a, accounting for approximately 67% of mixture 6. Thus, we initially investigated the SAR in the mixed isomer form. The introduction of an R-configured methyl group at the 5-position increased hydrophobic interactions with the side chain of W146 and increased conformational constraints on the benzyl group (Figure 4e), leading to a significant increase in enzymatic potency compared to ONC212 (EC50 of ONC212 = 131.05 ± 17.75 nM and EC50 of mixture 6 = 21.77 ± 2.18 nM). However, mixture 7 exhibited slightly decreased activity (EC50 of mixture 7 = 138.95 ± 10.55 nM), indicating that the S-configured methyl group at this site positively impacted ClpP activator activity. Introduction of larger alkyl groups (such as ethyl or isopropyl) at this site would lead to further decreases in enzyme activity (EC50 of 8 = 300.60 ± 37.80 nM and EC50 of 9 > 1000 nM). Meanwhile, at a concentration of 20 μM, mixture 6 exhibited a significant increase in activation fold by 15.42-fold (Figure S9). Hence mixture 6 was selected as the lead compound for subsequent optimization.
Table 1. Effects of Different Substitution Sites on ClpP Proteolytic Activitya.

Data are presented as the mean value of triplicate samples ±standard deviation (n = 3).
Figure 4.

Effects of different substitution sites on ClpP proteolytic activity. (a) Overview of methyl substitution sites on ONC212 and the electrostatic surface of its binding site. Methylated modification at 1-position (b), 3-position (c), 4-position (d), 5-position (e), and 2-position (f).
Scheme 1. Chemical Synthesis of 6a, 6b, 21a–42a, 21b–42b, 46a–60a, and 46b–60b.

Reagents and conditions: (a) DBU, TEA, DCM, 40 °C, 24 h, 87% yield; (b) Boc2O, TEA, DCM, r.t., 24 h, 75% yield; (c) t-BuOK, THF, 0 °C, 4 h, 85% yield; (d) 3-cyanobenzaldehyde, NaBH(OAc)3, AcOH, DCM, r.t., 47% yield; (e) 14a + 14b, MeONa, MeOH, reflux, 18 h, 78% yield; (f) TFA, DCM, r.t., 2 h; (g) Cs2CO3, MeCN, reflux, overnight, 65–88% yield; (h) THF, 40 °C, overnight; (i) 16a + 16b, MeONa, MeOH, reflux, 18 h, 50–74% yield.
After confirming the agonistic effect of mixture 6 on ClpP, we conducted structural optimization by introducing different substituents on both sides of the scaffold (Scheme 1). On the one hand, an asymmetric intermediate 12 was obtained by adding commercially available (R)-3-methyl aminobutyrate hydrochloride 10 to methyl acrylate 11 catalyzed by DBU. Compound 13 was then obtained through further reaction of 12 with (Boc)2O in the presence of triethylamine. Subsequently, compound 13 underwent a Dieckmann condensation reaction in the presence of potassium tert-butoxide in an ice bath to form mixture 14 containing 14a and 14b, followed by a continuous nucleophilic substitution reaction with compound 17 to get mixture 18 with 78% yield. In the presence of trifluoroacetic acid, mixture 18 was deprotected to give mixture 19. Subsequently, mixture 19 reacted with substituted benzyl bromo 20 in the presence of cesium carbonate to produce imipridones 6 and 21–42 in 65–88% yield. On the other hand, compound 12 and 3-cyanobenzaldehyde underwent Borch reduction to form compound 15, which then underwent Dieckmann condensation to form mixture 16. Simultaneously, substituted benzylamine 43 and compound 44 synthesized compound 45 in THF, followed by intermolecular cyclization with mixture 16 in the presence of sodium methoxide to produce imipridones 46–60 with 50–74% yield.
After determining the optimal scaffold, we initiated a screening of the left-side groups (Table 2). Mixture 6, with a benzene ring side chain similar to that of ONC212 (1), exhibited a 5-fold increase in enzyme activity (EC50 of 6 = 21.77 ± 2.18 nM) and a comparable cell activity (IC50 of 6 = 59.33 ± 4.38 nM). Subsequent substitution of the benzene ring revealed that meta-position substitution was more crucial than ortho- or para-position substitution, as evidenced by compounds 21–23 (EC50 of 21 > 250 nM, EC50 of 22 = 21.28 ± 2.32 nM, and EC50 of 23 > 250 nM). Additionally, the introduction of other halogens such as chlorine (24, EC50 = 14.94 ± 4.22 nM), bromine (25, 40.33 ± 10.01 nM), and iodine (26, 55.35 ± 20.38 nM) had not shown better activator activity and cell inhibitory activity. Similar to the case for 21–23, the introduction of a cyano group at different positions on the left benzene ring also led to significant differences in activity. Among them, when the cyano group was positioned at the ortho-position (28) and para-position (30), both enzyme activity and cellular activity exceeded 250 nM. However, when the cyano group was located at the meta-position (29), it exhibited good enzyme activity (EC50 = 33.89 ± 2.21 nM) and excellent cellular activity (IC50 = 3.59 ± 0.86 nM). This result reaffirmed the importance of meta-substitution. Introduction of electron-withdrawing trifluoromethyl (27, EC50 = 29.97 ± 0.61 nM) and electron-donating methyl (31, EC50 = 172.65 ± 13.65 nM) or methoxy (32, EC50 = 133.15 ± 5.45 nM) showed no significant difference in activity at this position. Suggesting minimal electronic effect from substituents on compound activity. In addition, larger volumes of 4-tert-butylphenyl (33) or naphthalene ring (34) led to significant decreases in activity; their EC50 were all higher than 250 nM. Introduction of multiple fluorine atoms on the phenyl ring demonstrated that high activity could still be maintained when two fluorine atoms were present (35–40). We synthesized compounds 41 and 42 by introducing both fluorine and cyano groups as double substitutions on the benzene ring; however, their potencies were both higher than 250 nM. Based on the above results, we selected 22 and 29 for further study and named them CLPP-1060 and CLPP-1061, respectively.
Table 2. Investigation of SAR on the R1 Groupa,b.
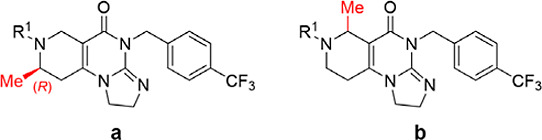

Data are presented as the mean value of triplicate samples ±standard deviation (n = 3).
The ratio of a and b was obtained by 1H Nuclear magnetic resonance (NMR).
After screening, we found that compounds CLPP-1060 and CLPP-1061 exhibited high sensitivity to ClpP protein and HL60 cell. Therefore, we further compared their liver microsomal stability and pharmacokinetic properties (Table 3). As shown in Table 3, CLPP-1060 and CLPP-1061 exhibited similar half-life (T1/2 = 6.24 min vs 6.21 min) and clearance (Clint = 0.556 mL/min/kg vs 0.558 mL/min/kg in human microsomes). On the other hand, pharmacokinetic parameters showed that CLPP-1060 and CLPP-1061 have comparable T1/2 and Tmax, but CLPP-1061 has higher Cmax, AUC0–t, and AUC0–∞ than CLPP-1061 (CLPP-1061: Cmax = 813 ± 145 ng/mL, AUC0–t = 1000 ± 184 h·ng/mL, and AUC0–∞ = 1030 ± 187 h·ng/mL). Nonetheless, these parameters of CLPP-1061 are inferior to those of ONC212 reported in the literature,38 the pharmacokinetic properties of the activators still need to be improved. Therefore, we conducted further structure optimization based on the left-side group of CLPP-1061.
Table 3. Liver Microsomal Stability and Pharmacokinetic Parameters of 22 (CLPP-1060), 29 (CLPP-1061), and ONC212.
| compds | 22 (CLPP-1060) | 29 (CLPP-1061) | testosterone | ONC212b |
|---|---|---|---|---|
| liver microsomal stabilitya | ||||
| T1/2 (min) H/M | 6.24/2.95 | 6.21/2.70 | 8.69/0.723 | 10.24/- |
| Clint (mL/min/kg) H/M | 0.556/1.17 | 0.558/1.28 | 0.399/4.80 | 0.338/- |
| Clapp (mL/min/kg) H/M | 650/4653 | 653/5074 | 467/8634 | 396/- |
| Clh (mL/min/kg) H/M | 20.1/88.3 | 20.1/88.4 | 19.8/54.8 | 19.7/- |
| Eh (%) | 96.9/98.1 | 96.9/98.3 | 95.8/99.4 | 95.0/- |
| PO PK Parameters at50 mg/kgin Ratc,d | ||||
| T1/2 (h) | 1.20 ± 0.24 | 0.92 ± 0.10 | 4.31 | |
| Tmax (h) | 0.58 ± 0.38 | 0.42 ± 0.14 | 0.5 | |
| Cmax (ng/mL) | 374 ± 114 | 813 ± 145 | 1460 | |
| AUC0–t (h·ng/mL) | 460 ± 122 | 1000 ± 184 | 8010 | |
| AUC0–∞ (h·ng/mL) | 1.85 ± 0.26 | 1030 ± 187 | ||
| MRT (h) | 1.85 ± 0.26 | 1.35 ± 0.08 | ||
Testosterone as a reference compound. Abbreviations: Cl: plasma clearance, T1/2 = terminal half-life (H/M = human/mice).
Pharmacokinetic Parameters derived from ref (38).
Data are presented as the mean value of triplicate samples ±standard deviation (n = 3).
CLPP-1060 and CLPP-1061 compounds were administered orally at 50 mg/kg (0.5% CMC-Na) in ICR female mice. Plasma samples were collected at 0.083, 0.25, 0.5, 1, 2, 4, 6, and 24 h from three mice at each time point and analyzed by LC–MS/MS.
After completing the structural optimization on the left-side moieties, we proceeded to optimize the right-side moieties (Table 4). Initially, the introduction of cyclopropyl (46) led to a significant decrease in both enzyme activity and cell viability (EC50 > 250 nM, IC50 > 250 nM). Subsequently, we explored monosubstituted phenyl derivatives (47–52) and found that para-position substitution exhibited superior properties. Importantly, the enzymatic and cellular activities of the para-bromo substituted product 52 (EC50 = 50.61 ± 9.59 nM, IC50 = 11.11 ± 0.36 nM) were approximately 2-fold and 8-fold higher than those of ONC212, respectively. Introducing two substituents onto the phenyl ring (53–57) did not yield further optimization of the potency. Additionally, incorporating fused ring structures such as the 1,3-dioxolane ring (58, EC50 = 222.65 ± 109.65 nM) and naphthalene rings (EC50 of 59 = 178.60 ± 1.00 nM and EC50 of 60 = 77.40 ± 0.91 nM) did not demonstrate improved activity. Finally, compound 52 was identified as the most optimized compound.
Table 4. Investigation of SAR on the R2 Groupa,b.


Data are presented as the mean value of triplicate samples ±standard deviation (n = 3).
The ratio of a and b was obtained by 1H NMR.
Due to 52 having superior enzyme and cell activity, we have isolated the mixture through chiral supercritical fluid chromatography (SFC) separation and have obtained three isomers, as shown in Figure 5a. We identified the chemical structures of individual components through 1D and 2D NMR spectra. The 1D and 2D NMR spectra of 52b′ and 52b″ exhibit high similarity, while the spectrum of 52a shows significant differences. The protons of methyl in 52a are strongly correlated to C-2 and C-3, whereas the protons of methyl in 52b′ and 52b″ show strong correlation to C-5 and C-6 (Figure 5b). Therefore, we determined that the methyl group in 52a was located at the 2-position of the piperidine ring, while the methyl group in both 52b′ and 52b″ was positioned at the 6-position of the piperidine ring. After comparing the NMR spectra and optical, we suggested that 52b′ and 52b″ were a pair of enantiomers, and they were produced by racemization in the final step of the synthesis process, and the possible reaction mechanism was shown in Figure S3. Subsequently, we further tested the activity of the three isomers; the activity data showed that only 52a exhibited comparable enzyme activity and cellular activity to the mixture 52 (EC50 of mixture 52 = 50.61 ± 9.59 nM, IC50 of mixture 52 = 11.11 ± 0.36 nM, EC50 of 52a = 23.49 ± 0.14 nM, and IC50 of 52a = 4.61 ± 0.21 nM). Meanwhile, the EC50 values of the two enantiomers were 252.15 ± 1.25 and 739.25 ± 10.45 nM, and the IC50 values were all greater than 250 nM. Because of the poor activity of 52b′ and 52b″, we did not further explore the relationship between the configuration and activity. Finally, we confirmed that compound 52a was the main bioactive component of mixture 52.
Figure 5.

(a) Isomers of 52 on ClpP activation activity, HL60 cell growth inhibition. (b) HMBC spectrum of compounds 52a (CLPP-1071), 52b′, and 52b″.
Considering that 52a was obtained by SFC separation and the efficiency of this process was not favorable for drug discovery, we developed a regioselective synthetic method. The synthesis route of 52a is shown in Scheme 2. First, we obtained 62 containing two different ester groups by the Michael addition of commercially available 10 and tert-butyl acrylate 61 under the catalysis of DBU with 84% yield. Then nucleophilic substitution of 62 with 3-(bromomethyl)benzonitrile 63 affords compound 64 at reflux with 74% yield. After screening reaction conditions (Table S1), compound 64 underwent a Dieckmann condensation reaction under the action of potassium tert-butoxide at −70 °C to generate 65 with 55% yield. In a low-temperature environment, differences in the ester groups on either side of 64 enable intramolecular Dieckmann condensation to achieve high regioselectivity, resulting in the formation of single-isomer compound 65. Notably, intermediate 65 can be synthesized on a gram scale, allowing for the preparation of large quantities of the final product. Meanwhile, this method can provide a strategy for the synthesis of similar chiral compounds. Finally, 65 was bonded with 66 through continuous nucleophilic substitution to afford compound 52a with yield. Therefore, 52a could be synthesized on a gram scale through four steps, achieving an overall yield of 25%.
Scheme 2. Gram Scale Position-Specific Synthesis of CLPP-1071.

Reagents and conditions: (a) DBU, TEA, DCM, 40 °C, 24 h, 84% yield; (b) DIPEA, MeCN, reflux, overnight, 74% yield; (c) t-BuOK, THF, −70 °C, 4 h, 55% yield; (d) MeONa, MeOH, reflux, overnight, 18 h, 74% yield.
We also applied this chemoselectivity synthesis method to the representative compounds in Table 4, obtained their corresponding single-isomer compounds, and further evaluated their activity and liver microsomal stability (Table S2). From the activity data, these single-isomer compounds showed a consistent trend compared to the mixtures (Figure S8); meanwhile, 52a exhibited a better microsomal stability profile across two species. Therefore, we selected 52a for subsequent mechanistic and pharmacodynamic studies and named it CLPP-1071.
PK Profile of CLPP-1071 and ONC212
Encouraged by the above results, we compared the liver microsomal stability and PK properties of CLPP-1071 and ONC212 (Table 5). First, CLPP-1071 had a slightly longer half-life than ONC212 in human microsome preparations (t1/2 = 13.7 min vs 10.24 min) and was slightly weaker than ONC212 in all clearance parameters. Overall, similar to ONC212, CLPP-1071 still possessed high clearance. We subsequently evaluated the pharmacokinetic properties of CLPP-1071 and ONC212 through oral administration at a dose of 50 mg/kg, consistent with reference doses. Notably, CLPP-1071 exhibited a lower T1/2, Tmax and MRT0–∞ compared to ONC212, but it exhibited better total exposure and higher Cmax, AUC0–t, and AUC0–∞. Taken together, these findings proved that CLPP-1071 is a promising compound for further development.
