

Abstract
The tumor microenvironment is highly complex owing to its heterogeneous composition and dynamic nature. This makes tumors difficult to replicate using traditional 2D cell culture models that are frequently used for studying tumor biology and drug screening. This often leads to poor translation of results between in vitro and in vivo and is reflected in the extremely low success rates of new candidate drugs delivered to the clinic. Therefore, there has been intense interest in developing 3D tumor models in the laboratory that are representative of the in vivo tumor microenvironment and patient samples. 3D bioprinting is an emerging technology that enables the biofabrication of structures with the virtue of providing accurate control over distribution of cells, biological molecules, and matrix scaffolding. This technology has the potential to bridge the gap between in vitro and in vivo by closely recapitulating the tumor microenvironment. Here, a brief overview of the tumor microenvironment is provided and key considerations in biofabrication of tumor models are discussed. Bioprinting techniques and choice of bioinks for both natural and synthetic polymers are also outlined. Lastly, current bioprinted tumor models are reviewed and the perspectives of how clinical applications can greatly benefit from 3D bioprinting technologies are offered.
Keywords: biofabrication of 3D tumor models, bioinks, bioprinting, drug screening, personalized medicine, tumor microenvironments
3D bioprinting enables the creation of 3D tumor‐like constructs in the laboratory by loading a mixture of cells and biomaterials. This review provides an overview of key considerations of the complex tumor microenvironment and matrix design when developing 3D tumor models. In this review, 3D bioprinted tumor models that are developed are highlighted and the main challenges for their clinical application discussed.
![]()
1. Introduction
Cancer is a leading cause of death worldwide and its incidence and mortality continue to grow.[ 1 ] Advances have been made in our understanding of cancer biology and treatment for the past decades. Yet, the success rate of a new candidate drug that can pass all clinical trial phases in oncology is estimated to be only 3.4 %.[ 2 ] One reason for the poor translation rate of drugs to the clinic is the discrepancy of their efficacy between in vitro and in vivo. The current understanding of tumor biology is predominately based on 2D monolayer cell culture systems due to the simplicity of the models. However, cancer is a complex disease that involves uncontrolled physical and biological interactions between multiple cell types and their surrounding heterogeneous 3D extracellular matrix (ECM) within the tumor microenvironment.[ 3 ] Therefore, the tumor microenvironment has emerged as a significant determinant of tumor growth and progression. Animal models are better able to represent the tumor microenvironment than 2D plastic cultures. However, the clinical translatability of animal models has been questioned as the efficacy and toxicity of drugs evaluated in animal studies often fail to predict the response in human patients, especially when considering subcutaneous or orthotopic implantation of human tumor cells into immune‐compromised animals.[ 4 ] Further, there are ethical concerns regarding the use of animals in cancer research. Hence, to overcome these challenges, there is an urgent need to develop novel tumor model systems and technologies that can recreate the in vivo tumor microenvironment as closely as possible for better understanding of tumor biology and to bridge the gap between in vitro and in vivo systems to predict drug responses. In this context, fabrication of tumor models in 3D is evolving as an innovative approach for appropriately mimicking the tumor microenvironment.[ 5 , 6 ] 3D tumor models, such as spheroids, organoids, biopolymer scaffolds, and tumor‐on‐a‐chip, are more reflective of human tumor‐like features, including hypoxic regions and gradient distribution of chemical/biological factors.[ 7 , 8 ] In addition, these 3D models offer a higher chance to represent the genomic diversity, biomarker expression and pathological properties of the human tumors compared to 2D cell culture.[ 6 ] Thus, 3D models are able to exhibit reliable responses in tumor drug screening and predict chemoresistance.[ 9 , 10 ]
3D bioprinting is emerging as a widely used biofabrication technology in many areas, including tissue engineering and cancer research. 3D bioprinting is an automated process that involves precise positioning of bioinks on the layer‐by‐layer basis to build functional 3D living human constructs. The main bioprinting technologies include extrusion, droplet, laser‐based and stereolithography bioprinting.[ 11 , 12 , 13 , 14 , 15 ] Bioinks are cell‐laden solutions, containing living cells and biomaterials with biological compatibility, with various compositions and accessible bioprinting methods.[ 16 , 17 ] By utilizing bioprinting technologies and bioink compositions with diverse biological and mechanical properties, numerous 3D tumor models that can mimic the naïve tumor microenvironment have been developed for the study of tumor cell biology.
This review provides an overview on the current understanding of physicochemical and biological features of the tumor microenvironment, essential factors to be considered for the 3D bioprinting process. We further present critical discussion on the current uses of biomaterials, mimicking tumor microenvironment components, as well as 3D bioprinted tumor models and their limitations.
2. Key Characteristics of Tumor Microenvironment for 3D Bioprinting Tumor Models
One of the most important design parameters in fabrication of bioprinted tumor models is creating an appropriate environment, mimicking the naïve tumor microenvironment, as a tumor is not simply a mass of tumor cells. The tumor microenvironment is a highly complex realm where tumor cells are surrounded by various types of non‐malignant cells, including stromal cells, such as fibroblasts, endothelial cells, and adipocytes, and immune cells embedded within ECM (Figure 1 ).[ 18 , 19 ] Although the tumor microenvironment is heterogeneous between tumors, the major characteristics are conserved across different tumor types. In order to develop a reliable tool for cancer research, it is important to preserve the key characteristics of the tumor microenvironment when designing 3D tumor models. Each component of tumor microenvironment has previously been described in detail elsewhere and therefore we will provide a brief overview of those key characteristics.
Figure 1.
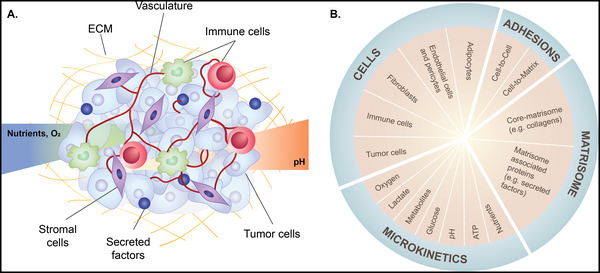
Tumor microenvironment components. A) Schematic illustration of tumor microenvironment and B) its components and characteristics. The tumor microenvironment is a highly complex realm where tumor cells are surrounded by stromal cells (fibroblasts, adipocytes, endothelial cells and pericytes) and immune cells within ECM. Tumor microenvironment characteristics that contribute to tumorigenesis, tumor progression and metastasis include altered expression of ECM and ECM‐associated proteins (matrisome), interactions of cell‐cell and cell‐matrix, nutrient availability, and gradient of metabolic waste, pH and oxygen (O2).
2.1. Cellular Components and Physiological Environments
Tumor cells coexist in a complex microenvironment with a diversity of stromal cell types, including fibroblasts, immune cells that can respond to infection and injury (e.g. lymphocytes, T‐cells, macrophages and mast cells), and cells comprising the vasculature (e.g. endothelial cells, pericytes and smooth muscle cells).[ 20 ] Although cancer is initiated by genetic mutations within the tumor cells, the reciprocal interaction between tumor cells and the stromal components plays a pivotal role in tumor progression and metastasis as well as in patient response to therapies.[ 18 , 21 ] Tumor cells release stimulatory chemoattractants to recruit non‐malignant cells that in turn secrete tumor‐promoting signaling molecules. These molecules regulate the expression of genes and proteins related to tumor metabolic pathways and remodel the ECM to create the tumor microenvironment.[ 3 , 22 ] Rapidly proliferating tumor cells increasingly demand a supply of oxygen and nutrients, resulting in hypoxic and acidic niches. This in turn activates angiogenesis, the abnormal blood vessel formation from the existing vasculature, in response to tumor cells and endothelial cells.[ 23 , 24 ] The inadequate supply or demand of oxygen then cause hypoxia.[ 25 ] Hypoxic niche is thus one of the prominent hallmarks of the tumor microenvironment, along with other specialized microenvironments, including acidic niche, suppressive immune microenvironment, metabolism reprogramming, innervated niche and mechanical microenvironment.[ 26 , 27 ] Tumor cells adapt to a hypoxic environment primarily through the transcriptional activity of hypoxia‐inducible factors that recruit the stromal cell components to the tumor microenvironment.[ 28 ] This leads to increased ECM remodeling and degradation, and acquisition of the epithelial‐mesenchymal transition phenotype in a number of tumors where tumor cells gain metastatic properties, including increased cell mobility, invasion and resistance to apoptotic stimuli.[ 29 , 30 ]
2.2. Extracellular Matrix (ECM)
Another key characteristic of the tumor microenvironment is the ECM, noncellular components present in all tissues. The ECM is composed of hundreds of proteins that provide not only structural and mechanical support (stiffness and elasticity) for the cellular constituents but also crucial biophysical and biochemical cues, influencing all aspects of cell biology in tissues.[ 31 , 32 ] ECM components actively bind cells via their adhesion receptors that transduce signaling pathways into cells from ECM and also various growth factors, cytokines and chemokines, affecting cell behaviors.[ 33 ] In healthy tissue, the loss of cellular contact from the ECM can be rapidly detrimental to cells, where cells die through a process known as “anoikis” or programmed cell death, due to the loss of adherence.[ 34 ] However, in tumor cells that can metastasize (spread to distant tissues), the loss of cell–cell and cell–matrix contacts can be a survival advantage as instead of dying, tumor cells would migrate and invade.[ 35 ] In tumor tissues, the dynamic interaction of the cells with ECM disrupts the balance between matrix deposition, remodeling, and crosslinking, which leads towards an increased quantity of ECM proteins and modifies the tensile and compressive strength of ECM. Consequently, tumors are typically stiffer than the surrounding healthy tissues.[ 36 , 37 , 38 ]
The advent of bioinformatic and proteomic strategies has enabled, over the years, to unravel the components of ECMs. In silico ECM proteome or matrisome, defined as an ensemble of ECM and ECM‐associated proteins, provides a platform for the analysis of physiological and disease‐specific patterns of ECM protein expression.[ 39 , 40 ] The matrisome components are classified into “core matrisome” encompassing the collagens, proteoglycans and glycoproteins and the “matrisome‐associated” components, including the regulators, affiliated proteins and secreted factors. The matrisome is commonly deregulated and expressed at significantly higher levels in human tumors compared with their normal tissue counterparts. Recent pan‐cancer matrisome analysis studies demonstrated that different tumor types exhibit similar but distinct matrisome gene expression.[ 41 , 42 , 43 ] Of those, Izzi et al. evaluated the levels of matrisome gene expression in 10 487 patients across 32 tumor types using The Cancer Genome Atlas (TCGA) data and showed that gene expression of the matrisome can distinguish different tumor types.[ 41 ] Thus, compositions of the ECM are markedly heterogenous and tissue and disease specific. Together, for biofabrication of 3D tumor models, it is important to incorporate appropriate cell types and understand the composition of the ECM in different tumor types as well as their disease stages to accurately capture tumor‐type specific ECM components in vitro.
3. Simulating the Matrix Components for Biofabrication: Bioprinting Technologies and Biomaterials
In the following section, we discuss bioprinting technologies and biomaterials currently available and their limitations for the biofabrication of 3D bioprinted tumor models.
