

Significance
Myalgic encephalomyelitis/chronic fatigue syndrome (ME) is a serious disabling chronic illness that lacks FDA-approved therapies. Comprehensive transcriptomic, epigenomic, and flow cytometric profiles of primary CD8+ T cell subsets implicate T cell exhaustion in pathophysiology. We show that T cells in ME cases are epigenetically predisposed toward terminal exhaustion and that exhaustion markers are upregulated following exercise challenge. Using single-cell genomics, we provide important information about the role of CD8+ T cell exhaustion development and progression. Our findings are consistent with the hypothesis that chronic viral infection is a factor in ME; by dissecting the molecular basis of T cell dysfunction in ME, we offer potential avenues for treatment.
Keywords: single-cell RNAseq, ATACseq, T cell exhaustion, flow cytometry, ME/CFS
Abstract
Myalgic encephalomyelitis/chronic fatigue syndrome (ME) is a severe, debilitating disease, with substantial evidence pointing to immune dysregulation as a key contributor to pathophysiology. To characterize the gene regulatory state underlying T cell dysregulation in ME, we performed multiomic analysis across T cell subsets by integrating single-cell RNA-seq, RNA-seq, and ATAC-seq and further analyzed CD8+ T cell subpopulations following symptom provocation. Specific subsets of CD8+ T cells, as well as certain innate T cells, displayed the most pronounced dysregulation in ME. We observed upregulation of key transcription factors associated with T cell exhaustion in CD8+ T cell effector memory subsets, as well as an altered chromatin landscape and metabolic reprogramming consistent with an exhausted immune cell state. To validate these observations, we analyzed expression of exhaustion markers using flow cytometry, detecting a higher frequency of exhaustion-associated factors. Together, these data identify T cell exhaustion as a component of ME, a finding which may provide a basis for future therapies, such as checkpoint blockade, metabolic interventions, or drugs that target chronic viral infections.
Myalgic encephalomyelitis (ME), also known as chronic fatigue syndrome (CFS), is a devastating disease that afflicts millions of people globally (1, 2). Patients suffer from exhaustion, cognitive deficits, flu-like symptoms, gastrointestinal issues, muscular pain, orthostatic intolerance, and post-exertional malaise (PEM), a hallmark trait in which symptoms are exacerbated following exertion. Studies undertaken during past outbreaks of ME suggest that one or more viruses may act as the trigger for ME, but the causative agent(s) has not been identified (3, 4). Our study focuses on ME cases that were triggered before the emergence of SARS-CoV-2. While some individuals with Long COVID (LC) fulfill the phenotypic diagnostic criteria for ME, it is presently unknown how similar ME and LC are at the molecular level.
Although the etiology of ME remains unknown, it is clear that immune system dysregulation plays a critical role in the development and/or progression of ME, including alterations in the frequencies of effector CD8+ T cells (Teff) and regulatory T cells (Treg), decreased cytotoxicity in natural killer (NK) cells and CD8+ T cells, as well as decreased proliferation and IFN-γ production in CD4+ T cells (5–9). Nonetheless, it is not clear which T cell subset abnormalities are most associated with ME.
While alterations in gene expression likely contribute to T cell dysregulation in ME, little is known regarding the nature of such dysregulation, nor are the identities of the underlying regulatory factors known. We recently reported a single-cell RNA sequencing (scRNA-seq) transcriptomic atlas of ME leukocytes in a large cohort of patients and sedentary controls (10). Although we found the strongest dysregulation in classical monocytes in ME, we also uncovered substantial dysregulation in T cells, including increased cytokine and chemokine signaling from classical monocytes to Teff in ME. These findings suggest that ME T cells may exhibit aberrant activation due to an inflammatory milieu.
As CD8+ T cells differentiate and acquire effector or memory fates, they undergo transcriptional and epigenetic reprogramming to elicit changes in their metabolic program and responsivity (11). In contrast, following prolonged antigen exposure or inflammatory cues, such as in chronic infections or cancer, CD8+ T cells enter exhaustion, a dysfunctional state where metabolic requirements are permanently altered to favor a suppressed immune response (12, 13). Exhaustion follows a trajectory progressively controlled by multiple transcriptional and epigenetic checkpoints, including CD69 and the transcription factors (TFs) TCF1, T-BET, EOMES, and TOX (14, 15). Exhausted cells are also characterized by increased expression of PD-1 and other inhibitory receptors (IRs), resulting in reduced proliferation, survival, and impaired effector function (16).
Several pieces of evidence are consistent with exhaustion as a potential mode of T cell dysfunction in ME. CD8+ T cells in ME exhibit higher fatty acid oxidation rates, lower glycolytic rates, and decreased mitochondrial membrane potential (17, 18), observations consistent with metabolic reprogramming under T cell exhaustion. Despite inconsistencies across cytokine studies, there is strong evidence that cytokine networks are altered in ME progression, exhibiting both upregulated proinflammatory cytokines and certain immunosuppressive signatures (19–21). Importantly, a recent report found reduced production of TNF-α and IFN-γ by CD8+ T cells, indicative of a hypofunctional state (22). Furthermore, analysis of ME CD8+ T cells in cerebrospinal fluid revealed increased expression of PD-1 (23), while increased plasma levels of TIM-3, an IR expressed by exhausted cells, have also been reported in ME (24). Collectively, these findings hint at the involvement of T cell exhaustion in ME, but this theory remains inconclusive due to the overlapping nature of the various types of dysfunctional states.
To systematically interrogate the transcriptional and epigenetic states of T cells in ME, and to investigate whether T cell exhaustion occurs in ME, we analyzed scRNA-seq data (10), focusing on T lymphoid cells. Our initial goal was to identify the cell subsets most dysregulated in ME. To validate and extend observations made using the scRNA-seq data, we generated transcriptome and accessible chromatin profiles for purified subsets of interest. Using this integrated approach, we found that ME effector CD8+ T cells upregulate key exhaustion factors and display epigenetic signatures indicative of exhaustion. To validate these observations, we performed flow cytometry of circulating CD8+ subpopulations in an expanded cohort.
As key players in viral clearance and autoimmunity, and with mounting support for their role in ME pathology, T cells present an attractive yet underexplored target for therapeutics. This study provides a more definitive understanding of ME T cell dysfunction and identifies T cell exhaustion as a possible mechanism of ME development and progression, thus providing a potential treatment approach via reversal of this pathway.
Results
Gene Expression Across T and NK Cell Subsets in ME.
To examine gene dysregulation in T and NK cells, we extracted and analyzed 336,269 T lymphoid and related cells from the ME scRNA-seq atlas, consisting of 28 patients and 30 sedentary controls at baseline and after symptom provocation (10). We performed high-resolution reclustering of these data, yielding 21 clusters (Fig. 1A; and SI Appendix, Fig. S1A). Evaluating the expression profiles of each cluster using known markers, we assigned cell identities to almost all clusters, including CD8+ T cells across distinct stages of differentiation (Fig. 1A; and SI Appendix, Fig. S1B). We observed a slight decrease in the number of early and intermediate effector memory cells (Teff; clusters 16 and 12, respectively) in ME patients at baseline, while proportions of all other cell subsets were minimally altered either at baseline or after symptom provocation (SI Appendix, Fig. S1C).
Fig. 1.