Table 5. Liver Microsomal Stability and Pharmacokinetic Parameters of CLPP-1071 and ONC212 in Mice.
| compds | CLPP-1071 | ONC212 | testosterone |
|---|---|---|---|
| liver microsomal stabilitya | |||
| T1/2 (min) H/R | 13.7/6.89 | 10.24/7.12 | 8.69/0.723 |
| Clint (mL/min/kg) H/R | 0.253/0.503 | 0.338/0.487 | 0.399/4.80 |
| Clapp (mL/min/kg) H/R | 296/906 | 396/876 | 467/8634 |
| Clh (mL/min/kg) H/R | 19.3/52.0 | 19.7/51.9 | 19.8/54.8 |
| Eh (%) | 93.5/94.3 | 95.0/94.1 | 95.8/99.4 |
| PO PK parameters at50 mg/kgin miceb | |||
| T1/2 (h) | 1.45 ± 0.33 | 2.03 ± 0.30 | |
| Tmax (h) | 0.50 ± 0.43 | 0.58 ± 0.38 | |
| Cmax (ng/mL) | 3423 ± 570 | 2020 ± 344 | |
| AUC0–t (h*ng/mL) | 9010 ± 1322 | 5715 ± 472 | |
| AUC0–∞ (h*ng/mL) | 9839 ± 990 | 6841 ± 57 | |
| MRT (h) | 2.61 ± 0.41 | 3.26 ± 0.00 | |
Testosterone as a reference compound. Abbreviations: Cl: plasma clearance and T1/2 = terminal half-life (H/R = human/rat).
Data are presented as the mean value of triplicate samples ±standard deviation (n = 3). ONC212 and CLPP-1071 compounds were administered orally at 50 mg/kg (0.5% CMC-Na) in ICR female mice. Plasma samples were collected at 0.083, 0.25, 0.5, 1, 2, 4, 6, and 24 h from three mice at each time point and analyzed by LC–MS/MS.
From the binding mode analysis of CLPP-1071, the introduction of an R-configured methyl substitution enhances the hydrophobic interaction between CLPP-1071 and the indole side chain of W146. The introduction of bromine on the phenyl ring increases hydrophobic interactions with the hydrophobic pocket on the right side. In addition, the cyano group can form hydrogen bonding interactions with T135 (Figure 6). Compared to ONC201, CLPP-1071 shows a closer proximity to key residues Y138 and Q107, thereby facilitating stable π stacking and hydrogen bonding interactions with the protein. All of these interactions collectively contribute to enhancing the activity of CLPP-1071.
Figure 6.

Binding mode of CLPP-1071. Comparison of the binding modes of ONC201 (a) and CLPP-1071 (b).
CLPP-1071 Interacts and Activates with Mitochondrial ClpP
At the molecular level, enzyme activity measurements indicate that the compound CLPP-1071 can activate ClpP with an EC50 of 23.49 ± 0.14 nM. Furthermore, the thermal stability of the CLPP-1071 complex was evaluated by using thermal shift assay (TSA), as described previously.52 Briefly, 0.5 mg/mL CLPP-1071 was incubated with 10m SYPRO Orange (Thermo Fisher) for 20 min at room temperature. The reaction was performed in 96-well plates with a final volume of 10 μL. The thermal melting curve was monitored using a LightCycler 480 II Real-Time PCR System (Roche Diagnostics) with a ramp rate of 4.4 °C per minute from 25 to 98 °C to reveal that CLPP-1071, at a concentration of 0.67 mM, exhibits good thermal stability with the protein (ΔTm = 17.7 °C) (Figure 7a,b). Subsequently, biolayer interferometry techniques were used to detect the binding capacity of small molecule compounds to the ClpP protein, demonstrating that CLPP-1071 binds to ClpP in a dose-dependent manner with a Kd of 26.9 nM (Figure 7c). Finally, the cellular thermal shift assay (CETSA) was employed to further confirm the engagement of CLPP-1071 with ClpP in the HL60 and MOLM13 cells. After the cells were treated with 10 μM CLPP-1071 for 6 h, the shift in thermal stability of ClpP was assessed by measuring the amount of soluble target protein remaining at different temperatures using Western blotting. As shown in Figure 7d, CLPP-1071 effectively stabilized ClpP protein at higher temperatures compared to the control group, indicating that compound CLPP-1071 can still bind to ClpP protein within cells, consistent with the results obtained in vitro (Figure 7a–d).
Figure 7.

CLPP-1071 binds to ClpP (n = 3). (a) Effect of CLPP-1071 on peptidase activity. (b) TSA experiments were used to investigate the effect of compounds on the thermal stability of ClpP, in which the concentration of compounds CLPP-1071 was 0.67 mM. (c) Biolayer interferometry techniques detect the binding of ClpP with different concentrations of CLPP-1071. (d) Cellular thermal shift analysis of the thermal stability of ClpP in HL60 and MOLM13 cells treated with CLPP-1071 at 10 μM for 6 h.
CLPP-1071 Induces Mitochondrial Dysfunction
Drawing upon previous studies,9,39 which have shown that abnormal activation of mitochondrial ClpP in both solid tumors and AML can lead to downregulation of oxidative respiratory chain complex proteins, simultaneously, we found that CLPP-1071 exhibited favorable IC50 values in HL60 and MOLM13 cells (Figure S10). Therefore, we selected these two cell lines for further investigation of its effects.
First, we utilized Western blotting analysis to detect the reduction of key proteins belonging to respiratory chain complexes I and II in HL60 and MOLM13 cells following treatment with CLPP-1071. Both SDHB and NDUFA12 proteins decreased at 1 nM CLPP-1071 treatment and even mostly disappeared at 10 nM CLPP-1071. In contrast, 10 nM ONC212 treatment showed little effect on the SDHB and NDUFA12 protein levels, indicating that CLPP-1071 has significantly stronger ClpP activation than ONC212 (Figure 8a,b).
Figure 8.

CLPP-1071 induces mitochondrial dysfunction (n = 3). (a,b) Western blotting analysis of the degradation levels of SDHB and NDUFA12 in HL60 and MOLM13 cells treated with CLPP-1071 for 36 h at the indicated concentrations. (c–f) Flow cytometry was used to analyze the elevation of reactive oxygen species (ROS) in HL60 and MOLM13 cells treated with CLPP-1071 for 38 h, and the statistical analysis revealed the mean fluorescence intensity of ROS in both cell lines. (g,h) Laser confocal microscopy detected the reduction of mitochondrial membrane potential (MMP) in HL60 and MOLM13 treated with CLPP-1071 for 48 h. (i–l) Flow cytometry was used to analyze the reduction of MMP in HL60 and MOLM13 cells treated with CLPP-1071 for 48 h, and the statistical mean fluorescence intensity for both cell lines was calculated. The results are presented as the mean ± standard deviation, *p < 0.05, **p < 0.01, and #p < 0.001 vs vehicle.
Oxidative phosphorylation damage triggers the accumulation of ROS and a concomitant decrease in MMP.53 To elucidate the effects of this damage on mitochondrial ROS production, we employed MitoSOX Red (Invitrogen) as a fluorescent indicator and treated HL60 and MOLM13 with CLPP-1071 for 38 h. Subsequently, cytometry was utilized to acquire and analyze the data. Our findings revealed a pronounced elevation in ROS accumulation in HL60 following CLPP-1071 induction at a concentration of 5 nM, as compared to the DMSO group (Figure 8c,d). Analogously, in MOLM13, CLPP-1071 treatment also resulted in a significant increase in the ROS levels (Figure 8e,f). Notably, the interference of CLPP-1071 with ROS production in both cell types surpassed that of ONC212 (Figure S11), suggesting that CLPP-1071 exhibited a heightened sensitivity toward modulating ROS generation and possessed superior biological activity within these cellular contexts.
Subsequently, to evaluate the impact on MMP, we employed the tetramethyl rhodamine (TMRM) fluorescence probe. Following the treatment of HL60 and MOLM13 with varying concentrations of CLPP-1071 for 48 h, alterations in red fluorescence were visualized by using a laser confocal microscope in the RFP channel (Figure 8g,h). Our observations indicated that CLPP-1071 significantly decreased MMP in cells at a concentration of 10 nM, as compared to the DMSO group. To further quantify these findings, we performed cell flow cytometry (Figure 8i–l), and the results corroborated the trends observed by confocal microscopy. Specifically, CLPP-1071 treatment led to a notable reduction in the MMP in both HL60 and MOLM13. Notably, the extent of MMP reduction induced by CLPP-1071 surpassed that of ONC212 (Figure S11), underscoring the stronger effect of CLPP-1071 on mitochondrial damage.
Collectively, these findings suggest that CLPP-1071 exerts a more potent effect on mitochondrial function, evidenced by its ability to significantly decrease the level of MMP and interfere with ROS production to a greater degree than ONC212. This enhanced mitochondrial damage may play a pivotal role in the antitumor activity of CLPP-1071, further validating its potential as a promising therapeutic agent (Figure 8a–l).
CLPP-1071 Induces Cell Cycle G1-Phase Arrest and Apoptosis
The data from flow cytometry experiments demonstrate that CLPP-1071 treatment led to an increase in the percentage of cells arrested in the G1 phase of the cell cycle in HL60 and MOLM13. The data from Figure 9a,b, demonstrate that CLPP-1071 treatment results in a dose-dependent increase in the percentage of cells arrested in the G1 phase of the cell cycle. In HL60, the percentage of cells arrested in G1 increased from 50% at 10 nM to 60% at 100 nM. Similarly, in MOLM13, the percentage of cells arrested in G1 increased from 65% to 73% with increasing dose (Figure 9c,d). These results suggest that CLPP-1071 causes cell cycle arrest in a dose-dependent manner, which could contribute to its potential antitumor activity. The dose-dependent effect further supports the conclusion that CLPP-1071 causes cell cycle arrest. At the same time, we also evaluated cell apoptosis (Figure 9c,d), demonstrating that the CLPP-1071 treatment results in a dose-dependent increase in apoptosis in HL60 and MOLM13. Notably, CLPP-1071 interfered significantly better with cell cycle G1-Phase arrest and apoptosis than did ONC212 (Figure S12).
Figure 9.

CLPP-1071 induces cell cycle arrest and apoptosis (n = 3). (a,b) Cell cycle analysis of the HL60 and MOLM13 cells treated with CLPP-1071 for 24 h. (c,d) Apoptosis analysis of the HL60 and MOLM13 cells treated with CLPP-1071 for 36 h. The results are presented as the mean ± standard deviation. *p < 0.05, **p < 0.01, and #p < 0.001 vs vehicle.
Additionally, it is worth noting that these results are consistent with the literature on ClpP dysregulation and its effect on cell cycle arrest and apoptosis in pancreatic and colon cancer cells.25,28 The use of cell flow cytometry to study apoptosis and cell cycle progression is a valid method for assessing the biological activity of compounds like CLPP-1071. Therefore, the existing data support the conclusion that CLPP-1071 induces cell cycle arrest in a dose-dependent manner in HL60 and MOLM13 cells. This arrest and apoptosis may contribute to the compound’s potential antitumor activity.
CLPP-1071 Exerts Antitumor Effects In Vivo
Based on a comprehensive evaluation of in vitro activity and pharmacokinetic data, CLPP-1071 was further investigated in the MOLM13 and HL60-disseminated NSG mice model. Female NSG mice were maintained in a pathogen-free animal facility. Tumors were established via tail vein injection of MOLM13 cells (2 × 104 cells/mice) and HL60 (5 × 106 cells/mice). On the first day after inoculation MOLM13, the mice were randomly divided into three groups and received ONC212 (n = 10, 10 mg/kg, p.o, qd), CLPP-1071 (n = 10, 10 mg/kg, p.o, qd), or vehicle (n = 20, 0.5% CMC-Na, p.o, qd) daily for 18 days, CLPP-1071 extended the survival of female NSG mice at 10 mg/kg (Figure 10a). Furthermore, Six days after HL60 vaccination, the mice were randomly divided into five groups and received ONC212 (n = 9, 10 mg/kg, p.o, qd), CLPP-1071 (n = 9, 10 mg/kg, p.o, qd), CLPP-1071 (n = 9, 5 mg/kg, p.o, qd), CLPP-1071 (n = 9, 2.5 mg/kg, p.o, qd), or vehicle (n = 18, 0.5% CMC-Na, p.o, qd) daily for 19 days. CLPP-1071 dose-dependently extended the survival of female NSG mice at 5 and 10 mg/kg (Figure 10b). These results showed that CLPP-1071 exerted antitumor activity in vivo.
Figure 10.

Antitumor activity of CLPP-1071 in MOLM13 (a) and HL60 (b) leukemia models in NSG mice. Each group had nine-ten mice. *p < 0.05, **p < 0.01, and #p < 0.001 vs vehicle.
Conclusions
Through the analysis of cocrystal results of ADEP-28 and ONC201 with ClpP proteins, we noted that there are still some flexible spaces in the binding pocket that can accommodate smaller groups. Hence, methyl groups were introduced at various positions in ONC212, and compound 6 with an R-configuration methyl group at the 5-position of the piperidine ring showed approximately a 5-fold increase in enzymatic activity. Subsequent SAR studies led to the identification of compound 29 (EC50 = 33.89 ± 2.21 nM and IC50 = 3.59 ± 0.86 nM in HL60) as a potent activator of ClpP; however, its pharmacokinetic properties were still inferior to that of ONC212. Further SAR optimization resulted in the discovery of compound 52 derived from CLPP-1071 (EC50 = 23.49 ± 0.14 nM, IC50 = 4.61 ± 0.21 nM in HL60, and Kd = 26.9 nM) with high activity after fractionation by SFC. Pharmacokinetic testing revealed that CLPP-1071 had an AUC0–∞ approximately 1.5 times higher than that of ONC212. To further explore the biological function and potential as a candidate compound for CLPP-1071, optimization of synthesis conditions allowed for gram-scale chiral synthesis. CLPP-1071 achieved protein degradation downstream from ClpP at lower concentrations and induced cell cycle arrest and apoptosis in cancer cells more effectively. In vivo efficacy evaluation on disseminated models including HL60 and MOLM13 showed that administration of CLPP-1071 at a dose of 5 mg/kg resulted in stronger effects compared to 10 mg/kg ONC212 and significantly prolonged survival periods in mice.
Collectively, these data demonstrate that CLPP-1071 represents a highly promising ClpP activator for further advanced preclinical development for the treatment of acute myeloid leukemia and other types of human cancers through ClpP hyperactivation.
Experimental Section
Chemistry
General information: starting materials, reagents, and solvents were purchased from Bide Pharmatech, Adamas-beta, Energy Chemical, and J&K, and were used without further purification. Flash chromatography was performed on Biotage Isolera One with the Sfär Bio C18 column. 1H and 13C NMR spectroscopy was performed on a Bruker Ascend 500 MHz NMR (IS as TMS) and a Bruker Ascend 600 MHz NMR (IS as TMS). Chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Proton coupling patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). High-resolution mass spectra (HRMS) and low-resolution mass spectra were obtained by electrospray ionization (ESI) using a Thermo Exactive Plus and Agilent 6125C MS. HPLC data analysis of compounds was performed on an Agilent 1290 with a quaternary pump and diode-array detector, and the peak purity was verified by UV spectra. All compounds are >95% pure by HPLC analysis. The NMR and MS spectra, HPLC methods, and HPLC traces are contained in Supporting Information.
Methyl (R)-3-((tert-Butoxycarbonyl)(3-methoxy-3-oxopropyl)amino)butanoate (12)
To a mixture of (R)-methyl 3-aminobutanoate hydrochloride (10, 5.0 g, 33.0 mmol) and methyl acrylate (3.2 mL, 36.0 mmol) in DCM (60 mL), were added TEA (4.5 mL, 33.0 mmol) and DBU (0.5 mL, 3.3 mmol). The mixture was stirred for 24 h at 40 °C. TEA (9.0 mL, 66.0 mmol) and Boc2O (9.0 mL, 39.0 mmol) were added, and the mixture was stirred for an additional 24 h at 40 °C. The mixture was cooled to room temperature and concentrated under reduced pressure. t-BuOMe (50 mL) was added, and the mixture was stirred for 30 min. The mixture was filtered and washed with t-BuOMe (2 × 50 mL), and the filtrate was concentrated under reduced pressure. The residue was purified via silica gel chromatography (PE/EA = 4:1) to afford the title compound (12, 7.3 g, 75% yield) as a colorless liquid.