3.1. Bioprinting Technologies
To fabricate 3D tumor models, four major bioprinting techniques including droplet‐based bioprinting (DBB), extrusion‐based bioprinting (EBB), laser‐based bioprinting (LBB), and stereolithography bioprinting (SLB) have been used. Each bioprinting technique has previously been described in detail elsewhere.[ 11 , 12 , 13 , 14 , 15 ] Here, we will thus summarize the advantages and limitations of each bioprinting technique. EBB uses a pneumatic‐, mechanical‐, or solenoid‐based system to deposit cells with high density in the form of a cylindrical filament.[ 15 ] This modality has been the most used for fabricating 3D cellular structure, due to its affordability and availability of a wide range of compatible biomaterials. However, high cell death could occur due to high extrusion pressure and shear stress during extrusion.[ 14 ] DBB employs the use of viable cells together with low viscosity bioinks to deposit droplets of cells with great control over the deposition pattern and bioink volumes. The advantages of DBB include high cell viability and the ability to generate 3D constructs in a high throughput (HTP) manner, despite the potential non‐homogenous droplet size and nozzle‐clogging issues with high density of cells and bioinks.[ 13 , 14 ] Choice of biomaterials is also limited due to viscosity constraints. LBB, on the other hand, is a nozzle free bioprinting technology that utilizes a pulsed laser for precise patterning and deposition of cells.[ 44 , 45 ] Laser energy is used to irradiate a ribbon coated with a laser absorbing layer that is covered with liquid bioink.[ 46 ] The bioink at the site of irradiation evaporates, which results in the generation of an expanding bubble on the surface between the ribbon and the liquid bioink. This expansion creates a droplet of bioink to fall on the receiving substrate. While LBB has the advantages of high‐spatial resolution patterning, the overall precision and ability to dispense viscous or small volumes is slower than DBB and EBB.[ 45 ] SLB utilizes a micromirror array to photopolymerize a polymer resin in the presence of cells.[ 47 ] There are no limitations on the polymer's viscosity as long as it is light sensitive, photo‐crosslinkable and has a melting temperature lower than room temperature or the processing temperature.[ 47 , 48 ] As a printing method, SLB boasts good accuracy, fast fabrication speeds, and high cell viability (>85 % viability).[ 49 , 50 , 51 ] However, this method often leaves residual photo‐initiators, such as Irgacure 2959, Irgacure 184, and Irgacure 651.[ 47 ] Furthermore, the harmful effects of UV light on cells, which is common in stereolithography, cannot be ignored.
3.2. Biomaterials
The primary roles of biomaterials in biofabrication are to maintain cellular homeostasis and to provide cues for cell adhesion, proliferation and differentiation, as well as the structure and mechanical properties for cell migration and polarity, and morphogenesis.[ 52 ] Such biomaterials used in bioinks for 3D bioprinting include natural polymers and synthetic hydrogels. In order to select biomaterials for bioprinting, its degradation rate should not be faster than the rate of cell proliferation and ECM synthesis, allowing it to be replaced without compromising the structural integrity. Stiffness mimicking original tissues (102–107 Pa as determined using shear rheology, compression testing, or tensile testing; Table 1 and Figure 2 ) is also an integral factor that has been established to play a role in cell function and must be considered when selecting a biomaterial. In general, their stiffness can be easily tuned by adjusting concentration. The biomaterials should be able to mimic the mechanical properties of the tumor tissues in simple 3D tumor models. As the ECM stiffness can change during tumor progression, there is an opportunity to develop complex 3D tumor models to study tumor metastasis where the tumor cells grow in the stiff tumor‐like matrix and are surrounded by a normal tissue‐like matrix. In these 3D metastasis models, when cancer cells are cultured within the matrix representing the in vivo tumor‐like environment, cells will proliferate within the matrix. As cells digest the surrounding matrix and replace it with their own matrix, tumor cells should be placed in a matrix with a stiffness that mimics the native tissue. For 3D bioprinting fabrication, the printability and structure fidelity are determined by the viscosity, speed of gelation and crosslinking methods. These are closely tied to the concentration and type of components of the biopolymer.[ 53 ] The method of cell integration prior to the final structure formation should also be properly designed. Another important factor to be considered when selecting biomaterials is hydrogel crosslinking methods, such as chemical, physical, ionic, visible light and UV crosslinking as each bioprinting method has different requirements.[ 53 , 54 ] In addition, thermal and shear properties of a polymer must also be considered as the printability of a biomaterial depends highly on the chosen bioprinting method.[ 16 ] The various types of polymers and their compatible printing methods have been well documented in the literature.[ 55 , 56 ] Here, we will briefly describe natural and synthetic polymers.
Table 1.
Tissue stiffness reported in the literature
| Tissue | Storage Modulus [Pa] | Young's modulus [Pa] | Refs. |
|---|---|---|---|
| Brain | 200 | 1000 | [57, 58, 59] |
| Lung | 800 | 3000 | [60, 61] |
| Breast | 1000 | 1000 | [62, 63] |
| Liver | 3000 | 3000 | [64, 65] |
| Heart | 10 000 | 100 000 | [66, 67] |
| Skeletal muscle | 15 000 | 25 000 | [67, 68, 69] |
| Cartilage | 900 000 | 1 200 000 | [70, 71] |
| Bone | 12 000 000 | 14 000 000 | [70, 72, 73] |
Figure 2.
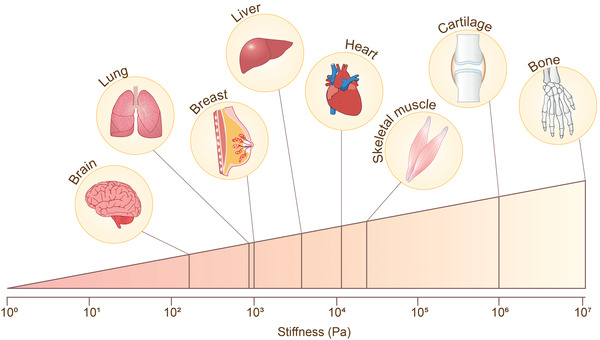
Stiffness of human tissues.
3.2.1. Natural Polymers
Many natural polymers have been used in 3D bioprinted tumor models. These include collagen, fibrinogen, alginate, gelatin and hyaluronic acid.[ 74 ] Collagen is commonly used for many tissue fabrication applications due to the advantage of its biomimetic properties, such as cytocompatibility, low‐antigenicity when in the high purity form and native viscoelastic properties (0.5–8 mg mL‐1 has 20–1000 Pa storage modulus).[ 75 , 76 , 77 ] While many collagen types can also be found commercially (e.g. type I, II, III, IV and V), the majority of printable collagen bioinks are produced from collagen type I.[ 51 , 78 ] However, collagen gels with high stiffnesses are difficult to generate without substantial crosslinking that impacts their degradability.[ 79 ] Collagen is also temperature‐dependent, has limited long‐term stability, and suffers from batch‐to‐batch variability.[ 79 ] Alginate is a cytocompatible anionic polymer with low viscosity derived from brown algae and is commonly used in biofabrication technology. It is an inert passive gel, meaning that it does not support cell adhesion or proliferation and has no biodegradability. However, its commercial availability and wide range of stiffness (up to 3 mPa) makes it a popular bioink choice for 3D bioprinting. Due to its passivity, the polymer is often modified or mixed with other biomaterials, such as gelatin that have good cell adhesion and biodegradable properties.[ 74 , 80 ] Gelatin is another widely used biopolymer for bioprinting. It is a water soluble protein derived from collagen that is denatured through irreversible hydrolysis.[ 81 ] As gelatin is thermo‐sensitive, the viscosity of gelatin‐based gels can be easily modified by altering temperature or concentrations of the polymer. Gelatin is commonly reacted to methacrylic anhydride to produce GelMA that is a popular choice in bioprinting.[ 82 , 83 , 84 ] GelMA is generally more compatible with EBB bioprinting than other natural polymers as it has superior control over its crosslinkability, due its on‐demand photo‐crosslinking mechanism.
Natural polymers can also be extracted from tissue that are especially rich in ECM components. Decellularized ECMs (dECMs) are harvested by removing all cellular components from various tissue sources, such as the heart, skin, intestine, and tendons using physical and chemical methods.[ 74 ] The dECM can better offer intrinsic native properties of the tumor ECM microenvironment than synthetic hydrogels. In 3D bioprinting, they can be extruded through the deposition nozzles in filamentary form, which then undergo gelation at physiological temperature.[ 85 ] Several 3D bioprinted tumor models using dECM bioinks have been developed to recapitulate the complexity of the tumor microenvironment, including hepatocellular carcinoma using liver dECM bioinks and glioblastoma‐on‐a‐chip bioprinted with the dECM bioink extracted from normal porcine skin or brain.[ 86 , 87 , 88 ] Despite the benefits of the decellularized ECM mimicking the original tumor matrix with high complexity, they may disproportionately lose some of their components, such as the glycosaminoglycans during common decellularization techniques.[ 89 ] Furthermore, hydrogels formed from dECM is also known to have poor mechanical properties and therefore physical, photochemical or chemical crosslinking methods have been implemented to tune their mechanical properties, such as the physical stability and stiffness.[ 86 , 90 ] It is also important to use appropriate dECM (e.g., tumor tissue rather than healthy tissue and tumor type specific tissue), as tumor cells can behave differently under the influence of different ECMs, which may subsequently dramatically alter the behavior of the cell signal network.[ 52 ] Matrigel is a basement membrane extract from mouse Engelbreth‐Holm‐Swarm tumor cells, primarily composed of laminin, collagen IV, and proteoglycans with a reported porosity of ≈2 µm (50 % Matrigel) and storage modulus of 10–50 Pa (50–100 % Matrigel).[ 91 ] Nearly 2000 unique protein components have been identified in a collection of batches of Matrigel.[ 92 ] This level of complexity is comparable to the composition of in vivo tissues. Thus, Matrigel has widely been used as substrate for 3D tumor models, providing more accurate representation of tumor tissues than conventional 2D monolayer culture. However, the physical properties of Matrigel hinder its printability. Matrigel crosslinks at room temperature, which means that a temperature regulated system is required to prevent unwanted gelation. Due to the low storage modulus values, Matrigel is not ideal for bioprinting complex structures and very poorly compatible with extrusion based printing.[ 93 ] Matrigel also suffers from lot‐to‐lot variations and ill‐defined compositions, a property also common to decellularized ECM. This hampers the reproducibility and the precise control over chemical and mechanical properties of those natural bioinks for bioprinting.[ 94 ]
3.2.2. Synthetic Hydrogels
Synthetic hydrogel‐based 3D matrices provide the unique advantage of being programmable to address distinct structural, biological, biochemical, porosity conditions and mechanical properties for specific cell growth and movement.[ 16 ] While the synthetic biomaterials lack the intrinsic cytocompatibility and complexity of natural polymers, the properties of synthetic hydrogels can be precisely tuned in terms of stiffness, porosity, and the expression of bioactive components, such as cell adhesive ligands, enzyme degradable linkers and growth factors. The tunable composition and properties of such biomaterials are particularly important for not only the efforts to standardize the cellular environment for 3D cell culture applications, but also to make possible a reductionist approach to evaluating the importance of different factors in the ECM on cellular responses. It is this tunability that allows synthetic hydrogels to often outperform natural polymers.[ 52 , 79 ]
Polyethylene glycol (PEG) is one of the most highly tunable synthetic hydrogels. It is biologically inert and non‐immunogenic, owing to the minimal adsorption of protein to PEG but can be rendered compatible with cell growth. This is generally achieved through chemical crosslinking to various user‐defined peptide chains necessary for cellular adhesion, such as collagen, fibrinogen and hyaluronic acids.[ 95 ] Moreover, these hydrogels can be polymerized using chain‐growth, step‐growth, or mixed mode processes, to generate linear and branched structures with 3,4 (commonly used as bioink) or 8 arms and offer more design flexibility.[ 79 , 96 ] PEG can also be modified to the photocrosslinkable PEG‐DA and methacrylated PEG (PEG‐MA) for better mechanical properties.[ 16 , 48 , 97 ] PEG has been shown to be useful in a wide range of cell culture applications. Ready‐made functionalized PEG polymers tailored to different tumor types are already available in the market in the form of assay kits for biology models and HTP drug screening by a Swiss group QGel bio Inc (www.qgelbio.com). However, cell seeding with these gels has been more oriented to liquid handling systems rather than 3D bioprinting systems. One of the limitations using PEG hydrogels is its non‐biodegradable nature by cells, which can be improved by modifications of the gels to have biodegradable segments.[ 98 ] In addition to the PEG‐based hydrogels, polyester‐based biodegradable polymers, including polycaprolactone (PCL), polylactic acid (PLA) and polylactic‐co‐glycolic acid (PLGA) have been used in bioprinting and described in detail elsewhere.[ 17 , 96 ] Of these, PLA and PLGA are also approved by the FDA and the European Medicine Agency in drug delivery systems.[ 99 ] Together, 3D tumor models can be fabricated with bioinks of living cells and biomaterials via various 3D bioprinting technologies (Figure 3 ).
