

Abstract
Caffeine, a primary flavor component in tea, has been the subject of intense research. With the goal of shedding light on the complex regulatory processes governing caffeine biosynthesis in tea plants, liquid chromatography coupled with mass spectrometry (LC–MS), transcriptomics, and small RNA analyses were employed on diverse tea cultivars such as ‘Jianghua Kucha’ [including ‘Xianghong 3’ (XH3H) and ‘Kucha 3’ (KC3H)], ‘Fuding Dabaicha’ (FDDB), ‘Yaoshan Xiulv’ (YSXL), and ‘Bixiangzao’ (BXZ). The results showed that the caffeine level in ‘Jianghua Kucha’ was significantly higher than that in other tea plant cultivars. In addition, weighted gene co-expression network analysis indicated that that the CsbHLH1 gene might play a pivotal role as a potential hub gene related to the regulation of caffeine biosynthesis. Subcellular localization analysis showed that the CsbHLH1 protein was localized in the nucleus of the cells. Moreover, CsbHLH1 suppresses the transcription of TCS1 by binding to the TCS1 promoter, as evidenced by a yeast one-hybrid assay, an electrophoretic mobility shift assay, and dual luciferase analysis. In addition, a microRNA, miR1446a, was identified that directly cleaves CsbHLH1, leading to an increase in caffeine levels. Therefore, our findings imply that CsbHLH1 binds to the TCS1 promoter (−971 to −1019 bp) to reduce its expression, thereby negatively regulating caffeine biosynthesis. On the other hand, miR1446a enhances the biosynthesis of caffeine by suppressing the expression of CsbHLH1. This work enhances our understanding of the molecular mechanisms of caffeine biosynthesis in tea plants and offers potential directions for manipulating caffeine levels in future tea cultivation.
Introduction
Kucha (Camellia sinensis), which is mainly distributed in Hunan, Guangdong, Guangxi, and Yunnan, is a special tea resource in China [1]. When brewed, its fresh leaves offer a distinctly bitter flavor, setting it apart from other teas. Notably, ‘Jianghua Kucha’ is an excellent local landrace of Kucha recognized in Hunan Province [2], and has attracted more and more attention from researchers due to its high level of purine alkaloids compared with other tea cultivars, such as ‘Bixiangzao’ (BXZ) [2–6].
Researchers have broadly mapped out the main pathways of alkaloid biosynthesis, donor pathways, and catabolic pathways in tea plant (Supplementary Data Fig. S1). The specific biosynthesis pathway is: adenosine → guanosine → xanthosine → 7-methylxanthosine → 7-methylxanthine → theobromine → caffeine → theacrine [7–9]. Within the donor pathway, there are multiple sources for the eventual biosynthesis of xanthine nucleosides, which provide the starting methyl acceptor for the core pathway [8]. At least four pathways have been identified: the adenine nucleotide route, the guanine nucleotide route, the purine ring de novo biosynthesis route, and the S-adenosylmethionine cycle route, the adenine nucleotide route being the most critical [10–12]. Genes encoding the key enzymes in the biosynthetic pathway of alkaloids have been identified, including 7-methylxanthine synthase (catalyzing the conversion of xanthine to 7-methylxanthine), theobromine synthase (3,7-dimethylxanthine synthase, catalyzing the formation of theobromine from 7-methylxanthine), caffeine synthase (1,3,7-trimethylxanthine synthase, catalyzing the formation of caffeine from theobromine), and theacrine synthase (1,3,7,9-tetramethyluric acid synthase, catalyzing the formation of theacrine from caffeine) [13–16]. The 7-methylxanthine synthase gene (CmXRS1) was first cloned from coffee leaves by Mizuno et al. [15]. Its coding cDNA sequence is 1307 bp long and encodes a protein with 372 amino acids. Suzuki and Takahashi isolated and identified theobromine synthase and caffeine synthase from crude tea extracts for the first time [17]. The CgcTS gene, which encodes theobromine synthase, was cloned from young leaves of the tea plant Camellia gymnogyna Chang of Danyao Mountain, Guangxi [16]. Additionally, the TCS1 gene, which has a 1483-bp open reading frame encoding 369 amino acids, was first cloned from tea shoots [18]. Recently, theacrine synthase CkTcS1 isolated from Kucha was identified as an N-methyltransferase with novel activity, which uses theacrine as substrate instead of caffeine, indicating that C8 oxidation occurs prior to N9 methylation [19].
Although the biosynthetic pathway of caffeine has been elucidated and the key enzymes and their coding genes involved in the caffeine biosynthetic pathway have been identified, the molecular regulation of caffeine biosynthesis is still unclear [20–23]. So far, only a few transcription factors (TFs) related to the regulation of caffeine levels in plants have been discovered. MYB184 has been reported to bind and activate the TCS1 promoter, as confirmed by yeast single-hybridization experiments and transactivation assays [24]. The binding of MYB1AT to the fused MYBPLANT motif was further validated in vitro by an electrophoretic mobility shift assay (EMSA), which established MYB184 as a key activating agent of TCS1, and the production of caffeine in tea [24].
The mechanism and function of microRNA (miRNA) in the secondary metabolism of the tea plant has gradually developed into a new direction of modern tea plant research [25–28]. At present, most of the studies on the regulation of caffeine biosynthesis are from the perspective of structural genes and TFs, and fewer studies have been reported on the involvement of miRNAs in caffeine biosynthesis in the tea plant. miRNA serves as a key inhibitor in gene expression, primarily managing gene methylation during transcription and guiding mRNA translation in the post-transcription phase to control gene expression [29–30]. Its role extends to complex biological functions, regulating numerous target mRNA molecules. In plants, miRNA is integral in shaping plant morphology, facilitating signal transduction, orchestrating hormonal responses, enhancing stress tolerance, overseeing nutrient metabolism, and playing a vital part in various facets of plant growth and development [26–35]. The miRNAs have been shown to negatively regulate catechin biosynthesis through the downregulation of their biosynthesis-related target genes [36]. Epigallocatechin gallate from green tea suppresses the growth of 3T3-L1 cells through the miRNA-143/MAPK7 pathways [34]. Melatonin significantly increased transcript levels of several miRNAs, including miR-171b. Importantly, the α-glucan water dikinase (GWD) gene was identified as miR171b’s target [37]. Chlorophyll and starch degradation were improved and GWD gene expression was suppressed by miR171b overexpression [37]. These results suggest that melatonin may be able to delay dark-induced leaf senescence in tomato by inducing the expression of miR171b. In another study, miR319b was shown to target CsTCP3 transcription to control the development of the shoot tip and the biosynthesis of catechins in the tea plant [38]. Moreover, miRNAs were found to cross-regulate N-methyltransferase genes in Coffea canephora, which are involved in the biosynthesis of caffeine [25]. Nonetheless, there is a notable scarcity of research focusing on the pathway of caffeine biosynthesis in the tea plant, specifically concerning the regulatory role of miRNA in caffeine biosynthesis. This area merits further exploration and study.
In our study, we identified the CsbHLH1 as a possible central gene involved in the biosynthesis of the purine alkaloids of the tea plant. Yeast one-hybrid assays, EMSA assays, antisense oligonucleotide (As ODN) assays, and dual luciferase reporter gene assays have shown that CsbHLH1 can bind to the TCS1 promoter, inhibit the transcription of the initiating TCS1, and reduce caffeine content in tea plant shoots. The 5′-RLM–RACE (RNA ligase-mediated rapid amplification of cDNA ends) assay showed that miR1446a could positively regulate TCS1 biosynthesis by repressing the target gene CsbHLH1.
Results
Analysis of the main alkaloid contents of different tea plant cultivars
The results of the detection of the main alkaloid substances of the five different tea plant cultivars are shown in Fig. 1. Compared with the tea plant cultivars ‘Fuding Dabaicha’ (FDDB), ‘Yaoshan Xiulv’ (YSXL) and BXZ, ‘Jianghua Kucha’ cultivars [‘Xianghong 3’ (XH3H) and ‘Kucha 3’ (KC3H)] were significantly higher in caffeine content than the conventional tea plant cultivars, with XH3H having the highest caffeine content, 41.3 mg g−1 (Fig. 1G). YSXL had the lowest caffeine content. The highest levels of theobromine were found in XH3H (one of the ‘Jianghua Kucha’ cultivars), while KC3H (another ‘Jianghua Kucha’ cultivar) displayed the highest level of theacrine (Fig. 1G). It is worth noting that theacrine was not detected in any of the cultivars except KC3H, which contained 7.5 mg g−1. This indicates that there is a large difference in alkaloid composition between ‘Jianghua Kucha’ and conventional tea plant cultivars.
Figure 1.

High performance liquid chromatograms of standards and major alkaloids of five tea plant cultivars. A Standard. B Chromatograms of the main purine alkaloid substances of BXZ. C Chromatograms of the main purine alkaloid substances of FDDB. D Chromatograms of the main purine alkaloid substances of KC3H. E Chromatograms of the main purine alkaloid substances of XH3H. F Chromatograms of the main purine alkaloid substances of YSXL. G Main alkaloid components of different tea plant cultivars. The data represent the mean from three replicates. ND, not detected. Different small letters indicate a significant different difference among different treatments (P < 0.05).