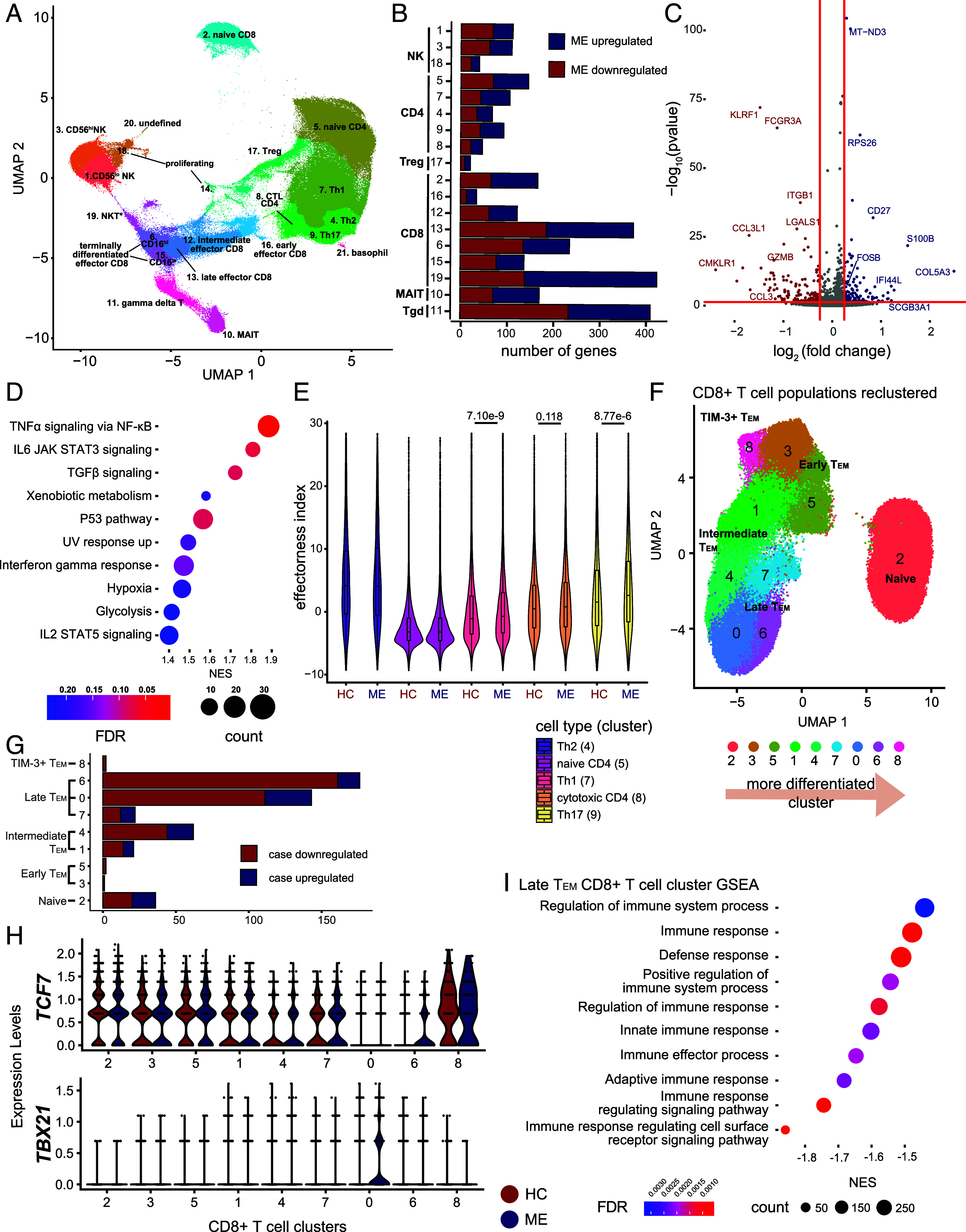
scRNA-seq analysis of T cells in ME. (A) scRNA-seq of T lymphoid cells from 28 cases and 30 controls, depicted by uniform manifold approximation and projection (UMAP). Cluster numbers assigned independent of cluster size. (B) Number of differentially expressed genes (y-axis) per cluster (x-axis), as log2 fold change > 0.25, adjusted P-value < 0.05 (FDR). (C) Volcano plot of differentially expressed genes in γδT cells (cluster 11). Colored dots indicate increased expression in case (blue) or control (red; P ≤ 0.05, log2 fold change > 0.25). (D) GSEA dot plot of γδT cells against hallmark pathways from Molecular Signature Database. Size indicates number of leading-edge genes; color indicates adjusted P-value (FDR). (E) Violin plot showing the distribution of effector index per cell in CD4+ T cell clusters and biological condition (control, ME). (F) scRNA-seq analysis of CD8+ T cells from 28 cases and 30 matched controls, depicted by UMAP. (G) Number of differentially expressed genes per CD8+ T cell cluster, defined as log2 fold change > 0.4 and adjusted P-value < 0.05 (FDR). (H) Violin plots showing differential expression between case and control in each cluster for two exhaustion-associated TFs—TCF7 and TBX21. (I) GSEA of cluster 6 against gene ontology: biological processes terms. Positive values (x-axis) indicate enrichment in cases and vice versa. Dot size and color indicate number of genes in the leading edge, and adjusted P-values (FDR), respectively.
We sought to interrogate the transcriptional programs of case and control lymphocytes at the single cell level at baseline. We identified dysregulation across multiple clusters, with the greatest signal in CD8+ T cell subsets and γδT cells (Fig. 1B and Dataset S1). We detected only subtle shifts in the transcriptome of NK and CD4+ T cell subsets, and virtually no differential expression in Treg cells. ME γδT cells exhibited decreased expression of effector genes, including GZMB, FCGR3A, and KLRF1 (Fig. 1C). However, these cells also exhibited divergent patterns of expression in genes associated with chemokine and inflammatory signaling, with monocyte-attracting chemokines CCL3 and CCL3L1 downregulated in ME despite the upregulation of proinflammatory TFs such as FOSB (Fig. 1D). Gene set enrichment analysis (GSEA) of γδT cells also revealed enrichment for activating pathways like TNFα signaling via NF-κB and IL-6 signaling, with positive trends for TGF-β signaling and IFNγ response in ME (Fig. 1D). Thus, dysregulation of γδT cells is likely a component of ME pathophysiology.
To survey signaling irregularities in ME γδT cells, we used CellChat (25) to infer intercellular communication networks between γδT cells and other lymphocytes and compared the interaction strengths between cases and controls. We focused on the female-only cohort, representing the majority of the data and allowing us to control for sex-dependent differences. In ME patients, we observed increased interactions in IL-2, TGF-β, and IL-16 signaling pathways, similar to our findings in GSEA (SI Appendix, Fig. S1 D and E). We detected elevated CCL5 (SI Appendix, Fig. S1E), another chemokine responsible for localization of lymphocytes to inflamed tissues (26) and previously implicated in ME pathology (10).
Given that ME γδT cells exhibit increased outgoing signaling of cytokines known to activate CD4+ T cells (27), we examined the transcriptional programs of memory CD4+ T cell subtypes. We used an established single-cell effector potential index that reflects T cell activation and cytokine response (28). As expected, naïve CD4+ T cells (cluster 5) have the lowest effector index. We found that Th1 and Th17 CD4+ T cells (clusters 7 and 9, respectively) have modestly elevated effector indices in ME patients (Fig. 1E), with no differences in other CD4+ subsets. In summary, although ME results in dysregulation in γδT cells and to a lesser extent in certain CD4+ T cell subsets, the strongest signal is found in CD8+ T cell subsets.
Antigen-Experienced CD8+ T Cell Subsets Are Impaired in ME.
Given that CD8+ T cells exhibit the most pronounced dysregulation in ME across T lymphoid cells (Fig. 1B), and also exhibit altered metabolic states (17, 18), we focused on these cells. To account for the complex nature of CD8+ T cell populations, we extracted the 88,052 CD8+ T cells in the scRNA-seq data and reclustered these cells at higher resolution, yielding 9 clusters (Fig. 1F). By examining established naïve, effector, memory, and exhaustion markers (29), we assigned identities to clusters, including a naïve subset (TN; cluster 2), early effector memory (TEMearly; 3 and 5), intermediate effector memory (TEMint; 1 and 4), late effector memory subsets (TEMlate; 0, 6, and 7), which differ in IL7R and CD16 expression, and a cluster of TIM-3+ effector memory T cells (TIM-3+ TEM; 8; Fig. 1F and SI Appendix, Fig. S2A).
Using pseudotime analysis to generate a developmental trajectory across the subsets, we established a framework of CD8+ T cells in different developmental states, facilitating analyses to determine the subsets whose dysregulation is most prevalent in ME (SI Appendix, Fig. S2B). We then interrogated the transcriptional programs of case and control CD8+ T cells at the single-cell level across each subset. We identified the most dysregulation in TEMlate cells (clusters 0 and 6; Fig. 1G), with modest signal in TEMint (cluster 4) and TN. In ME, cluster 0 demonstrated marked downregulation of many genes related to effector function, including GNLY, encoding a cytotoxic peptide (30), IKZF2 (HELIOS), a TF required for T cell activation (31), and IL18R1, a receptor downregulated in chronic infections, which normally interacts with IL-18 to activate downstream effectors (32; SI Appendix, Fig. S2C). Meanwhile, KLRB1, an IR with antagonistic roles in cytotoxic programs (33), is upregulated in case samples (SI Appendix, Fig. S1C), along with TBX21 (T-BET; Fig. 1H). Cluster 6 exhibited similar patterns of downregulation in ME T cells, including downregulation of GNLY, and additional repression of TFs related to T cell activation or effector function, including ID2, JUNB, FOS, and IRF1 (SI Appendix, Fig. S2D). Interestingly, ID2 functions to restrain Teff differentiation into TCF7- and EOMES-expressing memory cells (34), perhaps contributing to the retention of TCF7 expression in cluster 6 despite its late effector identity (Fig. 1H). GSEA of clusters 0 and 6 revealed downregulation of gene sets related to regulation of immune responses in cases (Fig. 1I; and SI Appendix, Fig. S2E), suggesting functional impairment. Although we did not detect substantial upregulation of PD-1 (PDCD1), more cells express this IR in TEMlate cells (cluster 0; SI Appendix, Fig. S2F). These data suggest that ME TEMlate have impaired effector function, with cells exhibiting signs concordant with exhaustion.
ME CD8+ Memory T Cells Exhibit Higher Expression of Exhaustion Markers.
To examine the gene expression program of CD8+ T cells in ME more comprehensively, we isolated naïve (TN) and memory (TM) CD8+ T cells from 7 female patients and 7 age- and BMI-matched sedentary healthy controls (Fig. 2A). The ME cohort, all diagnosed prepandemic, reported lower Bell Activity Scale and physical health scores in the SF-36 survey (Fig. 2A and SI Appendix, Table S1), indicating more severe physical debility in patients. From these new cohorts, we performed RNA-seq and ATAC-seq (assay for transposase accessible chromatin) to assess their transcriptomes and chromatin landscapes, respectively.
Fig. 2.