Figure 3.
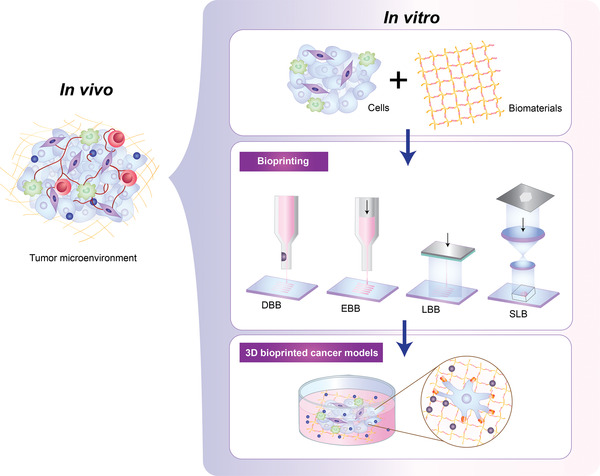
3D bioprinting process to generate a 3D tumor model mimicking the tumor microenvironment. 3D bioprinting enables the fabrication of 3D tumor models in the laboratory. A mixture of cells and biomaterials with high cytocompatibility and diverse compositions (a bioink) are loaded onto 3D bioprinting to fabricate tissue‐like structures via various technologies including, droplet‐based (DBB), extrusion‐based (EBB), laser‐based (LBB) and stereolithography (SLB) bioprinting.
4. Biofabrication and 3D Bioprinting Applications in Tumor
In this section, we describe some of important applications of bioprinting to fabricate tumor models.
4.1. 3D Bioprinted Tumors for the Study of Tumor Biology and Drug Discovery
Tumor spheroids or tumoroids are scaffold‐free 3D self‐assembled aggregates of tumor cells. In addition to immortal tumor cell lines, tumoroids are generated with tumor cells derived from patient‐specific tissue samples that mimic key pathophysiological features of the parent tumor.[ 100 ] These patient‐derived tumoroids are also known as tumor organoids. Currently, the tumoroid model is one of the most promising approaches to study personalized medicine. One aspect that must be considered for the successful generation of tumoroids is the quality of the 3D culture protocol, related to relevant variables, such as cell seeding density, culture environment (e.g., culture media, the amount of CO2 and O2 available), and the tumoroid formation technique. In tumoroid cultures, cells produce and secrete their own ECM components to reside and form a mini‐tumor like construct. This 3D tumor model resembles the complexities of tumors, including cell–cell and cell–ECM interactions, regions of hypoxia and zones of proliferating and quiescent cells.[ 101 ] Various methods have been developed for generating tumor spheroids. Simple and straightforward methods are the forced floating method, using round‐bottom or nonadherent culture plates, and the hanging drop method using gravitational force, both of which suppress cell–substrate interaction.[ 102 ] Despite the ease of use, these methods require the manual seeding of cells, which is labor intensive, can result in nonuniform cell seeding and spheroid shapes and lacks the throughput to ensure statistically significant amounts of experimental data. Moreover, these classic 3D tumoroid culture techniques do not allow for exact spatiotemporal control of various factors in the tumor microenvironment. Thus, new platforms based on biofabrication, such as 3D bioprinting, enable for the rapid generation of the 3D tumor models with more homogeneous size and shapes in a HTP manner for studies of tumor biology and development of anti‐tumor drugs.
In order to produce tumoroids with controlled sizes and shapes, various strategies of 3D bioprinting have been developed. One approach is to bioprint cell‐embedded hydrogel arrays. Ling et al. bioprinted sacrificial gelatin arrays with MCF‐7, a breast tumor cell line, and sequentially concave wells with PEG‐DA onto a culture petri‐dish through pressure‐assisted bioprinting based on EBB.[ 103 ] Similar concave PEG‐DA hydrogel structures for breast tumor spheroid culture were bioprinted and fabricated using nonlinear UV light exposure onto glass coverslips.[ 104 ] In both studies, uniform distribution of cells between concave wells was achieved. In long term culture, compacted single spheroids, displaying hypoxic cores with the presence of necrotic cells were observed. In another study conducted by Kingsley et al., LBB was applied to fabricate and spatially pattern cell‐containing chitosan‐shelled alginate structures (termed core‐shell microbeads in the study).[ 45 ] Within the chiton–shell alginate structures that were bioprinted in petri‐dishes, cells self‐assembled to from spheroids over a 14 day time course. The core‐shelled structures of different sizes, ranging from 200 to 400 µm produced spheroids of different sizes, which exhibited spatial heterogeneity in transferrin ligand uptake within larger tumor spheroids. Hakobyan et al. created square arrays of pancreatic cell spheroids from 10 by 10 cell suspension spots spaced by 750 µm using LBB.[ 105 ] In their study, the AR42J‐B‐13 rat acinar cell line, a classic model of acinar‐to‐ductal metaplasia was bioprinted in GelMA hydrogels and grew to form spheroids with a diameter around 30 µm. The rat model can address the full complexity of the pancreatic tumor initiation to facilitate development of new therapies for human tumor. The close resemblance of 3D bioprinted tumoroids with in vivo tumor conditions enables for such a model to be an efficient drug screening tool. However, the tumoroids bioprinted on petri‐dishes or glass coverslips still lack the suitability to be a HTP drug screening platform.
Recently, we developed a bioprinting strategy, in which alginate‐based cup structures with cell‐embedded hydrogels were bioprinted in a 96‐well plate via a drop‐on‐demand DBB, allowing for reliable production of 3D tumor spheroids in a HTP manner.[ 106 ] In the study, we successfully generated tumor spheroids of three different tumor cells of neuroblastoma, non‐small cell lung cancer and glioblastoma. Tumor spheroids grew to conform to the shape of the cups, which highlights the capability of the bioprinter to produce single spheroids with a controlled shape by matching cup size, shape, and cell volume, enabling HTP drug response analysis (Figure 4 ).[ 106 ] Another approach for the generation of HTP tumoroids developed by Maloney and co‐workers is an immerse printing technique using EBB, which bioprints tumor cells mixed with hyaluronic acid and collagen hydrogels into a viscous gelatin bath that provides structural support to form spheroids in 96‐well plates.[ 107 ] In their study, in addition to tumor cell lines, the capability of this printing technique to fabricate tumor organoids was demonstrated using glioblastoma and sarcoma patient biopsies for drug screening, underscoring the potential of bioprinting for tumor organoid generation. A simple method of 3D bioprinted multitumoroids of cervical cancer was generated using EBB with HeLa cells premixed with gelatin/alginate/fibrinogen where a grid structure with the HeLa/hydrogel construct was designed for improved nutrient, oxygen and metabolic waste transport.[ 108 ] Those HeLa cells formed multiple tumor spheroids in the ECM mimics showed higher matrix metalloproteases expression and chemoresistance than cells in 2D culture. Bioprinting of pre‐formed tumoroids for rapid production of a tumor model was investigated by Swaminathan and co‐workers.[ 109 ] Breast tumoroids manually prepared in Matrigel were directly bioprinted into a grid‐structure with interconnected channels made of alginate/collagen or alginate/gelatin via EBB. This study validated the capability of printing spheroids without loss of a viability or morphology in monoculture or co‐culture with HUVEC endothelial cells. However, additional biofabrication steps should be considered to control spheroid size, allowing spheroids to fall within a specific size range. Reid et al. demonstrated that breast tumoroids can be generated by bioprinting clusters of a small number of breast tumor cells (10–40 cells per structure) into equally spaced locations inside collagen gels that were precoated in 24 well plates using EBB.[ 110 ] Normal breast cells were bioprinted near tumor cells in separate print runs, which further fused to form a single layer of mammary cells and developed contiguous lumen circle, mimicking the in vivo scenario of primary breast cancer. The study showed the capability of the bioprinting platform to generate chimeric mammary organoids by precisely placing different types of cells.
Figure 4.
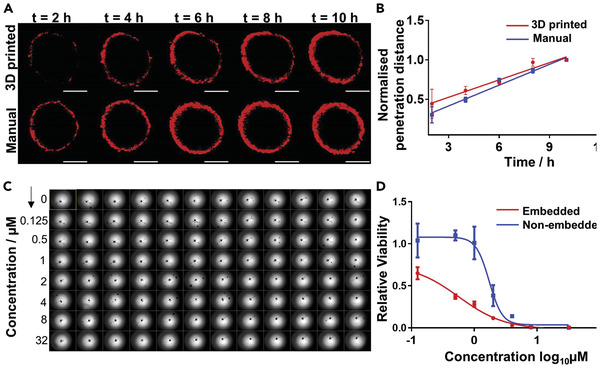
Bioprinted neuroblastoma SK‐N‐BE(2) cell spheroids for HTP drug discovery. A) Confocal microscopic images of the penetration of doxorubicin (red) into the bioprinted and manually prepared spheroids. Scale bars, 200 µm. B) Penetration depth profile of doxorubicin into bioprinted and manual spheroids over 10 h. Over time, doxorubicin slowly penetrates and accumulates in the spheroids in both recovered 3D bioprinted and manual samples. C) Image of a full 96‐well plate of 3D bioprinted embedded spheroids before doxorubicin treatment. D) Dose‐response curve of bioprinted spheroids, embedded in the hydrogel matrix and non‐embedded. The presence of a hydrogel matrix increased doxorubicin sensitivity of the spheroids compared with non‐embedded ones, possibly due to the hydrogel matrix accumulating the drugs from the media into the inner gel region and thus increasing the availability of the drug to the spheroid. Adapted with permission.[ 108 ] Copyright 2014, Elsevier.
4.2. Recapitulating Tumor Microenvironment: Coculture, Vascularization, and Metastasis
In addition to spheroids, the 3D bioprinting technology permits modeling the complexity of the tumor microenvironment by printing multiple cell types with precise control on their positioning. An ovarian tumor model was created using a simple DBB method, in which each droplet of ovarian tumor cells and normal fibroblasts were bioprinted within spatially controlled locations onto precoated Matrigel.[ 111 ] This method overcomes the limitation of manual coculture systems, which lack the precise cell position within a biologically relevant scale. Another recent study by Hwang et al. established a HTP bioprinting platform using GelMA hydrogels and a digital light processing‐based system.[ 112 ] This bioprinting platform was capable of printing dual cell‐type populations of Hep G2, a hepatocellular carcinoma cell line and HUVEC endothelial cells in a HTP manner. The authors showed that up to 96 constructs per batch can be generated in standard well plates, suggesting potential utility of 3D bioprinted tissue constructs in drug discovery process. Yi et al. incorporated microfluidic technology with EBB to bioprint glioblastoma multiform (GBM)‐on‐chips that combined a compartmentalized tumor‐stroma structure generated with GBM cells and endothelial cells, an oxygen‐gradient‐generating system, and porcine brain decellularized ECM.[ 88 ] The GBM cells grew in a highly dense spherical shape surrounded by endothelial cells within the printed concentric‐ring structure, mimicking the hypoxic tumor microenvironment. Importantly, the compartmentalized structure of the GBMs‐on‐chip printed with patient derived tumor cells showed differential therapy responses that replicated patient‐specific treatment responses to concurrent chemoradiation using temozolomide, the standard first‐line treatment. Such a microfluidics‐based tumor model allows for analyzing the dynamic interactions of cells to an oxygen gradient, rather than the functional analysis of entrapped cells.[ 113 ]
A plethora of recent work has shown that the advancement of 3D bioprinting technology can fabricate more complex, in vivo‐like tumor models. Tang et al. fabricated a tumor model of 3D GBM that recapitulates the biophysically heterogeneous ECM microenvironment of the disease using GBM patient‐derived xenograft (PDX) cells and GelMA‐hyaluronic acid hydrogels via SLB.[ 114 ] Tumor regions created with a diameter of 500 µm were separated from endothelial regions by a donut‐shape acellular ECM region that was soft (2 kPa) or stiff (21 kPa) (compressive modulus), representing healthy brain parenchyma and GBM stroma respectively (Figure 5 ). The authors observed that rapid cell proliferation predominantly occurred in soft ECM while in stiff conditions, the malignant phenotypes of tumor cells including hypoxia, stemness, invasive and angiogenic potentials, and drug resistance, were enhanced. Moreover, the 3D bioprinted GBM models showed a dramatically distant transcription profile when compared to the classic 3D sphere culture of GBM PDX, indicating the effects of the biophysical cues from the ECM on tumor development.