Transcriptomic data analysis and differentially expressed gene identification and validation
Sequencing of 15 tea samples yielded an average of 6.39 Gb of high-quality data for all cDNA libraries, characterized by an average Q30 level of 97.36%, indicating a high degree of accuracy (Supplementary Data Table S1). The read mapping ratio to the reference genome ranged from 82.47 to 85.09% (Supplementary Data Table S2) for each sample. The values of the coefficient of determination for the FPKM (fragments per kilobase million) between biological duplicates were 0.96 ≤ R2 ≤ 0.99, indicating a good degree of reproducibility (Supplementary Data Fig. S2A).
In addition, 48 984 unique genes were found, of which 35 273 were differentially expressed in the 10 groups (Supplementary Data Fig. S2B). Twelve differentially expressed genes (DEGs) were selected for real-time qRT–PCR analysis aimed at validating the different patterns of expression that were observed in the RNA-seq data (Supplementary Data Table S3). We found that changes in expression levels of those obtained from the RNA-seq data were consistent with changes in expression levels from qRT–PCR (Supplementary Data Fig. S3). These results reinforced the reliability of the RNA-seq data obtained in this study.
GO and KEGG enrichment analysis of the differentially expressed genes
In order to further reveal the DEGs specific to ‘Jianghua Kucha’ cultivars (group 1: XH3H and KC3H) and conventional tea plant cultivars (group 2: FDDB, BXZ, and YSXL), comparative analyses of the DEGs between the two groups of tea plant cultivars were carried out. A total of 7026 DEGs were screened in group 2 versus group 1. Among them, 3198 were upregulated and 3828 were downregulated (Fig. 2A). The DEGs of group 2 versus group 1 mapped to 4141 GO terms (Supplementary Data Table S4). Specifically with this comparison, we found 111 GO terms that are potentially associated with caffeine biosynthesis. Of these identified GO terms, 81 were characterized under the biological process (BP) category, 7 under cellular components (CC), and 23 were associated with molecular functions (MF) (Supplementary Data Table S5). These GO terms are mainly related to TFs, purine ribonucleotide metabolic process, methionine biosynthetic process, purine nucleotide binding, S-adenosylmethionine-dependent methyltransferase activity, and N-methyltransferase activity (Supplementary Data Table S6; Fig. 2B). In addition, the group 2 versus group 1 comparator group was enriched for 139 pathways according to KEGG analysis (Supplementary Data Table S7). Notably, a number of pathways, such as carbon metabolism (ko01200), purine metabolism (ko00230), photosynthetic biocarbon fixation (ko00710), circadian-plant (ko04712), pyrimidine metabolism (ko00240), cysteine and methionine metabolism (ko00270), biosynthesis of various alkaloids (ko00996), tropane, piperidine and pyridine alkaloid biosynthesis (ko00960), and caffeine metabolism (ko00232) are related to caffeine biosynthesis (Fig. 2C). Purine and caffeine metabolism pathways are essential for the production of caffeine alkaloids, and the data showed that 57 DEGs were enriched in purine metabolism in the group 2 versus group 1 comparator group. Additionally, two DEGs were notably enriched in caffeine metabolism (Supplementary Data Table S8). We found that the major differential GO terms and KEEG pathways enriched were broadly consistent regardless of the comparison method.
Figure 2.
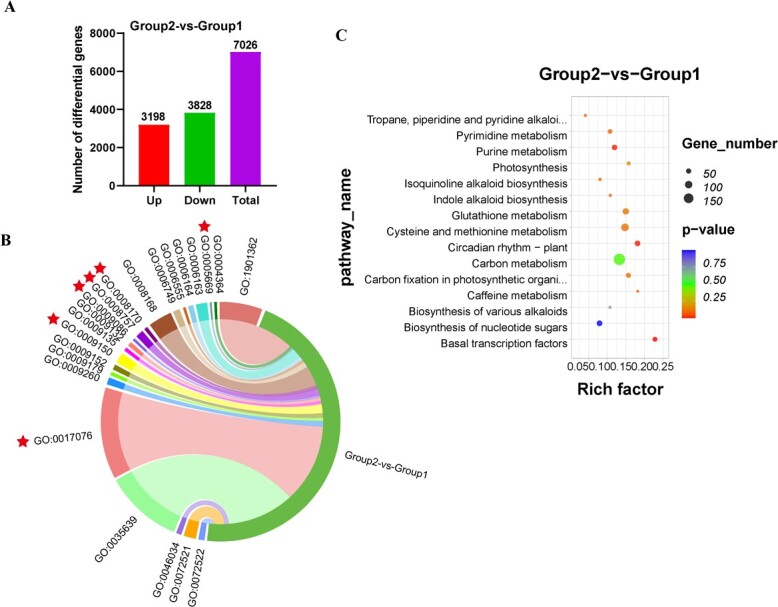
Pairwise comparisons of GO and KEGG enrichment analysis. A Analysis of DEGs in group 2 versus group 1. B Enrichment GO terms of DEGs from group 2 versus group 1. C KEGG enrichment terms of DEGs from group 2 versus group 1.
Differentially expressed genes involved in biosynthesis of purine alkaloids
Since the purine alkaloid content of ‘Jianghua Kucha’ tea differs from that of conventional tea cultivars, we then performed transcriptomic data analysis on these samples to look at the gene expression associated with them. Theobromine synthases (3-NMTs), caffeine synthases (TCS1), and theacrine synthases (9-NMTs) catalyze the last three N-methylations in the purine alkaloid biosynthetic pathway, respectively. Compared with other enzymes associated with this pathway, they are considered to be three key enzymes and have been extensively studied.
Expression levels of 3-NMTs and TCS1 in new shoots of KC3H and XH3H tea plants were much higher than those of YSXL (Fig. 3). The expression of 3-NMT and TCS1 genes followed the same trend as the metabolite content. Except for KC3H, transcription levels of two 9-NMT genes were zero or very low in the shoots of all other tea plant cultivars (Fig. 3). These results indicated that the alkaloid contents of five tea cultivars were consistent with the expression patterns of most 3-NMTs, 9-NMTs, and TCS1. Therefore, according to metabolite analysis and expression patterns of three key genes, 3-NMTs, 9-NMTs, and TCS1 positively regulate the biosynthesis of purine alkaloids.
Figure 3.

Biosynthesis pathways of purine alkaloids and expression of related genes. Biosynthesis pathways of purine alkaloids and expression of related genes. (1) SAMS: S-adenosine-l-methionine synthetase. (2) SAM-Mtases: S-adenosine- l-methionine dependent methyltransferase. (3) SAH hydrolase: S-adenosine-l-homocysteine hydrolase. (4) MTR: methionine synthase. (5) Adenosine nucleosidase. (6) APRT: adenine phosphoribosyl transferase. (7) ADK: adenosine kinase. (8) AMPD: adenosine 5′-monophosphate deaminase. (9) Inosine 5′-monophosphate dehydrogenase. (10) 5′-NT: 5′-nucleotidase. (11) GUD: guanosine deaminase. (12) 7-Methylxanthosine synthase. (13) 7-Methylxanthosine nucleosidase. (14) 3-NMTs: theobromine synthase. (15) TCS1: caffeine synthase. (16) 9-NMTs: theacrine synthases. (17) 7-Methyltransferase. (18) Theophylline synthetase. (19) 3-Methyltransferase. (20) 7-Methyltransferase. (21) 3-Methylxanthine synthetase. Group 1: ‘Jianghua Kucha’; group 2: conventional tea plant cultivars.
Several purine precursors of the purine alkaloids have been suggested (Fig. 3). SAMS catalyzes the conversion of methionine to S-adenosine-l-methionine (SAM). SAM is an important methyl donor in the biosynthesis of caffeine methylation. The expressions of SAMS (CSS0041489) and SAMS (CSS0037881) were the highest in the tender shoot of XH3H. The expressions of SAMS (CSS0016252) and SAMS (CSS0008561) were the highest in the tender shoot of KC3H (Fig. 3). S-Adenosine-l-methionine-dependent methyltransferases (SAM-Mtases) catalyze the conversion of SAM to S-adenosine homocysteine. The expressions of five SAM-Mtases, namely CSS0041977, CSS0034203, CSS0009658, CSS0027992, and CSS0014701 were the highest in KC3H (Fig. 3). The conversion of SAH to homocysteine and adenosine is catalyzed by S-adenosine-l-homocysteine hydrolase (SAHH). There was no difference in the expression of the SAHH gene among the five tea cultivars in this study. Adenosine kinases (ADKs) are catalysts for converting adenosine to adenosine 5′-monophosphate. Among these, ADK (CSS0018713) showed maximum expression in the KC3H variety, while ADK (CSS0012836) and ADK (novel G011025) were more prominently expressed in XH3H (Fig. 3). Adenosine riboside catalyzes the conversion of adenosine to adenine, and adenine phosphoribosyl transferase (APRT) catalyzes the conversion of adenine to adenosine 5′-monophosphate (AMP). All four APRT genes, novel_G008043, CSS0003599, CSS0029609, and CSS0038815, displayed the highest expression in one of the ‘Jianghua Kucha’ cultivars: XH3H or KC3H (Fig. 3). Furthermore, IMPDH, encoding inosine 5'-monophosphate dehydrogenase (IMPDH), catalyzes the conversion of inosine 5′-monophosphate (IMP) to xanthosine 5′-monophosphate (XMP). The 5′ nucleotide enzyme catalyzes the conversion of XMP to xanthosine. However, there was no difference in the gene expression level of IMPDH, among the five tea cultivars in this study. The 5′-nucleotide (5′-NT) is responsible for the conversion of guanosine 5′-monophosphate (GMP) to guanosine. The expression levels of 5′-NT and GUD genes were the highest in XH3H, followed by KC3H (Fig. 3).