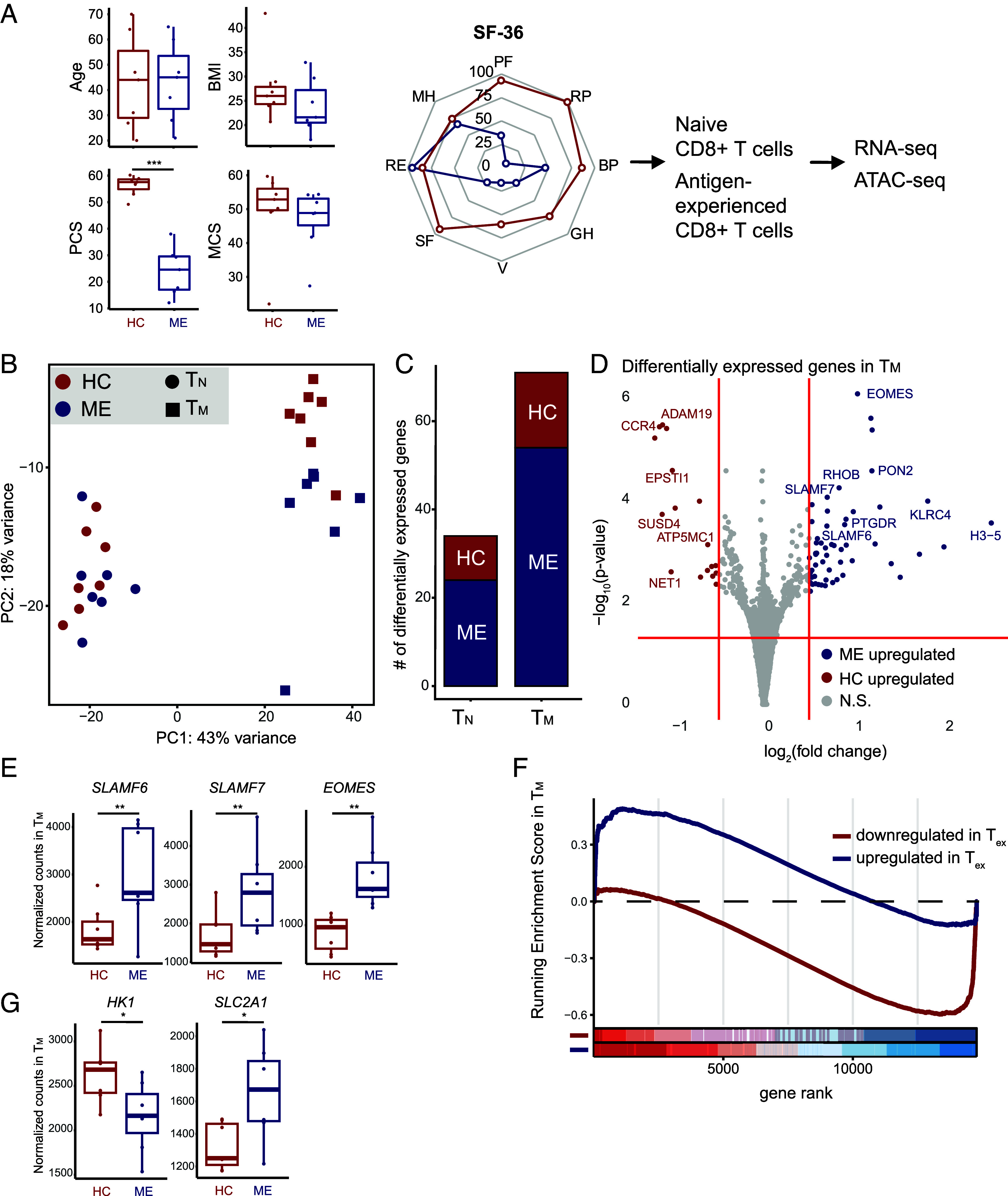
Higher expression of key exhaustion markers in ME TM. (A) Comparisons of cohorts and schematic of the experimental approach, including sample size, age (P = 0.95), BMI (P = 0.38), and SF-36 results, including box plots of physical (PCS, P = 0.0006) and mental component scores (MCS, P = 0.26). Comparisons performed with the Wilcoxon rank-sum test (n=7 controls; n = 7 ME samples) *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. (PF = physical functioning, RP = role: physical, BP = bodily pain, GH = general health, V = vitality, SF = social functioning, RE = role: emotional, MH = mental health). (B) PCA of RNA-seq of TN and TM from case (ME) and control (HC). (C) Bar plot of number of differentially expressed genes (y-axis) per cell type (x-axis). (D) Volcano plot highlighting differentially expressed genes in TM (P ≤ 0.05, colored dots). (E) Box plot of normalized counts of SLAMF6 (P = 5.7 × 10–4), SLAMF7 (P = 8.8 × 10–5), and EOMES (P = 8.2 × 10–7) in TM, comparing between case and control. *P ≤ 0.05 and **P ≤ 0.01 (F) GSEA enrichment plot of TM against genes differentially expressed in Tex. Positive values indicate enrichment in ME and vice versa. ME TM exhibited negative enrichment for genes downregulated in Tex (P = 1.1 × 10–12) and positive enrichment for genes upregulated in Tex (P = 8.7 × 10–3). Bottom: individual bars indicate a gene in the specific gene set; color shading indicates distribution of genes within each gene set, separated into 8 bins. (G) Box plot of normalized counts of HK1 (P = 0.024) and SLC2A1 (P = 0.017) in TM, comparing between case and control.
Principal component analysis (PCA) demonstrated clear separation between TN and TM populations along PC1, and separation of case and control TM along PC2 by RNA-seq (Fig. 2B). Consistent with the PCA, we detected 71 differentially expressed genes in TM between case and control cohorts, and only 34 in TN, indicating more extensive changes in the ME effector populations (Fig. 2C). Nevertheless, in TN, we observed higher expression of GNLY and TNFRSF1A in ME samples, suggesting that these cells may adopt a more effector-like transcriptional program (SI Appendix, Fig. S3A). Indeed, GSEA revealed that ME TN trended toward increased expression for gene modules associated with loss of the naïve state (35; SI Appendix, Fig. S3B). Thus, one aspect of CD8+ T cell abnormalities in ME involves perturbations to antigen-inexperienced cells.
In ME TM, we observed increased expression of multiple key regulators of T cell exhaustion, including the TF EOMES, and the IRs SLAMF6 and SLAMF7 (Fig. 2 D and E), although we did not observe significant upregulation of PDCD1 (SI Appendix, Fig. S3C). EOMES has dual roles in CD8+ T cells, enabling memory formation and also contributing to establishment of the exhaustion expression program (36). Concurrent upregulation of SLAMF6 and SLAMF7 indicates that ME TM are likely biased toward adopting a progenitor exhausted state (37). Importantly, we did not detect upregulation of genes indicative of T cell anergy (SI Appendix, Fig. S3D), and found little evidence of exhaustion-independent senescence (38, 39, SI Appendix, Fig. S3E), indicating that the dysfunctional state in ME TM is likely exhaustion, rather than other programs of suppression. Moreover, GSEA demonstrated that the transcriptional program in ME TM aligns closely with RNA-seq and ATAC-seq data on exhausted T cells (Tex) in a chronic infection (Fig. 2F; 40 and SI Appendix, Fig. S3F). Comparing genes with increased expression in both Tex and ME TM via leading edge analysis, we found enrichment for many genes associated with exhaustion, including EOMES and CD244; meanwhile, genes negatively enriched in both Tex and ME TM included effector genes, such as IL17RA, as well as multiple genes related to metabolic regulation.
Induction of T cell exhaustion in ME TM may lead to alterations in phenotype and function, including metabolic reprogramming and response to cytokine signaling. This possibility is supported by lower expression of HK1 (hexokinase) and higher expression of SLC2A1 (a glucose transporter) in ME TM (Fig. 2G). These alterations suggest impaired glycolytic potential, a phenomenon found in exhausted T cells (41), and is consistent with previous findings regarding T cell metabolism in ME. Meanwhile, pathways related to oxidative phosphorylation and MYC signaling were repressed, indicating reduction of metabolic activity and proliferative potential (SI Appendix, Fig. S3G). Finally, we observed significant downregulation of the chemokine receptor CCR4 in ME TM (Fig. 2D), indicating that these cells may become intransigent to tissue homing signals via chemokines (42). Overall, these findings support that ME TM undergo transcriptional reprogramming consistent with establishment of an exhausted state.
ME TM Develop Epigenetic Signatures of Exhaustion.
Establishment of the exhaustion transcriptional program entails rewiring of gene regulatory circuits. These epigenetic changes at cis-regulatory elements across the genome underlie the long-term nature of exhaustion (43). In cases of chronic antigen stimulation or sustained inflammatory milieu, epigenomic aberrations, or scars, persist after the initial stimulatory environment subsides (44, 45). We hypothesized that ME CD8+ T cells could be epigenetically scarred due to exhaustion, leading to the dysfunctional state observed via transcriptomics. Thus, we used ATAC-seq to compare chromatin accessibilities of TM (and TN) cells in the same cohort of patients and healthy controls profiled by RNA-seq, identifying 67,189 consensus chromatin-accessible regions (ChARs) across all conditions. Accessible regions were enriched at transcriptional start sites, as expected for reliable ATAC-seq profiles (SI Appendix, Fig. S4A).
Differential accessibility analysis found 471 upregulated and 1,477 downregulated ChARs in ME TM cells (Fig. 3A). We associated ChARs with their nearest genes and aggregated them to generate gene-level analyses. Using this approach, we found 43 upregulated genes and 192 downregulated genes between the two cohorts (Fig. 3A). ME TM cells exhibited decreased accessibility at genes associated with chemokine and cytokine signaling (CCR4, CCL2, CSF2, IL3; Fig. 3B), and decreased accessibility for genes involved in NF-κB/TNFα signaling and KRAS signaling (Fig. 3C), suggesting that ME TM cells are refractory to activation to facilitate peripheral tolerance (46). Alternatively, due to downregulation of NF-κB/TNFα signaling, the long-term ability of ME TM cells to survive in circulation may be impaired (47).