(R)-1-(tert-Butyl) 3-Methyl 2-Methyl-4-oxopiperidine-1,3-dicarboxylate and 1-(tert-Butyl) 3-methyl (R)-6-Methyl-4-oxopiperidine-1,3-dicarboxylate (13, Mixture of Position Isomer)
To a solution of the product from the previous step (10.6 g, 34.9 mmol) in anhydrous THF (50 mL) at 0 °C was added t-BuOK 1.0 M in THF (42 mL, 90.0 mmol) slowly down the sides of the flask submerged in an ice water bath, and the resulting mixture was stirred at 0 °C for 12 h. After the reaction was completed, the mixture was warmed to room temperature and concentrated under reduced pressure. Water (50 mL) was added, and the layers were separated. The aqueous layer was extracted with EA (4 × 50 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified via silica gel chromatography (PE/EA = 4:1) to afford the title compound (13, 9.6 g, 85% yield) as a yellow liquid.
N-(4-(Trifluoromethyl)benzyl)-4,5-dihydro-1H-Imidazole-2-amine Hydroiodide (16)
To a mixture of 2-(methylthio)-4,5-dihydro-1H-imidazole hydroiodide (24.1 g, 100.0 mmol) in anhydrous THF (100 mL) was added (4-(trifluoromethyl) phenyl)methanamine (14.0 g, 110.0 mmol), and the mixture was stirred for 24 h at room temperature. After the reaction was completed, the mixture was concentrated under reduced pressure. t-BuOMe (50 mL) was added, and the mixture was stirred for 5 min. The mixture was filtered and washed with t-BuOMe (2 × 10 mL). The filter cake was dried under high vacuum to yield the desired compound (16, 28.0 g, assumed quantitative yield) as a white powder.
tert-Butyl 6-Methyl-5-oxo-4-(4-(trifluoromethyl)benzyl)-1,2,4,5,8,9-hexahydro-imidazo[1,2-a]pyrido[3,4-e]pyrimidine-7(6H)-carboxylate and tert-Butyl (R)-8-Methyl-5-oxo-4-(4-(trifluoromethyl)benzyl)-1,2,4,5,8,9-hexahydroimidazo[1,2-a] Pyrido[3,4-e]pyrimidine-7(6H)-carboxylate (17, Mixture of Position Isomer)
To a mixture of 1-(tert-butyl) 3-methyl 2-methyl-4-oxopiperidine-1,3-dicarboxylate and 1-(tert-butyl) 3-methyl (R)-6-methyl-4-oxopiperidine-1,3-dicarboxylate (13, mixture of position isomer, 2.7 g, 10.0 mmol) and N-(4-(trifluoromethyl)benzyl)-4,5-dihydro-1H-imidazole-2-amine hydroiodide (16, 4.5 g, 12.0 mmol) in anhydrous MeOH (20.0 mL) were added to MeONa (2.7 g, 50.0 mmol), and the mixture was stirred for overnight at 80 °C. The mixture was cooled to room temperature and concentrated under reduced pressure. Water (50 mL) was added, and the layers were separated. The aqueous layer was extracted with DCM (4 × 50 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified via silica gel chromatography (DCM/MeOH = 95:5) to afford the title compound (17, 3.9 g, 83% yield) as a yellow solid.
6-Methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a] Pyrido[3,4-e]pyrimidin-5(1H)-one and (R)-8-Methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (18, Mixture of Position Isomer)
To a solution of the product from the previous step (17, 10.6 g, 34.9 mmol) in DCM (40 mL) was added TFA (20 mL), and the resulting mixture was stirred at room temperature. After the end of the reaction monitored by TLC, the solvent was removed by vacuum distillation, quenched by adding saturated NaHCO3 aqueous solution, and extracted with DCM (4 × 50 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to afford the title compound (18, 9.6 g, assumed quantitative yield) as a yellow solid.
General Procedure for the Preparation of 20–42
For preparation of 20–42, (R)-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one and (R)-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (18, 0.20 mmol, 1.0 equiv) was dissolved in anhydrous acetonitrile (2.0 mL) and was added alkyl halides (19, 0.24 mmol, 1.2 equiv), followed by the addition of cesium carbonate anhydrous (0.4 mmol, 2.0 equiv). The mixture was stirred overnight at 80 °C and filtered. The filtrate was concentrated and purified using silica gel column chromatography (MeOH in DCM = 0 to 4%) to get the desired compound in 50–70% yield.
(R)-7-Benzyl-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydro Imid-azo[1,2-a]pyrido[3,4-e]pyrimidin −5(1H)-One (6a) and 7-Benzyl-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydro Imidazo [1,2-a]Pyrido[3,4-e] Pyrimidin-5(1H)-one (6b), 6a/6b = 2.5:1
1H NMR (500 MHz, CDCl3): δ 7.63–7.57 (m, 1H), 7.54 (q, J = 8.4 Hz, 6H), 7.51–7.40 (m, 1H), 7.37–7.19 (m, 14H), 5.15–5.03 (m, 3H), 4.38 (t, J = 6.6 Hz, 1H), 3.92–3.82 (m, 7H), 3.81 (s, 1H), 3.78 (d, J = 5.2 Hz, 1H), 3.74 (d, J = 13.4 Hz, 1H), 3.71–3.57 (m, 3H), 3.51 (dd, J = 30.6, 13.2 Hz, 2H), 3.38–3.24 (m, 3H), 3.24–3.16 (m, 1H), 3.13–3.04 (m, 1H), 2.91 (ddd, J = 12.8, 10.4, 5.1 Hz, 1H), 2.73 (tq, J = 10.8, 6.9, 5.3 Hz, 1H), 2.68–2.58 (m, 2H), 2.58–2.35 (m, 2H), 2.14 (ddd, J = 17.0, 11.4, 4.5 Hz, 2H), 1.12 (d, J = 6.5 Hz, 3H); HRMS (ESI) calcd for C25H26F3N4O+ 455.2053 [M + H]+; found, 455.2050.
(R)-7-(2-Fluorobenzyl)-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexa-hydroimidazo [1,2-a]Pyrido[3,4-e]pyrimidin-5(1H)-one (21a) and 7-(2-Fluorobenzyl)-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido [3,4-e]Pyrimidin-5(1H)-one (21b), 21a/21b = 2.9:1
1H NMR (500 MHz, CDCl3): δ 7.67–7.47 (m, 4H), 7.37 (td, J = 7.5, 1.8 Hz, 1H), 7.22 (tdd, J = 7.5, 5.2, 1.8 Hz, 1H), 7.08 (td, J = 7.5, 1.2 Hz, 1H), 7.01 (ddd, J = 9.7, 8.2, 1.2 Hz, 1H), 5.07 (d, J = 2.5 Hz, 2H), 3.95–3.84 (m, 4H), 3.80 (dd, J = 13.3, 1.2 Hz, 1H), 3.62 (d, J = 13.4 Hz, 1H), 3.34 (d, J = 1.9 Hz, 2H), 3.11 (dtd, J = 11.5, 6.5, 4.6 Hz, 1H), 2.63 (ddt, J = 17.4, 5.5, 2.1 Hz, 1H), 2.19–2.10 (m, 1H), 1.25 (s, 1H), 1.15 (d, J = 6.6 Hz, 3H); HRMS (ESI) calcd for C25H25F4N4O+ 473.1959 [M + H]+; found, 473.1964.
(R)-7-(3-Fluorobenzyl)-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexa-hydroimidazo [1,2-a]Pyrido[3,4-e]pyrimidin-5(1H)-one (22a) and 7-(3-Fluorobenzyl)-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido [3,4-e]Pyrimidin-5(1H)-one (22b), 22a/22b = 3.1:1
1H NMR (500 MHz, CDCl3): δ 7.54 (q, J = 8.4, 7.6 Hz, 5H), 7.33–7.19 (m, 2H), 7.12–7.01 (m, 3H), 6.93 (ddd, J = 10.4, 8.1, 2.7 Hz, 1H), 5.09 (dd, J = 11.5, 2.6 Hz, 3H), 3.90 (dq, J = 5.4, 2.7 Hz, 5H), 3.82–3.74 (m, 1H), 3.54 (d, J = 13.5 Hz, 1H), 3.29 (s, 2H), 3.08 (ddd, J = 13.0, 6.5, 4.6 Hz, 1H), 2.65 (ddt, J = 17.3, 5.2, 2.2 Hz, 1H), 2.22–2.08 (m, 2H), 1.23 (d, J = 6.4 Hz, 1H), 1.11 (d, J = 6.5 Hz, 3H); HRMS (ESI) calcd for C25H25F4N4O+ 473.1959 [M + H]+; found, 473.1981.
(R)-7-(4-Fluorobenzyl)-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexa-hydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (23a) and 7-(4-fluorobenzyl)-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido [3,4-e]Pyrimidin-5(1H)-one (23b), 23a/23b = 2.8:1
1H NMR (500 MHz, CDCl3): δ 7.55 (p, J = 8.4 Hz, 6H), 7.28 (td, J = 8.0, 5.4 Hz, 3H), 6.98 (td, J = 8.7, 2.2 Hz, 3H), 5.09 (dd, J = 12.2, 2.7 Hz, 3H), 3.99–3.84 (m, 6H), 3.81–3.69 (m, 1H), 3.58–3.42 (m, 1H), 3.27 (d, J = 2.0 Hz, 2H), 3.14–3.00 (m, 1H), 2.79–2.35 (m, 2H), 2.23–2.06 (m, 2H), 1.29–1.20 (m, 3H), 1.11 (d, J = 6.5 Hz, 3H); HRMS (ESI) calcd for C25H25F4N4O+ 473.1959 [M + H]+; found, 473.1963.
(R)-7-(3-Chlorobenzyl)-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexa-hydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (24a) and 7-(3-Chlorobenzyl)-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido [3,4-e]pyrimidin-5(1H)-one (24b), 24a/24b = 3.3:1
1H NMR (500 MHz, CDCl3): δ 7.65–7.47 (m, 7H), 7.37 (dq, J = 8.0, 2.0 Hz, 1H), 7.27–7.21 (m, 3H), 7.17 (td, J = 7.7, 3.5 Hz, 1H), 5.09 (dd, J = 11.4, 2.4 Hz, 3H), 3.98–3.79 (m, 6H), 3.73 (dd, J = 23.5, 13.6 Hz, 1H), 3.51 (d, J = 13.5 Hz, 1H), 3.29 (s, 2H), 3.13–3.04 (m, 1H), 2.74–2.61 (m, 1H), 2.23–2.11 (m, 1H), 1.23 (d, J = 6.4 Hz, 1H), 1.11 (d, J = 6.5 Hz, 3H); HRMS (ESI) calcd for C25H25ClF3N4O+ 489.1664 [M + H]+; found, 489.1684.
(R)-7-(3-Bromobenzyl)-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexa-hydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (25a) and 7-(3-Bromobenzyl)-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido [3,4-e]Pyrimidin-5(1H)-one (25b), 25a/25b = 1.9:1
1H NMR (500 MHz, CDCl3): δ 7.67–7.48 (m, 6H), 7.37 (dtd, J = 23.5, 8.5, 6.6 Hz, 2H), 6.90–6.73 (m, 3H), 5.09 (dd, J = 11.0, 1.9 Hz, 3H), 3.98–3.84 (m, 6H), 3.83–3.73 (m, 2H), 3.73–3.62 (m, 1H), 3.58 (d, J = 13.5 Hz, 1H), 3.31 (d, J = 1.9 Hz, 2H), 3.10 (dtd, J = 11.5, 6.5, 4.7 Hz, 1H), 2.63 (ddt, J = 17.4, 5.5, 2.1 Hz, 1H), 2.23–2.11 (m, 2H), 1.25 (d, J = 6.4 Hz, 1H), 1.15 (d, J = 6.5 Hz, 3H); HRMS (ESI) calcd for C25H25BrF3N4O+ 533.1158 [M + H]+; found, 533.1157.
(R)-7-(3-Iodobenzyl)-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexa-hydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (26a) and 7-(3-Iodobenzyl)-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido [3,4-e]Pyrimidin-5(1H)-one (26b), 26a/26b = 2.9:1
1H NMR (500 MHz, CDCl3): δ 7.77–7.67 (m, 2H), 7.65–7.59 (m, 2H), 7.59–7.53 (m, 6H), 7.36–7.26 (m, 3H), 7.05 (td, J = 7.6, 3.7 Hz, 2H), 5.11 (dd, J = 11.3, 2.3 Hz, 3H), 3.93 (q, J = 3.3 Hz, 6H), 3.73 (dd, J = 22.5, 13.6 Hz, 2H), 3.51 (d, J = 13.4 Hz, 1H), 3.30 (d, J = 2.0 Hz, 2H), 3.10 (dtd, J = 11.2, 6.7, 3.3 Hz, 1H), 2.70–2.62 (m, 1H), 2.61–2.48 (m, 1H), 2.18 (tdd, J = 17.4, 5.6, 2.8 Hz, 2H), 1.39–1.23 (m, 4H), 1.13 (d, J = 6.6 Hz, 3H); HRMS (ESI) calcd for C25H25F3IN4O+ 581.1020 [M + H]+; found, 581.1044.
(R)-8-Methyl-7-(3-(trifluoromethyl)benzyl)-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8, 9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (27a) and 6-Methyl-7-(3-(trifluoromethyl)benzyl)-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimi-dazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (27b), 27a/27b = 3.1:1
1H NMR (500 MHz, CDCl3): δ 7.82–7.47 (m, 10H), 7.47–7.32 (m, 2H), 5.09 (dd, J = 12.7, 2.6 Hz, 3H), 4.03–3.87 (m, 5H), 3.81 (dd, J = 21.1, 13.7 Hz, 2H), 3.61 (d, J = 13.6 Hz, 1H), 3.29 (d, J = 1.7 Hz, 2H), 3.15–3.03 (m, 1H), 2.73–2.58 (m, 1H), 2.24–2.10 (m, 1H), 1.13 (d, J = 6.6 Hz, 3H); HRMS (ESI) calcd for C26H25F3N5O+ 480.2006 [M + H]+; found, 480.2012.
(R)-2-((8-Methyl-5-oxo-4-(4-(trifluoromethyl)benzyl)-1,2,4,5,8,9-hexahydroim-idazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (28a) and 2-((6-Methyl-5-oxo-4-(4-(trifluoromethyl)benzyl)-1,2,4,5,8,9-hexahydroimidazo[1,2-a] Pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (28b), 28a/28b > 20:1
1H NMR (500 MHz, CDCl3): δ 7.71–7.54 (m, 3H), 7.52 (dd, J = 8.6, 6.0 Hz, 5H), 7.48 (dd, J = 7.8, 1.5 Hz, 1H), 7.34 (td, J = 7.5, 1.5 Hz, 1H), 5.06 (s, 2H), 3.99–3.85 (m, 5H), 3.78 (d, J = 13.7 Hz, 1H), 3.25 (q, J = 2.3 Hz, 3H), 2.75 (ddt, J = 17.5, 5.1, 2.2 Hz, 1H), 2.18 (dt, J = 17.3, 2.7 Hz, 1H), 1.32–1.25 (m, 1H), 1.25–1.16 (m, 4H); HRMS (ESI) calcd for C26H25F6N4O+ 523.1927 [M + H]+; found, 523.1926.
(R)-3-((8-Methyl-5-oxo-4-(4-(trifluoromethyl)benzyl)-1,2,4,5,8,9-hexahydro-imidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (29a) and 3-((6-Methyl-5-oxo-4-(4-(trifluoromethyl)benzyl)-1,2,4,5,8,9-hexahydroimidazo[1,2-a] pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (29b), 29a/29b = 2.2:1
1H NMR (500 MHz, CDCl3): δ 7.66 (q, J = 2.1 Hz, 1H), 7.61–7.47 (m, 8H), 7.40 (td, J = 7.7, 4.4 Hz, 2H), 5.16–4.94 (m, 3H), 4.01–3.85 (m, 6H), 3.78 (dd, J = 19.1, 14.0 Hz, 2H), 3.58 (d, J = 13.8 Hz, 1H), 3.26 (d, J = 1.9 Hz, 2H), 3.09 (dtd, J = 11.4, 6.5, 4.7 Hz, 1H), 2.77–2.60 (m, 1H), 2.28–2.11 (m, 1H), 1.23 (d, J = 6.4 Hz, 1H), 1.13 (d, J = 6.6 Hz, 3H); HRMS (ESI) calcd for C26H25F3N5O+ 480.2006 [M + H]+; found, 480.2009.