Figure 5.
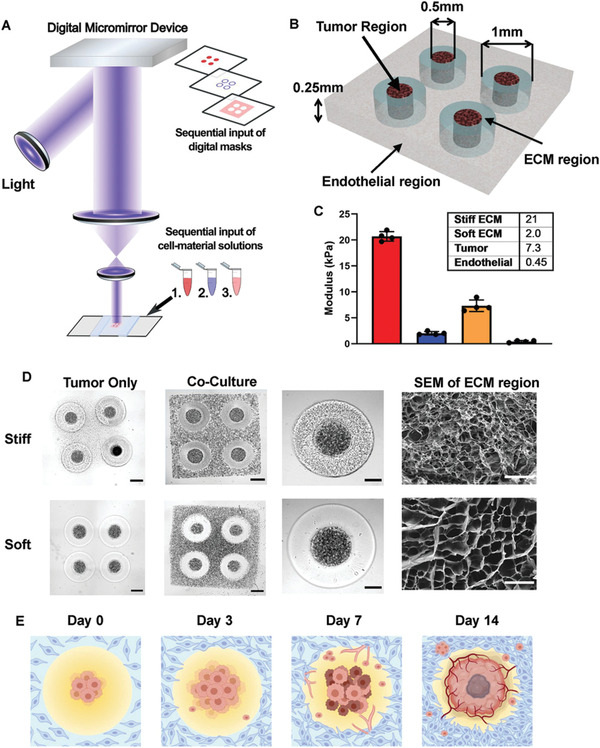
3D‐bioprinted GBM models with regionally varied biophysical properties. A) Schematic diagram of a multistep digital light processing‐based bioprinting approach. B) Illustration of the model dimensions. C) Stiffness of each region in the 3D‐printed model. Bar charts represent mean compressive modulus of each region ± standard deviation. n = 4 technical replicates per group. D) Representative bright field image and scanning electron microscopy of the 3D printed models with stiff (upper row) and soft conditions (lower row) on day 0. (Scale bars from left to right: 500 µm, 500 µm, 250 µm, 50 µm.) E) Illustration of the timeline of tumor development and angiogenesis event in the 3D‐bioprinted models. Reproduced with permission.[ 114 ] Copyright 2021, Wiley‐VCH GmbH.
In another study, a mini‐brain was fabricated with GelMA/gelatin hydrogels using an EBB method to model the brain tumor microenvironment. The miniaturized brain was created via a two‐step bioprinting process, in which a larger brain model encapsulating macrophages with an empty cavity was first bioprinted and GBM cells were subsequently bioprinted to fill the cavity.[ 115 ] Within the mini‐brain model, GBM cells actively recruited macrophages, which activated both cell types by upregulating GBM marker genes. Further testing of the inhibitory effect of drugs, including a common chemotherapy for GBM and two other immunomodulatory drugs, suggests that the model can act as a platform to test drugs in the context of 3D brain tumors. In seminal work by Langer et al., a tumor tissue, measuring approximately 2 mm × 2 mm × 1 mm, was bioprinted using EBB onto transwell membranes, in which a core tumor cell compartment was surrounded by a normal stromal cell region.[ 116 ] Alginate‐based hydrogels were used as a sacrificial scaffold, which was removed 48 h after printing, allowing for the formation of a scaffold‐free tumor model and the effective integration of additional cell types into the tumor region. They also showed that breast and pancreatic tumor models can be created by independently manipulating the microenvironment containing multiple stromal cell types (fibroblasts, endothelial cells, adipocytes or mesenchymal stem cells) relevant to their in vivo tumors, which could respond accordingly to extrinsic signals or therapeutic treatments. Moreover, the capability of bioprinting primary patient or PDX tumor cells demonstrated in the study offers great promise for improved personalized medicine. van Pel and co‐workers created a scaffold‐free tumor model for GBM using “Kenzan,”, a microneedle‐based method.[ 117 ] Neurospheres of pluripotent stem cells (iPSC)‐derived human neural progenitor cells were precultured in 96 well U‐bottom plates robotically placed in the microneedle arrays. Preformed U118 human glioma cell spheroids were subsequently printed on the top of the neural organoid to monitor glioma cell invasion into neural‐like tissue, mimicking the in vivo invasive niche. Despite the potential of the microneedling method for fabrication of various scaffold‐free tumor models, due to the fixed interneedle distance, the size of usable spheroids falls in a relative narrow range.[ 118 ]
Tumor vasculature is closely related to tumor growth and metastasis. 3D bioprinting techniques have offered new possibilities in advancing cancer research by creating vascularized tissues. A simple 3D bioprinted vascularized tumor for drug testing was designed by Han et al.[ 119 ] A blood vessel layer consisting of endothelial cells and fibroblasts was bioprinted with a mix of gelatin, alginate and fibrinogen via EBB. Once lumens were formed in the microvessels, glioblastoma spheroids (U87) that were pre‐assembled in concave wells were manually pipetted onto the vascularized tissues in this model. The authors observed tumor spheroid invasion into the vascularized tissues and neovascularized structures infiltrated into the tumor spheroids. While the printability of the structures within 24 well plates suggests the potential of the platform for HTP screening, the manual transfer of tumoroids to the bioprinted blood vessel layers may hinder its reproducibility. In another study conducted by Kim et al., a cancer‐vascular platform enabling precise positioning of tumoroids and vessel‐like structure was fabricated using EBB.[ 87 ] One‐step fabrication of 3D tumor spheroids was achieved by bioprinting a high density of melanoma cells with porcine skin‐derived decellularized ECM (SdECM) bioink at pre‐gel state in the predefined location. The printed melanoma tumoroids with 600 µm diameter could recapitulate the hypoxia and angiogenic signaling of tumor. The vessel‐like structure was fabricated with HUVEC cells in SdECM bioink, using a coaxial cell printing tube fabrication technique. A sophisticated vascularized tumor model was recently developed with precise spatial control via EBB to mimic key steps of metastasis including invasion, intravasation, and angiogenesis.[ 120 ] The model included the tumor cell droplet as the primary tumor and endothelialized microchannels, which were constructed onto the fibroblast‐laden fibrin matrix as tumor stroma (Figure 6 ). In the model, tumor cell invasion of the surrounding matrix and intravasation into the vasculature were guided by signaling molecule gradients, which were dynamically generated within 3D hydrogel matrices via controllable release after laser irradiation. They designed two built‐in chambers located at each end of the fibrin gels to measure the levels of growth signal molecules (EGF; epidermal growth factor and VEGF: vascular endothelial growth factor). The versatility and selectivity of the platform were demonstrated by testing multiple tumor cell lines derived from different tumor types, including lung carcinoma and melanoma. While the platform has great potential to be utilized to study fundamental biology related to metastasis, limitations of using tumor and stromal cell lines as well as the requirements for specially designed culture chambers and laser irradiation for capsule rupturing could hamper its applicability and practicality as a preclinical drug screening platform in the clinic.
Figure 6.
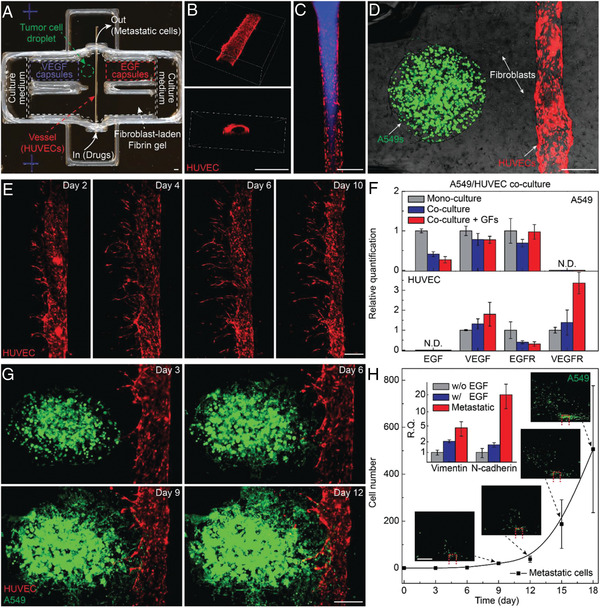
Metastatic tumor model. A) Photo of a 3D printed culture chamber for tests of guided tumor cell dissemination. B) Confocal images of the top view (upper panel) and cross section (lower panel) of a representative microchannel lined by HUVECs within a fibrin gel. C) Fluorescence images showing a vessel perfused by fluorescent fluid (blue). D) Composite image showing a representative tumor model before laser‐triggered rupture of EGF and VEGF capsules (green fluorescence: GFP‐expressing A549 lung carcinoma cells, red fluorescence: RFP‐expressing HUVECs, bright field: fibroblasts). E) Panoramic fluorescence images showing sprouts generated from a main vessel and their extension toward a single direction over. F) Bar chart showing the expression of EGF, VEGF, EGFR, and VEGFR of A549s (upper panel) and HUVECs (lower panel) when mono cell‐cultured and co‐cell‐cultured. G) Fluorescence images of a metastatic model, showing that A549s approach and enter the vasculature through the fibroblast‐laden fibrin gel. H) Plots of the population of disseminated A549s detected in the collection chamber versus time. Inset photos: fluorescence images showing the disseminated A549s in the collection chamber, with the red dash frame showing the vessel position. Inset bar chart: expression of vimentin and N‐cadherin of 3D bioprinted A549s in tumor models without (gray) and with (blue) EGF release, and metastatic tumor cells (red) harvested in collection. Scale bar: 500 µm. Reproduced with permission.[ 120 ] Copyright 2019, Wiley‐VCH GmbH.
Clinically, rapid drug screening and prediction of treatment options for patients requires 3D bioprinted tumor models that can support the viability and growth of patient‐derived cells, are easy to generate and straightforward. Neufeld et al. recently proposed a 3D‐bioprinted engineered tumor model incorporating a penta‐culture of patient‐derived glioblastoma tumor cells and stromal cells as well as a perfusable vascular network (Figure 7 ).[ 121 ] Their 3D engineered tissue constructs are composed of two compartments, tumor/stroma and blood vessels. The main tumor compartment in the tumor model was created by bioprinting natural polymers fibrin bioinks sharing similar mechanical properties of glioblastoma tumor and containing patient‐derived glioblastoma cells, astrocytes and microglia. The vascular bioink composed of the thermo‐reversible cytocompatible synthetic polymer, Pluronic F127 and HUVEC cells was bioprinted to create a pattern resembling 3D lumen vascular structure on top of the fibrin 3D glioblastoma‐stroma bioink. The drug responses and transcriptomic expression profiling of patient‐derived glioblastoma cells cultured in the perfusable 3D tumor model were shown to resemble the in vivo tumor settings better than 2D models.[ 121 ] Importantly, in this study the authors performed thorough calibration and optimizations for protocol standardization regarding the mechanical properties of the bioink, cell seeding density and viability for multiple cell types and origins as well as the ratio between tumor cells and microenvironment cells. The standardization protocol will provide the starting parameters for bioprinting, which can facilitate the use of the 3D bioprinted models in the clinic. While these recently developed tumor‐vasculature models were successfully fabricated to mimic key steps of tumor metastasis, all these bioprinted platforms still have challenges for HTP production and drug screening for the identification of novels drugs targeting metastatic processes. A summary of the 3D bioprinted tumor models discussed here is presented in Table 2 .
Figure 7.