Fig. 3.
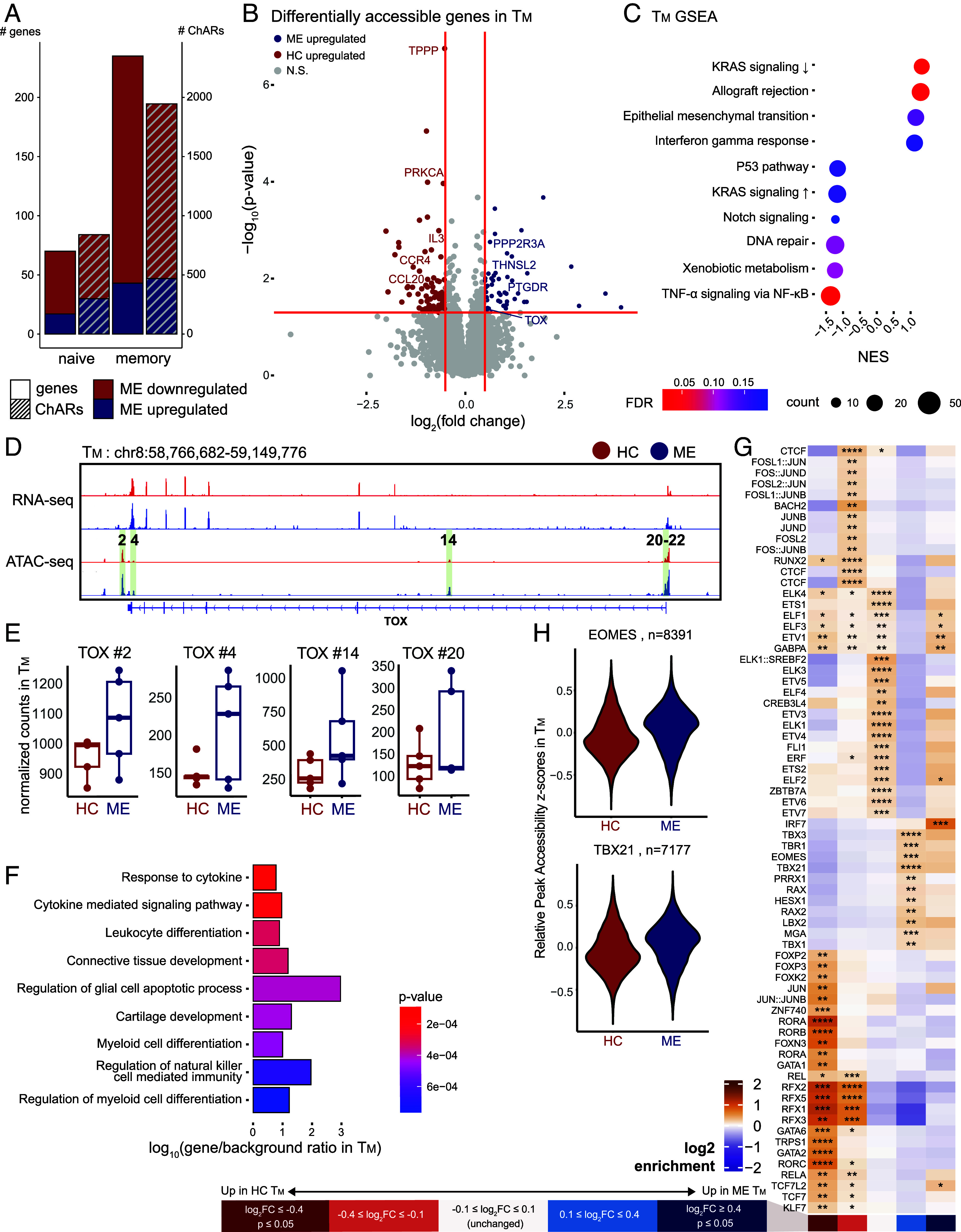
ME TM develop epigenetic signatures of exhaustion. (A) Bar plot of number of differentially accessible ChARs (right y-axis) and genes (left y-axis) per cell type, differentiating between down- and upregulated in ME (color). (B) Volcano plot highlighting differentially accessible genes identified by ATAC-seq in TM (P ≤ 0.05, colored dots, denoting direction of change as per panel A). (C) GSEA dot plot showing enrichment for hallmark pathway gene sets in TM. Dot size and color represent the number of core enrichment genes and log10 P-value, respectively. Positive NES values indicate enrichment in ME and vice versa. (D) Genome browser view of the TOX locus with RNA-seq and ATAC-seq tracks from TM. Numbered and highlighted regions denote relevant ChARs of interest. (E) Box plot of normalized counts at ChARs highlighted in panel (D), comparing between case and control. (F) Overrepresentation analysis of genes showing lower accessibility but no significant changes in expression level in ME TM against Gene Ontology: Biological Process pathways. Color indicates P-value and x-axis represents degree of enrichment (log10 gene-to-background ratio). (G) Heatmap of TF binding motifs significantly enriched in at least one bin of ChARs in TM. ChARs were grouped into five bins by differential accessibility in cases versus controls: significantly decreased (dark red), decreased (light red), unchanged (white), increased (light blue), and significantly increased (dark blue) accessibility in ME vs. controls. Color of each cell corresponds to log2 enrichment score of a motif in the respective bin, where positive values (orange) indicate enrichment and negative values (purple) indicate depletion. Significance of motif binding defined as q ≤ 0.0001. Enrichment scores normalized row-wise. (H) Distribution of peak accessibility z-scores in TM across peaks with at least one EOMES (Top) or T-BET (Bottom) motif.
ME TN cells also exhibited differentially accessible ChARs, showing reduced accessibility at genes associated with cell–cell communication pathways (CCR5, CLEC12A, IL17RB, CD93; SI Appendix, Fig. S4B). However, we also observed increased accessibility for CX3CR1, which is involved in inflammatory signaling, suggesting that intercellular signaling homeostasis in ME TN may be disrupted.
We compared our ME TM gene-level accessibility profiles against published RNA-seq and ATAC-seq datasets in Tex via GSEA (SI Appendix, Fig. S4C), which revealed enrichment for genes upregulated in Tex, demonstrating that epigenetic reprogramming in ME TM is concordant with that observed in an established T cell exhaustion paradigm. Notably, ME TM showed higher accessibility for TOX (Fig. 3 D and E), the key TF controlling an epigenetic checkpoint that governs progression into circulating progenitor and terminal Tex subsets (48). Given a lack of a corresponding significant change in TOX transcription (Fig. 3D), these data suggest that ME TM are differentially primed toward an exhausted T cell state. As exhausted T cells become epigenetically “scarred” long after induction, and enhancers proximal to TOX are known to exhibit such scarring, remaining more accessible (45); we asked whether changes in accessibility at TOX enhancers reflected wider epigenetic scarring in ME TM. Indeed, when we compared ChARs upregulated in our ME cohort to those found to be epigenetically scarred in exhaustion (45), we found significant overlap between the two sets of ChARs (SI Appendix, Fig. S4D). Thus, epigenetic hallmarks characteristic of exhaustion are evident in ME TM.
To examine genes with altered accessibility but without a corresponding change in expression, such as TOX, we collated expression levels of genes with altered accessibility in ME TM. In functional TM, many effector genes are situated in accessible chromatin, albeit with little transcriptional activity; such genes are poised for rapid upregulation upon activation (49). We identified 251 genes in ME TM associated with downregulated ChARs without concordant changes in transcript levels, indicating a loss of regulatory poising in these genes (Dataset S2). Overrepresentation analysis revealed that these genes were associated with effector pathways, such as response to cytokine, leukocyte differentiation, and natural killer cell-mediated immunity (Fig. 3F). These data indicate that the chromatin landscape in TM in ME is characterized by impairments in genes associated with T cell activation and survival (50), observations consistent with T cell exhaustion.
Progression of CD8+ T cells into an exhausted state is regulated by multiple TFs, whose activities specify gene expression programs that underlie exhaustion. To investigate how the altered epigenetic landscape in ME T cells may affect TF binding, we performed TF motif enrichment analysis in ChARs across bins grouped by differential accessibility. ChARs with significantly higher accessibility in ME TM exhibited strong enrichment for IRF7 (Fig. 3G), which activates interferon-alpha-inducible genes and facilitates viral latency in Epstein–Barr virus infections (51, 52). The more accessible ChARs were enriched for additional TFs, including EOMES and T-BET (TBX21; Fig. 3 G and H), suggesting that ME TM may be driven toward late-stage exhaustion. On the other hand, ChARs with lower accessibility in ME TM were enriched for TCF1 (TCF7) and TCF4 (TCF7L2), whose activities are associated with early exhaustion progression (53). Interestingly, immunomodulatory TFs such as those in the ROR family (RORA, RORB, RORC), as well as FOXP3, were also enriched in ChARs downregulated in ME TM. Though not commonly associated with exhaustion, repression of ROR family TFs in CD8+ T cells has been shown to attenuate activation and alter metabolic pathways, potentially contributing to functional impairment (54). These observations suggest that despite the multiple hallmarks of exhaustion in TM (Figs. 2 E–G and 3 B–F), an additional possibility exists where alterations could derive from a sustained inflammatory milieu. Indeed, it is possible that T cell dysregulation in ME derives from a composite of both antigen-specific exhaustion as well as from increased proinflammatory signals.