TFs have been considered to play a crucial role in the regulation of the biosynthesis of purine alkaloids in tea plants. To further understand the potential function of these TFs in purine alkaloid biosynthesis in tea plants, we systematically analyzed DEGs encoding TFs. We identified a total of 213 differentially expressed TFs from 13 different families, which included MYB, bHLH, AP2-EREBP, RAS, SBP, NAC, Trihelix, WRKY, TCP, ABI3VP1, bZIP, C2H2, and Tify (Supplementary Data Table S9). Correlation values were calculated using the R language based on FPKM values of 213 TFs and key genes for purine alkaloid biosynthesis. We specifically focused on TFs with correlation coefficients >0.65 (Supplementary Data Table S9). Cytoscape software was used to draw positive and negative correlation network interactions between TFs and key genes for purine alkaloid biosynthesis (Fig. 4). The TFs showing the most interactions belonged to the bHLH, AP2-EREBP, MYB, RAS, SBP, and ABI3VP1 families (Supplementary Data Table S9). Based on these findings, we speculate that the biosynthesis of alkaloids may involve these TFs.
Figure 4.
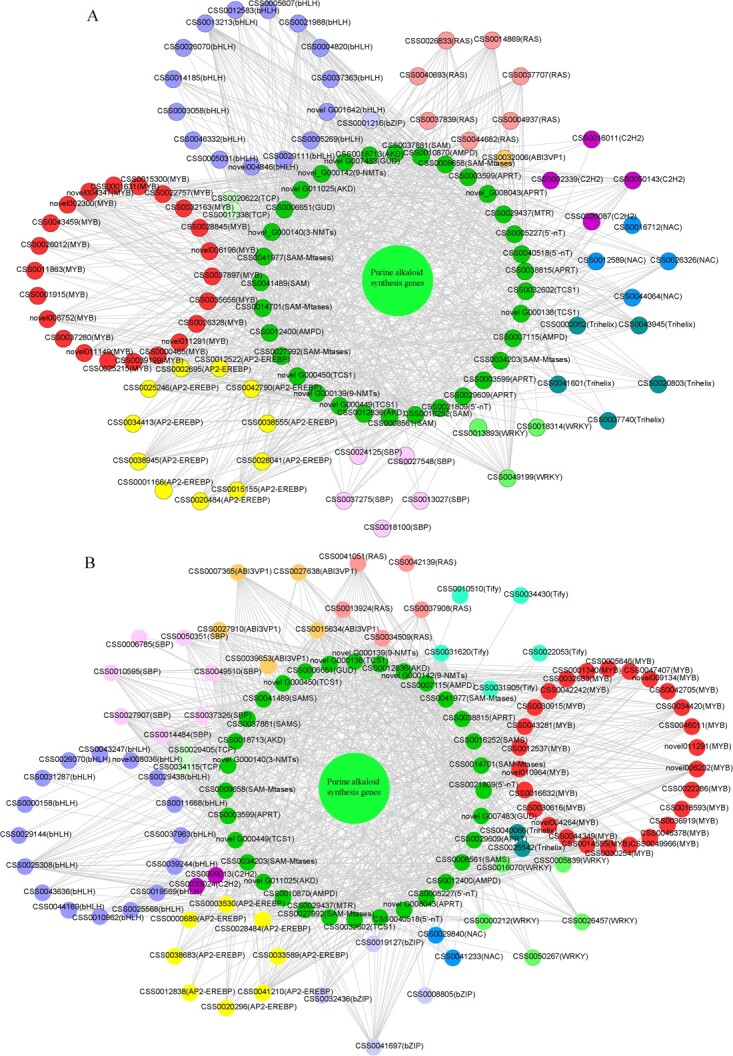
All identified TFs in the TF-mRNA network. A Network interaction of key genes of purine alkaloid biosynthesis positively correlated with TFs (R2 > 0.65). B Network interaction of key genes of purine alkaloid biosynthesis negatively correlated with TFs (R2 > 0.65).
Co-expressed genes related to purine alkaloid content
The application of weighted gene co-expression network analysis (WGCNA) enabled us to identify gene sets associated with purine alkaloid traits (theobromine (TB), caffeine (CAF), theacrine (TC), and Theophylline (TP)) in tea plants. This analysis used a dataset of 35 273 DEGs and associated phenotypes. Twenty-five different co-expression modules (marked by 25 different colors; gray modules are sets of genes not assigned to other modules) of WGCNA were produced, containing closely connected gene clusters with high correlation coefficients between genes in the same cluster (Fig. 5A). The turquoise module aggregated the most genes, with 5599 genes, and saddle brown had only 92 genes, averaging 1310 genes per module (Fig. 5B). The ME magenta module (682) was strongly correlated with TB (r = 0.6, P = 0.02), the ME brown module (1098) was significantly and positively correlated with TC (r = 0.69, P = 0.004), and the ME cyan module (390) with TP (r = 0.69, P = 0.004) (Fig. 5B). In addition, the ME green module (980) was significantly negatively correlated with CAF (r = −0.67, P = 0.03) and the ME midnight blue module (372) with TB (r = −0.82, P = 2e−04) (Fig. 5B). These results suggest that the genes most likely to regulate purine alkaloid formation in the tea plant are those belonging to these five modules.
Figure 5.

Co-expression network analysis of tea plant purine alkaloids. A WGCNA analysis-based hierarchical clustering tree of modules. B Relationships between modules and the characteristics of the samples tested. The numbers represent R and the numbers in parenthesis represent P values. C Co-expression network diagram of candidate genes in the ME magenta module. D Candidate gene co-expression network diagram in the ME green module. (E) ME brown module co-expression network diagram of candidate genes. F Co-expression network diagram of the candidate genes in the ME cyan module.
In addition, we selected the TFs and purine biosynthesis-related genes as key hub genes using the top 100 unigene pairs in the magenta, blue, brown, and cyan modules to construct the co-expression network. In all, 15 key hub genes have been identified, which include two ARF, two GRAS, bHLH, C3H, MYB, TUB, Alfin-like, zf-HD, and TCP TF genes in the ME magenta module (Fig. 5C). Additionally, we identified three pivotal genes (APRT, ATPS, and SAM) that play a central role in the purine alkaloid biosynthesis pathway (Fig. 5C). Eight genes were selected as key hub genes for the green ME module. Of these, seven were TF genes, including one C2C2-GATA, one Trihelix, one CPP, one GRAS, one mTERF, one RWP-RK, and one bHLH (Fig. 5D). In addition, there was one TCS gene in the purine alkaloid biosynthesis pathway (Fig. 5D). A total of 15 key hub genes were selected for the ME brown module. Of these, 11 were TFs. These included two GRAS, two ARF, one bHLH, one zf-HD, one TUB, one C3H, one TCP, one MYB, and one Alfin-like gene (Fig. 5E). In addition, two ATPS genes, one APRT gene, and one SAM gene were identified in the purine alkaloid biosynthesis pathway (Fig. 5E). In the cyan module of the ME, 24 genes were selected as the most important hub genes. Of these, 20 were TF genes. These included three AP2-EREBP, three MYB, two ABI3VP1, two TUB, one WRKY, one bHLH, one bZIP, one Trihelix, one C3H, one C2C2-Dof, one DBP, one LIM, one MADS, one FHA, and one GRAS (Fig. 5F). In addition, four ATPase genes related to the purine alkaloid biosynthesis pathway were included (Fig. 5F). These analyses suggest that the molecular regulatory mechanism of purine alkaloid formation in the tea plant is complex, involving many different functional genes related to purine alkaloid biosynthesis, such as the TF family.
Interaction analysis of miRNAs and target genes
In total, 123.08 Gb of clean tags were obtained from the five tea samples. After filtering, the total clean reads of each sample in this project were from 20.95 to 23.88 M. The genome mapping rate ranged from 86.92 to 89.22%. The sncRNA mapping rate accounted for 11.49–8.65%. Non-coding RNAs were identified as snoRNA (406), rRNA (321), sRNA (237), cis-reg (199), tRNA (94), lncRNA (66), CRISPR (59), Rfam_other_RNA (50), ribozyme (17), antisense (15), other (15), snRNA (12), antitoxin (6), misc_RNA (4), the predicted non-coding RNA as miRNA (6422), and siRNA (1015). In addition, 77 differential genes were common to the four comparator groups (Fig. 6A). Results of GO enrichment analysis showed that DEGs were mainly enriched in methyltransferase activity, purine nucleotide binding, S-adenosylmethionine-dependent methyltransferase activity, N-methyltransferase activity, and carbohydrate derivative binding in the four comparator groups (Fig. 6B). KEGG enrichment analysis showed that the main metabolic pathways enriched for differential genes in the four comparison groups were carbon metabolism, MAPK signaling pathway-vegetation, purine metabolism, glutathione metabolism, pyrimidine metabolism, scopolamine, caffeine metabolism, and indole alkaloid biosynthesis (Fig. 6C).