Fibrin brain‐mimicking 3D‐bioink integrated with 3D engineered printed perfusable vascular network. A) Schematic illustration of the 3D‐bioprinting model multistage process. B) 3D‐printed Pluronic‐based vascular bioink (in cyan) on top of 3D‐printed layers of fibrin 3D glioblastoma (GB)‐stroma bioink (in white). C) 3D‐bioprinted vascularized GB model sealed in a metal frame showing the complete perfusion chip. D) The vascularized 3D‐bioprinted GB model is connected to a peristaltic pump through a tubing system, placed in a designated incubator. E) Tiled Z‐stack confocal microscopy images of the 3D‐printed penta‐culture vascularized GB model. Blood vessels are lined with iRFP‐labeled hPericytes (in cyan) together with mCherry‐labeled HUVEC (in red) and surrounded by azurite‐labeled patient‐derived (PD)‐GB4 (in blue), GFP‐labeled human astrocytes (hAstro) (in green), and nonlabeled human microglia (hMG). The dashed box represents a coronal cross‐sectional plane of the vessel. F) Fluorescence microscopy images of the 3D‐bioprinted vascularized GB model before (top) and after (bottom) perfusion of 70‐kDa dextran‐FITC. The 3D‐bioprinted model is composed of a fluorescently labeled vascular network (mCherry‐labeled HUVEC and iRFP‐labeled hPericytes) surrounded by nonlabeled GB‐bioink (hAstro, PD‐GB4, and hMG). Reproduced with permission.[ 121 ] Copyright 2021, American Association for the Advancement of Science (AAAS).
Table 2.
3D bioprinted tumor models (Abbreviations: PEG‐DMA PEG‐dimethacrylate; GelMA methacrylated gelatin; PLGA poly(lactic‐co‐glycolic) acid; PEVA A poly(ethylene‐co‐vinyl acetate); NA not applicable
| 3D bioprinted model | Tumor model type | Cell type | Scaffold | Bioprinting modality | Printing structure | Printing substrate | Refs. |
|---|---|---|---|---|---|---|---|
| Tumoroids | Breast | MCF‐7 | Sacrificial gelatin and PEG‐DMA | EBB | Concave structure | Petri‐dish | [103] |
| Breast | BT474 | PEG‐MA | SLB | Concave structure | Glass‐cover slip | [104] | |
| Breast | MDA‐MB‐231 | Alginate/Chitosan | LBB | Microbeads | Petri‐dish/Glass bottom chamber plates | [45] | |
| Pancreatic cancer | AR42J‐B‐13 | GelMA | LBB | Microdroplet | Glass slides | [105] | |
| Neuroblastoma, Lung cancer and Glioblastoma | SK‐N‐BE(2), H460 & U87vIII | Alginate | DBB | Cup structure | Multi‐well plate | [106] | |
| Liver & colorectal cancers | HepG2 & Caco2 | HA/Collagen and gelatin bath | EBB | Immersion into a gelatin bath | Multi‐well plate | [107] | |
| Cervical cancer | HeLa | gelatin/alginate/fibrinogen | EBB | A grid structure | Petri‐dish | [108] | |
| Co‐culture tumor models | Ovarian cancer | OVCAR5 & MRC5 | Matrigel for pre‐coating petri‐dish | EBB | Two droplets of OVCAR5 (ovarian cancer) and MRC5 fibroblasts on Matrigel coated wells | Petri‐dish | [111] |
| Liver Cancer | HepG2 and/or HUVEC | PEG‐DA/GelMA | Digital Light‐based | Constructs of varying spatial geometries | Multi‐well plate | [112] | |
| Glioblastoma | U87 & HUVEC | Decellularized ECM | EBB | Tumor on‐a‐chip | Chamber | [88] | |
| Glioblastoma | RAW264.6 & GL261 | GelMA | EBB | Mini‐brain (Two‐step printing) | Glass slide | [115] | |
| Glioblastoma | TS576 (Glioblastoma patient derived cell line) & HUVEC | PEG‐HA & GelMA | SLB | A tumor region surrounded by a donut‐shape acellular ECM region. HUVEC cell layer encompass the ring | Glass slide | [114] | |
| Glioblastoma/Vasculature model | HUVEC, U87 & fibroblasts | Gelatin/Alginate/Fibrinogen | EBB | A cuboidal shape | Petri‐dish | [119] | |
| Pancreatic cancer/metastatic model | Primary human pancreatic tumor cells, fibroblasts and preadipocytes & HUVEC | Alginate as a sacrificial scaffold | EBB | Scaffold‐free tissue models to study metastasis | Transwell membrane | [116] | |
| Glioma | Human neural progenitor cells & U118 | NA | Kenzen, micro‐needling | Needle array | Needle array | [117] | |
| Melanoma & Gastric cancer/Vasculature model | SK‐MEL‐28 &HTB‐103 | Decellularized ECM | EBB | Cell droplets into PEVA structure & perfusable vessel | Chamber | [87] | |
| Lung cancer/Vasculature and metastatic model | A549 & HUVEC | Fibrin and GelMA/PLGA | EBB | Tumor cell droplet/perfusable vessel onto a fibroblast‐laden fibrin gel; Core/shell structured capsules with growth factors | Chamber | [120] | |
| Glioblastoma/vasculature and metastatic model | Patient‐derived glioblastoma cells, astrocytes, microglia, primary human microvascular brain pericytes & HUVEC | Fibrinogen/Gelatin and Pluronic F127 | EBB | Tumor compartment (Fibrin bioink with glioblastoma cells and stromal cell)s; Perfusable vessels on top of the fibrin bioinks | A thin coverslip, framed by a polydimethylsiloxane gasket | [121] |
4.3. Potential Clinical Applications: Biobanks and In Vitro Drug Screening for Guided Therapy
One of the important applications of 3D tumor models is as an in vitro drug screening platform for drug development and personalized medicine. However, the clinical studies in the current literature show the reliance of these studies on 2D cultures, manual spheroid formation and Matrigel cell embedding as well as the absence of bioprinting technology.[ 122 , 123 ] The major obstacles of in vitro drug screening using patient samples are the small volume of fresh tissue available (0.2–0.3 mm2 for biopsies and 0.5 mm2–0.5 cm2 for resections) and the low number of viable cells for direct drug sensitivity testing (only 7 % of specimens).[ 124 , 125 , 126 ] To overcome these limitations, the cells are either expanded manually in vitro or engraft in mouse models to develop PDX models. However, the development of PDXs takes a long time without any guarantees that the tumor would grow (52 % success rate of mouse engraftment).[ 127 , 128 , 129 ] Therefore, the great potential of in vitro tumor organoids has prompted the establishment of large collections of living tumor organoids derived from many individuals for biobanking. This possibility is highlighted by multiple studies in the literature developing organoid biobanks.[ 130 , 131 , 132 , 133 , 134 , 135 ] For instance, Pauli and co‐workers successfully established a living biobank of tumor organoids from 56 samples out of 152 received tissue biopsies by manually encapsulating cells within Matrigel.[ 124 ] Of those tumor organoids, four different patient‐derived organoids were subjected to HTP drug dose‐response screens for the identification of effective drugs and drug combinations. The success rate of tumor organoid establishment for biobanking can vary from 38.6 % to 100 % between studies (example studies are summarized in Table 3 ).[ 126 , 130 , 131 , 135 , 136 , 137 , 138 ] In addition, the time frame to complete tumor organoid in vitro expansion followed by drug screening can take 4 to 13 weeks, depending on patient‐to‐patient variations and different study designs. This timeline often fails to align with optimal treatment schedules for patients and needs to be addressed. Hence, despite the volume of organoids produced, current techniques for tumor organoid culture still faces technical challenges, such as the translation to rapid and mass production and organoid reproducibility.
Table 3.
Clinical studies using patient‐derived organoids for in vitro cell sensitivity drug screening guided therapy (Abbreviations: No, number; GFR, growth‐factor reduced; S, sensitivity; Sp, specificity; NPV, negative predictive value; PPV, positive predictive value)
| Cancer type | No. of patients | No. of drugs | Culture format | Bioink/scaffold | No. of cells per volume of bioink | Success rate of in vitro expansion | Concordance with clinical response | Ref |
|---|---|---|---|---|---|---|---|---|
| Uterine carcinosarcoma, endometrial adenocarcinoma & colorectal cancer | 4 | ≈160 drugs for 2D culture. Top hits for 3D culture | Cells embedded in the bioink | GFR‐ Matrigel | 1000–3000 cells per 10 µL bioink | 38.6 % | Yes | [124] |
| Pediatric cancers | 17 | 111 | 2D culture, spheroids | None (scaffold‐free) | 1000–4000 cells per well | 50 % | Yes (4/5 patients, predictive value 80 %) | [126] |
| Colorectal and gastroesophageal | 71 | 55 | Cells embedded in the bioink | GFR‐ Matrigel | 4500–6000 cells in 30 µL bioink | 71 % | Yes (S 100 %, Sp 93 %, PPV 88 %, and NPV 100 %) | [135] |
| Rectal cancer | 80 | Irradiation and 2 drugs | Cells embedded in the bioink | Matrigel | 200 ± 50 organoids in 15 µL bioink | 85 % | Yes (Sp 92 %, S 79 %, Accuracy 84 %) | [139] |
| Pancreatic ductal adenocarcinoma | 138 | 26 | Cells embedded in the bioink | Matrigel | 500 cells in 20 µL bioink | 75 % | Yes, with drug‐specific exceptions | [138] |
Bioprinting has the potential to overcome these limitations due to its highly reproducible and high throughput nature. One major advantage of bioprinting is the ability to collect large volumes of data which increases the chance of success in in vitro drug screening. Several research groups have developed bioprinting approaches for generating tumor models in a HTP manner. A simple and cost‐effective miniaturized cell culture system was developed to print cells on droplet microarrays (with hydrophilic‐hydrophobic patterning) either manually or using an acoustic dispenser (I‐DOT nanoliter dispenser (Dispendix GmbH, Germany)).[ 127 , 128 , 129 ] This culture system can print ≈100 cells in a 100 nL volume per spot, with a required cell seeding density of 0.75 to 1 × 106 cells mL‐1. Furthermore, recent studies have developed HTP bioprinting platforms, which permit printing a user‐defined number of cells and a nanoliter‐range droplets in multi‐well plates.[ 106 , 112 , 140 ] Another key factor for in vitro organoid systems is to preserve the original cellular heterogeneity and genomic stability.[ 100 , 141 ] Sachs and colleagues reported that some tumoroids acquired new genetic mutations especially through microsatellite instability after long‐term in vitro passages.[ 142 ] These automated bioprinting platforms can control the culture environment using highly tunable bioinks that aid cells to maintain their original heterogeneity. Therefore, the integration of bioprinting platforms for the production, expansion and biobanking, and drug screening of tumoroids could satisfy many essential requirements to ultimately improve clinical outcomes.
5. Future Perspectives
5.1. Potential Use of 3D Bioprinted Models as a Preclinical Drug Screening Platform for Personalized Medicine
As discussed above, a tumor is heterogeneous, which contributes to the variable therapeutic responses against the standard ‘one size fits all’ antitumor treatments and drives the transition toward personalized medicine for patients. This generates a demand for improving preclinical modeling. The high fidelity and precision offered by bioprinting provides a solution to the limitations of current organoid technology. 3D bioprinting technology is an invaluable tool to move to the next level in personalized tumor therapy as 1) the bioprinting of 3D tumor models enables appropriate mimicking of the native tumor environment and 2) the new HTP bioprinting technology enables not only HTP drug screening but also mass production of tumor organoids derived from PDX and fresh patient samples. The current application of in vitro drug screening is typically included in proof‐of‐concept clinical studies as listed in Table 3. Currently, no bioprinted tumor platform has yet to be approved in the clinical setting. Therefore, for bioprinting to advance to the clinic for tumor personalized medicine, standardized bioprinting protocols, tumor tissue collection and cell isolation methods, culture conditions, selection of bioprinting modalities and bioinks to support growth of different tumor types, and analytical methods to identify the choice of treatment from a list of clinically approved drugs. Finally, the data obtained from bioprinting and screening patient tumor organoids will need to validate in clinical trials to ensure that the treatment recommendations correlate with improved long‐term and overall survival of cancer patients.