(R)-4-((8-Methyl-5-oxo-4-(4-(trifluoromethyl)benzyl)-1,2,4,5,8,9-hexahydro-imidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (30a) and 4-((6-Methyl-5-oxo-4-(4-(trifluoromethyl)benzyl)-1,2,4,5,8,9-hexahydroimidazo[1,2-a] Pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (30b), 30a/30b = 2.7:1
1H NMR (500 MHz, CDCl3): δ 7.68–7.60 (m, 1H), 7.60–7.50 (m, 5H), 7.45 (t, J = 7.9 Hz, 2H), 5.08 (dd, J = 12.5, 3.4 Hz, 2H), 3.91 (dt, J = 3.9, 2.3 Hz, 4H), 3.80 (dd, J = 20.3, 14.3 Hz, 1H), 3.60 (d, J = 14.1 Hz, 1H), 3.26 (d, J = 1.8 Hz, 1H), 3.08 (dtd, J = 11.5, 6.5, 4.8 Hz, 1H), 2.76–2.60 (m, 1H), 2.18 (tdd, J = 17.4, 5.5, 1.7 Hz, 1H), 1.35–1.18 (m, 4H), 1.13 (d, J = 6.6 Hz, 2H); HRMS (ESI) calcd for C26H25F3N5O+ 480.2006 [M + H]+; found, 480.2009.
(R)-8-Methyl-7-(3-methylbenzyl)-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hex-ahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (31a) and 6-Methyl-7-(3-methylbenzyl)-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a] Pyrido[3,4-e]pyrimidin-5(1H)-one (31b), 31a/31b = 17:1
1H NMR (500 MHz, CDCl3): δ 7.59–7.48 (m, 5H), 7.26–7.10 (m, 4H), 7.08–7.03 (m, 1H), 5.15–5.02 (m, 2H), 3.99–3.83 (m, 5H), 3.77 (d, J = 13.1 Hz, 1H), 3.49 (d, J = 13.0 Hz, 1H), 3.30 (q, J = 1.8 Hz, 2H), 3.13–3.04 (m, 1H), 2.64 (ddt, J = 17.4, 5.6, 2.1 Hz, 1H), 2.32 (s, 4H), 2.14 (ddt, J = 17.4, 4.6, 1.6 Hz, 1H), 1.12 (d, J = 6.6 Hz, 3H); HRMS (ESI) calcd for C29H34F3N4O+ 469.2210 [M + H]+; found, 469.2213.
(R)-7-(3-Methoxybenzyl)-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (32a) and 7-(3-Methoxybenzyl)-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydro-imidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (32b), 32a/32b = 2.9:1
1H NMR (500 MHz, CDCl3): δ 7.54 (q, J = 8.3 Hz, 6H), 7.21 (dd, J = 8.6, 7.0 Hz, 1H), 6.94–6.86 (m, 3H), 6.78 (ddd, J = 8.2, 2.6, 1.0 Hz, 2H), 5.09 (dd, J = 10.6, 2.2 Hz, 3H), 3.98–3.89 (m, 3H), 3.89–3.80 (m, 4H), 3.80–3.66 (m, 6H), 3.52 (d, J = 13.2 Hz, 1H), 3.31 (dt, J = 4.3, 1.9 Hz, 2H), 3.14–3.04 (m, 1H), 2.64 (ddt, J = 17.4, 5.6, 2.1 Hz, 1H), 2.20–2.09 (m, 2H), 1.23 (d, J = 6.5 Hz, 1H), 1.10 (d, J = 6.6 Hz, 3H); HRMS (ESI) calcd for C26H28F3N4O2+ 485.2159 [M + H]+; found, 485.2161.
(R)-7-(4-(tert-Butyl)benzyl)-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (33a) and 7-(4-(tert-Butyl)benzyl)-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo [1,2-a]Pyrido[3,4-e]pyrimidin-5(1H)-one (33b), 33a/33b = 2.4:1
1H NMR (500 MHz, CDCl3): δ 7.63–7.47 (m, 6H), 7.32 (dd, J = 8.3, 1.9 Hz, 3H), 7.27–7.21 (m, 6H), 5.09 (dd, J = 11.4, 2.9 Hz, 3H), 3.94–3.81 (m, 6H), 3.74 (dd, J = 29.4, 13.2 Hz, 2H), 3.52 (d, J = 13.1 Hz, 1H), 3.36–3.25 (m, 2H), 3.11 (dq, J = 10.7, 6.3 Hz, 1H), 2.65 (dd, J = 16.9, 5.1 Hz, 1H), 2.20–2.10 (m, 1H), 1.31 (d, J = 2.8 Hz, 14H), 1.29–1.21 (m, 4H), 1.12 (d, J = 6.5 Hz, 3H); HRMS (ESI) calcd for C29H34F3N4O+ 511.2679 [M + H]+; found, 511.2679.
(R)-6-Methyl-7-(naphthalen-1-ylmethyl)-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (34a) and (R)-8-Methyl-7-(naphthalen-1-ylmethyl)-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydro-Imidazo [1,2-a]Pyrido[3,4-e]pyrimidin-5(1H)-one (34b), 34a/34b = 3.3:1
1H NMR (500 MHz, CDCl3): δ 8.34–8.18 (m, 1H), 7.84 (dt, J = 7.2, 2.6 Hz, 1H), 7.77 (dd, J = 7.7, 1.5 Hz, 2H), 7.66–7.35 (m, 12H), 5.18–4.98 (m, 3H), 4.26 (d, J = 13.0 Hz, 1H), 3.98–3.79 (m, 7H), 3.44 (dt, J = 15.5, 1.6 Hz, 1H), 3.31 (dt, J = 15.7, 2.1 Hz, 1H), 3.16 (td, J = 6.0, 3.8 Hz, 1H), 2.68–2.58 (m, 1H), 2.11 (ddd, J = 17.3, 3.6, 1.8 Hz, 2H), 1.33 (d, J = 6.4 Hz, 1H), 1.18 (d, J = 6.6 Hz, 3H); HRMS (ESI) calcd for C29H28F3N4O+ 505.2210 [M + H]+; found, 505.2212.
(R)-7-(2,3-Difluorobenzyl)-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (35a) and 7-(2,3-Difluorobenzyl)-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydro-imidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (35b), 35a/35b = 12.5:1
1H NMR (500 MHz, CDCl3): δ 7.62–7.48 (m, 5H), 7.14 (ddd, J = 7.6, 4.7, 1.7 Hz, 1H), 7.09–7.02 (m, 1H), 7.02–6.97 (m, 1H), 5.08 (d, J = 2.1 Hz, 2H), 3.96–3.84 (m, 4H), 3.81 (dd, J = 13.6, 1.5 Hz, 1H), 3.68–3.61 (m, 1H), 3.33 (d, J = 2.1 Hz, 2H), 3.16–3.07 (m, 1H), 2.69–2.60 (m, 1H), 2.16 (dt, J = 17.5, 3.1 Hz, 1H), 1.16 (d, J = 6.5 Hz, 3H); HRMS (ESI) calcd for C25H24F5N4O+ 491.1865 [M + H]+; found, 491.1866.
(R)-7-(3,4-Difluorobenzyl)-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (36a) and 7-(3,4-Difluorobenzyl)-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydro-imidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (36b), 36a/36b = 2:1
1H NMR (500 MHz, CDCl3): δ 7.61–7.47 (m, 6H), 7.22–7.11 (m, 2H), 7.09–6.97 (m, 3H), 5.09 (dd, J = 11.3, 1.9 Hz, 3H), 3.96–3.85 (m, 6H), 3.85–3.60 (m, 3H), 3.33 (d, J = 1.9 Hz, 2H), 3.12 (dt, J = 6.7, 5.0 Hz, 1H), 2.64 (ddt, J = 17.5, 5.2, 2.1 Hz, 1H), 2.19–2.11 (m, 1H), 1.26 (d, J = 6.5 Hz, 1H), 1.16 (d, J = 6.6 Hz, 3H); HRMS (ESI) calcd for C25H24F5N4O+ 491.1865 [M + H]+; found, 491.1869.
(R)-7-(2,5-Difluorobenzyl)-6-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (37a) and 7-(2,5-Difluorobenzyl)-8-methyl-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydro-imidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (37b), 37a/37b = 8.1:1
1H NMR (500 MHz, CDCl3): δ 7.55 (q, J = 8.3 Hz, 5H), 6.87 (qd, J = 6.6, 3.4 Hz, 2H), 6.68 (tt, J = 8.9, 2.4 Hz, 1H), 5.09 (d, J = 2.1 Hz, 2H), 3.92 (q, J = 3.3 Hz, 4H), 3.75 (d, J = 13.9 Hz, 1H), 3.54 (d, J = 13.9 Hz, 1H), 3.29 (d, J = 1.9 Hz, 2H), 3.14–3.05 (m, 1H), 2.66 (ddt, J = 17.3, 5.1, 2.1 Hz, 1H), 2.16 (ddt, J = 17.3, 3.5, 1.6 Hz, 1H), 1.11 (d, J = 6.6 Hz, 3H); HRMS (ESI) calcd for C25H24F5N4O+ 491.1865 [M + H]+; found, 491.1868.
(R)-8-Methyl-7-(2,4,6-trifluorobenzyl)-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (38a) and 6-Methyl-7-(2,4,6-trifluorobenzyl)-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo [1,2-a]Pyrido[3,4-e]pyrimidin-5(1H)-one (38b), 38a/38b = 3.2:1
1H NMR (500 MHz, CDCl3): δ 7.54 (q, J = 8.3 Hz, 6H), 7.02–6.91 (m, 3H), 5.09 (dd, J = 10.3, 2.8 Hz, 3H), 3.91 (q, J = 2.9 Hz, 6H), 3.77–3.63 (m, 2H), 3.48 (d, J = 14.0 Hz, 1H), 3.25 (d, J = 2.3 Hz, 2H), 3.08 (dt, J = 11.4, 5.7 Hz, 1H), 2.72–2.61 (m, 2H), 2.18 (td, J = 19.2, 17.3, 4.4 Hz, 2H), 1.11 (d, J = 6.6 Hz, 3H); HRMS (ESI) calcd for C25H23F6N4O+ 509.1771 [M + H]+; found, 509.1775.
(R)-8-Methyl-7-(3,4,5-trifluorobenzyl)-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (39a) and 6-Methyl-7-(3,4,5-trifluorobenzyl)-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydroimidazo [1,2-a]Pyrido[3,4-e]pyrimidin-5(1H)-one (39b), 39a/39b = 12.3:1
1H NMR (500 MHz, CDCl3): δ 7.66–7.53 (m, 1H), 7.53 (s, 3H), 6.71–6.59 (m, 2H), 5.08 (s, 2H), 3.97–3.87 (m, 3H), 3.87–3.81 (m, 1H), 3.80 (d, J = 12.6 Hz, 1H), 3.57 (d, J = 12.7 Hz, 1H), 3.34 (s, 2H), 3.12 (q, J = 5.5 Hz, 1H), 2.62 (ddt, J = 17.6, 5.3, 2.2 Hz, 1H), 2.15 (dt, J = 17.4, 3.1 Hz, 1H), 1.18 (d, J = 6.5 Hz, 3H); HRMS (ESI) calcd for C25H23F6N4O+ 509.1771 [M + H]+; found, 509.1776.
(R)-8-Methyl-7-((perfluorophenyl)methyl)-4-(4-(trifluoromethyl)benzyl)-2,4,6,7, 8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (40a) and 6-Methyl-7-((perfluorophenyl)methyl)-4-(4-(trifluoromethyl)benzyl)-2,4,6,7,8,9-hexahydro- imidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one (40b), 40a/40b = 1.8:1
1H NMR (500 MHz, CDCl3): δ 7.70–7.41 (m, 8H), 5.10 (d, J = 11.5 Hz, 3H), 4.01–3.80 (m, 9H), 3.68 (dd, J = 29.7, 13.0 Hz, 2H), 3.33 (s, 2H), 3.21–2.99 (m, 2H), 2.81–2.48 (m, 3H), 2.19 (ddd, J = 27.9, 17.6, 5.2 Hz, 2H), 1.19 (d, J = 6.5 Hz, 3H); HRMS (ESI) calcd for C25H21F8N4O+ 545.1582 [M + H]+; found, 545.1587.
(R)-2-Fluoro-4-((8-methyl-5-oxo-4-(4-(trifluoromethyl)benzyl)-1,2,4,5,8,9-hexa-hydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (41a) and 2-Fluoro-4-((6-methyl-5-oxo-4-(4-(trifluoromethyl)benzyl)-1,2,4,5,8,9-hexahydro-imidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (41b), 41a/41b = 8.3:1
1H NMR (500 MHz, CDCl3): δ 7.65–7.51 (m, 6H), 7.26–7.17 (m, 3H), 5.10 (s, 2H), 3.94 (q, J = 3.0 Hz, 5H), 3.82 (d, J = 14.5 Hz, 1H), 3.62 (d, J = 14.5 Hz, 1H), 3.28 (d, J = 1.9 Hz, 2H), 3.15–3.05 (m, 1H), 2.67 (dd, J = 17.3, 5.1 Hz, 1H), 2.18 (dd, J = 17.2, 4.6 Hz, 1H), 1.13 (d, J = 6.6 Hz, 3H); HRMS (ESI) calcd for C26H24F4N5O+ 498.1911 [M + H]+; found, 498.1917.
(R)-4-Fluoro-2-((8-methyl-5-oxo-4-(4-(trifluoromethyl)benzyl)-1,2,4,5,8,9-hexa-hydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (42a) and 4-fluoro-2-((6-methyl-5-oxo-4-(4-(trifluoromethyl)benzyl)-1,2,4,5,8,9-hexahydro-imidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (42b), 42a/42b = 2.4:1
1H NMR (500 MHz, CDCl3): δ 7.62 (dd, J = 8.6, 5.3 Hz, 2H), 7.58–7.48 (m, 6H), 7.39–7.28 (m, 1H), 7.25–7.14 (m, 1H), 7.04 (tt, J = 8.2, 2.9 Hz, 2H), 5.29 (s, 1H), 5.08 (d, J = 15.8 Hz, 3H), 3.92 (dt, J = 4.9, 3.0 Hz, 5H), 3.89–3.75 (m, 3H), 3.27 (d, J = 1.9 Hz, 2H), 3.24–3.18 (m, 1H), 2.85–2.54 (m, 2H), 2.19 (ddd, J = 17.3, 7.2, 4.2 Hz, 2H), 1.27 (d, J = 6.4 Hz, 1H), 1.17 (d, J = 6.6 Hz, 3H); HRMS (ESI) calcd for C26H24F4N5O+ 498.1911 [M + H]+; found, 498.1913.
General Procedure for the Preparation of 46–60
2-(Methylthio)-4,5-dihydro-1H-imidazole hydroiodide (15, 2.0 mmol, 1.0 equiv) was dissolved in anhydrous THF (5.0 mL), and alkyl amine was added (43, 2.4 mmol, 1.2 equiv). The mixture was kept stirring for 24 h at 40 °C. After the reaction was completed, the mixture was concentrated under reduced pressure. t-BuOMe (20 mL) was added, and the mixture was stirred for 5 min. The mixture was filtered and washed with t-BuOMe (2 × 10 mL). The filter cake was dried under a high vacuum to yield the desired compound (44) as a white powder. For preparation of 46–57, the compounds (44, 0.2 mmol, 1.0 equiv) were dissolved in anhydrous MeOH (2.0 mL) and methyl 1-(3-cyanobenzyl)-2-methyl-4-oxopiperidine-3-carboxylate and methyl (6R)-1-(3-cyanobenzyl)-6-methyl-4-oxopiperidine-3-carboxylate (45, 0.24 mmol, 1.2 equiv) were added, followed by the addition of MeONa (1.0 mmol, 5.0 equiv). The mixture was cooled to room temperature (rt) and concentrated under reduced pressure. Water (20 mL) was added, and the layers were separated. The aqueous layer was extracted with DCM (4 × 20 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified via silica gel chromatography (MeOH in DCM = 0 to 4%) to get the desired compound in 65–88% yield.