When we examined ME TN-upregulated ChARs, we found strong enrichment for NF-κB family TFs (REL, RELA, RELB, NFKB1, NFKB2; SI Appendix, Fig. S4 E and F), a family with well-established roles in TN survival, T cell activation, and effector function (47). Hence, ME TN may be primed toward increased activation and/or differentiation, a result that reinforces our observation that the transcriptomes of ME TN exhibit a partial loss of naïve identity. Collectively, our epigenomics data strongly suggest that ME TM are epigenetically predisposed to developing an exhaustion-associated gene regulatory program, while ME TN exhibit a regulatory program consistent with aberrantly increased activation. Potentially, alterations to both TN and TM share a common source.
Elevated Levels of SLAMF6 Expression in ME CD8+ T Cells.
Our multiomics data indicate that the transcriptome and chromatin landscape of ME TM exhibit multiple hallmarks of exhaustion, both in terms of individual relevant genes and the overall gene expression program. To validate these observations and investigate ME T cell exhaustion phenotypes in peripheral cells, we performed flow cytometric analysis of isolated circulating CD8+ T cells to assess cell surface markers and TFs that delineate exhausted T cell states. We performed these experiments on prepandemic samples obtained following an exercise challenge, to provoke PEM and potentially enhance detection of exhausted phenotypes in peripheral T cells. Onset of PEM following an exercise challenge induces oxidative stress in patients (55, 56), which, in chronic viral infections, promotes terminal CD8+ T cell exhaustion (57, 58). We extended our cohort to include 15 patients and 11 controls, all females and matched for age and BMI (SI Appendix, Fig. S5A). This ME cohort exhibited phenotypes consistent with our sequencing cohort, with significantly lower Bell Activity Score and SF-36 physical health scores (SI Appendix, Table S1). Following a maximal-effort CPET (cardio-pulmonary exercise test) to induce PEM, blood was drawn for PBMC isolation and subsequent T cell separation. After gating for single live CD8+ T cells, the frequency of PD-1, CD69, SLAMF6, and TOX-expressing cells was assessed using flow cytometry (SI Appendix, Fig. S5B). PD-1 and CD69 are markers of Tex subsets; exhausted cells express PD-1, whereas PD-1+CD69− cells correspond to a circulating progenitor exhausted state (15).
In total CD8+ T cells, we did not observe any significant changes in the frequency of PD-1+ T cells between case and control cohorts, although we observed a trend of increased PD-1 expression in the case cohort (Fig. 4A and SI Appendix, Fig. S5C). Similarly, abundance of the IR SLAMF6 in total, PD-1+, and PD-1+CD69− CD8+ T cells was slightly greater in the patient cohort compared to the healthy controls (Fig. 4B and SI Appendix, Fig. S5D). A subpopulation of SLAMF6+ T cells was detected in our cell populations, noted as SLAMF6hi T cells; this subset was present at significantly higher frequencies in ME (Fig. 4C and SI Appendix, Fig. S5E). This trend was also observed in PD-1+ CD8+ and PD-1+CD69-CD8+ T cells, indicating a higher proportion of cells with this IR present (Fig. 4D and SI Appendix, Fig. S5E). We saw no differences in TOX expression or TOX+ frequency between our two cohorts in total CD8+ T cells (SI Appendix, Fig. S6). Nonetheless, our observations of elevated PD-1 and SLAMF6 expression in total ME CD8+ T cells agree with our findings of increased IR expression at the transcript level.
Fig. 4.
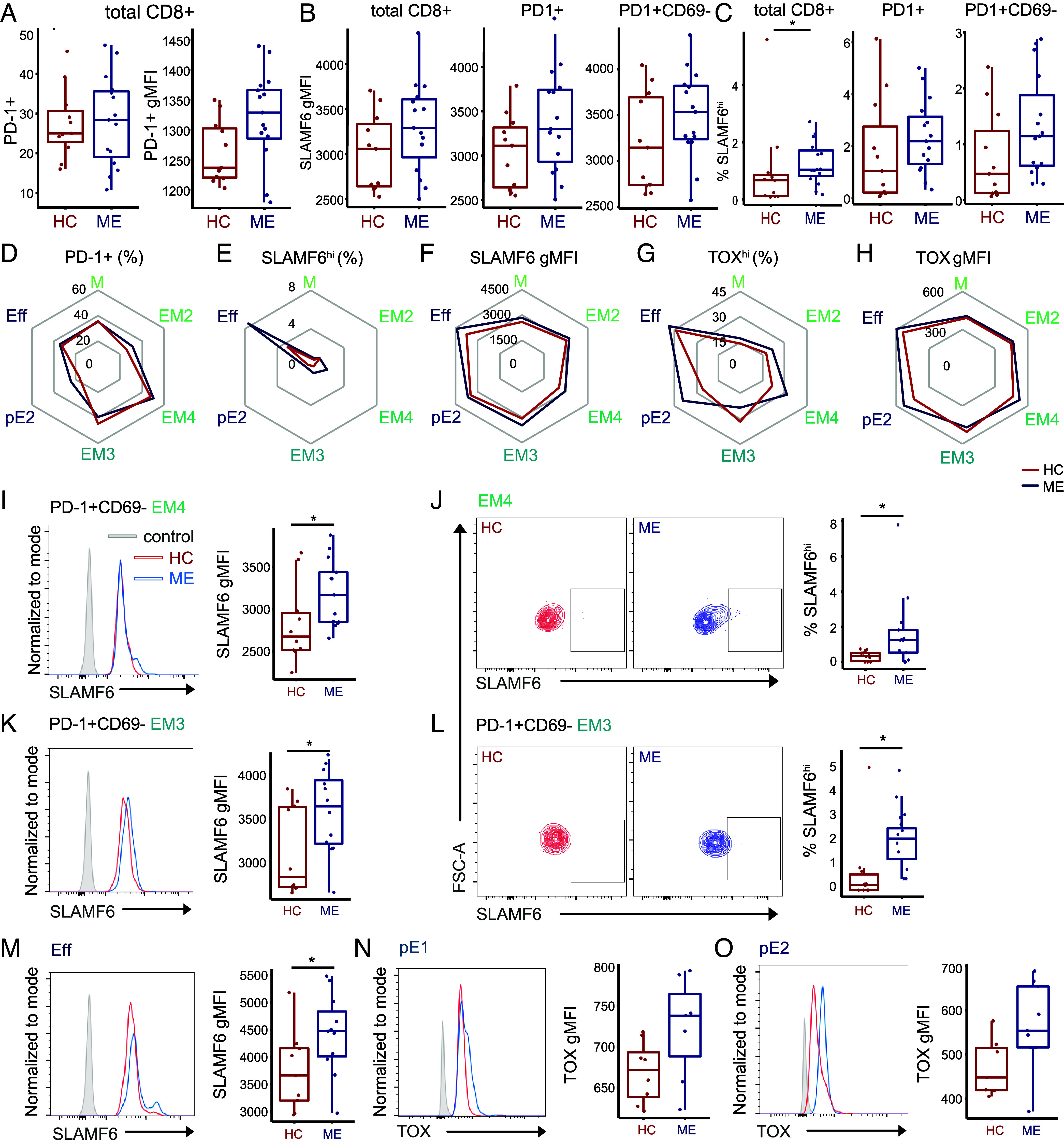
TOX and SLAMF6 are elevated in differentiated CD8+ T cell subsets. (A) Left: PD-1+ frequency (P = 0.92) in total CD8+ T cells. Right: gMFI expression levels of PD-1 (P = 0.055) in CD8+ T cells, compared between HC and ME. gMFI = geometric mean fluorescence intensity, FMO = fluorescent minus one negative control. (B) SLAMF6 expression levels in total CD8+ (P = 0.14), CD8+PD-1+ (P = 0.16), and CD8+PD-1+CD69− (P = 0.16) T cell subsets, compared between HC and ME. (C) SLAMF6hi frequencies in total CD8+ (P = 0.04), CD8+PD-1+ (P = 0.15), and CD69-SLAMF6hi frequency in CD8+PD-1+ (P = 0.055) T cell subsets, compared between HC and ME. (D) Spider plots of T cell subsets depicting PD-1+ frequency, (E) SLAMF6hi frequency, (F) SLAMF6 gMFI, (G) TOXhi frequency, and (H) TOX gMFI between HC and ME. Each red (HC) or blue (ME) line represents the mean percentage or gMFI per T cell subset (y axis). (I) Representative scatter plots of SLAMF6hi populations in EM4 T cells, and box plot of SLAMF6hi frequency in EM4 T cells (P = 0.02, n = 10/13), compared between HC and ME. (J) Representative histogram and box plot of SLAMF6 expression in PD-1+CD69− EM4 T cells (P = 0.049, n = 10/13), compared between HC and ME. (K) Representative scatter plots of SLAMF6hi populations in PD-1+CD69− EM3 T cells, and box plot of SLAMF6hi frequency in EM3 T cells (P = 0.036, n = 10/12), compared between HC and ME. (L) Representative histogram and box plot of SLAMF6 expression in PD-1+ CD69− EM3 T cells (P = 0.05, n = 10/12), compared between HC and ME. (M) Representative histogram and box plot of SLAMF6 expression in Eff (P = 0.046, n = 9/11), compared between HC and ME. (N) TOX expression levels in pE1 (P = 0.072, n = 7/7), and of (O) TOX expression in pE2 (P = 0.057, n = 7/9), compared between HC and ME.