5.2. 3D Bioprinted Tumor Models Can Minimize Animal Usages
Due to the limited availability of fresh tumor samples, PDX models have been widely used for preclinical drug evaluation, biomarker identification, biological studies, and personalized medicine strategies as the PDX cells mostly retain the histological and genetic characteristics of their primary patient tumors.[ 143 ] However, the generation of PDX models remains technically challenging, time‐consuming and costly. For instance, PDX models of glioblastoma, an aggressive type of brain tumor, are often generated through intracranial injection, which takes >45 d to establish and test drug efficacy, has limited engraftment success rates and also requires advanced imaging technologies to monitor tumor formation.[ 144 ] Moreover, to validate results generated with PDXs, a large number of mice would be required for every patient to test top hit drugs and for statistical relevance. The results of a particular patient can only be valid for a limited time though, as any surviving tumor cells could still acquire resistance, and new biopsy testing would then be required for a longitudinal patient follow‐up.[ 138 , 145 ] The high fidelity and precision offered by bioprinting provides a solution to the limitations of current tumor organoid production. The 3D bioprinting technology has the potential to replace and reduce the use of animals in tumor drug testing for patients. Moreover, by refining the number of drugs and procedures being used on animals we will minimize and reduce their suffering. Thus, this technology will facilitate researchers world‐wide to replace and reduce use of animals.
5.3. Challenges Remain
The bioprinting technologies, incorporated with advanced biomaterials, have offered incredibly intricate possibilities for the development of in vitro tumor models, closely mimicking the native tumor microenvironment. However, many challenges remain to be overcome in terms of designing 3D bioprinted tumor models and their potential to be used as a preclinical platform. First of all, when dealing with fresh patient‐derived tumor cells, it is critical to generate 3D models in an efficient, rapid and viable way. However, currently available bioprinting platforms and bioprinters can be complicated to set up, use and require highly skilled staff for application and maintenance. The development of simple, straightforward and time‐, as well as, cost‐effective bioprinting technology is necessary to bring the bioprinter as a preclinical platform for clinical use. Second, the cost of 3D bioprinters can range from $10,000 to as much as $300,000 according to manufacturer's pricing charts available online.[ 146 ] To expedite the adoption of the technology in medical research and the clinical setting, the bioprinting platform for 3D tumor models needs to be affordable and simple‐to‐use for everyone in laboratories. For future clinical application, guidelines for standardization and reproducibility of bioprinting protocols will be required. Third, as mentioned above, a level of HTP capability is required for the use of 3D bioprinted tumor models in a clinical set‐up. Despite the advancement of various sophisticated 3D bioprinted tumor models, it is still early stage for the technology to be utilized in HTP screening using patient‐derived tumor organoids. The technology requires high cell numbers to achieve the 3D tumor‐like constructs for large‐scale and higher throughput assays. This hinders the technology to be used in precision medicine platforms as tumor biopsies are often insufficient. Further development of more automated and HTP platform using a small number of patient‐derived cells would increase feasibility as a preclinical model. Last, absence of immune cells often fails to mimic the tumor microenvironment in 3D tumor models and therefore the addition of immune microenvironment components in the 3D bioprinted tumor models would improve the potential of the 3D bioprinting technology for personalized medicine.
6. Conclusions
3D bioprinting technology is a promising tool that enables the biofabrication of 3D tumor models with accurate control over distribution of cells, biological molecules, and bioinks. The 3D bioprinted tumor models have shown a huge potential to be used as a platform to study tumor biology as well as for antitumor drug screening, biobanking and personalized medicine. Future development of simple, time/cost‐effective and higher throughput platforms together with standardization protocols and bioinks will facilitate progress towards truly personalized cancer treatment.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the Children's Cancer Institute, which is affiliated with the University of New South Wales (UNSW Sydney), and the Sydney Children's Hospital Network and by grants from the National Health and Medical Research Council (NHMRC) Investigator (APP1196648 to J.J.G.), Cancer Institute New South Wales (2019/TPG2037 to M.K.), Cancer Australia (RG200076 to M.K., J.J.G., M.J.) and the Australian Research Council Linkage Grant (LP170100623 to J.J.G., M.K.). The authors thank Angie Davina Tjandra for assistance in drawing the schematics.
Open access publishing facilitated by University of New South Wales, as part of the Wiley ‐ University of New South Wales agreement via the Council of Australian University Librarians.
After initial online publication, the open access funding statement was added to the Acknowledgements on December 21, 2022.
Biographies
MoonSun Jung is a cancer researcher at the Children's Cancer Institute, Australia. Her research has advanced knowledge in cancer cell biology and clinical data analysis in both childhood and adult cancers. Dr. Jung's research led to the identification of prognostic markers and therapeutic targets in cancers. Dr. Jung leads a research project that aims to develop 3D bio‐printed cancer models to predict cancer therapeutic response and understand metastasis in this disease. Prior to joining the Children's Cancer Institute, Dr. Jung obtained her Ph.D. in biomedical engineering at the University of New South Wales.

Maria Kavallaris is a professor and director of the Australian Centre for Nanomedicine at the University of New South Wales and Head of Translational Cancer Nanomedicine at Children's Cancer Institute in Sydney. Her laboratory studies cancer biology, migration and invasion, and the application of nanotechnology for the development of therapeutics and devices. Research from her group and collaborators has led to the development of an award‐winning 3D bioprinter and extra‐cellular matrix hydrogel mimics to grow cancer cells. Professor Kavallaris completed her undergraduate degree at the University of Technology Sydney and Ph.D. at the University of New South Wales, Australia.

Jung M., Ghamrawi S., Du E. Y., Gooding J. J., Kavallaris M., Advances in 3D Bioprinting for Cancer Biology and Precision Medicine: From Matrix Design to Application. Adv. Healthcare Mater. 2022, 11, 2200690. 10.1002/adhm.202200690
References
- 1. Bray F., Ferlay J., Soerjomataram I., Siegel R. L., Torre L. A., Jemal A., Ca‐Cancer J. Clin. 2018, 68, 394. [DOI] [PubMed] [Google Scholar]
- 2. Wong C. H., Siah K. W., Lo A. W., Biostatistics 2019, 20, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baghban R., Roshangar L., Jahanban‐Esfahlan R., Seidi K., Ebrahimi‐Kalan A., Jaymand M., Kolahian S., Javaheri T., Zare P., Cell Commun., Signaling 2020, 18, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Benam K. H., Dauth S., Hassell B., Herland A., Jain A., Jang K.‐J., Karalis K., Kim H. J., MacQueen L., Mahmoodian R., Musah S., Torisawa Y.‐S., van der Meer A. D., Villenave R., Yadid M., Parker K. K., Ingber D. E., Annu. Rev. Pathol.: Mech. Dis. 2015, 10, 195. [DOI] [PubMed] [Google Scholar]
- 5. Pape J., Emberton M., Cheema U., Front. Bioeng. Biotechnol. 2021, 9, 660502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Law A. M. K., Rodriguez de la Fuente L., Grundy T. J., Fang G., Valdes‐Mora F., Gallego‐Ortega D., Front. Oncol. 2021, 11, 782766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drost J., Clevers H., Nat. Rev. Cancer 2018, 18, 407. [DOI] [PubMed] [Google Scholar]
- 8. Monteiro M. V., Zhang Y. S., Gaspar V. M., Mano J. F., Trends Biotechnol. 2022, 40, 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karkampouna S., La Manna F., Benjak A., Kiener M., De Menna M., Zoni E., Grosjean J., Klima I., Garofoli A., Bolis M., Vallerga A., Theurillat J.‐P., De Filippo M. R., Genitsch V., Keller D., Booij T. H., Stirnimann C. U., Eng K., Sboner A., Ng C. K. Y., Piscuoglio S., Gray P. C., Spahn M., Rubin M. A., Thalmann G. N., Kruithof‐de Julio M., Nat. Commun. 2021, 12, 1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Amaral R., Zimmermann M., Ma A.‐H., Zhang H., Swiech K., Pan C.‐X., Cancers 2020, 12, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Agarwal S., Saha S., Balla V. K., Pal A., Barui A., Bodhak S., Front. Mech. Eng. 2020, 6, 589171. [Google Scholar]
- 12. Murphy S. V., Atala A., Nat. Biotechnol. 2014, 32, 773. [DOI] [PubMed] [Google Scholar]
- 13. Gudapati H., Dey M., Ozbolat I., Biomaterials 2016, 102, 20. [DOI] [PubMed] [Google Scholar]
- 14. Datta P., Barui A., Wu Y., Ozbolat V., Moncal K. K., Ozbolat I. T., Biotechnol. Adv. 2018, 36, 1481. [DOI] [PubMed] [Google Scholar]
- 15. Ozbolat I. T., Hospodiuk M., Biomaterials 2016, 76, 321. [DOI] [PubMed] [Google Scholar]
- 16. Ramesh S., Harrysson O. L. A., Rao P. K., Tamayol A., Cormier D. R., Zhang Y., Rivero I. V., Bioprinting 2021, 21, e00116. [Google Scholar]
- 17. Ashammakhi N., Ahadian S., Xu C., Montazerian H., Ko H., Nasiri R., Barros N., Khademhosseini A., Mater. Today Bio 2019, 1, 100008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Quail D. F., Joyce J. A., Nat. Med. 2013, 19, 1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lyssiotis C. A., Kimmelman A. C., Trends Cell Biol. 2017, 27, 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bhowmick N. A., Neilson E. G., Moses H. L., Nature 2004, 432, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Valkenburg K. C., de Groot A. E., Pienta K. J., Nat. Rev. Clin. Oncol. 2018, 15, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giraldo N. A., Sanchez‐Salas R., Peske J. D., Vano Y., Becht E., Petitprez F., Validire P., Ingels A., Cathelineau X., Fridman W. H., Sautès‐Fridman C., Br. J. Cancer 2019, 120, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McDonald P. C., Chafe S. C., Dedhar S., Front. Cell, Dev. Biol. 2016, 4, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.A. Nejad E., Najafgholian S., Rostami A., Sistani A., Shojaeifar S., Esparvarinha M., Nedaeinia R., Haghjooy Javanmard S., Taherian M., Ahmadlou M., Salehi R., Sadeghi B., Manian M., Cancer Cell Int. 2021, 21, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krock B. L., Skuli N., Simon M. C., Genes Cancer 2011, 2, 1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin M.‐Z., Jin W.‐L., Signal Transduction Targeted Ther. 2020, 5, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hanahan D., Weinberg R. A., Cell 2011, 144, 646. [DOI] [PubMed] [Google Scholar]