(R)-3-((4-(Cyclopropylmethyl)-8-methyl-5-oxo-1,2,4,5,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (46a)
1H NMR (600 MHz, CDCl3): δ 7.69 (s, 1H), 7.60–7.51 (m, 2H), 7.42 (t, J = 7.7 Hz, 1H), 3.96–3.91 (m, 4H), 3.82 (d, J = 13.8 Hz, 1H), 3.76 (d, J = 7.1 Hz, 2H), 3.61 (d, J = 13.8 Hz, 1H), 3.29 (s, 2H), 3.12 (dtd, J = 11.5, 6.5, 4.7 Hz, 1H), 2.68 (dd, J = 17.3, 5.5 Hz, 1H), 2.19 (dd, J = 17.3, 4.2 Hz, 1H), 0.50–0.33 (m, 3H); 13C NMR (151 MHz, CDCl3): δ 161.58, 153.43, 144.38, 140.66, 132.98, 132.09, 130.89, 129.18, 118.95, 112.55, 100.98, 56.95, 50.82, 50.48, 46.73, 46.62, 44.99, 32.95, 14.28, 9.35, 3.84, 3.81; HRMS (ESI) calcd for C25H25FN5O+ 376.2132 [M + H]+; found, 376.2148; mp 56–58 °C; HPLC retention time = 4.4 min, purity = 98.9%, (λ = 254 nm).
(R)-3-((8-Methyl-4-(2-methylbenzyl)-5-oxo-1,2,4,5,8,9-hexahydroimidazo[1,2-a] pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (47a)
1H NMR (600 MHz, CDCl3): δ 7.69 (d, J = 1.9 Hz, 1H), 7.56 (dd, J = 11.8, 7.7 Hz, 2H), 7.42 (t, J = 7.7 Hz, 1H), 7.15–7.05 (m, 4H), 5.06 (s, 2H), 4.04–3.87 (m, 4H), 3.83 (d, J = 13.8 Hz, 1H), 3.61 (d, J = 13.8 Hz, 1H), 3.30 (d, J = 1.8 Hz, 2H), 3.13 (qd, J = 6.8, 3.2 Hz, 1H), 2.72 (dd, J = 17.4, 5.5 Hz, 1H), 2.42 (s, 3H), 2.23 (dd, J = 17.4, 4.3 Hz, 1H), 1.18 (d, J = 6.5 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 161.41, 153.15, 144.59, 140.62, 137.08, 133.93, 132.99, 132.09, 130.90, 129.19, 128.96, 128.86, 118.94, 112.54, 100.78, 56.94, 50.85, 50.58, 46.81, 45.12, 44.93, 32.90, 21.17, 14.33; HRMS (ESI) calcd for C26H24FN5O+ 426.2288 [M + H]+; found, 426.2290; mp 57–60 °C; HPLC retention time = 6.0 min, purity = 99.8%, (λ = 254 nm).
(R)-3-((8-Methyl-4-(4-methylbenzyl)-5-oxo-1,2,4,5,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (48a)
1H NMR (600 MHz, CDCl3): δ 7.67 (d, J = 1.7 Hz, 1H), 7.56 (t, J = 7.5 Hz, 2H), 7.42 (d, J = 7.7 Hz, 1H), 7.39 (d, J = 7.9 Hz, 2H), 7.11 (d, J = 7.8 Hz, 2H), 5.02 (s, 2H), 4.06–3.84 (m, 4H), 3.81 (d, J = 13.8 Hz, 1H), 3.60 (d, J = 13.7 Hz, 1H), 3.28 (s, 2H), 3.10 (dtd, J = 11.5, 6.5, 4.7 Hz, 1H), 2.66 (ddt, J = 17.4, 5.0, 2.1 Hz, 1H), 2.32 (s, 3H), 2.16 (dd, J = 17.3, 4.2 Hz, 1H), 1.14 (d, J = 6.6 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 161.41, 153.15, 144.59, 140.62, 137.08, 133.93, 132.99, 132.09, 130.90, 129.19, 128.96, 128.86, 118.94, 112.54, 100.78, 56.94, 50.85, 50.58, 46.81, 45.12, 44.93, 32.90, 21.17, 14.33; HRMS (ESI) calcd for C25H25FN5O+ 426.2288 [M + H]+; found, 426.2305; mp 64–66 °C; HPLC retention time = 6.7 min, purity = 97.2%, (λ = 254 nm).
(R)-3-((4-(4-Fluorobenzyl)-8-methyl-5-oxo-1,2,4,5,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (49a)
1H NMR (600 MHz, CDCl3): δ 7.67 (t, J = 1.7 Hz, 1H), 7.58–7.53 (m, 2H), 7.51–7.45 (m, 2H), 7.41 (t, J = 7.7 Hz, 1H), 7.00–6.93 (m, 2H), 5.00 (s, 2H), 4.00–3.86 (m, 4H), 3.81 (d, J = 13.8 Hz, 1H), 3.59 (d, J = 13.8 Hz, 1H), 3.27 (s, 2H), 3.10 (dtd, J = 11.3, 6.5, 4.8 Hz, 1H), 2.66 (dd, J = 17.3, 5.4 Hz, 1H), 2.17 (dd, J = 17.4, 4.4 Hz, 1H), 1.14 (d, J = 6.6 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 162.19 (d, J = 245.4 Hz), 161.35, 153.06, 144.84, 140.58, 132.99, 132.71 (d, J = 3.3 Hz), 132.08, 130.91, 130.74 (d, J = 8.1 Hz), 129.20, 118.93, 115.05 (d, J = 21.2 Hz) 112.54, 100.72, 56.95, 50.85, 50.57, 46.85, 44.89, 44.68, 32.97, 14.32; 19F NMR (565 MHz, CDCl3): δ −115.09; HRMS (ESI) calcd for C25H24FN5O 430.2038 [M + H]+; found, 430.2037; mp 60–62 °C; HPLC retention time = 4.6 min, purity = 100.0%, (λ = 254 nm).
(R)-3-((7-(2-Bromobenzyl)-8-methyl-5-oxo-1,2,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-4(5H)-yl)methyl)benzonitrile (50a)
1H NMR (600 MHz, CDCl3): δ 7.70 (s, 1H), 7.59–7.53 (m, 3H), 7.42 (t, J = 7.7 Hz, 1H), 7.23 (t, J = 7.6 Hz, 1H), 7.09 (t, J = 7.7 Hz, 1H), 7.02 (d, J = 7.8 Hz, 1H), 5.21–5.07 (m, 2H), 4.03–3.88 (m, 4H), 3.85 (d, J = 13.8 Hz, 1H), 3.61 (d, J = 13.8 Hz, 1H), 3.30 (s, 2H), 3.14 (dtd, J = 11.4, 6.5, 4.8 Hz, 1H), 2.78–2.68 (m, 1H), 2.25 (dd, J = 17.4, 4.6 Hz, 1H), 1.19 (d, J = 6.6 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 161.22, 153.08, 145.18, 140.51, 135.08, 133.02, 132.81, 132.14, 130.94, 129.21, 128.37, 127.40, 126.31, 122.73, 118.93, 112.59, 100.70, 57.02, 50.86, 50.65, 47.01, 46.05, 45.00, 33.20, 14.34; HRMS (ESI) calcd for C25H25BrN5O+ 490.1237 [M + H]+; found, 490.1240 and 492.1259; mp 73–77 °C; HPLC retention time = 4.9 min, purity = 99.1%, (λ = 254 nm).
(R)-3-((4-(3-Bromobenzyl)-8-methyl-5-oxo-1,2,4,5,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (51a)
1H NMR (600 MHz, CDCl3): δ 7.68 (s, 1H), 7.61 (d, J = 1.9 Hz, 1H), 7.56 (t, J = 7.4 Hz, 2H), 7.46–7.33 (m, 3H), 7.17 (t, J = 7.8 Hz, 1H), 5.10–4.93 (m, 2H), 3.94 (q, J = 3.6 Hz, 4H), 3.82 (d, J = 13.8 Hz, 1H), 3.61 (d, J = 13.8 Hz, 1H), 3.28 (s, 2H), 3.11 (dtd, J = 11.5, 6.5, 4.7 Hz, 1H), 2.71–2.64 (m, 1H), 2.19 (dd, J = 17.4, 4.3 Hz, 1H), 1.16 (d, J = 6.5 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 161.26, 153.02, 144.96, 140.56, 139.09, 132.98, 132.09, 131.43, 130.93, 130.58, 129.85, 129.21, 127.42, 122.36, 118.92, 112.57, 100.69, 56.93, 50.80, 50.58, 46.90, 44.91, 44.85, 33.00, 14.34; HRMS (ESI) calcd for C25H25BrN5O+ 490.1237 [M + H]+; found, 490.1240 and 492.1216; mp 74–77 °C; HPLC retention time = 6.1 min, purity = 99.1%, (λ = 254 nm).
(R)-3-((4-(4-Chloro-3-fluorobenzyl)-8-methyl-5-oxo-1,2,4,5,8,9-hexahydro-imidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (53a)
1H NMR (600 MHz, CDCl3): δ 7.67 (s, 1H), 7.59–7.52 (m, 2H), 7.42 (t, J = 7.7 Hz, 1H), 7.29 (d, J = 8.8 Hz, 2H), 7.20 (d, J = 8.2 Hz, 1H), 5.03–4.96 (m, 2H), 3.93 (dq, J = 5.2, 2.6, 1.9 Hz, 4H), 3.82 (d, J = 13.8 Hz, 1H), 3.60 (d, J = 13.8 Hz, 1H), 3.27 (s,2H), 3.11 (dtd, J = 11.5, 6.5, 4.7 Hz, 1H), 2.68 (dd, J = 17.4, 5.4 Hz, 1H), 2.19 (dd, J = 17.4, 4.1 Hz, 1H), 1.15 (d, J = 6.6 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 161.20, 157.84 (d, J = 248.7 Hz), 152.94, 145.12, 140.54, 137.70 (d, J = 6.5 Hz), 132.97, 132.07, 130.93, 130.31, 129.21, 125.28 (d, J = 3.6 Hz), 119.89 (d, J = 17.6 Hz), 118.92, 116.94 (d, J = 21.6 Hz), 112.56, 100.66, 56.95, 50.81, 50.56, 46.92, 44.85, 44.50, 33.03, 14.30; 19F NMR (565 MHz, CDCl3): δ −115.54; HRMS (ESI) calcd for C25H24ClFN5O+ 464.1648 [M + H]+; found, 464.1671; mp 59–61 °C; HPLC retention time = 7.2 min, purity = 97.7%, (λ = 254 nm).
(R)-3-((4-(4-Bromo-3-fluorobenzyl)-8-methyl-5-oxo-1,2,4,5,8,9-hexahydro-imidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (54a)
1H NMR (600 MHz, CDCl3): δ 7.68 (s, 1H), 7.57–7.54 (m, 2H), 7.46 (dd, J = 8.2, 7.1 Hz, 1H), 7.42 (t, J = 7.7 Hz, 1H), 7.28–7.26 (m, 1H), 7.15 (dd, J = 8.2, 2.0 Hz, 1H), 5.03–4.96 (m, 2H), 4.00–3.87 (m, 4H), 3.82 (d, J = 13.8 Hz, 1H), 3.60 (d, J = 13.8 Hz, 1H), 3.27 (s, 2H), 3.11 (h, J = 6.5 Hz, 1H), 2.68 (ddt, J = 17.4, 5.5, 2.1 Hz, 1H), 2.19 (dd, J = 17.5, 4.5 Hz, 1H), 1.15 (d, J = 6.6 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 161.19, 158.84 (d, J = 247.1 Hz), 152.94, 145.10, 140.53, 138.55 (d, J = 6.6 Hz), 133.21, 132.97, 132.08, 130.94, 129.21, 125.73 (d, J = 3.5 Hz), 118.92, 116.84 (d, J = 22.6 Hz), 112.57, 107.87 (d, J = 21.0 Hz), 100.68, 56.95, 50.80, 50.56, 46.92, 44.85, 44.52, 33.03, 14.29; 19F NMR (565 MHz, CDCl3): δ −107.45; HRMS (ESI) calcd for C25H24BrFN5O+ 508.1143 [M + H]+; found, 508.1165; mp 61–65 °C; HPLC retention time = 5.5 min, purity = 98.0%, (λ = 254 nm).
(R)-3-((4-(4-Fluoro-3-(trifluoromethyl)benzyl)-8-methyl-5-oxo-1,2,4,5,8,9-hexa hydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (55a)
1H NMR (600 MHz, CDCl3): δ 7.78 (dd, J = 6.9, 2.2 Hz, 1H), 7.71–7.66 (m, 2H), 7.59–7.54 (m, 2H), 7.42 (t, J = 7.7 Hz, 1H), 7.11 (dd, J = 10.2, 8.6 Hz, 1H), 5.08–4.99 (m, 2H), 3.99–3.90 (m, 4H), 3.82 (d, J = 13.8 Hz, 1H), 3.61 (d, J = 13.8 Hz, 1H), 3.27 (s, 2H), 3.11 (h, J = 6.5 Hz, 1H), 2.71–2.64 (m, 1H), 2.19 (dd, J = 17.5, 4.2 Hz, 1H), 1.15 (d, J = 6.6 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 161.21, 159.12 (d, J = 256.4 Hz), 152.94, 145.15, 140.52, 134.75 (d, J = 8.5 Hz), 133.15 (d, J = 3.8 Hz), 132.97, 132.07, 130.93, 129.21, 127.89 (q, J = 4.7 Hz), 122.56 (q, J = 272.4 Hz), 118.92, 118.02 (qd, J = 33.0, 12.6 Hz), 116.63 (d, J = 20.6 Hz), 112.56, 100.68, 56.92, 50.78, 50.53, 46.91, 44.83, 44.34, 33.03, 14.28. 19F NMR (565 MHz, CDCl3): δ −61.25, −116.44; HRMS (ESI) calcd for C26H24F4N5O+ 498.1911 [M + H]+; found, 498.1904; mp 60–65 °C; HPLC retention time = 6.2 min, purity = 96.7%, (λ = 254 nm).
(R)-3-((4-(2,4-Difluorobenzyl)-8-methyl-5-oxo-1,2,4,5,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (56a)
1H NMR (600 MHz, CDCl3): δ 7.68 (s, 1H), 7.56 (t, J = 6.4 Hz, 2H), 7.42 (t, J = 7.8 Hz, 1H), 7.33–7.24 (m, 1H), 6.79 (q, J = 8.7, 8.1 Hz, 2H), 5.09 (s, 2H), 4.00–3.91 (m, 4H), 3.83 (d, J = 13.7 Hz, 1H), 3.60 (d, J = 13.7 Hz, 1H), 3.28 (s, 2H), 3.12 (h, J = 5.9 Hz, 1H), 2.70 (dd, J = 17.4, 5.5 Hz, 1H), 2.21 (dd, J = 17.3, 4.5 Hz, 1H), 1.17 (d, J = 6.5 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 162.22 (dd, J = 227.0, 12.0 Hz), 161.26, 160.58 (dd, J = 229.3, 11.7 Hz), 153.01, 145.10 (d, J = 4.7 Hz), 140.53, 132.99, 132.11, 130.93, 129.93 (dd, J = 9.9, 5.5 Hz), 129.21, 119.67 (dd, J = 14.7, 3.8 Hz), 118.93, 112.58 (d, J = 3.3 Hz), 111.07 (dd, J = 21.2, 3.8 Hz), 103.75 (t, J = 25.3 Hz), 100.68 (d, J = 3.5 Hz), 57.00, 50.87, 50.63, 46.95, 44.92, 38.90, 33.11, 14.31; 19F NMR (565 MHz, CDCl3): δ −111.87, −113.04; HRMS (ESI) calcd for C25H24F2N5O+ 446.1742 [M + H]+; found, 446.1744; mp 69–74 °C; HPLC retention time = 4.6 min, purity = 100.0%, (λ = 254 nm).