Figure 6.

Pairwise comparisons of differential gene GO and KEGG enrichment analysis of miRNAs. A Venn diagram of co-expressed DEGs and uniquely expressed DEGs for the four comparisons. B Overall enrichment terms of DEGs of FDDB versus XH3H, FDDB versus KC3H, FDDB versus YSXL, and FDDB versus BXZ comparisons. C KEGG enrichment terms of DEGs of FDDB versus XH3H, FDDB versus KC3H, FDDB versus YSXL, and FDDB versus BXZ comparisons.
In addition, KEGG enrichment analysis allowed the identification of 140, 137, 137, and 138 metabolic pathways for the four comparator groups. Of these, four pathways, ko00270, ko00230, ko00232, and ko00480, were associated with purine alkaloid biosynthesis, comprising 870, 909, 979, and 839 differentially expressed target genes, respectively (Supplementary Data Fig. S4A). Analysis of differentially expressed target genes and miRNA interactions in the purine alkaloid biosynthesis pathway further showed that 203 miRNAs were localized to the comparator group, with 36, 42, 17, and 27 upregulating gene expression and 48, 60, 29, and 35 downregulating gene expression, respectively (Supplementary Data Fig. S4B). The correlation analysis between target genes in the transcriptome and DEGs suggested that the TCS1 gene (CSS0032602.3) associated with caffeine biosynthesis may be under the regulatory control of several miRNAs. These include novel-miRNA-2129-5p, novel-miRNA-2130-5p, novel-miRNA-1821-3p, novel-miRNA-1822-3p, novel-miRNA-1823-3p, novel-miRNA-2083-3p, and novel-miRNA-849-5p. These miRNAs have been identified as primary candidates for involvement in the regulation of purine alkaloids (Supplementary Data Fig. S4C and Supplementary Data Table S10).
In the above study, we calculated correlation values based on the FPKM values of 213 TFs and key genes for purine alkaloid biosynthesis. A total of 167 TFs with correlation coefficients >0.65 were screened. Among these, the TF CsbHLH1 (CSS0025308) showed a strong correlation, ranging from −0.97 to −0.65, with 10 critical genes involved in purine alkaloid biosynthesis (Supplementary Data Table S9). Based on 35 273 DEGs and phenotypic data, WGCNA captured the set of genes associated with purine alkaloid traits (TB, CAF, TC, and TP) in the tea plant. The correlation coefficient between CsbHLH1 and novel G000139 (TCS) in the green module was −0.87. In addition, using the JASPAR in-line software for promoter binding prediction, we found that the bHLH TF CsbHLH1 was able to bind to the TCS1 promoter, scoring 10, the highest score among all bHLH TFs predicted to bind the same promoter. Thus, CsbHLH1 was chosen for further investigation.
Subcellular localization analysis of CsbHLH1
In order to determine the subcellular localization of CsbHLH1, pSuper1300-CsbHLH1-GFP was transiently expressed in tobacco leaves, while pSuper1300-GFP was used as a control in the epidermal cells of tobacco leaves. The results indicated that CsbHLH1 was located in the nucleus (Fig. 7A).
Figure 7.

CsbHLH1 is localized in the nucleus of the cell and has transactivating activity, and it binds to the promoter of CsTCS1. A Subcellular location of CsbHLH1 in tobacco leaf; scale bars = 10 μm. B Transcriptional transactivation in tobacco. C Transcriptional activity of CsbHLH1 in yeast cells. D Protein purification to obtain a fusion protein of MBP-CsbHLH1. M, protein marker; 1 and 2, supernatant and precipitation of 0.5 mmol/l IPTG-induced protein; 3 and 4, supernatant and precipitation of 0.7 mmol/l IPTG-induced protein; 5, 6, and 8, purified CsbHLH1 recombinant protein washing solution. 7, concentrated CsbHLH1 recombinant protein washing solution. E EMSA. From left to right: negative control; positive control; CsTCS1 probe; CsTCS1 probe bound to CsbHLH1 protein; CsTCS1 mutant probe; CsTCS1 mutant probe and CsbHLH1 protein; CsbHLH1, CsTCS1 mutant probe, and 10× CsTCS1 competitive probe; CsTCS1 mutant probe and 50× CsTCS1 competitive probe; CsTCS1 mutant probe and 100× CsTCS1 competitive probe. The 10× and 50× CsTCS1 competitive probes can compete with part of the strip, and the 100× CsTCS1 competitive probes can compete with all the strip. F Gene expression of CsbHLH1 in leaves after As ODN_CsbHLH1 treatment expression. G Caffeine content in young shoots treated with water and As ODN_CsbHLH1. H Relative expression of CsTCS1 after inhibition of CsbHLH1.
CsbHLH1 represses transcription of the TCS1 promoter
CsbHLH1 transcription was assessed using a double luciferase reporter gene analysis system. The results showed that the LUC/REN values of the reporter gene co-injected with pGreenII 62-SK-CsbHLH1 were significantly (P < 0.05) lower than those of the negative control (pGreenII 62-SK-Empty) (Fig. 7B). This indicates that the CsbHLH1 protein is able to inhibit the transcription of the TCS1 promoter.
Yeast one hybrid assays with CsbHLH1
To further investigate the transcriptional activating activity of CsbHLH1, recombinant plasmids containing the corresponding sequences were transferred to yeast monohybrid (Y187) receptor cells and grown in the appropriate media. pHis2-bait plasmids were obtained by constructing the TCS1 promoter (−953 to −1556 bp) into the pHis2 vector and the complete coding sequence of CsbHLH1 was constructed into the pGADT7 vector to obtain the AD-prey plasmid. pHis2-bait and AD-prey plasmids were mixed as the experimental group, and pHis2-bait and empty pGADT7 plasmids were mixed as the self-activation group. On the SD/−His/−Leu/−Trp medium, the growth of the Y187 strain transformed by the recombined vectors harboring the promoters of either TCS1 was inhibited by 50 mM 3-amino-1,2,4-trisole (3-AT). After transformation with the prey vector pGADT7-CsbHLH1, the strains with TCS1 grew and produced few colonies compared with the pGADT7 empty vector control on the SD/−His/−Leu/−Trp plates containing 50 mM 3-AT at the same dilution (Fig. 7C).
Electrophoretic mobility shift analysis of the TCS1 promoter
The fusion protein MBP-CsbHLH1 was obtained by IPTG (isopropyl-d-1-thiogalactopyranoside) induction and protein purification, setting the stage for the EMSA assay (Fig. 7D). The promoter sequence of TCS1 (−971 bp to −1019 bp) contains a predicted MYC binding site (CATGTG) and the EMSA results showed that CsbHLH1 was able to bind to the MYC binding site (CATGTG) in the TCS1 promoter (Fig. 7E). The mutation probe was synthesized by mutating the sequence of the MYC site (CATGTG) in its entirety to the A base, ultimately identifying the TCS1 promoter binding region.
Impact of inhibition of CsbHLH1 on caffeine biosynthesis
The oligodeoxynucleotide As ODN_CsbHLH1, designed to target and suppress the expression of CsbHLH1, proved effective in achieving significant inhibition after 24 and 48 h in samples placed in centrifuge tubes (Fig. 7F). This is evidence of the effective inhibitory capability of As ODN_CsbHLH1. Furthermore, we observed a notable increase in the caffeine content in the tea plant shoots treated with As ODN_CsbHLH1 at the 24- and 48-h marks, compared with the control group treated with water (Fig. 7G). This suggests that the suppression of CsbHLH1 gene expression is directly associated with an uptick in caffeine concentration.
Characterization of miR1446a target gene
CsbHLH1 was predicted using psRNATarget software based on miRNA information in tea leaves obtained by bioinformatics and high-throughput sequencing. Thirty-three miRNAs were predicted to be able to cleave CsbHLH1 (Supplementary Data Table S11). Cleavage site validation was performed using modified RLM–RACE. Our findings unequivocally demonstrate that miR1446a specifically targets and cleaves the downstream TF CsbHLH1, an integral component in the regulation of caffeine biosynthesis (Supplementary Data Fig. S5). This key observation provides compelling evidence for the suppressive role of miR1446a in the regulation of CsbHLH1 expression.
Discussion
In the tea plant, the primary purine alkaloids include caffeine, theobromine, theophylline, and theacrine, caffeine being the most prevalent [4]. Typically, caffeine accounts for 2–5% of the dry matter content, making it a key quality and functional component of tea [39]. Consequently, the process of caffeine biosynthesis has garnered significant attention [6]. While the pathway of caffeine biosynthesis is now largely understood, the details of its regulatory mechanisms still require further exploration and study.