TOX and SLAMF6 are Elevated in Differentiated CD8+ T Cell Subsets.
To investigate Tex populations with more precision, we expanded our flow cytometric analysis to assay exhaustion phenotypes between different TM subsets, as analyzing T cells partitioned by differentiation status improves detection of exhausted phenotypes (59). We began by examining CD8+ T cell subpopulation frequencies, assessing naïve (TN), central and early effector memory (M), early intermediate (EM2), intermediate effector memory (EM4), late effector memory (EM3), effector memory reexpressing CD45RA (TEMRApE1, TEMRApE2) and terminally differentiated effector memory (Eff) populations (SI Appendix, Fig. S7). Subset frequencies were not significantly different compared between cohorts (SI Appendix, Fig. S8A). In each subset, we then analyzed the frequency and abundance of TOX, SLAMF6, and PD-1, as well as the presence of TOX and SLAMF6 within PD-1+ and PD-1+CD69– populations (Fig. 4 D−H and SI Appendix, Fig. S8, B–J).
Unexpectedly, in ME TN, we observed significantly higher abundances of TOX and frequencies of TOXhi populations compared to controls (SI Appendix, Fig. S9; A and B), with no significant differences seen in SLAMF6 measures ( SI Appendix, Fig. S9 C and D). As TOX is normally repressed in TN and TOX expression negatively correlates with naïve-related genes (48, 60), elevated TOX in ME TN may underlie the loss of naïve identity we observed by transcriptomics (SI Appendix, Fig. S3B). There are few studies investigating TOX in naïve T cells, although TOX+ TN have been identified in HBV and EBV, two chronic viral infections, and at higher frequencies compared to TN in acute viral infection paradigms such as influenza (61). Thus, elevated TOX in ME TN may result from chronic viral infection, a possibility that is consistent and supportive of T cell exhaustion in ME.
In ME effector memory populations, we detected significantly elevated frequencies of SLAMF6hi populations in EM4 and PD-1+CD69-EM3 T cells (Fig. 4 E, I and K and SI Appendix, Fig. S8F), with higher expression of SLAMF6 in PD-1+CD69- subsets of EM4 and EM3 (Fig. 4 F, J, and L; and SI Appendix, Fig. S8E) compared to controls. Moreover, SLAMF6 abundance was significantly higher in ME Eff T cell populations, (Fig. 4 F and M and SI Appendix, Figs. S8, C and E and S10 A and B). These changes in SLAMF6 expression were not observed in most early or late effector memory populations (M, pE1, and pE2 CD8+ T cells; SI Appendix, Fig. S10, C–H), suggesting that intermediate effector memory T cells are more likely to enter a progenitor exhausted state and drive SLAMF6 differences observed in total ME CD8+ T cells.
In TEMRA subsets (TEMRApE1 and TEMRApE2), which are terminally differentiated, we observed a trend toward higher TOX expression in cases (Fig. 4 H, N, and O). These results suggest a differentiation trajectory where intermediate effector memory T cells express precursor exhaustion markers such as SLAMF6, while the more differentiated TEMRA subsets displayed higher propensity for terminal exhaustion markers like TOX. Indeed, we did not observe any substantial difference in TOX expression or TOXhi frequency in M, EM4, or EM3 subsets (Fig. 4 G and H and SI Appendix, Figs. S8 G–J and S10 I–L). Considering these results together with our genomic data, these findings support a model where ME effector memory CD8+ T cells adopt a progenitor exhaustion program, characterized by upregulation of IRs such as SLAMF6. This dysregulation appears to be most pronounced in intermediate effector memory cells, which are poised to become terminally exhausted through increased chromatin accessibility of TOX. Meanwhile, we observed elevated TOX expression in more differentiated T cell subsets. Strikingly, this pattern mirrors findings in chronic viral infections, where immune activation drives cells toward terminal differentiation, ultimately resulting in exhaustion and depletion of these cells (62). Collectively, our results support a hypothesis that the poised regulatory state of ME TM drives transition into exhausted phenotypes.
Discussion
There is growing appreciation for the role of T cell dysfunction in ME, as evidenced by immunometabolism studies, functional assays, and cytokine profiling (17, 20, 22). However, the T cell compartment includes multiple different subsets, with specialized functions, and the identities of the cell types most relevant to ME pathobiology have not been systematically determined. Moreover, the regulatory mechanisms that contribute to T cell dysfunction in ME and how these dysregulated states could be reversed remain unknown. Here, we combined transcriptional, epigenetic, cell surface receptor, and marker gene analyses to comprehensively analyze CD8+ T cell subsets in ME. As a major study profiling the epigenetic landscape of purified ME T cell subsets, we provide a critical resource not only to understand immune dysfunction in ME, but to inform comparative studies on other infection-associated chronic conditions (IACC), including LC.
Our epigenomic profiling of ME CD8+ T cells revealed multiple lines of converging evidence indicative of T cell exhaustion in ME. Analysis of scRNA-seq and RNA-seq on purified subsets revealed upregulation of exhaustion-associated TFs in patient effector memory populations. Moreover, we show that the epigenome of these cells is rewired toward an exhaustion program (15). Integrating expression profiles and chromatin accessibility landscapes revealed a loss of regulatory poising in genes involved in cytokine signaling, proliferation, and cytotoxicity, indicating a hypofunctional state—exhaustion. Our flow cytometry and transcriptome analysis revealed elevated expression of SLAMF6, a key IR expressed by Tex. After symptom provocation, we observed elevated populations of exhausted T cells across various subsets, with TOX expressed at higher levels in these cells, implicating a progression into later Tex states (15). Of note, oxidative stress, observed in ME (56, 63, 64) can lead to reactive oxygen species accumulation, which exacerbates T cell exhaustion (57). Taken together, these results suggest that ME leaves epigenetic scars on TM, which are primed for progression into late exhaustion states. We propose that following induction by further stimuli such as an exercise challenge, these cells fail to upregulate effector programs, and instead become terminally exhausted, potentially contributing to inflammation and PEM (Fig. 5).
Fig. 5.

Model of proposed ME T cell mechanism of action via exhaustion.
While TM cells exhibited the most prominent dysfunction in ME, our analyses revealed additional T cell populations with notable regulatory and phenotypic distinctions between ME and healthy controls. Single-cell surveillance of intercellular communications revealed substantial dysregulation in γδT cells, which may contribute to an inflammatory milieu by producing tissue-homing signals such as CCL5 in ME. As major producers of cytokines and chemokines, γδT cell dysregulation may lead to improper crosstalk within the adaptive immune system, which has been recently characterized in LC (65) and implicated in our data, where some CD4+ T cell subsets have elevated expression of effector genes.
Notably, we observed disrupted gene regulatory states in CD8+ TN, which are directed toward a more activated transcriptional program. Thus, the dysfunctional state of immune cells in ME may derive from an environment conducive for ectopic activation, where excessive inflammatory signaling enhances the sensitivity of TN and thus prime them toward such a fate (66). Alternatively, when provoked by persistent stimulation and/or systemic inflammation, immune homeostasis could be disrupted, potentially leading to impairment of negative selection in the thymus (67). Thus, the transcriptional loss of naïve identity we observed in ME TN may be an indirect effect of the same chronic stimulatory environment that drives exhaustion in ME TM (68).
Our single-cell RNA-seq datasets include females and males at a 2:1 ratio, reflecting the increased incidence of ME in women (69). In order not to lose statistical power, we analyzed the RNA-seq data from both sexes together. All of our downstream analyses utilized female subjects only, given that sex differences in PBMC gene expression have been reported, including in a recent study that examined total PBMC RNA-seq before a maximal exercise test and 4 h later (70). Additionally, a prior gene expression study, using microarray technology, also reported sex differences in ME/CFS gene expression at baseline (71). Also, studies have demonstrated sex difference in the immune system across the general human population, noting variations in immune responses and peripheral immune cell characteristics between males and females, independent of specific diseases or contexts (72).
Although we gathered strong evidence for increased T cell exhaustion in ME, it remains unknown whether this state is a driver of pathology, or a consequence of prior triggering events. Thus, it is important to acknowledge that it is currently unclear whether the T cell dysregulation we observed derives from a chronic infection, such as a virus that triggered or is reactivated in ME, or alternatively, as a result of an inflammatory environment in lieu of antigen-specific stimulation. The induction of T cell exhaustion is a multifaceted process with various triggers. Chronic infections leave epigenetic scars on CD8+ T cells even after the initial infection has been eliminated (45, 61). Exhaustion may also be induced and maintained by stress responses or an inflammatory milieu (73–75). Recent findings have suggested links between the autonomous nervous system and exhaustion mediated by beta-adrenergic signaling (73), with important implications for understanding how mental or physical exertion may drive PEM, and how this can be prevented. Additional support for this notion comes from recent findings of disrupted catecholamine biosynthesis in the ME central nervous system (23), and similar findings of decreased serum catechols and their metabolites reported in LC (76). Additionally, monocytes, which have been shown to exhibit hallmarks of increased tissue homing in ME (10), can upregulate the coinhibitory PD-L1/2 and CD86-CTLA-4 pathways to suppress CD8+ T cell cytotoxicity (77). Future studies on T cell dysfunction in ME should also consider any crosstalk between the adaptive immune system and other compartments.