(R)-3-((4-(4-Bromo-2-methylbenzyl)-8-methyl-5-oxo-1,2,4,5,8,9-hexahydro-imidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (57a)
1H NMR (600 MHz, CDCl3): δ 7.68 (d, J = 1.7 Hz, 1H), 7.56 (t, J = 7.9 Hz, 2H), 7.42 (t, J = 7.7 Hz, 1H), 7.23 (dd, J = 8.2, 2.1 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 5.02–4.94, (m, 2H), 4.04–3.87 (m, 4H), 3.83 (d, J = 13.8 Hz, 1H), 3.61 (d, J = 13.8 Hz, 1H), 3.28 (s, 2H), 3.13 (h, J = 6.5 Hz, 1H), 2.72 (dd, J = 17.3, 5.5 Hz, 1H), 2.40 (s, 3H), 2.23 (dd, J = 17.3, 4.1 Hz, 1H), 1.17 (d, J = 6.5 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 161.34, 153.21, 145.08, 140.52, 138.04, 133.56, 133.01, 132.93, 132.12, 130.93, 129.21, 128.92, 127.50, 120.60, 118.93, 112.56, 100.74, 56.99, 50.84, 50.55, 46.99, 44.97, 42.75, 33.12, 19.11, 14.33; HRMS (ESI) calcd for C26H27BrN5O+ 503.1393 [M + H]+; found, 504.1394; mp 68–72 °C; HPLC retention time = 11.0 min, purity = 96.8%, (λ = 254 nm).
(R)-3-((4-(Benzo[d][1,3]dioxol-5-ylmethyl)-8-methyl-5-oxo-1,2,4,5,8,9-hexahydro- imidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (58a)
1H NMR (600 MHz, CDCl3): δ 7.67 (d, J = 1.8 Hz, 1H), 7.58–7.53 (m, 2H), 7.42 (t, J = 7.7 Hz, 1H), 7.04 (d, J = 1.7 Hz, 1H), 6.99 (dd, J = 7.9, 1.7 Hz, 1H), 6.73 (d, J = 7.9 Hz, 1H), 5.92 (s, 2H), 4.96 (s, 2H), 4.02–3.85 (m, 4H), 3.81 (d, J = 13.8 Hz, 1H), 3.60 (d, J = 13.8 Hz, 1H), 3.27 (s, 2H), 3.14–3.05 (m, 1H), 2.75–2.59 (m, 1H), 2.23–2.12 (m, 1H), 1.14 (d, J = 6.6 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 161.34, 153.14, 147.40, 146.85, 144.69, 140.60, 132.98, 132.08, 130.91, 130.72, 129.19, 122.50, 118.93, 112.55, 109.54, 108.02, 100.89, 100.79, 56.94, 50.83, 50.56, 46.84, 45.16, 44.90, 32.94, 14.32; HRMS (ESI) calcd for C26H26N5O3+ 456.2030 [M + H]+; found, 456.2051; mp 62–66 °C; HPLC retention time = 6.0 min, purity = 95.8%, (λ = 254 nm).
(R)-3-((8-Methyl-4-(naphthalen-1-ylmethyl)-5-oxo-1,2,4,5,8,9-hexahydroimidazo [1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (59a)
1H NMR (600 MHz, CDCl3): δ 8.23 (d, J = 8.5, 1H), 7.85 (d, J = 8.2, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.70 (s, 1H), 7.59–7.52 (m, 3H), 7.49 (ddd, J = 8.0, 6.8, 1.2 Hz, 1H), 7.41 (dt, J = 9.9, 7.9 Hz, 2H), 7.34 (d, J = 7.1, 1H), 5.62–5.54 (m, 2H), 4.02–3.88 (m, 4H), 3.83 (d, J = 13.8 Hz, 1H), 3.61 (d, J = 13.9 Hz, 1H), 3.32 (s, 2H), 3.13 (h, J = 6.5 Hz, 1H), 2.71 (dd, J = 17.4, 5.4 Hz, 1H), 2.23 (dd, J = 17.5, 4.4 Hz, 1H), 1.18 (d, J = 6.6 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 161.55, 153.28, 145.00, 140.59, 133.80, 133.01, 132.12, 131.41, 131.22, 130.91, 129.20, 128.65, 127.69, 126.03, 125.55, 125.28, 123.77, 123.44, 118.96, 112.56, 100.81, 56.96, 50.85, 50.60, 46.96, 45.05, 43.33, 33.08, 14.35; HRMS (ESI) calcd for C29H28N5O+ 462.2288 [M + H]+; found, 462.2306; mp 74–77 °C; HPLC retention time = 7.0 min, purity = 98.3%, (λ = 254 nm).
(R)-3-((8-Methyl-4-(naphthalen-2-ylmethyl)-5-oxo-1,2,4,5,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (60a)
1H NMR (600 MHz, CDCl3): δ 7.93 (s, 1H), 7.84–7.76 (m, 3H), 7.68 (s, 1H), 7.63 (dd, J = 8.4, 1.8 Hz, 1H), 7.55 (t, J = 8.1 Hz, 2H), 7.47–7.39 (m, 3H), 5.22 (d, J = 3.1 Hz, 2H), 4.01–3.85 (m, 4H), 3.81 (d, J = 13.8 Hz, 1H), 3.59 (d, J = 13.8 Hz, 1H), 3.30 (s, 2H), 3.09 (dtd, J = 11.5, 6.5, 4.7 Hz, 2H), 2.65 (ddt, J = 17.5, 5.6, 2.1 Hz, 2H), 2.23–2.11 (m, 2H), 1.14 (d, J = 6.5 Hz, 3H); 13C NMR (151 MHz, CDCl3): δ 161.47, 153.15, 144.81, 140.60, 134.38, 133.26, 133.01, 132.82, 132.10, 130.91, 129.20, 127.98, 127.94, 127.58, 126.91, 125.89, 125.73, 118.95, 112.54, 100.73, 56.93, 50.85, 50.61, 46.83, 45.56, 44.97, 32.94, 14.36; HRMS (ESI) calcd for C29H28N5O+ 462.2288 [M + H]+; found, 462.2305; mp 75–80 °C; HPLC retention time = 7.3 min, purity = 98.6%, (λ = 254 nm).
SFC-Preparation of CLPP-1071
Instrument: WATERS 150 preparative SFC(SFC-26) Column: ChiralPak AD, 250 × 30 mm I.D.,10 μM; Mobile phase: A for CO2 and B for Ethanol (0.1% NH3H2O) Gradient: B 25% (Figure S1).
(R)-3-((4-(4-Bromobenzyl)-8-methyl-5-oxo-1,2,4,5,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (CLPP-1071)
1H NMR (500 MHz, Chloroform-d): δ 7.67 (d, J = 1.7 Hz, 1H), 7.56 (dt, J = 8.0, 1.7 Hz, 2H), 7.49–7.32 (m, 5H), 4.99 (d, J = 1.5 Hz, 2H), 3.96–3.90 (m, 4H), 3.81 (d, J = 13.8 Hz, 1H), 3.60 (d, J = 13.8 Hz, 1H), 3.27 (d, J = 1.8 Hz, 2H), 3.16–3.04 (m, 1H), 2.67 (ddt, J = 17.4, 5.5, 2.1 Hz, 1H), 2.24–2.11 (m, 1H), 1.15 (d, J = 6.6 Hz, 3H); 13C NMR (126 MHz, chloroform-d): δ 161.28, 153.01, 144.90, 140.55, 135.87, 132.97, 132.08, 131.37, 130.92, 130.67, 129.20, 121.44, 118.92, 112.56, 100.74, 77.31, 77.05, 76.80, 56.95, 50.82, 50.54, 46.86, 44.80, 44.87, 32.99, 14.30.
(R)-3-((4-(4-Bromobenzyl)-6-methyl-5-oxo-1,2,4,5,8,9-hexahydroimidazo[1,2-a] pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (49b′)
[α]20D = −20.0° (c = 0.50, MeOH). 1H NMR (500 MHz, chloroform-d): δ 7.67 (s, 1H), 7.56 (dd, J = 12.9, 7.7 Hz, 2H), 7.45–7.35 (m, 5H), 5.04–4.97 (m, 2H), 3.93–3.89 (m, 4H), 3.77 (dd, J = 13.5, 5.1 Hz, 2H), 3.66 (d, J = 14.1 Hz, 1H), 3.02–2.94 (m, 1H), 2.69 (dd, J = 12.9, 6.7 Hz, 1H), 2.54 (ddd, J = 18.0, 10.8, 6.9 Hz, 1H), 2.28–2.15 (m, 1H), 1.24 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, Chloroform-d): δ 161.32, 152.79, 145.49, 140.69, 135.97, 132.92, 131.94, 131.36, 130.93, 130.64, 129.20, 121.38, 118.95, 112.48, 106.89, 57.01, 50.81, 50.47, 46.88, 44.81, 40.57, 24.67, 14.78.
(S)-3-((4-(4-Bromobenzyl)-6-methyl-5-oxo-1,2,4,5,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile (49b″)
[α]20D = +20.0° (c = 0.50, MeOH). 1H NMR (500 MHz, chloroform-d): δ 7.67 (s, 1H), 7.56 (dd, J = 12.3, 7.2 Hz, 2H), 7.44–7.33 (m, 5H), 5.04–4.98 (m, 2H), 3.92–3.89 (m, 4H), 3.77 (dd, J = 13.2, 5.6 Hz, 2H), 3.66 (d, J = 14.1 Hz, 1H), 3.04–2.93 (m, 1H), 2.70 (dd, J = 13.0, 6.7 Hz, 1H), 2.54 (ddd, J = 17.6, 10.6, 6.9 Hz, 1H), 2.23–2.17 (m, 1H), 1.24 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, chloroform-d): δ 161.32, 152.80, 145.49, 140.68, 135.97, 132.92, 131.94, 131.36, 130.93, 130.63, 129.20, 121.38, 118.95, 112.48, 106.90, 57.01, 50.82, 50.46, 46.88, 44.81, 40.57, 24.67, 14.78.
Regioselective Synthesis of 52a
Methyl (R)-3-((3-(tert-Butoxy)-3-oxopropyl)amino)butanoate (62)
To a mixture of (R)-methyl 3-aminobutanoate hydrochloride (10, 5.0 g, 33.0 mmol) and tert-butyl acrylate (61, 3.2 mL, 36.0 mmol) in DCM (60 mL) were added TEA (4.5 mL, 33.0 mmol) and DBU (0.5 mL, 3.3 mmol), and the mixture was stirred for 24 h at 40 °C. Water (50 mL) was added, and the layers were separated. The aqueous layer was extracted with DCM (4 × 50 mL). The combined organic layers were washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to afford the crude product (62, 6.8 g, 84% yield) as a colorless liquid.
Methyl (R)-3-((3-(tert-Butoxy)-3-oxopropyl)(3-cyanobenzyl)amino)butanoate (64)
To a mixture of methyl (R)-3-((3-(tert-butoxy)-3-oxopropyl)amino)butanoate (62, 6.8 g, 27.2 mmol) and 3-(bromomethyl)benzonitrile (63, 7.9 g, 40.0 mmol) in MeCN (60 mL) was added DIPEA (7.0 mL, 40.0 mmol), and the reaction mixture was stirred at reflux overnight. After the reaction was completed, the mixture was warmed to room temperature and concentrated under reduced pressure. Water (50 mL) was added, and the layers were separated. The aqueous layer was extracted with EA (4 × 50 mL). The combined organic layers were washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified via silica gel chromatography (PE/EA = 30:1) to afford the title compound (64, 7.4 g, 74% yield) as a colorless liquid.
tert-Butyl (6R)-1-(3-Cyanobenzyl)-6-methyl-4-oxopiperidine-3-carboxylate (65)
To a solution of the product from the previous step (64, 7.4 g, 20.1 mmol) in anhydrous THF (50 mL) at 0 °C was added t-BuOK 1.0 M in THF (42 mL, 42 mmol) slowly down the sides of the flask submerged in an ice water bath, and the resulting mixture was stirred at 0 °C for 12 h. After the reaction was completed, the mixture was warmed to room temperature and concentrated under reduced pressure. Water (50 mL) was added, and the layers were separated. The aqueous layer was extracted with EA (4 × 50 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified via silica gel chromatography (PE/EA = 15:1) to afford the title compound (65, 3.7 g, 55% yield) as a white solid.
Gram-Scale Position-Specific Synthesis of CLPP-1071
To a tert-butyl (6R)-1-(3-cyanobenzyl)-6-methyl-4-oxopiperidine-3-carboxylate (65, 3.7 g, 11.3 mmol) and N-(4-bromobenzyl)-4,5-dihydro-1H-imidazole-2-amine hydroiodide (66, 4.5 g, 12.0 mmol) in anhydrous MeOH (20.0 mL) was added MeONa (2.7 g, 50.0 mmol), and the mixture was stirred for overnight at 100 °C. The mixture was cooled to room temperature and concentrated under reduced pressure. Water (50 mL) was added, and the layers were separated. The aqueous layer was extracted with DCM (4 × 50 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified via silica gel chromatography (DCM/MeOH = 95:5) to afford the title compound (CLPP-1071, 4.1 g, 74% yield) as a yellow solid.
Protein Preparation
The preparation of ClpP protein was carried out as reported in previous literature. Generally, ClpP sequence 57–277 with an N-terminal SUMO tag was cloned and transformed in Escherichia coli. The bacteria were cultured with LB medium in the presence of 50 μg/mL ampicillin at 37 °C. The bacteria were harvested after treatment with 0.5 mM isopropyl β-d-1-thiogalactopyranoside for 4 h and sequentially lysed in buffer (25 mM Tris-2HCl pH 7.5, 0.5 M NaCl, 5 mM β-mercaptoethanol, 10 mM imidazole, 10% glycerol, 1% Triton X-100) by sonication. The pellet was removed by centrifugation, the supernatant was combined with Ni sepharose 6 Fast Flow for 2 h, and then purified using a gravity column, first with washing buffer (25 mM Tris-2HCl pH 7.5, 0.5 M NaCl, 5 mM β-mercaptoethanol, 10 mM imidazole, 10% glycerol) for the first impurity protein elution and finally with 400 mM imidazole for the protein of interest. The eluted protein was dialyzed in 4 L of dialysate for 6 h, quantified by BCA, and stored at −80 °C with 50% glycerol.
In Vitro Enzyme Assay
The ClpP protein was diluted with buffer (50 mM Tris-2HCl pH 8, 10 mM MgCl2, 100 mM KCl, 1 mM DTT, 5% glycerol, and 0.02% Triton X-100) solution to the final concentration of 0.7 μM and AC-WLA-AMC54 to the final concentration of 100 μM. In a 384 black plate, 20 μL of ClpP protein was first added, and then 10 μL of the compound was added. After mixing, it was incubated at room temperature for 10 min. Finally, 20 μL of AC-WLA-AMC was added. The first fluorescence signal was detected at 355 and 460 nm wavelengths using a multifunctional enzyme labeler After detection, the board was sealed and incubated in 37 incubators for 4 h, and the fluorescence signal was collected for a second time. The obtained data were fitted with the enzyme activity curve and EC50 value.
Cell Lines and Cell Culture
The HL60 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). MOLM13 was obtained from the Japanese Collection of Research Bioresources Cell Bank (JCRB, Osaka, Japan). All cells were maintained in the appropriate culture medium suggested by the suppliers. All cell lines were authenticated by STR profiling, tested for mycoplasma contamination, and used within 15 passages and less than 6 months.
Growth Inhibition Assay
Briefly, 80 μL of cells (1 × 104 cells/well) were seeded into 96-well plates. After 0.5 h of incubation, the cells were treated with 20 μL of 0.02% DMSO or varying concentrations of the test compounds for 72 h. Cell viability was measured using the CellTiter 96 AQueous nonradioactive cell proliferation assay (MTS; Promega, Madison, WI). The combined solution of MTS (20 μL) was pipetted into each well of the 96-well plates and incubated at 37 °C for 2–4 h. Optical density was determined at 490 nm (background values measured at 690 nm were subtracted) using a SpectraMax 340 microplate reader (Molecular Devices, Sunnyvale, CA, USA). Growth inhibition was calculated as % = [1 – (ODcompound – ODblank)/(ODDMSO – ODblank)] × 100.