In order to reveal the transcriptional regulation mechanism of caffeine in tea, we obtained five tea plant cultivars with gradient changes in caffeine level (ranging from 41.3 to 18.1 mg g−1), selected from a pool of >50 different resources. The five chosen tea cultivars served as excellent resources for isolating the key regulatory genes. This was achieved by analyzing the relationship between caffeine content and gene expression levels using transcriptome data and WGCNA. WGCNA has been widely used in mining transcriptionally regulated genes for secondary metabolite biosynthesis [40–42]. It also helps to identify highly related gene clusters and reveal their regulatory networks in plant secondary metabolite biosynthesis. In a previous study, three modules related to marker metabolites were identified, and key structural genes potentially influencing hypoxic germination were pinpointed from these modules through a WGCNA analysis of targeted metabolomics and transcriptomics [43]. Recently, the use of the WGCNA approach to establish a link between tea aroma accumulation and gene expression revealed 13 TF genes that may be involved in terpene metabolism [44]. Among these genes, CsOCS2 was verified in vitro to play a role in terpene biosynthesis, indicating the reliability of WGCNA for trait-related gene discovery [44]. In this study, our transcriptome analyses revealed a greater GO enrichment of methionine biosynthetic process, S-adenosylmethionine-dependent methyltransferase activity, and N-methyltransferase activity in ‘Jianghua Kucha’ cultivars compared with conventional tea cultivars (Fig. 2B and Supplementary Data Table S6). Furthermore, the KEGG pathways were more enriched in purine and caffeine metabolisms (Fig. 2C and Supplementary Data Table S8). The expression pattern of CsbHLH1 was negatively correlated with the expression pattern of TCS1 and showed an opposite trend to the caffeine content (Figs 1G and4B and Supplementary Data Table S9). Based on WGCNA analysis, we found a novel TF, CsbHLH1, which regulates the gene expression of TCS1 (Fig. 5).
The bHLH TFs, which have been identified across a wide range of cultivars, hold a crucial place in the biosynthetic regulation of plant polyphenols [45–50]. The overexpression of PabHLH in both callus and thallus boosted bisbibenzyl accumulation and upregulated PaPAL, Pa4CL1, PaSTCS11, and two P450 cytochrome-encoding genes [54]. The bHLH-related TF DcTT8, isolated from Dendrobium candidum was involved in anthocyanin biosynthesis in D. candidum stems, through regulation of the expression of DcF3′H and DcUFGT [55]. The bHLH TF AcB2 has been found to regulate anthocyanin biosynthesis in onion (Allium cepa L.) [51]. Luo et al. reported that CsbHLH62 negatively regulates EGCG3″Me biosynthesis in C. sinensis [52]. Furthermore, SlAN11 in tomato has been associated with the regulation of flavonoid biosynthesis through its interaction with bHLH proteins [53]. This study uncovered a novel function of the bHLH TF, revealing that CsbHLH1 plays a role in reducing caffeine levels in ‘Jianghua Kucha’ varieties, including KC3H and XH3H (Fig. 7G and H). An intriguing correlation was found between the expression pattern of CsbHLH1 and caffeine content when analyzing the major purine alkaloid content of five tea cultivars (Figs 1G and4B), suggesting that CsbHLH1 could act as a negative regulator of caffeine content.
In order to verify the function of CsbHLH1, we inhibited the expression of CsbHLH1 using As ODN technology. This suppression of CsbHLH1 led to an increase in caffeine level in the leaves of the tea plant (Fig. 7G). The As ODN method involves utilization of synthetic or constructed antisense expression vectors for the expression of oligonucleotide fragments [56]. These fragments, based on the principle of base complementarity, interfere with various aspects of gene functions, including replication, transcription, and mRNA splicing, processing, export, and translation. This interference can effectively regulate cellular processes such as growth and differentiation [56]. This technique is unique in that it focuses on the transient silencing of genes to study their function in plants [57], which can compensate to some extent for the shortcomings of other technologies by not altering plant traits when selecting for transgenic plants compared with marker genes, or by silencing genes in plants where transgenic challenges exist, such as tea plants [58]. The efficacy of As ODN approaches in transiently suppressing gene expression is well documented. CsFtsH5-AsODN plants have shown significantly higher relative electrolyte leakage values than CsFtsH5-AsODN plants under freezing stress, indicating increased cold sensitivity in tea plants as a result of downregulation of CsFtsH5 [59]. In our research, we observed that lowering the expression of CsbHLH1 directly led to an increase in both TCS1 expression and caffeine biosynthesis (Fig. 7G and H). This finding solidifies the conclusion that CsbHLH1 acts as a negative regulator of TCS1.
Additionally, this study delved into the interaction between CsbHLH1 and TCS1. The EMSA result showed that CsbHLH1 specifically binds to the MYC binding site in the promoter region of TCS1 (Fig. 7E). CsbHLH1 directly targets TCS1 to reduce its expression, which in turn influences caffeine biosynthesis in the tea plant. We transiently co-expressed the reporter (promoter) and effector (TF) by Agrobacterium-mediated transformation of tobacco leaves. The LUC/REN ratio of 0.079 indicated that the CsbHLH1 protein inhibits the transcription of TCS1 (Fig. 7B). Gene expression correlation analysis, gene function validation, and interaction analysis confirmed that CsbHLH1 is a TF of TCS1. Previous research has identified CsMYB184, CsNAC7, and CsS40 as TFs that regulate TCS1, suggesting that multiple TFs may simultaneously influence the regulation of the same gene [22–24]. This is corroborated by the fact that multiple TFs regulate transcription of the rpoE gene in Escherichia coli [60]. This was verified by the fact that two TFs, TaPpm1 and TaPpb1, co-regulate anthocyanin biosynthesis in purple pericarps of wheat [61].
While previous research has highlighted the significant role of bHLH TFs in plant metabolite production, the exact regulatory mechanisms of these bHLH factors remain largely unknown. The miRNA sequencing in our study revealed that miR1446a targets and cleaves CsbHLH1, thereby regulating the transcription of TCS1 (Supplementary Data Fg. S5), which leads to increased caffeine accumulation. The miRNAs, as key regulators in plants, significantly contribute to the orchestration of plant secondary metabolism pathways [62, 63], which influences the biosynthesis of secondary metabolites in plants mainly through two pathways. Firstly, miRNAs act directly on genes related to secondary metabolism pathways to cleave target genes and negatively regulate the accumulation of secondary metabolites. Secondly, miRNAs act on transcriptional regulators, further impacting plant secondary metabolism [64, 65]. miRNAs in Arabidopsis were found to target MYB114 and PAP2, playing a significant role in the biosynthesis of anthocyanin [65, 66]. Arabidopsis miRNAs can also cleave the target gene SPL (Squamosa promoter binding protein-like), a negative regulator of anthocyanin biosynthesis, thereby disrupting the WD40-bHLH-MYB complex. By targeting and cleaving the SPL, miR156 promotes anthocyanin accumulation [67]. Conversely, a reduction in miR156 expression bolsters anthocyanin accumulation [67]. Our finding implied a novel role for miR1446a in the regulation of caffeine biosynthesis in plants.
Conclusion
In conclusion, our findings indicate that the CsbHLH1 TF binds to the promoter region of TCS1, specifically between −971 and −1019 bp, effectively repressing the transcriptional activity of TCS1. Furthermore, we demonstrated that As ODN_CsbHLH1 effectively represses the expression of the CsbHLH1, which subsequently leads to an increase in caffeine content. In addition, we discovered that miR1446a precisely targets and cleaves CsbHLH1, consequently suppressing its expression and subsequently influencing caffeine biosynthesis in the tea plant. Based on these results, we propose a possible regulation model (Fig. 8) of caffeine in the tea plant, which provides new insights into the regulation of caffeine biosynthesis and lays the foundation for breeding tea plants with low or high caffeine content. Despite our progress, numerous questions remain about the regulatory mechanisms of caffeine biosynthesis. For instance, it is unclear whether allelic variations in CsbHLH1 correlate with the expression of TCS1 or other key genes involved in caffeine biosynthesis. Additionally, understanding how CsbHLH1 cooperates with other reported TFs of TCS1 to regulate caffeine production in tea plants requires further exploration.
Figure 8.
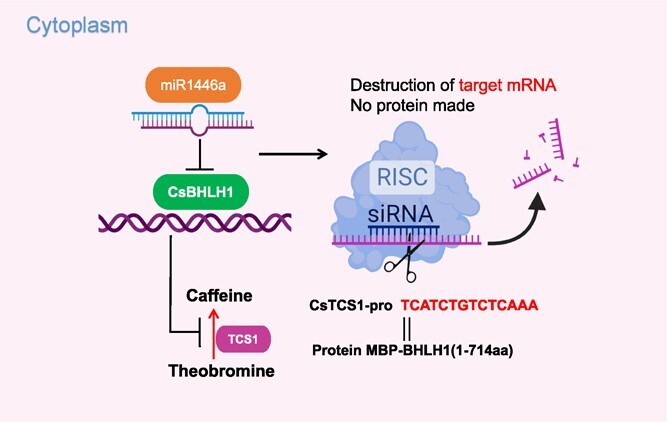
A possible model for the involvement of CsbHLH1 and miR1446 in the biosynthetic regulation of tea plant caffeine.