Much like ME, certain immunological signatures of LC point toward T cell exhaustion—SARS-CoV-2-reactive CD8+ T cells display features of exhaustion in severely ill patients (65, 78), and persistence of SARS-CoV-2 antigens has been observed in LC after the acute infection (79). LC patients also had higher levels of antibodies against SARS-CoV-2 spike proteins and latent viruses (80). Both viral persistence and latent virus reactivation could lead to exhaustion and functional impairment of T cells. Critically, however, while it is known that SARS-CoV-2 infection initiates LC, the infectious agent that ME patients report preceded their illness is usually unknown, a crucial distinction which means that not all interventions will be applicable to both conditions. Although our findings suggest the existence of a viral or infectious component that initiates or maintains ME, many open questions remain: What are the pathogens causing the initial priming event, and are patient T cell clonotypes altered in ME? Are members of a single pathogen family responsible for the onset of ME, or can multiple pathogens incite the illness? Is there a role of reactivation of herpesvirus infections or altered activity of endogenous retroviruses? What conditions lead to long-term scarring of CD8+ T cells and subsequently contribute to ME pathogenesis, and do different pathogens employ varying mechanisms, thereby influencing their propensity to induce T cell dysfunction? Future research in ME should continue to search for the specific viruses(es) involved in the disease, which will facilitate the analysis of virus-specific CD8+ T cells and improve the characterization of exhausted T cells. This includes testing for the presence of Tex within lymphoid tissues, where terminally exhausted T cells preferentially reside (81).
In conclusion, this study provides insight into a state of immune cell dysfunction in ME. Our high-resolution data encompass both the transcriptomes and epigenomes of diseased CD8+ T cells, serving as a critical resource for comparison studies to offer insights into the pathophysiological mechanisms underlying other IACCs. Crucially, our observations open numerous avenues for developing potential treatment options targeting T cell exhaustion and associated metabolic alterations, offering promising prospects for future research and, ultimately, therapeutic interventions in ME.
Materials and Methods
The experimental procedures and reagents utilized in this study are detailed in SI Appendix.
Study design.
Our goal was to identify T cell populations that may underlie ME pathology, characterize the epigenetic landscape of ME T cells, and establish whether T cell exhaustion is a feature of ME. To explore the heterogeneity of ME T cells, we first analyzed the scRNA-seq atlas of ME leukocytes, applying a combination of single-cell level differential gene expression, intercellular communication, and gene module analyses. We then comprehensively profiled purified TM and TN using RNA-seq and ATAC-seq. To validate our findings, we expanded our cohort for flow cytometric analysis, following symptom provocation. To remove confounding factors, cases and controls are matched for activity level, and participants who reported recent infectious events were excluded. Survey data were compared to ensure comparability between the genomics and flow cytometry cohorts. Importantly, all subjects included in this study were diagnosed prior to the 2020 SARS-CoV-2 pandemic.
The study populations for the scRNA-seq atlas and this project were collected as part of the NIH-funded Cornell Collaborative ME/CFS Center, as previously described (10). ME and healthy sedentary control participants were identified by experienced medical doctors, and all ME subjects fulfilled the IOM and Canadian Consensus Criteria for diagnosis (82). 70% of those ME cases reported sudden onset of illness. Onset of the illness occurred before the emergence of LC, and thus was not initiated by SARS-CoV-2. This cohort completed approved questionnaires to characterize their current health condition and medical history. Other information about the subjects can be found in SI Appendix, Table S1.
Study Approval.
All human subject protocols were approved by the Institutional Review Boards of Weill Cornell Medical College (Protocol 1708018518) and Ithaca College (IRB 1017-12Dx2). Written informed consent was received prior to sample collection.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Acknowledgments
We thank the Genomics Innovation Hub (RRID:SCR_022547), Transcriptional Regulation and Expression Facility (RRID:SCR_022532), and Flow Cytometry Facility (RRID:SCR_021740) in the Cornell Biotechnology Resource Center for providing key reagents, services, and for technology expertise. Visualizations were created using the iPad application Procreate, or Adobe Illustrator 2024 software v.28.0. We are grateful to the subjects who participated in this study and all those involved in diagnosis of ME subjects, performance of the exercise tests, and collection of samples. NIH grant U54NS105541, an initial grant provided to the Cornell ME/CFS Collaborative Research Center, was cofunded by the National Institute of Neurological Disorders and Stroke, National Institute of Allergy and Infectious Diseases (NIAID), National Institute on Drug Abuse, National Heart, Lung, and Blood Institute, National Human Genome Research Institute, and the Office of the Director. A renewal grant was received from NIH NIAID: U54AI178855. Funding was also provided by the Amar Foundation.
Author contributions
D.S.I., J.M., M.R.H., and A.G. designed research; D.S.I., J.M., L.T.V., E.A.F., and C.J.F. performed research; F.A. and P.R.M. contributed new reagents/analytic tools; D.S.I., J.M., L.T.V., A.J.M., J.K.G., M.R.H., and A.G. analyzed data; C.J.F. and M.R.H. data curation; and D.S.I., J.M., L.T.V., E.A.F., J.K.G., M.R.H., and A.G. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: L.N., Nova Southeastern University; and L.K.S., University of Massachusetts Medical School.
Contributor Information
Maureen R. Hanson, Email: maureen.hanson@cornell.edu.
Andrew Grimson, Email: agrimson@cornell.edu.
Data, Materials, and Software Availability
Sequence data have been deposited in GEO (GSE268212 (83) and GSE268223 (84)).
Supporting Information
References
- 1.Institute of Medicine, Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness (National Academies Press, 2015). [PubMed] [Google Scholar]
- 2.Valdez A. R., et al. , Estimating prevalence, demographics, and costs of ME/CFS using large scale medical claims data and machine learning. Front. Pediatr. 6, 412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Neal A. J., Hanson M. R., The enterovirus theory of disease etiology in myalgic encephalomyelitis/chronic fatigue syndrome: A critical review. Front. Med. 8, 688486 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanson M. R., The viral origin of myalgic encephalomyelitis/chronic fatigue syndrome. PLOS Pathog. 19, e1011523 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Visser J., et al. , CD4 T Lymphocytes from patients with chronic fatigue syndrome have decreased interferon-γ production and increased sensitivity to dexamethasone. J. Infect. Dis. 177, 451–454 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Caligiuri M., et al. , Phenotypic and functional deficiency of natural killer cells in patients with chronic fatigue syndrome. J. Immunol. 139, 3306–3313 (1987). [PubMed] [Google Scholar]
- 7.Brenu E. W., et al. , Role of adaptive and innate immune cells in chronic fatigue syndrome/myalgic encephalomyelitis. Int. Immunol. 26, 233–242 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Fletcher M. A., et al. , Biomarkers in chronic fatigue syndrome: evaluation of natural killer cell function and dipeptidyl peptidase IV/CD26. PLoS ONE 5, e10817 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strayer D., Scott V., Carter W., Low NK cell activity in Chronic Fatigue Syndrome (CFS) and relationship to symptom severity. J. Clin. Cell Immunol. 6, 4 (2015). [Google Scholar]
- 10.Vu L. T., et al. , Single-cell transcriptomics of the immune system in ME/CFS at baseline and following symptom provocation. Cell Rep. Med. 5, 101373 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corrado M., Pearce E. L., Targeting memory T cell metabolism to improve immunity. J. Clin. Invest. 132, e148546 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bettonville M., et al. , Long-term antigen exposure irreversibly modifies metabolic requirements for T cell function. eLife 7, e30938 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wherry E. J., et al. , Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007). [DOI] [PubMed] [Google Scholar]
- 14.McLane L. M., Abdel-Hakeem M. S., Wherry E. J., CD8 T cell exhaustion during chronic viral infection and cancer. Ann. Rev. Immunol. 37, 457–495 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Beltra J.-C., et al. , Developmental relationships of four exhausted CD8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 52, 825–841.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wherry E. J., Kurachi M., Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mandarano A. H., et al. , Myalgic encephalomyelitis/chronic fatigue syndrome patients exhibit altered T cell metabolism and cytokine associations. J. Clin. Invest. 130, 1491–1505 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maya J., Leddy S. M., Gottschalk C. G., Peterson D. L., Hanson M. R., Altered fatty acid oxidation in lymphocyte populations of myalgic encephalomyelitis/chronic fatigue syndrome. Int. J. Mol. Sci. 24, 2010 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hornig M., et al. , Distinct plasma immune signatures in ME/CFS are present early in the course of illness. Sci. Adv. 1, e1400121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montoya J. G., et al. , Cytokine signature associated with disease severity in chronic fatigue syndrome patients. Proc. Natl. Acad. Sci. U.S.A. 114, E7150–E7158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brenu E. W., et al. , Immunological abnormalities as potential biomarkers in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. J. Transl. Med. 9, 81 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gil A., et al. , Identification of CD8 T-cell dysfunction associated with symptoms in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and Long COVID and treatment with a nebulized antioxidant/anti-pathogen agent in a retrospective case series. Brain, Behavior, Immunity Health 36, 100720 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walitt B., et al. , Deep phenotyping of post-infectious myalgic encephalomyelitis/chronic fatigue syndrome. Nat. Commun. 15, 907 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Germain A., Levine S. M., Hanson M. R., In-depth analysis of the plasma proteome in ME/CFS exposes disrupted Ephrin-Eph and Immune System Signaling. Proteomes 9, 6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin S., et al. , Inference and analysis of cell-cell communication using cell chat. Nat Commun 12, 1088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardcastle S. L., et al. , Characterisation of cell functions and receptors in chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). BMC Immunol. 16, 35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huffman A. P., Lin J. H., Kim S. I., Byrne K. T., Vonderheide R. H., CCL5 mediates CD40-driven CD4+ T cell tumor infiltration and immunity JCI Insight 5, e137263 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cano-Gamez E., et al. , Single-cell transcriptomics identifies an effectorness gradient shaping the response of CD4+ T cells to cytokines. Nat. Commun. 11, 1801 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu J. E., et al. , In vitro modeling of CD8+ T cell exhaustion enables CRISPR screening to reveal a role for BHLHE40. Sci. Immunol. 8, eade3369 (2023). [DOI] [PubMed] [Google Scholar]