Biolayer Interferometry
The binding affinity of ClpP protein with compounds ONC212 and CLPP-1071 were determined using the GatorPrime biolayer interferometry system (Gator Bio, Palo Alto, CA, USA). All step were performed at 30 °C, 1000 rpm. Briefly, SA-XT sensors (Gator Bio) were dipped into the ClpP protein solutions for 15 min for loading and then dipped into various concentrations of ONC212 (0–200 μM) or CLPP-1071 (0–40 μM) in PBS, 0.002% Tween 20, 0.02% BSA, 2% DMSO buffer. Background binding controls used a duplicate set of sensors that were incubated in a buffer without proteins. All the data were analyzed by Gator Bio data analysis software. The equilibrium dissociation constant (KD) values were calculated from the ratio of Koff to Kon, based on global fitting of several curves generated from serial dilutions of the compound.
Cellular Thermal Shift Assay
6 × 106 cells were divided into the DMSO and compounds groups. In the compounds group, 10 μM of the compound was added, while an equal volume of DMSO was added to the DMSO group. Both groups were further incubated for 6 h. After the cells were centrifuged at 800 rpm for 3 min at 4 °C, 310 μL of precooled PBS was added to the cells. The resuspended cells were then divided into six PCR tubes and heated at different temperatures (53, 56, 59, 62, 65, 68 °C) for 3 min each, after freeze–thaw cycles.
Western Blot
Whole protein extracts were from cultured cells. Equal volumes of protein extracts were resolved on tricine SDS-PAGE gels and transferred onto nitrocellulose membranes (Amersham, 10600002). The blots were incubated for 1 h in a TBS-T solution supplemented with 5% nonfat dry bovine albumin. Membranes were probed with the specific primary antibodies overnight at 4 °C, followed by washing three times with a TBS-T solution and incubating with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. After being washed three times with a TBS-T solution, the membranes were incubated with ECL reagent and then were exposed to a ChemiDoc Touch Imaging System (Eblot). The primary antibodies are listed as follows: CLPP (CST, 14181S), NDUFA12 (Abcam, ab192617), and SDHB (Abcam, ab175225).
Mitochondrial Reactive Oxygen Species
5 × 105 cells/well were seeded in 12-well plates and stabilized in an incubator for 30 min, and the cells were treated with varying concentrations of the test compounds for 38 h. After drug treatment, the cells were washed twice with ice-cold PBS. PBS resuspension cells containing 500 nM MitoSOX Red (Invitrogen, M36008) were incubated at 37 °C for 1 h and washed once with precooled PBS. After 500 μL of PBS was added, the Agilent flow PE channel was used for data acquisition and processing.
Mitochondrial Membrane Potential
Flow Cytometry: 4 × 105 cells/well were seeded in 12-well plates and stabilized in an incubator for 30 min; the cells were treated with varying concentrations of the test compounds for 48 h. After treatment, the cells were washed twice with ice-cold PBS. 500 μL of PBS resuspension cells (500 μL) containing 100 μM tetramethylrhodamine (TMRM, Invitrogen, I34361) were incubated at 37 °C for 1 h and washed once with precooled PBS. After 500 μL of PBS was added, an Agilent flow PE channel was used for data acquisition and processing. Laser Confocal Microscopy: The cell handling and staining procedures are consistent with those described above, after TMRM staining is completed, cells were counted, and 2 × 105 – 4 × 105 cells were added to 100 μM Hoechst and incubated for 15 min cells were washed with ice-cold PBS. After resuspension in 500 μL of PBS, the cells were plated in a 12-well live cell plate, mixed, and incubated for 10 min. Image acquisition was then performed by using a laser confocal microscope (FV3000).
Cell Cycle
1 ×106 cells were added to the 6-well plate and treated with compounds of different concentrations for 24 h. After drug treatment, the cells were washed twice with ice-cold PBS, the cells were resuspended in 300 μL of PBS, and then 700 μL of anhydrous ethanol was slowly added while swirling. The cells were fixed overnight at 4 °C in a refrigerator. After overnight fixation, the cells were washed once with 1 mL of precooled PBS. Then, 1 mL of precooled PBS was added to the suspended cells, followed by the addition of 10 μg/mL RNase (Beyotime), and they were incubated at 37 °C for 15 min. After, 20 μg/mL propidium iodide (PI, Sigma) dye was added, and the cells were incubated at 4 °C for 30 min. Finally, the data were collected and processed by using flow cytometry (Agilent).
Apoptosis
4 ×105 cells were added to the 12-well plate and treated with compounds of different concentrations for 36 h. After drug treatment, the cells were washed twice with ice-cold PBS. 300 μL of 1× binding buffer (Apoptosis Detection Kit; Vazyme) was added to each sample. After 2.5 μL of PI and Annexin V-FITC solution was added to each sample, it was immediately analyzed using flow cytometry (Agilent).
Animals
ICR mice (female, 6–8 weeks old, 18–22 g) were purchased from Zhuhai Baishitong Biotechnology Co., Ltd. NSG mice (female, 6–8 weeks old, 18–22 g) were purchased from Biocytogen Pharmaceuticals (Beijing) Co., Ltd. These mice were bred at the Animal Facility of the Zhongshan Institute for Drug Discovery. The animals were maintained under a 12 h light–dark cycle with free access to water and food. All animal studies were performed in strict accordance with the institutional guidelines, as defined by the Institutional Animal Care and Use Committee (IACUC: 2023-01-LJ-01), approved by the Animal Care and Use Committee, Zhongshan Institute for Drug Discovery Animal Center (Zhongshan, China).
PK Studies
Three ICR mice were orally administered the compound with a dose of 50 mg/kg in the solvent (0.5% CMC-Na). Venous blood samples were collected at 0.083, 0.25, 0.5, 1, 2, 4, 6, and 24 h. Plasma was separated from whole blood by centrifugation and stored at −80 °C until analysis. Compound levels were determined using an LC/MS system.
Evaluation of Xenograft Tumor
Female NSG mice were maintained in a pathogen-free animal facility. Tumors were established via tail vein injection of MOLM13 cells (2 × 104 cells/mice) and HL60 (5 × 106 cells/mice). On the first day after inoculation MOLM13, the mice were randomly divided into three groups and received ONC212 (n = 10, 10 mg/kg, p.o, qd), CLPP-1071 (n = 10, 10 mg/kg, p.o, qd), or vehicle (n = 20, 0.5% CMC-Na, p.o, qd) daily for 18 days. Six days after HL60 vaccination, the mice were randomly divided into five groups and received ONC212 (n = 9, 10 mg/kg, p.o, qd), CLPP-1071 (n = 9, 10 mg/kg, p.o, qd), CLPP-1071 (n = 9, 5 mg/kg, p.o, qd), CLPP-1071 (n = 9, 2.5 mg/kg, p.o, qd) or vehicle (n = 18, 0.5% CMC-Na, p.o, qd) daily for 19 days. Compounds were dissolved in 0.5% CMC-Na. The body weight of mice was recorded every 3 or 4 days.
Statistical Analysis
Statistical analysis was performed by using GraphPad Prism software. Unless otherwise stated, the values for all samples under the different experimental conditions were averaged, and the standard deviation of the mean was calculated. Statistical differences between the two groups were assessed by Student’s t test. A P value of less than 0.05 was considered statistically significant.
Molecular Modeling
The crystal structure of ClpP/ONC201 (PDB ID: 6DL7)9 was used to construct the binding modes of compounds substituted with methyl groups at different positions. The initial ClpP structure was imported into the Protein Preparation Wizard (ProPrep)55 in Schrödinger software to rebuild the missing side chains and assign the protonation states of the amino acids at pH 7.0. We used Glide56 to perform docking calculations at standard precision, setting the binding site centered on ONC201 in ClpP with a radius similar in size to the ONC201. Each ligand retained up to 20 docking poses, and the top-ranked pose was selected for binding mode analysis.
Acknowledgments
This work was supported in part by funding from the Guangdong High-level New R&D Institute (2019B090904008), the Guangdong High-level Innovative Research Institute (2021B0909050003), the Creative Research Group of Zhongshan City (Lingnan Pharmaceutical Research and Innovation team CXTD2022011), the National Natural Science Foundation of China (82304539), Shanghai Science and Technology Development Funds (Yangfan program: 22YF1457600), the China Postdoctoral Science Foundation (2023M733629), and the Postdoctoral Fellowship Program of CPSF (GZC20232846).
Glossary
Abbreviations
- Ac-WLA-AMC
N-acetyl-Trp-Leu-Ala-7-amino-4-methylcoumarin
- AML
acute myelocytic leukemia
- AUC0–t
area under the plasma concentration–time curve from time 0 to last time of quantifiable concentration
- AUC0–∞
area under the plasma concentration–time curve from time 0 extrapolated to infinite time
- (Boc)2O
di-tert-butyl decarbonate
- ClpP
caseinolytic protease P
- Cmax
peak concentration
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCM
dichloromethane
- EC50
half maximal effect concentration
- H3 K27
histone H3 lysine 27
- HMBC
1H detected heteronuclear multiple bond correlation
- IC50
half maximal inhibitory concentration
- Kd
dissociation constant
- NMR
nuclear magnetic resonance
- NSG
NOD scid gamma
- PE
petroleum ether
- PK
pharmacokinetics
- ROS
reactive oxygen species
- SAR
structure–activity relationship
- SFC
supercritical fluid chromatography
- TEA
triethylamine
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TSA
thermal shift assay
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c01605.
Separation and identification of components in mixtures 52 and 29 (Figures S1–S6); possible mechanisms for the generation of the three isomers of compound 6 (Figure S7); the correlation analysis between the molecular enzyme activities of seven single isomers and seven mixtures (Figure S8); the effect of compound 6 on peptidase activity (Figure S9); CLPP-1071 on HL60 cell and MOML13 cell growth inhibition (Figure S10); ONC212 induces mitochondrial dysfunction (Figure S11). ONC212 induces cell cycle arrest and apoptosis (Figure S12); optimization of Dieckmann condensation reaction (Table S1); activity and liver microsomal stability data for representative single-isomer compounds (Table S2 and Table S3); synthesis method of 2–5; 1H/13C/19F NMR, HRMS spectra, SFC analysis and HPLC traces of representative compounds (PDF)
Predicted_binding_mode_compound_2 (PDB)
Predicted_binding_mode_compound_3 (PDB)
Predicted_binding_mode_compound_6a (PDB)
Predicted_binding_mode_compound_5 (PDB)
Predicted_binding_mode_compound_CLPP-1071 (PDB)
Molecular string files for all of the final target compounds (CSV)
Author Contributions
⧫ B.C., M.S., C.Z., and Q.H. contributed equally to this work. The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Song J. Y.; Herrmann J. M.; Becker T. Quality Control of the Mitochondrial Proteome. Nat. Rev. Mol. Cell Biol. 2021, 22 (1), 54–70. 10.1038/s41580-020-00300-2. [DOI] [PubMed] [Google Scholar]
- Vyas S.; Zaganjor E.; Haigis M. C. Mitochondria and Cancer. Cell 2016, 166 (3), 555–566. 10.1016/j.cell.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J.; Suomalainen A. Mitochondria: In Sickness and in Health. Cell 2012, 148 (6), 1145–1159. 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes C. M.; Petrova K.; Benedetti C.; Yang Y.; Ron D. ClpP Mediates Activation of a Mitochondrial Unfolded Protein Response in C. elegans. Dev. Cell 2007, 13 (4), 467–480. 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Topf U.; Wrobel L.; Chacinska A. Chatty Mitochondria: Keeping Balance in Cellular Protein Homeostasis. Trends Cell Biol. 2016, 26 (8), 577–586. 10.1016/j.tcb.2016.03.002. [DOI] [PubMed] [Google Scholar]
- Deepa S. S.; Bhaskaran S.; Ranjit R.; Qaisar R.; Nair B. C.; Liu Y. H.; Walsh M. E.; Fok W. C.; Van Remmen H. Down-Regulation of the Mitochondrial Matrix Peptidase ClpP in Muscle Cells Causes Mitochondrial Dysfunction and Decreases Cell Proliferation. Free Radical Biol. Med. 2016, 91, 281–292. 10.1016/j.freeradbiomed.2015.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F.; Weil A.; Hamann A.; Osiewacz H. D. Human CLPP Reverts the Longevity Phenotype of a Fungal ClpP Deletion Strain. Nat. Commun. 2013, 4, 1397. 10.1038/ncomms2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J. H.; Rivadeneira D. B.; Caino M. C.; Chae Y. C.; Speicher D. W.; Tang H. Y.; Vaira V.; Bosari S.; Palleschi A.; Rampini P.; Kossenkov A. V.; Languino L. R.; Altieri D. C. The Mitochondrial Unfoldase-Peptidase Complex ClpXP Controls Bioenergetics Stress and Metastasis. PLoS Biol. 2016, 14 (7), e1002507 10.1371/journal.pbio.1002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawa J.; Zarabi S. F.; Davis R. E.; Halgas O.; Nii T.; Jitkova Y.; Zhao R.; St-Germain J.; Heese L. E.; Egan G.; Ruvolo V. R.; Barghout S. H.; Nishida Y.; Hurren R.; Ma W.; Gronda M.; Link T.; Wong K.; Mabanglo M.; Kojima K.; Borthakur G.; MacLean N.; Ma M. C. J.; Leber A. B.; Minden M. D.; Houry W.; Kantarjian H.; Stogniew M.; Raught B.; Pai E. F.; Schimmer A. D.; Andreeff M. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35 (5), 721–737.e9. 10.1016/j.ccell.2019.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M.; Wallace D. C.; Burelle Y. The Rise of Mitochondria in Medicine. Mitochondrion 2016, 30, 105–116. 10.1016/j.mito.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andréasson C.; Ott M.; Büttner S. Mitochondria Orchestrate Proteostatic and Metabolic Stress Responses. EMBO Rep. 2019, 20 (10), e47865 10.15252/embr.201947865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L. Q.; Sang P.; Zhang R. P.; Liu S. Q. Substrate-Induced Changes in Dynamics and Molecular Motions of Cuticle-Degrading Serine Protease PL646: a Molecular Dynamics Study. RSC Adv. 2017, 7 (67), 42094–42104. 10.1039/C7RA07797A. [DOI] [Google Scholar]
- Bhandari V.; Wong K. S.; Zhou J. L.; Mabanglo M. F.; Batey R. A.; Houry W. A. The Role of ClpP Protease in Bacterial Pathogenesis and Human Diseases. ACS Chem. Biol. 2018, 13 (6), 1413–1425. 10.1021/acschembio.8b00124. [DOI] [PubMed] [Google Scholar]
- Suomalainen A.; Battersby B. J. Mitochondrial Diseases: The Contribution of Organelle Stress Responses to Pathology. Nat. Rev. Mol. Cell Biol. 2018, 19 (2), 77–92. 10.1038/nrm.2017.66. [DOI] [PubMed] [Google Scholar]
- Eisenberg-Bord M.; Schuldiner M. Ground Control to Major TOM: Mitochondria-Nucleus Communication. FEBS J. 2017, 284 (2), 196–210. 10.1111/febs.13778. [DOI] [PubMed] [Google Scholar]
- Javadov S.; Kozlov A. V.; Camara A. K. S. Mitochondria in Health and Diseases. Cells 2020, 9 (5), 1177. 10.3390/cells9051177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole A.; Wang Z. Z.; Coyaud E.; Voisin V.; Gronda M.; Jitkova Y.; Mattson R.; Hurren R.; Babovic S.; Maclean N.; Restall I.; Wang X. M.; Jeyaraju D. V.; Sukhai M. A.; Prabha S.; Bashir S.; Ramakrishnan A.; Leung E.; Qia Y. H.; Zhang N. X.; Combes K. R.; Ketela T.; Lin F. S.; Houry W. A.; Aman A.; Al-Awar R.; Zheng W.; Wienholds E.; Xu C. J.; Dick J.; Wang J. C. Y.; Moffat J.; Minden M. D.; Eaves C. J.; Bader G. D.; Hao Z. Y.; Kornblau S. M.; Raught B.; Schimmer A. D. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27 (6), 864–876. 10.1016/j.ccell.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M.; Zhang C.; Lee S.; Sjöstedt E.; Fagerberg L.; Bidkhori G.; Benfeitas R.; Arif M.; Liu Z. T.; Edfors F.; Sanli K.; von Feilitzen K.; Oksvold P.; Lundberg E.; Hober S.; Nilsson P.; Mattsson J.; Schwenk J. M.; Brunnström H.; Glimelius B.; Sjoblom T.; Edqvist P. H.; Djureinovic D.; Micke P.; Lindskog C.; Mardinoglu A.; Ponten F. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357 (6352), eaan2507 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- Cormio A.; Musicco C.; Gasparre G.; Cormio G.; Pesce V.; Sardanelli A. M.; Gadaleta M. N. Increase in Proteins Involved in Mitochondrial Fission, Mitophagy, Proteolysis and Antioxidant Response in type I Endometrial Cancer as an Adaptive Response to Respiratory Complex I Deficiency. Biochem. Biophys. Res. Commun. 2017, 491 (1), 85–90. 10.1016/j.bbrc.2017.07.047. [DOI] [PubMed] [Google Scholar]
- Cormio A.; Sanguedolce F.; Pesce V.; Musicco C. Mitochondrial Caseinolytic Protease P: A Possible Novel Prognostic Marker and Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22 (12), 6228. 10.3390/ijms22126228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K. S.; Houry W. A. Chemical Modulation of Human Mitochondrial ClpP: Potential Application in Cancer Therapeutics. ACS Chem. Biol. 2019, 14 (11), 2349–2360. 10.1021/acschembio.9b00347. [DOI] [PubMed] [Google Scholar]
- Gersch M.; Famulla K.; Dahmen M.; Göbl C.; Malik I.; Richter K.; Korotkov V. S.; Sass P.; Rübsamen-Schaeff H.; Madl T.; Brötz-Oesterhelt H.; Sieber S. A. AAA + Chaperones and Acyldepsipeptides Activate the ClpP Protease via Conformational Control. Nat. Commun. 2015, 6, 6320. 10.1038/ncomms7320. [DOI] [PubMed] [Google Scholar]
- Mirali S.; Schimmer A. D. The Role of Mitochondrial Proteases in Leukemic Cells and Leukemic Stem Cells. Stem Cells Transl. Med. 2020, 9 (12), 1481–1487. 10.1002/sctm.20-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurizi M. R.; Clark W. P.; Kim S. H.; Gottesman S. ClpP Represents a Unique Family of Serine Proteases. J. Biol. Chem. 1990, 265 (21), 12546–12552. 10.1016/S0021-9258(19)38379-6. [DOI] [PubMed] [Google Scholar]
- Nouri K.; Feng Y.; Schimmer A. D. Mitochondrial ClpP Serine Protease-Biological Function and Emerging Target for Cancer Therapy. Cell Death Dis. 2020, 11 (10), 841. 10.1038/s41419-020-03062-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W.; Li N.; Wang J. J.; Yu N. R.; Lei Z. C.; Zhang L. L.; Sun J. B.; Chen L. Design and Synthesis of Novel Mitochondria-Targeted CDDO Derivatives as Potential Anti-Cancer Agents. Bioorg. Chem. 2021, 115, 105249. 10.1016/j.bioorg.2021.105249. [DOI] [PubMed] [Google Scholar]
- Kim J. E.; Park H.; Choi S. H.; Kong M. J.; Kang T. C. CDDO-Me Selectively Attenuates CA1 Neuronal Death Induced by Status Epilepticus via Facilitating Mitochondrial Fission Independent of LONP1. Cells 2019, 8 (8), 833. 10.3390/cells8080833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers L. J.; John T.; Park J.; Tucker M.; Ma H. D.; Wu Y.; Rayalam S.; Wang X. Y. Growth Inhibition and Apoptosis of Human Multiple Myeloma Cells Induced by 2-cyano-3,12-dioxooleana-1,9-dien-28-oic Acid Derivatives. Anti-Cancer Drugs 2020, 31 (8), 806–818. 10.1097/CAD.0000000000000941. [DOI] [PubMed] [Google Scholar]
- ONC201 and Atezolizumab in Obesity-Driven Endometrial Cancer. (NCT05542407).