Materials and methods
Tea plant materials
Five tea plant cultivars, namely KC3H, XH3H, BXZ, FDDB, and YSXL, were used as experimental materials. XH3H and KC3H belong to ‘Jianghua Kucha’, FDDB originated in Southeast China's Fujian Province, YSXL in Southwest China’s Guangxi Province, and BXZ in central China’s Hunan Province. The five tea cultivars were collected from the tea gardens of the Tea Research Institute of the Hunan Academy of Agricultural Sciences in Changsha, Hunan Province, China. One bud and one leaf of healthy tea plants (KC3H, XH3H, BXZ, FDDB, and YSXL) were collected in spring (April 2021) from 8:30 to 11:00 a.m. Freshly collected samples were stored in liquid nitrogen at −80°C for transcriptome, metabolome, and small RNA analysis and subsequent experiments. Three biological replicates of each cultivar were used for each of the above samples.
Sample preparation for alkaloid determination
Samples were lyophilized and ground to a fine powder. About 10 mg of tea sample was weighed into a 2-ml centrifuge tube, and 0.5 ml of 70% hot methanol was added. Samples were treated for 10 min in a 70°C water bath. Samples were then centrifuged at 12 000 rpm for 10 min. The supernatant was used for analysis by LC–MS/MS.
LC–MS/MS analysis
Analyses were performed on a Shimadzu Nexera X2 LC-30 AD high-performance LC system (Kyoto, Japan). The separations were carried out on an Acquity BEH C18 column (2.1 × 100 mm) with 1.7-μm particles from Waters (Milford, MA, USA), which was maintained at 40°C throughout the separation. The optimum gradient used A (0.1% formylic acid (FA) in water)–B (0.1% FA in acetonitrile) as eluent. The gradient conditions are summarized in Supplementary Data Table S12. The injection volume was 1 μl and the flow rate was 0.3 ml/min.
Mass spectrometry conditions
Detection was carried out using an AB SCIEX 5500 series ion trap, which was equipped with an ESI source and operated in the positive ion mode. The source temperature was 550°C, ion source gas 1 (GS1) 55, ion source gas 2 (GS2) 55, curtain gas (CUR) 35, and ion spray voltage (IS) 5500 V. Detection of metabolites was performed in a positive multiple reaction monitoring (MRM) mode.
Transcriptome sequencing, RNA preparation, and qRT–PCR
Fifteen cDNA libraries were constructed using total RNA from BXZ, FDDB, XH3H, YSXL, and KC3H. Transcriptome sequencing was performed on triplicate samples, which was carried out by BGI Genomics (Shenzhen, China) using the DNBSEQ platform. FPKM values of key differential genes were analyzed in this study.
RNA preparation and qRT–PCR were performed according to previously described methodology with minor modifications [68]. Total RNA from the tea samples was isolated and purified using the RNAprep Pure Plant Kit (Tiangen Biotech, Beijing, China). Subsequently, cDNA was reverse-transcribed using a One-Step gDNA Removal kit (TransGen Biotech, Beijing, China) and was then used as template for qRT–PCR according to the Two-Step qRT-PCR Kit instructions (TransGen Biotech, Beijing, China), with the internal reference gene GAPDH. The analysis was performed using 2−ΔΔCT, and qRT–PCR primers are listed in Supplementary Data Table S9 [69].
Small RNA library construction and sequencing
Small RNA sequencing was performed by BGI Genomics on triplicate samples using the Solexa platform. The cleaned target sequences were classified and annotated by GenBank and Rfam database comparison, and the statistics of sequence length distribution as well as the statistics of public sequences among samples and the types and numbers of unique sequences were determined. Sequences were aligned to miRBase 21.0 using the nucleotide–nucleotide Basic Local Alignment Search Tool (BLASTn) to identify known conserved miRNAs. To identify new miRNAs we used the Mireap program (http://sourceforge.net/projects/mireap). Finally, the corresponding identified known miRNAs and new miRNAs were subjected to target gene prediction and functional analysis.
Subcellular localization of CsbHLH1
A bacterial broth containing 35S::CsbHLH1-GFP was injected into the leaves of tobacco plants and the leaves that were injected were used as the experimental group. At the same time, tobacco leaves were injected with pSuper1300-GFP empty vector as a control group. The injected tobacco plants were incubated for 36 h (23 ± 2°C, 85% relative humidity) and their leaves were cut into square pieces (2 mm × 2 mm) at the injection site [70]. The results of the experiments were observed using a laser scanning confocal microscope (LSCM; Nikon Corporation, Tokyo, Japan).
Dual-luciferase reporter assays
The dual-report vector included the smallest TATA region of firefly luciferase (LUC) driven by five GAL4 binding elements (5 × GAL4) and CaMV 35S. Renilla luciferase (REN) driven by the 35S promoter was used as the control. The full-length cDNA of CsbHLH1 was cloned into pGreenII 62-SK vector, and the recombinant plasmid pGreenII 62-SK-CsbHLH1 was constructed as effector. At the same time, the promoter sequence of TCS1 was cloned into pGreenII 0800-LUC dual reporter vector as the reporter. Next, these plasmids were introduced into Agrobacterium tumefaciens for transient expression and the preparation was cultured for 48–72 h [71]. The Dual-Luciferase® Reporter Assay System kit was used to determine the ratio of LUC to REN luciferin in tobacco leaves.
Yeast one-hybrid assays
The full-length sequence of CsbHLH1 was linked to the yeast expression vector pGADT7, leading to creation of recombinant plasmid pGADT7-CsbHLH1 (1–714 aa). Simultaneously, the promoter sequence of TCS1 (−953 to −1556 bp) was ligated into the pHis2 vector, resulting in the recombinant plasmid pHis2-TCS1. The pHis2-TCS1 and pGADT7-CsbHLH1 plasmids were mixed as the experimental group. The pHis2-TCS1 and empty pGADT7 plasmids were mixed as the self-activating group. The yeast strain Y187 was then transformed with these plasmids. The yeast cells were then inoculated on triple-deficient (SD/−His/−Leu/−Trp) medium with or without 3-AT in serial dilutions and incubated at 28°C for 3 days [72]. The formation of white colonies in the experimental group and lack thereof in the self-activation group would suggest successful transactivation.
Electrophoretic mobility shift assay
The pMAI-C6T-CsbHLH1 fusion construct was formed by inserting the full-length CsbHLH1 sequence into the pMAI-C6T expression vector. The recombinant plasmid was inserted into BL21 receptor cells and the addition of 0.7 mM IPTG induced protein expression [73]. Protein purification was then carried out using the Ni-NTA Sefinose Resin Kit (Sangon Biotech, Shanghai, China).
For the biosynthesis of the universal probe, a 102-bp DNA fragment containing the promoter region of an MYC element at the 5′ end of TCS1 was used. The sequence of the competing probes was identical to that of the universal probe. The mutant probe was synthesized by replacing the sequence of the MYC site (CATGTG) with the base A, while retaining the rest of the sequence identical to the universal probe [74]. DNA probes for EMSA were prepared at Wuhan Jinkairui Bioengineering Co. Ltd (Wuhan, China). Probes and samples were loaded on 6% polyacrylamide gels and run in TBE buffer.
Antisense oligonucleotide experiment
Several optimal As ODNs were designed and selected based on the sequences of the tea plant gene CsbHLH1 using the online software Soligo [75] (http://sfold.wadsworth.org/cgi-bin/index.pl). Tea plant shoots (a bud and two leaves) were placed in a centrifuge tube with 20 μM CsbHLH1-related As ODNs (placed in water as control). Samples were taken after 6, 24, and 48 h to detect changes in CsbHLH1 gene expression and caffeine content.
Validation of cleavage sites using modified RLM–RACE
A quantity of 1 μl from each of the qualified RNA samples was mixed, and was then used for modified RLM–RACE reverse transcription. The GeneRacer kit (Invitrogen, USA) was used for RLM–RACE following the specific test procedure described elsewhere [76]. The target gene cleavage primers were designed using DNAMAN and nested PCR amplification was performed using specific and universal primers (Supplementary Data Table S3). TA cloning of these isolated products was performed, followed by the selection of multiple positive single colonies for sequencing. Sequencing was carried out by T&J Bioengineering (Shanghai) Co. Ltd.
Acknowledgements
This work was supported by the Natural Science Foundation of Hunan Province (2021JC0007), the National Natural Science Foundation of China (U22A20500, U19A2030, 32172629), and the key Science and Technology Research Projects of Hunan Province (2021NK0008, 2021NK1020).