- 28. Muz B., de la Puente P., Azab F., Azab A. K., Hypoxia 2015, 3, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sánchez‐Tilló E., Liu Y., de Barrios O., Siles L., Fanlo L., Cuatrecasas M., Darling D. S., Dean D. C., Castells A., Postigo A., Cell. Mol. Life Sci. 2012, 69, 3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mittal V., Annu. Rev. Pathol.: Mech. Dis. 2018, 13, 395. [DOI] [PubMed] [Google Scholar]
- 31. Bonnans C., Chou J., Werb Z., Nat. Rev. Mol. Cell Biol. 2014, 15, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hynes R. O., Science 2009, 326, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Theocharis A. D., Skandalis S. S., Gialeli C., Karamanos N. K., Adv. Drug Delivery Rev. 2016, 97, 4. [DOI] [PubMed] [Google Scholar]
- 34. Fouquet S., Lugo‐Martíne V.‐H., Faussat A.‐M., Renaud F., Cardot P., Chambaz J., Pinçon‐Raymond M., Thenet S., J. Biol. Chem. 2004, 279, 43061. [DOI] [PubMed] [Google Scholar]
- 35. Roche J., Cancers 2018, 10, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jang I., Beningo K. A., Cancers 2019, 11, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sahai E., Astsaturov I., Cukierman E., DeNardo D. G., Egeblad M., Evans R. M., Fearon D., Greten F. R., Hingorani S. R., Hunter T., Hynes R. O., Jain R. K., Janowitz T., Jorgensen C., Kimmelman A. C., Kolonin M. G., Maki R. G., Powers R. S., Puré E., Ramirez D. C., Scherz‐Shouval R., Sherman M. H., Stewart S., Tlsty T. D., Tuveson D. A., Watt F. M., Weaver V., Weeraratna A. T., Werb Z., Nat. Rev. Cancer 2020, 20, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Belhabib I., Zaghdoudi S., Lac C., Bousquet C., Jean C., Cancers 2021, 13, 3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hynes R. O., Naba A., Cold Spring Harbor Perspect. Biol. 2012, 4, a004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Naba A., Clauser K. R., Hoersch S., Liu H., Carr S. A., Hynes R. O., Mol. Cell. Proteomics 2012, 11, M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Izzi V., Davis M. N., Naba A., Cancers 2020, 12, 2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bin Lim S., Chua M. L. K., Yeong J. P. S., Tan S. J., Lim W.‐T., Lim C. T., npj Precis. Oncol. 2019, 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yuzhalin A. E., Urbonas T., Silva M. A., Muschel R. J., Gordon‐Weeks A. N., Br. J. Cancer 2018, 118, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ovsianikov A., Gruene M., Pflaum M., Koch L., Maiorana F., Wilhelmi M., Haverich A., Chichkov B., Biofabrication 2010, 2, 014104. [DOI] [PubMed] [Google Scholar]
- 45. Kingsley D. M., Roberge C. L., Rudkouskaya A., Faulkner D. E., Barroso M., Intes X., Corr D. T., Acta Biomater. 2019, 95, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guillotin B., Ali M., Ducom A., Catros S., Keriquel V., Souquet A., Remy M., Fricain J.‐C., Guillemot F., in Biofabrication (Eds: Forgacs G., Sun) W., 2013, William Andrew Publishing, Boston, pp. 95–118. [Google Scholar]
- 47. Mondschein R. J., Kanitkar A., Williams C. B., Verbridge S. S., Long T. E., Biomaterials 2017, 140, 170. [DOI] [PubMed] [Google Scholar]
- 48. Magalhaes L., Santos F. E. P., Elias C. M. V., Afewerki S., Sousa G. F., Furtado A. S. A., Marciano F. R., Lobo A. O., J. Funct. Biomater. 2020, 11, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Samadian H., Jafari S., Sepand M. R., Alaei L., Sadegh Malvajerd S., Jaymand M., Ghobadinezhad F., Jahanshahi F., Hamblin M. R., Derakhshankhah H., Izadi Z., Mater. Today Adv. 2021, 12, 100160. [Google Scholar]
- 50. Wang Z., Abdulla R., Parker B., Samanipour R., Ghosh S., Kim K., Biofabrication 2015, 7, 045009. [DOI] [PubMed] [Google Scholar]
- 51. Unagolla J. M., Jayasuriya A. C., Appl. Mater. Today 2020, 18, 100479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gentilin E., D'Angelo E., Agostini M., Astolfi L., Cancer Gene Ther. 2022, 29, 879. [DOI] [PubMed] [Google Scholar]
- 53. Augustine R., Kalva S. N., Ahmad R., Zahid A. A., Hasan S., Nayeem A., McClements L., Hasan A., Transl. Oncol. 2021, 14, 101015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lowe S. B., Tan V. T. G., Soeriyadi A. H., Davis T. P., Gooding J. J., Bioconjugate Chem. 2014, 25, 1581. [DOI] [PubMed] [Google Scholar]
- 55. Hospodiuk M., Dey M., Sosnoski D., Ozbolat I. T., Biotechnol. Adv. 2017, 35, 217. [DOI] [PubMed] [Google Scholar]
- 56. Mobaraki M., Ghaffari M., Yazdanpanah A., Luo Y., Mills D. K., Bioprinting 2020, 18, e00080. [Google Scholar]
- 57. Budday S., Nay R., de Rooij R., Steinmann P., Wyrobek T., Ovaert T. C., Kuhl E., J. Mech. Behav. Biomed. Mater. 2015, 46, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kaster T., Sack I., Samani A., J. Biomech. 2011, 44, 1158. [DOI] [PubMed] [Google Scholar]
- 59. Tabet A., Mommer S., Vigil J. A., Hallou C., Bulstrode H., Scherman O. A., Adv. Healthcare Mater. 2019, 8, 1900068. [DOI] [PubMed] [Google Scholar]
- 60. Hinz B., Proc. Am. Thorac. Soc. 2012, 9, 137. [DOI] [PubMed] [Google Scholar]
- 61. Polio S. R., Kundu A. N., Dougan C. E., Birch N. P., Aurian‐Blajeni D. E., Schiffman J. D., Crosby A. J., Peyton S. R., PLoS One 2018, 13, e0204765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Levental K. R., Yu H., Kass L., Lakins J. N., Egeblad M., Erler J. T., Fong S. F. T., Csiszar K., Giaccia A., Weninger W., Yamauchi M., Gasser D. L., Weaver V. M., Cell 2009, 139, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Samani A., Zubovits J., Plewes D., Phys. Med. Biol. 2007, 52, 1565. [DOI] [PubMed] [Google Scholar]
- 64. Clarke E. C., Cheng S., Green M., Sinkus R., Bilston L. E., J. Biomech. 2011, 44, 2461. [DOI] [PubMed] [Google Scholar]
- 65. Yeh W.‐C., Li P.‐C., Jeng Y.‐M., Hsu H.‐C., Kuo P.‐L., Li M.‐L., Yang P.‐M., Lee P. H., Ultrasound Med. Biol. 2002, 28, 467. [DOI] [PubMed] [Google Scholar]
- 66. Jiao T., Clifton R. J., Converse G. L., Hopkins R. A., Tissue Eng., Part A 2011, 18, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ogneva I. V., Lebedev D. V., Shenkman B. S., Biophys. J. 2010, 98, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Brauck K., Galbán C. J., Maderwald S., Herrmann B. L., Ladd M. E., Eur. J. Endocrinol. 2007, 156, 673. [DOI] [PubMed] [Google Scholar]
- 69. Feng Y. N., Li Y. P., Liu C. L., Zhang Z. J., Sci. Rep. 2018, 8, 17064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chizhik S. A., Wierzcholski K., Trushko A. V., Zhytkova M. A., Miszczak A., Adv. Tribol. 2010, 2010, 243150. [Google Scholar]
- 71. Peters A. E., Akhtar R., Comerford E. J., Bates K. T., Sci. Rep. 2018, 8, 5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Morgan E. F., Unnikrisnan G. U., Hussein A. I., Annu. Rev. Biomed. Eng. 2018, 20, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rho J. Y., Ashman R. B., Turner C. H., J. Biomech. 1993, 26, 111. [DOI] [PubMed] [Google Scholar]
- 74. Benwood C., Chrenek J., Kirsch R. L., Masri N. Z., Richards H., Teetzen K., Willerth S. M., Bioengineering 2021, 8, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Diamantides N., Dugopolski C., Blahut E., Kennedy S., Bonassar L. J., Biofabrication 2019, 11, 045016. [DOI] [PubMed] [Google Scholar]
- 76. Yang Y. l., Kaufman L. J., Biophys. J. 2009, 96, 1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lai G., Li Y., Li G., Int. J. Biol. Macromol. 2008, 42, 285. [DOI] [PubMed] [Google Scholar]
- 78. Osidak E. O., Kozhukhov V. I., Osidak M. S., Domogatsky S. P., Int. J. Bioprint. 2020, 6, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Caliari S. R., Burdick J. A., Nat. Methods 2016, 13, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Emami Z., Ehsani M., Zandi M., Foudazi R., Carbohydr. Polym. 2018, 198, 509. [DOI] [PubMed] [Google Scholar]
- 81. Bello A. B., Kim D., Kim D., Park H., Lee S. H., Tissue Eng., Part B 2020, 26, 164. [DOI] [PubMed] [Google Scholar]
- 82. Zhang Y., Wang Z., Hu Q., Luo H., Lu B., Gao Y., Qiao Z., Zhou Y., Fang Y., Gu J., Zhang T., Xiong Z., Small 2022, 18, e2200364. [DOI] [PubMed] [Google Scholar]
- 83. Liu W., Zhong Z., Hu N., Zhou Y., Maggio L., Miri A. K., Fragasso A., Jin X., Khademhosseini A., Zhang Y. S., Biofabrication 2018, 10, 024102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shao L., Gao Q., Xie C., Fu J., Xiang M., He Y., Biofabrication 2020, 12, 035014. [DOI] [PubMed] [Google Scholar]
- 85. Pati F., Jang J., Ha D.‐H., Won Kim S., Rhie J.‐W., Shim J.‐H., Kim D.‐H., Cho D.‐W., Nat. Commun. 2014, 5, 3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ma X., Yu C., Wang P., Xu W., Wan X., Lai C. S. E., Liu J., Koroleva‐Maharajh A., Chen S., Biomaterials 2018, 185, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kim B. S., Cho W.‐W., Gao G., Ahn M., Kim J., Cho D.‐W., Small Methods 2021, 5, 2100072. [DOI] [PubMed] [Google Scholar]
- 88. Yi H.‐G., Jeong Y. H., Kim Y., Choi Y.‐J., Moon H. E., Park S. H., Kang K. S., Bae M., Jang J., Youn H., Paek S. H., Cho D.‐W., Nat. Biomed. Eng. 2019, 3, 509. [DOI] [PubMed] [Google Scholar]
- 89. Uhl F. E., Zhang F., Pouliot R. A., Uriarte J. J., Rolandsson Enes S., Han X., Ouyang Y., Xia K., Westergren‐Thorsson G., Malmstrom A., Hallgren O., Linhardt R. J., Weiss D. J., Acta Biomater. 2020, 102, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Amirazad H., Dadashpour M., Zarghami N., J. Biol. Eng. 2022, 16, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zaman M. H., Trapani L. M., Sieminski A. L., Mackellar D., Gong H., Kamm R. D., Wells A., Lauffenburger D. A., Matsudaira P., Proc. Natl. Acad. Sci. USA 2006, 103, 10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hughes C. S., Postovit L. M., Lajoie G. A., Proteomics 2010, 10, 1886. [DOI] [PubMed] [Google Scholar]
- 93. Fan R., Piou M., Darling E., Cormier D., Sun J., Wan J., J. Biomater. Appl. 2016, 31, 684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Aisenbrey E. A., Murphy W. L., Nat. Rev. Mater. 2020, 5, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yu J., Park S. A., Kim W. D., Ha T., Xin Y. Z., Lee J., Lee D., Polymers 2020, 12, 2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Valot L., Martinez J., Mehdi A., Subra G., Chem. Soc. Rev. 2019, 48, 4049. [DOI] [PubMed] [Google Scholar]
- 97. Aduba D. C., Margaretta E. D., Marnot A. E. C., Heifferon K. V., Surbey W. R., Chartrain N. A., Whittington A. R., Long T. E., Williams C. B., Mater. Today Commun. 2019, 19, 204. [Google Scholar]
- 98. Liu F., Wang X., Polymers 2020, 12, 204.31941163 [Google Scholar]