- ONC201 in H3 K27 M mutant Diffuse Glioma Following Radiotherapy (NCT05580562).
- Luo B. Z.; Ma Y.; Zhou Y. Z.; Zhang N. N.; Luo Y. F. Human ClpP Protease, a Promising Therapy Target for Diseases of Mitochondrial Dysfunction. Drug Discovery Today 2021, 26 (4), 968–981. 10.1016/j.drudis.2021.01.007. [DOI] [PubMed] [Google Scholar]
- Song R.; Qiao W. L.; He J.; Huang J. S.; Luo Y. F.; Yang T. Proteases and Their Modulators in Cancer Therapy: Challenges and Opportunities. J. Med. Chem. 2021, 64 (6), 2851–2877. 10.1021/acs.jmedchem.0c01640. [DOI] [PubMed] [Google Scholar]
- Wong K. S.; Mabanglo M. F.; Seraphim T. V.; Mollica A.; Mao Y. Q.; Rizzolo K.; Leung E.; Moutaoufik M. T.; Hoell L.; Phanse S.; Goodreid J.; Barbosa L. R. S.; Ramos C. H. I.; Babu M.; Mennella V.; Batey R. A.; Schimmer A. D.; Houry W. A. Acyldepsipeptide Analogs Dysregulate Human Mitochondrial ClpP Protease Activity and Cause Apoptotic Cell Death. Cell Chem. Biol. 2018, 25 (8), 1017–1030.e9. 10.1016/j.chembiol.2018.05.014. [DOI] [PubMed] [Google Scholar]
- Stahl M.; Korotkov V. S.; Balogh D.; Kick L. M.; Gersch M.; Pahl A.; Kielkowski P.; Richter K.; Schneider S.; Sieber S. A. Selective Activation of Human Caseinolytic Protease P (ClpP). Angew. Chem., Int. Ed. 2018, 57 (44), 14602–14607. 10.1002/anie.201808189. [DOI] [PubMed] [Google Scholar]
- Venneti S.; Kawakibi A. R.; Ji S.; Waszak S. M.; Sweha S. R.; Mota M.; Pun M.; Deogharkar A.; Chung C.; Tarapore R. S.; Ramage S.; Chi A.; Wen P. Y.; Arrillaga-Romany I.; Batchelor T. T.; Butowski N. A.; Sumrall A.; Shonka N.; Harrison R. A.; de Groot J.; Mehta M.; Hall M. D.; Daghistani D.; Cloughesy T. F.; Ellingson B. M.; Beccaria K.; Varlet P.; Kim M. M.; Umemura Y.; Garton H.; Franson A.; Schwartz J.; Jain R.; Kachman M.; Baum H.; Burant C. F.; Mottl S. L.; Cartaxo R. T.; John V.; Messinger D.; Qin T. T.; Peterson E.; Sajjakulnukit P.; Ravi K.; Waugh A.; Walling D.; Ding Y. J.; Xia Z. Y.; Schwendeman A.; Hawes D.; Yang F. S.; Judkins A. R.; Wahl D.; Lyssiotis C. A.; de la Nava D.; Alonso M. M.; Eze A.; Spitzer J.; Schmidt S. V.; Duchatel R. J.; Dun M. D.; Cain J. E.; Jiang L.; Stopka S. A.; Baquer G.; Regan M. S.; Filbin M. G.; Agar N. Y. R.; Zhao L. L.; Kumar-Sinha C.; Mody R.; Chinnaiyan A.; Kurokawa R.; Pratt D.; Yadav V. N.; Grill J.; Kline C.; Mueller S.; Resnick A.; Nazarian J.; Allen J. E.; Odia Y.; Gardner S. L.; Koschmann C. Clinical Efficacy of ONC201 in H3K27M-Mutant Diffuse Midline Gliomas Is Driven by Disruption of Integrated Metabolic and Epigenetic Pathways. Cancer Discovery 2023, 13 (11), 2370–2393. 10.1158/2159-8290.cd-23-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves P. R.; Aponte-Collazo L. J.; Fennell E. M. J.; Graves A. C.; Hale A. E.; Dicheva N.; Herring L. E.; Gilbert T. S. K.; East M. P.; McDonald I. M.; Lockett M. R.; Ashamalla H.; Moorman N. J.; Karanewsky D. S.; Iwanowicz E. J.; Holmuhamedov E.; Graves L. M. Mitochondrial Protease ClpP is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem. Biol. 2019, 14 (5), 1020–1029. 10.1021/acschembio.9b00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennell E. M. J.; Aponte-Collazo L. J.; Wynn J. D.; Drizyte-Miller K.; Leung E.; Greer Y. E.; Graves P. R.; Iwanowicz A. A.; Ashamalla H.; Holmuhamedov E.; Lang H.; Karanewsky D. S.; Der C. J.; Houry W. A.; Lipkowitz S.; Iwanowicz E. J.; Graves L. M. Characterization of TR-107, a Novel Chemical Activator of the Human Mitochondrial Protease ClpP. Pharmacol. Res. Perspect. 2022, 10 (4), 00993. 10.1002/prp2.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J.; Kline C. L.; Ralff M. D.; Lev A.; Lulla A.; Zhou L. L.; Olson G. L.; Nallaganchu B. R.; Benes C. H.; Allen J. E.; Prabhu V. V.; Stogniew M.; Oster W.; El-Deiry W. S. Preclinical Evaluation of The Imipridone Family, Analogs of Clinical Stage Anti-Cancer Small Molecule ONC201, Reveals Potent Anti-Cancer Effects of ONC212. Cell Cycle 2017, 16 (19), 1790–1799. 10.1080/15384101.2017.1325046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. Y.; Zhang T.; Wang X. J.; Xiao H. Y.; Li H. T.; Zhou L. L.; Yang T.; Wei B. Y.; Zhu Z. Y.; Zhou L.; Yang S.; Lu X. X.; Zhang Y. H.; Huang Y.; Gan J. H.; Yang C. G. Aberrant Human ClpP Activation Disturbs Mitochondrial Proteome Homeostasis to Suppress Pancreatic Ductal Adenocarcinoma. Cell Chem. Biol. 2022, 29 (9), 1396–1408.e8. 10.1016/j.chembiol.2022.07.002. [DOI] [PubMed] [Google Scholar]
- Zhang R. R.; Wang P. Y.; Wei B. Y.; Chen L.; Song X. M.; Pan Y. H.; Li J. H.; Gan J. H.; Zhang T.; Yang C. G. Assessment of the Structure-Activity Relationship and Antileukemic Activity of Diacylpyramide Compounds as Human ClpP Agonists. Eur. J. Med. Chem. 2023, 258, 115577. 10.1016/j.ejmech.2023.115577. [DOI] [PubMed] [Google Scholar]
- Zhou L. L.; Zhang T.; Xue Y.; Yue C.; Pan Y. H.; Wang P. Y.; Yang T.; Li M. X.; Zhou H.; Ding K.; Gan J. H.; Ji H. B.; Yang C. G. Selective Activator of Human ClpP Triggers Cell Cycle Arrest to Inhibit Lung Squamous Cell Carcinoma. Nat. Commun. 2023, 14 (1), 7069. 10.1038/s41467-023-42784-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. S.; Zhang J. N.; Luo B. Z.; Qiao W. L.; Qiu Z. Q.; Song R.; Dai Z. Y.; Sui J.; Xu X.; Ruan S. H.; Li C. W.; Luo Y. F.; Yang T. Discovery of a Novel Series of Imipridone Compounds as Homo sapiens Caseinolytic Protease P Agonists with Potent Antitumor Activities In Vitro and In Vivo. J. Med. Chem. 2022, 65 (11), 7629–7655. 10.1021/acs.jmedchem.1c02071. [DOI] [PubMed] [Google Scholar]
- Zhang J. N.; Qiu Z. Q.; Liu S.; Huang J. S.; Luo B. Z.; Sui J.; Dai Z. Y.; Xiang X. R.; Yang T.; Luo Y. F. Discovery of a Novel Series of Homo sapiens Caseinolytic Protease P Agonists for Colorectal Adenocarcinoma Treatment via ATF3-Dependent Integrated Stress Response. J. Med. Chem. 2024, 67 (4), 2812–2836. 10.1021/acs.jmedchem.3c01950. [DOI] [PubMed] [Google Scholar]
- Xiang X. R.; Dai Z. Y.; Luo B. Z.; Zhao N. L.; Liu S.; Sui J.; Huang J. S.; Zhou Y. Z.; Gu J. L.; Zhang J. N.; Yang T.; Bao R.; Luo Y. F. Rational Design of a Novel Class of Human ClpP Agonists through a Ring-Opening Strategy with Enhanced Antileukemia Activity. J. Med. Chem. 2024, 67 (8), 6769–6792. 10.1021/acs.jmedchem.4c00338. [DOI] [PubMed] [Google Scholar]
- Liu S.; Sui J.; Luo B. Z.; Zhang J. N.; Xiang X. R.; Yang T.; Luo Y. F.; Liu J. Discovery of 5-(Piperidin-4-yl)-1,2,4-oxadiazole Derivatives as a New Class of Human Caseinolytic Protease P Agonists for the Treatment of Hepatocellular Carcinoma. J. Med. Chem. 2024, 67 (13), 10622–10642. 10.1021/acs.jmedchem.4c00080. [DOI] [PubMed] [Google Scholar]
- Jiang J. X.; Xie G. J.; Li T.; Ding H.; Tang R.; Luo J. J.; Li Q. N.; Lu W. G.; Xiao Y. B.; Sun H. Y. Discovery of Dehydrogenated Imipridone Derivatives as Activators of Human Caseinolytic Protease P. J. Med. Chem. 2024, 67 (17), 15328–15352. 10.1021/acs.jmedchem.4c00798. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Z.; Jiang J. X.; Ding H.; Li Q. N.; Xiao Y. B.; Sun H. Y. Development of novel imipridone derivatives with potent anti-cancer activities as human caseinolytic peptidase P (hClpP) activators. Bioorg. Chem. 2024, 153, 107765. 10.1016/j.bioorg.2024.107765. [DOI] [PubMed] [Google Scholar]
- Barreiro E. J.; Kümmerle A. E.; Fraga C. A. M. The Methylation Effect in Medicinal Chemistry. Chem. Rev. 2011, 111 (9), 5215–5246. 10.1021/cr200060g. [DOI] [PubMed] [Google Scholar]
- Leung C. S.; Leung S. S. F.; Tirado-Rives J.; Jorgensen W. L. Methyl Effects on Protein-Ligand Binding. J. Med. Chem. 2012, 55 (9), 4489–4500. 10.1021/jm3003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönherr H.; Cernak T. Profound Methyl Effects in Drug Discovery and A Call for New C-H Methylation Reactions. Angew. Chem., Int. Ed. 2013, 52 (47), 12256–12267. 10.1002/anie.201303207. [DOI] [PubMed] [Google Scholar]
- Popović-Djordjević J.; Stepanović S.; Došen-Mićović L.; Ivanović E.; Ivanović M. D. High-yielding method for preparation of carbocyclic or N-containing heterocyclic β-keto esters using in situ activated sodium hydride in dimethyl sulfoxide. Green Chem. Lett. Rev. 2016, 9 (1), 61–68. 10.1080/17518253.2016.1145744. [DOI] [Google Scholar]
- Yin W. C.; Mao C. Y.; Luan X. D.; Shen D. D.; Shen Q. Y.; Su H. X.; Wang X. X.; Zhou F. L.; Zhao W. F.; Gao M. Q.; Chang S. H.; Xie Y. C.; Tian G. H.; Jiang H. W.; Tao S. C.; Shen J. S.; Jiang Y.; Jiang H. L.; Xu Y. C.; Zhang S. Y.; Zhang Y.; Xu H. E. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368 (6498), 1499–1504. 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde K. R.; Taanman J. W.; Schapira A. H. A LON-ClpP Proteolytic Axis Degrades Complex I to Extinguish ROS Production in Depolarized Mitochondria. Cell Rep. 2016, 17 (10), 2522–2531. 10.1016/j.celrep.2016.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gersch M.; Stahl M.; Poreba M.; Dahmen M.; Dziedzic A.; Drag M.; Sieber S. A. Barrel-shaped ClpP Proteases Display Attenuated Cleavage Specificities. ACS Chem. Biol. 2016, 11 (2), 389–399. 10.1021/acschembio.5b00757. [DOI] [PubMed] [Google Scholar]
- Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27 (3), 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47 (7), 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.