Author contributions
Q.J. designed and performed the experiments and drafted the paper. Z.W. analyzed the data and prepared the figures and tables. DS performed data analysis and reviewed and revised the manuscript. L.C., C.S., F.S., S.X., F.H., Z.C., X.Z., J.H., G.L., Q.S., and M.H. participated in the experiments and data analysis. Z.L., J.H., N.T., and S.L. conceived the study, participated in the coordination, data analysis, and interpretation, and drafted and reviewed the manuscript. All authors read and approved the final manuscript.
Data availability statement
All the data supporting our findings are contained within the manuscript. All raw transcriptome data reported in this article have been deposited in the Sequence Read Archive (SRA) under accession number PRJNA986690 and PRJNA1056246.
Conflict of interests
The authors declare they have no competing interests.
Supplementary information
Supplementary data is available at Horticulture Research online.
Supplementary Material
Contributor Information
Qifang Jin, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Zhong Wang, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Devinder Sandhu, United States Salinity Laboratory, United States Department of Agriculture, Agricultural Research Service, Riverside, CA 92504, USA.
Lan Chen, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Chenyu Shao, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Fanghuizi Shang, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Siyi Xie, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Feiyi Huang, Tea Research Institute, Hunan Academy of Agricultural Sciences/National Small and Medium Leaf Tea Plant Germplasm Resource Nursery, Changsha 410128, China.
Zhenyan Chen, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Xiangqin Zhang, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Jinyu Hu, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Guizhi Liu, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Qin Su, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Mengdi Huang, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Zhonghua Liu, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Jianan Huang, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Na Tian, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
Shuoqian Liu, Key Laboratory of Tea Science of Ministry of Education, Hunan Agricultural University, Changsha 410128, China; National Research Center of Engineering and Technology for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China; CoInnovation Center of Education Ministry for Utilization of Botanical Functional Ingredients, Hunan Agricultural University, Changsha 410128, China.
References
- 1. Wang S, Chen J, Ma J. et al. Novel insight into theacrine metabolism revealed by transcriptome analysis in bitter tea (Kucha, Camellia sinensis). Sci Rep. 2020;10:6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wu W, Lu M, Peng J. et al. Nontargeted and targeted metabolomics analysis provides novel insight into nonvolatile metabolites in Jianghua Kucha tea germplasm (Camellia sinensis var. assamica cv Jianghua). Food Chem X. 2022;13:100270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jin JQ, Jiang CK, Yao MZ. et al. Baiyacha, a wild tea plant naturally occurring high contents of theacrine and 3″-methyl-epigallocatechin gallate from Fujian, China. Sci Rep. 2020;10:9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ying C, Chen J, Chen J. et al. Differential accumulation mechanisms of purine alkaloids and catechins in Camellia ptilophylla, a natural caffeine-free tea. Beverage Plant Res. 2023;3:15 [Google Scholar]
- 5. Zheng XQ, Ye CX, Kato M. et al. Theacrine (1,3,7,9-tetramethyluric acid) synthesis in leaves of a Chinese tea, kucha (Camellia assamica var. kucha). Phytochemistry. 2002;60:129–34 [DOI] [PubMed] [Google Scholar]
- 6. Zhang S, Jin J, Chen J. et al. Purine alkaloids in tea plants: component, biosynthetic mechanism and genetic variation. Beverage Plant Res. 2022;2:1–9 [Google Scholar]
- 7. Zhu B, Chen LB, Lu M. et al. Caffeine content and related gene expression: novel insight into caffeine metabolism in Camellia plants containing low, normal, and high caffeine concentrations. J Agric Food Chem. 2019;67:3400–11 [DOI] [PubMed] [Google Scholar]
- 8. Perrois C, Strickler SR, Mathieu G. et al. Differential regulation of caffeine metabolism in Coffea arabica (Arabica) and Coffea canephora (Robusta). Planta. 2015;241:179–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen X, Shao S, Yang R. et al. Identification of co-expressed genes related to theacrine synthesis in tea flowers at different developmental stages. Int J Mol Sci. 2021;22:13394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ashihara H, Gillies FM, Crozier A. Metabolism of caffeine and related purine alkaloids in leaves of tea (Camellia sinensis L.). Plant Cell Physiol. 1997;38:413–9 [Google Scholar]
- 11. Suzuki T, Ashihara H, Waller GR. Purine and purine alkaloid metabolism in Camellia and Coffea plants. Phytochemistry. 1992;31:2575–84 [Google Scholar]
- 12. Koshiishi C, Kato A, Yama S. et al. A new caffeine biosynthetic pathway in tea leaves: utilisation of adenosine released from the S-adenosyl-L-methionine cycle. FEBS Lett. 2001;499:50–4 [DOI] [PubMed] [Google Scholar]
- 13. Zhou MZ, Yan CY, Zeng Z. et al. N-Methyltransferases of caffeine biosynthetic pathway in plants. J Agric Food Chem. 2020;68:15359–72 [DOI] [PubMed] [Google Scholar]
- 14. Kato M, Mizuno K, Crozier A. et al. Caffeine synthase gene from tea leaves. Nature. 2000;406:956–7 [DOI] [PubMed] [Google Scholar]
- 15. Mizuno K, Kato M, Irino F. et al. The first committed step reaction of caffeine biosynthesis: 7-methylxanthosine synthase is closely homologous to caffeine synthases in coffee (Coffea arabica L.). FEBS Lett. 2003;547:56–60 [DOI] [PubMed] [Google Scholar]
- 16. Teng J, Yan C, Zeng W. et al. Purification and characterization of theobromine synthase in a theobromine-enriched wild tea plant (Camellia gymnogyna Chang) from Dayao Mountain, China. Food Chem. 2020;311:125875 [DOI] [PubMed] [Google Scholar]
- 17. Suzuki T, Takahashi E. Biosynthesis of caffeine by tea-leaf extracts. Enzymic formation of theobromine from 7-methylxanthine and of caffeine from theobromine. Biochem J. 1975;146:87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jin JQ, Chai YF, Liu YF. et al. Hongyacha, a naturally caffeine-free tea plant from Fujian. China J Agric Food Chem. 2018;66:11311–9 [DOI] [PubMed] [Google Scholar]
- 19. Zhang YH, Li YF, Wang Y. et al. Identification and characterization of N9-methyltransferase involved in converting caffeine into non-stimulatory theacrine in tea. Nat Commun. 2020;11:1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jin JQ, Yao MZ, Ma CL. et al. Association mapping of caffeine content with TCS1 in tea plant and its related species. Plant Physiol Biochem. 2016;105:251–9 [DOI] [PubMed] [Google Scholar]
- 21. Zhong H, Wang Y, Qu FR. et al. A novel TcS allele conferring the high-theacrine and low-caffeine traits and having potential use in tea plant breeding. Hortic Res. 2022;9:uhac191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yao X, Chen H, Ai A. et al. The transcription factor CsS40 negatively regulates TCS1 expression and caffeine biosynthesis in connection to leaf senescence in Camellia sinensis. Hortic Res. 2023;9:uhad162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma W, Kang X, Liu P. et al. The NAC-like transcription factor CsNAC7 positively regulates the caffeine biosynthesis-related gene yhNMT1 in Camellia sinensis. Hortic Res. 2022;9:uhab046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li P, Ye Z, Fu J. et al. CsMYB184 regulates caffeine biosynthesis in tea plants. Plant Biotechnol J. 2022;20:1012–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mohanan S, Gowda K, Kandukuri SV. et al. Involvement of a novel intronic microRNA in cross regulation of N-methyltransferase genes involved in caffeine biosynthesis in Coffea canephora. Gene. 2013;519:107–12 [DOI] [PubMed] [Google Scholar]
- 26. Krishnatreya DB, Agarwala N, Gill SS. et al. Understanding the role of miRNAs for improvement of tea quality and stress tolerance. J Biotechnol. 2021;328:34–46 [DOI] [PubMed] [Google Scholar]
- 27. Zhou C, Tian C, Zhu C. et al. Hidden players in the regulation of secondary metabolism in tea plant: focus on non-coding RNAs. Beverage Plant Res. 2022;2:1–12 [Google Scholar]
- 28. Djami-Tchatchou AT, Sanan-Mishra N, Ntushelo K. et al. Functional roles of microRNAs in agronomically important plants—potential as targets for crop improvement and protection. Front. Plant Sci. 2017;8:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang K, Han H, Li Y. et al. Significance of miRNA in enhancement of flavonoid biosynthesis. Plant Biol (Stuttg). 2022;24:217–26 [DOI] [PubMed] [Google Scholar]
- 30. Sharma D, Tiwari M, Pandey A. et al. microRNA858 is a potential regulator of phenylpropanoid pathway and plant development. Plant Physiol. 2016;171:944–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Y, Zhu X, Chen X. et al. Identification and characterization of cold-responsive microRNAs in tea plant (Camellia sinensis) and their targets using high-throughput sequencing and degradome analysis. BMC Plant Biol. 2014;14:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu SC, Xu YX, Ma JQ. et al. Small RNA and degradome profiling reveals important roles for microRNAs and their targets in tea plant response to drought stress. Physiol Plant. 2016;158:435–51 [DOI] [PubMed] [Google Scholar]