- 99. Bee S.‐L., Hamid Z. A. A., Mariatti M., Yahaya B. H., Lim K., Bee S.‐T., Sin L. T., Polym. Rev. 2018, 58, 495. [Google Scholar]
- 100. LeSavage B. L., Suhar R. A., Broguiere N., Lutolf M. P., Heilshorn S. C., Nat. Mater. 2022, 21, 143. [DOI] [PubMed] [Google Scholar]
- 101. Lazzari G., Couvreur P., Mura S., Polym. Chem. 2017, 8, 4947. [Google Scholar]
- 102. Fennema E., Rivron N., Rouwkema J., van Blitterswijk C., de Boer J., Trends Biotechnol. 2013, 31, 108. [DOI] [PubMed] [Google Scholar]
- 103. Ling K., Huang G., Liu J., Zhang X., Ma Y., Lu T., Xu F., Engineering 2015, 1, 269. [Google Scholar]
- 104. Hribar K. C., Finlay D., Ma X., Qu X., Ondeck M. G., Chung P. H., Zanella F., Engler A. J., Sheikh F., Vuori K., Chen S. C., Lab Chip 2015, 15, 2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Hakobyan D., Médina C., Dusserre N., Stachowicz M.‐L., Handschin C., Fricain J.‐C., Guillermet‐Guibert J., Oliveira H., Biofabrication 2020, 12, 035001. [DOI] [PubMed] [Google Scholar]
- 106. Utama R. H., Atapattu L., O'Mahony A. P., Fife C. M., Baek J., Allard T., O'Mahony K. J., Ribeiro J. C. C., Gaus K., Kavallaris M., Gooding J. J., iScience 2020, 23, 101621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Maloney E., Clark C., Sivakumar H., Yoo K., Aleman J., Rajan S. A. P., Forsythe S., Mazzocchi A., Laxton A. W., Tatter S. B., Strowd R. E., Votanopoulos K. I., Skardal A., Micromachines 2020, 11, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhao Y., Yao R., Ouyang L., Ding H., Zhang T., Zhang K., Cheng S., Sun W., Biofabrication 2014, 6, 035001. [DOI] [PubMed] [Google Scholar]
- 109. Swaminathan S., Hamid Q., Sun W., Clyne A. M., Biofabrication 2019, 11, 025003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Reid J. A., Palmer X.‐L., Mollica P. A., Northam N., Sachs P. C., Bruno R. D., Sci. Rep. 2019, 9, 7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Xu F., Celli J., Rizvi I., Moon S., Hasan T., Demirci U., Biotechnol. J. 2011, 6, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hwang H. H., You S., Ma X., Kwe L., Victorine G., Lawrence N., Wan X., Shen H., Zhu W., Chen S., Biofabrication 2021, 13, 025007. [DOI] [PubMed] [Google Scholar]
- 113. Bhatia S. N., Ingber D. E., Nat. Biotechnol. 2014, 32, 760. [DOI] [PubMed] [Google Scholar]
- 114. Tang M., Tiwari S. K., Agrawal K., Tan M., Dang J., Tam T., Tian J., Wan X., Schimelman J., You S., Xia Q., Rana T. M., Chen S., Small 2021, 17, 2006050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Heinrich M. A., Bansal R., Lammers T., Zhang Y. S., Schiffelers R. Michel, Prakash J., Adv. Mater. 2019, 31, 1806590. [DOI] [PubMed] [Google Scholar]
- 116. Langer E. M., Allen‐Petersen B. L., King S. M., Kendsersky N. D., Turnidge M. A., Kuziel G. M., Riggers R., Samatham R., Amery T. S., Jacques S. L., Sheppard B. C., Korkola J. E., Muschler J. L., Thibault G., Chang Y. H., Gray J. W., Presnell S. C., Nguyen D. G., Sears R. C., Cell Rep. 2019, 26, 608.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. van Pel D. M., Harada K., Song D., Naus C. C., Sin W. C., J. Cell Commun. Signaling 2018, 12, 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Moldovan N. I., Hibino N., Nakayama K., Tissue Eng., Part B 2016, 23, 237. [DOI] [PubMed] [Google Scholar]
- 119. Han S., Kim S., Chen Z., Shin H. K., Lee S.‐Y., Moon H. E., Paek S. H., Park S., Int. J. Mol. Sci. 2020, 21, 2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Meng F., Meyer C. M., Joung D., Vallera D. A., McAlpine M. C., Panoskaltsis‐Mortari A., Adv. Mater. 2019, 31, 1806899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Neufeld L., Yeini E., Reisman N., Shtilerman Y., Ben‐Shushan D., Pozzi S., Madi A., Tiram G., Eldar‐Boock A., Ferber S., Grossman R., Ram Z., Satchi‐Fainaro R., Sci. Adv. 2021, 7, eabi9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Popova A. A., Levkin P. A., Adv. Ther. 2020, 3, 1900100. [Google Scholar]
- 123. Veninga V., Voest E. E., Cancer Cell 2021, 39, 1190. [DOI] [PubMed] [Google Scholar]
- 124. Pauli C., Hopkins B. D., Prandi D., Shaw R., Fedrizzi T., Sboner A., Sailer V., Augello M., Puca L., Rosati R., McNary T. J., Churakova Y., Cheung C., Triscott J., Pisapia D., Rao R., Mosquera J. M., Robinson B., Faltas B. M., Emerling B. E., Gadi V. K., Bernard B., Elemento O., Beltran H., Demichelis F., Kemp C. J., Grandori C., Cantley L. C., Rubin M. A., Cancer Discovery 2017, 7, 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Rajer M., Kmet M., Radiol. Oncol. 2005, 39, 269. [Google Scholar]
- 126. Lau L. M. S., Mayoh C., Xie J., Barahona P., MacKenzie K. L., Wong M., Kamili A., Tsoli M., Failes T. W., Kumar A., Mould E. V. A., Gifford A., Chow S. O., Pinese M., Fletcher J. I., Arndt G. M., Khuong‐Quang D. A., Wadham C., Eden G., Trebilcock P., Joshi S., Alfred S., Gopalakrishnan A., Khan A., Grebert Wade D., Strong P. A., Manouvrier E., Morgan L. T., Cadiz R., Ung C., et al., EMBO Mol. Med. 2021, 14, 14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Neto A. I., Demir K., Popova A. A., Oliveira M. B., Mano J. F., Levkin P. A., Adv. Mater. 2016, 28, 7613. [DOI] [PubMed] [Google Scholar]
- 128. Popova A. A., Dietrich S., Huber W., Reischl M., Peravali R., Levkin P. A., SLAS Technol. 2021, 26, 274. [DOI] [PubMed] [Google Scholar]
- 129. Popova A. A., Reischl M., Kazenmaier D., Cui H., Amberger T., Levkin P. A., SLAS Technol. 2022, 27, 44. [DOI] [PubMed] [Google Scholar]
- 130. Fan H., Demirci U., Chen P., J. Hematol. Oncol. 2019, 12, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Fujii M., Shimokawa M., Date S., Takano A., Matano M., Nanki K., Ohta Y., Toshimitsu K., Nakazato Y., Kawasaki K., Uraoka T., Watanabe T., Kanai T., Sato T., Cell Stem Cell 2016, 18, 827. [DOI] [PubMed] [Google Scholar]
- 132. Sachs N., de Ligt J., Kopper O., Gogola E., Bounova G., Weeber F., Balgobind A. V., Wind K., Gracanin A., Begthel H., Korving J., van Boxtel R., Duarte A. A., Lelieveld D., van Hoeck A., Ernst R. F., Blokzijl F., Nijman I. J., Hoogstraat M., van de Ven M., Egan D. A., Zinzalla V., Moll J., Boj S. F., Voest E. E., Wessels L., van Diest P. J., Rottenberg S., Vries R. G. J., Cuppen E., et al., Cell 2018, 172, 373.e10. [DOI] [PubMed] [Google Scholar]
- 133. Tiriac H., Belleau P., Engle D. D., Plenker D., Deschênes A., Somerville T. D. D., Froeling F. E. M., Burkhart R. A., Denroche R. E., Jang G.‐H., Miyabayashi K., Young C. M., Patel H., Ma M., LaComb J. F., Palmaira R. L. D., Javed A. A., Huynh J. C., Johnson M., Arora K., Robine N., Shah M., Sanghvi R., Goetz A. B., Lowder C. Y., Martello L., Driehuis E., LeComte N., Askan G., Iacobuzio‐Donahue C. A., et al., Cancer Discovery 2018, 8, 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. van de Wetering M., Francies H. E., Francis J. M., Bounova G., Iorio F., Pronk A., van Houdt W., van Gorp J., Taylor‐Weiner A., Kester L., McLaren‐Douglas A., Blokker J., Jaksani S., Bartfeld S., Volckman R., van Sluis P., Li V. S. W., Seepo S., Pedamallu C. S., Cibulskis K., Carter S. L., McKenna A., Lawrence M. S., Lichtenstein L., Stewart C., Koster J., Versteeg R., van Oudenaarden A., Saez‐Rodriguez J., Vries R. G. J., et al., Cell 2015, 161, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Vlachogiannis G., Hedayat S., Vatsiou A., Jamin Y., Fernandez‐Mateos J., Khan K., Lampis A., Eason K., Huntingford I., Burke R., Rata M., Koh D. M., Tunariu N., Collins D., Hulkki‐Wilson S., Ragulan C., Spiteri I., Moorcraft S. Y., Chau I., Rao S., Watkins D., Fotiadis N., Bali M., Darvish‐Damavandi M., Lote H., Eltahir Z., Smyth E. C., Begum R., Clarke P. A., Hahne J. C., et al., Science 2018, 359, 920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. van de Wetering M., Francies H. E., Francis J. M., Bounova G., Iorio F., Pronk A., van Houdt W., van Gorp J., Taylor‐Weiner A., Kester L., McLaren‐Douglas A., Blokker J., Jaksani S., Bartfeld S., Volckman R., van Sluis P., Li V. S., Seepo S., Sekhar Pedamallu C., Cibulskis K., Carter S. L., McKenna A., Lawrence M. S., Lichtenstein L., Stewart C., Koster J., Versteeg R., van Oudenaarden A., Saez‐Rodriguez J., Vries R. G., et al., Cell 2015, 161, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Sachs N., de Ligt J., Kopper O., Gogola E., Bounova G., Weeber F., Balgobind A. V., Wind K., Gracanin A., Begthel H., Korving J., van Boxtel R., Duarte A. A., Lelieveld D., van Hoeck A., Ernst R. F., Blokzijl F., Nijman I. J., Hoogstraat M., van de Ven M., Egan D. A., Zinzalla V., Moll J., Boj S. F., Voest E. E., Wessels L., van Diest P. J., Rottenberg S., Vries R. G. J., Cuppen E., et al., Cell 2018, 172, 373. [DOI] [PubMed] [Google Scholar]
- 138. Tiriac H., Belleau P., Engle D. D., Plenker D., Deschenes A., Somerville T. D. D., Froeling F. E. M., Burkhart R. A., Denroche R. E., Jang G. H., Miyabayashi K., Young C. M., Patel H., Ma M., LaComb J. F., Palmaira R. L. D., Javed A. A., Huynh J. C., Johnson M., Arora K., Robine N., Shah M., Sanghvi R., Goetz A. B., Lowder C. Y., Martello L., Driehuis E., LeComte N., Askan G., Iacobuzio‐Donahue C. A., et al., Cancer Discovery 2018, 8, 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Yao Y., Xu X., Yang L., Zhu J., Wan J., Shen L., Xia F., Fu G., Deng Y., Pan M., Guo Q., Gao X., Li Y., Rao X., Zhou Y., Liang L., Wang Y., Zhang J., Zhang H., Li G., Zhang L., Peng J., Cai S., Hu C., Gao J., Clevers H., Zhang Z., Hua G., Cell Stem Cell 2020, 26, 17. [DOI] [PubMed] [Google Scholar]
- 140. Utama R. H., Tan V. T. G., Tjandra K. C., Sexton A., Nguyen D. H. T., O'Mahony A. P., Du E. Y., Tian P., Ribeiro J. C. C., Kavallaris M., Gooding J. J., Macromol. Biosci. 2021, 21, 2100125. [DOI] [PubMed] [Google Scholar]
- 141. Letai A., Nat. Med. 2017, 23, 1028. [DOI] [PubMed] [Google Scholar]
- 142. Nuciforo S., Fofana I., Matter M. S., Blumer T., Calabrese D., Boldanova T., Piscuoglio S., Wieland S., Ringnalda F., Schwank G., Terracciano L. M., Ng C. K. Y., Heim M. H., Cell Rep. 2018, 24, 1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Hidalgo M., Amant F., Biankin A. V., Budinská E., Byrne A. T., Caldas C., Clarke R. B., de Jong S., Jonkers J., Mælandsmo G. M., Roman‐Roman S., Seoane J., Trusolino L., Villanueva A., Cancer Discovery 2014, 4, 998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Lee J., Jo D. H., Kim J. H., Cho C. S., Han J. E., Kim Y., Park H., Yoo S. H., Yu Y. S., Moon H. E., Park H. R., Kim D. G., Kim J. H., Paek S. H., Exp. Mol. Med. 2019, 51, 1. [Google Scholar]
- 145. Yamada S., Hongo T., Okada S., Watanabe C., Fujii Y., Ohzeki T., Leukemia 2001, 15, 1892. [DOI] [PubMed] [Google Scholar]
- 146. Goldstein T. A., Epstein C. J., Schwartz J., Krush A., Lagalante D. J., Mercadante K. P., Zeltsman D., Smith L. P., Grande D. A., Tissue Eng., Part C 2016, 22, 1071. [DOI] [PubMed] [Google Scholar]