- 30.Dotiwala F., Lieberman J., Granulysin: Killer lymphocyte safeguard against microbes. Curr Opin. Immunol. 60, 19–29 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akimova T., Beier U. H., Wang L., Levine M. H., Hancock W. W., Helios expression is a marker of T cell activation and proliferation. PLoS One 6, e24226 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ingram J. T., Yi J. S., Zajac A. J., Exhausted CD8 T cells downregulate the IL-18 receptor and become unresponsive to inflammatory cytokines and bacterial co-infections. PLOS Pathog. 7, e1002273 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathewson N. D., et al. , Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 184, 1281–1298.e26 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masson F., et al. , Id2-mediated inhibition of E2A represses memory CD8+ T cell differentiation. J. Immunol. 190, 4585–4594 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luckey C. J., et al. , Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc. Natl. Acad. Sci. U.S.A. 103, 3304–3309 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McLane L. M., et al. , Role of nuclear localization in the regulation and function of T-bet and Eomes in exhausted CD8 T cells. Cell Rep 35, 109120 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yigit B., et al. , SLAMF6 as a regulator of exhausted CD8+ T cells in cancer. Cancer Immunol. Res. 7, 1485–1496 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Liu X., et al. , Blockades of effector T cell senescence and exhaustion synergistically enhance antitumor immunity and immunotherapy. J. Immunother Cancer 10, e005020 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toledo L. I., Murga M., Gutierrez-Martinez P., Soria R., Fernandez-Capetillo O., ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes. Dev. 22, 297–302 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bengsch B., et al. , Epigenomic-guided mass cytometry profiling reveals disease-specific features of exhausted CD8 T cells. Immunity 48, 1029–1045.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patsoukis N., et al. , PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 6, 6692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshie O., Matsushima K., CCR4 and its ligands: From bench to bedside. Int. Immunol. 27, 11–20 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Khan O., et al. , TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abdel-Hakeem M. S., et al. , Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat. Immunol. 22, 1008–1019 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yates K. B., et al. , Epigenetic scars of CD8+ T cell exhaustion persist after cure of chronic infection in humans. Nat. Immunol. 22, 1020–1029 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barnabei L., Laplantine E., Mbongo W., Rieux-Laucat F., Weil R., NF-κB: At the borders of autoimmunity and inflammation. Front. Immunol. 12, 716469 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerondakis S., Siebenlist U., Roles of the NF-κB Pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2, a000182 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sekine T., et al. , TOX is expressed by exhausted and polyfunctional human effector memory CD8+ T cells. Sci. Immunol. 5, eaba7918 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Henning A. N., Roychoudhuri R., Restifo N. P., Epigenetic control of CD8+ T cell differentiation. Nat. Rev. Immunol. 18, 340–356 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mognol G. P., et al. , Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc. Natl. Acad. Sci. U.S.A. 114, E2776–E2785 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ning S., Pagano J. S., Barber G. N., IRF7: Activation, regulation, modification and function. Genes. Immun. 12, 399–414 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang L., Pagano J. S., IRF-7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol. Cell Biol. 17, 5748–5757 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang J., Lyu T., Cao Y., Feng H., Role of TCF-1 in differentiation, exhaustion, and memory of CD8+ T cells: A review. FASEB J. 35, e21549 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Lee I. K., et al. , RORα regulates cholesterol metabolism of CD8+ T cells for anticancer immunity. Cancers (Basel) 12, 1733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miwa K., Fujita M., Fluctuation of serum vitamin E (α-tocopherol) concentrations during exacerbation and remission phases in patients with chronic fatigue syndrome. Heart Vessels 25, 319–323 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Jammes Y., Steinberg J. G., Delliaux S., Chronic fatigue syndrome: Acute infection and history of physical activity affect resting levels and response to exercise of plasma oxidant/antioxidant status and heat shock proteins. J. Internal Med. 272, 74–84 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Wu H., et al. , Mitochondrial dysfunction promotes the transition of precursor to terminally exhausted T cells through HIF-1α-mediated glycolytic reprogramming. Nat. Commun. 14, 6858 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vardhana S. A., et al. , Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 21, 1022–1033 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jubel J. M., Barbati Z. R., Burger C., Wirtz D. C., Schildberg F. A., The role of PD-1 in acute and chronic infection. Front. Immunol. 11, 487 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim K., et al. , Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 12, 22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heim K., et al. , TOX defines the degree of CD8+ T cell dysfunction in distinct phases of chronic HBV infection. Gut 70, 1550–1560 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guo L., Liu X., Su X., The role of TEMRA cell-mediated immune senescence in the development and treatment of HIV disease. Front. Immunol. 14, 1284293 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shungu D. C., et al. , Increased ventricular lactate in chronic fatigue syndrome. III. Relationships to cortical glutathione and clinical symptoms implicate oxidative stress in disorder pathophysiology. NMR in Biomedi. 25, 1073–1087 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shankar V., et al. , Oxidative Stress is a shared characteristic of ME/CFS and Long COVID. bioXriv [Preprint] (2024). Available at: https://www.biorxiv.org/content/10.1101/2024.05.04.592477v1 (Accessed 21 July 2024).
- 65.Yin K., et al. , Long COVID manifests with T cell dysregulation, inflammation and an uncoordinated adaptive immune response to SARS-CoV-2. Nat. Immunol. 25, 218–225 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Richer M. J., Nolz J. C., Harty J. T., Pathogen-specific inflammatory milieux tune the antigen sensitivity of CD8(+) T cells by enhancing T cell receptor signaling. Immunity 38, 140–152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elsaesser H. J., et al. , Chronic virus infection drives CD8 T cell-mediated thymic destruction and impaired negative selection. Proc. Natl. Acad. Sci. U.S.A. 117, 5420–5429 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barnstorf I., et al. , Chronic viral infections impinge on naive bystander CD8 T cells. Immunity, Inflammation and Dis. 8, 249–257 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vahratian A., Lin J.-M.S., Bertolli J., Unger E. R. “Myalgic Encephalomyelitis/Chronic Fatigue Syndrome in Adults: United States, 2021–2022” (National Center for Health Statistics (U.S.). National Health Interview Survey (NHIS), 2023). [Google Scholar]
- 70.Gamer J., et al. , Sex-dependent transcriptional changes in response to stress in patients with myalgic encephalomyelitis/chronic fatigue syndrome: A pilot project. Int. J. Mol. Sci. 24, 10255 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kerr J. R., et al. , Gene expression subtypes in patients with chronic fatigue syndrome/myalgic encephalomyelitis. J. Infect. Dis. 197, 1171–1184 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Jacobsen H., Klein S. L., Sex differences in immunity to viral infections. Front. Immunol. 12, 720952 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Globig A.-M., et al. , The β1-adrenergic receptor links sympathetic nerves to T cell exhaustion. Nature 662, 383–392 (2023). 10.1038/s41586-023-06568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scharping N. E., et al. , Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22, 205–215 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alatrakchi N., et al. , Hepatitis C virus (HCV)-specific CD8+ cells produce transforming growth factor β that can suppress HCV-specific T-cell responses. J. Virol. 81, 5882–5892 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thomas T., et al. , COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight. 5, e140327 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Padgett L. E., Araujo D. J., Hedrick C. C., Olingy C. E., Functional crosstalk between T cells and monocytes in cancer and atherosclerosis. J. Leukocyte Biol. 108, 297–308 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kusnadi A., et al. , Severely ill COVID-19 patients display impaired exhaustion features in SARS-CoV-2-reactive CD8+ T cells. Sci. Immunol. 6, eabe4782 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peluso M. J., et al. , Plasma-based antigen persistence in the post-acute phase of COVID-19. Lancet Infect. Dis. 24, e345–e347 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Klein J., et al. , Distinguishing features of long COVID identified through immune profiling. Nature 623, 139–148 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Im S. J., Konieczny B. T., Hudson W. H., Masopust D., Ahmed R., PD-1+ stemlike CD8 T cells are resident in lymphoid tissues during persistent LCMV infection. Proc. Natl. Acad. Sci. U.S.A. 117, 4292–4299 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carruthers B. M., et al. , Myalgic encephalomyelitis/chronic fatigue syndrome: Clinical working case definition, diagnostic and treatment protocols. J. Chronic Fatigue Syndrome 11, 7–115 (2003). [Google Scholar]
- 83.Iu D. S., et al. , RNA-seq data from “Transcriptional reprogramming primes CD8+ T cells towards exhaustion in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome.” Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE268212. Deposited 23 May 2024.
- 84.Iu D. S. et al., ATAC-seq data from “Transcriptional reprogramming primes CD8+ T cells towards exhaustion in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome.” Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE268223. Deposited 23 May 2024.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Data Availability Statement
Sequence data have been deposited in GEO (GSE268212 (83) and GSE268223 (84)).