- 33. Zhang F, Yang J, Zhang N. et al. Roles of microRNAs in abiotic stress response and characteristics regulation of plant. Front. Plant Sci. 2022;13:919243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen WT, Yang MJ, Tsuei YW. et al. Green tea epigallocatechin gallate suppresses 3T3-L1 cell growth via microRNA-143/MAPK7 pathways. Mol Nutr Food Res. 2023;247:1670–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou C, Zhu C, Fu H. et al. Genome-wide investigation of superoxide dismutase (SOD) gene family and their regulatory miRNAs reveal the involvement in abiotic stress and hormone response in tea plant (Camellia sinensis). PLoS One. 2019;14:e0223609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao LQ, Shan CM, Shan TY. et al. Comparative transcriptomic analysis reveals the regulatory mechanisms of catechins synthesis in different cultivars of Camellia sinensis. Food Res Int. 2022;157:111375 [DOI] [PubMed] [Google Scholar]
- 37. Wang K, Cai S, Xing Q. et al. Melatonin delays dark-induced leaf senescence by inducing miR171b expression in tomato. J Pineal Res. 2022;72:e12792 [DOI] [PubMed] [Google Scholar]
- 38. Yu S, Li P, Zhao X. et al. CsTCPs regulate shoot tip development and catechin biosynthesis in tea plant (Camellia sinensis). Hortic Res. 2021;8:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wan XC. Tea Biochemistry. Beijing: China Agricultural Press; 2003: [Google Scholar]
- 40. Cheng H, Wu W, Liu X. et al. Transcription factor CsWRKY40 regulates L-theanine hydrolysis by activating the CsPDX2.1 promoter in tea leaves during withering. Hortic Res. 2022;9:uhac025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu Y, Chen S, Chen J. et al. Comprehensive analysis and expression profiles of the AP2/ERF gene family during spring bud break in tea plant (Camellia sinensis). BMC Plant Biol. 2023;23:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sun S, Wang D, Li J. et al. Transcriptome analysis reveals photoperiod-associated genes expressed in rice anthers. Front. Plant Sci. 2021;12:621561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li D, Liu K, Zhao C. et al. GWAS combined with WGCNA of transcriptome and metabolome to excavate key candidate genes for rice anaerobic germination. Rice (N Y). 2023;16:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu Q, He Y, Yan X. et al. Unraveling a crosstalk regulatory network of temporal aroma accumulation in tea plant (Camellia sinensis) leaves by integration of metabolomics and transcriptomics. Environ Exp Bot. 2018;149:81–94 [Google Scholar]
- 45. Tarczewska A, Greb-Markiewicz B. The significance of the intrinsically disordered regions for the functions of the bHLH transcription factors. Int J Mol Sci. 2019;20:5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hao Y, Zong X, Ren P. et al. Basic helix-loop-helix (bHLH) transcription factors regulate a wide range of functions in Arabidopsis. Int J Mol Sci. 2021;22:7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li X, Duan X, Jiang H. et al. Genome-wide analysis of basic/helix-loop-helix transcription factor family in rice and Arabidopsis. Plant Physiol. 2006;141:1167–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lu R, Li Y, Zhang J. et al. The bHLH/HLH transcription factors GhFP2 and GhACE1 antagonistically regulate fiber elongation in cotton. Plant Physiol. 2022;189:628–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sun F, Ding L, Feng W. et al. Maize transcription factor ZmBES1/BZR1-5 positively regulates kernel size. J Exp Bot. 2021;72:1714–26 [DOI] [PubMed] [Google Scholar]
- 50. Sun X, Wang Y, Sui N. Transcriptional regulation of bHLH during plant response to stress. Biochem Biophys Res Commun. 2018;503:397–401 [DOI] [PubMed] [Google Scholar]
- 51. Li X, Cao L, Jiao B. et al. The bHLH transcription factor AcB2 regulates anthocyanin biosynthesis in onion (Allium cepa L.). Hortic Res. 2022;9:uhac128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Luo Y, Yu SS, Li J. et al. Characterization of the transcriptional regulator CsbHLH62 that negatively regulates EGCG3″me biosynthesis in Camellia sinensis. Gene. 2019;699:8–15 [DOI] [PubMed] [Google Scholar]
- 53. Gao Y, Liu J, Chen Y. et al. Tomato SlAN11 regulates flavonoid biosynthesis and seed dormancy by interaction with bHLH proteins but not with MYB proteins. Hortic Res. 2018;5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu YF, Zhao Y, Liu XY. et al. A bHLH transcription factor regulates bisbibenzyl biosynthesis in the liverwort Plagiochasma appendiculatum. Plant Cell Physiol. 2018;59:1187–99 [DOI] [PubMed] [Google Scholar]
- 55. Jia N, Wang JJ, Liu J. et al. DcTT8, a bHLH transcription factor, regulates anthocyanin biosynthesis in Dendrobium candidum. Plant Physiol Biochem. 2021;162:603–12 [DOI] [PubMed] [Google Scholar]
- 56. Wdowikowska A, Janicka M. Antisense oligonucleotide technology as a research tool in plant biology. Funct Plant Biol. 2021;49:1–12 [DOI] [PubMed] [Google Scholar]
- 57. Krasnodębski C, Sawuła A, Kaźmierczak U. et al. Oligo—not only for silencing: overlooked potential for multidirectional action in plants. Int J Mol Sci. 2023;24:4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xie Z, Sundström JF, Jin Y. et al. A selection strategy in plant transformation based on antisense oligodeoxynucleotide inhibition. Plant J. 2014;77:954–61 [DOI] [PubMed] [Google Scholar]
- 59. Di T, Wu Y, Peng J. et al. CsCIPK11-regulated metalloprotease CsFtsH5 mediates the cold response of tea plants. Int J Mol Sci. 2023;24:6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Klein GA-O, Stupak A, Biernacka D. et al. Multiple transcriptional factors regulate transcription of the rpoE gene in Escherichia coli under different growth conditions and when the lipopolysaccharide biosynthesis is defective. J Biol Chem. 2016;291:22999–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jiang W, Liu T, Nan W. et al. Two transcription factors TaPpm1 and TaPpb1 co-regulate anthocyanin biosynthesis in purple pericarps of wheat. J Exp Bot. 2018;69:2555–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li M, Yu B. Recent advances in the regulation of plant miRNA biogenesis. RNA Biol. 2021;18:2087–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jeena GS, Singh N, Shikha.. et al. An insight into microRNA biogenesis and its regulatory role in plant secondary metabolism. Plant Cell Rep. 2022;41:1651–71 [DOI] [PubMed] [Google Scholar]
- 64. Samad AFA, Rahnamaie-Tajadod R, Sajad M. et al. Regulation of terpenoid biosynthesis by miRNA in Persicaria minor induced by Fusarium oxysporum. BMC Genomics. 2019;20:586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tirumalai V, Swetha C, Nair A. et al. miR828 and miR858 regulate VvMYB114 to promote anthocyanin and flavonol accumulation in grapes. J Exp Bot. 2019;70:4775–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang X, Wang W, Chen S. et al. Tropaeolum majus R2R3 MYB transcription factor TmPAP2 functions as a positive regulator of anthocyanin biosynthesis. Int J Mol Sci. 2022;23:12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zheng C, Ye M, Sang M. et al. A regulatory network for miR156-SPL module in Arabidopsis thaliana. Int J Mol Sci. 2019;20:6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jin Q, Wang Z, Chen Y. et al. Transcriptomics analysis reveals the signal transduction mechanism of brassinolides in tea leaves and its regulation on the growth and development of Camellia sinensis. BMC Genomics. 2022;1:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hellemans J, Mortier G, De Paepe A. et al. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yang X, Luo Y, Bai H. et al. DgMYB2 improves cold resistance in chrysanthemum by directly targeting DgGPX1. Hortic Res. 2022;9:uhad028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gao Y, Wei W, Zhao X. et al. A NAC transcription factor, NOR-like1, is a new positive regulator of tomato fruit ripening. Hortic Res. 2018;5:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fang Y, Liu J, Zheng M. et al. SbMYB3 transcription factor promotes root-specific flavone biosynthesis in Scutellaria baicalensis. Hortic Res. 2023;10:uhac266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jiang B, Wu Z, Yang G. et al. Cloning and functional analysis of flavanone 3-hydroxylase gene in Rhododendron hybridum Hort. Sheng Wu Gong Cheng Xue Bao. 2023;39:653–69 [DOI] [PubMed] [Google Scholar]
- 74. Han YC, Kuang JF, Chen JY. et al. Banana transcription factor MaERF11 recruits histone deacetylase MaHDA1 and represses the expression of MaACO1 and expansins during fruit ripening. Plant Physiol. 2016;171:1070–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ding Y, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res. 2003;31:7280–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fang Z-Z, Zhang Y-L, Lai C-C. et al. Developmental regulation of Ran 3' untranslated region during somatic embryogenesis in Dimocarpus longan Lour. Sci Hortic. 2014;176:297–302 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data supporting our findings are contained within the manuscript. All raw transcriptome data reported in this article have been deposited in the Sequence Read Archive (SRA) under accession number PRJNA986690 and PRJNA1056246.